

Abstract
Due to their very good chemical and proteolytic stability, ability to penetrate cell membranes, and resemblance to a peptide bond, carbamate derivatives have received much attention in recent years and got an important role in modern drug discovery and medicinal chemistry. Today, carbamates make structural and/or functional part of many drugs and prodrugs approved and marketed for the treatment of various diseases such as cancer, epilepsy, hepatitis C, HIV infection, and Alzheimer’s disease. In drugs they can play a role in drug-target interaction or improve the biological activity of parent molecules. In prodrugs they are mainly used to delay first-pass metabolism and enhance the bioavailability and effectiveness of compounds. This brief review takes a look at the properties and use of carbamates in various fields of medicine and provides quick insights into the mechanisms of action for some of them.
Key words: anticonvulsants, antiepileptics, cholinesterase inhibitors, neurodegenerative diseases, prodrugs, protease inhibitors
Abstract
Zbog svoje vrlo dobre kemijske i proteolitičke stabilnosti, sposobnosti prodiranja kroz stanične membrane i sličnosti s peptidnom vezom, derivati karbamata posljednjih godina sve više privlače pozornost medicinskih kemičara i dobivaju važniju ulogu u modernom načinu otkrivanja lijekova. Tako je u današnje vrijeme karbamatna skupina strukturni i funkcionalni element mnogih odobrenih lijekova, a mnogi se već i koriste kao lijekovi za liječenje raznih vrsta bolesti poput raka, epilepsije, hepatitisa C, infekcije HIV-om, Alzheimerove bolesti i mnogih drugih. U lijekovima, karbamatna skupina može biti važan dio molekule koji ima ulogu u interakciji lijek-meta ili je umetnuta u strukturu spoja kako bi se poboljšala biološka aktivnost temeljne molekule. U protulijekovima, karbamatna se skupina koristi uglavnom zbog mogućnosti smanjenja osjetljivosti spoja na metaboličke enzime, odnosno povećanja hidrolitičke stabilnosti samoga spoja. U ovom je radu dan ne samo kratki pregled karbamata koji se koriste kao lijekovi u raznim područjima primjene, kao i karbamata koji se koriste kao protulijekovi, nego i uvid u mehanizam djelovanja nekih od njih.
Ključne Riječi: antiepileptici, antikonvulzanti, inhibitori kolinesteraza, inhibitori proteaza, neurodegenerativne bolesti, protulijekovi
Carbamates are derivatives of carbamic acid, whose amino and carboxyl termini are substituted by a variety of structurally diverse alkyl, aryl, or alkyl-aryl substituents and are identified by the presence of the -O-CO-NH- linkage (1). Carbamates are integral part of many drugs and prodrugs approved by the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). Many compounds that contain a carbamate group are currently in various stages of preclinical and clinical trials (1, 2). An example of a carbamate compound recently approved by the FDA is cenobamate (3), indicated for the treatment of partial-onset seizures in adults (4).
The research of carbamates as potential drugs began with the discovery of physostigmine, a methyl carbamate ester isolated in pure form from the seeds of Calabar beans (Physostigma venenosum) in 1864 (5). Initially, physostigmine was used to treat high eye pressure and glaucoma, but today, it is also used to treat delayed gastric emptying and myasthenia gravis (5, 6). Broader utilisation of carbamate compounds began in 1959, when carbaryl was registered as first carbamate pesticide for use in the USA (5). Today, carbamate compounds are widely used as pesticides (insecticides, fungicides, and herbicides), as starting materials in the production of paints and polyurethanes, and as protecting groups of amines in organic synthesis (7).
Studies have shown that the carbamate group increases the biological activity of active pharmacophores of structurally different natural or synthesised compounds (7, 8). By varying the substituents at the amino and carboxyl termini of the carbamate group, it is possible to modulate their biological and pharmacokinetic properties and improve their stability (7, 8).
The chemical properties of carbamates, their conformational and metabolic stability, the ability to pass through cell membranes, and some through the blood-brain barrier, have made the carbamate group a desirable part of the structure of many pharmacologically important compounds and a structural motif of many drugs and prodrugs.
Moreover, strong growth of carbamate use in the pharmaceutical industry is additionally boosted by the fact that the carbamate group can be considered a structural analogue of the amide bond (8, 9, 10). Amide-based molecules make a very good starting point for developing many drugs, enzyme inhibitors in particular. However, their inadequate pharmacokinetic properties, most notably poor in vivo stability and low bioavailability, limit their broader use and development. These limitations of amide-based compounds have recently been addressed by the use of carbamates as amido- or peptidomimetics to improve drug potency, duration of action, or target specificity (2, 9, 10, 11).
Today, the carbamate group makes part of many approved drugs which act as chemotherapeutic agents (mitomycin C, irinotecan), cholinesterase inhibitors in the treatment of neurodegenerative disorders (rivastigmine, neostigmine, physostigmine, pyridostigmine), human immunodeficiency virus (HIV) protease inhibitors (ritonavir, amprenavir, atazanavir, darunavir), anticonvulsants (felbamate, retigabine, cenobamate), anthelmintics (febendazole , albendazole, febantelmebendazole), and muscle relaxants (methocarbamol, metoxalone) (Figure 1). They also make part of prodrugs with different therapeutic applications (irinotecan, bambuterol, gabapentin encabril, capecitabine) (2, 7, 12) (Figure 1).
Figure 1.

Structures of carbamate-based drugs and prodrugs of different application (carbamate group is presented in blue; active substance of prodrugs is presented in red)
The aim of this review is to take a brief look at carbamate proprieties that make them useful in drug design, discuss some of the carbamate-based drugs and prodrugs, and explain the mechanisms of action for some of them.
Carbamate properties and applications
Chemical properties of carbamates
The structure of biologically active carbamates is shown in Figure 2. The carbamate group consists of a carbonyl group (C=O) to which an alkoxyl group (OR1 in Figure 2, marked blue) and an amino group (R2NR3 in Figure 2, marked red) are attached. R1, R2, and R3 may be different alkyl, aryl, and alkyl-aryl or substituted alkyl, aryl, and alkyl-aryl groups (2, 7). Replacing the carbonyl oxygen atom with sulphur generates thiocarbamates, while additional replacement of the alkoxy oxygen with a sulphur atom generates dithiocarbamates (2).
Figure 2.

Possible resonance structures for the carbamate group (amino group is presented in red, and alkoxy group in blue) (adopted from ref. 14)
The carbamate group owes its functionality to the structural similarity between amides (R2NR3-CO-R1) and carbamates. Namely, carbamates can be structurally considered as “amide-ester” hybrids with chemical reactivity comparable to these two functional groups. Due to this amide-ester combination, carbamates are chemically stable and able to modify inter- and intramolecular interactions (13).
Carbamate stability stems from the resonance between the amide and carboxyl group, which has been studied theoretically and experimentally by estimating the C-N bond rotational barriers (14). Figure 2 shows three resonance structures obtained from carbamate group stabilisation. The carbamate rotational barrier of the C-N bond is about 3–4 kcal/mol (15–20 %) lower than the rotational barrier of structurally analogue amides due to steric and electron perturbations which result from the presence of additional oxygen of the carboxyl group (14, 15, 16) and which make carbamates more electrophilic than amides and sufficiently reactive to spontaneously react with nucleophiles (9).
Another important feature of carbamates is their conformation. Due to a pseudo double bond in their structure, carbamate molecules can exist as cis and trans isomers (Figure 3) (17, 18), but they show no preference for either isomeric form, as the difference in free energy of the isomers is small, about 1–1.5 kcal/mol due to the steric and electrostatic properties of the substituents.
Figure 3.

Cis and trans conformations of carbamates (adopted from ref. 14)
However, this difference in free energy and consequently the ratio of the two conformations can change with conditions like type and/or composition of the solvents, concentration of salts, and pH of the reaction mixture (17, 18). For example, decreasing the pH of a reaction mixture in which amino acids are protected by the tert-butyl carbamate (Boc) group, increases the concentration of cis isomers, while increasing the temperature increases the concentration of trans isomers (19). One study (20) which used a carbamate group to connect different ammonium groups to the entrance of the gramicidin ion channel, demonstrated that the ratio of cis/trans isomers depended on combined electrostatic and steric effects of the carbamate linker on cation flux through the channel. This suggests that a compound able to change the carbamate cis/trans ratio may also be used to regulate ion channel flux (13). R and R1 (Figure 3) substituents must also be taken into account, as the steric effects of R and electronegativity of R1 influence the difference in free energy and therefore the cis/trans ratio.
