

Host‐microbe interactions are highly dynamic in space and time. Recent approaches – combining experiments using neutral genetic tags with stochastic population dynamic models – allow precise quantification of biologically relevant parameters that govern the interaction between microbe and host cell populations. In this review, we present tools to study population dynamics in vivo using genetic tags, explain examples for their implementation, and discuss future applications.
Keywords: host–microbe interaction, in vivo models, population dynamics
Summary
Host–microbe interactions are highly dynamic in space and time, in particular in the case of infections. Pathogen population sizes, microbial phenotypes and the nature of the host responses often change dramatically over time. These features pose particular challenges when deciphering the underlying mechanisms of these interactions experimentally, as traditional microbiological and immunological methods mostly provide snapshots of population sizes or sparse time series. Recent approaches – combining experiments using neutral genetic tags with stochastic population dynamic models – allow more precise quantification of biologically relevant parameters that govern the interaction between microbe and host cell populations. This is accomplished by exploiting the patterns of change of tag composition in the microbe or host cell population under study. These models can be used to predict the effects of immunodeficiencies or therapies (e.g. antibiotic treatment) on populations and thereby generate hypotheses and refine experimental designs. In this review, we present tools to study population dynamics in vivo using genetic tags, explain examples for their implementation and briefly discuss future applications.
Abbreviations
- CI
Competitive index
- DC
dendritic cell
- E. coli
Escherichia coli
- H. influenzae
Haemophilus influenzae
- K. michiganensis
Klebsiella michiganensis
- mLN
mesenteric lymph nodes
- MMM
mechanistic mathematical model
- S. pneumoniae
Streptococcus pneumoniae
- S. Typhimurium
Salmonella Typhimurium
- STM
Signature tag mutagenesis
- V. cholerae
Vibrio cholerae
- WITS
wild‐type isogenic tagged strains
- Y. pseudotuberculosis
Yersinia pseudotuberculosis
SPATIOTEMPORAL DYNAMICS OF HOST–MICROBE INTERACTIONS
Our bodies are exposed to billions of microbes every day. Most of them are harmless and some even beneficial. A few pathogenic microbes however can invade multicellular organisms and cause life‐threatening infections. To block systemic proliferation of harmless, commensal microbes after accidental entry (and thereby enable a long‐lasting symbiotic relationship with commensals), and to fend off pathogenic microbes, multicellular organisms have evolved sophisticated systems of immune defences. 1
The interaction of the immune system with microbes is highly complex. Innate immune responses are triggered by tissue damage and the recognition of conserved molecular patterns associated with beneficial and pathogenic microbes. 2 Host responses to microbe exposure must therefore be highly context‐dependent and have evolved to minimize immune reactions to commensals while providing efficient defence against invading pathogens. Localization, duration and intensity of the microbial stimulus influence the outcome of the host response. 3 , 4 , 5 Adaptive immune responses can further modify these pathogen–host interactions at later phases of the infection or after a second encounter with the pathogen. The immune response affects the microbial population, which, in turn, feeds back onto the immune response. The final state of this dynamic system, in which the microbial population and immune responses mutually affect each other, is difficult to predict and can range from microbial clearance to persistent infection (Figure 1A).
Figure 1.
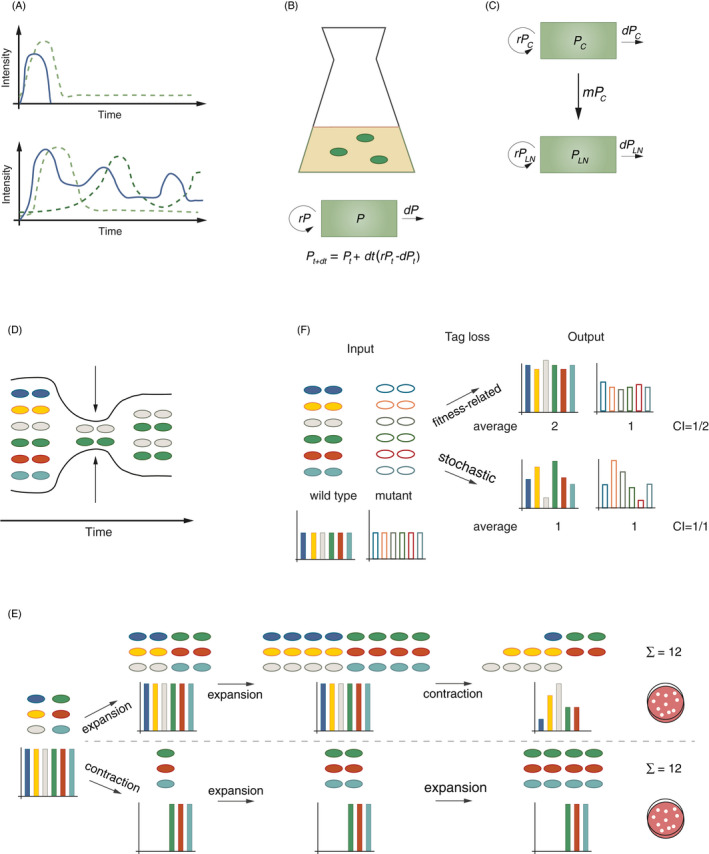
(A) Intensity of a microbial trigger (blue line) and the induced host response(s) (dashed green line(s)) vary over time. Examples for accidental spillover of a commensal into the host body (upper panel) and for prolonged colonization of a host by a pathogenic bacterium (lower panel). (B) Compartmental model describing a bacterial population P growing in an Erlenmeyer flask, defined by the parameters replication rate (r) and death rate (d). (C) Compartmental model describing the migration (m) of a bacterium from the caecum (PC) to the mLN (PLN) (similar to ref. 39). (D) Bottlenecks represent constraints (arrows) on a population leading to a reduction in population size. Loss of genetic diversity during bottleneck passage remains imprinted on the population even after re‐expansion. (E) Schematic drawing of two hypothetical population dynamic trajectories of one starting population. In both panels (upper and lower), the population undergoes two expansion events (doubling population size) and one contraction event (reducing population size by 50%). The final population size is equal, while the population structure (genetic diversity, different colours) differs. (F) Tracing of genetic diversity by neutral genetic tags can be used to differentiate stochastic from fitness‐related effects on populations. CI experiments allow the fitness assessment of bacterial mutants in comparison with wild‐type bacteria during a co‐infection. The CI of a mutant is calculated by dividing the number of mutant bacteria by the number of wild‐type bacteria after correcting for the abundance of both strains in the inoculum (CI > 1: mutant has a fitness advantage; CI < 1: mutant has a fitness disadvantage; CI = 1: no fitness difference). Using a pool of genetically tagged wild‐type and mutant bacteria allows the assessment of the nature of genetic tag loss: if the genetic tag loss is fitness related, the genetic diversity within one bacterial population (wild type or mutant, respectively) should remain similar (high evenness). Stochastic genetic tag loss, by contrast, leads to substantial variation in genetic diversity within one bacterial population (wild type or mutant, respectively; low evenness).
Deciphering these complex interactions is one of the major challenges in the field of host–microbe studies. 6 In systems with two or more interacting populations, the spatiotemporal dynamics make it challenging to dissect the underlying reciprocal interactions and to identify and quantify contributions of different components (Table 1).
Table 1.
Examples for experimental systems in the context of host–microbe interaction. Please note that the listed options are examples and represent incomplete lists
| System | Components (examples) | Environments (examples) | Interactions (examples) | Parameters (examples) |
|---|---|---|---|---|
| In vitro bacterial growth assay | Strain a, strain b | Culture medium | Competition, inhibition, cooperation | Replication rate, death rate (strain a, b) |
| In vitro T‐cell proliferation assay | T cells, Dendritic cells (DCs) | Culture medium | Activation, inhibition | Replication rate, death rate, activation (T cells,DCs) |
| Mouse associated with SPF microbiota | Microbiota, various immune cells | Intestinal lumen, mucosa, distant body sites | Tolerance, killing, symbiosis, activation, silencing | Replication rate, death rate, activation, migration (microbiota, immune cells) |
| Oral S. Typhimurium infection | Microbiota, S. Typhimurium, various immune cells | Intestinal lumen, mucosa, distant body sites | Competition, inhibition, cooperation, tolerance, killing, activation, silencing | Replication rate, death rate, activation, migration (microbiota, S. Typhimurium, immune cells) |
The traditional approach of knocking out single host genes to assess their contribution to microbial defence often neglects the complexity of host–microbial interactions. Many powerful immune effectors show surprisingly small effects when knocked out in models of pathogenic infection of a host. 7 We hypothesize that the redundancy of response pathways, their time‐dependent functions during pathogen defence and the pathogen's ability to adapt its phenotype (i.e. in case of surviving pathogen subpopulations) in response to the environment (i.e. the type of immune defence mechanism faced by a particular pathogen cell at a particular time‐point) contribute to the difficulty to connect phenotypes to single immune effectors. Additional challenges arise from pathogens expressing inhibitors of or resistance against particular immune effectors. Given the importance of constant immune surveillance, backup mechanisms that maintain functions even in the absence of a particular immune effector are crucial. Therefore, if the absence of a single immune effector in an infection set‐up does not produce a phenotype, it does not necessarily mean that this effector is irrelevant. Instead, it could also point to a specific relevance of the context of this effector for the analysed system, which favoured the evolution of redundant mechanisms. Nevertheless, the lack of single immune effector mechanisms can shift the dynamics of the microbe–host interaction. These can be detected by advanced methods as discussed below.
