

Abstract
The RAS signaling pathway regulates cell growth, survival and differentiation, and its inappropriate activation is associated with disease in humans. The RASopathies, a set of developmental syndromes, arise when the pathway is overactive during development. Patients share a core set of symptoms, including congenital heart disease, craniofacial anomalies, and neurocognitive delay. Due to the conserved nature of the pathway, animal models are highly informative for understanding disease etiology, and zebrafish and Xenopus are emerging as advantageous model systems. Here we discuss these aquatic models of RASopathies, which recapitulate many of the core symptoms observed in patients. Craniofacial structures become dysmorphic upon expression of disease-associated mutations, resulting in wider heads. Heart defects manifest as delays in cardiac development and changes in heart size, and behavioral deficits are beginning to be explored. Furthermore, early convergence and extension defects cause elongation of developing embryos: this phenotype can be quantitatively assayed as a readout of mutation strength, raising interesting questions regarding the relationship between pathway activation and disease. Additionally, the observation that RAS signaling may be simultaneously hyperactive and attenuated suggests that downregulation of signaling may also contribute to etiology. We propose that models should be characterized using a standardized approach to allow easier comparison between models, and a better understanding of the interplay between mutation and disease presentation.
Keywords: craniofacial anomalies, heart defects, RASopathies, signaling, Xenopus, zebrafish
Introduction
The development of an organism from a single cell into a well-formed individual requires precise coordination. This coordination relies upon the integration of information from many sources. Each cell may receive multiple signals from surrounding tissues, and these inputs are transduced to control fundamental processes. Disruptions to either the signal or the transduction pathway can drastically alter the output of the cell, causing irregularities such as premature differentiation or unrestrained growth. These disruptions to signaling pathways frequently manifest as developmental disease. Determining how altered signaling dynamics leads to disease is critical for developing new therapeutic strategies, and can be informative for understanding normal developmental processes.
A range of model organisms are informative in such studies. Recently, technological advances have made aquatic models increasingly attractive. The external development and high fecundity of Danio rerio (zebrafish) and Xenopus laevis (African clawed frog), coupled with their new genetic tractability, are leading to progressively more aquatic models of human development disease. In this review, we focus on the insights gained from using aquatic models to study RASopathy syndromes.
RAS signaling and RASopathies
A prime example of aberrant signaling leading to developmental disease comes from the RAS signaling pathway. RAS signaling is activated by extracellular ligands, such as Fibroblast growth factor (FGF), which bind to a receptor tyrosine kinases (RTK; Figure 1). Ligand engagement results in autophosphorylation of the RTK. The activated RTK then recruits Tyrosine-protein phosphatase non-receptor type 11 (SHP2) and Growth factor receptor-bound protein 2 (GRB2) to the intracellular portion of the receptor. GRB2 in turn recruits a guanine exchange factor (GEF), such as Son of sevenless (SOS), RasGEF Domain Family Member 1A (RASGEF1A), or one of the Ras Protein Specific Guanine Nucleotide Releasing Factor (RASGRF) proteins. This membrane localization of the GEF facilitates its activation of the RAS GTPases (HRAS, NRAS or KRAS). RAS activation initiates a phosphorylation cascade involving mitogen-activated protein kinase kinase kinases (BRAF and RAF1), mitogen-activated protein kinase kinases (MEK1 and MEK2) and mitogen-activated protein kinases (ERK1 and ERK2). Activated ERK phosphorylates substrates in the cytoplasm and nucleus to regulate proliferation, differentiation and apoptosis (Simanshu et al., 2017).
Figure 1: Mutations in the RAS signaling pathway are associated with RASopathies.
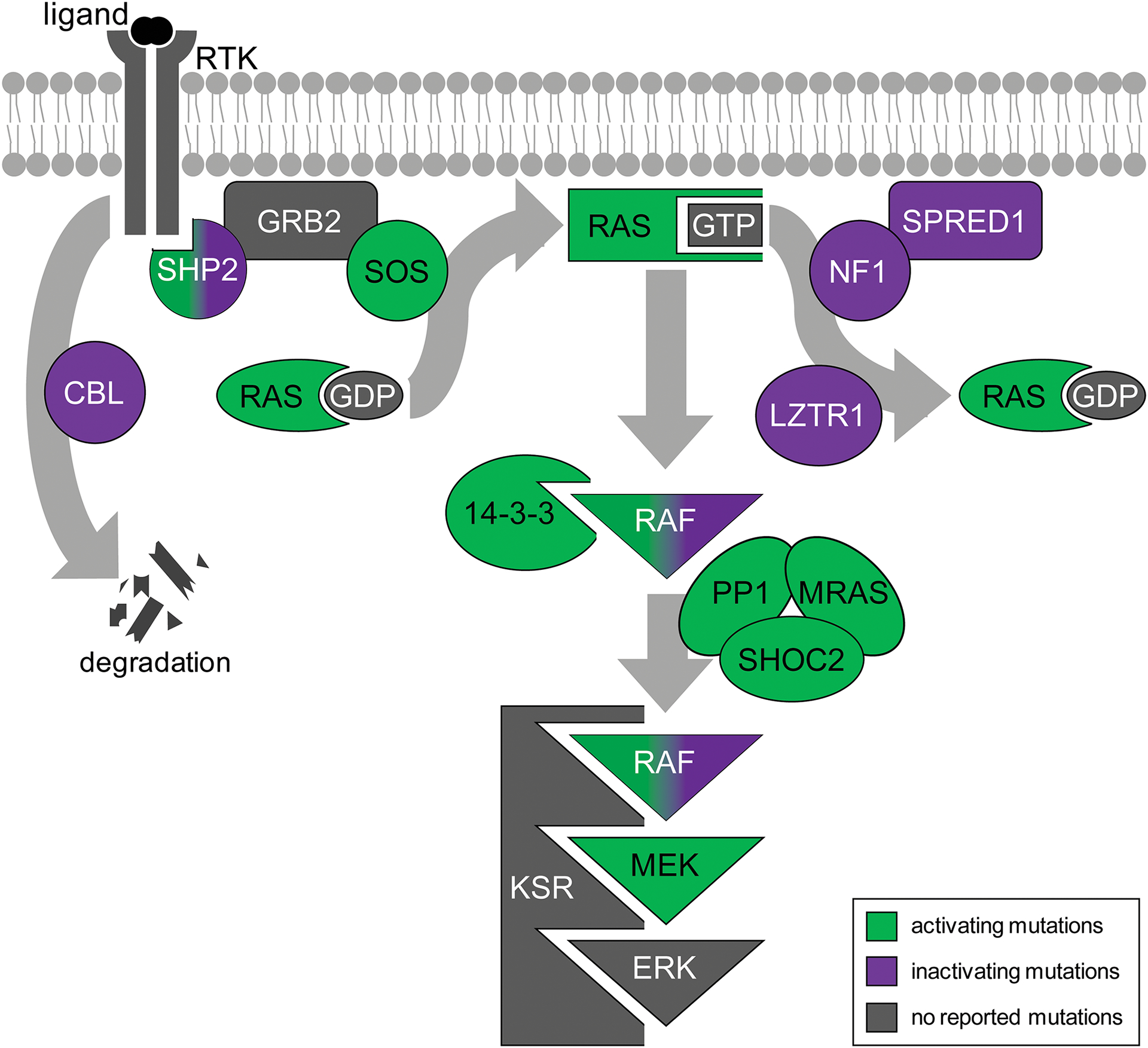
Signaling is triggered by binding of extracellular ligand to a receptor tyrosine kinase (RTK). Activation of the RTK recruits Tyrosine-protein phosphatase non-receptor type 11 (SHP2) and Growth factor receptor-bound protein 2 (GRB2), which subsequently recruits RAS guanine exchange factors such as Son of sevenless homolog 1 (SOS) to the membrane. SOS promotes the exchange of GDP for GTP, activating RAS. RAS-GTP initiates the phosphorylation cascade involving Mitogen-activated protein kinase kinase kinases (RAF), mitogen-activated protein kinase kinases (MEK) and mitogen-activated protein kinases (ERK). Meanwhile, feedback loops modulate signaling. The E3 ubiquitin protein ligase CBL mediates degradation of the RTK. Sprouty-related EVH1 domain-containing protein 1 (SPRED1) recruits the GTPase activating protein Neurofibromin 1 (NF1) to the membrane, returning RAS to a GDP-bound inactive state. Leucine-zipper-like transcriptional regulator 1 (LZTR1) ubiquitinates RAS to prevent its membrane localization. A complex of Protein phosphatase 1 (PP1), Leucine-rich repeat protein SHOC2, and MRAS GTPase dephosphorylates RAF, allowing 14-3-3 protein to switch from stabilizing the inactive form of RAF to facilitating active RAF dimer formation. Both activating mutations (green) in pathway components and inactivating mutations (purple) in negative regulators cause activation of the signaling pathway. Components that do not cause RASopathies are not shown unless no alternative protein is associated with disease (gray).
Additional layers of both positive and negative control are superimposed on this core pathway through the use of modulating proteins. For example, the E3 ubiquitin protein ligase CBL (CBL) targets the RTK for proteolytic degradation (Levkowitz et al., 1998). Sprouty-related EVH1 domain-containing protein 1 (SPRED1) is thought to recruit Neurofibromin 1 (NF1), itself a GTPase activating protein and negative regulator of RAS signaling, to the membrane (Martin et al., 1990; Stowe et al., 2012; Xu et al., 1990). Several other GTPase activating proteins, including the RAS p21 Protein Activator (RASA) proteins and the RAS protein activator like (RASAL) proteins, also inactivate RAS signaling (Vigil et al., 2010). RAS is also ubiquitinated by Leucine-zipper-like transcriptional regulator 1 (LZTR1), which prevents its association with the membrane (Steklov et al., 2018).
Other components mediate protein interactions that facilitate or attenuate signaling. For example, the scaffolding protein Kinase Suppressor of Ras (KSR) facilitates signaling by bringing RAF, MEK and ERK together (Denouel-Galy et al., 1998; Yu et al., 1998). By contrast, 14-3-3 proteins have a dual role, depending on the phosphorylation state of RAF. When RAF bears an inhibitory phosphate, 14-3-3 protein binds to sites in both the N- and C-terminus of RAF and promotes the closed, inactive conformation. In the absence of this inhibitory phosphate, 14-3-3 protein binds to the C-terminal sites in separate monomers, favoring the dimeric, active form of RAF (Lavoie and Therrien, 2015; Tzivion et al., 1998). This inhibitory group is removed by Protein Phosphatase 1 (PP1) in complex with the MRAS GTPase and Leucine-rich repeat protein SHOC2 (Rodriguez-Viciana et al., 2006a).
