

Abstract
Purpose:
Clear cell renal cell carcinoma (ccRCC) is frequently associated with inactivation of the von Hippel Lindau tumor suppressor, resulting in activation of HIF-1α and HIF-2α. The current paradigm, established using mechanistic cell-based studies, supports a tumor promoting role for HIF-2α, and a tumor suppressor role for HIF-1α. However, few studies have comprehensively examined the clinical relevance of this paradigm. Furthermore, the hypoxia associated factor (HAF), which regulates the HIFs, has not been comprehensively evaluated in ccRCC.
Experimental design:
To assess the involvement of HAF/HIFs in ccRCC, we analyzed their relationship to tumor grade/stage/outcome using tissue from 380 patients, and validated these associations using tissue from 72 additional patients and a further 57 patients treated with antiangiogenic therapy for associations with response. Further characterization was performed using single cell mRNA sequencing (scRNA-seq), RNA-in situ hybridization (RNA-ISH) and immunohistochemistry.
Results:
HIF-1α was primarily expressed in tumor-associated macrophages (TAMs), whereas HIF-2α and HAF were expressed primarily in tumor cells. TAM-associated HIF-1α was significantly associated with high tumor grade and increased metastasis and was independently associated with decreased overall survival. Further, elevated TAM HIF-1α was significantly associated with resistance to antiangiogenic therapy. By contrast, high HAF or HIF-2α were associated with low grade, decreased metastasis and increased overall survival. ScRNA-seq, RNA-ISH and Western blotting confirmed the expression of HIF-1α in M2-polarized CD163 expressing TAMs.
Conclusions:
These findings highlight a potential role of TAM HIF-1α in ccRCC progression and support the re-evaluation of HIF-1α as a therapeutic target and marker of disease progression.
Keywords: HIF-1α, HIF-2α, HAF, macrophage, CD163, C1q
Introduction
Clear cell renal cell carcinoma (ccRCC) is the most common and aggressive form of kidney cancer, and is also one of the most resistant of solid tumors to conventional chemotherapy (1). The etiology of ccRCC is uniquely linked to loss of the von Hippel-Lindau (VHL) tumor suppressor, which occurs in more than 90% of ccRCC (2). pVHL loss results in the constitutive stabilization of HIF-1α and HIF-2α that drive the expression of hundreds of genes associated with tumor progression (3–6). Despite sharing many transcriptional targets, the HIFs also drive unique transcriptional targets. For example, anerobic glycolysis appears to be predominantly controlled by HIF-1α, whereas erythropoietin synthesis is largely HIF-2α-regulated (7,8). Hypoxia- or pVHL-independent HIF-α stabilization can also occur through increased mRNA transcription and/or translation, such as through cytokine- and growth factor receptor-mediated signaling, particularly within inflammatory mediators such as macrophages (9). We have previously proposed the notion of a HIF switch, which orchestrates the complementary roles of HIF-1α and HIF-2α during acute versus chronic hypoxia, and during vascular and bone development in the embryo (10). The hypoxia-associated factor (HAF), acts as a mediator of the HIF switch, by ubiquitinating and degrading HIF-1α, while enhancing HIF-2α transactivation through hypoxia-dependent HAF SUMOylation (11–13).
The current paradigm for the HIFs in ccRCC describes a driving role for HIF-2α and a tumor suppressor role for HIF-1α that is unique to ccRCC. This is based on studies showing that overexpression of HIF-2α promotes, whereas overexpression of HIF-1α inhibits the growth of ccRCC xenografts in mice (14). Consistent with this view, homozygous deletion of HIF1A has been reported in approximately 50% of high-grade ccRCC, implicating HIF-1A as a ccRCC tumor suppressor gene (15). Although lost in high-grade ccRCC, HIF-1α is believed to play an important role during ccRCC initiation and is elevated in the earliest pre-neoplastic lesions in VHL patients. The appearance of HIF-2α protein in these early lesions is associated with increased dysplasia and cellular atypia (16,17). The HIF-2 bias observed in ccRCC has been attributed to the increased potency of HIF-2α compared to HIF-1α in promoting expression of pro-tumorigenic factors such as Cyclin D1, TGF-α and VEGFA (14,18). HIF-2α also potentiates pro-tumorigenic c-Myc and β-catenin activity, which are inhibited by HIF-1α (19,20). Consequently, there has been increasing enthusiasm for the selective targeting of HIF-2α in ccRCC (21–24). However, HIF-1α protein has been detected via immunohistochemistry in most ccRCC cases, which is inconsistent with its proposed tumor suppressor role (15,25,26). In view of these conflicting observations, we recognized a need for additional studies to clarify the involvement of the HIFs in ccRCC progression (14,19,27,28). Here, we report a previously unanticipated role of HIF-1α in tumor associated macrophages (TAMs), particularly in M2-polarized TAMs, as a marker of ccRCC morbidity, whereas ccRCC cell-specific HIF-2α and HAF levels indicate better patient prognosis.