We thus postulate that, while traditional tools can help to dissect fundamental relations in defined conditions, more holistic approaches are needed to decipher the complex dynamics underlying host–microbe interactions and other complex disease scenarios. Omics and single‐cell analysis methods are certainly important tools to achieve this task, and together with increasing integration of different omic approaches, will continue to play a central role. 8 , 9 , 10 , 11 , 12 In this context, phenomenological mathematical models are used extensively to dissect patterns from big data sets. These models however do not explicitly account for molecular and cellular mechanisms. 13
Complementary to this, mechanistic mathematical models (MMMs) are powerful tools to dissect the functions of essential components within complex systems. These models capture the changing population sizes of microbes and host cells during an infection. Despite being often highly simplified, in a given system, MMMs can identify and quantify the relative contributions of its components to the dynamics of the system (Table 1). This facilitates mechanistic insights, reveals interaction networks, challenges assumptions or identifies gaps in our understanding. 10 , 13 , 14 , 15 For a detailed, easy‐to‐read overview on mathematical modelling of immune responses, we direct the reader to Handel et al., (2020). 13 MMM approaches have been successfully applied to quantify the dynamics of viral infections, 16 , 17 the population dynamics of T cells 17 , 18 and to understand the generation and maintenance of immune memory. 19 , 20
More recently, MMMs have been extended to describe experiments in which pathogens or host cells have been tagged genetically. 14 , 21 , 22 By accounting for the stochastic dynamics of tagged subpopulations, such mathematical models allow dissection of the intricate relations underlying host–microbe interactions, and therefore represent a promising extension of traditional methods (further described below). A major obstacle is the requirement of detailed knowledge of the analysed system to set up MMMs. Especially in complex systems, it can be challenging to identify relevant processes and parameters to include into the MMM. To verify hypotheses based on mathematical models, experimentation is required. 10 , 13 , 14 , 15 The crosstalk between modellers and wet laboratory scientists is therefore of critical importance. With this review, we aim to make approaches combining MMMs with tag‐based population dynamics for analysis of host–microbe relationships easily accessible to wet laboratory scientists.
COMPARTMENTAL MODELS TO DESCRIBE HOST–MICROBE INTERACTIONS
A large fraction of models applied to the field of host–microbe interaction are compartmental models. In these models, a group of individuals is classified into compartments that subdivide the population according to traits of interest, such as spatial location or differentiation state. 13 This can be as simple as one bacterial strain growing in liquid culture (Figure 1B). The replication rate r of this population and its death rate d define the population size at any given time, as described by the differential equation in Figure 1B. Figure 1C displays a compartmental model of a more complex situation, describing bacterial growth (r) in the caecal lumen (PC), migration (m) from that compartment to the mesenteric lymph nodes (mLN) and microbial growth (r) at that site (PLN).
MMMs are based on a priori information on the biological system. For example, the model in Figure 1C is built on knowing that the bacteria first colonize the intestine and from there spread to the mLN. The model parameters quantitatively characterize processes that shape the dynamics of the respective compartment (here: a population within a specific anatomical site), for example replication, death and migration. Computer simulations can be run with different values for these parameters. Comparing the output of simulations to empirical data then allows the estimation of parameter values that are most consistent with observations. In simulations, the validity of different experimental or mechanistic scenarios can be tested against experimental data. Once set up, MMMs enable the prediction of the behaviour of a system under altered conditions, for example the effect of antibiotic treatment on a bacterial population during infection. Thus, besides the quantification of biologically relevant parameters, modelling can provide a formal, quantitative, cost‐efficient and fast way of hypothesis generation and the design of optimal follow‐up experiments. Iterative improvement is achieved by feedback between wet laboratory experimentation and modellers to fully exploit the potential of MMMs. 13
POPULATION DYNAMICS OF NEUTRAL GENETIC TAGS AS A TOOL TO DECIPHER SPATIOTEMPORAL KINETICS OF HOST–MICROBE INTERACTION
Population dynamics describe the kinetics of changes in the structure of a population, for example regarding age, developmental stage, disease state, phenotypic manifestation or localization. In the following, we will focus on approaches analysing the distribution of neutral genetic tags within a population (‘population structure’). When using the term ‘population dynamics’, we refer to the dynamics of the distribution of a set of genetic neutral tags within a population. This serves as a readout for changes within that population.
To characterize a host–microbe interaction, compartmental MMMs allow quantification of parameters describing a population, for example its replication rate, death rate and pheno‐ or genotypic subpopulations of the microbe and/or the affected host cells. 14 , 22 This approach is especially fruitful for well‐characterized experimental models. Here, the analysis of population structures by genetic tagging enables precise descriptions of the underlying population dynamics by MMMs. These approaches can be applied to host cell and microbial populations. Below, we review the tools available in the different fields and give examples for their application.
STUDYING BACTERIAL POPULATION DYNAMICS IN VIVO
In studies of host–bacterial interactions, the success of microbial colonization is traditionally assessed by selective plating of homogenized organs on agars, which are permissive for the growth of the bacterium of interest. In some cases, qPCR or 16S‐sequencing have also been employed. However, all these methods share an important shortcoming: they only provide a snapshot of the current size of the microbial population, but lack information about its past changes.
A widely applied approach in population dynamics of host–bacterial interactions is the analysis of bottlenecks. A bottleneck describes internal and external factors (constraints, e.g. intraspecies competition, effects of immune responses) that restrict the capacity (e.g. population size) of a component (e.g. a pathogen population in a host organ) (Figure 1D, Table 1). 14 The total population size can ‘easily’ be measured. For bacterial infections, enumeration of colony‐forming units provides information about the size of a population at a given time in a certain organ. For immune cells, for example, flow cytometric methods yield cell numbers. These measures integrate events of migration, replication and cell death. Disentangling those parameters can help to identify immune defences or intervention strategies, even if these fail to completely control the infection. This is however highly challenging, especially in complex in vivo settings where pathogen populations can shrink and grow and host defences respond in a dynamic fashion (Figure 1A, bottom panel). Here, continuous sampling is often not possible, which renders the experimental analysis of population sizes a snapshot analysis integrating all events that affected the population until then. In multistep processes like infections, it becomes especially difficult to assign bottlenecks to certain events. By contrast, analysing changes in the diversity of neutral genetic tags in a population provides information about the population structure at earlier time‐points. Thereby, bottleneck‐inflicted changes in a population can be traced with the help of genetic tags.
Approaches employing neutral genetic tags for the studies of population dynamics assume that the manifestation of genetic diversity (i.e. the genetic tags) is fitness neutral in the studied condition. Given this, changes in the diversity of a population are typically a direct consequence of a reduction in population size. 14 This altered diversity will remain imprinted on the pathogen population, no matter how much it expands after passing a particular bottleneck (Figure 1E). In principle, this approach employs the evolutionary concept of genetic drift, which is particularly pronounced in events of dramatic population reduction or the founding of a new, spatially separated population by a low number of founder organisms. 23 Importantly, these changes reflect stochastic sampling events and are fitness independent (as the analysed organisms are isogenic and equally fit, see above). By contrast, an expansion of population size in this context usually stably maintains diversity. 14 Figure 1E illustrates a scenario in which the same starting population of cells with equal fitness in a given system undergoes one contraction and two expansion events in a different order, and how this affects population structure. Notably, the number of cells at the start as well as at the end of the experiment is equal in both scenarios. This example illustrates how two scenarios, indistinguishable by conventional methods of cell counting, derive from two different population dynamics and result in a very different population (genetic tag) structure. This principle can also be employed as a quality control for experiments analysing the competitive index (CI) of bacterial mutants, in which the number of mutant bacteria in an organ of interest is divided by the number of wild‐type bacteria to assess fitness differences. Using a batch of tagged isogenic mutant strains, and a batch of tagged wild‐type strains allows the detection of stochastic loss in the infection model and thereby enables the differentiation between fitness‐related and stochastic effects (Figure 1F). 24 , 25
POPULATION DYNAMICS APPROACHES TO STUDY HOST–PATHOGEN INTERACTIONS
Population dynamics approaches successfully defined and quantified parameters of bacterial infection in in vivo experimental set‐ups. Illustrative published examples using genetic tagging approaches are summarized in Table 2.
Table 2.
