

Abstract
Some genes have repeatedly been found to control diverse adaptations in a wide variety of organisms. Such gene reuse reveals not only the diversity of phenotypes these unique genes control but also the composition of developmental gene networks and the genetic routes available to and taken by organisms during adaptation. However, the causes of gene reuse remain unclear. A small number of large-effect Mendelian loci control a huge diversity of mimetic butterfly wing color patterns, but reasons for their reuse are difficult to identify because the genetic basis of mimicry has primarily been studied in two systems with correlated factors: female-limited Batesian mimicry in Papilio swallowtails (Papilionidae) and non-sex-limited Müllerian mimicry in Heliconius longwings (Nymphalidae). Here, we break the correlation between phylogenetic relationship and sex-limited mimicry by identifying loci controlling female-limited mimicry polymorphism Hypolimnas misippus (Nymphalidae) and non-sex-limited mimicry polymorphism in Papilio clytia (Papilionidae). The Papilio clytia polymorphism is controlled by the genome region containing the gene cortex, the classic P supergene in Heliconius numata, and loci controlling color pattern variation across Lepidoptera. In contrast, female-limited mimicry polymorphism in Hypolimnas misippus is associated with a locus not previously implicated in color patterning. Thus, although many species repeatedly converged on cortex and its neighboring genes over 120 My of evolution of diverse color patterns, female-limited mimicry polymorphisms each evolved using a different gene. Our results support conclusions that gene reuse occurs mainly within ∼10 My and highlight the puzzling diversity of genes controlling seemingly complex female-limited mimicry polymorphisms.
Keywords: adaptation, genetic constraint, convergent evolution, Lepidoptera, mimicry, genome-wide association
Introduction
Convergent evolution occurs when populations evolve similar phenotypes in the face of similar environmental pressures, strongly suggesting that natural selection produces predictable phenotypic outcomes (Endler 1986; Losos 2011). Cases of phenotypic convergence have been critical model systems for understanding the genetic basis of adaptations and whether natural selection also produces predictable genetic outcomes (Gompel and Prud’homme 2009; Elmer and Meyer 2011). Genetic mapping, quantitative trait locus, and genome-wide association (GWA) studies have all been effectively used to show that convergent phenotypes are indeed often controlled by mutations in orthologous genes in the convergent lineages (reviewed in Martin and Orgogozo [2013] and Stern [2013]). Such gene reuse may occur through mutations that arise independently in the convergent lineages, or mutations that arise once and are shared between the convergent lineages due to incomplete lineage sorting or introgression (genetic convergence and parallelism, respectively; Colosimo et al. 2005; Reed et al. 2011; Song et al. 2011; Davies et al. 2012; Jones et al. 2012; Gallant et al. 2014; Van Belleghem et al. 2018).
However, candidate gene and association studies of a wide variety of adaptive phenotypes in diverse and distantly related organisms suggest that gene reuse also frequently occurs in the absence of obvious phenotypic convergence (Martin and Orgogozo 2013). For instance, sexually selected plumage color in birds, adaptive coat color in mice, adaptive skin color in lizards and frogs, and several other vertebrate color patterns have all been associated with genetic variation in Melanocortin 1 receptor (reviewed in Mundy [2005], Manceau et al. [2010], and Kronforst et al. [2012]). Thus, these studies identify the genes and genetic networks that control adaptive phenotypes, but they also reveal the variety of phenotypes those genes control and the conditions under which gene reuse occurs (Martin and Orgogozo 2013; Stern 2013).
Butterfly wing color patterns provide especially useful models for understanding how and when gene reuse occurs during adaptation. A rich body of natural historical and theoretical work has clearly described the adaptive values and the wide variety of roles that color patterns serve, from predator avoidance to signaling potential mates (Bates 1862; Müller 1878; Fisher 1958; Mallet and Joron 1999; Beldade and Brakefield 2002; McMillan et al. 2002; Ruxton et al. 2004). In particular, many species have evolved color patterns resembling those of toxic species to gain protection from visual predators, that is, mimicry (Bates 1862; Müller 1878; Mallet and Barton 1989; Mallet et al. 1990). The genetics of mimicry has been studied in two main systems: phenotypic convergence between toxic species (Müllerian mimicry) of Heliconius longwings (Nymphalidae:Heliconiinae) and Batesian mimicry polymorphisms in palatable Papilio swallowtails (Papilionidae:Papilioninae).
Heliconius species have provided key insight into the genetic bases of phenotypic convergence on short time scales. This genus comprises two major clades that diverged ∼12 Mya (Kozak et al. 2015). Many species pairs, usually one from each clade, have converged on the same mimetic color patterns through the reuse of just four major-effect Mendelian loci and a number of small-effect modifier loci (Sheppard et al. 1985; Mallet 1989; Joron et al. 2006; Kronforst et al. 2006; Reed et al. 2011; Martin et al. 2012; Martin and Reed 2014; Kronforst and Papa 2015; Nadeau et al. 2016; Westerman et al. 2018). Major-effect loci containing WntA and cortex establish broad scale melanic patterns or act as melanic shutters, while optix and aristaless 1 determine scale color (Gilbert 2003). Convergent evolution is also mediated by sharing of alleles of these color patterning loci via hybridization and introgression between species in the same clade (Kronforst 2008; Dasmahapatra et al. 2012; Pardo-Diaz et al. 2012). These results suggest that gene reuse in Heliconius is due in large part to their close relationships. Recent work, however, has shown that WntA and cortex also control melanic variation across Nymphalidae and Lepidoptera, respectively, although in different ways and in different wing regions in various species (Gallant et al. 2014; Ito et al. 2016; Nadeau et al. 2016; van’t Hof et al. 2016; Mazo-Vargas et al. 2017).
In contrast to Heliconius, many Papilio species have evolved female-limited mimicry in which males develop a single, nonmimetic color pattern, whereas females develop one or more mimetic color patterns (Wallace 1865; Clarke et al. 1968; Clarke and Sheppard 1972; Kunte 2009a). Each of the four female-limited mimicry polymorphisms studied to date is controlled by a single, different switch locus. Female-limited mimicry polymorphism in Papilio polytes, P. memnon and their close relatives is controlled by alleles of the autosomal transcription factor gene doublesex (Kunte et al. 2014; Nishikawa et al. 2015; Iijima et al. 2018; D. H. Palmer and M. R. Kronforst, unpublished data). The switch in Papilio dardanus maps to the autosomal transcription factor genes engrailed and invected (Timmermans et al. 2014). Finally, the Papilio glaucus switch maps to an unknown sex-linked locus (Scriber et al. 1996; Koch et al. 2000). It is not clear if dsx reuse in the P. polytes group is due to genetic convergence or parallelism, but these species diverged only within the last ∼10 My (Wu et al. 2015). Thus, Papilio dsx, similar to Heliconius optix and aristaless 1, appears to be reused only within 10–15 My. Heliconius numata is one of the only species in its genus that displays mimicry polymorphism, but the polymorphism is not sex-limited and is controlled by the P supergene locus, which contains cortex but is not homologous to any known Papilio switch (Joron et al. 2006; Joron et al. 2011; Jay et al. 2018). Thus, despite the complexity of phenotype switching, a variety of loci are used to produce distinct mimetic color patterns.
Reuse of Heliconius optix and aristaless 1 and P. polytes dsx therefore suggest that gene reuse occurs over short time scales (within 10–15 My). However, the two sets of results suggest different patterns of gene reuse over longer time scales: WntA and cortex have been repeatedly used across 80–120 My of lepidopteran evolution, whereas no Papilio switch locus has been reused outside 10 My. However, these patterns are confounded by the fact that all studied Heliconius are sexually monomorphic Müllerian mimics, while all studied Papilio are sexually dimorphic Batesian mimics. Here, we break this correlation between mimicry mode and phylogeny by studying the genetic basis of mimicry polymorphisms in two unique species: Hypolimnas misippus (Nymphalidae) and Papilio clytia (Papilionidae). Hypolimnas misippus evolved female-limited mimicry polymorphism like many Papilio but is in the same family as Heliconius. In contrast, P. clytia evolved non-sex-limited mimicry polymorphism like He. numata, but is ∼25 My diverged from P. polytes. We use GWA, synteny, phylogenetic, and population genetic analyses to identify the mimicry switch loci in H. misippus and P. clytia and compare our results to other known Lepidopteran mimicry and color patterning loci.
