

Summary
Naturally occurring antibodies are abundant in human plasma, but their importance in the defence against bacterial pathogens is unclear. We studied the role of the most abundant of such antibodies, the antibody against terminal galactose‐α‐1,3‐galactose (anti‐αGal), in the protection against pneumococcal infections (Streptococcus pneumonia). All known pneumococcal capsular polysaccharides lack terminal galactose‐α‐1,3‐galactose, yet highly purified human anti‐αGal antibody of the IgG class reacted with 48 of 91 pneumococcal serotypes. Anti‐αGal was found to contain multiple antibody subsets that possess distinct specificities beyond their general reactivity with terminal galactose‐α‐1,3‐galactose. These subsets in concert targeted a wide range of microbial polysaccharides. We found that anti‐αGal constituted up to 40% of the total antibody reactivity to pneumococci in normal human plasma, that anti‐αGal drives phagocytosis of pneumococci by human neutrophils and that the anti‐αGal level was twofold lower in patients prone to pneumococcal infections compared with controls. Moreover, during a 48‐year period in Denmark, the 48 anti‐αGal‐reactive serotypes caused fewer invasive pneumococcal infections (n = 10 927) than the 43 non‐reactive serotypes (n = 18 107), supporting protection on the population level. Our findings explain the broad‐spectrum pathogen reactivity of anti‐αGal and support that these naturally occurring polyreactive antibodies contribute significantly to human protective immunity.
Keywords: antibodies, epitopes, flow cytometry, microbiota
The importance of naturally occurring antibodies in the human defence against bacterial pathogens is unclear. The work investigates anti‐αGal, the most abundant of such antibodies. Anti‐αGal binds common pathogens by broad‐spectrum polyreactivity and likely protects humans from invasive bacterial infections.
Abbreviations
- 95%CI
95% confidence interval
- anti‐αGal
Antibody against terminal galactose‐α‐1,3‐galactose
- CW
Cell wall
- EcO86
Escherichia coli O86
- EDTA
Ethylenediaminetetraacetic acid
- FI
Fluorescent intensity
- Galα3Gal
Galactose‐alpha‐1,3‐galactose
- HSA
Human serum albumin
- IgA
Immunoglobulin of class A
- IgG
Immunoglobulin of class G
- IgM
Immunoglobulin of class M
- MFI
Median fluorescence intensity
- MFIrel
Relative median fluorescence intensity
- nhIgG
Normal human IgG pool
- PBS
Phosphate‐buffered saline
- ROS
Reactive oxygen species
- TBS
Tris‐buffered saline
- TRIFMA
Time‐resolved immunofluorometric assay
INTRODUCTION
Human plasma is rich in various naturally occurring antibodies, 1 , 2 , 3 but their contribution to host protection against bacterial pathogens remains largely speculative. One of the most abundant of these antibodies, anti‐αGal, possesses reactivity for the xenocarbohydrate terminal galactose‐α‐1,3‐galactose (Galα3Gal). 4 The anti‐αGal antibody may have played a critical role in human evolution. Ancestors of the apes and Old World monkeys lost the α1,3 galactosyl transferase activity required for formation of terminal Galα3Gal because of inactivating mutations in the gene encoding the α1,3‐galactosyl transferase, 5 , 6 , 7 which abruptly ended immunological tolerance to the terminal Galα3Gal structure. This may have provided a strong selective advantage 8 by allowing for production of the anti‐αGal antibody capable of protecting against enveloped viruses originating from species that express Galα3Gal. 9 , 10 , 11
It is not clear what drives the production of the anti‐αGal antibody. The prevailing theory is that enteric bacteria present terminal Galα3Gal to our immune system and therefore continuously stimulate the production of the antibody. 12 Many enteric bacteria (~25% of strains) do indeed react with the anti‐αGal antibody; 13 however, few commensals carry a gene encoding an α1,3 galactosyl transferase, 14 which suggests that the antibody binds bacteria by polyreactivity. A polyreactive antibody has biologically relevant affinities for at least two distinct epitopes. 15 Indeed, polyreactivity of the anti‐αGal antibody is reported for various structures, 16 , 17 albeit not of microbial origin. Polyreactive antibodies could be important in human first‐line defence against invading pathogens, 15 , 18 , 19 but direct evidence is lacking.
Anti‐αGal antibody of all immunoglobulin classes is found in human plasma, and concentrations can amount to as much as 1% of total IgG2 and even larger percentages of total IgM. 20 The average levels are lower, approximating 10 mg/L for anti‐αGal of the IgG class in the plasma of healthy adults, 21 , 22 , 23 but levels vary more than 400‐fold between individuals. 23 Part of the variation relates to the presence or absence of the antigens of the ABO blood group system: Persons carrying the B‐antigen have lower concentrations of anti‐αGal antibody, 23 , 24 , 25 likely because their repertoire of anti‐αGal clones is restricted by the similarity of the B‐antigen to terminal Galα3Gal. 26
The involvement of the anti‐αGal antibody in combatting bacterial pathogens in humans is largely unexplored. In general, antibodies of the IgG class are particularly important in humans, and IgG deficiency predominantly manifests as recurrent airway infections. 27 , 28 Recurrent lower airway infections are especially devastating because of the resulting structural lung damage, respiratory failure and early death. The most serious lung pathogens are encapsulated bacteria with Streptococcus pneumoniae (pneumococci) being a dominating species. 27 , 29 In general, pneumococci are major pathogens in humans and a leading cause of pneumonia, 30 which is the most deadly communicable disease, causing 3·0 million deaths worldwide in 2016. 31
The pneumococcal capsule is a pivotal virulence factor that, among other functions, shields the subcapsular antigens from the immune system. 32 The capsules themselves are targets for human antibodies, which are crucial components in human protection against pneumococcal disease. 33 , 34 Specific antibodies in synergy with complement opsonize pneumococci for killing by phagocytes. However, pneumococcal capsule structures are exceptionally diverse, and 100 different serotypes have been identified. 35 As a consequence, broad‐spectrum protection against pneumococci requires many different antibody specificities. Anti‐αGal could be a candidate for providing broad‐spectrum pneumococcal reactivity, as the antibody reacts with most of approximately 200 different bacterial pathogens. 13 , 36 , 37 Such broad pathogen reactivity likely involves polyreactivity. We recently demonstrated that anti‐αGal reacts with pneumococci of serotype 9V, 38 which does not contain terminal Galα3Gal in its capsule polysaccharide. 39 Broad‐spectrum bacterial reactivity is not unique to the anti‐αGal antibody among the naturally occurring antibodies. As early as 1961, Springer and co‐workers reported this phenomenon for the antibodies to ABO antigens. 1 However, the mechanism behind the broad reactivity is an unsolved puzzle.
Another mystery is how anti‐αGal‐reactive bacterial pathogens can establish infection in humans who have natural occurrence of the anti‐αGal antibody. Nearly three decades ago, Hamadeh and co‐worker proposed that anti‐αGal binding to some bacteria paradoxically promotes infection by preventing a complement attack on the pathogens. 13 In contrast, we recently documented that anti‐αGal is fully capable of activating complement on both pneumococci of serotype 9V and Escherichia coli O86 (EcO86), 38 an often used model bacterium for studying anti‐αGal reactivity. 12 , 37 , 40 , 41 However, the capability of the antibody to opsonize reactive bacteria is undocumented, although opsonization is a fundamental feature of antibodies. Thus, the involvement of anti‐αGal in human defence against bacteria has not been established.
We therefore examined the basis for the broad pathogen reactivity of anti‐αGal and the implications for protective immunity in humans. We found that anti‐αGal displayed broad polyreactivity, which allows reactions with most serotypes of pneumococci. Our results support that this confers important protection to humans.
MATERIALS AND METHODS
Human samples
Blood samples were obtained from healthy blood donors (n = 60, mean age = 41 years) or patients (n = 375) with a history of increased burden of infection referred for diagnosis of immunodeficiency (Department of Clinical Immunology, Aarhus University Hospital, Denmark). Plasma samples from blood donors, which served as controls for anti‐αGal measurements, were collected from routine plasma portions with an equal representation of the ABO blood types. Plasma samples from patients were obtained between May 2011 and August 2016. Prior to measurements, patient samples were categorized into two main groups: I) patients with idiopathic infections (n = 289) and II) patient controls (n = 86) as outlined below.
When establishing the idiopathic infections group, we excluded persons with known likely secondary immunodeficiency (treated with steroids, rituximab, etc.). The group was further subdivided according to the predominant site of infection: I) lower airways (n = 118), II) upper airways (n = 53) and III) other sites (non‐airways) (n = 118). Due to compliance of information privacy, detailed data on the frequency and severity of infections for each patient were not available for this study. However, for each patient, the burden of infections was sufficiently serious to invoke a suspicion of immunodeficiency by clinicians experienced in the field. A patient control group, lung damage (n = 34), were candidates for lung transplantation due to severe structural lung damage. Their lung diseases were I) chronic obstructive pulmonary disease (n = 16), II) pulmonary fibrosis (n = 12), III) cystic fibrosis (n = 4), IV) lymphangioleiomyomatosis (n = 1) and V) unspecified disease (n = 1). Human hypogammaglobulinaemia serum was separated from whole blood by centrifugation. The blood was obtained from a person with hypogammaglobulinaemia (low IgG (<0·8 g/L), low IgA (<0·16 g/L) and low IgM (0·10 g/L)) but with normal activity of the complement system. 38 Blood cells were prepared from samples of 4 ml heparin‐stabilized freshly drawn blood from random blood donors. The cells were washed thrice by centrifugation (200g, 5 min) and twice in phosphate‐buffered saline (PBS, pH 7·4) with heparin, 17 IU/ml, followed by a final wash in RPMI‐1640 (Thermo Fisher Scientific) with human serum albumin (HSA; CSL Behring), 5 g/L). Then, 2 µl dihydrorhodamine‐123 (Sigma‐Aldrich, St. Louis, MO, USA) at 1 g/L in dimethyl sulfoxide was added to the final volume of 4 ml to enable detection of cellular production of ROS. Neutrophil concentrations were geomean 2700/µl (95% CI: 2000/µl, 3800/µl), n = 4, by Sysmex measurement.
Pig erythrocytes
Venous blood from anaesthetized pigs undergoing experimental surgery at Institute of Clinical Medicine, Aarhus University, Denmark, was drawn into ethylenediaminetetraacetic acid (EDTA)‐containing tubes. Fixation of the pig erythrocytes was done as follows: EDTA‐stabilized blood (3 ml) was washed twice by centrifugation (200g, 5 min) in PBS, resuspended in 45 ml PBS with 2% glucose (w/v) and 0·25% glutaraldehyde (v/v) and rotated gently end over end for 1 h at ambient temperature. Residual aldehyde groups were inactivated by ethanolamine (2·5 ml, 1 M, pH 8, rotation for 15 min). The erythrocytes were washed twice by centrifugation (200g, 5 min), first in Tris‐buffered saline (TBS) and then in PBS before resuspension in PBS with 0·1% (w/v) HSA and 0·1% sodium azide (w/v).