Materials and Methods
Human subjects:
Studies were conducted in accordance with the Declaration of Helsinki. Human ccRCC and uninvolved kidney tissue were obtained from archival samples from patients who had provided written informed consent according to protocols approval by institutional review boards at their respective institutions. Fresh tumor and uninvolved kidney for scRNA-Seq were obtained after informed consent from patients undergoing nephrectomies at Huntsman Cancer Institute (HCI), approved under IRB#67518 and #10924. A separate cohort of 57 patients with metastatic ccRCC from HCI who were treated with antiangiogenic therapy were retrospectively analyzed for HIF-1/2α protein levels and correlated to radiographic disease progression as determined by the local radiologist. Brief tissue descriptions: Analytic cohort: Multistage TMA (M.D. Anderson Cancer Center [MDACC], 380 patients), Stage 1 tissue (MDACC, 12 patients). Validation cohort: Multi-grade TMA (Mayo Clinic, 24 patients), multi-grade sections (Mayo Clinic, 25 patients). Immune analysis: multi-grade sections (HCI, 23 patients). More detailed descriptions are provided in Supplemental Methods.
Antibodies and IHC.
TMAs and tissue were stained for HAF, HIF-2α and HIF-1α after initial antibody workup and validation (Supplemental Figures 2, 8). Monoclonal antibody for HAF has been previously described, whereas all other antibody details, staining and statistical methods are described in Supplemental Methods (13).
Single cell sequencing and analysis:
Tumors were dissociated using the Human Tumor Dissociation Kit (Macs Milteny Biotech, Cambridge, MA) according to the manufacturer’s guidelines. Three ccRCC and paired uninvolved kidney samples showing >40% cell viability were selected for sequencing. Assessment of gene expression in single cells was achieved using the 10X Genomics Chromium single-cell gene expression solution (10x Genomics, Pleasanton, CA). Sequencing data was processed using the 10X Genomics Cell Ranger pipeline, which uses the Spliced Transcripts Alignment to a Reference (STAR) aligner for genome and transcriptome alignment. Further processing was conducted using the Seurat (3.0.0) package for R (3.5.3). Further detail is provided in Supplemental Methods.
RNA in situ hybridization.
This was performed to assess RNA levels of HIF1A, NDUFA4L2, CD163 and SLC2A1 using RNAscope®Multiplex Fluorescent Assay V2 (Advanced Cell Diagnostics, Newark, CA). Staining of tissue using Opal 520, Opal 570, Opal 620, Opal 690 fluorescent dyes (Akoya Biosciences, Menlo Park, CA) was performed according to the manufacturer’s guidelines. Confocal images were captured on a Leica SP8 confocal microscope equipped with UV, 488nm, and white lasers and using an oil emersion 40x objective. For quantification, the average number of HIF1A or SLC2A1 mRNA molecules present within a 5μM radius of NDUFA4L2 and CD163 molecules was determined. At least 200 NDUFA4L2 and CD163 molecules were assessed across all patients within multiple fields of view. Statistical significance was determined by t-test using GraphPad Prism.
Cell Lines.
THP-1, ACHN, Caki-1, A498 and 786–0 cells were from ATCC (Manassas, VA), whereas RCC4, RCC10, UMRC2 and UOK101 were kind gifts from M. Celeste Simon (University of Pennsylvania). All commercially available cell lines were authenticated using STR fingerprinting upon receipt and stored in frozen aliquots. Fresh vials were thawed for use in experiments and discarded after 30–35 passages or approximately 2 months. Mycoplasma testing using the MycoAlert detection kit (Lonza, Ben OR) was performed every 2 months with the last test performed on February 2020. Cells were maintained at 37°C 5% CO2 in Dulbecco’s MEM (Life Technologies, ThermoFisher Scientific, Waltham MA) or RPMI 1640 (THP-1 cells) with 10% FBS. Hypoxia incubations were performed using a Whitley H35 Hypoxystation (HypOxygen, Frederick, MO). THP-1 cells were activated to M0 by incubation with 50nM Phorbol 12-myristate 13-acetate (Sigma-Aldrich, St Louis, MO) for 24 hours. Differentiation from M0 to M1 cells was performed by treatment for 48 hours with 15ng/ml lipopolysaccharides (Sigma-Aldrich) and 50ng/ml interferon-γ (R&D Systems, Bio-Techne, Minneapolis, MN)). For M2 differentiation, cells were incubated with 5ng/ml interleukin-13 and 5ng/ml interleukin-4 (both from R&D Systems) for 48 hours.