Results
Papilio clytia Mimicry Polymorphism Is Associated with the Genome Region Containing cortex
Similar to He. numata, P. clytia has evolved a mimicry polymorphism in which both males and females can develop alternate mimetic color patterns (fig. 1a) . The papilio clytia clytia color pattern consists of a uniform field of brown pigmented scales with white submarginal crescents and a dark brown body mimicking the toxic common crow, Euploea core (Nymphalidae:Danaine). In contrast, the dissimilis color pattern is characterized by white pigmented and black melanic scales and white and black striped body that mimic the toxic tigers in the genera Tirumala and Parantica (Nymphalidae:Danainae).
Fig. 1.

The Papilio clytia mimicry switch is associated with the cortex region. (a) Papilio clytia males and females develop one of two main wing color patterns. (b) Phylogenetic relationships between P. clytia and other mimetic Papilio species (Materials and Methods, supplementary table S3, Supplementary Material online). Male morphs, female morphs, and the identities of the switch loci are shown to the right. Papilio dardanus and P. polytes females may develop several additional color patterns. (c) GWA for the switch between P. clytia forms dissimilis and clytia. Significance thresholds were calculated using FDR (Benjamini and Hochberg 1995). GWA results for all scaffolds are shown in supplementary figure S1, Supplementary Material online. (d) Papilio clytia GWA results, FST between dissimilis and clytia, and linkage disequilibrium (D′) around the peak of association. PclyML is highlighted. Mean FST and D′ were calculated in 10-kb nonoverlapping windows. Papilio glaucus scaffolds and gene models are shown on the x axis. Full information is in supplementary table S2, Supplementary Material online. Images: P. glaucus: M. McCarty, CC BY-SA 3.0; P. dardanus: London National History Museum, CC BY-NC-SA 3.0; Papilio memnon: Accassidy, CC BY-SA 4.0.
We found a single region on chromosome 13 associated with the switch between forms clytia and dissimilis using GWA with 10 clytia and 17 dissimilis individuals (fig. 1 and supplementary figs. S1 and S2 and supplementary table S1, Supplementary Material online). This P. clytia mimicry locus (PclyML) is a ∼500-kb region containing three distinct peaks of association, FST, and linkage disequilibrium and 31 protein-coding genes (fig. 1 and supplementary fig. S3, Supplementary Material online). Although PclyML is not homologous to any known Papilio switch loci, it overlaps the He. numata supergene P and loci associated with color pattern variation in at least seven additional lepidopteran species (fig. 2 and supplementary table S2, Supplementary Material online; Beldade et al. 2009; Ferguson et al. 2010; Counterman et al. 2010; Ito et al. 2016; Nadeau et al. 2016; van’t Hof et al. 2016). PclyML and many of these other loci contain cortex, the gene that controls industrial melanism in the peppered moth, acts as a melanic shutter in Heliconius, and underlies melanic Bombyx mori mutants (Ito et al. 2016; Nadeau et al. 2016; van’t Hof et al. 2016). Our finding that the cortex locus controls the switch between P. clytia dissimilis and clytia color patterns is therefore consistent with the known roles of cortex in He. numata and other lepidopterans but provides the first evidence that this region controls any color pattern variation in Papilionidae (figs. 1 and 2).
Fig. 2.

The cortex region controls wing color pattern variation across Lepidoptera. Syntenic relationships near PclyML in Bombyx mori (Bombycidae) and Heliconius melpomene (Nymphalidae). Color patterning loci, derived from fine mapping experiments or GWA, are underlined in representative reference genomes. Divergence times taken from Wahlberg et al. (2013) are shown to the left. Gene information is found in supplementary table S2, Supplementary Material online. Bbet carbonaria: Biston betularia industrial melanization (van’t Hof et al. 2016); Bmor Ws and Bm: B. mori Wing Spot and Black Moth mutations (Ito et al. 2016); PclyML: Papilio clytia mimicry locus (here); Hnum P: Heliconius numata supergene P, showing P1 and P2 inversions (Joron et al. 2011); Hmel Yb and Sb: H. melpomene melanic shutters (Ferguson et al. 2010); Hera Cr: Heliconius erato Yb and Sb (Counterman et al. 2010).
We next examined the evolutionary history of PclyML haplotypes. The 22 single-nucleotide polymorphism (SNPs) most significantly associated with the P. clytia mimicry switch were homozygous for one allele in all dissimilis individuals but heterozygous in all clytia individuals, suggesting that the clytia allele is completely dominant to the dissimilis allele. Consistent with this observation, maximum likelihood reconstructions using phased PclyML haplotypes revealed two well-supported groups: 1) a group containing one haplotype from each clytia individual and 2) a group containing all haplotypes from dissimilis individuals and one haplotype from each clytia individual (fig. 3a). Each haplotype group is subtended by a long internal branch, suggesting PclyML alleles have been maintained by balancing selection for some period of time. This pattern was not found using haplotypes immediately flanking PclyML suggesting a sharp distinction between PclyML and the surrounding sequence (fig. 3b). Last, dissimilis individuals varied in the width of their melanic stripes, creating both light (thin stripes) and dark (thick stripes) dissimilis (supplementary table S1, Supplementary Material online). We did not observe structure among dissimilis haplotypes that distinguished dark and light dissimilis, suggesting this quantitative variation is caused either by small differences between dissimilis alleles or by an unknown modifier locus (fig. 3b).
Fig. 3.
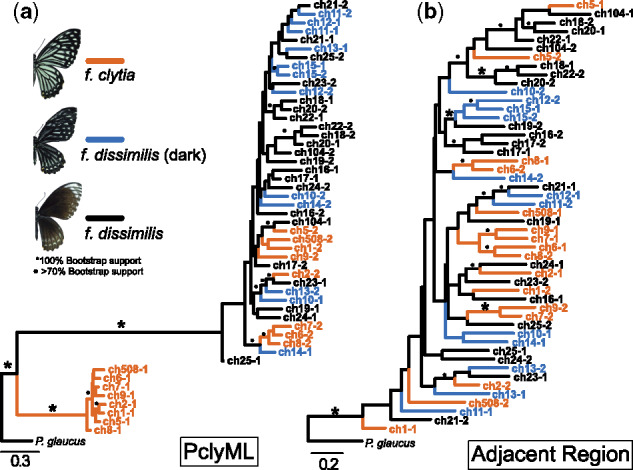
Relationships between phased Papilio clytia haplotypes within and outside the mimicry locus. (a) Relationships between haplotypes in the 500-kb PclyML region. Branches are colored according to the individual’s color pattern. Leaves indicate the individual and haplotype, for example, ch8-1 and ch8-2 are the haplotypes from individual ch8 (supplementary table S1, Supplementary Material online). (b) Relationships between haplotypes in the 500 kb immediately adjacent to PclyML.
Altogether, our results show that the P. clytia mimicry polymorphism is controlled by alternate haplotypes in the genome region containing cortex.
Hypolimnas misippus Female-Limited Mimicry Polymorphism Is Associated with a Novel Color Patterning Locus
Like many Papilio species, H. misippus evolved female-limited mimicry polymorphism in which males develop a single nonmimetic color pattern while females develop several mimetic color patterns (fig. 4; Smith 1976; Smith and Gordon 1987; Gordon et al. 2010). Female forms are primarily distinguished by the presence or absence of a melanic patch and white bar on the forewing apex that mimic morphs of the toxic African queen, Danaus chrysippus (Nymphalidae:Danainae; fig. 4a). The switch between black/white and orange apexes is controlled by alleles at a single autosomal locus, M, that we sought to identify using GWA (Gordon and Smith 1989).
Fig. 4.

The Hypolimnas misippus mimicry switch is associated with a single genome region on chromosome 26. (a) Hypolimnas misippus male and female color patterns. (b) Phylogenetic relationships between H. misippus and other nymphalids (Materials and Methods; supplementary table S3, Supplementary Material online). Additional Hypolimnas bolina forms exist but are not shown here. (c) GWA for the switch between the female forms misippus and immima/inaria. GWA results for all scaffolds are shown in supplementary figure S4, Supplementary Material online. The top SNPs are located on H. misippus scaffold 84 (supplementary fig. S4, Supplementary Material online). (d) Scaffold 84 GWA results. Hypolimnas misippus gene models are shown on the x axis, with Sox5/6 and pink marked in green and pink, respectively. Gene information is in supplementary table S5, Supplementary Material online. Melitaea cinxia: D. Descouens CC BY-SA 4.0.