Microorganisms
For convenience, throughout this paper we use the abbreviations EcO86, Rough1, Rough2, Rough4, and C‐mutant for some bacterial strains as indicated. Strains from American Type Culture Collection, LGC standards, were Escherichia coli O86 strain ATCC 12701 (‘EcO86’), Streptococcus pneumoniae strain ATCC 12213 (‘Rough1’, unencapsulated, mutated from an original serotype 1) and Streptococcus pneumoniae strain ATCC 27336 (‘Rough2’, unencapsulated, mutated from an original serotype 2). Additional strains of Streptococcus pneumoniae were from the Kilian collection, the bacterial culture collection at Department of Biomedicine, Aarhus University, Denmark. These were 91 encapsulated strains of serotypes 1, 2, 3, 4, 5, 6A, 6B, 6C, 7F, 7A, 7B, 7C, 8, 9A, 9L, 9N, 9V, 10F, 10A, 10B, 10C, 11F, 11A, 11B, 11C, 11D, 12F, 12A, 12B, 13, 14, 15F, 15A, 15B, 15C, 16F, 16A, 17F, 17A, 18F, 18A, 18B, 18C, 19F, 19A, 19B, 19C, 20, 21, 22F, 22A, 23F, 23A, 23B, 24F, 24A, 24B, 25F, 25A, 27, 28F, 28A, 29, 31, 32F, 32A, 33F, 33A, 33B, 33C, 33D, 34, 35F, 35A, 35B, 35C, 36, 37, 38, 39, 40, 41F, 41A, 42, 43, 44, 45, 46, 47F, 47A and 48. Also, two additional unencapsulated strains were included: CSR SCS‐2 clone I (‘C‐mutant’) 42 , 43 and strain SK1443 (‘Rough4’, mutated from an original serotype 4). Bacteria were cultured in 50 ml Todd–Hewitt broth overnight at 35°C in an incubator (5% CO2). The bacteria were collected by centrifugation (2000g, 30 min) and resuspended in PBS containing 1% formaldehyde (v/v). The next day, the formaldehyde‐fixed bacteria were washed twice by centrifugation in 50 ml PBS before resuspension in 10 ml TBS to block residual aldehyde groups. The bacteria were centrifuged, and the supernatant was discarded before resuspension in TBS and storage at 4°C until use.
Affinity‐purified anti‐αGal antibody
Highly purified anti‐αGal antibody of the IgG class was prepared as previously described in detail. 37 Briefly, a pool of therapeutic‐grade normal human IgG (nhIgG) (Beriglobulin, CSL Behring, King of Prussia, PA) diluted to a protein concentration of 30 g/L was passed through a column containing Galα3Gal‐derivatized beads. The Galα3Gal‐derivatized beads had been prepared by coupling Galα1‐3Gal (G203, Dextra Laboratories, Reading, UK) to divinyl sulphone‐activated SK75 resin beads (HW75F, Tosoh Bioscience GmbH, Stuttgart, Germany) as previously described in detail. 38 After the passage of the nhIgG, the beads were washed extensively. Subsequently, bound antibodies were eluted with glycine buffer (0·1 M, pH 2·5) directly into pH‐neutralizing buffer. To remove antibodies reacting with the column matrix without Galα3Gal, the eluate was passed over a column containing uncoupled beads. The flow‐through was subjected to a second positive selection on Galα3Gal‐derivatized beads performed as described above to obtain the final preparation of anti‐αGal antibody.
αGalactosidase treatment of cells
Formaldehyde‐fixed pig erythrocytes or bacteria were diluted in PBS/HSA (pH adjusted to 6·5), and 5 µl cell suspension (with approximately 106 bacteria or 2 × 105 erythrocytes) was mixed with 5 µl PBS/HSA, pH 6·5, with or without 0·3 Units αGalactosidase (G8507, Sigma‐Aldrich). Reactions were allowed to proceed for 3 h at 25°C before the cells were washed and resuspended in TBS at their original concentration.
Fluorescent labelling of pneumococci
A 100 µl suspension of formaldehyde‐fixed pneumococci of serotype 9V (106/µl) was washed twice in PBS by centrifugation. The pneumococci were resuspended in 1·5 ml PBS with Cell Proliferation Dye eFluor 670 (Thermo Fisher Scientific) at 150 µM. After 30 min at 37°C, the reaction was stopped by five wash steps in 37°C warm TBS/HSA (centrifugations at 2000g, 10 min). An estimated 30% of pneumococci were lost during the preparation.
Quantification of anti‐αGal antibody by time‐resolved immunofluorometric assay (TRIFMA)
The protocol is described elsewhere. 23 Briefly, to detect binding of antibodies onto relevant antigens, the surfaces in microtitre wells were coated with Galα(1‐3)Gal‐HSA or HSA. Antigens were diluted in carbonate buffer (pH 9·4) to 1 mg/L and coated in wells of microtitre plates overnight at 4°C. Unoccupied binding sites were blocked with TBS/Tween/HSA (TBS with 0·1% Tween and HSA at 1 g/L). Each of the subsequent steps was separated by three wash cycles in TBS/Tween. A volume of 100 µl sample was loaded per well. Sample was plasma‐diluted (by default to 1%) in TBS/Tween/HSA with 10 mM EDTA. All samples were analysed in duplicates. Antibody binding took place overnight at 4°C. A total of 100 µl biotin‐labelled anti‐human IgG at 1 mg/L in TBS/Tween/HSA was added and reacted for 1 h at room temperature. A volume of 100 µl europium‐labelled streptavidin diluted 1:1000 in TBS/Tween with 25 µM EDTA was added and reacted for 1 h at room temperature. A volume of 200 µl enhancement solution was loaded per well, and after shaking of the plates, the signals were read as time‐resolved fluorescence. IgG binding to the carbohydrate moieties of glycoconjugate was determined indirectly as the signal from the glycoconjugate‐coated surface minus the signal from the HSA‐coated surface (background). To quantify antibody reactivity, logarithmically transformed standard curve data were used to approximate formulae (third‐degree polynomial fit) for the concentration as function of TRIFMA signal (Microsoft Excel 2016 spreadsheets, Microsoft Corporation, WA, USA). Samples yielding signals higher or lower than standard curve samples were re‐examined at appropriate dilutions. To enable quantification of anti‐αGal in mg/l, we related the signals of the standard sample to those obtained from serial dilution of purified anti‐αGal in known concentration.
Quantification of antibodies to pneumococcal capsular polysaccharides
A commercial bead‐based multiplex assay (xMAP® Pneumo 14: Pneumococcal Immunity Panel; Luminex Corporation, Austin, TX, USA) was used according to the manufacturer´s instructions for estimation of antibodies to 14 different pneumococcal polysaccharides. Read‐out was done on a Luminex 100 reader (Luminex Corporation).
Flow cytometry
Antibody reactivity with cells was determined by flow cytometry. 37 Target cells were diluted in PBS containing HSA at 1 mg/L (PBS/HSA) to reach acquisition event rates of approximately 100 s−1 for pig erythrocytes or 500 s−1 for bacteria. Ten µl cell suspension was mixed with 10 µl PBS/HSA solutions with and without primary antibodies. Unless otherwise stated, concentrations of the primary antibodies were as follows: purified anti‐αGal, 5 mg/L; IgG anti‐CD20 (control of unspecific reactivity, rituximab, monoclonal, chimeric mouse/human anti‐hCD20, Roche, Switzerland), 10 mg/L; and nhIgG (Subcuvia, Baxter, Deerfield, IL, USA), 500 mg/L. The primary incubation (60 min at 37°C) was terminated by washing the cells in 1 ml PBS/HSA by centrifugation (2000g, 10 min for bacteria or 200g, 10 min for erythrocytes). For experiments containing plasma or serum, an additional wash was performed. Cells were resuspended in 20 µl PBS/HSA with 1% (v/v) fluorescein isothiocyanate‐coupled polyclonal rabbit F(ab’)2 anti‐human IgG (F0315, DAKO). This incubation proceeded in the dark at room temperature for 30 min. A total of 200 µl sterile‐filtered (0·45 µm) flow buffer (BD FACSFlow, BD Biosciences, San Jose, CA, USA) was added, and samples were analysed by flow cytometry on a standard configuration, 2‐laser (488 nm/633 m) BD FACSCanto II Cell analyser (BD Biosciences). The instrument performance was monitored with BD CS&T beads throughout. Before each analysis run, settings were adjusted according to the following strategy: (a) forward‐scatter and side‐scatter acquisition thresholds were assigned to their minimum set points. (b) Sterile‐filtered flow buffer was then sampled at medium flow rate while adjusting the voltage of forward‐ and side‐scatter detectors to obtain an acquisition (background) event rate between 0 and 4 s−1. These settings were maintained for the subsequent analysis of samples. Signal height was acquired for forward‐scatter, side‐scatter and fluorescein isothiocyanate fluorescence emission (excitation 488 nm, emission bandpass filter 530/30 nm). Each sample was pre‐sampled (5 s) before data were recorded for 20 s. Instrument rinse was done between all samples. To control for bacterial spillover, samples of sterile‐filtered flow buffer were run between each bacterial strain (event rate below 1% of that from samples containing bacteria). Observed final event rates for bacteria ranged between 170 s−1 and 980 s−1. Event rates correlated with bacterial concentration based on optical density at 600 nm (P(H0) < 0·0001, R 2 = 0·59 by linear regression on linear scales of 22 pneumococcal serotypes). One event on average represented 10 bacterial cells, based on the regression analysis, the sample rate of the flow cytometer and the assumption that an OD600 of 1 corresponds to 1·8 × 109 bacterial cells/ml. Data were analysed using FlowJo software (version 9.7.6; FlowJo LLC, Ashland, OR, USA) and exported to Microsoft Excel spreadsheets. MFIrel was median fluorescence intensities for a cell incubated with primary antibodies relative to parallel incubation in PBS/HSA only. A positive antibody reaction was defined as a mean of two separately determined MFIrel at least two standard deviations above 1·10. This value (1·10) was the maximal observed MFIrel observed for the IgG anti‐CD20 (reactivity of an irrelevant control antibody) and each of the 91 pneumococcal serotypes. Quantification of antibody reactivity by flow cytometry was done for each strain by comparing sample MFI with the similar signals from a concurrent standard curve (twofold serial dilutions). Data handling was done as described for quantification by TRIFMA. Compounds used for inhibition were Galα3Gal (Dextra Laboratories), Glcα2Fru (Merrild Professional ApS, Denmark), melibiose (Galα6Glc, Sigma‐Aldrich) and pneumococcal capsular polysaccharides of serotypes 3, 6B, 7F, 9V, 10A, 12F, 15B, 17F, 18C, 19A, 22F and 33F (vaccine quality, LCG standards, Teddington, United Kingdom), and cell wall (CW) polysaccharide (Statens Serum Institut, Copenhagen, Denmark). Compounds were incubated with antibody solutions for 60 min at 37°C prior to incubation with cells. For tests of the inhibitory effect of Glcα2Fru and Galα3Gal on nhIgG reactivity with selected serotypes, nhIgG was titrated for each serotype to produce uninhibited MFIrel approximating 4. The nhIgG concentrations used were as follows: 6·3 mg/L for serotypes 3 and 12F, 10 mg/L for serotype 7F, 25 mg/L for serotypes 6B, 9V, 15B, 18C, 19A and 33F, and 50 mg/L for serotype 17F. Serotype 10A was omitted from these experiments because monotonic standard curves of sufficient range for quantification could not be established. For tests of the inhibitory effect of Glcα2Fru and Galα3Gal on plasma IgG anti‐7F and IgG anti‐9V, plasma was diluted in PBS/HSA to 0·3% and 1%, respectively. To assess pneumococcal reactivity of isolectin B4, biotinylated IB4 lectin from Bandeiraea simplicifolia (L2140, Sigma‐Aldrich), at 3·5 mg/L, in PBS/HSA, or merely PBS/HSA, was incubated with bacteria in volumes of 20 µl for 60 min at 37°C. Bacteria were washed in 1 ml PBS/HSA by centrifugation (2000g, 10 min). Next, 20 µl streptavidin–APC–eFluor 780 (eBioscience, 47‐4317) at 0·67 mg/L in PBS/HSA was allowed to react in the dark for 30 min. Finally, 200 µl filtered flow buffer was added prior to analysis.