Disrupting the function of either the core transduction molecules or the modulating proteins can affect pathway output, resulting in either increased or decreased RAS signaling. Decreases in levels of RAS signaling are not common in disease, presumably since it causes embryonic lethality as a result of placental defects and aberrant mesoderm specification (Deng et al., 1994; Yamaguchi et al., 1994). Conversely, hyperactive RAS signaling in somatic cells is accepted as a major contributor to cancer; indeed, mutations in the RAS proteins were the first to be associated with cancer and continue to be among the most prevalent mutations. However, inheriting similar activating mutations from a parent causes developmental disease. The resultant developmental syndromes are collectively termed the RASopathies (Tidyman and Rauen, 2009). RASopathies can arise from any mutation that has the net effect of activating RAS signaling. These include gain of function mutations in any of components that facilitate or transduce signaling, as well as loss of function mutations in negative regulators of the pathway (Grant et al., 2018). However, not all signaling components have been associated with RASopathies in patients. Figure 1 outlines the components of the pathway currently known to be associated with disease. While the underlying genetic causes have been determined for many patients, ongoing efforts continue to identify novel causative genes and mutations.
Although the incidence of individual syndromes varies, RASopathies collectively occur in 1 in 1000 births (Rauen, 2013). Each syndrome is caused by mutations in specific subsets of RAS pathway components (Table 1). Patients present a core set of symptoms that include congenital heart defects (CHD) and craniofacial anomalies (CFA). Cardiofaciocutaneous (CFC) syndrome patients present with thickened skin and macrocephaly (Roberts et al., 2006). Macrocephaly and skin defects in the form of papilloma are features of Costello syndrome (CS) (Abe et al., 2012; Rauen, 2007). Noonan syndrome (NS) patients suffer growth retardation and neurocognitive delay, while patients with one of two subsets of NS exhibit these symptoms in addition to more unique pathologies. Noonan Syndrome with Multiple Lentigines (NSML) patients have additional sensorineural deafness and lentigines (Roberts et al., 2013; Sarkozy et al., 2008), while Noonan Syndrome with Loose Anagen Hair (NSLAH) patients have coarse, easily pluckable hair (Mazzanti et al., 2003). Legius syndrome (LS) patients present café-au-lait spots and axillary freckling, which are also symptoms of Neurofibromatosis type 1 (NF1) (Brems et al., 2007; NIH, 1988). In addition, NF1 patients suffer skeletal malformations and neurofibromas.
Table 1:
RAS component mutations and associated phenotypes.
| Syndrome and incidence | Genes | Patient Symptoms | Phenotypes in RASopathy models | References |
|---|---|---|---|---|
| Noonan Syndrome (NS): 1:1500 | CBL, PTPN11, SOS, KRAS, NRAS, MRAS, RRAS, RRAS2, SHOC2, RAF1, BRAF, LZTR1 | CHD, CFA, short stature, neurocognitive delay | oval embryo, CHD (delayed morphogenesis, changes to heart size, randomized laterality, reduced cell migration, slower heart rate), short body, CFA: (wider head), KV (fewer cilia, shorter cilia, smaller lumen, reduced flow) | (Bonetti et al., 2014a; Capri et al., 2019; Grant et al., 2018; Jopling et al., 2007; Langdon et al., 2012; Niihori et al., 2019; Rauen, 2013; Roberts et al., 2013; Runtuwene et al., 2011) |
| Noonan Syndrome with Multiple Lentigines (NSML)* | PTPN11, RAF1, BRAF, MAP2K1 | CHD, CFA, short stature, neurocognitive delay, hyperpigmentation, sensorineural deafness | oval embryo, CHD (edema, randomized laterality, reduced CPC migration, slower heart rate), shorter body, CFA: (wider head), KV (fewer cilia, shorter cilia, smaller lumen, reduced flow), more sympathetic neurons, increased pigmentation | (Bonetti et al., 2014a; Jopling et al., 2007; Sarkozy et al., 2008; Stewart et al., 2010b) |
| Noonan Syndrome with Loose Anagen Hair (NS-LAH)* | SHOC2, PPP1CB | CHD, CFA, short stature, neurocognitive delay, hyperpigmentation, macrocephaly, loose anagen hair | oval embryo | (Gripp et al., 2016; Gripp et al., 2013; Kota et al., 2019; Mazzanti et al., 2003; Motta et al., 2019) |
| Neurofibromatosis type 1 (NF1): 1:3000 | NF1 | Neurofibromas, gliomas, Lisch nodules, skeletal malformation, neurocognitive delay, hyperpigmentation | CHD (pericardial edema, thinning of ventricular myocardium, vasculature defects), learning and memory deficits, disrupted myelination, disrupted pigmentation | (Lee et al., 2010; NIH, 1988; Padmanabhan et al., 2009; Shin et al., 2012; Williams et al., 2009; Wolman et al., 2014) |
| Legius Syndrome (LS): 1:150,000** | SPRED1 | Hyperpigmentation, macrocephaly | No aquatic models available | (Brems et al., 2007; Messiaen et al., 2009; Muram-Zborovski et al., 2010) |
| Cardiofacio-cutaneous Syndrome (CFC): 1:810,000 | KRAS, BRAF, MAP2K1, MAP2K2, YWHAZ | CHD, CFA, short stature, ectodermal defects, neurocognitive delay, macrocephaly | oval embryo, CHD (larger heart), shorter body, KV (fewer cilia, smaller lumen), increased pigmentation | (Abe et al., 2012; Anastasaki et al., 2009; Anastasaki et al., 2012; Goyal et al., 2017; Jindal et al., 2017b; Popov et al., 2019; Roberts et al., 2006) |
| Costello Syndrome (CS): 1:1,290,000 | HRAS | CHD, CFA, short stature, neurocognitive delay, ectodermal defects, macrocephaly, hypotonia | CHD (delayed morphogenesis, small heart, thicker myocardium, edema), CFA (wider head), shorter body, scoliosis, sterility, increased cancer risk | (Abe et al., 2012; Rauen, 2007; Santoriello et al., 2009) |
Purple: loss-of-function mutations, green: gain-of-function mutations, red: both.
incidence data are not available,
estimated from misdiagnosis of NF1, CHD: congenital heart defects, CFA: craniofacial anomalies, KV: Kupffer’s vesicle.
The highly conserved nature of the RAS signaling pathway facilitates the study of pathway dynamics across model organisms. Animal models have proven informative for elucidating mechanisms underlying symptoms (Jindal et al., 2015). Mice that have been engineered to express RASopathy-associated mutations in genes encoding RAS pathway components display phenotypes that are reminiscent of human symptoms: for example, expression of NS alleles display reduced body length, craniofacial abnormalities and cardiac defects (Araki et al., 2004). These mouse models have recently been reviewed in detail (Hernandez-Porras and Guerra, 2017). Such tools allow the comparison of phenotype and genotype, but are limited by the relatively low-throughput techniques involved in generating modified mice. Conversely, overexpression of proteins encoding RASopathy-associated mutations in Drosophila allows precise, quantitative analysis of signaling pathway activity in a higher-throughput manner. However, the resultant phenotypes are less informative for the human condition: activation of RAS signaling during development causes ectopic wing veins, a rough eye phenotype, and lethality (Oishi et al., 2009).
Aquatic organisms offer a compromise between ease of manipulation and relevance for disease progression. Both zebrafish, Danio rerio, and Xenopus laevis have been utilized to study RASopathy-associated mutations in vivo. Expression of variant proteins in zebrafish and Xenopus is quick and easy compared to mouse, allowing multiple alleles to be screened in a high-throughput way. At the same time, the phenotypes observed in these vertebrate models are more reminiscent of the human condition than those of Drosophila. Additionally, the external development of transparent embryos make zebrafish an ideal system for studying how phenotypes emerge over time and testing novel therapeutic strategies, while their fecundity allows statistical confidence in results.
Expressing RASopathy-associated mutations in zebrafish and Xenopus
Generating Xenopus and zebrafish models of RASopathies has, to date, relied on two main approaches: overexpressing activating mutations in proteins that facilitate signaling, and depleting proteins that act as negative regulators of signaling (Table 2). Overexpression of activating proteins is achieved either by injection of synthesized RNA or the integration of transgenes encoding RASopathy-associated alleles (Anastasaki et al., 2009; Anastasaki et al., 2012; Bonetti et al., 2014a; Goyal et al., 2017; Jindal et al., 2017b; Jopling et al., 2007; Kota et al., 2019; Langdon et al., 2012; Miura et al., 2013; Niihori et al., 2019; Popov et al., 2019; Razzaque et al., 2012; Runtuwene et al., 2011; Santoriello et al., 2009; Stewart et al., 2010b). Negative regulators are depleted by generating null alleles or using morpholino-mediated knockdown of proteins (Lee et al., 2010; Padmanabhan et al., 2009; Shin et al., 2012; Wolman et al., 2014).
Table 2:
Zebrafish and Xenopus models of RASopathies.
| Organism | Method | RASopathy mutations tested | Reported phenotypes | References | |
|---|---|---|---|---|---|
| NS | zebrafish | RNA | SHP2: D61G, T73I | oval embryos, C&E defects, edema, jogging randomization, wider head, shorter body | (Jopling et al., 2007) |
| zebrafish | RNA | SHP2: D61G, T73I | oval embryos, jogging randomization, slower cardiac migration, slower heart rate, left-right patterning defects, fewer, shorter cilia in Kupffer’s vesicle | (Bonetti et al., 2014a) | |
| Xenopus | RNA | SHP2: D61G, Q79R, N308D | delayed heart morphogenesis, smaller heart | (Langdon et al., 2012) | |
| zebrafish | RNA | KRAS: N116S | larger heart, reduced ventricular thickness | (Razzaque et al., 2012) | |
| zebrafish | RNA | NRAS: I24N | oval embryos, C&E defects, wider head | (Runtuwene et al., 2011) | |
| zebrafish | RNA | RRAS2: G24_G26dup, Q72H*, F75C, Q72L | oval embryos, pericardial edema, wider head, shorter body | (Niihori et al., 2019) | |
| zebrafish | RNA | SHOC2: S2G | oval embryos | (Motta et al., 2019) | |
| NSML | zebrafish | RNA | SHP2: Y280C, A462T, G465A, T469M | elongated embryos, C&E defects, edema, jogging randomization, wider head, shorter body, increased pigmentation, increased sympathetic neurons | (Jopling et al., 2007; Stewart et al., 2010a) |
| zebrafish | RNA | SHP2: A462T, G465A | oval embryos, jogging randomization, slower cardiac migration, slower heart rate, left-right patterning defects, fewer, shorter cilia in Kupffer’s vesicle | (Bonetti et al., 2014a) | |
| NF1 | zebrafish | morpholino | NF1: knockdown | edema, thinner myocardium, heart valve function impaired, vasculature defects, increased oligodendrocyte and Schwann cell lineages | (Lee et al., 2010; Padmanabhan et al., 2009) |
| zebrafish | null allele | NF1: nf1a−/−;nf1b−/− | decreased pigmentation, increased oligodendrocyte and Schwann cell lineages, motor and learning deficits | (Shin et al., 2012; Wolman et al., 2014) | |
| CFC | zebrafish | RNA | BRAF: A246P, Q257R, G464V, S467A, K499E, G534R, N581D, G596V*, V600E, D638E. MEK1: F53S, T55P, G128V, Y130C. MEK2: F57C, A62P, G132V, Y134C, K273R | oval embryos, C&E defects, shorter body, lack of tail formation | (Anastasaki et al., 2009; Anastasaki et al., 2012) |
| zebrafish | RNA | MEK1: L42F, E44G, F53S, T55P, D67N, P124L, P124Q, G128N, G128V, Y130C, Y130H, Y130N, Q164K, E203K*, E203Q | oval embryos, larger heart | (Jindal et al., 2017b) | |
| zebrafish | RNA | MEK1: G128V | fewer cilia in Kupffer’s vesicle | (Goyal et al., 2017) | |
| Xenopus | RNA | YWHAZ: S230W | shorter body, defective head structures, clustered pigment cells | (Popov et al., 2019) | |
| CS | zebrafish | transgenic integration | HRAS: G12V | delayed heart morphogenesis, small heart, thicker myocardium, edema, wider head, shorter body, scoliosis, sterility, increased cancer risk | (Santoriello et al., 2009) |
mutation also associated with cancer.