We first generated a high-quality H. misippus genome assembly. The final assembly comprised 1,580 scaffolds, with an N50 of 1.01 Mb and total length of 408.8 Mb (table 1 and supplementary table S4, Supplementary Material online). This genome size is consistent with the 435 Mb predicted for its sister species Hypolimnas bolina (Hanrahan and Johnston 2011). The H. misippus genome’s completeness, measured using BUSCO, is similar to or higher than most other Nymphalidae (table 1; Waterhouse et al. 2018). Furthermore, we predicted 14,525 H. misippus gene models using publicly available data and MAKER, similar to other nymphalids like Danaus plexippus (15,130), Heliconius erato (14,613), and Melitaea cinxia (16,571; Holt and Yandell 2011; Zhan et al. 2011; Ahola et al. 2014; Campbell et al. 2014; Lewis et al. 2016). Finally, we assigned H. misippus scaffolds to M. cinxia chromosomes using BLAT; M. cinxia represents the ancestral nymphalid chromosome configuration (Kent 2002; Ahola et al. 2014).
Table 1.
Assembly Statistics for the Hypolimnas misippus and Other Publicly Available Nymphalidae Reference Genomes.
| Species a | No. Scaffolds | N 50 (Mb)b |
BUSCO Results (%)
c
|
||
|---|---|---|---|---|---|
| Complete (Dup.) | Frag. | Missing | |||
| Bicyclus anynana | 10,800 | 0.6 | 89.6 (0.7) | 3.9 | 6.5 |
| Danaus plexippus | 5,395 | 0.7 | 96.6 (1.9) | 2.3 | 1.1 |
| Heliconius erato | 196 | 10.7 | 85.5 (0.7) | 4.5 | 10.0 |
| Heliconius melpomene | 332 | 14.3 | 86.2 (0.4) | 4.1 | 9.7 |
| Hypolimnas misippus | 1,580 | 1.0 | 88.9 (0.3) | 3.8 | 7.3 |
| Junonia coenia | 1,136 | 1.6 | 96.9 (14.7) | 1.4 | 1.7 |
| Melitaea cinxia | 8,261 | 0.1 | 57.1 (0.2) | 11.8 | 31.1 |
| Vanessa tameamea | 1,558 | 3.0 | 96.3 (0.4) | 2.2 | 1.5 |
See Materials and Methods for assembly identifiers
Half of the assembly is contained in scaffolds at least this long.
Statistics calculated using BUSCO v3.01.01-beta and endopterygota gene set from OrthoDBv9 (2,442 single copy orthologs [SCOs]). Dup. and Frag. - duplicated and fragmented SCOs, respectively (Waterhouse et al. 2018; Zdobnov et al. 2017).
We identified a large region on chromosome 26 associated with H. misippus female-limited mimicry polymorphism using GWA with 14 misippus, 5 immima, and 15 inaria females (fig. 4 and supplementary figs. S4 and S5 and supplementary table S1, Supplementary Material online). The low resolution and stratified P values are probably a result of high relatedness between individuals, which were derived from four families (supplementary table S1, Supplementary Material online). However, the genomic inflation factor (λ) was only 0.87, suggesting these association test P values were not inflated and that we effectively accounted for this family structure in our linear mixed models, which included relatedness and the first three principal components as covariates (Materials and Methods; supplementary fig. S5, Supplementary Material online).
Despite this family structure, a clear peak of association was apparent on H. misippus scaffold 84 (443,878–453,307 bp) and this strongly differentiated 10-kb region likely corresponds to the M locus for four reasons (supplementary fig. S4, Supplementary Material online; Gordon and Smith 1989). First, the 102 most significantly associated SNPs, including 10 of the 51 total SNPs in the M locus, were all located on scaffold 84. All of these 102 SNPs were homozygous in all inaria and immima individuals, but heterozygous in all misippus individuals, consistent with the results from Gordon and Smith’s experimental crosses. Second, we used polymerase chain reaction (PCR) and Sanger sequencing in a lab-reared cross to confirm that alleles in M segregated according to female wing color pattern (supplementary fig. S6, Supplementary Material online). Third, the M locus exhibits high FST between misippus and inaria or misippus and immima, but low FST between inaria and immima (supplementary fig. S7, Supplementary Material online). Fourth, the phylogenetic relationships between phased haplotypes in the M locus reflect female forewing phenotype, rather than the relationships between individuals, except for misippus individuals misippus 1 and misippus 52 (fig. 5b and supplementary fig. S5 and supplementary table S1, Supplementary Material online). Individuals misippus 1 and misippus 52 harbored recombinant haplotypes that were homozygous for the misippus haplotype along most of 10-kb region (fig. 5b).
Fig. 5.
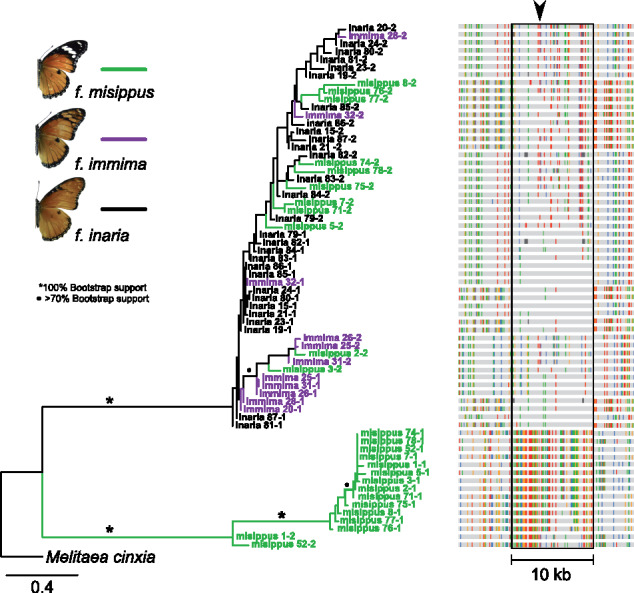
Relationships between phased Hypolimnas misippus M locus haplotypes. Relationships between phased haplotypes in the 10-kb region that is strongly differentiated between forms misippus and immima/inaria females (the M locus). Branches are colored according to the individual’s color pattern, and leaves indicate the individual and haplotype, for example, individual misippus 1 haplotypes 1 and 2 (supplementary table S1, Supplementary Material online). Corresponding haplotypes are shown to the right, the M locus is boxed. The most significantly associated SNP is indicated by an arrow. Only variant sites are shown and gray nucleotides match the majority consensus sequence.
Importantly, the H. misippus M locus is located near no known lepidopteran color patterning loci. Instead, M is located in an intergenic region 48-kb upstream of the nearest gene, Hmis009060 (Sox5/6), suggesting that the causative mutation(s) may reside in a cis-regulatory element of Sox5/6 or another nearby gene such as Hmis009058 (pink; supplementary table S5, Supplementary Material online).
Discussion
Identifying the genetic bases of adaptive phenotypes in a variety of organisms provides deep insight into the composition of developmental gene networks, how those networks evolve over time, and the conditions in which genes are reused during adaptation. Mimetic butterfly color pattern variation is controlled by a diverse but small set of large-effect Mendelian loci that have been repeatedly modified throughout the evolution of Lepidoptera. Here, we identified Mendelian switch loci controlling mimicry polymorphisms in P. clytia and H. misippus to assess the influence of phylogenetic relationship and mimicry mode on gene reuse during the evolution of diverse adaptive wing color patterns.
Unlike other swallowtails studied to date, we find that the P. clytia mimicry polymorphism is controlled by alternate alleles of a ∼500-kb genome region containing cortex. This region, and particularly the gene cortex, is well known to control color pattern variation across Lepidoptera. cortex expression levels, expression patterns, and splicing are correlated with melanic wing color patterns: Melanism in the peppered moth is caused by general upregulation of cortex in larval wing discs and melanic regions of Heliconius hindwings are prefigured by cortex expression patterns in larval wing discs (Nadeau et al. 2016; van’t Hof et al. 2016; Saenko et al. 2019). It therefore seems plausible that the switch between melanic/white scales and brown scales in P. clytia forms is controlled at least in part by expression pattern variation in cortex between the two forms (fig. 1a). This hypothesis is supported by the large amount of genetic differentiation between PclyML alleles, especially high FST and top switch GWA variants being located just upstream of cortex (fig 1d). Precisely how cortex, a cell division cycle regulator, affects color patterns is still unclear, but there is a strong link between the rate of scale development and final scale color; one hypothesis is that cortex alters color patterns by altering scale maturation rates (Gilbert et al. 1988; Koch et al. 2000; Nadeau et al. 2016; van’t Hof et al. 2016).