Five-, six-, and seven-membered cyclic carbamates can only exist as trans conformers. Five- or six-membered carbamates are quite stable because they generally do not undergo metabolic ring opening (17).
Furthermore, carbamates are semi-polar compounds that can form hydrogen bonds, both as hydrogen donors and as hydrogen acceptors, and various interactions can take place at their O- and N-termini. Carbamates contain C=O and N-C dipoles arising from covalent bonding of electronegative oxygen and nitrogen atoms with electroneutral carbon atoms. Because of the π-bonding arrangement of carbonyl and greater electronegativity of oxygen, carbonyl is a stronger dipole than the N-C dipole. The presence of a C=O dipole allows carbamates to act as H-bond acceptors, whereas the N-C dipole allows them to act as H-bond donors, but to a lesser extent (9).
Unlike amides, carbamates are proteolytically stable against various proteases (9), and can even inhibit them as discussed later. Thanks to these properties – proteolytic, chemical, and conformational stability and ability to pass through the cell membrane and blood brain barriers (not all) – carbamates are increasingly replacing peptides in pharmaceuticals (7).
Their pharmacological activity mostly depends on the speed and intensity of their hydrolysis (21), and their major hydrolysis pathway in physiological conditions is base hydrolysis. The mechanisms of base hydrolysis of monosubstituted and disubstituted carbamates are shown in Figure 4 (22). The difference between these two mechanisms is in the intermediate, which is an isocyanate anion in monosubstituted carbamates and a carbonate anion in disubstituted carbamates. Following basic hydrolysis, parent alcohol and carbamic acid are released, and carbamic acid rapidly decomposes to the corresponding amine and carbon dioxide (21, 22). Carbamate derivatives could be used as prodrugs for amines or alcohols and phenols to delay first-pass metabolism and enhance hydrolytic stability of compounds (23).
Figure 4.

Alkaline hydrolysis of monosubstituted (A) and disubstituted (B) carbamates (adopted from ref. 22)
A recent review by Vacondio et al. (21) has compiled substantial data from recent studies of metabolic stability of therapeutic carbamates to evaluate the qualitative relationship between molecular structure of carbamates and their susceptibility to metabolic hydrolysis and has proposed the following order in metabolic resistance: aryl-OCO-NHalkyl ˃˃ alkyl-OCO-NHalkyl ~alkyl-OCO-N(alkyl)2 ≥ alkyl-OCO-N(endocyclic) ≥ aryl-OCO-N(alkyl)2 ~ aryl-OCO-N(endocyclic) ≥ alkyl-OCONHAryl ~ alkyl-OCO-NHacyl ˃˃ alkyl-OCO-NH2 > cylclic carbamates (21).
Application of carbamates
Carbamate derivatives have a wide range of applications. Their first massive use in agriculture as pesticides, fungicides, and insecticides in various crops all around the world started in the 1950s (25, 26, 27). They have been the second most common pesticides and a welcome replacement of poisonous organochloride pesticides since the 1970s. Even so, some of them are assumed to be potentially carcinogenic and mutagenic or display acute toxicity for mammals and aquatic organisms (28, 29, 30). Most carbamates used in agriculture represent an issue for the environment with more than 10,000 tonnes deployed each year, which makes their clean-up one of environmental priorities (30).
Mass use of carbamates in everyday life was expanded by the discovery of polyurethanes (PUs) – compounds with polymerised carbamate groups whose biological, chemical, and physical properties enabled carbamate application in surface coatings, synthetic fibres, elastomers, foams, and packaging (31). It must be mentioned that ethyl carbamate, also known as urethane, has no connection with polyurethanes in any way. Urethanes are commonly synthesised from an alcohol and an isocyanate, while carbamate synthesis usually does not involve isocyanate as reactant (32).
Carbamates are also used to protect amino groups, most notably tert-butoxycarbonyl (Boc), benzyloxycarbonyl (Cbz), and allyloxycarbonyl (Alloc) (33). As starting material, carbamates have also a leading role in the paint industry (8, 34).
In recent years, carbamate derivatives, including urethanes (especially five- or six-membered cyclic and bicyclic fused carbamates) have seen an expansion to pharmaceutical industry as important structural and functional elements in the design of drugs and prodrugs (32, 35).
Carbamates as drugs
The biological activity of a carbamate was first reported by European missionaries in West Africa in the 19th century. There a local tribe was using a white extract from Calabar beans (Physostigma venenosum) as an ordeal poison in witchcraft trials (36). These beans were imported to Great Britain in 1840, and in 1864, Jobst and Hesse isolated an active alkaloid component they named physostigmine (37). Physostigmine is a naturally occurring methyl carbamate ester initially used as a medicine to treat glaucoma, but its application broadened over the years. Physostigmine was reported to markedly improve muscle strength in patients with myasthenia gravis and had been the drug of choice for this condition for three decades, until it was replaced by a longer-acting oral agent pyridostigmine. Today, physostigmine is also used to treat delayed gastric emptying and anticholinergic poisoning caused by overdosing with atropine, scopolamine, and other anticholinergic drugs. Furthermore, pyridostigmine seems to improve long-term memory, which is why it was clinically studied for treatment of Alzheimer’s disease but was abandoned due to moderate to severe side effects in favour of other acetylcholinesterase inhibitors (38, 39).
Because of their chemical and proteolytic stability, ability to create inter- and intramolecular interactions, and bond resembling that of amides, carbamates soon piqued the interest of pharmaceutical industry (2, 7, 9). Recent studies (2, 8) have shown that incorporating the carbamate group in a molecule increases the biological activity of active pharmacophores of structurally different natural or synthesised compounds. For example, replacing the unsaturated ester chain at C-6 in fumagillin (a natural antibiotic and endothelial cell proliferation inhibitor) with the O-(chloroacetyl) carbamoyl moiety resulted in a 50 times more potent antitumor compound. Another example is betulinic acid, a very promising anticancer drug (8), whose imidazole and triazole carbamate derivatives were 12 times more potent and less cytotoxic (2). This possibility to change the biological and pharmacokinetic properties and improve the stability of parent compounds by varying the substituents on the O- and/or N- termini of a carbamate group has recently boosted the interest in developing efficient and safe synthesis of carbamate esters (8). Today, many drugs incorporate the carbamate moiety, whose roles in some drugs and prodrugs are listed in Table 1.
Table 1.
Roles of the carbamate moiety in drugs and prodrugs
| Drug | The role of the carbamate moiety in the drug | Reference |
|---|---|---|
| Docetaxel | prolongs drug action, increases drug potency, improves water solubility | 39 |
| Mytomicin C | participates in the formation of an alkylating compound during reaction with target | 40 |
| Rivastigmine, neostigmine, physostigmine, pyridostigmine | key element for interaction with the target | 38 |
| Ritonavir, amprenavir, atazanavir, darunavir | improves drug bioavailability and potency, engaged in a backbone interaction with protease | 8 |
| Ombitasvir, elbasvir, daclatasavir | improves drug stability and lipophilicity | 41 |
| Febendazole, mebendazole, febantel, albendazole | improves aqueous solubility and bioavailability, increases cytotoxicity | 42 |
| Mehocarabamol, metaxalone | inhibits acetylcholinesterase at synapses in the autonomic nervous system, neuromuscular junction, and central nervous system | 43 |
| Felbamate | improves drug stability and bioavailability | 44, 45 |
| Retigabine | major pharmacophore responsible for interacting with residues in the KCNQ2–5 channels | 46 |
| Gabapentin enacarbil | improves bioavailability | 47 |
| Capecitabine | improves selectivity and bioavailability | 47 |
| Bambuterol | delays first-pass metabolism | 47 |
| Irinotecan | improves aqueous solubility | 47 |
Carbamates in tumour treatment
Two carbamate drugs, mitomycin C and docetaxel, have so far been approved for the treatment of various types of tumours (Figure 1). Both can be used alone or in combination to other antitumor drugs (48, 49, 50). Their antitumor activity stems from their ability to selectively inhibit the synthesis of DNA in a tumour cell or to inhibit the tubulin polymerisation, both resulting in the arrest of mitotic phase of cell division (48).