These techniques lead to phenotypes that phenocopy the core symptoms of the human disease: embryos are shorter than siblings, exhibit defects in cardiac development and develop CFA. In several cases, quantitative assays are used to assess the severity of these phenotypes (Fig. 2). Early convergence and extension defects are assayed by measuring the angle between the most anterior and posterior extent of the embryonic tissue, or by measuring the aspect ratio of the embryo. Craniofacial anomalies are quantified by measuring the angle between the ceratohyal cartilage or the distance between the eyes. And although behavioral tests for zebrafish are limited, learning and memory can be assayed by testing habituation to acoustic or visual cues.
Figure 2: Assaying RASopathy mutations in zebrafish embryos.
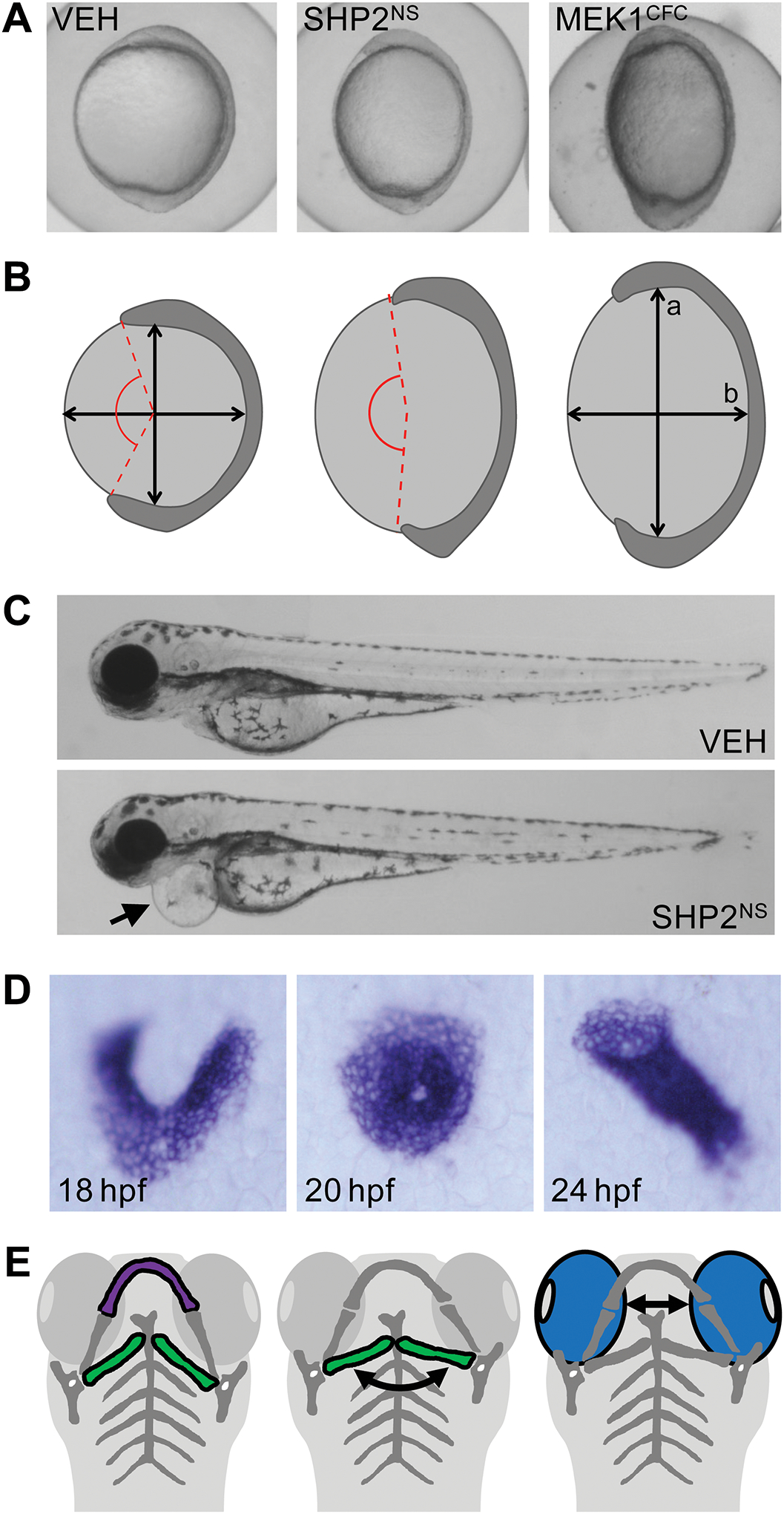
(A) Embryos expressing SHP2NS and MEK1CFC exhibit lengthening of the major axis, forming an elongated embryo compared to vehicle injected controls (VEH). (B) Embryo elongation is quantified by measuring the ratio of the major axis (a) to the minor axis (b). Alternatively, the angle between the most rostral and caudal limits of the embryonic tissue (red lines) can be used to quantify extension of the anterior-posterior axis. (C) Embryos expressing SHPNS are shorter than vehicle injected controls at 3 dpf (arrowhead) and display pericardial edema (arrow). (D) In situ hybridization for the myocardial marker myl7 illustrates steps during heart morphogenesis. Bilateral populations migrate medially to fuse into a cone in the midline. The cone undergoes rotation and involution to form a linear heart tube. (E) Craniofacial structures including Meckel’s cartilage (purple) and the ceratohyal cartilage (green) are hypoplastic in RASopathy models. Widening of the head is measured as an increase in the ceratohyal angle (curved arrow) or the distance between the eyes (blue).
For the purposes of this review, groups of mutations will be referred to in the format PROTEINDISEASE, while individual mutations will be represented as PROTEINMUTATION. For example, SHP2NS refers collectively to all variant forms of SHP2 that cause Noonan syndrome, while SHP2D61G refers specifically to the D61G variant of SHP2.
Early patterning events
In the zebrafish zygote, the nucleus and cytoplasm are situated at the animal pole, with large amounts of yolk at the vegetal pole. Rounds of cell cleavages increase cell number, but the blastomeres remain localized at the animal pole, where they form a mound of cells on top of the yolk. These cells radially intercalate and spread vegetally during epiboly (Roszko et al., 2009). Although many factors are involved in driving epiboly, RAS-mediated FGF signaling at the blastoderm margin is required for its successful execution (Krens et al., 2008). The ectodermal cells proceed vegetally to enclose the yolk, but the mesendoderm population involutes during gastrulation. After involution, convergence and extension (C&E) movements lengthen the anterior-posterior axis. Cells move toward the dorsal midline of the developing embryo, resulting in the mediolateral narrowing of the tissue and its extension along the AP axis. These movements are regulated by the ventral to dorsal gradient of bone morphogenetic protein (BMP) signaling (Myers et al., 2002; von der Hardt et al., 2007). The BMP gradient is itself regulated by FGF signaling in the dorsal embryo, since FGF signaling represses BMP dorsally (Furthauer et al., 1997; Furthauer et al., 2004).
Considering its roles in gastrulation, it is unsurprising that activating RAS signaling by introducing RASopathy-associated mutations perturbs the early events that pattern the embryo. MEK1CFC-, BRAFCFC-, SHP2NS-, NRASNS-, SHPNSML, SHP2NSLAH, and RRASNS-expressing embryos are all elongated by 10–12 hours post fertilization (hpf) (Anastasaki et al., 2009; Bonetti et al., 2014a; Jindal et al., 2017b; Jopling et al., 2007; Kota et al., 2019; Niihori et al., 2019; Runtuwene et al., 2011; Stewart et al., 2010b). This altered morphology is a consequence of impaired convergence and extension movements, which results in reduced extension of the anterior-posterior axis (Jopling et al., 2007; Runtuwene et al., 2011). Similar effects can be seen upon introducing activated SHP2 variants into Xenopus, which cause elongation of the animal cap (O’Reilly et al., 2000). Although the SHP2 mutations introduced in Xenopus are not specifically associated with RASopathies, they exert similar effects in causing activation of signaling. These data point toward conserved mechanisms in Xenopus.
Embryonic elongation induced in zebrafish by activating RAS signaling can be rescued using drugs to inhibit either MEK or the FGF receptor (Anastasaki et al., 2009; Anastasaki et al., 2012; Bonetti et al., 2014a; Jindal et al., 2017b; Kota et al., 2019; Runtuwene et al., 2011). Indeed, treatment from 4.5 to 5.5 hpf was shown to be necessary and sufficient to prevent embryo elongation, demonstrating the underlying cause of elongation to be activated RAS signaling during epiboly (Anastasaki et al., 2009). The fact that inhibition of the FGF receptor prevents embryo elongation indicates that there is still a requirement for endogenous signal to initiate the RAS signaling cascade, presumably from FGF at the blastoderm margin. This is further supported by the observation that MEKCFC variants that cannot be activated by phosphorylation do not cause embryonic elongation in zebrafish (Jindal et al., 2017a).
The anterior-posterior axis is also shorter at later points in development. Expression of a CFC-associated variant of YWHAZ, a 14-3-3 protein that regulates RAF activity, shortens body length in Xenopus (Popov et al., 2019). Similarly, expression of KRASCS, BRAFCFC, SHP2NS or SHP2NSML in zebrafish reduces larval body length by 4 days post fertilization (dpf) (Fig. 2C) (Anastasaki et al., 2012; Jopling et al., 2007; Santoriello et al., 2009). Continuous exposure to low doses of MEK inhibitor restored the zebrafish body axis to normal proportions (Anastasaki et al., 2012). However, a minimal time window using higher drug doses could not be established without observing drug-induced axis defects (Anastasaki et al., 2009; Grzmil et al., 2007). This reduction in body length is similar to the short stature characteristic of CS, CFC, NS and NSML patients. Together, these data suggest that inhibiting the RAS pathway may be an attractive therapeutic approach to treating patients.