A second, nonmutually exclusive possibility is that the P. clytia mimicry polymorphism is controlled by multiple linked genes in the cortex region (i.e., a supergene), similar to the He. numata mimicry polymorphism. It is clear that the cortex region does contain multiple color patterning genes because, for example, genetic variation in the He. numata P2 inversion, which does not contain cortex, produces a large amount of wing pattern diversity in this species (fig. 2; Ferguson et al. 2010; Joron et al. 2011; Ito et al. 2016; Nadeau et al. 2016). A recent analysis also found at least five other genes in this region are significantly differentially expressed between He. numata morphs and may contribute to this polymorphism (Saenko et al. 2019). Supergene alleles may be maintained by a variety of mechanisms, including selection and inversions, that effectively repress recombination between them (reviewed by Schwander et al. [2014]). The three peaks of association, D′, and FST in PclyML could indicate strong selection for beneficial combinations of alleles in the different peaks or that an inversion distinguishes PclyML alleles (fig. 1d). The elevated FST, D′, and nucleotide diversity in the outermost PclyML peaks closely resemble the patterns expected and observed near the breakpoints of old polymorphic inversions, suggesting that PclyML alleles may also be distinguished by an inversion (fig. 1d and supplementary fig. S3, Supplementary Material online; Navarro et al. 2000; Andolfatto et al. 2001; Corbett-Detig and Hartl 2012; Guerrero et al. 2012). Interestingly, the cortex region in particular has gained at least three independent inversions in Heliconius (fig. 2; Joron et al. 2011; Jay et al. 2018; Edelman NB, Frandsen PB, Miyagi M, Clavijo B, Davey J, Dikow R, Garcia-Accinelli G, Van Belleghem S, Patterson N, Neafsey DE, Challis R, Kumar S, Moreira G, Salazar C, Chouteau M, Counterman B, Papa R, Blaxter M, Reed RD, Dasmahapatra K, Kronforst MR, Joron M, Jiggins CD, McMillan WO, Di Palma F, Blumberg AJ, Wakeley J, Jaffe D, Mallet J, unpublished data). However, we did not find sequencing reads or de novo scaffolds physically supporting the presence of an inversion in P. clytia (Materials and Methods). Furthermore, FST, D′, and nucleotide diversity drop to background levels just outside cortex and its flanking intergenic regions, suggesting that the three peak regions are roughly in linkage equilibrium. A proper P. clytia genome assembly is required to determine the presence of an inversion and if species outside Heliconius have converged on similar genome structures during the evolution of diverse mimicry polymorphisms. The strength of selection for Batesian mimicry depends on a number factors, including the relative abundance of the model (Charlesworth and Charlesworth 1975), and we know little about P. clytia natural history, so it is difficult to estimate the origins of or strength of selection for mimicry in this species.
Thus, the cortex region has been repeatedly involved in the evolution of diverse nonmimetic and mimetic melanic color patterns across Lepidoptera. In particular, P. clytia and He. numata converged on similar genetic architectures for their genetic switches despite diverging ∼120 Ma and evolving distinct color patterns that mediate Batesian or Müllerian mimicry. One commonality between all of this color pattern variation, beyond melanism, is that it is all sexually monomorphic. These observations may suggest that the cortex region cannot control sexually dimorphic color pattern variation. However, wing color patterns are shaped by a complex interplay between natural selection, sexual selection, and demographic processes that make it difficult to precisely identify the causes of gene reuse. Species that help separate some of these confounding factors, like P. clytia, will obviously be critical for identifying these causes.
In contrast to non-sex-limited mimicry polymorphism in P. clytia, we found that female-limited mimicry polymorphism in H. misippus is associated with a novel color patterning locus (fig. 4). Thus, the only known case of gene reuse in the evolution of female-limited mimicry occurs with dsx in P. polytes and its close relatives (Kunte et al. 2014; Nishikawa et al. 2015; Zhang et al. 2017; Iijima et al. 2018; D. H. Palmer and M. R. Kronforst, unpublished data). It is not clear if dsx reuse is caused by genetic convergence or parallelism—the inversions that distinguish dsx alleles in different species appear to be independently derived (Iijima et al. 2018; Palmer and Kronforst, unpublished data). In any case, these results and those from Heliconius reinforce the general conclusion that the probability of gene reuse is high within ∼10–15 My (Conte et al. 2012; Dasmahapatra et al. 2012; Pardo-Diaz et al. 2012; Van Belleghem et al. 2018). In contrast, we have no evidence that the probability of gene reuse is affected by the type of mimicry (Batesian/Müllerian, sex-limited or not).
Despite the seeming complexity of female-limited mimicry polymorphisms, each polymorphism that has been studied, in both Papilionidae and our work here in Nymphalidae, is controlled by a different locus. It is important to note that female-limited mimicry itself is diverse: in some species, such as P. polytes, females develop a male-like form in addition to one or more mimetic forms, whereas in others, like H. misippus or Papilio polyxenes, females never resemble males and develop one or more mimetic forms (Kunte 2009a). However, any locus controlling a female-limited mimicry polymorphism must carry multiple alleles that produce distinct adaptive color patterns and also limit their effects to one sex. All of the known genes are transcription factors that control diverse developmental programs (Kunte et al. 2014; Timmermans et al. 2014). Although we can at least envision how dsx, a master regulator of somatic cell sex differentiation, could limit its effects to one sex, it is still not clear how it switches between developmental programs that produce multiple distinct color patterns (Kunte et al. 2014; Nishikawa et al. 2015; Deshmukh et al. 2018; Iijima et al. 2018). Identification of the mimicry switch in H. misippus’ sister species H. bolina will be important for elucidating the time scales over which genetic convergence and parallelism occur in the evolution of female-limited mimicry polymorphisms. The diverse genetic bases of female-limited mimicry polymorphisms are therefore likely a result of the diversity of sex-limited mimicry and the natural selection, sexual selection, and demographic forces that drive their evolution (Kunte 2009b).
We suggest Sox5/6 and pink as reasonable candidates for the H. misippus mimicry switch. Sox5/6 is member of the SoxD transcription factor family and its Drosophila melanogaster ortholog Sox102F is required for proper sensory neuron development (Pevny and Lovell-Badge 1997; Stolt et al. 2006; Lefebvre 2010). SoxD contains the primary vertebrate sex-determination genes, whereas butterfly wing scales are modified sensory bristles (Galant et al. 1998). Other Sox genes have also been implicated in color pattern development in fish, frogs, and mice, including Sox10 (Dutton et al. 2001; Aoki et al. 2003; Harris et al. 2010). Similarly, mutations in pink, which control melanosome development in D. melanogaster, are associated with albinism in mice, fish, and Bombyx, consistent with the presence/absence of the melanic forewing patch in H. misippus (fig. 4 and supplementary table S4, Supplementary Material online; Zhang et al. 2003; Falcón-Pérez et al. 2007; Fujii et al. 2012; Daly et al. 2013). It will therefore be critical to experimentally test the functions of Sox5/6, pink, and other genes near the M locus in future work to understand how these adaptive phenotypes are produced.
Materials and Methods
Butterfly Care
Hypolimnas misippus and P. clytia pupae were obtained through LPS imports (www.lpsimports.com; last accessed August 30, 2019). Papilio clytia and H. misippus adults were kept in 2-m3 cages in a greenhouse with 65% humidity, constant 27 °C, light:dark cycle of 16 h:8 h and fed Birds Choice Butterfly Nectar. Hypolimnas misippus larvae were raised on Portulaca oleracea.
Sequencing, Assembly, and Annotation of the H. misippus Genome
DNA was isolated from thorax of a single H. misippus f. misippus female using phenol–chloroform extraction. We constructed Illumina paired-end (PE) libraries with insert sizes 250 and 500 bp using the KAPA Hyper Prep Kit (KR0961 – v1.14) and 1 μg genomic DNA. We constructed mate pair (MP) libraries with insert sizes of 2, 6, and 15 kb using the Nextera Mate Pair Library Prep kit (FC-132-1001) and 4 μg genomic DNA. We pooled libraries in a ratio of 50:18:10:17:4 and sequenced them 2× 150 bp on a single lane of Illumina HiSeq 4000. We performed additional 2× 100-bp HiSeq 4000 sequencing of the PE libraries. We trimmed low-quality regions and remaining adapters from raw PE reads using Trimmomatic v0.36 (Bolger et al. 2014) and from MP libraries using the platanus_internal_trim tool from Platanus v1.2.4 (Kajitani et al. 2014). Trimmed libraries were assembled using Platanus v1.2.4 (default settings) and the assembly polished using Redundans v0.13a (default settings; Pryszcz and Gabaldón 2016). We removed scaffolds <5 kb from the final assembly. Finally, we generated species-specific repeat libraries and masked repeats using RepeatScout 1.0.5 and RepeatMasker 4.0.8, respectively (Price et al. 2005; Smit et al. 2015).