Phagocytosis
General opsonization of pneumococci was done as follows: bacteria (with or without eFluor 670 label) at approximately 20 000/µl (based on measurement of optical density at a wavelength of 600 nm) in RPMI‐1640 with 10% human hypogammaglobulinaemia serum as complement source and additional HSA (5 g/L) were supplemented with purified anti‐αGal to 20 mg/L and mixed. Suspensions were incubated for 2 h at 37°C. Where indicated, the opsonization was conducted in the presence of inhibitors of complement function. These were single‐domain antibodies (nanobodies) against complement factors. Nanobodies were derived from heavy‐chain antibodies produced in llamas, selected by phage‐display, expressed in bacteria and purified as described previously. 44 Four nanobodies were used: (i) C1qNb75 binds the globular head of C1q and therefore blocks C1q docking to immunoglobulin Fc domains; 45 (ii) hC3Nb1 binds C3 and therefore blocks the action of the alternative pathway C3 convertase; 44 (iii) hC3Nb2 binds C3 and therefore blocks the action of C3 convertases from both classical pathway and alternative pathway; 46 and (iv) KRA152, used as control, binds an intracellular yeast kinase, that is represents irrelevant reactivity. C1qNb75 concentration was 3·5 mg/L, and hC3Nb1, hC3Nb2 and KRA152 were 12 mg/L. Opsonized bacteria in 30 µl volumes were gently mixed with 50 µl dihydrorhodamine‐123‐labelled, plasma‐depleted blood cells (the ratio of bacteria to neutrophils was approximately 4:1) in round‐bottomed polystyrene FACS tubes and placed in the dark at 37°C on an orbital shaker (500 rpm) for 1 h. Phagocytosis was terminated, and erythrocytes were lysed by resuspension in 2 ml, 0°C lysis buffer (155 mM ammonium chloride, 10 mM potassium hydrogen carbonate, 0·1 mM EDTA, pH 7·3). After 15 min at 0°C, cells were pelleted and washed once in 0°C flow buffer with 10 mM EDTA by centrifugation (4°C, 300g, 10 min). Cells were resuspended in 300 µl 0°C flow buffer with 3% formaldehyde (v/v) and left at 0°C for 30 min. Cell suspensions were analysed on a NovoCyte 3000 Flow Cytometer (ACEA Biosciences, CA, USA). Rhodamine‐123 was excited at 488 nm and emission detected behind a 530/30‐nm bandpass filter. eFluor 670 was excited at 640 nm and emission detected behind a 675/30‐nm bandpass filter. MFIrel in phagocytosis experiments was the MFI measured in an experiment with opsonization in serum and added anti‐αGal relative to the MFI in the same experiment but without added anti‐αGal. To inhibit phagocytosis, neutrophils were fed opsonized bacteria in the presence of 50 µM cytochalasin D (from Zygosporium mansonii, Sigma‐Aldrich, product C8273, solubilized in dimethyl sulfoxide), which inhibits the contractility of the cytoskeleton by inhibiting actin polymerization and ultimately phagocytosis.
For detection of bound IgG on bacteria used in the phagocytosis assay, identical primary incubations were done in parallel, except they contained 10 mM EDTA. EDTA was added to avoid masking of bound IgG by deposited C3 fragments. 38 Bacteria were washed twice and analysed by flow cytometry as described above but on a NovoCyte 3000 flow cytometer.
Confocal microscopy
The set‐up was as described for ‘Phagocytosis’ above with the following modifications. Prior to formaldehyde fixation, cells were resuspended in 500 µL, 0°C flow buffer with HSA at 1·0 g/L. Four similar incubations were pooled. Cells were pelleted by centrifugation (4°C, 300g, 10 min) and resuspended in flow buffer/HSA and 5% (v/v) BV421‐labelled anti‐hCD45 (BD Biosciences, product 563879). After 10 min in the dark at 0°C, the cells were washed in flow buffer by centrifugation (4°C, 300g, 10 min) and fixed in formaldehyde as above. Supernatants were discarded (300g, 10 min) and replaced by mounting medium (DAKO). A 10‐µl cell suspension was placed on slides with coverslip and sealed with nail polish. Confocal microscopy for blue, green, and red fluorescence was performed on a Zeiss LSM780, confocal microscope system (ZEISS, Germany).
Data on invasive pneumococcal disease in Denmark
The data were provided by Statens Serum Institut, Copenhagen, Denmark. This institute serotypes clinical pneumococcal isolates that are identified locally as the cause of invasive infections at departments of clinical microbiology from all parts of Denmark. Participation in this surveillance protocol is mandatory according to Danish legislation. Data from the period between 1966 and 2014 totalling 29 034 cases were included.
Statistics
Data were generally log10‐transformed before analysis. Comparison of plasma anti‐αGal levels between groups and calculation of confidence intervals was done based on bootstrap sampling distributions. 47 Otherwise, confidence intervals were calculated using the t‐distribution (continuous variables) or the binomial distribution (dichotomous variables). To compare the inhibition by homologous and heterologous capsules on the reactivity of anti‐αGal with each serotype, we calculated the probability of observing an inhibition level similar to that of the homologous polysaccharide with a heterologous polysaccharide, based on the observed data set for all heterologous polysaccharides and the particular cell. We made no corrections for multiple comparisons (to limit risk of type II errors), arguing that the single tests were only exploratory in an overall context. We only performed tests we found directly relevant for the context (to limit risk of type I errors). Data analyses were performed in GraphPad PRISM v. 6.07 (GraphPad Software, CA, USA), Estimation Statistics (www.estimationstats.com) and STATA 11 (StataCorp LP, TX, USA). The level of significance was chosen at 0·05.
Ethics
The anonymized blood samples were collected from voluntary blood donors in accordance with the Danish legislation. The studies on excess material from clinical tests of patients referred for laboratory investigations were approved by The Danish Data Protection Agency (reference number 1‐16‐02‐40‐12/2007‐58‐0010) and the Ethics Committee in Central Denmark Region (reference number 1‐10‐72‐127‐12).
Results
Low plasma levels of the anti‐αGal antibody in persons with recurrent lower airway infections
Solid‐phase immunoassay was used to measure the levels of the IgG class of anti‐αGal antibody in plasma from patients with increased burdens of idiopathic infections (n = 289, Figure S1) and in healthy individuals (n = 60). The latter group consisted of 15 persons of each ABO blood group type. The patients suffered from numerous infections to an extent prompting clinical assessment of immunodeficiency. Overall, anti‐αGal ranged between 98 µg/L and 240 mg/L (i.e. 2000‐fold) (Figure 1A). Patients were categorized according to their predominant type of infection into non‐airway infections (n = 118), upper airway infections (n = 53) or lower airway infections (n = 118). In healthy persons, the mean anti‐αGal level was 7·5 mg/L (95% confidence interval (95%CI): 5·4–10) (Figure 1A). In comparison with that of healthy persons, the mean anti‐αGal level was 64% (95%CI: 44%–95%) in patients with lower airway infections, 66% (95%CI: 41%–110%) in patients with upper airway infections and 140% (95%CI: 93%–215%) in patients with non‐airway infections (Figures 1A and S2). Idiopathic airway infections were thus associated with decreased levels of anti‐αGal.
FIGURE 1.
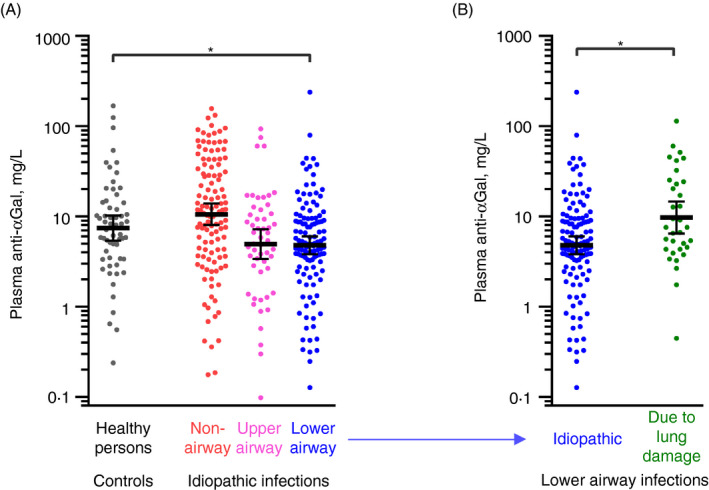
Low plasma levels of anti‐αGal antibody in humans with increased burden of lower airway infections. Anti‐αGal antibody was quantified in plasma samples by solid‐phase immunoassay. (A) Comparison of anti‐αGal levels in healthy persons (control, n = 60) and in patients suffering from recurrent infections (n = 289) to a degree prompting experienced medical specialist to suspect causative primary immunodeficiency. Patients were categorized according to their dominating site of infection: (i) non‐airways (n = 118), (ii) upper airways (n = 53) and (iii) lower airways (n = 118). The black bars show the geometric means with 95% confidence intervals. The control group was compared with each of the patient subgroups based on bootstrap sampling distributions (Figure S2), and significant group difference (P < 0·05) in the anti‐αGal levels is marked by a grey horizontal line and asterisk. (B) As in panel A but for patients with idiopathic lower‐airway infections (from panel A) compared with an additional control group comprised of patients with severe lung damage (lung transplantation candidates) and thus highly increased tendency to acquire lower‐airway infections (n = 34)
The level of anti‐αGal antibody in plasma varies with gender, age and ABO blood group, 23 so group differences in these parameters might bias the comparisons. In our cohort, we found that the anti‐αGal level was associated with both age and B‐antigen expression but not gender (Figure S3). A repeated comparison in a regression model with adjustments for age and B‐antigen expression showed that mean anti‐αGal level, relative to that of healthy individuals, was 48% (95%CI: 24%–95%) in patients with lower airway infections, 170% (95%CI: 40%–750%) in patients with upper airway infections and 130% (95%CI: 52%–320%) in patients with non‐airway infections. The corrections thus obliterated the tendency of lower anti‐αGal antibody level to associate with upper airway infections but strengthened the association between decreased anti‐αGal levels and idiopathic infections in the lower airways.
Decreased anti‐αGal level may dispose to lower airway infections. Alternatively, lower airway infections may lead to reduction in the anti‐αGal level. To clarify the causal direction, we examined a patient control group of lung transplantation candidates. This group suffered from various primary lung diseases with severe structural damage (n = 34), which disposes heavily to recurrent lower airway infections. The mean level of anti‐αGal in this group was 200% (95% CI: 130%–310%) of that in patients with idiopathic lower airway infections (Figures 1B and S4) and 130% (95%CI: 78%–220%) of that in healthy persons. Recurrent lower airway infections per see are thus unlikely to cause low anti‐αGal levels, which favours the interpretation that decreased anti‐αGal levels dispose to recurrent lower airway infections.
The anti‐αGal antibody reacts with most serotypes of pneumococci
We investigated anti‐αGal for reactivity with bacterial pathogens that cause lower airway infections. Clinical strains from the individual patients were not available. Instead, we studied reactivity to various serotypes of pneumococci, which is considered the leading cause of community‐acquired pneumonia in humans. 48 Pneumococcal capsules are comprised of capsular polysaccharide, which are long chains of repeating oligosaccharide units. Different oligosaccharide units give rise to 100 known distinct serotypes. Structures of most capsular polysaccharides are reported, and none of these contains terminal Galα3Gal. 48 As antibody source, we used a well‐characterized and highly purified preparation of human anti‐αGal antibody of the IgG class 37 , 38 and examined its reactivity to 91 serotypes of S. pneumoniae by flow cytometry. A positive reaction was defined as more binding signal than the maximal signal observed with a control antibody of irrelevant specificity (IgG anti‐CD20 antibody). Positive reactions were observed with 48 (53%) of the 91 serotypes (Figures 2A–C and S5). Of the 48 reactive serotypes, the capsular polysaccharides of 37 are characterized and known free of terminal Galα3Gal, which supports binding by polyreactivity to at least these serotypes. Several of the reactive serotypes displayed higher levels of reactivity than the positive control strain EcO86 (Figure 2C), which carries an unknown epitope for anti‐αGal. 12 Reactivity was also observed with four unencapsulated pneumococcal strains: Rough1, Rough2, Rough4 and C‐mutant. The C‐mutant lacks a genuine capsule but possesses an unusually thick capsule‐like layer of the cell wall (CW) polysaccharide, 32 which is also free of terminal Galα3Gal. 49 Thus, the reactivity with the C‐mutant strain strongly indicated polyreactivity for the CW polysaccharide. CW polysaccharide is produced by all pneumococci (and some other streptococci), 43 but the capsules of encapsulated strains shield the CW polysaccharide and other subcapsular antigens from reactive antibodies. 32 , 50 In accordance, the unencapsulated strains Rough1, Rough2 and Rough4 all reacted, whereas their capsulated progenitors (serotypes 1, 2 and 4) displayed poor (serotype 2) or no reactivity (serotypes 1 and 4) (Figure 2C).