Mitomycin C is a miscellaneous antibiotic that selectively inhibits DNA synthesis in a tumour cell and is indicated for chemotherapy of gastrointestinal, anal, and breast cancers (49). Its mechanism of action is given in detail in Figure 5 (40). All starts with an in situ bioreductive activation of quinone that involves two consecutive reduction steps to the corresponding hydroquinone. The elimination of methanol from hydroquinone produces a reactive imine. Deprotonation of imine results in an indole derivative that undergoes rearrangement to produce a quinone methide, which then reacts with nucleophilic DNA groups, yielding an unstable intermediate that binds to complementary strands of double-stranded DNA coils. This binding inhibits DNA replication and causes tumour cell death. At high concentrations, mitomycin C has also been shown to suppress RNA and protein synthesis, inhibit proliferation of B-cells, T-cells, and macrophages in vitro, and to reduce the secretion of interferon gamma, tumour necrosis factor alpha (TNFα), and interleukin 2 (IL-2) (40, 51).
Figure 5.

Mechanism of action of mitomycin C (adopted from ref. 40)
Docetaxel (Figure 1) is a chemotherapeutic used to treat breast, head/neck, stomach, prostate, and lung cancers. It binds to the β-subunit of tubulin and forms the docetaxel-tubulin complex, which interferes with tubulin polymerisation and, in turn, leads to tumour cell cycle arrest and apoptosis, including the apoptosis of B-cells affected by leukaemia (50, 52).
Carbamates in HIV infection treatment
HIV-1 protease is essential for viral maturation, as it cleaves newly synthesised polyproteins Gag and Gag-Po to create mature protein components of the HIV virion, the infectious form of the virus outside the host cell (53). There are ten HIV protease inhibitors approved by the FDA, four of which have the carbamate group in their structure (ritonavir, atazanavir, amprenavir, and darunavir). These bind directly to the active site of HIV-1 protease to prevent Gag and Gag-Po cleavage (53, 54, 55, 56, 57, 58). One of them, ritonavir, was later found that not only it inhibits HIV protease but can also boost blood concentrations of other HIV protease inhibitors by inhibiting cytochrome P450 3A4, which would otherwise metabolise them and render inefficient (54, 55, 56). Another HIV protease inhibitor, atazanavir, shows good oral bioavailability allowing a once-a-day dosing. It is used only in combination with ritonavir and/or other antiviral drugs (55, 56) and as such currently makes the first-line antiretroviral treatment (56, 57).
Carbamates as antiepileptics and anticonvulsants
Carbamate anticonvulsants used in epilepsy treatment are felbamate and retigabine (Figure 1). Their exact mechanism of action is still unclear but what is known is that they inhibit N-methyl-D-aspartate (NMDA) receptors to some extent and slightly enhance gamma-aminobutyric acid (GABA) activity (59, 60).
Felbamate is potent and very effective anticonvulsant approved by FDA in 1993 for the management of focal seizures and Lennox-Gastaut syndrome. It is effective as monotherapy or add-on to phenytoin and carbamazepine in patients with uncontrolled focal epilepsy (61, 62). However, its approval has now been limited because its use is associated with the development of aplastic anaemia and hepatic failure. It is now available in the USA only for a very limited use, principally by neurologists in patients for whom potential benefit outweighs the risk (60). It is believed that felbamate acts as NMDA receptor-ionophore complex antagonist at the strychnine-insensitive glycine-recognition site (62), as it blocks the effects of excitatory amino acids, suppresses neuronal firing, and prevents seizure. As for GABA receptors, some studies suggest that felbamate weakly inhibits GABAA-receptor binding sites and thus enhances GABA-elicited Cl- currents in cultured cortical neurons. This may explain how felbamate dampens neuronal excitation and inhibits voltage-gated sodium and calcium channels (Figure 6) producing a barbiturate-like effect (63, 64). However, one in vitro receptor-binding study reported that felbamate did not enhance GABA evoked 36Cl- influx in cultured spinal cord neurons (65). These discrepancies between reports may lie in varying regional and ontogenetic expression of GABA receptor subunits and varying properties of GABA receptors between central nervous system (CNS) regions and stages of development (66).
Figure 6.

Proposed targets and mechanism of action of felbamate and retigabine in postsynaptic neuron (adopted from refs. 62 and 63)
Retigabine is an anticonvulsant used as adjunct in the treatment of partial seizures in adult patients, tinnitus, migraine, and neuropathic pain (45, 46, 67). The mechanism of its action is unique among antiepileptic drugs and represents a new approach in the treatment of neurological conditions. Retigabine works primarily by opening a particular group of voltage-regulated potassium ion channels in brain cells – KCNQ2 and KCNQ3 (Figure 6) – which stabilises the resting membrane potential and regulates electrical neuron excitation to keep it below the threshold. This prevents the onset of epileptiform discharges (46, 67, 68).
Cenobamate is indicated for the treatment of partial-onset seizures in adults (3, 4). It selectively blocks the persistent sodium current of voltage-gated sodium channels (VGSCs) (3, 4). It also acts as a positive allosteric modulator of high-affinity GABAA receptors to stabilise neural circuits of the epileptic hippocampus (69).
Carbamates in the treatment of neurodegenerative diseases
Neurodegenerative diseases are characterised by progressive structural and functional degeneration of the central and/or peripheral nervous system (70). One of their clinical manifestations is the depletion of acetylcholine (ACh), a neurotransmitter in cholinergic synapses, caused by its excessive metabolism to choline and acetic acid mediated by cholinesterases. In humans, there are two cholinesterases: acetylcholinesterase (AChE), whose physiological role is to hydrolyse ACh, and butyrylcholinesterase (BChE), whose physiological role is still unclear, except that it hydrolyses ACh and other esters and scavenges some toxins by reacting with them before they reach AChE (70, 71, 72). Treatment of patients with neurodegenerative diseases who have low ACh levels (Alzheimer’s disease) and disorders of the neuromuscular system (Parkinson’s disease, myasthenia gravis) is focused on alleviating symptoms. Restoring the concentration of ACh by inhibiting AChE (Figure 7) is the primary treatment for cognitive deficits, although more recent studies point to BChE as a new possible target of cholinesterase inhibitors (73, 74, 75). To date, different types of cholinesterase inhibitors have been identified, designed, and synthesised. The first such cholinesterase inhibitor to be clinically approved was a natural carbamate physostigmine (76). Since then, many carbamates have been developed and tested for the treatment of various disorders of cholinergic neurotransmission. Four of them are currently approved for treating neurodegenerative diseases: physostigmine, pyridostigmine, rivastigmine, and neostigmine (Figure 1) (77).
Figure 7.

A proposed simplified mechanism for AChE inhibition by carbamates. Rapid formation of the covalent enzyme-carbamate intermediates, followed by slow regeneration of a free AChE prevents breaking down of acetylcholine in postsynaptic cleft by AChE (adopted from ref. 81)
Physostigmine acts by inhibiting AChE activity and preventing ACh hydrolysis, which in turn increases ACh levels at the synapse and indirectly stimulates nicotinic and muscarinic receptors. As it crosses the blood-brain barrier, it can also treat the effects of atropine and other anticholinergic drug overdoses on the CNS (78, 79).
Pyridostigmine and neostigmine are parasympathomimetics used in the treatment of myasthenia gravis and both inhibit AChE (78, 80). Rivastigmine inhibits both AChE and BChE and is used to treat mild to moderate Alzheimer’s and Parkinson’s dementia (77).
Cymserine is a physostigmine-based compound with a isopropylphenyl instead of a methyl group at the N-4’ position, which makes it 15 times more selective for BChE than AChE (81, 82, 83, 84). In clinical trials it has shown highly promising results in patients with Alzheimer’s disease, but also unacceptable side effects caused by its toxic metabolites (85).