We annotated the reference using MAKER v3.01.02 (Holt and Yandell 2011; Campbell et al. 2014). We used predicted transcripts from Junonia coenia (NCBI BioBroject PRJNA237755; Daniels et al. 2014), Vanessa tameamea (GCF_002938995.1), and V. cardui (Zhang et al. 2017) as evidence for transcription. We used protein sequences from the UniProt/SwissProt protein database (UniProt Consortium 2016), and RefSeq protein models for Danaus plexippus, Papilio xuthus, Bombyx mori, Vanessa tameamea, Pieris rapae, and D. melanogaster as evidence for protein-coding regions. We trained SNAP using this evidence, then used SNAP, Augustus v3.2 with Heliconius melpomene parameters, and GeneMark-ES 4 with MAKER to generate the final gene models (Korf 2004; Haussler et al. 2008; Ter-Hovhannisyan et al. 2008). We functionally annotated predicted proteins using BlastP against the UniProt/SwissProt database and combined that information using scripts included in MAKER.
We used data from the following assemblies for the annotation and BUSCO pipelines: Bany v1.2 (GenBank GCA_900239965.1), Dple v3 (GenBank GCA_000235995.2), Hera demophoon v1 (LepBase v4), Hmel v2.5 (LepBase v4), Jcoe v1.0 (LepBase v4), Mcin (GenBank GCA_000716385.1), and Vtam (RefSeq GCF_002938995.1).
Whole-Genome Resequencing
We isolated DNA from thorax of 34 H. misippus and 27 adult P. clytia butterflies using chloroform extractions (supplementary table S1, Supplementary Material online). Illumina PE libraries were constructed using a KAPA Hyper Prep Kit and sequenced using 2× 100-bp Illumina HiSeq 2500. Low-quality regions and adapters were trimmed from raw reads using Trimmomatic v0.36 before mapping.
Papilio clytia GWA
Trimmed P. clytia reads were mapped to the P. glaucus genome assembly (all scaffolds >5 kb from GenBank Accession GCA_000931545.1; Cong et al. 2015) using Stampy v 1.0.31 (Lunter and Goodson 2011). We assigned 1,252/2,796 (271 Mb/325 Mb, 83.4%) P. glaucus scaffolds to the chromosome-level P. xuthus reference genome assembly (Li et al. 2015) using a custom BLAT-based pipeline (available at https://github.com/nwvankuren/scripts/). PCR duplicates were marked with Picard 2.18 (http://broadinstitute.github.io/picard). We realigned reads around putative indels using the Genome Analysis ToolKit’s (GATK 3.8) RealignerTargetCreator and IndelRealigner, then called SNPs using the UnifiedGenotyper with default settings except the heterozygosity prior was set to 0.02 and minimum allowable base quality scores was set to 30 (McKenna et al. 2010). We removed genotypes with read depth below 3, sites with more than 4/27 missing genotypes, and sites with minor allele frequency below 0.1. We removed sites in high linkage disequilibrium using Plink’s indep-pairwise function (–indep-pairwise 10 2 0.2), then performed a principal components analysis (PCA) to identify covariates (Chang et al. 2015). Association tests were performed using a linear mixed model in gemma v0.94b and Wald’s test P values (Zhou and Stephens 2012). We included gemma’s genetic relatedness matrix and all PCs that explained >1.5% of the variance as covariates. False discovery rates (FDRs) were determined using Benjamini–Hochberg correction (Benjamini and Hochberg 1995). GWA results using all P. glaucus scaffolds, rather than just those assigned to chromosomes, are shown in supplementary figure S1, Supplementary Material online. We used gene models for P. glaucus and applied P. xuthus RefSeq gene models (GCF_000836235.1; supplementary table S2, Supplementary Material online) to the P. glaucus assembly using BlastP because RefSeq is the most comprehensive annotation set available. We calculated average D′ and FST in nonoverlapping 10-kb windows using VCFtools 0.1.13 (Danecek et al. 2011). D′ was calculated using all samples. FST was calculated between all dissimilis and all clytia. The average number of differences within and between dissimilis and clytia shown in supplementary figure S3, Supplementary Material online, was calculated using VCFtools 0.1.13.
PclyML Synteny across Lepidoptera
We identified syntenic regions between P. xuthus, P. glaucus, Heliconius melpomene v2.5, and the Bombyx mori chromosome-level assembly using BlastP (supplementary table S2, Supplementary Material online). Coordinates of color patterning loci and genes were taken from previous studies (Counterman et al. 2010; Ferguson et al. 2010; Joron et al. 2011; Ito et al. 2016; van’t Hof et al. 2016).
Papilio clytia Phylogenetic Analyses
We placed P. clytia within the broader context of its family by aligning whole-genome sequencing reads from other Papilionidae to the P. glaucus genome and calling and filtering SNPs following the above pipeline (supplementary table S3, Supplementary Material online). We used consensus sequences for each sample, including ambiguities, as input to maximum likelihood reconstructions using RAxML 8.2.11 under the GTRCAT model (Stamatakis 2014). Of the papilionids with publicly available sequencing data, P. clytia is most closely related to the female-limited mimic P. glaucus, consistent with single- to three-gene phylogenies (fig. 1 and supplementary table S3, Supplementary Material online; Tsao and Yeh 2008; Wu et al. 2015). We then phased P. clytia SNPs using SHAPEIT2 supported by read-backed phasing (Delaneau et al. 2013) and used these haplotypes and the P. glaucus consensus as input to RAxML reconstructions for smaller regions. We excluded P. clytia samples ch3 and ch4 because phasing was poor from low coverage. Phylogenetic support was determined using 1,000 bootstraps.
Hypolimnas misippus GWA
Hypolimnas misippus data were processed identically to the P. clytia data, except they were mapped to the new H. misippus assembly. We assigned H. misippus scaffolds to the M. cinxia chromosome-level assembly (Ahola et al. 2014) using our BLAT pipeline. This pipeline ordered 531/1,580 (87.0% of the total length) H. misippus scaffolds, including all scaffolds with sites associated with the mimicry polymorphism at FDR < 0.01. GWA results using all 1,580 scaffolds are shown in supplementary figure S4, Supplementary Material online. The GWA was performed similarly to the P. clytia GWA, including linkage disequilibrium-based pruning and principal components analysis. All PCs explaining >3% of the variance (1 and 2) were included as covariates in the GWA.
Hypolimnas misippus Genotyping
We performed PCR and Sanger sequencing of a ∼700-bp region containing two of the top 100 GWA SNPs in one of our families, a cross between a male (C507) and a f. inaria female (C302). This cross yielded 98 offspring, including 36 females (21 f. misippus, 8 f. immima, and 7 f. inaria). We extracted DNA from 1/2 of each female’s thorax using a phenol–chloroform protocol and used this in PCR with forward primer 5′-TCTTCTGGACGGCACAACTC-3′ and reverse primer 5′-CGTCAGCGGTTTA-GAATGCG-3′, 6.5 μl GoTaq Colorless Master Mix, 6.4 μl water, 0.3 μl 10 μmol each primer, and 0.5 μl DNA per reaction. Reactions were run on a Bio-Rad C1000 Touch Thermal Cycler using an initial denaturation at 95 °C for 1 min, followed by 35 cycles of 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 2 min. Products were purified with Applied Biosystems ExoSAP-IT and sequenced by the University of Chicago Comprehensive Cancer Center DNA Sequencing & Genotyping Facility on an Applied Biosystems 3730XL 96-capillary sequencer. Sequences were aligned to H. misippus scaffold84 using Geneious v9.1.3.