Studies analysing pathogen population dynamics in vivo. bp = base pairs
| Bacterium | Method | Number of unique identifiers | Analysed parameters | Main finding | References |
|---|---|---|---|---|---|
| Vibrio cholerae (V. cholerae) | STAMP – 30 bp barcodes, DNA sequencing | ~500 | Bottleneck size, genetic distance between subpopulations | Upstream migration of V. cholerae within the intestine | 26 |
| L. monocytogenes | STAMP – 30 bp barcodes, DNA sequencing | 200 | Bottleneck size, genetic distance between subpopulations | Identification of gall bladder as critical reservoir for host‐to‐host transmission; role for Gr1+ immune cells and microbiota in pathogen restriction | 27 |
| L. monocytogenes | 40 bp barcodes, qPCR/sequencing | 20 | Migration and replication rate of L. monocytogenes to/in various organs | Identification of small intestinal villi as reservoir for bacterial replication; identification of direct and indirect pathways of dissemination | 28 |
| Yersinia pseudotuberculosis (Y. pseudotuberculosis) | 40 bp barcodes, qPCR | 33 | Bottleneck size, genetic distance between subpopulations | Intestinal Y. pseudotuberculosis critical for dissemination; establishment of independent subpopulations | 29 |
| Haemophilus influenzae (H. influenzae) | Antibiotic resistances, differential plating | 2 | Bottleneck size | H. influenzae dissemination is independent of bacterial cooperation | 30 |
| H. influenzae | antibiotic resistances, differential plating | 2 | Bottleneck size | Bacteremia is caused by single dissemination events of H. influenzae | 31 |
| Streptococcus pneumoniae (S. pneumoniae) | OVA/AVO surface tags, qPCR | 2 | Migration, replication and death rate of nasal S. pneumoniae population | Small founding population required for stable nasal colonization by S. pneumoniae | 32 |
| Salmonella Typhimurium (S. Typhimurium) | Antibiotic resistances, differential plating | 2 | Bottleneck size | S. Typhimurium dissemination from the intestine is a rare event and independent of bacterial cooperation | 33 |
| S. Typhimurium | Serotypes, metabolic functions | 3, 2 | Bottleneck size | Independent host invasion by few Salmonella cells | 34 |
| S. Typhimurium | Fluorescent proteins | 2 | Clumping dynamics of S. Typhimurium in intestinal content | Protection by vaccination‐induced IgA is mediated via enchainment of dividing bacteria, resulting in clonal elimination of S. Typhimurium from intestinal lumen | 35 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 8 | Migration, replication and death rate of S. Typhimurium to/in various organs | Independent organ subpopulations during early infection, mixing via haematogenous spread at later stages | 21 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 7 | Bottleneck size during intestinal colonization, ‘evenness’ score | Identification of a Gr1+‐cell, inflammation dependent contraction of the intestinal S. Typhimurium population at 2 dpi | 36 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 8 | Genetic distance between organ subpopulations | Intraspecies competition for intestinal colonization impacts host‐to‐host transmission | 37 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 8 | Genetic distance between organ subpopulations | Antibiotic treatment efficiently targets fast‐dividing S. Typhimurium cells | 38 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 7 | Migration to and replication in mLN | mLN colonization during oral S. Typhimurium infection depends on immune cell migration from the intestinal mucosa to mLN. Restriction of replication of S. Typhimurium in the mLN depends on NADPH oxidase | 39 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 7 | Migration to and replication in mLN | Intestinal epithelial NAIP/NLRC4 restricts S. Typhimurium migration to the mLN and thereby protects from systemic dissemination | 40 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 7 | Migration to and replication in mLN | Slow‐growing intracellular S. Typhimurium persist after antibiotic treatment and can cause relapse | 41 |
| S. Typhimurium | 40 bp barcodes on plasmids, qPCR | 5 | Plasmid transfer rates, intestinal luminal replication | Systemic S. Typhimurium persisters can reseed to the gut lumen and promote spread of antibiotic resistance by plasmid transfer | 42 |
| S. Typhimurium | WITS – 40 bp barcodes, qPCR | 7 | Bottleneck size in intestinal lumen as quality control for STM screen | S. Typhimurium uses microbiota‐derived hydrogen as energy source in the intestinal lumen | 25 |
As described above, changes in genetic diversity help to deduce changes in population dynamics retrospectively. In an experimental set‐up, diversity – as a readout for population structure – should ideally be traceable. Traditional tools for tracing of genetic diversity are different antibiotic resistances, 30 , 33 serotypes 34 or the tagging with fluorescent proteins. 35 As these markers potentially interfere with infection kinetics and might skew population dynamics by introducing different fitness costs, new approaches were developed to label bacteria genetically with phenotypically neutral sequence tags (‘barcodes’). 14 , 21 Strains resulting from this approach are often referred to as wild‐type isogenic tagged strains (WITS) 21 (Figure 2A) and are phenotypically identical. Besides being fitness neutral, a large variety of these barcodes can be introduced into the population. This results in a higher number of distinguishable markers (i.e. genetic variation) and thereby increases the sensitivity for detecting large bottlenecks in the history of a given pathogen population (see refs. 26 and 27 for a detailed analysis of the relation between number of barcodes and bottleneck sensitivity). qPCR or sequence counting techniques by next‐generation sequencing allow precise and fast quantification of the relative distribution of these barcodes within the population. Absolute numbers of each barcoded bacterium are deduced by combining the relative barcode distributions with bacterial numbers obtained by conventional methods. These data serve as input for the MMMs. More recently, the usage of hundreds of barcodes in combination with high‐throughput DNA sequencing methods has helped to further increase the sensitivity of this approach. 14 , 25
Figure 2.
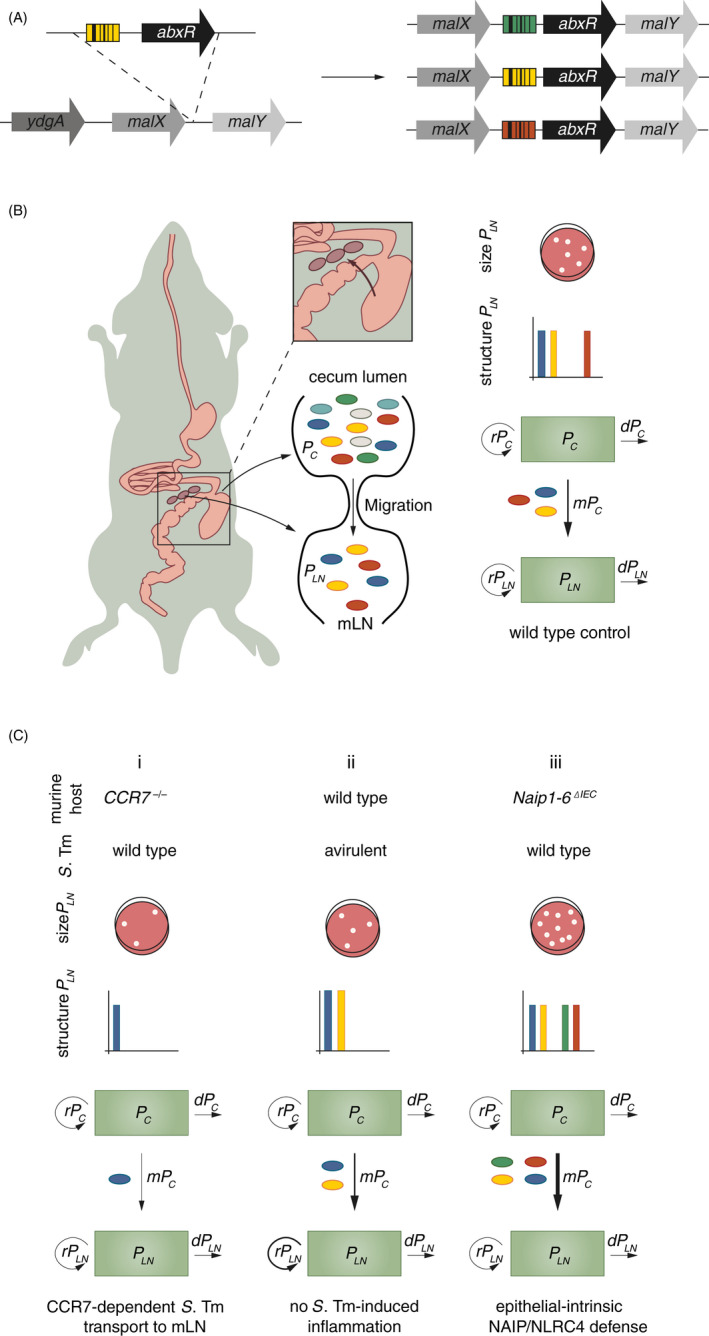
(A) Genetic tagging approach to obtain S. Typhimurium WITS. 21 A 40‐bp DNA sequence tag (coloured barcode) coupled to an antibiotic resistance cassette (abxR) is introduced into a neutral locus in the S. Typhimurium genome between malX and malY. (B) Schematic of S. Typhimurium migration from the caecal lumen to the mLN during oral infection (left panel). ~300 S. Typhimurium cells per day migrate from the caecal lumen to the mLN during oral infection (wild‐type mouse, right panel), contributing to the bacterial mLN population PLN as described by the compartmental model shown in Figure 1C. 39 Compared to the population in the caecum (~109 S. Typhimurium cells/g content) this number is small. The migration to the mLN thus represents a bottleneck, which only a small number of bacteria can pass. In conclusion, genetic diversity (different colours, see also graphs ‘structure PLN’) of the mLN population (PLN) is reduced in comparison to the caecal population (PC) (middle panel). Using the size and structure of PLN as input for the MMM enables calculation of m and r of PLN. (C) Migration of S. Typhimurium to the mLN (m) is reduced in the absence of CCR7 (scenario i, CCR7‐/‐ mouse). 39 Replication of S. Typhimurium within the mLN (rPLN) is controlled by S. Typhimurium‐induced inflammation (scenario ii, infection with an S. Typhimurium mutant that is unable to trigger early inflammation). 39 Migration of S. Typhimurium to the mLN (m) is increased in the absence of NAIP/NLRC4‐mediated expulsion of infected epithelial cells (scenario iii, Naip1‐6ΔIEC mouse). 40