Cardiac abnormalities
Cardiac development is a highly dynamic process involving multiple precisely regulated steps that effect successful morphogenesis of the heart. Disruption of any one of these events can result in CHD, the most common structural birth defect in the U.S. (Mozaffarian et al., 2016). The zebrafish heart is simpler than the human heart, consisting of a single atrium and ventricle. The Xenopus heart is more complex, consisting of two atria and one ventricle. While the human heart has four chambers, the morphogenetic processes and signaling cues that direct cardiac development are well conserved across vertebrates (Grant et al., 2017; Warkman and Krieg, 2007). In zebrafish, cardiac precursor cells originate in bilateral sheets that migrate towards the embryonic midline (Stainier et al., 1993). These populations fuse into a shallow cone, which undergoes clockwise rotation in response to asymmetric Nodal signaling from the left lateral plate mesoderm (Baker et al., 2008; de Campos-Baptista et al., 2008; Lenhart et al., 2013; Smith et al., 2008; Yelon et al., 1999). The concomitant involution of the most right-posteriorly situated cells allows formation of a linear heart tube, which bends to situate the atrium to the left and anterior of the ventricle (Fig. 2D) (Rohr et al., 2008). In Xenopus, the process is very similar; bilateral cardiac populations fuse in the ventral midline, forming a trough of myocardium that fuses into a linear heart tube. The linear tube then forms into an anticlockwise spiral that positions the atrium dorsal to the ventricle, subsequently forming chambers and asymmetrically dividing the atrium (Mohun et al., 2000).
RAS-mediated FGF signaling regulates both precursor cell specification and accretion of secondary cardiac cell populations to the heart (Cavanaugh et al., 2015; Marques et al., 2008). The importance of this pathway in heart development is underscored by the prevalence of cardiac defects in RASopathy models. Every syndrome modeled presents some form of heart defect, manifesting as reduced heart rate, reduced blood oxygenation or presence of pericardial edema (Fig. 2C) (Anastasaki et al., 2012; Bonetti et al., 2014a; Jopling et al., 2007; Niihori et al., 2019; Padmanabhan et al., 2009; Santoriello et al., 2009). This is to be expected, as any perturbation in the earlier events of cardiac morphogenesis will impact cardiac function.
Cardiac hypertrophy is a common phenotype in RASopathy patients. In fact, cardiac cells induced from RASopathy patient iPSC cell lines are larger in size than those derived from unaffected controls (Carvajal-Vergara et al., 2010; Cashman et al., 2016; Josowitz et al., 2011). Consistent with the prevalence of this symptom in patients, the size of the heart is altered in aquatic models of CS, CFC and NS. Expression of MEK1CFC increases heart size (Jindal et al., 2017b). Conversely, HRASCS leads to a reduction in heart size with an associated reduction in proliferation (Santoriello et al., 2009). However, this difference may be accounted for by the observation that there is no detected increase in pathway output in the HRASCS model (Santoriello et al., 2009). The effect of NS-associated mutations on heart size is less clear; no change is reported for zebrafish, while a reduction in heart size is observed in Xenopus when expressing the same mutation (SHP2D61G) (Bonetti et al., 2014a; Langdon et al., 2012). KRASNS zebrafish embryos actually developed a larger heart (Razzaque et al., 2012). Together, these data suggest that the same developmental event may be differentially impacted depending on the specific allele expressed, or the organism studied. Additionally, the timing of the assay may be important; for example, heart size was assayed at 6 weeks for HRASCS, but 19.5 hpf for MEK1CFC model. Examining the heart at a standard time point, or several time points, would provide more directly comparable results.
Aberrant cardiomyocyte migration may also contribute to cardiac defects. Cardiac morphogenesis is delayed in aquatic models of NS and CS. HRASCS expression in zebrafish results in hearts remaining at the earlier cone stage when they should have formed linear heart tubes (Santoriello et al., 2009). Similarly, expression of SHP2NS causes delayed cardiac development in Xenopus, with cardiac troughs persisting (Langdon et al., 2012). In zebrafish models of NS, cardiac progenitor cells migrate more slowly than in wildtype siblings, suggesting a possible mechanism for this delayed development (Bonetti et al., 2014a).
Beyond delayed development, expression of SHP2NS or SHP2NSML also causes randomization of jogging laterality, such that the heart tube may be displaced to the right or remain at the embryonic midline instead of being positioned correctly to the left (Bonetti et al., 2014b; Jopling et al., 2007). This randomization is a consequence of disrupted left-right patterning, resulting from abnormalities in the zebrafish left-right organizer, Kupffer’s vesicle (KV). KV is lined with motile cilia, which generate an asymmetric fluid flow that specifies the left side of the embryo (Grimes and Burdine, 2017). Promigratory cues from the left lateral plate mesoderm subsequently instruct the heart tube to jog towards the left as a result of clockwise rotation of the cardiac cone (Baker et al., 2008; de Campos-Baptista et al., 2008; Lenhart et al., 2013; Rohr et al., 2008; Smith et al., 2008). SHP2NS and SHP2NSML cause a reduction in cilia length and number, which translates into loss of asymmetric patterning (Bonetti et al., 2014b). The randomization of heart laterality in this case is therefore a readout of this earlier defect, rather than a bone fide cardiac defect.
Interestingly, cilia number is also reduced upon expression of MEK1CFC, although embryonic lethality precluded characterization of the heart (Jindal et al., 2017b). This suggests that disruption of KV patterning may be a relatively common feature of inappropriate activation of RAS signaling, and is consistent with reports that FGF signaling is required for KV formation, ciliogenesis and regulating cilia length (Albertson and Yelick, 2005; Hong and Dawid, 2009; Neugebauer et al., 2009).
Neural crest derived structures
Neural crest cells (NCC) arise at the border between the neural and epidermal ectoderm. Following neural tube closure, NCC delaminate and migrate throughout the embryo. NCC contribute to a wide range of structures and cell populations, including skeletal elements, the developing heart, pigment cells, neurons and glia (Cooper and Raible, 2009; Cordero et al., 2011; Klymkowsky et al., 2010; Stewart et al., 2010a). Although the induction and migration of NCC is controlled by multiple pathways, RAS-mediated FGF signaling has been demonstrated to be involved in both (Abu-Issa et al., 2002; Cavanaugh et al., 2015; Frank et al., 2002; Kubota and Ito, 2000; Mayor et al., 1997). The prevalence of CFA and pigmented macules in patients is therefore unsurprising, and similar NCC-related phenotypes are recapitulated in zebrafish and Xenopus, where the emergence of the phenotype can be studied to elucidate the underlying mechanisms.
In zebrafish, RASopathy-associated CFA generally manifest as widening of the head. This widening has been observed upon HRASCS, SHP2NSML, SHP2NS, NRASNS and RRAS2NS expression (Jopling et al., 2007; Niihori et al., 2019; Runtuwene et al., 2011; Santoriello et al., 2009). In some cases, the ceratohyal cartilage is also more posteriorly positioned with respect to the Meckel’s cartilage (Jopling et al., 2007). There are as yet no reports of CFAs in CFC models, but the observed lethality rates may preclude such analysis in many cases (Jindal et al., 2017b). Mutations that cause syndromes that lack CFAs in the clinical setting, such as NF1, are not reported to cause craniofacial malformations in zebrafish (Padmanabhan et al., 2009; Shin et al., 2012). Thus, mutations that are associated with those syndromes that include a CFA in humans, also cause craniofacial malformations in zebrafish.
The development of pigment patterns is also disrupted in RASopathies. The pathologies of NF1, LS, NSML and NSLAH include pigmented macules. In zebrafish, expression of SHP2NSML leads to an increased number of pigment cells that is highly reminiscent of the multiple lentigines observed in patients (Stewart et al., 2010b). This increase in number was associated with increased expression of sox10 and foxd3, transcription factors that maintain the NCC in an undifferentiated state. In addition to the increased number, aberrant migration patterns were observed. Cranial NCC migrated laterally rather than into ventral regions of the head, while trunk NCC were delayed in migrating posteriorly over the yolk (Stewart et al., 2010b). In contrast, no changes in foxd3 or sox10 levels were detected upon knockdown of NF1 (Padmanabhan et al., 2009). Pigmentation defects in NF1 null embryos were observed at later time points, but comprised a decrease in melanocyte number rather than the expected increase (Shin et al., 2012). This is surprising considering the café-au-lait spots observed in NF1 patients. Melanocytes were initially specified correctly, but the regular pattern of melanocytes along the lateral stripes was disrupted, suggesting defects in the regeneration or migration of melanophores that develop from melanocyte stem cells (Hultman and Johnson, 2010). Expression of YWHAZCFC caused clustering of pigment cells in Xenopus, although the authors note this may be a consequence of changes in cell shape (Popov et al., 2019).
The sympathetic neurons also develop from NCC precursors, and this population is increased upon expression of SHP2NS mutations (Stewart et al., 2010a; Stewart et al., 2010b). Since the sympathetic nervous system innervates the myocardium to regulate heart rate, and is responsible for the flight-or-fight response, this change in cell number may also impact cardiac and neurocognitive development by altering the stimulus these tissues receive. In addition, if the increased numbers of sympathetic neurons are due to unrestrained proliferation, expression of these variants may increase the risk of developing tumors derived from these cells, such as neuroblastomas.
Neurocognitive impairment
Neurocognitive delay is a feature common to several RASopathy syndromes, but remains relatively understudied in aquatic models. Using behavioral testing, nf1a−/−:nf1b−/− fish were demonstrated to have learning and memory deficits (Shin et al., 2012; Wolman et al., 2014). NF1 depletion was also associated with increased numbers of Schwann cells and oligodendrocyte precursor cells (Lee et al., 2010; Padmanabhan et al., 2009; Shin et al., 2012). Consistent with disrupted Schwann and oligodendrocyte development, the myelin sheaths surrounding neurons in the nervous system were malformed in NF1-deficient embryos, presumably with negative effects on the conduction of electrical impulses and suggesting a possible mechanism for the emergence of the learning and memory deficits (Shin et al., 2012). The memory deficits could be rescued by inhibiting RAS signaling (Wolman et al., 2014), confirming a role for RAS signaling in memory formation.
Phenotype severity and mutation strength
Although many of the same tissues and morphogenetic events are disrupted by expression of RASopathy associated mutations, the precise phenotypic presentation can be quite disparate. When comparing models of different RASopathy syndromes, we expect to see different phenotypes since each syndrome is diagnosed according to a set of criteria that define the disease, and distinguish it from other RASopathies (Abe et al., 2012; Brems et al., 2007; Grant et al., 2018; NIH, 1988; Rauen, 2007; Roberts et al., 2006; Roberts et al., 2013; Sarkozy et al., 2008). It is therefore to be expected that models of CFC will not completely phenocopy models of NS. For example, the presence of pigmentation defects in models of NF1 and NSML reflects the pathology of these diseases (Shin et al., 2012; Stewart et al., 2010b). Similarly, the shortened body length in NS, NSML, CS and CFC models reflects the reduced stature of patients. The concordance of human symptoms and model phenotypes validates the use of zebrafish and Xenopus in studying the emergence of phenotypes.