FIGURE 2.
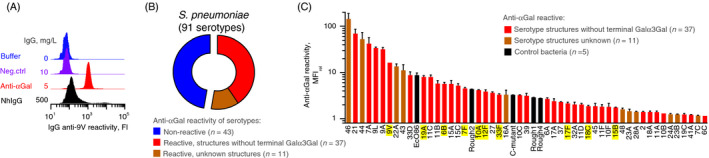
The anti‐αGal antibody binds most pneumococcal serotypes. Flow cytometry analyses showing antibody reactivity with various bacteria. (A) Example plot showing antibody reactivity with serotype 9V pneumococci after incubation with buffer only, negative control IgG (IgG anti‐CD20, i.e. irrelevant specificity) at 10 mg/L, purified human anti‐αGal at 5 mg/L or normal human IgG (nhIgG) at 500 mg/L. NhIgG is expected to contain antibodies to pneumococci and served as positive control. IgG on the pneumococci was assayed with fluorescently labelled rabbit F(ab’)2 anti‐human IgG. ‘FI’: Fluorescence intensity. (B) Pie‐chart summarizing reactivity of anti‐αGal (5 mg/L) with 91 serotypes of S. pneumoniae. (C) Level of reactivity with the reactive pneumococcal serotypes and five control bacterial strains. Control strains were E. coli O86 (EcO86) and four unencapsulated pneumococcal strains: C‐mutant (covered by a thick layer of cell wall (CW) polysaccharide), Rough1, Rough2 and Rough4. The columns are the mean reactivity (MFIrel) and standard deviation of two separate experiments. MFIrel was the MFI in experiments with anti‐αGal relative to the MFI in the same experiment but without primary antibody (fluorescently labelled anti‐hIgG was present in both experiments). A positive antibody reaction was defined as a mean MFIrel at least two standard deviations above 1·10 (corresponding to the highest reactivity observed in parallel and equal experiments but with the negative control IgG anti‐CD20 as primary antibody). The figure shows that anti‐αGal reacts with numerous serotypes although they do not possess terminal Galα3Gal in their polysaccharide structure (red columns) (see also Figure 3). Several serotypes demonstrate higher reactivity than the positive control strain, EcO86. The serotypes highlighted in yellow were selected for further experiments
Collectively, our results indicate that anti‐αGal possesses polyreactivity for numerous different microbial polysaccharides, including 48 distinct pneumococcal capsular polysaccharides and the CW polysaccharide.
The anti‐αGal antibody is polyreactive and binds multiple distinct microbial polysaccharides
To establish whether anti‐αGal antibody possess polyreactivity for the pneumococcal capsules, we challenged the reactivity between the antibody and each of ten pneumococcal serotypes (yellow highlight in Figure 2C) by soluble capsular polysaccharide of the same serotype (i.e. the homologous polysaccharide). The 10 serotypes were chosen because they constitute important pathogens, 51 their defining polysaccharides are known to be free of terminal Galα3Gal (Figure 3A), 48 and they displayed variable levels of reactivity with the anti‐αGal antibody (Figure 2C). Similar experiments were performed for the C‐mutant using the CW polysaccharide as competitor for binding. The influence on pneumococcal reactivity was quantified by flow cytometry by the use of standard curves (Figures S6 and S7). Each of the homologous polysaccharides abolished the reactivity with the associated pneumococcus (Figures 3B,C, and S8). As a control, capsular polysaccharide serotype 3 at similar concentration caused no inhibition for any of the pneumococci (in agreement, anti‐αGal had not reacted with pneumococci of serotype 3). In additional control experiments, soluble Galα3Gal also inhibited reactivity, whereas an irrelevant control disaccharide, Glcα2Fru, at similar concentration did not (Figure 3B). This established that anti‐αGal caused the reactions, ruling out the hypothetical possibility that small amounts of contaminating antibodies in the preparation were responsible. Anti‐αGal antibody thus possesses polyreactivity for multiple distinct microbial polysaccharides.
FIGURE 3.
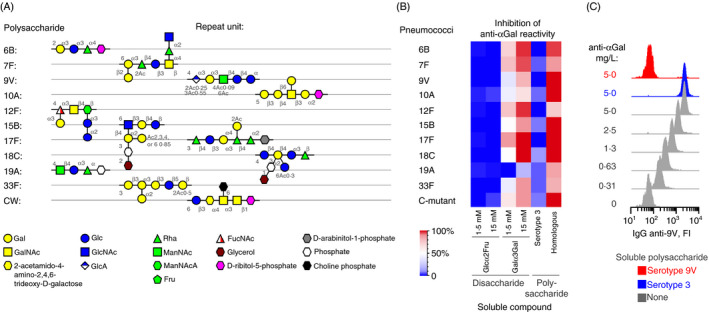
The anti‐αGal antibody is polyreactive and binds distinct capsular polysaccharides. (A) Schematic structures of the repeat unit in selected pneumococcal polysaccharides (capsular polysaccharide serotypes and CW polysaccharide). (B) Heatmap showing the inhibitory effect of various soluble compounds on the reaction between anti‐αGal and each of 10 pneumococcal serotypes and the C‐mutant strain. The concentration of the used anti‐αGal was 5 mg/L. For each strain, inhibition by the following soluble compounds was tested: Glcα2Fru (negative control), Galα3Gal, capsular polysaccharide of serotype 3 (negative control, intact pneumococci of serotype 3 did not react with anti‐αGal), and the homologous polysaccharide (i.e. capsular polysaccharide of the same serotype as the examined strain and CW polysaccharide for the C‐mutant strain). The polysaccharide concentration was 125 mg/L. The level of inhibition was determined by flow cytometry as the mean of two separate experiments. (C) Example plots of raw data sampled for serotype 9V pneumococci for quantification of anti‐αGal reactivity and inhibition by homologous serotype 9V polysaccharide (red) and heterologous serotype 3 polysaccharide (blue). The reactivity was calculated by means of standard curves as exemplified in Figure S7
The capsular polysaccharides of some reactive serotypes are only marginally different from some unreactive serotypes. For instance, the unreactive polysaccharide 33A contains two acetyl groups that are not found in the otherwise identical but reactive polysaccharide 33F 48 (Figure S9), which suggests that the two acetyl groups block anti‐αGal binding. We exploited observations of dissimilarities in reactivity between only marginally different serotypes to predict critical constituents in various epitopes for anti‐αGal (Figure S9).
Terminal αGalactose is present in some of the reactive polysaccharides (e.g. serotypes 12F, 15B, 17F and 33F; Figure 3A). To explore the involvement of such residues, we repeated the binding experiments with the antibody for the pneumococci with and without pretreatment by αGalactosidase, which can cleave off terminal αGalactose. As a control for enzyme activity, similar experiments were performed with pig erythrocytes, which carry terminal Galα3Gal on their surfaces 5 and binds anti‐αGal. 38 As expected, αGalactosidase treatment abrogated reactivity with the pig erythrocytes (Figure S10). However, αGalactosidase treatment did not decrease reactivity with the pneumococci (Figure S10), suggesting that the terminal αGalactose residues in the polysaccharides were not essential parts of the antibody epitopes.
Previously reported polyreactivity of anti‐αGal with human mucin peptides coincided with the binding of the αGalactosyl‐reactive plant lectin Isolectin B4, 16 suggesting that the antibody targets αGalactosyl‐mimotopes. We tested the reactivity of the lectin to 23 serotypes of pneumococci, but observed no correlation with anti‐αGal reactivity (Figure S11). Isolectin B4 merely bound pneumococci of serotype 10A, although several other serotypes presented αGalactosyl. We concluded that Isolectin B4 and anti‐αGal display different specificity for bacteria.
The collective results establish that the anti‐αGal antibody possess polyreactivity for a broad range of pneumococcal antigens.
Anti‐αGal antibody constitute significant parts of human anti‐pneumococcal antibodies
Each pneumococcal serotype presents a great number of different epitopes, so a single antibody is likely to account for only a small proportion of total human antibody reactivity against a given serotype. For the various serotypes, we estimated the percentage of reactive total human antibodies that was comprised by anti‐αGal antibody. A pool of normal human IgG (nhIgG) was used as a source of anti‐pneumococcal antibody. The reactivity with intact pneumococci was quantified with and without Galα3Gal disaccharide as competitor for binding. The decrease in reactivity with Galα3Gal addition was regarded as the reactivity caused by anti‐αGal. Reactivity was quantified by flow cytometry for serotypes 3 (negative control, unreactive with purified anti‐αGal), 6B, 7F, 9V, 12F, 15B, 17F, 18C, 19A and 33F. Anti‐αGal antibody comprised measurable percentages of the total IgG reactivity to all of the tested serotypes, except for serotypes 3 and 17F (Figure 4A). The largest percentage was observed for serotype 7F, where anti‐αGal comprised 18% of the anti‐7F reactivity in nhIgG. The percentages varied for serotypes (serotypes 3 and 17F excluded, Kruskal–Wallis test, P = 0·01). As control, Glcα2Fru disaccharide at similar concentration caused no loss of reactivity (Figure 4B). The percentages of inhibited reactivity are estimates of the percentages that anti‐αGal comprise of human anti‐pneumococcal antibodies. These estimates are however conservative as the concentration of Galα3Gal used (37 mM) did not completely saturate inhibition (Figure S12).
FIGURE 4.
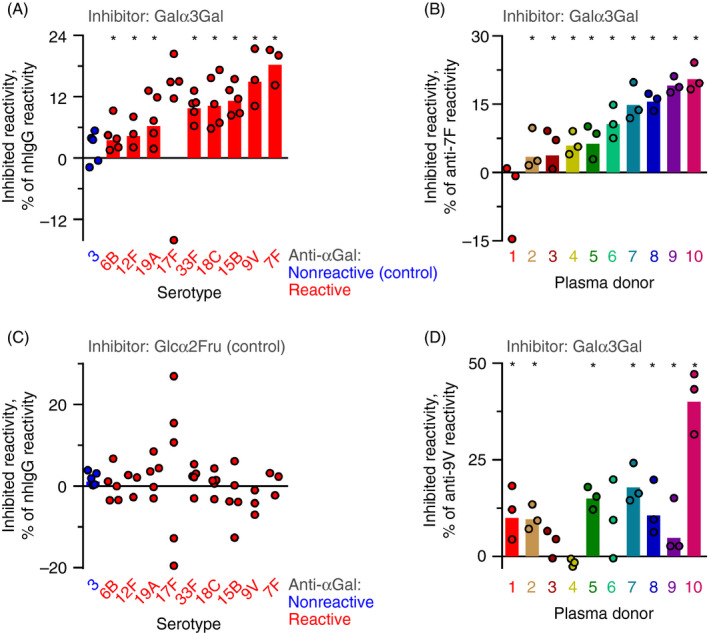
The anti‐αGal antibody comprises significant parts of human anti‐pneumococcal antibody reactivities. Inhibition of IgG reactivity with different serotypes of pneumococci by the presence of different disaccharides. Reactivity was quantified by flow cytometry. (A) Normal human IgG (nhIgG) was used as source of anti‐pneumococcal IgG. The reactivity with each of ten serotypes was challenged by Galα3Gal at 37 mM. Circles show the results of separate experiments, and the bars are the geometric mean for reactivity with each serotype (note that geometric means are not defined for datasets including negative values). Results are marked by an asterisk when the 95% confidence interval of the mean does not contain 0 (and therefore is considered significantly different from 0). The inhibited antibodies reacted with the soluble Galα3Gal and are therefore anti‐αGal. Thus, the level of lost reactivity represents the percentage of anti‐αGal that comprised the anti‐pneumococcal reactivity of nhIgG. (B) As in the previous panel, except that the reactions were challenged by Glcα2Fru. Glcα2Fru did not inhibit the reactivity of nhIgG with any serotype. (C) As in panel A, except that the antibody source was plasma samples from different healthy persons tested against serotype 7F pneumococci. (D) As in the previous panel, except serotype 9V pneumococci were used as target pneumococci
To examine whether individuals vary with respect to the percentage of total anti‐pneumococcal antibody that is comprised by anti‐αGal antibody, we performed similar experiments using individual plasma samples as sources of anti‐pneumococcal antibodies. Target cells in these experiments were pneumococci of serotypes 7F and 9V. In preliminary tests of 30 plasma samples, we selected 10 samples with comparable levels of IgG anti‐7F and of IgG anti‐9V (Figure S13). Anti‐αGal antibody explained up to 21% of anti‐7F reactivity and up to 40% of anti‐9V reactivity (Figure 4C,D). For both serotypes, the contribution of anti‐αGal to the total antibody reactivity varied among individuals (Kruskal–Wallis test, P < 0·02). In these plasma samples, the anti‐αGal contribution to the anti‐7F reactivity did not correlate with the contribution to the anti‐9V reactivity (Figure S14). As control, Glcα2Fru disaccharide at similar concentration caused no loss of reactivity (Figure S15).