Salmonella Typhimurium (S. Typhimurium) infections in mice have served as an important model for developing MMMs in the context of genetic tag dynamics, that is for describing the infection dynamics and identifying critical host barriers during infection. Here, we focus on an MMM describing S. Typhimurium migration from the intestinal lumen to the mLN during oral infection (quantified as migration events per day) (Box 1, Figure 2B, wild‐type mouse, ~300 bacterial cells/day migrate to the mLN 39 ). By combining population dynamics approaches with mouse knockout lines, S. Typhimurium migration was shown to heavily depend on CCR7‐mediated migration of immune cells from the lamina propria to the mLN (Figure 2C, scenario i: CCR7‐/‐ mouse). The application of this model to infections with mutant S. Typhimurium also revealed a role for inflammation in the restriction of bacterial replication in the mLN (Figure 2C, scenario ii). 39 Recently, we have extended the application of the above‐described model to identify the cell type responsible for NAIP/NLRC4‐mediated restriction of S. Typhimurium infection of the mLN. While epithelial NAIP/NLRC4 is highly relevant for the control of S. Typhimurium loads within the epithelium, 43 , 44 immune cell NAIP/NLRC4 can also be involved in bacterial restriction. 45 , 46 , 47 It remained unclear, whether S. Typhimurium loads in the mLN of NAIP/NLRC4‐deficient mice were increased due to an impaired antibacterial response at the epithelial level, or due to the inability of immune cells to restrict intracellular S. Typhimurium replication via NAIP/NLRC4. Assessing migration to the mLN and replication rates of S. Typhimurium within the mLN, we could exclude NAIP/NLRC4 mediated restriction of bacterial replication within immune cells in the mLN. Much rather, we identified NAIP/NLRC4 as a critical migration barrier for S. Typhimurium on their way to the mLN, protecting from the increased systemic spread. The combination of this modelling approach with cell type‐specific knockout mice revealed that NAIP/NLRC4 within the gut epithelium controls S. Typhimurium migration to systemic compartments (Figure 2C, scenario iii: Naip1‐6ΔIEC mouse), while immune cell NAIP/NLRC4 is dispensable for controlling bacterial loads during early infection. 40
Figure 3.
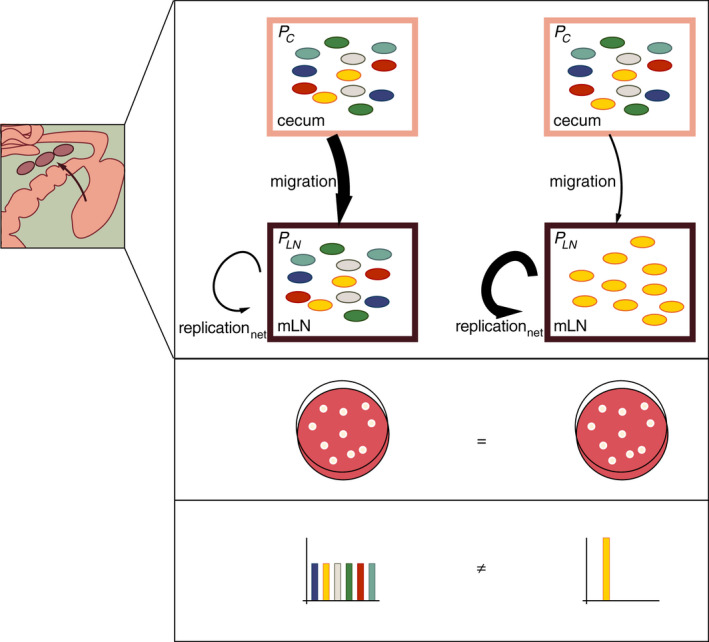
In the model described in Figure 1C, the size of the bacterial population in the mLN (PLN) depends on bacterial migration to the mLN (m), and bacterial replication (r) and death rate (d) within the mLN. For simplicity, we have here summarized replication and death rate as net replication rate (replicationnet = replication – death), as described previously. 39 PC (pink box) represents the bacterial population within the caecum, and PLN (purple box) the bacterial population within the mLN. The differentially coloured bacteria represent differentially genetically tagged bacteria (WITS).
The migration event in this model represents a bottleneck. As all bacteria are phenotypically identical and therefore have the same likelihood of migrating to the mLN (which depends on the size of the migration bottleneck), this bottleneck represents a random sampling event. A certain number of bacteria, carrying random tags, pass through this migration bottleneck and arrive in the mLN to found PLN. Consequently, information on this bottleneck remains imprinted on PLN. Stochastic loss of tags in PLN compared to PC enables the estimation of the size of the migration bottleneck and with this, the migration rate.
Under the premise that all bacteria are phenotypically identical, the same replicationnet rate is assumed for all bacteria in the mLN. Thus, the replicationnet rate does not affect the frequency of the tags, but their absolute abundance. In consequence, the number of bacteria per tag provides information on bacterial replicationnet in the mLN.
To illustrate the utility of disentangling the effect of migration to and replicationnet in the mLN on PLN, we present two extreme scenarios here: Left side: high bacterial migration to the mLN, low replicationnet in the mLN. Right side: low bacterial migration to the mLN, high replicationnet within the mLN. These two scenarios can potentially result in the same number of bacteria within the mLN at the time‐point of sampling, but derive from very different population dynamics (similarly to Figure 1E). This is reflected in the population structure in the two scenarios, which differ significantly with regard to the distribution of genetic tags (different colours). In the left scenario, many bacteria are able to migrate to the mLN and moderately replicate there. As the bottleneck of migration to the mLN is large, genetic diversity between PC and PLN is maintained and all tags are equally represented in PLN. By contrast, in the right scenario, where the migration rate is low, only few bacteria are able to migrate to the mLN, but can rapidly replicate at this site. This leads to stochastic loss of tags (i.e. genetic diversity) in PLN. Detectable tags are, however, present in high numbers.
In conclusion, the genetic tagging and modelling approach extends beyond the simple information about population size by providing retrospective information on the dynamics of PLN. In the given example, the above‐described approach allows pinpointing, for example, the effect of a certain treatment (e.g. immune cell activation) to/on bacterial migration or replicationnet and thereby hints towards mechanistic details affecting PLN.
Similarly, the elegant combination of genetic barcodes with mouse and bacterial knockout strains deciphered transmission routes of Listeria monocytogenes (L. monocytogenes) within and between hosts. 27 The comparison of the genetic (barcode) relatedness of bacterial subpopulations in spleen and liver revealed distinct initial founding events of organ subpopulations during early infection, and bacterial exchange between these organs via the bloodstream during later stages of the infection. Surprisingly, and in contrast to previous assumptions, this analysis revealed the gall bladder as the main reservoir for faecal host‐to‐host transmission. 27
Taken together, the above‐described examples illustrate the great potential of population dynamics and mathematical modelling in the identification of critical host barriers and prediction of intervention strategies during bacterial infections. This approach is especially powerful in combination with experimentation on host and bacterial knockout strains to pinpoint critical interactions for pathogen restriction during different disease stages and/or transmission at the molecular and cellular level.
POPULATION DYNAMICS APPROACHES TO STUDY HOST–MICROBIOTA INTERACTIONS
Similar to host–pathogen relationships, host–microbiota interactions are highly complex and dynamic. Deciphering the kinetics of microbiota colonization, response to perturbations and behaviour in disease conditions holds the key to the assessment of the role of microbiota members in these scenarios and to manipulation of microbiota contributions to disease development. Quantitative and causal microbiota analysis is facing three major challenges: i) the difficulty to set up a precise, overarching definition of ‘the’ microbiota; ii) the difficulty to culture and therefore manipulate a large fraction of typical microbiota members; and iii) the complexity of interactions within the microbiota community and with its host. In the past years, approaches were developed to tackle these challenges. Defined murine microbiota consortia enable studies of community interactions and contributions of specific strains to phenotypes. 48 , 49 , 50 Studies on culturable microbiota members such as Escherichia coli (E. coli) and Bacteroides thetaiotaomicron, 51 , 52 as well as longitudinal studies on microbiota composition, 53 , 54 have shed light on general concepts of intestinal colonization and interspecies competition. Despite these efforts, to date most studies modelling microbial community dynamics are based on in vitro or in silico systems, 55 , 56 general patterns extracted from sequencing data 57 , 58 or analysis of the microbiota as a metaorganism (see Table 3 for a summary of selected in vivo studies). 59
Table 3.