The distinction of diagnostic criteria between syndromes does not explain the variation in phenotype manifestation within models of the same syndrome. While the effects of RASopathy mutations are pleiotropic, and the presentation of a specific symptom is not guaranteed, much of this variability is presumably associated with the unique genetic and environmental factors encountered by individual patients. In a model organism, where genetic background and environmental exposures are more strictly controlled, the effects of activating RAS signaling might be expected to generate more consistent phenotypes. Part of this variability within syndromes may be due to differences in causative genes. CFC, NS and NSML are associated with mutations in multiple components of the RAS signaling pathway (Table 1). These genes represent distinct steps in the pathway sequence, and may thus experience different levels of regulation. Mutations in those components that are more downstream in the sequence may then have stronger effects, simply because there are fewer regulatory events that can impact them. For example, CFC is associated with mutations in KRAS, BRAF, MEK1 and MEK2. Perhaps those mutations in MEK1 and MEK2 might be more severe than those in KRAS due to their position in the pathway. Indeed, this appears to be the case at the syndrome level. CFC mutations, which occur in components of the phosphorylation cascade, seem to have stronger effects on morphogenetic events than do NS mutations, which are more prevalent in earlier steps of the pathway (Anastasaki et al., 2009; Bonetti et al., 2014a; Jindal et al., 2017b; Jopling et al., 2007; Runtuwene et al., 2011).
The variability that may arise from mutation of different genes does not account for the differences in phenotype severity observed for different alleles of the same gene. Although there are components for which both activating and inactivating mutations cause RASopathies, most mutations in the same gene exert an effect in the same direction. However, the amplitude of this effect may be variable and depend upon the specific mutation. It was recently shown that NRAS mutations can differentially activate RAS signaling, as assayed by the extent of embryo elongation at 11 hpf (Runtuwene et al., 2011). The same can be demonstrated for MEK1 (Jindal et al., 2017b). Furthermore, MEK1 mutations could be ranked by ‘strength’, and this rank was predictive of the inhibitor dose required to restore normal embryo morphology. Since this rank was generated based on a single developmental event, it would be interesting to test whether it is predictive of the severity of other phenotypes, or whether mutations differentially impact distinct phenotypes. Such insights could be crucial for developing effective treatment regimens using pharmacological inhibitors of the signaling pathway (Anastasaki et al., 2012; Jindal et al., 2017b).
The phenomenon of mutations displaying a range of strengths does not appear to be confined to MEK1 and NRAS. Although similar studies have not yet been performed for all other signaling components, there are indications that SHP2 and RRAS2 mutations may also have disparate strengths. A higher level of expression was required to generate phenotypes for the NS-associated mutation SHP2D61G than SHP2T73I, suggesting SHP2D61G has a weaker effect in activating signaling (Jopling et al., 2007). Additionally, by this metric the NSML-associated mutations SHP2G465A and SHP2A462T appear to be stronger than NS-associated mutations, despite having reduced phosphatase activity (Bonetti et al., 2014a; Jopling et al., 2007). Similarly, RRAS2Q72L caused embryonic lethality, while the same amounts of RRAS2Q72H or RRAS2G24-G25dup did not (Niihori et al., 2019). Systematic investigation of mutation strength across other pathway components may yield further insights into disease etiology.
These observations do raise an interesting question; are all RASopathy mutations fundamentally similar, differing only in extent of pathway activation? There is certainly overlap in causative genes; BRAF and KRAS alleles can cause either NS or CFC depending on the specific mutation. Perhaps NS and CFC represent different points along a spectrum of pathway activation. After all, RASopathy patients display overlapping symptoms. However, the relative dissimilarity in symptoms between RASopathies caused by activating signaling components (NS, NSML, NSLAH, CS, CFC) and those arising from inactivating negative regulators (NF1, LS) may suggest that there are different mechanisms at play. In any case, the discovery that there is a spectrum of mutation strength provides the opportunity to use such mutations as tools to incrementally increase pathway activation when investigating the role of RAS signaling in developmental events.
Activation and attenuation: dual effects of RASopathy mutations
While RASopathies arise from net activation of the RAS signaling pathway, some loss-of-function mutations have been identified in proteins that usually activate signaling. A subset of disease-associated alleles of BRAF have been characterized as kinase inactivating mutations that would be predicted to attenuate signaling. Both kinase activating and inactivating mutations, however, manifest as CFC in patients (Garnett et al., 2005; Niihori et al., 2006; Rodriguez-Viciana et al., 2006b; Wan et al., 2004). How then do mutations that have opposing effects cause the same outcome?
One explanation is that all mutations are, in fact, activating. The BRAF mutations were characterized as kinase inactivating using in vitro assays. An in vitro assay, which is an isolated system by its nature, may not fully reflect the signaling status in vivo. Indeed, it is known that mutations in MEK1 act differently in vitro when compared to in vivo effects (Jindal et al., 2017a). When tested in zebrafish embryos, both kinase activating and inactivating mutations in BRAF cause pathway activation, as assayed both by the levels of dually phosphorylated ERK (dpERK) and embryo elongation at 12 hpf (Anastasaki et al., 2009). Furthermore, combining suboptimal doses of activating and inactivating mutations results in pathway activation, indicating the mutations exert the same effect on signaling. This is likely because BRAF can activate signaling via interacting with C-RAF, despite reduced kinase activity (Garnett et al., 2005).
Intriguingly, this is not necessarily the same for all signaling components. SHP2 mutations can either activate or inactivate phosphatase activity. Although both subsets of mutation cause RASopathies in humans, activating mutations cause NS while mutations that decrease phosphatase activity are associated with NSML (Fragale et al., 2004; Hanna et al., 2006; Keilhack et al., 2005; Kontaridis et al., 2006; Tartaglia et al., 2006). SHP2NS and SHP2NSML cause overlapping phenotypes in zebrafish, but their effects on signaling are less clear. SHP2NS causes increased dpERK levels as a result of active signaling, but SHP2NSML variants have been reported to both decrease and increase dpERK levels (Bonetti et al., 2014a; Stewart et al., 2010b). Combining suboptimal doses of activating (NS) and inactivating (NSML) mutations did not cause phenotypes, suggesting that the mutations may not act in precisely the same way in vivo (Jopling et al., 2007). However, the early phenotypes of embryos expressing SHP2NSML variants could be rescued by inhibition of MEK, indicating that RAS signaling is activated in SHP2NSML-expressing embryos (Bonetti et al., 2014a). This is consistent with recent data that suggest SHP2NSML mutations activate signaling despite reduced phosphatase activity, by destabilizing the closed conformation such that the protein is more sensitive to pathway stimulus (Yu et al., 2013).
An alternative explanation for opposing mutations having the same effects, is that there is a critical level of pathway activation that allows proper embryonic development: therefore, either increasing or decreasing pathway activity will similarly disrupt normal morphogenesis. Although the effect of reduced signaling in humans is not well documented, there are phenotypic similarities between zebrafish models of signaling loss and signaling activation. Pharmacological inhibition of RAS signaling results in shorter embryos and craniofacial malformation, while knockdown of KRAS causes cardiac defects and craniofacial malformations (Anastasaki et al., 2012; Razzaque et al., 2012). Loss of SHP2 function, either through morpholino knockdown or null mutations, leads to shorter embryos that present with craniofacial malformations (Bonetti et al., 2014b; Jopling et al., 2007; Stewart et al., 2010b). Pigment cells in the lateral stripes are lost upon activating the pathway by removing the negative regulator NF1, and upon inhibition by loss of the RTK ErbB (Hultman et al., 2009; Shin et al., 2012). And in Xenopus, inhibiting SHP2 function leads to a smaller heart (Langdon et al., 2007).
However, there are also differences between models of increased signaling and those where signaling is attenuated. Strong inhibition of RAS signaling causes a reduction in the embryonic aspect ratio at 11 hpf rather than the increase observed upon pathway activation (Jindal et al., 2017b). Additionally, knockdown of SHP2 causes apoptosis in the brain and neural crest derived tissues, while SHP2NSML causes increased pigmentation (Stewart et al., 2010b). These differences may reflect morphogenetic events that do not require a specific threshold of activation to properly pattern embryonic structures. Instead, the morphogenesis may respond to varying levels of signaling in the manner of a rheostat, with weak, intermediate and strong activation translating into different effects along a continuous spectrum.
A third possibility is that it is a combination of pathway activation and attenuation within a single embryo that induces the observed phenotypes in zebrafish. As with most signaling events, the RAS pathway is finely tuned by positive and negative feedback loops. Hyperactive signaling at early time points may desensitize the pathway to later input, generating a system where there is an early gain-of-function in one set of tissues, and later loss-of-function in a separate set of tissues. MEK1CFC reduces pathway output in regions that receive endogenous signal, such as the blastoderm margin, even while ectopic signaling is detected in the animal cap (Goyal et al., 2017). Structures descended from the blastoderm margin may therefore be expected to present phenotypes associated with loss of signaling, while tissues derived from the cells in the animal cap may develop phenotypes associated with hyperactive RAS signaling. Consistent with this role of negative regulation of the pathway over time, transgenic integration of HRASCS did not result in increased pathway activity at 6 dpf, while inducible expression of the same variant did increase signaling, suggesting the pathway activation may be transient (Santoriello et al., 2009). Thus, it may be that a subset of phenotypes arises from pathway activation in regions not usually exposed to endogenous ligand, and others arise from attenuated signaling in regions where RAS signaling is normally active. Those phenotypes that are conserved between gain- and loss-of-signaling models may then be attributed to the loss of signaling output. Recent developments in harnessing optogenetics to activate RAS signaling in zebrafish will allow researchers to dissect these spatially disparate consequences of hyperactive signaling in vivo (Patel et al., 2019).
Conclusion
The disruptions to development caused by RASopathy-associated mutations can be effectively modeled in zebrafish and Xenopus embryos. The phenotypes observed in these vertebrate models are highly reminiscent of the symptoms of patients. CHD and CFA are present upon expression of many RASopathy-associated mutations, while less common phenotypes, such as disrupted pigmentation, are found only upon expression of mutations that cause similar symptoms in patients. The conserved nature of the developmental disruptions arising from RASopathy mutations allows mechanistic insight into how disease develops. However, the transient nature of models relying on injection of morpholinos or RNA makes longer term analysis of development more challenging. The advent of CRISPR and TALEN mediated gene editing will likely lead to increasing numbers of zebrafish lines carrying RASopathy mutations. Most patients are heterozygous for their causative alleles and knock-in fish carrying RASopathy mutations will provide more insight into the human condition, especially over time frames beyond early development.