Hypolimnas misippus Genomic Analyses
We generated genome-wide phylogenies for H. misippus using sequencing data for seven additional nymphalid species following the protocols outlined for P. clytia, including phasing for focal scaffolds 84 and 25 (supplementary table S3, Supplementary Material online). We excluded hmis72 from this analysis due to high missingness (∼0.8), which interfered with phasing.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank A. Kopp and an anonymous reviewer for their helpful comments on the manuscript. We thank Márcio Z. Cardoso for help constructing sequencing libraries, Iris Mire for help with H. misippus genotyping, the University of Chicago greenhouse staff and Carlos Sahagun for help with butterfly husbandry, and the University of Chicago Functional Genomics Facility for their help generating sequencing data. This work was supported by funding from the National Science Foundation (grant IOS-1452648), the National Institutes of Health (grant GM108626), and the Pew Biomedical Scholars program (to M.R.K.).
Author Contributions
N.W.V. analyzed the data and wrote the manuscript; D.M. collected samples, performed sequencing, analyzed data, and wrote the manuscript; S.N. performed sequencing and data analysis; M.R.K. conceived the project and wrote the manuscript.
References
- Ahola V, Lehtonen R, Somervuo P, Salmela L, Koskinen P, Rastas P, Välimäki N, Paulin L, Kvist J, Wahlberg N. 2014. The Glanville fritillary genome retains an ancient karyotype and reveals selective chromosomal fusions in Lepidoptera. Nat Commun. 5:4737.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andolfatto P, DePaulis F, Navarro A. 2001. Inversion polymorphisms and nucleotide variability in Drosophila. Genet Res. 771:1–8. [DOI] [PubMed] [Google Scholar]
- Aoki Y, Saint-Germain N, Gyda M, Magner-Fink E, Lee YH, Credidio C, Saint-Jeannet JP. 2003. Sox10 regulates the development of neural crest-derived melanocytes in Xenopus. Dev Biol. 2591:19–33. [DOI] [PubMed] [Google Scholar]
- Bates HW. 1862. Contributions to an insect fauna of the Amazon valley. Lepidoptera: Heliconidae. Trans Linn Soc Lond. 23:495–566. [Google Scholar]
- Beldade P, Brakefield PM. 2002. The genetics and evo-devo of butterfly wing patterns. Nat Rev Genet. 36:442–452. [DOI] [PubMed] [Google Scholar]
- Beldade P, Saenko SV, Pul N, Long AD. 2009. A gene-based linkage map for Bicyclus anynana butterflies allows for a comprehensive analysis of synteny with the lepidopteran reference genome. PLoS Genet. 52:e1000366.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 57:289–300. [Google Scholar]
- Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 3015:2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MS, Holt C, Moore B, Yandell M. 2014. Genome annotation and curation using MAKER and MAKER-P. Curr Protoc Bioinformatics 48:4.11.1–4.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier L, Vattikuti S, Purcell SM, Lee JJ. 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 41:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth D, Charlesworth B. 1975. Theoretical genetics of Batesian mimicry I. Single-locus models. J Theor Biol. 552:283–303. [DOI] [PubMed] [Google Scholar]
- Clarke CA, Sheppard PM. 1972. The genetics of the mimetic butterfly Papilio polytes L. Philos Trans R Soc Lond B Biol Sci. 263855:431–458. [DOI] [PubMed] [Google Scholar]
- Clarke CA, Sheppard PM, Thornton IWB. 1968. The genetics of the mimetic butterfly Papilio memnon L. Philos Trans R Soc B 254:37–89. [DOI] [PubMed] [Google Scholar]
- Colosimo PF, Hosemann KE, Balabhadra S, Villarreal G, Dickson M, Grimwood J, Schmutz J, Myers RM, Schluter D, Kingsley DM. 2005. Widespread parallel evolution in sticklebacks by repeated fixation of ectodysplasin alleles. Science 3075717:1928–1933. [DOI] [PubMed] [Google Scholar]
- Cong Q, Borek D, Otwinowski Z, Grishin NV. 2015. Tiger swallowtail genome reveals mechanisms for speciation and caterpillar chemical defense. Cell Rep. 106:910–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte GL, Arnegard ME, Peichel CL, Schluter D. 2012. The probability of genetic parallelism and convergence in natural populations. Proc Biol Sci. 2791749:5039–5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett-Detig R, Hartl DL. 2012. Population genomics of inversion polymorphisms in Drosophila melanogaster. PLoS Genet. 812:e1003056.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counterman BA, Araujo-Perez F, Hines HM, Baxter SW, Morrison CM, Lindstrom DP, Papa R, Ferguson L, Joron M, Ffrench-Constant RH. 2010. Genomic hotspots for adaptation: the population genetics of Müllerian mimicry in Heliconius erato. PLoS Genet. 62:e1000796.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly CMS, Willer J, Gregg R, Gross JM. 2013. snow white, a zebrafish model of Hermansky–Pudlak Syndrome type 5. Genetics 1952:481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker R, Lunter G, Marth G, Sherry ST. 2011. The Variant Call Format and VCFtools. Bioinformatics 2715:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels EV, Murad R, Mortazavi A, Reed RD. 2014. Extensive transcriptional response associated with seasonal plasticity of butterfly wing patterns. Mol Ecol. 2324:6123–6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasmahapatra KK, Walters JR, Briscoe AD, Davey JW, Whibley A, Nadeau NJ, Zimin AV, Hughes DST, Ferguson LC, Martin SH, et al. 2012. Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature 487:94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KT, Cotton JA, Kirwan JD, Teeling EC, Rossiter SJ. 2012. Parallel signatures of sequence evolution among hearing genes in echolocating mammals: an emerging model of genetic convergence. Heredity (Edinb). 1085:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Howie B, Cox AJ, Zagury J-F, Marchini J. 2013. Haplotype estimation using sequencing reads. Am J Hum Genet. 934:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh R, Baral S, Gandhimathi A, Kuwalekar M, Kunte K. 2018. Mimicry in butterflies: co-option and a bag of magnificent developmental genetic tricks. Wiley Interdiscip Rev Dev Biol. 7:1–21. [DOI] [PubMed] [Google Scholar]
- Dutton KA, Pauliny A, Lopes SS, Elworthy S, Carney TJ, Rauch J, Geisler R, Haffter P, Kelsh RN. 2001. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 12821:4113–4125. [DOI] [PubMed] [Google Scholar]
- Elmer KR, Meyer A. 2011. Adaptation in the age of ecological genomics: insights from parallelism and convergence. Trends Ecol Evol (Amst). 266:298–306. [DOI] [PubMed] [Google Scholar]
- Endler JA. 1986. Natural selection in the wild. Princeton: Princeton University Press. p. 354. [Google Scholar]
- Falcón-Pérez JM, Romero-Calderón R, Brooks ES, Krantz DE, Dell’Angelica EC. 2007. The Drosophila pigmentation gene pink (p) encodes a homologue of human Hermansky–Pudlak Syndrome 5 (HPS5). Traffic 82:154–168. [DOI] [PubMed] [Google Scholar]
- Ferguson L, Lee SF, Chamberlain N, Nadeau N, Joron M, Baxter S, Wilkinson P, Papanicolaou A, Kumar S, Kee T-J, et al. 2010. Characterization of a hotspot for mimicry: assembly of a butterfly wing transcriptome to genomic sequence at the HmYb/Sb locus. Mol Ecol. 19:240–254. [DOI] [PubMed] [Google Scholar]
- Fisher RA. 1958. The genetical theory of natural selection. New York: Dover Publications, Inc. p. 291. [Google Scholar]
- Fujii T, Banno Y, Abe H, Katsuma S, Shimada T. 2012. A homolog of the human Hermansky–Pudlak syndrome-5 (HPS5) gene is responsible for the oa larval translucent mutants in the silkworm, Bombyx mori. Genetica 140(10-12):463–468. [DOI] [PubMed] [Google Scholar]
- Galant R, Skeath JB, Paddock S, Lewis DL, Carroll SB. 1998. Expression pattern of a butterfly achaete-scute homolog reveals the homology of butterfly wing scales and insect sensory bristles. Curr Biol. 814:807–813. [DOI] [PubMed] [Google Scholar]
- Gallant JR, Imhoff VE, Martin A, Savage WK, Chamberlain NL, Pote BL, Peterson C, Smith GE, Evans B, Reed RD, et al. 2014. Ancient homology underlies adaptive mimetic diversity across butterflies. Nat Commun. 51:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LE. 2003. Adaptive novelty through introgression in Heliconius wing patterns: evidence for a shared genetic “tool box” from synthetic hybrid zones and a theory of diversification. In: Boggs CL, Watt WB, Ehrlich PR, editors. Ecology and evolution taking flight: butterflies as model systems. Chicago (IL: ): University of Chicago Press. p. 281–318. [Google Scholar]