Studies analysing microbiota population dynamics in vivo
| Bacterium | Method | Number of unique identifiers | Analysed parameters | Main finding | References |
|---|---|---|---|---|---|
| E. coli, Klebsiella michiganensis (K. michiganensis) | Fluorescent proteins | 2 | Intestinal colonization | K. michiganensis provides colonization resistance against E. coli | 52 |
| E. coli | Fluorescent proteins | 2 | Niche competition in intestinal colonization | Horizontal gene transfer promotes niche adaptation | 60 |
| Microbiota | 16S sequencing, qPCR quantification | n.a. | Replication and death rate of microbiota exposed to bile salts | Diet‐evoked increased bile salt concentrations perturb microbiota composition and lead to decreased colonization resistance to S. Typhimurium | 59 |
| Several microbiota members (mouse) | qPCR (16S) | n.a. | Replication rate, interstrain interactions | Colonization dynamics in the murine intestine | 58 |
| Several microbiota members (fruitfly) | Plating | n.a. | Transition time through intestine, intra‐intestinal replication and death rate | Colonization dynamics in the intestine of Drosophila melanogaster | 57 |
bp = base pairs, n.a. = not applicable.
These studies are fundamental in gaining a basic understanding of the formation of community structures and can potentially be extended to perturbation scenarios and disease settings. Precise quantification and identification of causalities in vivo on a species/strain level and in specific intestinal niches however remains difficult. In the future, it will therefore be extremely valuable to implement precise quantitative set‐ups offered by population dynamics using genetic barcodes in the field of host–microbiota interactions. Experimental models of defined microbiota, in which the community members are known, 48 , 49 will be an essential tool in this context. Specifically, combining defined microbiota models with neutral genetic barcoding tools as described above for host–pathogen interaction analysis will be a powerful approach to decipher those intricate interactions.
STUDYING HOST RESPONSE DYNAMICS IN VIVO
During infection, both pathogen and host cell numbers vary over time, in parallel with changes in the infection environment that the respective components are exposed to. This, in turn, affects phenotypes of the interaction partners, which again affects the infection environment. Given the complexity of these systems, studying host response dynamics to pathogen infection in vivo is a challenging task. As outlined above, mathematical modelling can help to decipher these complex interactions. A set of well‐established tools exists to study host cell population dynamics in this context.
In the field of host cell biology, population dynamics approaches based on genetic tags have been applied especially to lineage tracing. Lineage tracing is used to study cell proliferation and differentiation during development 20 , 61 , 62 , 63 , 64 and in the adult, 65 under homeostatic and disease conditions. 19 , 66 , 67 Lineage tracing tools include microscopy‐based techniques such as the Brainbow/Confetti construct, in which stochastic Cre‐mediated recombination results in cells tagged with different fluorescent markers. 63 , 65 This tool is well suited to study clonality, proliferation and migration dynamics within a specific tissue(Figure 3A). 63 , 65 , 66 , 67 A higher variety in possible unique markers to increase sensitivity was a rationale for the development of neutral genetic barcoding strategies similarly to the WITS approach in bacteria. These barcodes are classically introduced via retroviral vectors ex vivo, 62 , 68 , 69 and the manipulated cells are subsequently transferred into hosts (Figure 3B). This approach is well suited for studies of circulating immune cells such as T cells. 69 , 70 To allow instead tracing of tissue‐resident cells in their native environment, in vivo barcoding tools have been developed recently. These approaches use the CRISPR/Cas9 system for the labelling of embryos, 64 or inducible CRISPR, 71 transposase 72 and Cre recombinase 73 systems for barcoding of cells in the adult organism in vivo. The latter approaches allow time‐, site‐ and/or cell‐specific inducible introduction of barcodes, which makes them exceptionally powerful tools to resolve complex dynamic processes in particular target tissues of the adult host. Illustrative studies applying different genetic tag‐based population dynamic approaches are summarized in Table 4.
Figure 3.
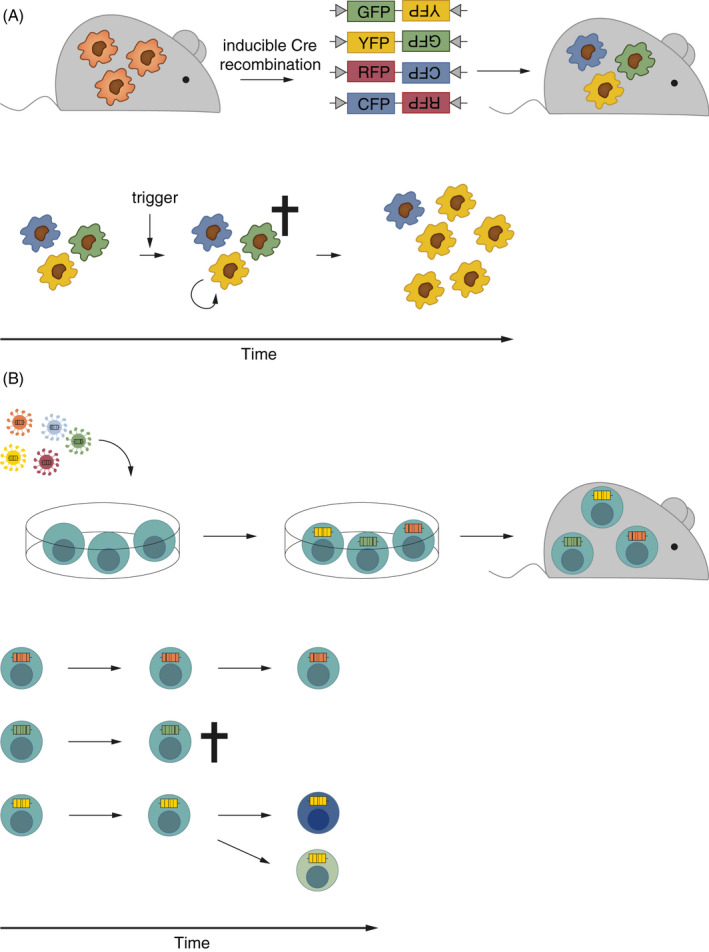
(A) Schematic of the Confetti approach for fluorescent labelling of immune cells. 63 Inducible Cre‐mediated recombination of the Confetti locus leads to differential labelling of cells with GFP, YFP, RFP or CFP (upper panel). This experimental approach can, for example, be used for analysis of local replication, death and migration rates of immune cells as observed upon exposure to a microbial trigger (lower panel). (B) Schematic of a neutral genetic barcoding approach of T cells. Genetic barcodes are introduced ex vivo by retroviruses. The barcoded cells can be transplanted into recipient hosts (upper panel) and employed to study cell survival, death, replication, differentiation and migration (lower panel). 69
Table 4.