The results collectively show that the anti‐αGal antibody comprise a significant percentage of human anti‐pneumococcal antibodies, which varies for different serotypes and between individuals.
The anti‐αGal antibody contains distinct subsets of polyreactive antibodies
We were puzzled by the broad reactivity of the anti‐αGal antibody. To investigate this further, we challenged the reactivity of anti‐αGal with each of ten reactive serotypes by adding soluble capsular polysaccharide from the other heterologous serotypes. Residual reactivity was quantified by flow cytometry. Compared with the effect of the homologous polysaccharide, each of the heterologous polysaccharides demonstrated markedly less inhibitory potential for each target cell (Figures 5A and S16) (the strain of serotype 12F is an exception, which is addressed below). The antibodies that reacted with one serotype thus possessed little or no reactivity for the heterologous serotypes. Our interpretation is that distinct antibody subsets within anti‐αGal possess specificity for distinct antigens, which allows reaction with distinct serotypes. Thus, in terms of antigen specificity, anti‐αGal is comprised of numerous antibodies of distinct antigen polyreactivity. Below we therefore use the plural anti‐αGal antibodies.
FIGURE 5.
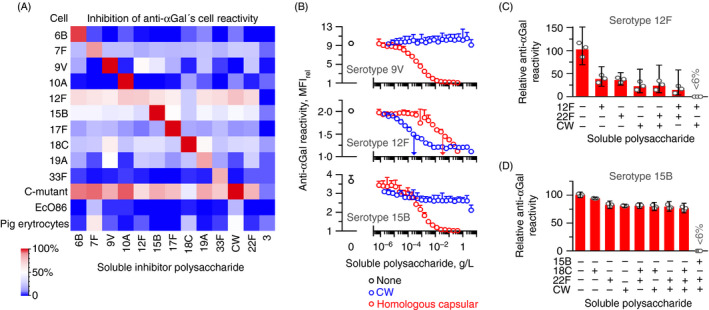
The anti‐αGal antibody contains antibody subsets of distinct polyreactivity. Flow cytometry analyses of anti‐αGal reactivity with target cells in the presence of soluble polysaccharides. (A) Heatmap showing results for various cells. Anti‐αGal was added at 5 mg/L and polysaccharides at 125 mg/L. Target cells were ten different serotypes of pneumococci, C‐mutant, EcO86 and pig erythrocytes (carries surface terminal Galα3Gal). The level of inhibition was determined as the mean of two separate experiments. The left downward diagonal of red squares shows that the homologous polysaccharide inhibited reactivity better than the heterologous polysaccharides. (B) Reactivity of anti‐αGal with each of three encapsulated pneumococcal strains in the presence of increasing concentration of either CW polysaccharide or homologous capsular polysaccharide. Anti‐αGal was added at 2·0 mg/L (serotypes 9V and 12F) or 10 mg/L (serotype 15B). Mean and standard deviation of two separate experiments. Centre (serotype 12F): vertical arrows indicate half‐maximal inhibitory concentration of each polysaccharide. The panel shows that CW polysaccharide was a more potent inhibitor than the homologous capsular polysaccharide for this serotype 12F strain. Also, neither substance alone caused complete inhibition. Bottom (serotype 15B): CW polysaccharide inhibited at most half of the reactivity, indicating that only a subset of the anti‐αGal antibodies that reacted with this strain possessed specificity for CW polysaccharide. (C) Residual reactivity of anti‐αGal at 2·0 mg/L with the strain of serotype 12F in the presence of soluble polysaccharides, each added at 125 mg/L. Mean with 95% confidence intervals of three separate experiments. (D) As in the previous panel, but with the strain of serotype 15B as target cells and anti‐αGal at 10 mg/L
The model requires that each anti‐αGal subset only constitutes minor parts of the total anti‐αGal antibodies. In agreement, the capsular polysaccharides generally failed to inhibit quantifiable reactivity with pig erythrocytes, which carries terminal Galα3Gal on their surfaces (Figure 5A). The same was true for EcO86. However, for C‐mutant, most capsular polysaccharides inhibited almost all reactivity. As C‐mutant presents dense CW polysaccharide, the inhibitory effect of the capsular polysaccharides with this strain likely reflects the presence of contaminating CW polysaccharide in the preparations of capsular polysaccharide. 52 , 53 This relates to covalent linkage between the CW polysaccharide and the capsule on most serotypes. 54 To explore the contribution of individual anti‐αGal subsets to the total anti‐αGal antibodies, we quantified the reactivity of total anti‐αGal to each of 14 different capsular polysaccharides in a bead‐based multiplex assay. Six serotypes that reacted in our cell‐based assay bound an average 2·0% (95%CI: 1·2%–3·4%) of total anti‐αGal in the bead‐based assay (Figure S17). Eight serotypes that did not react in our cell‐based assay bound an average 1·0% (95%CI: 0·56%–1·8%) of total anti‐αGal in the bead‐based assay. The latter may represent binding to contaminating CW polysaccharide or other unspecific reactivity in the bead‐based assay. Regardless, an individual anti‐αGal subset is likely to comprise only a small part of the total anti‐αGal (in the order of 1%).
Indeed, heterologous capsular polysaccharides caused partial inhibition of the reactivity with several serotypes (Figure 5A). For the encapsulated pneumococci, the level of partial inhibition seemed more related to the serotype of the target pneumococci than the serotype of the polysaccharide used for inhibition (the horizontal data are more homogenous than the vertical data in Figure 5A). For each pneumococcal strain, CW polysaccharide inhibited reactivity to a level very similar to that of the heterologous capsular polysaccharides (Figure 5A), supporting the assumption mentioned above that contaminating CW polysaccharide caused the partial inhibitory effect of the heterologous capsular polysaccharides. In agreement, polysaccharide of serotype 22F inhibited reactivity to a level similar to that observed for most heterologous capsular polysaccharides, although the strain of serotype 22F did not react with anti‐αGal. The CW polysaccharide is thus likely shielded by the unreactive serotype 22F capsule polysaccharide on serotype 22F pneumococci yet accessible for antibody binding in a solution of the 22F polysaccharide preparation. As a rarity among pneumococci, the capsular polysaccharide of serotype 3 is not covalent linked to CW polysaccharide, 54 so capsular polysaccharide preparations of this serotype should be free of contaminating CW polysaccharide. As control, the preparation of capsular polysaccharide serotype 3 failed to inhibit reactivity with the anti‐αGal‐reactive cells (Figure 5A).
We compared the inhibition of anti‐αGal reactivity by increasing concentrations of soluble CW polysaccharide and homologous capsular polysaccharide for three serotypes of pneumococci. Serotypes 12F and 15B were selected as they were most sensitive to inhibition by CW polysaccharide, and serotype 9V was included as control as its reactivity did not seem affected by CW polysaccharide (Figure 5A). For the serotype 9V pneumococci, even very high concentrations of CW polysaccharide (>0·1% w/v) did not show any signs of inhibition, whereas the homologous capsular polysaccharide potently inhibited reactivity in a concentration‐dependent manner (Figure 5B, top). For the serotype 12F pneumococci, CW polysaccharide proved a markedly more efficient inhibitor than the homologous capsular polysaccharide (approximately 100 times more efficient based on half‐maximal inhibitory concentrations) (Figure 5B, centre). Interestingly, neither polysaccharide alone was sufficient to completely inhibit reactivity with the serotype 12F pneumococci. Complete inhibition was achieved when the two substances were combined (Figure 5C), demonstrating that the anti‐αGal antibodies that bound this 12F strain is composed of antibodies of different specificity. For the serotype 15B pneumococci, the reactivity was eliminated dose‐dependently by the homologous capsular polysaccharide (Figure 5B, bottom), whereas CW polysaccharide inhibited less than half of the reactivity (Figure 5B, bottom). Thus, as for the serotype 12F pneumococci, the anti‐αGal antibodies that bind the serotype 15B pneumococci are composed of antibodies of different specificity. No further inhibition was achieved by adding additional heterologous capsular polysaccharides (Figure 5D), so contaminating CW polysaccharide is sufficient to explain the partial inhibition observed with the heterologous capsular polysaccharides for the serotype 15B pneumococci. We made the same experiments for serotype 18C pneumococci as described for the serotype 15B pneumococci and obtained similar results (Figure S18). Together, these results strongly support that contaminating CW polysaccharide explains the partial inhibition observed for preparations of heterologous capsular polysaccharides. Moreover, the anti‐αGal antibodies that display polyreactivity for a given pathogen may also be a mixture of subsets with different specificities.
The collective results in this section demonstrate that anti‐αGal contains numerous distinct polyreactive antibodies and explain the broad pathogen reactivity of the anti‐αGal antibodies.
Polyreactive anti‐αGal antibodies drive phagocytosis
Hamadeh and co‐workers claim that anti‐αGal reactivity enables the survival of some pathogens in humans by inhibiting complement attack. 13 Complement alone is insufficient to kill pneumococci and other Gram‐positive bacteria due to their peptidoglycan‐rich protective cell walls. Phagocytosis is therefore the principal mechanism for the elimination of such microorganisms. In general, phagocytosis of pneumococci requires opsonization by antibodies and complement fragments, especially fragments of C3. We recently reported that anti‐αGal bound on serotype 9V pneumococci activated the classical pathway of complement. 38 Here, we examine the opsonic effect of anti‐αGal bound on this pneumococcal serotype. Serum from a person with hypogammaglobulinaemia was used as a source of complement. Primary human blood leucocytes were used as phagocytes. As a read‐out of phagocytosis, we used flow cytometry to detect the production of reactive oxygen species (ROS) in the leucocytes, a hallmark of ongoing phagocytosis. 55 The suitability of the assay was supported by a strong association between uptake of fluorescently labelled pneumococci and ROS production in the leucocytes (Figure S19), and by the observation that the phagocytosis inhibitor, cytochalasin D, eliminated both pneumococcal uptake and ROS production in the leucocytes (Figure 6A). The predominant phagocytes were granulocytes (Figure S20).
FIGURE 6.