Perhaps the most problematic aspect of comparing models is the lack of consistent phenotyping. Although embryo elongation, cardiac and craniofacial phenotypes are well reported, there is no consensus in the timing and nature of the assays. It would be useful to have a standard phenotyping stratagem, such as the example pipeline outlined in Fig. 3. This would allow more comparable studies between models.
Figure 3.
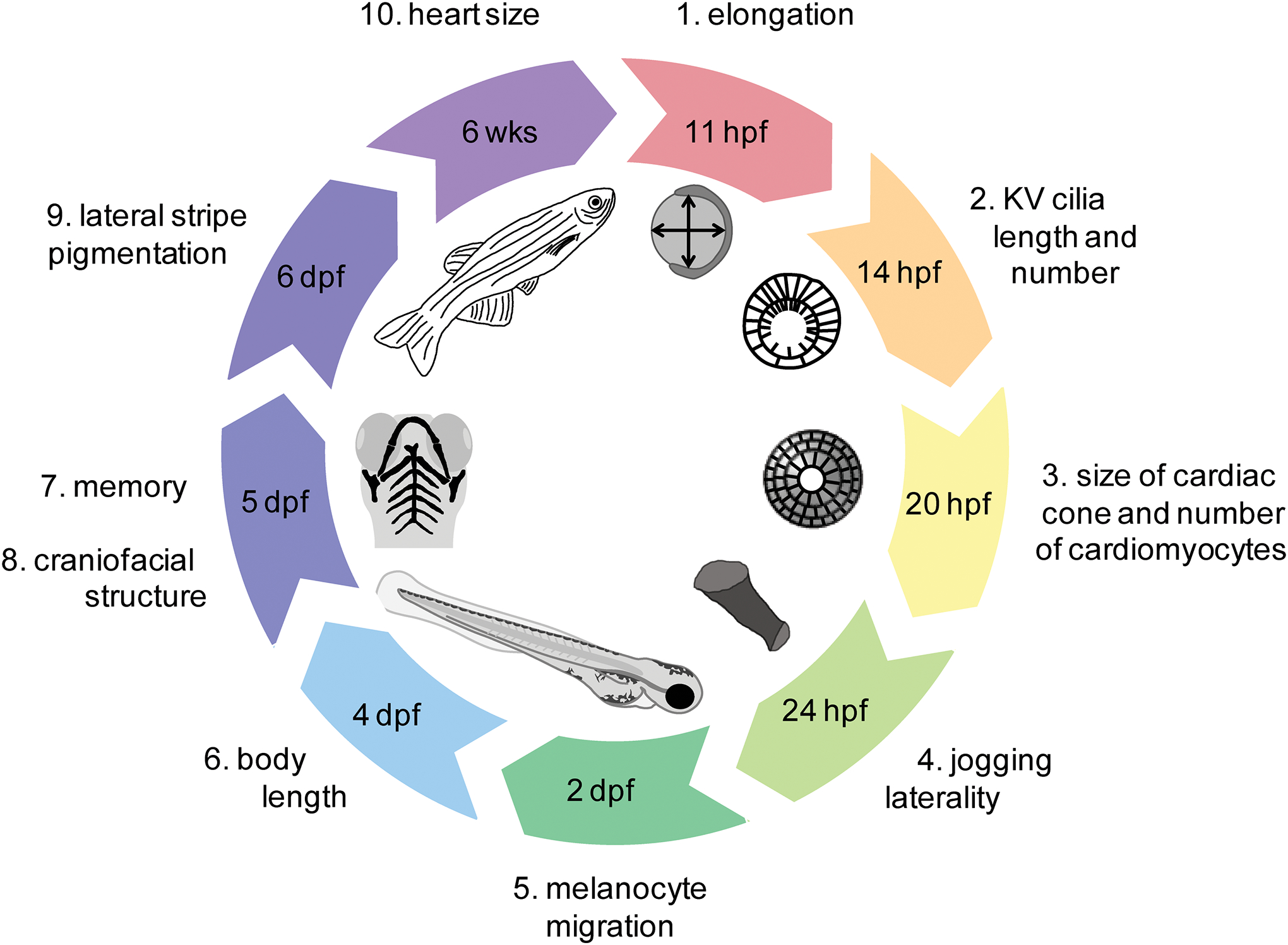
Schematic of an example phenotyping pipeline for zebrafish
Overall, zebrafish and Xenopus are attractive organisms in which to study RASopathies. The conserved morphogenetic events of vertebrate development allow investigation into the primary causes of symptoms. The accessibility of embryos, coupled with the wide range of tools allowing in vivo imaging of cell populations, provides the opportunity to evaluate changes in the developmental program over time. By understanding how symptoms emerge, we may be able to identify novel sites of intervention for patients. External embryonic development is especially attractive for conducting large scale pharmacological studies. The efficacy of drug treatments can be rapidly tested in a high-throughput manner using zebrafish and Xenopus embryos, and the effect on each phenotype can be quantitatively analyzed. Indeed, studies have already demonstrated critical time windows for pharmacological intervention with MEK inhibitors, and provided strategies for overcoming disadvantageous drug-induced side effects (Anastasaki et al., 2009; Anastasaki et al., 2012). Furthermore, when combined with quantitative analysis of mutation strength, the drug dose required to restore normal development can be predicted (Jindal et al., 2017b). These insights can only improve our understanding of the etiology and pathology of these debilitating diseases, and improve treatment strategies. Extending such studies will not only allow the rapid assessment of novel therapeutic strategies, but may facilitate treatment plans that are personalized to individual patients, according to genetic cause.
Acknowledgements
We are grateful to SY Shvartsman, GA Jindal and DT Grimes for helpful discussions and proofreading, and Marvin Cortez title suggestions.
Funding: This work was supported by the National Institutes of Health under award numbers R01 GM086537 and R03 HD092694. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- Abe Y, Aoki Y, Kuriyama S, Kawame H, Okamoto N, Kurosawa K, Ohashi H, Mizuno S, Ogata T, Kure S, Niihori T, Matsubara Y, Costello, Japan CFCssgi. 2012. Prevalence and clinical features of Costello syndrome and cardio-facio-cutaneous syndrome in Japan: findings from a nationwide epidemiological survey. Am J Med Genet A 158A(5):1083–1094. [DOI] [PubMed] [Google Scholar]
- Abu-Issa R, Smyth G, Smoak I, Yamamura K, Meyers EN. 2002. Fgf8 is required for pharyngeal arch and cardiovascular development in the mouse. Development 129(19):4613–4625. [DOI] [PubMed] [Google Scholar]
- Albertson RC, Yelick PC. 2005. Roles for fgf8 signaling in left-right patterning of the visceral organs and craniofacial skeleton. Dev Biol 283(2):310–321. [DOI] [PubMed] [Google Scholar]
- Anastasaki C, Estep AL, Marais R, Rauen KA, Patton EE. 2009. Kinase-activating and kinase-impaired cardio-facio-cutaneous syndrome alleles have activity during zebrafish development and are sensitive to small molecule inhibitors. Hum Mol Genet 18(14):2543–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki C, Rauen KA, Patton EE. 2012. Continual low-level MEK inhibition ameliorates cardio-facio-cutaneous phenotypes in zebrafish. Dis Model Mech 5(4):546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T, Mohi MG, Ismat FA, Bronson RT, Williams IR, Kutok JL, Yang W, Pao LI, Gilliland DG, Epstein JA, Neel BG. 2004. Mouse model of Noonan syndrome reveals cell type- and gene dosage-dependent effects of Ptpn11 mutation. Nat Med 10(8):849–857. [DOI] [PubMed] [Google Scholar]
- Baker K, Holtzman NG, Burdine RD. 2008. Direct and indirect roles for Nodal signaling in two axis conversions during asymmetric morphogenesis of the zebrafish heart. Proc Natl Acad Sci U S A 105(37):13924–13929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetti M, Paardekooper Overman J, Tessadori F, Noel E, Bakkers J, den Hertog J. 2014a. Noonan and LEOPARD syndrome Shp2 variants induce heart displacement defects in zebrafish. Development 141(9):1961–1970. [DOI] [PubMed] [Google Scholar]
- Bonetti M, Rodriguez-Martinez V, Paardekooper Overman J, Overvoorde J, van Eekelen M, Jopling C, Hertog J. 2014b. Distinct and overlapping functions of ptpn11 genes in Zebrafish development. PLoS One 9(4):e94884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brems H, Chmara M, Sahbatou M, Denayer E, Taniguchi K, Kato R, Somers R, Messiaen L, De Schepper S, Fryns JP, Cools J, Marynen P, Thomas G, Yoshimura A, Legius E. 2007. Germline loss-of-function mutations in SPRED1 cause a neurofibromatosis 1-like phenotype. Nat Genet 39(9):1120–1126. [DOI] [PubMed] [Google Scholar]
- Capri Y, Flex E, Krumbach OHF, Carpentieri G, Cecchetti S, Lissewski C, Rezaei Adariani S, Schanze D, Brinkmann J, Piard J, Pantaleoni F, Lepri FR, Goh ES, Chong K, Stieglitz E, Meyer J, Kuechler A, Bramswig NC, Sacharow S, Strullu M, Vial Y, Vignal C, Kensah G, Cuturilo G, Kazemein Jasemi NS, Dvorsky R, Monaghan KG, Vincent LM, Cave H, Verloes A, Ahmadian MR, Tartaglia M, Zenker M. 2019. Activating Mutations of RRAS2 Are a Rare Cause of Noonan Syndrome. Am J Hum Genet 104(6):1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal-Vergara X, Sevilla A, D’Souza SL, Ang YS, Schaniel C, Lee DF, Yang L, Kaplan AD, Adler ED, Rozov R, Ge Y, Cohen N, Edelmann LJ, Chang B, Waghray A, Su J, Pardo S, Lichtenbelt KD, Tartaglia M, Gelb BD, Lemischka IR. 2010. Patient-specific induced pluripotent stem-cell-derived models of LEOPARD syndrome. Nature 465(7299):808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman TJ, Josowitz R, Johnson BV, Gelb BD, Costa KD. 2016. Human Engineered Cardiac Tissues Created Using Induced Pluripotent Stem Cells Reveal Functional Characteristics of BRAF-Mediated Hypertrophic Cardiomyopathy. PLoS One 11(1):e0146697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavanaugh AM, Huang J, Chen JN. 2015. Two developmentally distinct populations of neural crest cells contribute to the zebrafish heart. Dev Biol 404(2):103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper CD, Raible DW. 2009. Mechanisms for reaching the differentiated state: Insights from neural crest-derived melanocytes. Semin Cell Dev Biol 20(1):105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero DR, Brugmann S, Chu Y, Bajpai R, Jame M, Helms JA. 2011. Cranial neural crest cells on the move: their roles in craniofacial development. Am J Med Genet A 155A(2):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Campos-Baptista MI, Holtzman NG, Yelon D, Schier AF. 2008. Nodal signaling promotes the speed and directional movement of cardiomyocytes in zebrafish. Dev Dyn 237(12):3624–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. 1994. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev 8(24):3045–3057. [DOI] [PubMed] [Google Scholar]
- Denouel-Galy A, Douville EM, Warne PH, Papin C, Laugier D, Calothy G, Downward J, Eychene A. 1998. Murine Ksr interacts with MEK and inhibits Ras-induced transformation. Curr Biol 8(1):46–55. [DOI] [PubMed] [Google Scholar]
- Fragale A, Tartaglia M, Wu J, Gelb BD. 2004. Noonan syndrome-associated SHP2/PTPN11 mutants cause EGF-dependent prolonged GAB1 binding and sustained ERK2/MAPK1 activation. Hum Mutat 23(3):267–277. [DOI] [PubMed] [Google Scholar]
- Frank DU, Fotheringham LK, Brewer JA, Muglia LJ, Tristani-Firouzi M, Capecchi MR, Moon AM. 2002. An Fgf8 mouse mutant phenocopies human 22q11 deletion syndrome. Development 129(19):4591–4603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furthauer M, Thisse C, Thisse B. 1997. A role for FGF-8 in the dorsoventral patterning of the zebrafish gastrula. Development 124(21):4253–4264. [DOI] [PubMed] [Google Scholar]
- Furthauer M, Van Celst J, Thisse C, Thisse B. 2004. Fgf signalling controls the dorsoventral patterning of the zebrafish embryo. Development 131(12):2853–2864. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Rana S, Paterson H, Barford D, Marais R. 2005. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell 20(6):963–969. [DOI] [PubMed] [Google Scholar]
- Goyal Y, Jindal GA, Pelliccia JL, Yamaya K, Yeung E, Futran AS, Burdine RD, Schupbach T, Shvartsman SY. 2017. Divergent effects of intrinsically active MEK variants on developmental Ras signaling. Nat Genet 49(3):465–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant AR, Cushman BJ, Cave H, Dillon MW, Gelb BD, Gripp KW, Lee JA, Mason-Suares H, Rauen KA, Tartaglia M, Vincent LM, Zenker M. 2018. Assessing the gene-disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum Mutat 39(11):1485–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant MG, Patterson VL, Grimes DT, Burdine RD. 2017. Modeling Syndromic Congenital Heart Defects in Zebrafish. Curr Top Dev Biol 124:1–40. [DOI] [PubMed] [Google Scholar]