Studies analysing host cell population dynamics in vivo
| Cell population | Method | Number of unique identifiers | Analysed parameters | Main finding | References |
|---|---|---|---|---|---|
| Neurons | Cre/LoxP‐mediated tagging by fluorescent proteins in vivo, ‘Brainbow’ reporter | ~90 | Cellular interactions in the brain | Visualization of neurons and cellular interactions in the mouse brain | 65 |
| Intestinal epithelial stem cells | Cre/LoxP‐mediated tagging by fluorescent proteins in vivo, ‘Confetti’ reporter | 4 | Cell differentiation, replication rate | Intestinal epithelial stem cells divide symmetrically | 63 |
| Microglia | Cre/LoxP‐mediated tagging by fluorescent proteins in vivo, ‘Confetti’ reporter | 4 | Replication and death rate, longevity | Stochastic, high self‐renewal capacity of microglia in steady‐state; selected clonal expansion in situ during disease conditions | 67 |
| DCs | Cre/LoxP‐mediated tagging by fluorescent proteins in vivo, ‘Confetti’ reporter | 4 | Replication rate, longevity, migration | Mucosal dcs highly depend on homeostatic replenishment from HSC‐derived precursors; clonal expansion in situ during disease conditions | 66 |
| HSCs | Cre/LoxP‐mediated tagging by fluorescent protein in vivo | 1 | Cell differentiation, replication rate | HSC‐derived progenitors self‐renew and mainly contribute to steady‐state hematopoiesis | 74 |
| CD4+ T cells | Congenic surface markers, flow cytometry | 2 | Replication rate, longevity | Naïve CD4+ T‐cell pool impacts CD4+ T memory cell lifetime and replication rate | 70 |
| CD8+ T cells | Congenic surface markers, flow cytometry | 8 | Cell differentiation, replication rate | Multiple precursors are required for induction of a robust effector and memory CD8+ T‐cell response | 19 |
| CD8+ T cells | Lentiviral barcoding ex vivo, microarray | ~102 | Differentiation and migration of antigen‐specific CD8+ T cells | Antigen‐specific CD8+ T cells in different organs derive from a common precursor pool | 69 |
| CD8+ T cells | Lentiviral barcoding ex vivo, DNA sequencing | ~103 | Cell differentiation, replication rate | T cells with identical T‐cell receptors display heterogenous expansion and differentiation patterns | 20 |
| CD8+ T cells | Lentiviral barcoding ex vivo, microarray | ~102 | Genetic distance between organ subpopulations | Both low‐ and high‐avidity T cells can differentiate into T effector and memory cells | 75 |
| HSCs | 33 bp tags, lentiviral barcoding ex vivo, DNA sequencing | ~102–103 | Cell differentiation | Distinct HSC differentiation patterns | 62 |
| HSCs | Lentiviral barcoding ex vivo, DNA sequencing | ~102 | Cell differentiation | Graded commitment model of hematopoiesis | 68 |
| CD8+ T cells | Potentially ~600 bp Cre/LoxP‐mediated random barcodes in vivo, DNA sequencing; here analysed in situ | Theoretically ~1012 | Not applicable | In situ simulation of barcode generation by optimized Cre/loxp construct | 73 |
| HSCs | Doxycycline‐inducible transposon barcoding in vivo, DNA sequencing | Theoretically unlimited | Cell differentiation | Long‐lived progenitors rather than hscs mainly contribute to steady‐state hematopoiesis | 72 |
| Germ cells (embryogenesis of zebrafish) | LINNAEUS – CRISPR/Cas9‐induced genetic scars in vivo, scRNAseq | Theoretically unlimited | Cell differentiation | Detailed cell differentiation map during zebrafish embryogenesis | 64 |
| HSCs | CARLIN – CRISPR/Cas9‐induced barcoding in vivo (sequential induction possible), DNA sequencing (transcriptomics) | ~104; Barcodes are transcribed | Cell differentiation and single‐cell gene expression | Unbiased lineage tracing and transcriptional profiling of hscs in vivo, expansion potential of foetal liver hscs is imprinted by the cellular niche | 71 |
The recently developed CARLIN mouse line 71 allows CRISPR‐mediated, large‐scale, cell type independent, inducible barcoding. Importantly, transcription of the CARLIN barcodes allows for a combined read‐out of barcodes and gene expression at the single‐cell level. This enables an unbiased analysis of cell populations, as well as correlation of clonality with gene expression. Specifically, the application of the CARLIN system revealed skewed proliferative responses of hematopoietic stem cells (HSCs) under stress conditions. Surprisingly, only a small fraction of HSCs contributed to hematopoiesis upon exposure to, for example, irradiation or chemotherapy. Differential gene expression analysis of inactive versus active HSCs revealed a potential regulator of HSC stress responses.
As exemplified above, the abstraction of complex scenarios by MMMs will help to gain more refined and holistic understanding of the processes underlying host responses to microbe exposure. This approach is especially valuable as complementation of traditional approaches including single knockout/transgenic mouse models. To extend current knowledge, it will be helpful to extend the application of the available tools to further cell types and models. The analysis of myeloid populations, for example, is highly complicated by the vast plasticity of this cell group, overlapping marker expression and longevity of specific subpopulations. 76 , 77 The above‐described Confetti construct has contributed to deciphering dynamics of regional turnover, contribution of bone marrow‐derived precursors, and response to insults of brain microglia 67 and mucosal dendritic cells (DCs). 66 The number of markers required for cell type identification however complicates the straightforward characterization of myeloid cell subsets involved in tissue homeostasis and immune responses. Recently developed inducible barcoding tools like the CARLIN mouse, 71 which allow read‐out of barcodes and high‐resolution cell type identification via single‐cell transcriptomics in parallel, have great potential in this context: i) the higher resolution due to a higher variety of barcodes allows more refined tracing of lineage dynamics during development and in the adult, and ii) the induction of barcode labelling before, during or after pathogen exposure or disease conditions can reveal contributions of different (myeloid) cell subsets to immune responses. Thus, in vivo labelling strategies in combination with MMMs hold promise for dissecting reciprocal, dynamic relationships underlying host–microbe interactions.
DISCUSSION AND OUTLOOK
Combining MMMs with experiments using genetic tagging techniques holds great potential for deciphering the intricate dynamics underlying host–microbe interactions. A couple of aspects however need to be considered for the application of this approach. One critical factor is the number of unique tags used in an experiment. This determines the sensitivity for bottleneck detection. The detection of wide bottlenecks requires the use of large tag libraries, 26 , 27 which can be achieved by genetic barcoding. 14 The calculation of population bottlenecks with the help of genetic barcodes relies on the assumption that the fitness of each barcoded cell is equal in the given conditions, as they are isogenic. This assumption however is often an over‐simplification, particularly when phenotypic heterogeneity occurs within bacterial subpopulations. Thereby, different cells carrying identical barcodes may express different virulence factors in a stochastic fashion and thereby interact differentially with host cells. Thus, also isogenic bacteria might have fitness differences. In scenarios with a low variety of barcodes (i.e. a high number of cells/barcode), population averages might equal this effect out. In scenarios in which a high variety of barcodes is used (i.e. a low number of cells/barcode), phenotypical diversity of isogenic organisms might however skew genetic barcode distribution. Besides that, especially when working with large barcode libraries that are randomly inserted, great care needs to be taken to exclude fitness effects by the insertion. Finally, while barcoding strategies allow a large‐scale analysis of populations, they require dissociation of the tissue. Thus, spatial resolution in these approaches is lost. This caveat could be overcome by combining barcoding strategies with fluorescence in situ hybridization of the barcodes. Finally, detailed a priori knowledge about the experimental system is required for setting up MMMs, which makes this method unsuitable for new and/or understudied experimental set‐ups.
Taken together, we see potential in extending the use of neutral barcodes to further fields, especially for i) microbiota members in the context of defined microbiota models and ii) a variety of host cells, for example the myeloid immune cell compartment to decipher the contribution of certain cell subsets to responses to microbial triggers. For the analysis of host cells, in vivo labelling approaches 64 , 71 , 72 , 73 prove especially useful, as they allow studying population dynamics in the absence of strong technically induced disturbances. These approaches hold promise to facilitate deciphering the complexity of host–microbe interaction and help to reveal contributions of different immune effectors.
Recent advances in sequencing technology provide a platform for cost‐efficient, high‐throughput read‐out of genetic barcodes. The introduction of barcodes that can be quantified by next‐generation sequencing techniques relieves constraints on the number of tags that can be used in an experiment, allowing the introduction of large barcode libraries and high‐resolution quantification of population parameters. By tagging of host cells and bacteria with distinct barcodes, sequencing techniques in combination with MMMs enable analysis of large‐scale population interaction dynamics within one host. Combined with single‐cell RNA sequencing, 71 this experimental set‐up promises to provide information about host–pathogen interaction at single‐cell level, for example in the context of intracellular infections.
GLOSSARY
| Phenomenological mathematical models | Quantitative mathematical models for extraction of patterns from large data sets (e.g. regression analysis) |
|---|---|
| Mechanistic mathematical models | Quantitative mathematical models to mechanistically describe the relationship between different components and parameters of a system |
| Population dynamics | Kinetics of changes of a population (e.g. composition, size), including parameters that describe these changes (e.g. replication, death). In this review, we focus on experimental approaches that use phenotypically neutral genetic tags to study pathogen and/or host cell populations |
| Compartmental models | Assigns populations to compartments. Members of the respective population can enter the compartment (e.g. by birth/replication, immigration) and exit from it (e.g. by death, emigration). Compartments can, for example, represent disease states, anatomical sites or stages in a pathogen’s life cycle |
| Bottleneck | Describes a reduction in population size due to environmental constraints. In a genetically diverse population, this will decrease its genetic diversity |
| Genetic diversity | Describes the number and frequency of genetic variants within a population |
| Genetic drift | Describes a selectively neutral change in allele frequencies in a population after a bottleneck event |
| Wild‐type isogenic tagged strains | Are strains that are genetically identical except for a short genetic tag that does not affect the phenotype and fitness of the strain |
| Metaorganism | A community of interdependent organisms often used in the context of complex microbial communities (and their hosts) |
ACKNOWLEDGEMENTS
We are grateful to Roland R. Regoes, Daniel Hoces, Erik Bakkeren and Roman Spörri for critical reading of the manuscript and helpful suggestions. We would like to thank the members of the Hardt laboratory for discussions and input. WDH was supported by the Swiss National Science Foundation (SNF, 310030_192567) and the NCCR Microbiomes. The authors declare no conflicts of interests.
Contributor Information
Annika Hausmann, Email: hannika@ethz.ch.