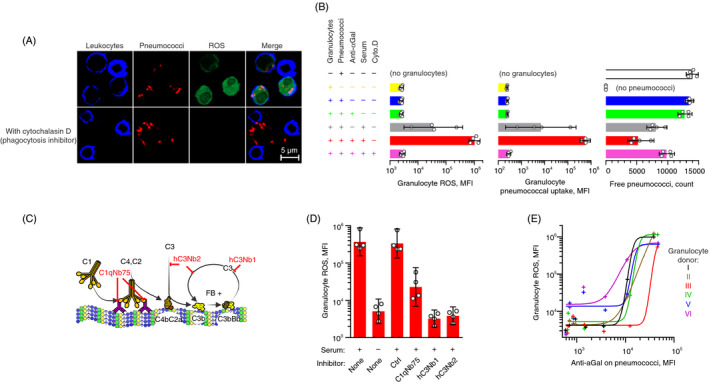
Polyreactive anti‐αGal antibodies drive phagocytosis of serotype 9V pneumococci in a complement‐dependent manner. Fluorescently labelled serotype 9V pneumococci were opsonized in 10% human hypogammaglobulinaemia serum (‘serum’) supplemented with anti‐αGal at 20 mg/L (unless otherwise is stated) before feeding to plasma‐depleted human blood cells labelled with dihydrorhodamine‐123. Dihydrorhodamine‐123 (non‐fluorescent) is oxidized to rhodamine‐123 (fluorescent) by intra‐phagosomal reactive oxygen species (ROS) produced in phagocytosing granulocytes. Confocal microscopy in A and flow cytometry data in B, D and E. (A) Experiments with and without the phagocytosis inhibitor cytochalasin D. The panel shows that pneumococci and ROS were inside the leucocytes when the phagocytosis inhibitor was omitted, whereas no ROS or pneumococci were inside the leucocytes when the phagocytosis inhibitor was present. (B) Effect of constituents in the experiments on granulocyte ROS (left), pneumococcal uptake (centre) and clearance of free pneumococci from the supernatant (right). Bars represent mean with 95% confidence intervals for different leucocyte donors (n = 4). (C) Illustration of complement activation by IgG antibodies bound on antigen and the points of action for the specific complement inhibitors used in the experiments shown in the following panel. ‘C1qNb75’, blocks C1q binding to immunoglobulin; ‘hC3Nb1’, blocks C3 cleavage by alternative pathway; and ‘hC3Nb2’, blocks C3 cleavage by classical and alternative pathways. (D) Granulocyte ROS induced by pneumococci, opsonized with anti‐αGal at 5 mg/L in the presence of inhibitors of selective complement factors. ‘Ctrl’, nanobody of irrelevant specificity. Mean with 95% confidence intervals for different leucocyte donors (n = 4). (E) Granulocyte ROS as function of the density of anti‐αGal on opsonized pneumococci. For each of six leucocyte donors, experiments were performed with the following concentrations of anti‐αGal: 0, 0·020, 0·078, 0·31, 1·3, 5 and 20 mg/L. Curves were fitted in sigmoid models (R 2 ≥ 0·92)
To examine the contribution of anti‐αGal antibodies to phagocytosis, we varied the assay constituents. Exclusion of anti‐αGal antibodies reduced the phagocytosis signal 30‐fold and increased the concentration of non‐phagocytosed pneumococci (Figures 6B and S21). The effect of anti‐αGal required the presence of serum as exclusion essentially abrogated phagocytosis. The serum conveyed some background phagocytosis (Figure 6B), likely initiated by its small amounts of complement‐activating endogenous anti‐9V antibodies. 38
To determine the involvement of complement for the phagocytic effect of anti‐αGal, we included various selective complement blockers in the opsonization process. The blockers were nanobodies generated against specific complement proteins. Their points of action are shown in Figure 6C. Blockade of C1q docking to immunoglobulins with C1qNb75 (Figure 6C) decreased the phagocytosis signal by 94% (Figure 6D), establishing that the classical pathway of complement was essential. Inhibition of C3 cleavage by both classical and alternative pathway C3 convertases with hC3Nb2 (Figure 6C) decreased the phagocytosis signal by 99% (Figure 6D), demonstrating a pivotal role of C3 fragments. Interestingly, specific inhibition of C3 cleavage by the alternative pathway C3 convertase with hC3Nb1 (Figure 6C) decreased the phagocytosis signal with equal efficiency (Figure 6D). This indicates that the alternative pathway delivers crucial amplification of the classical pathway activation by anti‐αGal on the pneumococci in agreement with our previous findings. 38 A control nanobody of irrelevant specificity, applied in a concentration equivalent to the highest used concentration of the functional nanobodies, did not affect the phagocytosis (Figure 6D). Together, these results show that opsonization of serotype 9V pneumococci by anti‐αGal is complement‐dependent, and the initiation is via the classical pathway of the complement system.
To assess the relationship between anti‐αGal density on the pneumococci and phagocytosis of the pneumococci, we performed phagocytosis experiments with leucocytes from six different donors and increasing concentrations of anti‐αGal during the opsonization. The density of anti‐αGal on the pneumococci was measured in parallel experiments in the presence of EDTA to quench complement activation and therefore prevent masking of bacterial‐bound antibodies by deposited C3 fragments. 38 Phagocytosis was not induced before the anti‐αGal density exceeded a threshold occurring at an anti‐αGal concentration between 1·3 mg/L and 5 mg/L (Figure 6E). Thereafter, phagocytosis increased with the density of the anti‐αGal antibodies.
The results collectively demonstrate that the subsets of anti‐αGal that possesses polyreactivity for serotype 9V promote phagocytosis of this serotype in synergy with complement.
Anti‐αGal antibodies opsonize reactive pneumococcal serotypes in general
To enable a more general conclusion on the role anti‐αGal antibodies play in the opsonization of pneumococci, we studied ten additional serotypes: 3 (negative control), 6B, 7F, 10A, 12F, 15B, 17F, 18C, 19A and 33F. IgG antibody binding on the pneumococci and pneumococcal phagocytosis were examined by flow cytometry. Experiments were performed with each serotype in i) buffer, ii) human hypogammaglobulinaemia serum and iii) human hypogammaglobulinaemia serum with anti‐αGal antibodies added to 20 mg/L. Although the endogenous level of total immunoglobulin in the serum was low, it contained IgG antibodies against all the serotypes (Figure S22). Addition of anti‐αGal increased IgG binding on the pneumococci compared with incubation in serum alone, except for serotypes 3, 19A and 33F (Figure S22). For each of the other serotypes, addition of anti‐αGal increased the point estimates of phagocytosis (Figure 7A). To clarify the general effect, the phagocytosis signal was compared with the antibody binding signal for each of the pneumococci. A clear positive association was observed (Figure 7B), supporting that the general effect of anti‐αGal binding on pneumococci is initiation of phagocytosis.
FIGURE 7.
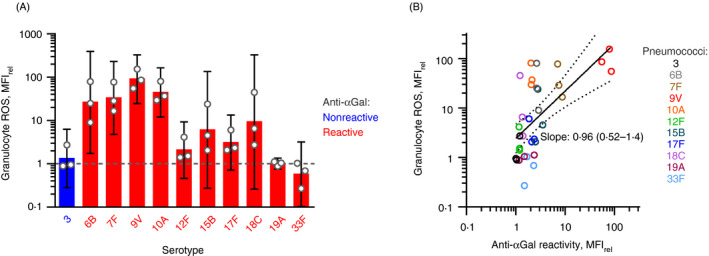
Binding of polyreactive anti‐αGal antibodies in general conveys phagocytosis of pneumococcal serotypes. (A) The opsonic effect of anti‐αGal for each 11 different pneumococcal serotypes. Flow cytometry data (MFIrel) for opsonization with hypogammaglobulinaemia serum (‘serum’) with added anti‐αGal compared with opsonization with hypogammaglobulinaemia serum only. Serum concentration was 10%. Concentration of added anti‐αGal was 20 mg/L. Mean with 95% confidence intervals for three leucocyte donors. (B) The general opsonic effect of anti‐αGal for reactive pneumococci. Granulocyte ROS (from previous panel, on y‐axis) as function of antibody binding on pneumococci after opsonization with anti‐αGal and hypogammaglobulinaemia serum relative to hypogammaglobulinaemia serum alone. The regression curve with 95% confidence interval, determined on log10–log10 scale, is shown together with the point estimate of the curve´s slope (with 95% confidence interval). For these serotypes, the data show a positive correlation between the level of anti‐αGal reactivity and the phagocytosis signal
Our data thus support that the polyreactive subsets within anti‐αGal enable phagocytosis of reactive pneumococci.
Protective effects of the anti‐αGal antibodies in the human population
To assess the net contribution of anti‐αGal to anti‐pneumococcal defences, we compared the prevalence of pneumococcal serotypes as invasive pathogens in humans to the level of anti‐αGal reactivity each of these serotypes displayed. To this end, we obtained data on the number of occasions each serotype has been diagnosed as the cause of invasive pneumococcal infections in Denmark between 1966 and 2014 (n = 29 034) (Table S1). An inverse relationship was observed (Figure 8A); that is, the most frequent pathogenic serotypes reacted poorly with anti‐αGal (Figure 8A,B) and the most anti‐αGal‐reactive serotypes rarely (or never) caused invasive pneumococcal disease (Figure 8A,C). In a simple statistical model where each serotype was weighted according to the number of times it had been isolated, the frequency of the serotypes correlated negatively with their anti‐αGal reactivity (rho = −0·33, P < 0·0001).
FIGURE 8.
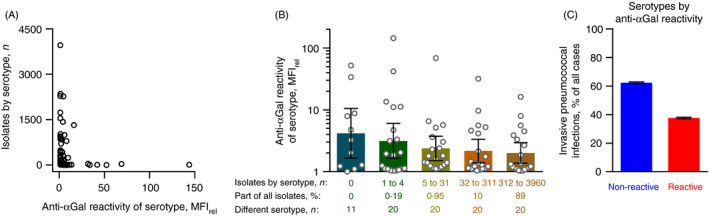
Relationship between anti‐αGal reactivity of pneumococcal serotypes and their occurrence among invasive infections in humans. All recorded cases of invasive infections due to encapsulated pneumococci in Denmark between 1966 and 2014 (n = 29 034) were included. (A) The number of isolates of each serotype is shown as a function of the anti‐αGal reactivity of that serotype (as determined in the present study). (B) Anti‐αGal reactivity for serotypes grouped according to the number of recorded isolates. For each group, the accumulated contribution to invasive pneumococcal infections and number of different serotypes included in the group are shown. Geometric mean with 95% confidence intervals. Anti‐αGal reactivity tended to be lower for the more prevalent serotypes. (C) The percentage of invasive pneumococcal infections caused by serotypes grouped according to their anti‐αGal reactivity. Error bars are 95% confidence interval. The non‐reactive serotypes were more prevalent than the reactive serotypes
These results are consistent with the notion that the polyreactivity of anti‐αGal provides natural protection of humans against invasive pneumococcal infections.
DISCUSSION
We studied the cause of the broad‐spectrum pathogen reactivity of anti‐αGal, 13 , 36 , 37 naturally occurring human antibodies and their possible role in human immunity. Naturally occurring antibodies have long been speculated to form a valuable first‐line defence against invading pathogens, but this assumption has not been firmly established previously in humans. Overall, our results confirm the broad‐spectrum pathogen reactivity of the anti‐αGal antibodies, establish that the reactivity is indeed caused by polyreactivity and support a significant contribution of these antibodies to the human defence against bacterial pathogens. We propose that the anti‐αGal antibodies act in synergy with the many other molecules and cells involved in the shaping of human immune defences.
To investigate the reported broad‐spectrum pathogen reactivity of anti‐αGal, we applied a well‐characterized preparation of human anti‐αGal of the IgG class 37 , 38 on a large collection of encapsulated pneumococci and observed reactivity with most serotypes (Figure 2). Because the anti‐αGal preparation originated from the IgG of thousands of individuals, the reactivity of the preparation reflects a general human potential for generation of anti‐αGal antibodies. We also confirmed that anti‐αGal can comprise a large part of the anti‐pneumococcal antibodies on the level of individuals (Figure 4). We used encapsulated pneumococci as model pathogens for three main reasons. First, pneumococci are major pathogens in humans, that is a leading cause of pneumonia, 30 which is the most deadly communicable disease, causing 3·0 million deaths worldwide in 2016. 31 Second, specific antibodies are key players in human protection against pneumococcal disease. 33 , 34 Third, pneumococcal capsules are very diverse, with 100 unique serotypes, 35 and all characterized serotypes lack terminal Galα3Gal, 48 which allows studies of reactivity to defined polysaccharides that are biologically relevant.