- Grimes DT, Burdine RD. 2017. Left-Right Patterning: Breaking Symmetry to Asymmetric Morphogenesis. Trends Genet. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Aldinger KA, Bennett JT, Baker L, Tusi J, Powell-Hamilton N, Stabley D, Sol-Church K, Timms AE, Dobyns WB. 2016. A novel rasopathy caused by recurrent de novo missense mutations in PPP1CB closely resembles Noonan syndrome with loose anagen hair. Am J Med Genet A 170(9):2237–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Zand DJ, Demmer L, Anderson CE, Dobyns WB, Zackai EH, Denenberg E, Jenny K, Stabley DL, Sol-Church K. 2013. Expanding the SHOC2 mutation associated phenotype of Noonan syndrome with loose anagen hair: structural brain anomalies and myelofibrosis. Am J Med Genet A 161A(10):2420–2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzmil M, Whiting D, Maule J, Anastasaki C, Amatruda JF, Kelsh RN, Norbury CJ, Patton EE. 2007. The INT6 cancer gene and MEK signaling pathways converge during zebrafish development. PLoS One 2(9):e959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna N, Montagner A, Lee WH, Miteva M, Vidal M, Vidaud M, Parfait B, Raynal P. 2006. Reduced phosphatase activity of SHP-2 in LEOPARD syndrome: consequences for PI3K binding on Gab1. FEBS Lett 580(10):2477–2482. [DOI] [PubMed] [Google Scholar]
- Hernandez-Porras I, Guerra C. 2017. Modeling RASopathies with Genetically Modified Mouse Models. Methods Mol Biol 1487:379–408. [DOI] [PubMed] [Google Scholar]
- Hong SK, Dawid IB. 2009. FGF-dependent left-right asymmetry patterning in zebrafish is mediated by Ier2 and Fibp1. Proc Natl Acad Sci U S A 106(7):2230–2235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultman KA, Budi EH, Teasley DC, Gottlieb AY, Parichy DM, Johnson SL. 2009. Defects in ErbB-dependent establishment of adult melanocyte stem cells reveal independent origins for embryonic and regeneration melanocytes. PLoS Genet 5(7):e1000544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultman KA, Johnson SL. 2010. Differential contribution of direct-developing and stem cell-derived melanocytes to the zebrafish larval pigment pattern. Dev Biol 337(2):425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal GA, Goyal Y, Burdine RD, Rauen KA, Shvartsman SY. 2015. RASopathies: unraveling mechanisms with animal models. Dis Model Mech 8(8):769–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal GA, Goyal Y, Humphreys JM, Yeung E, Tian K, Patterson VL, He H, Burdine RD, Goldsmith EJ, Shvartsman SY. 2017a. How activating mutations affect MEK1 regulation and function. J Biol Chem 292(46):18814–18820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jindal GA, Goyal Y, Yamaya K, Futran AS, Kountouridis I, Balgobin CA, Schupbach T, Burdine RD, Shvartsman SY. 2017b. In vivo severity ranking of Ras pathway mutations associated with developmental disorders. Proc Natl Acad Sci U S A 114(3):510–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jopling C, van Geemen D, den Hertog J. 2007. Shp2 knockdown and Noonan/LEOPARD mutant Shp2-induced gastrulation defects. PLoS Genet 3(12):e225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josowitz R, Carvajal-Vergara X, Lemischka IR, Gelb BD. 2011. Induced pluripotent stem cell-derived cardiomyocytes as models for genetic cardiovascular disorders. Curr Opin Cardiol 26(3):223–229. [DOI] [PubMed] [Google Scholar]
- Keilhack H, David FS, McGregor M, Cantley LC, Neel BG. 2005. Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J Biol Chem 280(35):30984–30993. [DOI] [PubMed] [Google Scholar]
- Klymkowsky MW, Rossi CC, Artinger KB. 2010. Mechanisms driving neural crest induction and migration in the zebrafish and Xenopus laevis. Cell Adh Migr 4(4):595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontaridis MI, Swanson KD, David FS, Barford D, Neel BG. 2006. PTPN11 (Shp2) mutations in LEOPARD syndrome have dominant negative, not activating, effects. J Biol Chem 281(10):6785–6792. [DOI] [PubMed] [Google Scholar]
- Kota P, Terrell EM, Ritt DA, Insinna C, Westlake CJ, Morrison DK. 2019. M-Ras/Shoc2 signaling modulates E-cadherin turnover and cell-cell adhesion during collective cell migration. Proc Natl Acad Sci U S A 116(9):3536–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krens SF, He S, Lamers GE, Meijer AH, Bakkers J, Schmidt T, Spaink HP, Snaar-Jagalska BE. 2008. Distinct functions for ERK1 and ERK2 in cell migration processes during zebrafish gastrulation. Dev Biol 319(2):370–383. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Ito K. 2000. Chemotactic migration of mesencephalic neural crest cells in the mouse. Dev Dyn 217(2):170–179. [DOI] [PubMed] [Google Scholar]
- Langdon Y, Tandon P, Paden E, Duddy J, Taylor JM, Conlon FL. 2012. SHP-2 acts via ROCK to regulate the cardiac actin cytoskeleton. Development 139(5):948–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langdon YG, Goetz SC, Berg AE, Swanik JT, Conlon FL. 2007. SHP-2 is required for the maintenance of cardiac progenitors. Development 134(22):4119–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie H, Therrien M. 2015. Regulation of RAF protein kinases in ERK signalling. Nat Rev Mol Cell Biol 16(5):281–298. [DOI] [PubMed] [Google Scholar]
- Lee JS, Padmanabhan A, Shin J, Zhu S, Guo F, Kanki JP, Epstein JA, Look AT. 2010. Oligodendrocyte progenitor cell numbers and migration are regulated by the zebrafish orthologs of the NF1 tumor suppressor gene. Hum Mol Genet 19(23):4643–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenhart KF, Holtzman NG, Williams JR, Burdine RD. 2013. Integration of nodal and BMP signals in the heart requires FoxH1 to create left-right differences in cell migration rates that direct cardiac asymmetry. PLoS Genet 9(1):e1003109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levkowitz G, Waterman H, Zamir E, Kam Z, Oved S, Langdon WY, Beguinot L, Geiger B, Yarden Y. 1998. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev 12(23):3663–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques SR, Lee Y, Poss KD, Yelon D. 2008. Reiterative roles for FGF signaling in the establishment of size and proportion of the zebrafish heart. Dev Biol 321(2):397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P, Cawthon RM, et al. 1990. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63(4):843–849. [DOI] [PubMed] [Google Scholar]
- Mayor R, Guerrero N, Martinez C. 1997. Role of FGF and noggin in neural crest induction. Dev Biol 189(1):1–12. [DOI] [PubMed] [Google Scholar]
- Mazzanti L, Cacciari E, Cicognani A, Bergamaschi R, Scarano E, Forabosco A. 2003. Noonan-like syndrome with loose anagen hair: a new syndrome? Am J Med Genet A 118A(3):279–286. [DOI] [PubMed] [Google Scholar]
- Messiaen L, Yao S, Brems H, Callens T, Sathienkijkanchai A, Denayer E, Spencer E, Arn P, Babovic-Vuksanovic D, Bay C, Bobele G, Cohen BH, Escobar L, Eunpu D, Grebe T, Greenstein R, Hachen R, Irons M, Kronn D, Lemire E, Leppig K, Lim C, McDonald M, Narayanan V, Pearn A, Pedersen R, Powell B, Shapiro LR, Skidmore D, Tegay D, Thiese H, Zackai EH, Vijzelaar R, Taniguchi K, Ayada T, Okamoto F, Yoshimura A, Parret A, Korf B, Legius E. 2009. Clinical and mutational spectrum of neurofibromatosis type 1-like syndrome. JAMA 302(19):2111–2118. [DOI] [PubMed] [Google Scholar]
- Miura K, Wakayama Y, Tanino M, Orba Y, Sawa H, Hatakeyama M, Tanaka S, Sabe H, Mochizuki N. 2013. Involvement of EphA2-mediated tyrosine phosphorylation of Shp2 in Shp2-regulated activation of extracellular signal-regulated kinase. Oncogene 32(45):5292–5301. [DOI] [PubMed] [Google Scholar]
- Mohun TJ, Leong LM, Weninger WJ, Sparrow DB. 2000. The morphology of heart development in Xenopus laevis. Dev Biol 218(1):74–88. [DOI] [PubMed] [Google Scholar]
- Motta M, Giancotti A, Mastromoro G, Chandramouli B, Pinna V, Pantaleoni F, Di Giosaffatte N, Petrini S, Mazza T, D’Ambrosio V, Versacci P, Ventriglia F, Chillemi G, Pizzuti A, Tartaglia M, De Luca A. 2019. Clinical and functional characterization of a novel RASopathy-causing SHOC2 mutation associated with prenatal-onset hypertrophic cardiomyopathy. Hum Mutat 40(8):1046–1056. [DOI] [PubMed] [Google Scholar]
- Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, Stroke Statistics S. 2016. Executive Summary: Heart Disease and Stroke Statistics--2016 Update: A Report From the American Heart Association. Circulation 133(4):447–454. [DOI] [PubMed] [Google Scholar]
- Muram-Zborovski TM, Stevenson DA, Viskochil DH, Dries DC, Wilson AR, Rong M. 2010. SPRED 1 mutations in a neurofibromatosis clinic. J Child Neurol 25(10):1203–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers DC, Sepich DS, Solnica-Krezel L. 2002. Bmp activity gradient regulates convergent extension during zebrafish gastrulation. Dev Biol 243(1):81–98. [DOI] [PubMed] [Google Scholar]
- Neugebauer JM, Amack JD, Peterson AG, Bisgrove BW, Yost HJ. 2009. FGF signalling during embryo development regulates cilia length in diverse epithelia. Nature 458(7238):651–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NIH. 1988. Neurofibromatosis: Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 45(5):575–578. [PubMed] [Google Scholar]