Wolf‐Dietrich Hardt, Email: wolf-dietrich.hardt@micro.biol.ethz.ch.
DATA AVAILABILITY STATEMENT
Not applicable.
References
- 1. Murphy K, Weaver C. Janeway’s Immunobiology. New York: Garland Science; 2016;925. [Google Scholar]
- 2. Medzhitov R, CharleSJ J. Innate immune recognition: mechanisms and pathways. Immunol Rev. 2000;173(1):89–97. [DOI] [PubMed] [Google Scholar]
- 3. Chen K, Geng S, Yuan R, Diao N, Upchurch Z, Li L. Super‐low dose endotoxin pre‐conditioning exacerbates sepsis mortality. EBioMedicine. 2015;2(4):324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pradeu T, Jaeger S, Vivier E. The speed of change: towards a discontinuity theory of immunity? Nat Rev Immunol. 2013;13(10):764–9. [DOI] [PubMed] [Google Scholar]
- 5. Tateda K, Matsumoto T, Miyazaki S, Yamaguchi K. Lipopolysaccharide‐induced lethality and cytokine production in aged mice. Infect Immun. 1996;64(3):769–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Germain RN. The art of the probable: system control in the adaptive immune system. Science 2001;293(5528):240–5. [DOI] [PubMed] [Google Scholar]
- 7. Abeler‐Dörner L, Laing AG, Lorenc A, Ushakov DS, Clare S, Speak AO, et al. High‐throughput phenotyping reveals expansive genetic and structural underpinnings of immune variation. Nat Immunol. 2020;21(1):86–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gardy JL, Lynn DJ, Brinkman FSL, Hancock REW. Enabling a systems biology approach to immunology: focus on innate immunity. Trends Immunol 2009;30(6):249–62. [DOI] [PubMed] [Google Scholar]
- 9. Gottschalk RA, Martins AJ, Sjoelund VH, Angermann BR, Lin B, Germain RN. Recent progress using systems biology approaches to better understand molecular mechanisms of immunity. Semin Immunol 2013;25(3):201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jansen JE, Gaffney EA, Wagg J, Coles MC. Combining mathematical models with experimentation to drive novel mechanistic insights into macrophage function. Front Immunol. 2019;10:1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pulendran B, Li S, Nakaya HI. Systems vaccinology. Immunity 2010;33(4):516–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zak DE, Tam VC, Aderem A. Systems‐level analysis of innate immunity. Annu Rev Immunol 2014;32(1):547–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Handel A, La Gruta NL, Thomas PG. Simulation modelling for immunologists. Nat Rev Immunol 2020;20(3):186–95. [DOI] [PubMed] [Google Scholar]
- 14. Abel S, Abel zur Wiesch P, Davis BM, Waldor MK. Analysis of Bottlenecks in experimental models of infection. PLOS Pathogens 2015;11(6):e1004823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gunawardena J. Models in biology: ‘accurate descriptions of our pathetic thinking’. BMC Biol. 2014;12(1):29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Davenport MP, Ribeiro RM, Zhang L, Wilson DP, Perelson AS. Understanding the mechanisms and limitations of immune control of HIV. Immunol Rev. 2007;216:164–75. [DOI] [PubMed] [Google Scholar]
- 17. Perelson AS. Modelling viral and immune system dynamics. Nat Rev Immunol. 2002;2(1):28–36. [DOI] [PubMed] [Google Scholar]
- 18. Borghans JAM, Boer RJD. Quantification of T‐cell dynamics: from telomeres to DNA labeling. Immunol Rev. 2007;216(1):35–47. [DOI] [PubMed] [Google Scholar]
- 19. Buchholz VR, Flossdorf M, Hensel I, Kretschmer L, Weissbrich B, Gräf P, et al. Disparate individual fates compose robust CD8+ T cell immunity. Science. 2013;340(6132):630–5. [DOI] [PubMed] [Google Scholar]
- 20. Gerlach C, Rohr JC, Perié L, van Rooij N, van Heijst JWJ, Velds A, et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science. 2013;340(6132):635–9. [DOI] [PubMed] [Google Scholar]
- 21. Grant AJ, Restif O, McKinley TJ, Sheppard M, Maskell DJ, Mastroeni P. Modelling within‐host spatiotemporal dynamics of invasive bacterial disease. PLoS Biol. 2008;6(4):e74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Höfer T, Barile M, Flossdorf M. Stem‐cell dynamics and lineage topology from in vivo fate mapping in the hematopoietic system. Curr Opin Biotechnol. 2016;39:150–6. [DOI] [PubMed] [Google Scholar]
- 23. Arnold J. Genetic Drift. In: Brenner S, Miller JH, eds. Encyclopedia of Genetics [Internet]. New York: Academic Press, 2001: 832–4. [Google Scholar]
- 24. Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. Simultaneous identification of bacterial virulence genes by negative selection. Science. 1995;269(5222):400–3. [DOI] [PubMed] [Google Scholar]
- 25. Nguyen BD, Cuenca VM, Hartl J, Gül E, Bauer R, Meile S, et al. Import of aspartate and malate by DcuABC Drives H2/Fumarate respiration to promote initial salmonella gut‐lumen colonization in mice. Cell Host Microbe 2020;27(6):922–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abel S, Abel zur Wiesch P, Chang H‐H, Davis BM, Lipsitch M, Waldor MK. Sequence tag‐based analysis of microbial population dynamics. Nat Methods. 2015;12(3):223–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang T, Abel S, Abel Zur Wiesch P, Sasabe J, Davis BM, Higgins DE, et al. Deciphering the landscape of host barriers to Listeria monocytogenes infection. Proc Natl Acad Sci USA. 2017;114(24):6334–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Melton‐Witt JA, Rafelski SM, Portnoy DA, Bakardjiev AI. Oral infection with signature‐tagged Listeria monocytogenes reveals organ‐specific growth and dissemination routes in guinea pigs. Infect Immun. 2012;80(2):720–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Barnes PD, Bergman MA, Mecsas J, Isberg RR. Yersinia pseudotuberculosis disseminates directly from a replicating bacterial pool in the intestine. J Exp Med. 2006;203(6):1591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Moxon ER, Murphy PA. Haemophilus influenzae bacteremia and meningitis resulting from survival of a single organism. Proc Natl Acad Sci USA. 1978;75(3):1534–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Margolis E, Levin BR. Within‐host evolution for the invasiveness of commensal bacteria: an experimental study of bacteremias resulting from Haemophilus influenzae nasal carriage. J Infect Dis. 2007;196(7):1068–75. [DOI] [PubMed] [Google Scholar]
- 32. Li Y, Thompson CM, Trzciński K, Lipsitch M. Within‐host selection is limited by an effective population of Streptococcus pneumoniae during nasopharyngeal colonization. Infect Immun. 2013;81(12):4534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Meynell GG. The Applicability of the Hypothesis of Independent Action to Fatal Infections in Mice given Salmonella typhimurium by Mouth. Microbiology 1957;16(2):396–404. [DOI] [PubMed] [Google Scholar]
- 34. Meynell GG, Stocker BAD. Some hypotheses on the aetiology of fatal infections in partially resistant hosts and their application to mice challenged with Salmonella paratyphi‐B or Salmonella typhimurium by intraperitoneal injection. Microbiology 1957;16(1):38–58. [DOI] [PubMed] [Google Scholar]
- 35. Moor K, Diard M, Sellin ME, Felmy B, Wotzka SY, Toska A, et al. High‐avidity IgA protects the intestine by enchaining growing bacteria. Nature 2017;544(7651):498–502. [DOI] [PubMed] [Google Scholar]
- 36. Maier L, Diard M, Sellin ME, Chouffane E‐S, Trautwein‐Weidner K, Periaswamy B, et al. Granulocytes impose a tight bottleneck upon the gut luminal pathogen population during Salmonella typhimurium colitis. PLoS Pathog. 2014;10(12):e1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lam LH, Monack DM. Intraspecies competition for niches in the distal gut dictate transmission during persistent salmonella infection. PLoS Pathog. 2014;10(12):e1004527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossi O, Dybowski R, Maskell DJ, Grant AJ, Restif O, Mastroeni P. Within‐host spatiotemporal dynamics of systemic Salmonella infection during and after antimicrobial treatment. J Antimicrob Chemother. 2017;72(12):3390–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kaiser P, Slack E, Grant AJ, Hardt W‐D, Regoes RR. Lymph node colonization dynamics after oral Salmonella Typhimurium infection in mice. PLoS Pathog. 2013;9(9):e1003532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hausmann A, Böck D, Geiser P, Berthold DL, Fattinger SA, Furter M, et al. Intestinal epithelial NAIP/NLRC4 restricts systemic dissemination of the adapted pathogen Salmonella Typhimurium due to site‐specific bacterial PAMP expression. Mucosal Immunol. 2020;13(3):530–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kaiser P, Regoes RR, Dolowschiak T, Wotzka SY, Lengefeld J, Slack E, et al. Cecum lymph node dendritic cells harbor slow‐growing bacteria phenotypically tolerant to antibiotic treatment. PLoS Biol 2014;12(2):e1001793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bakkeren E, Huisman JS, Fattinger SA, Hausmann A, Furter M, Egli A, et al. Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature 2019;573(7773):276–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rauch I, Deets KA, Ji DX, von Moltke J, Tenthorey JL, Lee AY, et al. NAIP‐NLRC4 Inflammasomes Coordinate Intestinal Epithelial Cell Expulsion with Eicosanoid and IL‐18 Release via Activation of Caspase‐1 and ‐8. Immunity 2017;46(4):649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sellin ME, Müller AA, Felmy B, Dolowschiak T, Diard M, Tardivel A, et al. Epithelium‐intrinsic NAIP/NLRC4 inflammasome drives infected enterocyte expulsion to restrict Salmonella replication in the intestinal mucosa. Cell Host Microbe. 2014;16(2):237–48. [DOI] [PubMed] [Google Scholar]