- Gilbert LE, Forrest HS, Schultz TD, Harvey DJ. 1988. Correlations of ultrastructure and pigmentation suggest how genes control development of wing scales of Heliconius butterflies. J Res Lepid. 26:141–160. [Google Scholar]
- Gompel N, Prud’homme B. 2009. The causes of repeated genetic evolution. Dev Biol. 3321:36–47. [DOI] [PubMed] [Google Scholar]
- Gordon IJ, Edmunds M, Edgar JA, Lawrence J, Smith D. 2010. Linkage disequilibrium and natural selection for mimicry in the Batesian mimic Hypolimnas misippus (L.) (Lepidoptera: Nymphalidae) in the Afrotropics. Biol J Linn Soc. 1001:180–194. [Google Scholar]
- Gordon IJ, Smith D. 1989. Genetics of the mimetic African butterfly Hypolimnas misippus: hindwing polymorphism. Heredity 633:409–425. [Google Scholar]
- Guerrero RF, Rousset F, Kirkpatrick M. 2012. Coalescent patterns for chromosomal inversions in divergent populations. Philos Trans R Soc B 3671587:430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanrahan SJ, Johnston JS. 2011. New genome size estimates of 134 species of arthropods. Chromosome Res. 196:809–823. [DOI] [PubMed] [Google Scholar]
- Harris ML, Baxter LL, Loftus SK, Pavan WJ. 2010. Sox proteins in melanocyte development and melanoma. Pigment Cell Melanoma Res. 234:496–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haussler D, Stanke M, Diekhans M, Baertsch R. 2008. Using native and syntenically mapped cDNA alignments to improve de novo gene finding. Bioinformatics 245:637–644. [DOI] [PubMed] [Google Scholar]
- Holt C, Yandell M. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima T, Kajitani R, Komata S, Lin C-P, Sota T, Itoh T, Fujiwara H. 2018. Parallel evolution of Batesian mimicry supergene in two Papilio butterflies, P. polytes and P. memnon. Sci Adv. 44:eaao5416.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Katsuma S, Kuwazaki S, Jouraku A, Fujimoto T, Sahara K, Yasukochi Y, Yamamoto K, Tabunoki H, Yokoyama T, et al. 2016. Mapping and recombination analysis of two moth colour mutations, Black moth and Wild wing spot, in the silkworm Bombyx mori. Heredity (Edinb). 1161:52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay P, Whibley A, Frézal L, Rodríguez de Cara MÁ, Nowell RW, Mallet J, Dasmahapatra KK, Joron M. 2018. Supergene evolution triggered by the introgression of a chromosomal inversion. Curr Biol. 2811:1839–1845. [DOI] [PubMed] [Google Scholar]
- Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J, Swofford R, Pirun M, Zody MC, White S, et al. 2012. The genomic basis of adaptive evolution in threespine sticklebacks. Nature 4847392:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joron M, Frezal L, Jones RT, Chamberlain NL, Lee SF, Haag CR, Whibley A, Becuwe M, Baxter SW, Ferguson L, et al. 2011. Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 4777363:203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joron M, Papa R, Beltrán M, Chamberlain N, Mavárez J, Baxter S, Abanto M, Bermingham E, Humphray SJ, Rogers J, et al. 2006. A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol. 410:e303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajitani R, Toshimoto K, Noguchi H, Toyoda A, Ogura Y, Okuno M, Yabana M, Harada M, Nagayasu E, Maruyama H, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 248:1384–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. 2002. BLAT—the BLAST-like alignment tool. Genome Res. 124:656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch PB, Lorenz U, Brakefield PM, Ffrench-Constant RH. 2000. Butterfly wing pattern mutants: developmental heterochrony and co-ordinately regulated phenotypes. Dev Genes Evol. 21011:536–544. [DOI] [PubMed] [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5:59.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KM, Wahlberg N, Neild AFE, Dasmahapatra KK, Mallet J, Jiggins CD. 2015. Multilocus species trees show the recent adaptive radiation of the mimetic Heliconius butterflies. Syst Biol. 643:505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronforst MR. 2008. Gene flow persists millions of years after speciation in Heliconius butterflies. BMC Evol Biol. 8:98.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronforst MR, Barsh GS, Kopp A, Mallet J, Monteiro A, Mullen SP, Protas M, Rosenblum EB, Schneider CJ, Hoekstra HE. 2012. Unraveling the thread of nature’s tapestry: the genetics of diversity and convergence in animal pigmentation. Pigment Cell Melanoma Res. 254:411–433. [DOI] [PubMed] [Google Scholar]
- Kronforst MR, Papa R. 2015. The functional basis of wing patterning in Heliconius butterflies: the molecules behind mimicry. Genetics 2001:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronforst MR, Young LG, Kapan DD, McNeely C, O’Neill RJ, Gilbert LE. 2006. Linkage of butterfly mate preference and wing color preference cue at the genomic location of wingless. Proc Natl Acad Sci U S A. 10317:6575–6580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunte K. 2009a. The diversity and evolution of Batesian mimicry in Papilio swallowtail butterflies. Evolution 6310:2707–2716. [DOI] [PubMed] [Google Scholar]
- Kunte K. 2009b. Female-limited mimetic polymorphism: a review of theories and a critique of sexual selection as balancing selection. Anim Behav. 785:1029–1036. [Google Scholar]
- Kunte K, Zhang W, Tenger-Trolander A, Palmer DH, Martin A, Reed RD, Mullen SP, Kronforst MR. 2014. doublesex is a mimicry supergene. Nature 5077491:229–232. [DOI] [PubMed] [Google Scholar]
- Lefebvre V. 2010. The SoxD transcription factors—Sox5, Sox6, and Sox13—are key cell fate modulators. Int J Biochem Cell Biol. 423:429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JJ, van der Burg KRL, Mazo-Vargas A, Reed RD. 2016. ChIP-Seq-annotated Heliconius erato genome highlights patterns of cis-regulatory evolution in Lepidoptera. Cell Rep. 1611:2855–2863. [DOI] [PubMed] [Google Scholar]
- Li X, Fan D, Zhang W, Liu G, Zhang L, Zhao L, Fang X, Chen L, Dong Y, Chen Y, et al. 2015. Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat Commun. 6:8212.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB. 2011. Convergence, adaptation, and constraint. Evolution 657:1827–1840. [DOI] [PubMed] [Google Scholar]
- Lunter G, Goodson M. 2011. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 216:936–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J. 1989. The genetics of warning colour in Peruvian hybrid zones of Heliconius erato and H. melpomene. Proc R Soc Lond B Biol Sci. 2361283:163–185. [Google Scholar]
- Mallet J, Barton N. 1989. Strong natural selection in a warning-color hybrid zone. Evolution 432:421–431. [DOI] [PubMed] [Google Scholar]
- Mallet J, Barton N, Lamas G, Santisteban J, Muedas M, Eeley H. 1990. Estimates of selection and gene flow from measures of cline width and linkage disequilibrium in Heliconius hybrid zones. Genetics 1244:921–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J, Joron M. 1999. Evolution of diversity in warning color and mimicry: polymorphisms, shifting balance, and speciation. Annu Rev Ecol Syst. 301:201–233. [Google Scholar]
- Manceau M, Domingues VS, Linnen CR, Rosenblum EB, Hoekstra HE. 2010. Convergence in pigmentation at multiple levels: mutations, genes and function. Philos Trans R Soc Lond B Biol Sci. 3651552:2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Orgogozo V. 2013. The loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution 675:1235–1250. [DOI] [PubMed] [Google Scholar]
- Martin A, Papa R, Nadeau NJ, Hill RI, Counterman BA, Halder G, Jiggins CD, Kronforst MR, Long AD, McMillan WO, et al. 2012. Diversification of complex butterfly wing patterns by repeated regulatory evolution of a Wnt ligand. Proc Natl Acad Sci U S A. 10931:12632–12637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin A, Reed RD. 2014. Wnt signaling underlies evolution and development of the butterfly wing pattern symmetry systems. Dev Biol. 3952:367–378. [DOI] [PubMed] [Google Scholar]