- Niihori T, Aoki Y, Narumi Y, Neri G, Cave H, Verloes A, Okamoto N, Hennekam RC, Gillessen-Kaesbach G, Wieczorek D, Kavamura MI, Kurosawa K, Ohashi H, Wilson L, Heron D, Bonneau D, Corona G, Kaname T, Naritomi K, Baumann C, Matsumoto N, Kato K, Kure S, Matsubara Y. 2006. Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet 38(3):294–296. [DOI] [PubMed] [Google Scholar]
- Niihori T, Nagai K, Fujita A, Ohashi H, Okamoto N, Okada S, Harada A, Kihara H, Arbogast T, Funayama R, Shirota M, Nakayama K, Abe T, Inoue SI, Tsai IC, Matsumoto N, Davis EE, Katsanis N, Aoki Y. 2019. Germline-Activating RRAS2 Mutations Cause Noonan Syndrome. Am J Hum Genet 104(6):1233–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly AM, Pluskey S, Shoelson SE, Neel BG. 2000. Activated mutants of SHP-2 preferentially induce elongation of Xenopus animal caps. Mol Cell Biol 20(1):299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi K, Zhang H, Gault WJ, Wang CJ, Tan CC, Kim IK, Ying H, Rahman T, Pica N, Tartaglia M, Mlodzik M, Gelb BD. 2009. Phosphatase-defective LEOPARD syndrome mutations in PTPN11 gene have gain-of-function effects during Drosophila development. Hum Mol Genet 18(1):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padmanabhan A, Lee JS, Ismat FA, Lu MM, Lawson ND, Kanki JP, Look AT, Epstein JA. 2009. Cardiac and vascular functions of the zebrafish orthologues of the type I neurofibromatosis gene NFI. Proc Natl Acad Sci U S A 106(52):22305–22310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AL, Yeung E, McGuire SE, Wu AY, Toettcher JE, Burdine RD, Shvartsman SY. 2019. Optimizing photoswitchable MEK. Proc Natl Acad Sci U S A 116(51):25756–25763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popov IK, Hiatt SM, Whalen S, Keren B, Ruivenkamp C, van Haeringen A, Chen MJ, Cooper GM, Korf BR, Chang C. 2019. A YWHAZ Variant Associated With Cardiofaciocutaneous Syndrome Activates the RAF-ERK Pathway. Front Physiol 10:388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauen KA. 2007. HRAS and the Costello syndrome. Clin Genet 71(2):101–108. [DOI] [PubMed] [Google Scholar]
- Rauen KA. 2013. The RASopathies. Annu Rev Genomics Hum Genet 14:355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razzaque MA, Komoike Y, Nishizawa T, Inai K, Furutani M, Higashinakagawa T, Matsuoka R. 2012. Characterization of a novel KRAS mutation identified in Noonan syndrome. Am J Med Genet A 158A(3):524–532. [DOI] [PubMed] [Google Scholar]
- Roberts A, Allanson J, Jadico SK, Kavamura MI, Noonan J, Opitz JM, Young T, Neri G. 2006. The cardiofaciocutaneous syndrome. J Med Genet 43(11):833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AE, Allanson JE, Tartaglia M, Gelb BD. 2013. Noonan syndrome. Lancet 381(9863):333–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. 2006a. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell 22(2):217–230. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, McCormick F, Rauen KA. 2006b. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science 311(5765):1287–1290. [DOI] [PubMed] [Google Scholar]
- Rohr S, Otten C, Abdelilah-Seyfried S. 2008. Asymmetric involution of the myocardial field drives heart tube formation in zebrafish. Circ Res 102(2):e12–19. [DOI] [PubMed] [Google Scholar]
- Roszko I, Sawada A, Solnica-Krezel L. 2009. Regulation of convergence and extension movements during vertebrate gastrulation by the Wnt/PCP pathway. Semin Cell Dev Biol 20(8):986–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runtuwene V, van Eekelen M, Overvoorde J, Rehmann H, Yntema HG, Nillesen WM, van Haeringen A, van der Burgt I, Burgering B, den Hertog J. 2011. Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Model Mech 4(3):393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoriello C, Deflorian G, Pezzimenti F, Kawakami K, Lanfrancone L, d’Adda di Fagagna F, Mione M. 2009. Expression of H-RASV12 in a zebrafish model of Costello syndrome causes cellular senescence in adult proliferating cells. Dis Model Mech 2(1–2):56–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkozy A, Digilio MC, Dallapiccola B. 2008. Leopard syndrome. Orphanet J Rare Dis 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Padmanabhan A, de Groh ED, Lee JS, Haidar S, Dahlberg S, Guo F, He S, Wolman MA, Granato M, Lawson ND, Wolfe SA, Kim SH, Solnica-Krezel L, Kanki JP, Ligon KL, Epstein JA, Look AT. 2012. Zebrafish neurofibromatosis type 1 genes have redundant functions in tumorigenesis and embryonic development. Dis Model Mech 5(6):881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simanshu DK, Nissley DV, McCormick F. 2017. RAS Proteins and Their Regulators in Human Disease. Cell 170(1):17–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KA, Chocron S, von der Hardt S, de Pater E, Soufan A, Bussmann J, Schulte-Merker S, Hammerschmidt M, Bakkers J. 2008. Rotation and asymmetric development of the zebrafish heart requires directed migration of cardiac progenitor cells. Dev Cell 14(2):287–297. [DOI] [PubMed] [Google Scholar]
- Stainier DY, Lee RK, Fishman MC. 1993. Cardiovascular development in the zebrafish. I. Myocardial fate map and heart tube formation. Development 119(1):31–40. [DOI] [PubMed] [Google Scholar]
- Steklov M, Pandolfi S, Baietti MF, Batiuk A, Carai P, Najm P, Zhang M, Jang H, Renzi F, Cai Y, Abbasi Asbagh L, Pastor T, De Troyer M, Simicek M, Radaelli E, Brems H, Legius E, Tavernier J, Gevaert K, Impens F, Messiaen L, Nussinov R, Heymans S, Eyckerman S, Sablina AA. 2018. Mutations in LZTR1 drive human disease by dysregulating RAS ubiquitination. Science 362(6419):1177–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RA, Lee JS, Lachnit M, Look AT, Kanki JP, Henion PD. 2010a. Studying peripheral sympathetic nervous system development and neuroblastoma in zebrafish. Methods Cell Biol 100:127–152. [DOI] [PubMed] [Google Scholar]
- Stewart RA, Sanda T, Widlund HR, Zhu S, Swanson KD, Hurley AD, Bentires-Alj M, Fisher DE, Kontaridis MI, Look AT, Neel BG. 2010b. Phosphatase-dependent and -independent functions of Shp2 in neural crest cells underlie LEOPARD syndrome pathogenesis. Dev Cell 18(5):750–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stowe IB, Mercado EL, Stowe TR, Bell EL, Oses-Prieto JA, Hernandez H, Burlingame AL, McCormick F. 2012. A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev 26(13):1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, Zampino G, Burgt I, Palleschi A, Petrucci TC, Sorcini M, Schoch C, Foa R, Emanuel PD, Gelb BD. 2006. Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am J Hum Genet 78(2):279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidyman WE, Rauen KA. 2009. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev 19(3):230–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivion G, Luo Z, Avruch J. 1998. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature 394(6688):88–92. [DOI] [PubMed] [Google Scholar]
- Vigil D, Cherfils J, Rossman KL, Der CJ. 2010. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer 10(12):842–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Hardt S, Bakkers J, Inbal A, Carvalho L, Solnica-Krezel L, Heisenberg CP, Hammerschmidt M. 2007. The Bmp gradient of the zebrafish gastrula guides migrating lateral cells by regulating cell-cell adhesion. Curr Biol 17(6):475–487. [DOI] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R, Cancer Genome P. 2004. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 116(6):855–867. [DOI] [PubMed] [Google Scholar]
- Warkman AS, Krieg PA. 2007. Xenopus as a model system for vertebrate heart development. Semin Cell Dev Biol 18(1):46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. 2009. Neurofibromatosis type 1 revisited. Pediatrics 123(1):124–133. [DOI] [PubMed] [Google Scholar]
- Wolman MA, de Groh ED, McBride SM, Jongens TA, Granato M, Epstein JA. 2014. Modulation of cAMP and ras signaling pathways improves distinct behavioral deficits in a zebrafish model of neurofibromatosis type 1. Cell Rep 8(5):1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GF, O’Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, et al. 1990. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62(3):599–608. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. 1994. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes Dev 8(24):3032–3044. [DOI] [PubMed] [Google Scholar]
- Yelon D, Horne SA, Stainier DY. 1999. Restricted expression of cardiac myosin genes reveals regulated aspects of heart tube assembly in zebrafish. Dev Biol 214(1):23–37. [DOI] [PubMed] [Google Scholar]
- Yu W, Fantl WJ, Harrowe G, Williams LT. 1998. Regulation of the MAP kinase pathway by mammalian Ksr through direct interaction with MEK and ERK. Curr Biol 8(1):56–64. [DOI] [PubMed] [Google Scholar]
- Yu ZH, Xu J, Walls CD, Chen L, Zhang S, Zhang R, Wu L, Wang L, Liu S, Zhang ZY. 2013. Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J Biol Chem 288(15):10472–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]