One of the pathological features of Alzheimer’s is the formation of β-amyloid (Aβ) peptides in the cortex of patients with Alzheimer’s. Aβ-peptides are generated by sequential cleavage of β-amyloid precursor protein (APP) by β-secretase and γ-secretase. These two enzymes have therefore become important targets in designing drugs to treat Alzheimer’s disease (8).To overcome the limitations imposed by cymserine metabolite induced side effects in clinical trials, several cymserine derivatives (Figure 8) have been tested in vivo and shown to increase ACh levels in the brain, produce nootropic effects, and reduce APP and Aβ-peptide levels. They have the potential to become the first drugs capable of stopping and even reversing the progression of Alzheimer’s disease (86).
Figure 8.
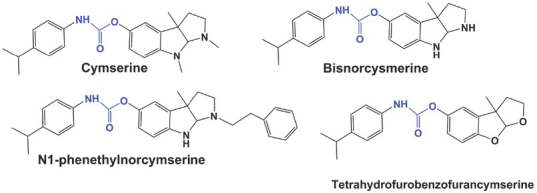
Cymserine and its derivatives (carbamate group in blue) (adopted from ref. 82)
Many other carbamate compounds have been designed as potential secretase inhibitors, but none have yet been approved for medical use. Some have shown great β-secretase inhibition potential, like the 16-membered macrocycle (compound A in Figure 9) containing a trans-olefin, amide, and carbamate functionalities. Others, like sulphonamide derivatives (B in Figure 9) have shown strong γ-secretase inhibition (8).
Figure 9.

Chemical structure of secretase inhibitors tested with potential to be used in treatment of Alzheimer’s disease A – a 16-membered macrocycle compound; B – sulphonamide compound (adopted from ref. 8)
Carbamates in the treatment of hepatitis C
Hepatitis C virus (HCV) causes acute and chronic mild to severe hepatitis. With time, a significant number of chronic patients develop cirrhosis or liver cancer (87).
Recent years have seen the development of a number of new and effective hepatitis C drugs which specifically target viral NS3/4A protease, non-structural protein 5A (NS5A), or NS5A RNA polymerase to inhibit viral replication, and counter NS5A-associated interferon-resistance as the common cause of treatment failure (87, 88, 89). Today, there are ten carbamates targeting NS5A. Six have been approved for use (89) and the rest are in phase III of clinical trials (89, 90). They inhibit NS5A by blocking signalling interactions, redistributing NS5A from the endoplasmic reticulum to the surface of lipid droplets, and by inhibiting HCV replication (9, 92). Figure 1 shows the structures of three approved carbamate HCV inhibitors: daclatasavir, elbasvir, and ombitsavir (93).
Antiviral action is further increased by combining NS5A with NS3/4A protease inhibitors, as is the case with daclatasavir and sofosbuvir (92, 93, 94, 95). Both daclatasavir and sofosbuvir and their combination have been included in the WHO model list of the most efficacious, safe, and cost-effective medicines for a specific condition, the so called essential medicines list (EML) (96).
Carbamates as muscle relaxants
The best known carbamate muscle relaxants are methocarbamol and metaxalon (Figure 1) (43). Although the exact mechanism of action of methocarbamol is still not clarified, it is believed to involve AChE inhibition at synapses in the autonomic nervous system, neuromuscular junction, and the CNS (97). Metaxalone is usually prescribed as adjuvant therapy that accompanies rest, physical therapy, and other measures to relieve acute musculoskeletal pain. Its mode of action is also not clear, but is most likely related to general CNS depression (98).
Carbamates as anthelmintics
The carbamates that act against parasitic worms or helminths in humans and animals are fenbendazole, febantel, mebendazole, and albendazole (Figure 1) (99). Mebendazole and albendazole are benzimidazole carbamate anthelmintics used to treat a broad range of parasitic infections (100, 101). Their mechanism of action is analogous to that of docetaxel: they selectively inhibit microtubule synthesis by binding to β-tubulin and blocking the polymerisation of tubulin dimers in intestinal parasite cells. This disruption of cytoplasmic microtubules results in blocking glucose and other nutrient transport in parasites cells and their gradual immobilisation and death (101).
Carbamates as prodrugs
Prodrugs are designed to undergo enzymatic and/or chemical metabolism in the body to release the active drug with a desired pharmacological effect (Figure 10) (47, 102). They serve as carriers to overcome physicochemical, biopharmaceutical, or pharmacokinetic limitations of active drugs by increasing their solubility in water and absorption, by delaying their first-pass metabolism, by delivering them to the brain, or by reducing their toxicity and local irritation (103). Prodrugs generally consist of a parent (active) drug and a promoiety that masks the functional group within the parent drug (Figure 10). Typically, a promoiety is attached to the active drug with bond(s) that break in certain conditions, such as the presence of an enzyme or a change in pH and releasing the active substance to do its job (103). Esters are used as prodrugs to improve lipophilicity and permeability through cell membranes, while phosphate esters, oximes, and amides are used to improve drug solubility in water and oral absorption (47, 103).
Figure 10.

A simplified illustration of the prodrug concept
Carbamates are used in the design of prodrugs of carboxyl, hydroxyl, or amine functionalities to delay first-pass metabolism and enhance systemic hydrolytic stability (103) (Table 1). Their bioconversion for active drug release involves rapid hydrolysis mediated by metabolic enzymes, mainly cytochrome P450 and esterases, which usually occurs in the liver (22, 47,103).
Carbamate prodrugs with a phenol as active substance
Carbamates are used as prodrugs of alcohols and phenols to achieve systemic hydrolytic stability and protection from first-pass metabolism (23). Carbamates of N-monosubstituted and N, N-disubstituted alcohols are chemically stable against hydrolysis (21), as are the carbamates of N, N-disubstituted phenols but not as much those of N-monosubstituted phenols (21, 24). Examples of carbamate prodrugs whose active substance is alcohol or phenol are irinotecan and bambuterol (Figure 1).
Irinotecan (CPT-11) is a semisynthetic analogue of the natural alkaloid camptothecin, commonly used for the treatment of colon, rectal, and ovarian cancers (104, 105). In human body, irinotecan is metabolised by tissue and serum carboxylesterases to an active compound SN-38 (106), which has a 100–1,000 times higher antitumour activity than irinotecan. SN-38 inhibits topoisomerase I activity by stabilising the cleavable complex between topoisomerase I and DNA, which leads to DNA breaks, inhibition of DNA replication, and triggers apoptotic cell death (Figure 11) (106).
Figure 11.

Irinotecan metabolism by carboxylesterases hCE-1 and hCE-2 (adopted from ref. 105)
Bambuterol (Figure 1) is a bis-dimethyl carbamate prodrug of terbutaline, used to treat asthma and chronic obstructive pulmonary disease. Bambuterol belongs to long-acting drugs due to catecholic hydroxyl groups in its structure that are quite resistant to hydrolysis and first-pass metabolism. In the lung tissue, bambuterol is hydrolysed to terbutaline by BChE (Figure 12), and in the liver, it is metabolised to terbutaline under the influence of cytochrome P-450-dependent oxidases (24, 107, 109). Terbutaline is an adrenergic agonist that predominantly stimulates ß-2 receptors to relax the smooth muscle of the bronchus and dilate the airways (107, 108, 109).
Figure 12.

Bambuterol metabolism into tertbutaline by cytochrome p450 and butyrylcholinesterase (BChE) (adopted from ref. 106)
Carbamate prodrugs with an amine as active substance
At physiological pH amine-based drugs or drugs having an amine group in the structure can undergo protonation and may not always be optimally distributed in the body (47, 102). Because of that, polar amino groups are often derivatised to make the compounds neutral or hydrophobic, i.e. more soluble in lipids (47, 102). One of the derivatisation strategies involves introduction of carbamate moiety into drug structure. Currently used amine-based carbamate prodrugs gabapentin enacarbil and capecitabine are presented in Figure 1.
Gabapentin enacarbil is a prodrug designed to increase oral bioavailability of gabapentin used to treat the restless legs syndrome (RLS) and postherpetic neuralgia (PHN) in adults (110). After oral administration gabapentin enacarbil gets strongly hydrolysed by non-specific carboxylesterases, primarily in enterocytes and to a lesser extent in the liver, to form the active drug gabapentin, carbon dioxide, acetaldehyde, and isobutyric acid (110).