We found that anti‐αGal antibodies reacted with 48 out of 91 serotypes including 37 serotypes that are known to be free of terminal Galα3Gal in their capsular structures (Figure 2). By selective inhibition with soluble capsular polysaccharides, we determined that the reactions were caused by polyreactivity of anti‐αGal (Figure 3). Moreover, our results support that different antibodies within anti‐αGal are responsible for the reactivity (Figure 5). We therefore propose that human anti‐αGal is not a single antibody in terms of specificity but instead contains multiple antibody subsets, each with one or more additional reactivities beyond their general reactivity with terminal Galα3Gal. Collectively, the distinct polyreactive subsets can bind a plethora of distinct structures, including numerous microbial polysaccharides. Based on our data, each of the subsets may comprise in the order of 1% of total anti‐αGal (Figure S17), but some may account for a considerably larger proportion. For example, we discovered that CW polysaccharide inhibited nearly half the reactivity with the terminal Galα3Gal on pig erythrocytes (Figure S16), suggesting that a significant proportion of anti‐αGal reacts with this antigen. CW polysaccharide also partly inhibited reactivity to several capsulated pneumococci, unravelling an additional layer of complexity in the pathogen reactivity of anti‐αGal. Not only do distinct anti‐αGal subsets target different pathogens, but also a given pathogen‐reactive subset may in fact contain several further subsets that bind distinct antigens on the pathogen by polyreactivity. The polyreactivity with CW polysaccharide may be explained by triple specificity of some anti‐αGal clones allowing binding to (i) terminal Galα3Gal (reactant used to purify), (ii) the capsular polysaccharide on the pneumococci and (iii) the CW polysaccharide used as competitor for pneumococcal binding. An alternative explanation is that some of the capsulated strains presented ‘naked’ CW polysaccharide for direct antibody binding. Along the same line of thinking, we cannot rule out that direct binding to CW polysaccharide contributes a part of the reactivity observed for some of the 48 encapsulated strains. But in general, the capsule is the primary target on reactive pneumococci, based on our findings for the ten strains that we examined in details (Figure 5).
We eluted our affinity‐purified anti‐αGal preparation from Galα3Gal‐derivatized beads by brief exposure to low pH. It has been proposed that such treatment can induce polyreactivity. 56 We find it unlikely that low pH caused the polyreactivity we observed as the anti‐αGal present in the nhIgG preparation, which had not been exposed to low pH, also were polyreactive for pneumococcal polysaccharides (Figure 4). The same applied to the anti‐αGal in human plasma samples (Figure 4). Affinity‐purified anti‐αGal antibodies eluted by 0·5 M melibiose (instead of low pH) also displayed broad‐spectrum bacterial reactivity 13 to a degree that strongly supports natural polyreactivity.
It will be interesting to learn whether broad reactivity is unique to anti‐αGal or, as we envision, a general phenomenon for human antibodies. We speculate the same mechanism can explain the reported broad bacterial reactivity of naturally occurring antibodies to ABO antigens. 1
Four independent lines of evidence supported a positive and significant contribution of anti‐αGal antibodies to human protective immunity against bacterial pathogens. First, we found a reduced plasma anti‐αGal level in humans with recurrent pneumonia (Figure 1). Second, anti‐αGal constituted significant percentages of human IgG antibodies to pneumococcal capsules (Figure 4) and such antibodies are essential for protection against pneumococcal infections. 33 , 34 Third, anti‐αGal in synergy with complement opsonized pneumococci for phagocytosis by neutrophils in a dose‐dependent manner (Figures 6 and 7), which is the primary way to eradicate invading pneumococci. Fourth, the level of IgG anti‐αGal reactivity with various pneumococcal serotypes correlated inversely with their prevalence in pneumococcal invasive diseases in an entire nation (Figure 8), which supports protection on the population level.
A major strength in our study is the strict application of human source material, including antibodies, primary phagocytic cells and serum. Humans are the natural host of pneumococci, and spontaneous infection in other animals is rare. Furthermore, typical animal models do not express FcγRIIA, 57 the main phagocyte receptor for human IgG2, 58 which is the predominant subclass of IgG anti‐αGal. 21 , 37 Animal models in this setting therefore require considerable genetic manipulation and non‐physiological pathogen dosages with uncertain additional consequences and uncertain relevance to humans. Future studies of the anti‐bacterial effect of anti‐αGal antibodies in animal models must account for such challenges to provide knowledge relevant to human immunology. Here, we applied nation‐wide data on pneumococcal infections, which also incorporates the influence of heterogeneities in host factors and environmental factors in a habitat relevant to contemporary man. Our data support an unequivocal and measurable protective effect, in stark contrast to earlier claims. 13
Our results introduce additional, novel perspectives on anti‐αGal antibodies. Our ancestral loss of the α1,3‐galactosyltransferase may be more important than merely blocking transmission of pathogens that carry terminal Galα3Gal. 9 , 10 , 11 We propose that the lost tolerance for terminal Galα3Gal unleashed a multitude of polyreactive antibodies with a combined reactivity far beyond this structure. If we still produced terminal Galα3Gal, all these antibody clones would be deleted according to the tolerance theory. Moreover, our observations may challenge the notion that enteric bacteria deliver the antigenic stimulation for anti‐αGal generation. 12 The essential immunogen for generation of a polyreactive anti‐αGal antibody clone may be any antigen carrying an epitope of that clone. Thus, the CW polysaccharide on commensal and pathogenic Streptococci may be the antigenic source for the anti‐αGal clones that bind this polysaccharide. On the other hand, terminal Galα3Gal delivered by bacteria or dietary mammal meat or milk could also be an antigenic source for generation of human antibodies against the CW polysaccharide. Future studies may resolve this, but we speculate that a person’s repertoire of anti‐αGal antibodies is the consequence of stimulation by many different immunogens. Finally, we propose an explanation for the long‐standing puzzle of how anti‐αGal‐reactive pathogens can pose infectious threats to humans with naturally occurring anti‐αGal. The present study highlights four reasons. First, the concentration of anti‐αGal in plasma, approximately 10 mg/L (present study and Refs. 21, 22, 23), is markedly lower than the originally reported level. 2 Second, levels of anti‐αGal antibodies vary more than 2000‐fold between humans, and individuals with concentrations far below 10 mg/L are common (Figure 1A). Third, only a small proportion of anti‐αGal (~1%) may possess polyreactivity for a reactive pathogen, thereby reducing the level of actual protective antibodies in the order of 100‐fold. Fourth, the proportion of anti‐αGal with reactivity for a given pathogen is likely to vary for different pathogens and between persons (Figure 4). The four reasons in combination likely explain why the concentration of anti‐αGal that target a given pathogen through polyreactivity can be low and insufficient for protection in some individuals.
It may be of interest to explore polyreactivity of anti‐αGal antibodies with other types of pathogens. For instance, some parasites are targeted by anti‐αGal antibodies (review 59) and low anti‐αGal levels may be associated with some parasitic infections. 60 In the light of the present study, it may be that similar polyreactivity is involved in the binding of anti‐αGal antibodies to parasites, which may be addressed in future studies.
The increasing microbial resistance to conventional antibiotics leads to failing treatment of infections and rising mortality, 61 and calls for novel approaches. In this light, the broad‐spectrum pathogen reactivity of naturally occurring anti‐αGal and the data supporting a significant contribution of these polyreactive antibodies to human protective immunity are of interest. Whether these antibodies can be harnessed for novel antimicrobial therapeutics must be explored in future studies.
CONFLICT OF INTEREST
The authors declare no competing financial and commercial interests in relation to the manuscript.
AUTHOR CONTRIBUTION
JMBJ conceptualized the study, designed experiments, conducted experiments, analysed data and drafted the manuscript. SS conducted experiments and analysed data. MSM advised on flow cytometry and statistical analyses. UBSS, JCJ and ST contributed reagents and analysed data. SH collected and contributed data. All authors contributed to the writing of the manuscript and reviewed and approved the final version of the manuscript.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We thank Mette Bjerre (Aarhus University) for performing Luminex measurements and Rasmus Kjeldsen and Nick S. Laursen (Aarhus University) for kindly providing nanobodies. We also thank Anette Tarp Hansen (Aalborg University Hospital) and Anne Færch Nielsen (Aarhus University) for suggestions to the manuscript. This work was supported by an Aarhus University grant to JMBJ, Bloddonorernes Forskningsfond and Forskningstræningspuljen, Region Midt.
Funding informationThis work was supported by an Aarhus University grant to J.M.B.J., Bloddonorernes Forskningsfond and Forskningstræningspuljen, Region Midt.
DATA AVAILABILITY STATEMENT
Data relating to the manuscript will be available upon reasonable request to the corresponding author.