- 45. Carvalho FA, Nalbantoglu I, Aitken JD, Uchiyama R, Su Y, Doho GH, et al. Cytosolic flagellin receptor NLRC4 protects mice against mucosal and systemic challenges. Mucosal Immunol. 2012;5(3):288–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Franchi L, Amer A, Body‐Malapel M, Kanneganti T‐D, Özören N, Jagirdar R, et al. Cytosolic flagellin requires Ipaf for activation of caspase‐1 and interleukin 1β in salmonella‐infected macrophages. Nat Immunol. 2006;7(6):576–82. [DOI] [PubMed] [Google Scholar]
- 47. Lara‐Tejero M, Sutterwala FS, Ogura Y, Grant EP, Bertin J, Coyle AJ, et al. Role of the caspase‐1 inflammasome in Salmonella typhimurium pathogenesis. J Exp Med 2006;203(6):1407–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Brugiroux S, Beutler M, Pfann C, Garzetti D, Ruscheweyh H‐J, Ring D, et al. Genome‐guided design of a defined mouse microbiota that confers colonization resistance against Salmonella enterica serovar Typhimurium. Nat Microbiol. 2016;2:16215. [DOI] [PubMed] [Google Scholar]
- 49. Maier L, Vyas R, Cordova CD, Lindsay H, Schmidt TSB, Brugiroux S, et al. Microbiota‐derived hydrogen fuels Salmonella typhimurium invasion of the gut ecosystem. Cell Host Microbe. 2013;14(6):641–51. [DOI] [PubMed] [Google Scholar]
- 50. Stecher B, Chaffron S, Käppeli R, Hapfelmeier S, Freedrich S, Weber TC, et al. Like will to like: abundances of closely related species can predict susceptibility to intestinal colonization by pathogenic and commensal bacteria. PLoS Pathog 2010;6(1):e1000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cremer J, Segota I, Yang C, Arnoldini M, Sauls JT, Zhang Z, et al. Effect of flow and peristaltic mixing on bacterial growth in a gut‐like channel. Proc Natl Acad Sci USA 2016;113(41):11414–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Oliveira RA, Ng KM, Correia MB, Cabral V, Shi H, Sonnenburg JL, et al. Klebsiella michiganensis transmission enhances resistance to Enterobacteriaceae gut invasion by nutrition competition. Nature Microbiology. 2020;5(4):630–41. [DOI] [PubMed] [Google Scholar]
- 53. Furman O, Shenhav L, Sasson G, Kokou F, Honig H, Jacoby S, et al. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat Commun. 2020;11(1):1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ji BW, Sheth RU, Dixit PD, Tchourine K, Vitkup D. Macroecological dynamics of gut microbiota. Nat Microbiol. 2020;5(5):768–75. [DOI] [PubMed] [Google Scholar]
- 55. Arnoldini M, Cremer J, Hwa T. Bacterial growth, flow, and mixing shape human gut microbiota density and composition. Gut Microbes 2018;1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Labarthe S, Polizzi B, Phan T, Goudon T, Ribot M, Laroche B. A mathematical model to investigate the key drivers of the biogeography of the colon microbiota. J Theor Biol 2019;462(7):552–81. [DOI] [PubMed] [Google Scholar]
- 57. Inamine H, Ellner SP, Newell PD, Luo Y, Buchon N, Douglas AE. Spatiotemporally heterogeneous population dynamics of gut bacteria inferred from fecal time series data. mBio 2018;9(1):e01453‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marino S, Baxter NT, Huffnagle GB, Petrosino JF, Schloss PD. Mathematical modeling of primary succession of murine intestinal microbiota. PNAS. 2014;111(1):439–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wotzka SY, Kreuzer M, Maier L, Arnoldini M, Nguyen BD, Brachmann AO, et al. Escherichia coli limits Salmonella Typhimurium infections after diet shifts and fat‐mediated microbiota perturbation in mice. Nat Microbiol. 2019;4(12):2164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frazão N, Sousa A, Lässig M, Gordo I. Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proc Natl Acad Sci USA. 2019;116(36):17906–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Guiu J, Hannezo E, Yui S, Demharter S, Ulyanchenko S, Maimets M, et al. Tracing the origin of adult intestinal stem cells. Nature 2019;570(7759):107–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lu R, Neff NF, Quake SR, Weissman IL. Tracking single hematopoietic stem cells in vivo using high‐throughput sequencing in conjunction with viral genetic barcoding. Nat Biotechnol 2011;29(10):928–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Snippert HJ, van der Flier LG, Sato T, van Es JH, van den Born M, Kroon‐Veenboer C, et al. Intestinal crypt homeostasis results from neutral competition between symmetrically dividing Lgr5 stem cells. Cell 2010;143(1):134–44. [DOI] [PubMed] [Google Scholar]
- 64. Spanjaard B, Hu B, Mitic N, Olivares‐Chauvet P, Janjuha S, Ninov N, et al. Simultaneous lineage tracing and cell‐type identification using CRISPR‐Cas9‐induced genetic scars. Nat Biotechnol. 2018;36(5):469–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Livet J, Weissman TA, Kang H, Draft RW, Lu J, Bennis RA, et al. Transgenic strategies for combinatorial expression of fluorescent proteins in the nervous system. Nature 2007;450(7166):56–62. [DOI] [PubMed] [Google Scholar]
- 66. Cabeza‐Cabrerizo M, van Blijswijk J, Wienert S, Heim D, Jenkins RP, Chakravarty P, et al. Tissue clonality of dendritic cell subsets and emergency DCpoiesis revealed by multicolor fate mapping of DC progenitors. Sci Immunol. 2019;4(33):eaaw1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tay TL, Mai D, Dautzenberg J, Fernández‐Klett F, Lin G, Sagar XX, et al. A new fate mapping system reveals context‐dependent random or clonal expansion of microglia. Nat Neurosci. 2017;20(6):793–803. [DOI] [PubMed] [Google Scholar]
- 68. Naik SH, Perié L, Swart E, Gerlach C, van Rooij N, de Boer RJ, et al. Diverse and heritable lineage imprinting of early haematopoietic progenitors. Nature 2013;496(7444):229–32. [DOI] [PubMed] [Google Scholar]
- 69. Schepers K, Swart E, van Heijst JWJ, Gerlach C, Castrucci M, Sie D, et al. Dissecting T cell lineage relationships by cellular barcoding. J Exp Med. 2008;205(10):2309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gossel G, Hogan T, Cownden D, Seddon B, Yates AJ. Memory CD4 T cell subsets are kinetically heterogeneous and replenished from naive T cells at high levels. Elife. 2017;6:e23013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bowling S, Sritharan D, Osorio FG, Nguyen M, Cheung P, Rodriguez‐Fraticelli A, et al. An engineered CRISPR‐Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell. 2020;181(6):1410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sun J, Ramos A, Chapman B, Johnnidis JB, Le L, Ho Y‐J, et al. Clonal dynamics of native haematopoiesis. Nature. 2014;514(7522):322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Weber TS, Dukes M, Miles DC, Glaser SP, Naik SH, Duffy KR. Site‐specific recombinatorics: in situ cellular barcoding with the Cre Lox system. BMC Syst Biol. 2016;10(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Busch K, Klapproth K, Barile M, Flossdorf M, Holland‐Letz T, Schlenner SM, et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature. 2015;518(7540):542–6. [DOI] [PubMed] [Google Scholar]
- 75. Gerlach C, van Heijst JWJ, Swart E, Sie D, Armstrong N, Kerkhoven RM, et al. One naive T cell, multiple fates in CD8+ T cell differentiation. J Exp Med. 2010;207(6):1235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Guilliams M, Ginhoux F, Jakubzick C, Naik SH, Onai N, Schraml BU, et al. Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat Rev Immunol. 2014;14(8):571–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Joeris T, Müller‐Luda K, Agace WW, Mowat AM. Diversity and functions of intestinal mononuclear phagocytes. Mucosal Immunol. 2017;10(4):845–64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.