Capecitabine is a fluoropyrimidine carbamate of the antimetabolite class of chemotherapeutics (111) that is selectively activated by tumour cells to produce cytotoxic 5-fluorouracil (5-FU) (112), which is then metabolised to its active components 5-fluoro-2-deoxyuridine monophosphate (FdUMP) and 5-fluorouridine triphosphate (FUTP). The first inhibits DNA synthesis and cell division by reducing normal thymidine production, and the second inhibits RNA and protein synthesis by competing with uridine triphosphate for incorporation into the RNA strand (111, 112).
Acknowledgment
This article was supported by the Croatian Science Foundation (project no. IP-2018-01-7683) and the Institute for Medical Research and Occupational Health (project no. IMI-IP-2017-2).
Footnotes
Conflict of interests
None to declare.
References
- 1.Ghosh AK, Brindisi M. Urea derivatives in modern drug discovery and medicinal chemistry. J Med Chem. 2020;63:2751–88. doi: 10.1021/acs.jmedchem.9b01541. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaturvedi D. Brahmachari G. Chemistry and pharmacology of naturally occurring bioactive compounds. 1st ed. Boca Raton (FL): CRC Press; 2013. Role of organic carbamates in anticancer drug design; pp. 117–40. editor. p. doi. [DOI] [Google Scholar]
- 3.Strzelczyk A, Mann C, Willems LM, Rosenow F, Bauer S. Cenobamate for the treatment of focal epilepsies. Expert Opin Pharmacother. 2020 doi: 10.1080/14656566.2020.1803830. doi. [DOI] [PubMed] [Google Scholar]
- 4.Keam SJ. Cenobamate: First approval. Drugs. 2020;80:73–8. doi: 10.1007/s40265-019-01250-6. doi. [DOI] [PubMed] [Google Scholar]
- 5.Adams P, Baron FA. Esters of carbamic acid. Chem Rev. 1965;65:567–602. doi: 10.1021/cr60237a002. doi. [DOI] [Google Scholar]
- 6.Eddleston M, Clark RF. Hoffman RS, Howland MA, Lewin NA, Nelson LS. Goldfrank’s toxicologic emergencies. 9th ed. New York: McGraw-Hill; 2011. Insecticides: organic phosphorus compounds and carbamates; pp. 1450–60. p. [Google Scholar]
- 7.Chaturvedi D. Perspectives on the synthesis of organic carbamates. Tetrahedron. 2012;68:15–45. doi: 10.1016/j.tet.2011.10.001. doi. [DOI] [Google Scholar]
- 8.Ghosh AK, Brindisi M. Organic carbamates in drug design and medicinal chemistry. J Med Chem. 2015;58:2895–940. doi: 10.1021/jm501371s. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeRuiter J.. Amides and related functional groups. Principles of Drug Action 1. 2005 http://webhome.auburn.edu/~deruija/pda1_amides.pdf [displayed 20 April 2019] Available at. [Google Scholar]
- 10.Vagner J, Qu H, Hruby VJ. Peptidomimetics, a synthetic tool of drug discovery. Curr Opin Chem Biol. 2008;12:292–6. doi: 10.1016/j.cbpa.2008.03.009. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Karaman R. Prodrugs design based on inter- and intramolecular chemical processes. Chem Biol Drug Des. 2013;82:643–68. doi: 10.1111/cbdd.12224. doi. [DOI] [PubMed] [Google Scholar]
- 12.Yılmaz S, Akbaba J, Özgeriş B, Polat Köse L, Göksu S, Gülçin I, Alwasel SH, Supuran CT. Synthesis and inhibitory properties of some carbamates on carbonic anhydrase and acetylcholine esterase. J Enzyme Inhib Med Chem. 2016;31:1484–91. doi: 10.3109/14756366.2016.1149477. doi. [DOI] [PubMed] [Google Scholar]
- 13.Moraczewski AL, Banaszynski LA, From AM, White CE, Smith BD. Using hydrogen bonding to control carbamate C-N rotamer equilibria. J Org Chem. 1998;63:7258–62. doi: 10.1021/jo980644d. doi. [DOI] [PubMed] [Google Scholar]
- 14.Kaur D, Sharma P, Bharatam PV. Amide resonance in thioand seleno- carbamates: A theoretical study. J Mol Struct. 2005;757:149–53. doi: 10.1016/j.theochem.2005.09.019. doi. [DOI] [Google Scholar]
- 15.Deetz MJ, Forbes CC, Jonas M, Malerich JP, Smith BD, Wiest O. Unusually low barrier to carbamate C-N rotation. J Org Chem. 2002;67:3949–52. doi: 10.1021/jo025554u. doi. [DOI] [PubMed] [Google Scholar]
- 16.Jung T, Do HJ, Son J, Song JH, Cha W, Kim YJ, Lee KK, Kwak K. Hindered C-N bond rotation in triazinyl dithiocarbamates. J Mol Struct. 2018;1152:215–22. doi: 10.1016/j.molstruc.2017.09.063. doi. [DOI] [Google Scholar]
- 17.Dugave C, Demange L. Cis-trans isomerization of organic molecules and biomolecules: implications and applications. Chem Rev. 2003;103:2475–532. doi: 10.1021/cr0104375. doi. [DOI] [PubMed] [Google Scholar]
- 18.Lauvergnat D, Hiberty PC. Role of conjugation in the stabilities and rotational barriers of formamide and thioformamide. An ab initio valence-bond study. J Am Chem Soc. 1997;119:9478–82. doi: 10.1021/ja9639426. doi. [DOI] [Google Scholar]
- 19.Marcovici-Mizrahi D, Gottlieb HE, Marks V, Nudelman A. On the stabilization of the syn-rotamer of amino acid carbamate derivatives by hydrogen bonding. J Org Chem. 1996;61:8402–6. doi: 10.1021/jo961446u. doi. [DOI] [Google Scholar]
- 20.Woolley GA, Jaikaran ASI, Zhang Z, Peng S. Design of regulated ion channels using measurements of cis-trans isomerization in single molecules. J Am Chem Soc. 1995;117:4448–54. doi: 10.1021/ja00121a002. doi. [DOI] [Google Scholar]
- 21.Vacondio F, Silva C, Mor M, Testa B. Qualitative structure-metabolism relationship in the hydrolysis of carbamates. Drug Metab Rev. 2010;42:551–89. doi: 10.3109/03602531003745960. doi. [DOI] [PubMed] [Google Scholar]
- 22.Reiner E, Škrinjarić-Špoljar M. Enzimska razgradnja karbamata [Carbamate metabolism, in Croatian] Arh Hig Rada Toksikol. 1968;19:251–8. https://hrcak.srce.hr/176452 [displayed 20 March 2019]. Available at. [PubMed] [Google Scholar]
- 23.Mattarei A, Azzolini M, Zoratti M, Biasutto L, Paradisi C.. N-monosubstituted methoxy-oligo(ethylene glycol) carbamate ester prodrugs of resveratrol. Molecules. 2015;20:16085–102. doi: 10.3390/molecules200916085. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parise Filho R, Polli MC, Garcia M, Barberato-Filho S. Prodrugs available on the Brazilian pharmaceutical market and their corresponding bioactivation pathways. Braz J Pharm Sci. 2010;46:393–420. doi: 10.1590/S1984-82502010000300003. doi. [DOI] [Google Scholar]
- 25.King AM, Aaron CK. Organophosphate and carbamate poisoning. Emerg Med Clin North Am. 2015;33:133–51. doi: 10.1016/j.emc.2014.09.010. doi. [DOI] [PubMed] [Google Scholar]
- 26.Maki T, Tsuritani T, Yasukata T. A mild method for the synthesis of carbamateprotected guanidines using the Burgess reagent. Org Lett. 2014;16:1868–71. doi: 10.1021/ol5002208. doi. [DOI] [PubMed] [Google Scholar]
- 27.Hong JY, Seo UR, Chung YK. Synthesis of carbamates from amines and N-tosylhydrazones under atmospheric pressure of carbon dioxide without an external base. Org Chem Front. 2016;3:764–7. doi: 10.1039/c6qo00111d. doi. [DOI] [Google Scholar]