- Mazo-Vargas A, Concha C, Livraghi L, Massardo D, Wallbank RWR, Zhang L, Papador JD, Martinez-Najera D, Jiggins CD, Kronforst MR, et al. 2017. Macroevolutionary shifts of WntA function potentiate butterfly wing-pattern diversity. Proc Natl Acad Sci U S A. 11440:10701–10706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 209:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan O, Monteiro A, Kapan D. 2002. Development and evolution on the wing. Trends Ecol Evol. 173:125–133. [Google Scholar]
- Müller F. 1878. Über die Vortheile der Mimicry bei Schmetterlingen. Zool Anz. 1:54–55. [Google Scholar]
- Mundy N. 2005. A window on the genetics of evolution: mC1R and plumage colouration in birds. Proc Biol Sci. 2721573:1633–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau NJ, Pardo-Diaz C, Whibley A, Supple MA, Saenko SV, Wallbank RWR, Wu GC, Maroja L, Ferguson L, Hanly JJ, et al. 2016. The gene cortex controls mimicry and crypsis in butterflies and moths. Nature 5347605:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro A, Barbadilla A, Ruiz A. 2000. Effect of inversion polymorphism on the neutral nucleotide variability of linked chromosomal regions in Drosophila. Genetics 1552:685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H, Iijima T, Kajitani R, Yamaguchi J, Ando T, Suzuki Y, Sugano S, Fujiyama A, Kosugi S, Hirakawa H, et al. 2015. A genetic mechanism for female-limited Batesian mimicry in Papilio butterfly. Nat Genet. 474:405–409. [DOI] [PubMed] [Google Scholar]
- Pardo-Diaz C, Salazar C, Baxter SW, Merot C, Figueiredo-Ready W, Joron M, McMillan WO, Jiggins CD. 2012. Adaptive introgression across species boundaries in Heliconius butterflies. PLoS Genet. 86:e1002752.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevny LH, Lovell-Badge R. 1997. Sox genes find their feet. Curr Opin Genet Dev. 73:338–344. [DOI] [PubMed] [Google Scholar]
- Price AL, Jones NC, Pevzner PA. 2005. De novo identification of repeat families in large genomes. In: Proceedings of the 13th Annual International conference on Intelligent Systems for Molecular Biology (ISMB-05). Detroit (MI). https://bix.ucsd.edu/repeatscout/
- Pryszcz LP, Gabaldón T. 2016. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 4412:e113.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed RD, Papa R, Martin A, Hines HM, Counterman BA, Pardo-Diaz C, Jiggins CD, Chamberlain NL, Kronforst MR, Chen R, et al. 2011. optix drives the repeated convergent evolution of butterfly wing pattern mimicry. Science 3336046:1137–1141. [DOI] [PubMed] [Google Scholar]
- Ruxton GD, Sherratt TN, Speed MP. 2004. Avoiding attack: the evolutionary ecology of crypsis, warning signals and mimicry. Oxford: Oxford University Press. p. 260. [Google Scholar]
- Saenko SV, Chouteau M, Piron-Prunier F, Blugeon C, Joron M, Llaurens V. 2019. Unravelling the genes forming the wing pattern supergene in the polymorphic butterfly Heliconius numata. EvoDevo 10:16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwander T, Libbrecht R, Keller L. 2014. Supergenes and complex phenotypes. Curr Biol. 247:R288–R294. [DOI] [PubMed] [Google Scholar]
- Scriber JM, Hagen RH, Lederhouse RC. 1996. Genetics of mimicry in the tiger swallowtail butterflies, Papilio glaucus and P. canadensis (Lepidoptera: Papilionidae). Evolution 501:222–236. [DOI] [PubMed] [Google Scholar]
- Sheppard PM, Turner JRG, Brown KS, Benson WW, Singer MC. 1985. Genetics and the evolution of Müllerian mimicry in Heliconius butterflies. Philos Trans R Soc Lond B 3081137:433–610. [Google Scholar]
- Smit AFA, Hubley R, Green P. 2015. RepeatMasker Open-4.0. Available http://www.repeatmasker.org/faq.html#faq3; last accessed February 7, 2019.
- Smith D. 1976. Phenotypic diversity, mimicry and natural selection in the African butterfly Hypolimnas misippus L. (Lepidoptera: Nymphalidae). Biol J Linn Soc. 83:183–204. [Google Scholar]
- Smith DAS, Gordon JS. 1987. The genetics of the butterfly Hypolimnas misippus (L.): the classification of phenotypes and the inheritance of forms misippus and inaria. Heredity 593:467–475. [Google Scholar]
- Song Y, Endepols S, Klemann N, Richter D, Matuschka F-R, Shih C-H, Nachman MW, Kohn MH. 2011. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Curr Biol. 2115:1296–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 309:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern DL. 2013. The genetic causes of convergent evolution. Nat Rev Genet. 1411:751.. [DOI] [PubMed] [Google Scholar]
- Stolt CC, Schlierf A, Lommes P, Hillgärtner S, Werner T, Kosian T, Sock E, Kessaris N, Richardson WD, Lefebvre V, et al. 2006. SoxD proteins influence multiple stages of oligodendrocyte development and modulate SoxE Protein Function. Dev Cell 115:697–709. [DOI] [PubMed] [Google Scholar]
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. 2008. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Res. 1812:1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmermans M, Baxter SW, Clark R, Heckel DG, Vogel H, Collins S, Papanicolaou A, Fukova I, Joron M, Thompson MJ, et al. 2014. Comparative genomics of the mimicry switch in Papilio dardanus. Proc R Soc B 2811787:20140465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao W-C, Yeh W-B. 2008. DNA-based discrimination of subspecies of swallowtail butterflies (Lepidoptera: Papilioninae) from Taiwan. Zool Stud. 47:633–643. [Google Scholar]
- UniProt Consortium. 2016. UniProt: the universal protein knowledgebase. Nucleic Acids Res. 45:D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Belleghem SM, Vangestel C, De Wolf K, De Corte Z, Möst M, Rastas P, De Meester L, Hendrickx F. 2018. Evolution at two time frames: polymorphisms from an ancient singular divergence event fuel contemporary parallel evolution. PLoS Genet. 1411:e1007796.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van’t Hof AE, Campagne P, Rigden DJ, Yung CJ, Lingley J, Quail MA, Hall N, Darby AC, Saccheri IJ. 2016. The industrial melanism mutation in British peppered moths is a transposable element. Nature 534:102–105. [DOI] [PubMed] [Google Scholar]
- Wahlberg N, Wheat CW, Peña C. 2013. Timing and patterns in the taxonomic diversification of Lepidoptera (Butterflies and Moths). PLoS One 811:e80875.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace AR. 1865. On the phenomena of variation and geographical distribution as illustrated by the Papilionidae of the Malayan region. Trans Linn Soc. 251:1–71. [Google Scholar]
- Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Klioutchnikov G, Kriventseva EV, Zdobnov EM. 2018. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol. 353:543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerman EL, VanKuren NW, Massardo D, Tenger-Trolander A, Zhang W, Hill RI, Perry M, Bayala E, Barr K, Chamberlain NL, et al. 2018. Aristaless controls butterfly wing color variation used in mimicry and mate choice. Curr Biol. 2821:3469–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L-W, Yen S-H, Lees DC, Lu C-C, Yang P-S, Hsu Y-F. 2015. Phylogeny and historical biogeography of Asian Pterourus butterflies (Lepidoptera: Papilionidae): a case of intercontinental dispersal from North America to East Asia. PLoS One 1010:e0140933.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdobnov EM, Tegenfeldt F, Kuznetsov D, Waterhouse RM, Simão FA, Ioannidis P, Seppey M, Loetscher A, Kriventseva EV. 2017. OrthoDB v9.1: cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 45(D1):D744–D749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan S, Merlin C, Boore JL, Reppert SM. 2011. The monarch butterfly genome yields insights into long-distance migration. Cell 1475:1171–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Mazo-Vargas A, Reed RD. 2017. Single master regulatory gene coordinates the evolution and development of butterfly color and iridescence. Proc Natl Acad Sci U S A. 11440:10707–10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Zhao B, Li W, Oiso N, Novak EK, Rusiniak ME, Gautam R, Chintala S, O’Brien EP, Zhang Y, et al. 2003. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky–Pudlak syndrome types 5 and 6. Nat Genet. 332:145.. [DOI] [PubMed] [Google Scholar]
- Zhang W, Westerman E, Nitzany E, Palmer S, Kronforst MR. 2017. Tracing the origin and evolution of supergene mimicry in butterflies. Nat Commun. 81:1269.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Stephens M. 2012. Genome-wide efficient mixed-model analysis for association studies. Nat Genet. 447:821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.