REFERENCES
- 1. Springer GF, Williamson P, Brandes WC. Blood group activity of gram‐negative bacteria. J Exp Med. 1961; 113:1077–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galili U, Rachmilewitz EA, Peleg A, Flechner I. A unique natural human IgG antibody with anti‐alpha‐galactosyl specificity. J Exp Med. 1984; 160:1519–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tangvoranuntakul P, Gagneux P, Diaz S, Bardor M, Varki N, Varki A, et al. Human uptake and incorporation of an immunogenic nonhuman dietary sialic acid. Proc Natl Acad Sci USA. 2003; 100:12045–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galili U. Anti‐Gal: an abundant human natural antibody of multiple pathogeneses and clinical benefits. Immunology. 2013; 140:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Galili U, Clark MR, Shohet SB, Buehler J, Macher BA. Evolutionary relationship between the natural anti‐Gal antibody and the Gal alpha 1–3Gal epitope in primates. Proc Natl Acad Sci USA. 1987; 84:1369–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Galili U, Shohet SB, Kobrin E, Stults CL, Macher BA. Man, apes, and Old World monkeys differ from other mammals in the expression of alpha‐galactosyl epitopes on nucleated cells. J Biol Chem. 1988; 263:17755–62. [PubMed] [Google Scholar]
- 7. Koike C, Fung JJ, Geller DA, Kannagi R, Libert T, Luppi P, et al. Molecular basis of evolutionary loss of the alpha 1,3‐galactosyltransferase gene in higher primates. J Biol Chem. 2002; 277:10114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Galili U, Swanson K. Gene sequences suggest inactivation of alpha‐1,3‐galactosyltransferase in catarrhines after the divergence of apes from monkeys. Proc Natl Acad Sci USA. 1991; 88:7401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rother RP, Fodor WL, Springhorn JP, Birks CW, Setter E, Sandrin MS, et al. A novel mechanism of retrovirus inactivation in human serum mediated by anti‐alpha‐galactosyl natural antibody. J Exp Med. 1995; 182:1345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Takeuchi Y, Porter CD, Strahan KM, Preece AF, Gustafsson K, Cosset FL, et al. Sensitization of cells and retroviruses to human serum by (alpha 1–3) galactosyltransferase. Nature. 1996; 379:85–8. [DOI] [PubMed] [Google Scholar]
- 11. Galili U. Natural anti‐carbohydrate antibodies contributing to evolutionary survival of primates in viral epidemics? Glycobiology. 2016; 26:1140–50. [DOI] [PubMed] [Google Scholar]
- 12. Galili U, Mandrell RE, Hamadeh RM, Shohet SB, Griffiss JM. Interaction between human natural anti‐alpha‐galactosyl immunoglobulin G and bacteria of the human flora. Infect Immun. 1988; 56:1730–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hamadeh RM, Jarvis GA, Galili U, Mandrell RE, Zhou P, Griffiss JM. Human natural anti‐Gal IgG regulates alternative complement pathway activation on bacterial surfaces. J Clin Investig. 1992; 89:1223–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montassier E, Al‐Ghalith GA, Mathe C, Le Bastard Q, Douillard V, Garnier A, et al. Distribution of Bacterial alpha1,3‐Galactosyltransferase Genes in the Human Gut Microbiome. Front Immunol. 2019; 10:3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dimitrov JD, Planchais C, Roumenina LT, Vassilev TL, Kaveri SV, Lacroix‐Desmazes S. Antibody polyreactivity in health and disease: statu variabilis. J Immunol. 2013; 191:993–9. [DOI] [PubMed] [Google Scholar]
- 16. Sandrin MS, Vaughan HA, Xing PX, McKenzie IF. Natural human anti‐Gal alpha(1,3)Gal antibodies react with human mucin peptides. Glycoconj J. 1997; 14:97–105. [DOI] [PubMed] [Google Scholar]
- 17. Satapathy AK, Ravindran B. Naturally occurring alpha‐galactosyl antibodies in human sera display polyreactivity. Immunol Lett. 1999; 69:347–51. [DOI] [PubMed] [Google Scholar]
- 18. Casali P, Notkins AL. CD5+ B lymphocytes, polyreactive antibodies and the human B‐cell repertoire. Immunol Today. 1989; 10:364–8. [DOI] [PubMed] [Google Scholar]
- 19. Notkins AL. Polyreactivity of antibody molecules. Trends Immunol. 2004; 25:174–9. [DOI] [PubMed] [Google Scholar]
- 20. McMorrow IM, Comrack CA, Sachs DH, DerSimonian H. Heterogeneity of human anti‐pig natural antibodies cross‐reactive with the Gal(alpha1,3)Galactose epitope. Transplantation. 1997; 64:501–10. [DOI] [PubMed] [Google Scholar]
- 21. Yu PB, Holzknecht ZE, Bruno D, Parker W, Platt JL. Modulation of natural IgM binding and complement activation by natural IgG antibodies: a role for IgG anti‐Gal alpha1‐3Gal antibodies. J Immunol. 1996; 157:5163–8. [PubMed] [Google Scholar]
- 22. Barreau N, Blancho G, Boulet C, Martineau A, Vusio P, Liaigre J, et al. Natural anti‐Gal antibodies constitute 0.2% of intravenous immunoglobulin and are equally retained on a synthetic disaccharide column or on an immobilized natural glycoprotein. Transpl Proc. 2000; 32:882–3. [DOI] [PubMed] [Google Scholar]
- 23. Bernth‐Jensen JM, Moller BK, Jensenius JC, Thiel S. Biological variation of anti‐alphaGal‐antibodies studied by a novel Time‐Resolved ImmunoFluorometric Assay. J Immunol Methods. 2011; 373:26–35. [DOI] [PubMed] [Google Scholar]
- 24. McMorrow IM, Comrack CA, Nazarey PP, Sachs DH, DerSimonian H. Relationship between ABO blood group and levels of Gal alpha,3Galactose‐reactive human immunoglobulin G. Transplantation. 1997; 64:546–9. [DOI] [PubMed] [Google Scholar]
- 25. Neethling FA, Cooper DK. Serum cytotoxicity to pig cells and anti‐alphaGal antibody level and specificity in humans and baboons. Transplantation. 1999; 67:658–65. [DOI] [PubMed] [Google Scholar]
- 26. Galili U, Buehler J, Shohet SB, Macher BA. The human natural anti‐Gal IgG. III. The subtlety of immune tolerance in man as demonstrated by crossreactivity between natural anti‐Gal and anti‐B antibodies. J Exp Med. 1987; 165:693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X‐linked agammaglobulinemia: report on a United States registry of 201 patients. Medicine. 2006; 85:193–202. [DOI] [PubMed] [Google Scholar]
- 28. Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2015; 136:1186–205.e1‐78. [DOI] [PubMed] [Google Scholar]
- 29. Oksenhendler E, Gerard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008; 46:1547–54. [DOI] [PubMed] [Google Scholar]
- 30. Bartlett JG, Mundy LM. Community‐acquired pneumonia. N Engl J Med. 1995; 333:1618–24. [DOI] [PubMed] [Google Scholar]
- 31. The top 10 causes of death. 2018. https://www.who.int/news‐room/fact‐sheets/detail/the‐top‐10‐causes‐of‐death
- 32. Skov Sorensen UB, Blom J, Birch‐Andersen A, Henrichsen J. Ultrastructural localization of capsules, cell wall polysaccharide, cell wall proteins, and F antigen in pneumococci. Infect Immun. 1988; 56:1890–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smit P, Oberholzer D, Hayden‐Smith S, Koornhof HJ, Hilleman MR. Protective efficacy of pneumococcal polysaccharide vaccines. JAMA. 1977; 238:2613–6. [PubMed] [Google Scholar]
- 34. Klugman KP, Madhi SA, Huebner RE, Kohberger R, Mbelle N, Pierce N, et al. A trial of a 9‐valent pneumococcal conjugate vaccine in children with and those without HIV infection. N Engl J Med. 2003; 349:1341–8. [DOI] [PubMed] [Google Scholar]
- 35. Ganaie F, Saad JS, McGee L, van Tonder AJ, Bentley SD, Lo SW, et al. A new pneumococcal capsule Type, 10D, is the 100th serotype and has a large cps fragment from an oral streptococcus. MBio. 2020; 11.e00937‐20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wetter LA, Hamadeh RM, Griffiss JM, Oesterle A, Aagaard B, Way LW. Differences in outer membrane characteristics between gallstone‐associated bacteria and normal bacterial flora. Lancet. 1994; 343:444–8. [DOI] [PubMed] [Google Scholar]
- 37. Bernth Jensen JM, Petersen MS, Ellerman‐Eriksen S, Moller BK, Jensenius JC, Sorensen UBS, et al. Abundant human anti‐Galalpha3Gal antibodies display broad pathogen reactivity. Sci Rep. 2020; 10:4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bernth Jensen JM, Laursen NS, Jensen RK, Andersen GR, Jensenius JC, Sørensen UBS, et al. Complement activation by human IgG antibodies to galactose‐α‐1,3‐galactose. Immunology. 2020; 161:66–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Calix JJ, Saad JS, Brady AM, Nahm MH. Structural characterization of Streptococcus pneumoniae serotype 9A capsule polysaccharide reveals role of glycosyl 6‐O‐acetyltransferase wcjE in serotype 9V capsule biosynthesis and immunogenicity. J Biol Chem. 2012; 287:13996–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Posekany KJ, Pittman HK, Bradfield JF, Haisch CE, Verbanac KM. Induction of cytolytic anti‐Gal antibodies in alpha‐1,3‐galactosyltransferase gene knockout mice by oral inoculation with Escherichia coli O86:B7 bacteria. Infect Immun. 2002; 70:6215–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yilmaz B, Portugal S, Tran TM, Gozzelino R, Ramos S, Gomes J, et al. Gut microbiota elicits a protective immune response against malaria transmission. Cell. 2014; 159:1277–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bornstein DL, Schiffman G, Bernheimer HP, Austrian R. Capsulation of pneumococcus with soluble C‐like (Cs) polysaccharide. I. Biological and genetic properties of Cs pneumococcal strains. J Exp Med. 1968; 128:1385–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sorensen UB, Henrichsen J. Cross‐reactions between pneumococci and other streptococci due to C polysaccharide and F antigen. J Clin Microbiol. 1987; 25:1854–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jensen RK, Pihl R, Gadeberg TAF, Jensen JK, Andersen KR, Thiel S, et al. A potent complement factor C3‐specific nanobody inhibiting multiple functions in the alternative pathway of human and murine complement. J Biol Chem. 2018; 293:6269–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Laursen NS, Pedersen DV, Gytz H, Zarantonello A, Bernth Jensen JM, Hansen AG, et al. Functional and structural characterization of a potent C1q inhibitor targeting the classical pathway of the complement system. Front Immunol. 2020; 11:1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pedersen H, Jensen RK, Hansen AG, Gadeberg TAF, Thiel S, Laursen NS, et al. A C3‐specific nanobody that blocks all three activation pathways in the human and murine complement system. J Biol Chem. 2020; 295:8746–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ho J, Tumkaya T, Aryal S, Choi H, Claridge‐Chang A. Moving beyond P values: data analysis with estimation graphics. Nat Methods. 2019; 16:565–6. [DOI] [PubMed] [Google Scholar]
- 48. Geno KA, Gilbert GL, Song JY, Skovsted IC, Klugman KP, Jones C, et al. Pneumococcal capsules and their types: past, present, and future. Clin Microbiol Rev. 2015; 28:871–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Karlsson C, Jansson PE, Skov Sorensen UB. The pneumococcal common antigen C‐polysaccharide occurs in different forms. Mono‐substituted or di‐substituted with phosphocholine. Eur J Biochem. 1999; 265:1091–7. [DOI] [PubMed] [Google Scholar]
- 50. Vitharsson G, Jonsdottir I, Jonsson S, Valdimarsson H. Opsonization and antibodies to capsular and cell wall polysaccharides of Streptococcus pneumoniae . J Infect Dis. 1994; 170:592–9. [DOI] [PubMed] [Google Scholar]
- 51. Robbins JB, Austrian R, Lee CJ, Rastogi SC, Schiffman G, Henrichsen J, et al. Considerations for formulating the second‐generation pneumococcal capsular polysaccharide vaccine with emphasis on the cross‐reactive types within groups. J Infect Dis. 1983; 148:1136–59. [DOI] [PubMed] [Google Scholar]
- 52. Sørensen UB, Henrichsen J. C‐polysaccharide in a pneumococcal vaccine. Acta Pathol Microbiol Immunol Scand C. 1984; 92:351–6. [DOI] [PubMed] [Google Scholar]
- 53. Xu Q, Abeygunawardana C, Ng AS, Sturgess AW, Harmon BJ, Hennessey JP Jr. Characterization and quantification of C‐polysaccharide in Streptococcus pneumoniae capsular polysaccharide preparations. Anal Biochem. 2005; 336:262–72. [DOI] [PubMed] [Google Scholar]
- 54. Sorensen UB, Henrichsen J, Chen HC, Szu SC. Covalent linkage between the capsular polysaccharide and the cell wall peptidoglycan of Streptococcus pneumoniae revealed by immunochemical methods. Microb Pathog. 1990; 8:325–34. [DOI] [PubMed] [Google Scholar]
- 55. Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G, et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature. 2002; 416:291–7. [DOI] [PubMed] [Google Scholar]
- 56. Djoumerska‐Alexieva IK, Dimitrov JD, Voynova EN, Lacroix‐Desmazes S, Kaveri SV, Vassilev TL. Exposure of IgG to an acidic environment results in molecular modifications and in enhanced protective activity in sepsis. FEBS J. 2010; 277:3039–50. [DOI] [PubMed] [Google Scholar]
- 57. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004; 172:2731–8. [DOI] [PubMed] [Google Scholar]
- 58. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood. 2009; 113:3716–25. [DOI] [PubMed] [Google Scholar]
- 59. Hodžić A, Mateos‐Hernández L, de la Fuente J, Cabezas‐Cruz A. α‐Gal‐based vaccines: advances, opportunities, and perspectives. Trends Parasitol. 2020; 36:992–1001. [DOI] [PubMed] [Google Scholar]
- 60. Cabezas‐Cruz A, Mateos‐Hernández L, Alberdi P, Villar M, Riveau G, Hermann E, et al. Effect of blood type on anti‐α‐Gal immunity and the incidence of infectious diseases. Exp Mol Med. 2017; 49:e301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Antimicrobial resistance: global report on surveillance 2014. 2014. https://www.who.int/antimicrobial‐resistance/publications/surveillancereport/en/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Data relating to the manuscript will be available upon reasonable request to the corresponding author.