- 28.Sogorb MA, Vilanova E. Enzymes involved in the detoxification of organophosphorus, carbamate and pyrethroid insecticides through hydrolysis. Toxicol Lett. 2002;128:215–28. doi: 10.1016/s0378-4274(01)00543-4. doi. [DOI] [PubMed] [Google Scholar]
- 29.Nunes G, Barceló D. Analysis of carbamate insecticides in foodstuffs using chromatography and immunoassay techniques. Trends Anal Chem. 1999;18:99–107. doi: 10.1016/S0165-9936(98)00076-4. doi. [DOI] [Google Scholar]
- 30.Wang Q, Lemley AT. Competitive degradation and detoxification of carbamate insecticides by membrane anodic fenton treatment. J Agric Food Chem. 2003;51:5382–90. doi: 10.1021/jf034311f. doi. [DOI] [PubMed] [Google Scholar]
- 31.Plastics Europe. Plastics-the facts. An analysis of European plastics production, demand and waste data. 2014/2015 https://www.plasticseurope.org/application/files/5515/1689/9220/2014plastics_the_facts_PubFeb2015.pdf [displayed 17 May 2020]. Available at. [Google Scholar]
- 32.Akindoyo JO, Beg MDH, Ghazali S, Islam MR, Jeyaratnam N, Yuvaray AR. Polyurethane types, synthesis and applications-a review. RSC Adv. 2016;6:114453–82. doi: 10.1039/C6RA14525F. doi. [DOI] [Google Scholar]
- 33.Pittelkow M, Lewinsky R, Christensen JB. Selective synthesis of carbamate protected polyamines using alkyl phenyl carbonates. Synthesis. 2002;15:2195–2202. doi: 10.1055/s-2002-34859. doi. [DOI] [Google Scholar]
- 34.Dhanapal D, Rebheka G, Palanivel S, Srinivasan AK. A comparative study on modified epoxy and glycidyl carbamate coatings for corrosion and fouling prevention. Surf Innov. 2015;3:127–39. doi: 10.1680/si.13.00025. doi. [DOI] [Google Scholar]
- 35.Grube A, Donaldson D, Kiely T, Wu L. Pesticides industry sales and usage 2006 and 2007 market estimates. https://www.epa.gov/pesticides/pesticides-industry-sales-and-usage-2006-and-2007-market-estimates [displayed 15 May 2020]. Availabile at. [Google Scholar]
- 36.Wharfe J. Thompson KC, Wadhia K, Loibner AP. Environmental toxicity testing. 1st ed. Oxford: Blackwell Publishing Ltd; 2005. Historical perspective and overview; pp. 1–32. p. [Google Scholar]
- 37.Proudfoot A. The early toxicology of physostigmine. Toxicol Rev. 2006;25:99–138. doi: 10.2165/00139709-200625020-00004. doi. [DOI] [PubMed] [Google Scholar]
- 38.Gupta RC. Gupta RC. Toxicology of oganophosphate & carbamate compounds. 1st ed. Waltham (MA): Academic Press; 2006. Classification and uses of organophosphates and carbamates; pp. 5–24. editor. p. doi. [DOI] [Google Scholar]
- 39.Sun B, Straubinger RM, Lovell JF. Current taxane formulations and emerging cabazitaxel delivery systems. Nano Res. 2018;11:5193–218. doi: 10.1007/s12274-018-2171-0. doi. [DOI] [Google Scholar]
- 40.Avendaño C, Menéndez JC. Avendaño C, Menéndez JC. Medicinal chemistry of anticancer drugs. 2nd ed. Elsevier Science; 2015. Anticancer drugs that interact with the DNA minor groove; pp. 243–71. p. doi. [DOI] [Google Scholar]
- 41.Meanwell NA, Belema M. The discovery and development of daclatasvir: an inhibitor of the hepatitis C virus NS5A replication complex. Top Med Chem. 2019;32:27–56. doi: 10.1007/7355_2018_47. doi. [DOI] [Google Scholar]
- 42.Daniel-Mwambete K, Torrado S, Cuesta-Bandera C, Ponce-Gordo F, Torrado J. The effect of solubilization on the oral bioavailability of three benzimidazole carbamate drugs. Int J Pharm. 2004;272:29–36. doi: 10.1016/j.ijpharm.2003.11.030. doi. [DOI] [PubMed] [Google Scholar]
- 43.See S, Ginzburg R. Choosing a skeletal muscle relaxant. Am Fam Physician. 2008;78:365–70. PMID: 18711953. [PubMed] [Google Scholar]
- 44.Kung CH, Kwon CH. Carbamate derivatives of felbamate as potential anticonvulsant agents. Med Chem Res. 2009;19:498–513. doi: 10.1007/s00044-009-9208-6. doi. [DOI] [Google Scholar]
- 45.Aícua-Rapún I, André P, Rossetti AO, Ryvlin P, Hottinger AF, Decosterd LA, Buclin T, Novy J. Therapeutic drug monitoring of newer antiepileptic drugs: a randomized trial for dosage adjustment. Ann Neurol. 2020;87:22–9. doi: 10.1002/ana.25641. doi. [DOI] [PubMed] [Google Scholar]
- 46.Flynn S, Babi A. Dowd F, Johnson B, Mariotti A. Pharmacology and therapeutics for dentistry. 7th ed. Chapter 12. St. Louis: Elsevier Inc; 2017. Anticonvulsants; pp. 176–92. authors. p. [Google Scholar]
- 47.Rautio J, Meanwell N, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 2018;17:559–87. doi: 10.1038/nrd.2018.46. doi. [DOI] [PubMed] [Google Scholar]
- 48.Ray S, Chaturvedi D. Application of organic carbamates in drug design. Part 1: anticancer agents - recent reports. Drugs Fut. 2004;29:343. doi: 10.1358/dof.2004.029.04.787236. doi. [DOI] [Google Scholar]
- 49.Kim RY, Yau MC, Galpin JD, Seebohm G, Ahern CA, Pless SA, Kurata HT. Atomic basis for therapeutic activation of neuronal potassium channels. Nat Commun. 2015;6:8116. doi: 10.1038/ncomms9116. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Montero A, Fossella F, Hortobagyi G, Valero V. Docetaxel for treatment of solid tumours: a systematic review of clinical data. Lancet Oncol. 2005;6:229–39. doi: 10.1016/S1470-2045(05)70094-2. doi. [DOI] [PubMed] [Google Scholar]
- 51.Wolkenberg SE, Boger DL. Mechanisms of in situ activation for DNA-targeting antitumor agents. Chem Rev. 2002;102:2477–96. doi: 10.1021/cr010046q. doi. [DOI] [PubMed] [Google Scholar]
- 52.Verweij J. Docetaxel (TaxotereTM a new anti-cancer drug with promising potential? Br J Cancer. 1994;70:183–4. doi: 10.1038/bjc.1994.276. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl) 2015;7:95–104. doi: 10.2147/HIV.S79956. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zeldin RK, Petruschke RA. Pharmacological and therapeutic properties of ritonavir-boosted protease inhibitor therapy in HIV-infected patients. J Antimicrob Chemother. 2004;53:4–9. doi: 10.1093/jac/dkh029. doi. [DOI] [PubMed] [Google Scholar]
- 55.Achenbach CJ, Darn KM, Murphy RL. Atazanavir/ritonavir-based combination antiretroviral therapy for treatment of HIV-1 infection in adults. Future Virol. 2011;6:157–77. doi: 10.2217/fvl.10.89. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hull MW, Montaner JS. Ritonavir-boosted protease inhibitors in HIV therapy. Ann Med. 2011;43:375–88. doi: 10.3109/07853890.2011.572905. doi. [DOI] [PubMed] [Google Scholar]
- 57.Croom KF, Dhilloh S, Keam SJ. Atazanavir: a review of its use in the management of HIV-1 infection. Drugs. 2009;69:1107–40. doi: 10.2165/00003495-200969080-00009. doi. [DOI] [PubMed] [Google Scholar]
- 58.Ghosh AK, Dawson ZL, Mitsuya H. Darunavir, a conceptually new HIV-1 protease inhibitor for the treatment of drug-resistant HIV. Bioorg Med Chem. 2007;15:7576–80. doi: 10.1016/j.bmc.2007.09.010. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shi LL, Dong J, Ni H, Geng J, Wu T. Felbamate as an add-on therapy for refractory partial epilepsy. Cochrane Database Syst Rev. 2017;7(7):CD008295. doi: 10.1002/14651858.CD008295.pub4. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glue P, Banfield CR, Perhach JL, Mather GG, Racha JK, Levy RH. Pharmacokinetic interactions with felbamate: In vitro-in vivo correlation. Clin Pharmacokinet. 1997;33:214–24. doi: 10.2165/00003088-199733030-00004. doi. [DOI] [PubMed] [Google Scholar]
- 61.Swinyard EA, Sofia RD, Kupferberg HJ. Comparative anticonvulsant activity and neurotoxicity of felbmate and four prototype antiepileptic drugs in mice and rats. Epilepsia. 1986;27:27–34. doi: 10.1111/j.1528-1157.1986.tb03497.x. doi. [DOI] [PubMed] [Google Scholar]
- 62.Rho JM, Donevan SD, Rogawski MA. Barbiturate-like actions of the propanediol dicarbamates felbamate and meprobamate. J Pharmacol Exp Ther. 1997;280:1383–91. PMID: 9067327. [PubMed] [Google Scholar]
- 63.Rho JM, Donevan SD, Rogawski MA. Mechanism of action of the anticonvulsant felbamate: opposing effects on N-methyl-D-aspartate and γ-aminobutyric acidA receptors. Ann Neurol. 1994;35:229–34. doi: 10.1002/ana.410350216. doi. [DOI] [PubMed] [Google Scholar]
- 64.Kume A, Greenfield Jr LJ, Macdonald RL, Albin RL. Felbamate inhibits [3H]t-butylbicycloorthobenzoate (TBOB) binding and enhances Cl- current at the gamma-aminobutyric AcidA (GABAA) receptor. J Pharmacol Exp Ther. 1996;277:1784–92. PMID: 8667250. [PubMed] [Google Scholar]
- 65.Ticku MK, Kamatchi GL, Sofia RD. Effect of anticonvulsant felbamate on GABAA receptor system. Epilepsia. 1991;32:389–91. doi: 10.1111/j.1528-1157.1991.tb04667.x. doi. [DOI] [PubMed] [Google Scholar]
- 66.Wisden W, Laurie UJ, Monyer H, Seeburg PH. The distribution of thirteen GABAA receptor subunit mRNAs in the rat brain. I. Telencephalon, diencephalon, mesencephalon. J Neurosci. 1992;12:1040–62. doi: 10.1523/JNEUROSCI.12-03-01040.1992. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Porter RJ, Nohria V, Rundfeldt C. Retigabine. Neurotherapeutics. 2007;4:149–54. doi: 10.1016/j.nurt.2006.11.012. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wuttke TV, Seebohm G, Bail S, Maljevic S, Lerche H. The new anticonvulsant retigabine favors voltage-dependent opening of the Kv7.2 KCNQ2 channel by binding to its activation gate. Mol Pharmacol. 2005;67:1009–17. doi: 10.1124/mol.104.010793. doi. [DOI] [PubMed] [Google Scholar]
- 69.Sharma R, Nakamura M, Neupane C, Jeon BH, Shinc H, Melnick SM, Glenn KJ, Jang IS, Park JB. Positive allosteric modulation of GABAA receptors by a novel antiepileptic drug cenobamate. Eur J Pharmacol. 2020;879:173117. doi: 10.1016/j.ejphar.2020.173117. doi. [DOI] [PubMed] [Google Scholar]
- 70.Haake A, Nguyen K, Friedman L, Chakkamparambil B, Grossberg GT. An update on the utility and safety of cholinesterase inhibitors for the treatment of Alzheimer’s disease. Expert Opin Drug Saf. 2020;19:147–57. doi: 10.1080/14740338.2020.1721456. doi. [DOI] [PubMed] [Google Scholar]
- 71.Giacobini E. Cholinesterases and Cholinesterase Inhibitors. London: Martin Dunitz Ltd; 2000. editor. [Google Scholar]
- 72.Bosak A, Katalinić M, Kovarik Z. Cholinesterases: structure, role, and inhibition. Arh Hig Rada Toksikol. 2011;62:175–90. doi: 10.2478/10004-1254-62-2011-2107. doi. [DOI] [PubMed] [Google Scholar]
- 73.Plata-Salaman CR, Zhao B, Teyman RE. Carbamate compounds for use in preventing or treating neurodegenerative disorders. Unites States Patent Application Publication 2002;US 2002/0165273 A1. https://patents.google.com/patent/US20020165273A1/en [displayed 23 November 2020]. Available at. [Google Scholar]
- 74.Darvesh S, Darvesh KV, McDonald RS, Mataija D, Walsh R, Mothana S, Lockridge O, Martin E. Carbamates with differential mechanism of inhibition toward acetylcholinesterase and butyrylcholinesterase. J Med Chem. 2008;51:4200–12. doi: 10.1021/jm8002075. doi. [DOI] [PubMed] [Google Scholar]
- 75.Prince M, Comas-Herrera A, Knapp M, Guerchet M, Karagiannidou M. World Alzheimer Report 2016: improving helatchare for people living with dementia: coverage, quality and costs now and in the future, Alzheimer’s disease International (ADI) 2016 https://www.alz.co.uk/research/WorldAlzheimerReport2016.pdf [displayed 23 November 2020]. Availabile at. [Google Scholar]
- 76.Kovacs GG, Adle-Biassette H, Milenkovic I, Cipriani S, van Scheppingen J, Aronica E. Linking pathways in the developing and aging brain with neurodegeneration. Neuroscience. 2014;269:152–72. doi: 10.1016/j.neuroscience.2014.03.045. doi. [DOI] [PubMed] [Google Scholar]
- 77.Camps P, Muñoz-Torrero D. Cholinergic drugs in pharmacotherapy of Alzheimer’s disease. Mini Rev Med Chem. 2002;2:11–25. doi: 10.2174/1389557023406638. doi. [DOI] [PubMed] [Google Scholar]
- 78.Bitzinger DI, Gruber M, Tummler S, Malsy M, Seyfried T, Weber F, Redel A, Graf BM, Zausig YA.. In vivo effects of neostigmine and physostigmine on neutrophil functions and evaluation of acetylcholinesterase and butyrylcholinesterase as inflammatory markers during experimental sepsis in rats. Mediat Inflamm. 2019;4:ID8274903. doi: 10.1155/2019/8274903. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Frascogna N. Physostigmine: is there a role for this antidote in pediatric poisonings? Curr Opin Pediatr. 2007;19:201–5. doi: 10.1097/MOP.0b013e32802c7be1. doi. [DOI] [PubMed] [Google Scholar]
- 80.Trevisani GT, Hyman NH, Church JM. Neostigmine: safe and effective treatment for acute colonic pseudo-obstruction. Dis Colon Rectum. 2000;43:599–603. doi: 10.1007/BF02235569. doi. [DOI] [PubMed] [Google Scholar]
- 81.Moghul S, Wikinson D. Use of acetylcholinesterase inhibitors in Alzheimer’s disease. Expert Rev Neurother. 2001;1:61–9. doi: 10.1586/14737175.1.1.61. doi. [DOI] [PubMed] [Google Scholar]
- 82.Kamal MA, Klein P, Luo WM, Li YZ, Holloway HW, Tweedie D, Greig NH. Kinetics of human serum butyrylcholinesterase inhibition by a novel experimental Alzheimer therapeutic, dihydrobenzodioxepine cymserine. Neurochem Res. 2008;33:745–53. doi: 10.1007/s11064-007-9490-y. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yu QS, Holloway HW, Utsuki T, Brossi A, Greig NH. Synthesis of novel phenserine-based-selective inhibitors of butyrylcholinesterase for Alzheimer’s disease. J Med Chem. 1999;42:1855–61. doi: 10.1021/jm980459s. doi. [DOI] [PubMed] [Google Scholar]
- 84.Greig NH, Utsuki T, Ingram DK, Wang Y, Pepeu G, Scali C, Yu Q-S, Mamczarz J, Holloway HW, Giordano T, Chen DM, Furukawa K, Sambamurti K, Brossi A, Lahiri DK. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proc Natl Acad Sci USA. 2005;102:17213–8. doi: 10.1073/pnas.0508575102. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guo T, Gu H, Hobbs DW, Rokosz LL, Stauffer TM, Jacob B, Clader JW. Design, synthesis, and evaluation of tetrahydroquinoline and pyrrolidine sulfonamide carbamates as γ-secretase inhibitors. Bioorg Med Chem Lett. 2007;17:3010–3. doi: 10.1016/j.bmcl.2007.03.055. doi. [DOI] [PubMed] [Google Scholar]
- 86.Kamal MA, Qu X, Yu Q, Tweedie D, Holloway HW, Li Y, Tan Y, Greig NH. Tetrahydrofurobenzofuran cymserine, a potent butyrylcholinesterase inhibitor and experimental Alzheimer drug candidate, enzyme kinetic analysis. J Neural Transm (Vienna) 2008;115:889–98. doi: 10.1007/s00702-008-0022-y. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alter MJ. Epidemiology of hepatitis C virus infection. World J Gastroenterol. 2007;13:2436–41. doi: 10.3748/wjg.v13.i17.2436. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chary A, Holodniy M. Recent advances in hepatitis C virus treatment: review of HCV protease inhibitor clinical trials. Rev Recent Clin Trials. 2010;5:158–73. doi: 10.2174/157488710792007293. doi. [DOI] [PubMed] [Google Scholar]
- 89.Hwang J, Huang L, Cordek DG, Vaughan R, Reynolds SL, Kihara G, Raney KD, Kao CC, Cameron CE. Hepatitis C virus nonstructural protein 5A: biochemical characterization of a novel structural class of RNA-binding proteins. J Virol. 2010;84:12480–91. doi: 10.1128/JVI.01319-10. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shi ST, Polyak SJ, Tu H, Taylor DR, Gretch DR, Lai MM. Hepatitis C virus NS5A colocalizes with the core protein on lipid droplets and interacts with apolipoproteins. Virology. 2002;292:198–210. doi: 10.1006/viro.2001.1225. doi. [DOI] [PubMed] [Google Scholar]
- 91.Kohler JJ, Nettles JH, Amblard F, Hurwitz SJ, Bassit L, Stanton RA, Ehteshami M, Schinazi RF. Approaches to hepatitis C treatment and cure using NS5A inhibitors. Infect Drug Resist. 2014;7:41–56. doi: 10.2147/IDR.S36247. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee C. Daclatasvir: potential role in hepatitis C. Drug Des Devel Ther. 2013;7:1220–33. doi: 10.2147/DDDT.S40310. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cada DJ, Kim AP, Baker DE. Elbasvir/Grazoprevir. Hosp Pharm. 2016;51:665–86. doi: 10.1310/hpj5108-665. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Keating GM. Ombitasvir/Paritaprevir/Ritonavir: a review in chronic HCV genotype 4 infection. Drugs. 2016;76:1203–11. doi: 10.1007/s40265-016-0612-1. doi. [DOI] [PubMed] [Google Scholar]
- 95.Belema M, Nguyen VN, Bachand C, Deon DH, Goodrich JT, James CA, Lavoie R, Lopez OD, Martel A, Romine JL, Ruediger EH, Snyder LB, St Laurent DR, Yang F, Zhu J, Wong HS, Langley DR, Adams SP, Cantor GH, Chimalakonda A, Fura A, Johnson BM, Knipe JO, Parker DD, Santone KS, Fridell RA, Lemm JA, O’Boyle DR, Colonno RJ, Gao M, Meanwell NA, Hamann LG. Hepatitis C virus NS5A replication complex inhibitors: the discovery of daclatasvir. J Med Chem. 2014;57:2013–32. doi: 10.1021/jm401836p. doi. [DOI] [PubMed] [Google Scholar]
- 96.World Health Organization. World Health Organization model list of essential medicines: 21st list 2019. Geneva: World Health Organization. https://apps.who.int/iris/handle/10665/325771 [displayed 8 December 2020]. Available at. [Google Scholar]
- 97.O’Doherty DS, Shields CD. Methocarbamol-new agent in treatment of neurological and neuromuscular diseases. J Am Med Assoc. 1958;167:160–3. doi: 10.1001/jama.1958.02990190014003. doi. [DOI] [PubMed] [Google Scholar]
- 98.Anthelmintic benzimidazole carbamates. https://patents.google.com/patent/US4512998A/en [displayed 20 September 2020]. Available at. [Google Scholar]
- 99.Köhler P. The biochemical basis of anthelmintic action and resistance. Int J Parasitol. 2001;31:336–45. doi: 10.1016/s0020-7519(01)00131-x. doi. [DOI] [PubMed] [Google Scholar]
- 100.Campbell WC. The chemotherapy of parasitic infections. J Parasitol. 1986;72:45–61. doi: 10.2307/3281795. doi. [DOI] [PubMed] [Google Scholar]
- 101.Giordani C, Marin GH, Perez D, Soraci A, Errecalde J. Mechanism of action of drugs with activity against multicellular parasites. Parazitologija. 2017;51:294–316. http://sedici.unlp.edu.ar/bitstream/handle/10915/98772/Mechanism_of_action_of_drugs_with_activity_against_multicellular_parasites.pdf-PDFA.pdf?sequence=1&isAllowed=y [displayed 23 November 2020]. Available at. [Google Scholar]
- 102.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T, Savolainen J. Prodrugs: design and clinical applications. Nat Rev Drug Discov. 2008;7:255–70. doi: 10.1038/nrd2468. doi. [DOI] [PubMed] [Google Scholar]
- 103.Hahn KK, Wolff JJ, Kolaser JM. Pharmacogenetics and irinotecan therapy. Am J Health Syst Pharm. 2006;63:2211–7. doi: 10.2146/ajhp060155. doi. [DOI] [PubMed] [Google Scholar]
- 104.Frese S, Diamond B. Structural modification of DNA therapeutic option in SLE. Nat Rev Rheumatol. 2011;7:733–8. doi: 10.1038/nrrheum.2011.153. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mathijessen RH, van Alphen RJ, Verweij J, Loos WJ, Nooter K, Stoter G, Sparreboom A. Clinical pharmacokinetics and metabolism of irinotecan (CPT-11) Clin Cancer Res. 2001;7:2182–94. PMID:11489791. [PubMed] [Google Scholar]
- 106.Sitar DS. Clinical pharmacokinetics of bambuterol. Clin Pharmacokinet. 1996;31:246–56. doi: 10.2165/00003088-199631040-00002. doi. [DOI] [PubMed] [Google Scholar]
- 107.Zhou T, Liu S, Zhao T, Zeng J, He M, Xu B, Qu S, Xu L, Tan W. Chiral analysis of bambuterol, its intermediate and active drug in human plasma by liquid chromatography-tandem mass spectrometry: application to a pharmacokinetic study. J Chromatogr B Analyt Technol Biomed Life Sci. 2015;997:38–44. doi: 10.1016/j.jchromb.2015.05.024. doi. [DOI] [PubMed] [Google Scholar]
- 108.Svensson LA, Tunek A. The design and bioactivation of presystemically stable prodrugs. Drug Metab Rev. 1998;19:165–94. doi: 10.3109/03602538809049622. doi. [DOI] [PubMed] [Google Scholar]
- 109.Yaltho TC, Ondo WG. The use of gabapentin enacarbil in the treatment of restless legs syndrome. Ther Adv Neurol Disord. 2010;3:269–75. doi: 10.1177/1756285610378059. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guerreiro C, Albuquerque L, Reimão S. Radiation recall myelitis following capecitabine: first case report. Clin Neurol Neurosurg. 2020;196:105978. doi: 10.1016/j.clineuro.2020.105978. doi. [DOI] [PubMed] [Google Scholar]
- 111.Terranova-Barberio M, Roca M, Zotti A, Leone A, Bruzzese F, Vitagliano C, Scogliamiglio G, Russo D, D’Angelo G, Franco R, Budillon A, Gennaro E. Valproic acid potentiates the anticancer activity of capecitabine in vitro and in vivo in breast cancer models via induction of thymidine phosphorylase expression. Oncotarget. 2016;7:7715–31. doi: 10.18632/oncotarget.6802. doi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Longley DB, Harkin DP, Johnston PG. 5-Fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330–8. doi: 10.1038/nrc1074. doi. [DOI] [PubMed] [Google Scholar]