Immunoblotting.
Membranes were probed with primary antibodies for HIF-1α (BD Biosciences, 610959), HIF-2α (Cell Signaling Technologies, 70862), CD206 (CST #91992), HLA-DR (Abcam #ab92511) and HAF as described in (11–13). Images were acquired using Fluorochem M imaging system (Protein Simple, San Jose, CA). Blot quantification was performed using ImageJ.
For all other methods, please refer to Supplemental Methods.
Results
HIF-1α protein positively correlates with high-grade tumors and increased metastasis.
We stained tumor microarrays (TMAs) containing tumor core samples from 380 patients for HAF, HIF-1α or HIF-2α. Patients had a median age of 59 years, were primarily white (75%) and male (69%) with good performance status (ECOG 0=74%). Representative images of cores after initial antibody validation and workup are shown in Supplemental Figures S1 and S2. Patient characteristics overall and by TMA block are shown in Supplemental Table 1. Total HIF-1α staining, HIF-1α (total) was associated with higher T stage (Table 1A and Supplemental Table 2A) (28). Similarly, cores from patients with metastatic tumors had higher HIF-1α levels than those without, and showed more variability within patients (given by higher standard error (SE) of staining within patients), which may indicate increased HIF-1α tumor heterogeneity within metastatic tumors. Higher levels of HIF-1α were also associated with higher grade and showed more variability within patients. Thus, high HIF-1α levels were associated with high tumor grade, T stage and increased metastasis.
Table 1A:
Levels of HIF-1α (total), HIF-2α and HAF within Patients by Clinical Characteristics. Included in table are p-values for whether HIF-1α, HIF-2α or HAF values differ significantly by T stage, M stage or grade (using ANOVA or Kruskall-Wallis test if ANOVA assumptions were violated)
| Patient Characteristics | Within Patient HIF-1α (total) Values | Within Patient HIF-2α Values | Within Patient HAF Values | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Staining intensity | SE | Staining Intensity | SE | Staining Intensity | SE | |||||||
| Mean | P-value | Median | P-value | Mean | P-value | Median | P-value | Mean | P-value | Median | P-value | |
| All | 0.026 | 0.007 | 0.192 | 0.037 | 0.095 | 0.023 | ||||||
| T Stage | 0.005 | 0.01 | <0.001 | <0.001 | <0.001 | 0.25 | ||||||
| T1 | 0.028 | 0.007 | 0.419 | 0.065 | 0.164 | 0.029 | ||||||
| T2 | 0.04 | 0.013 | 0.195 | 0.034 | 0.112 | 0.026 | ||||||
| T3 | 0.02 | 0.006 | 0.099 | 0.025 | 0.055 | 0.019 | ||||||
| T4 | 0.055 | 0.019 | 0.166 | 0.052 | 0.012 | 0.004 | ||||||
| M Stage | 0.02 | 0.02 | 0.01 | 0.37 | 0.001 | 0.18 | ||||||
| No | 0.024 | 0.006 | 0.219 | 0.037 | 0.113 | 0.027 | ||||||
| Yes | 0.033 | 0.009 | 0.137 | 0.032 | 0.049 | 0.017 | ||||||
| Tumor Grade | 0.04 | 0.01 | 0.01 | 0.64 | <0.001 | 0.14 | ||||||
| 1 | 0.02 | 0.005 | 0.473 | 0.068 | 0.21 | 0.024 | ||||||
| 2 | 0.022 | 0.005 | 0.245 | 0.038 | 0.167 | 0.031 | ||||||
| 3 | 0.025 | 0.007 | 0.188 | 0.037 | 0.104 | 0.024 | ||||||
| 4 | 0.035 | 0.011 | 0.121 | 0.031 | 0.038 | 0.015 | ||||||
HIF-2α and HAF are significantly associated with low-grade tumors and decreased metastasis.
Summaries for HIF-2α and HAF in relation to tumor stage, grade and metastasis are shown in Table 1A with full tables in Supplemental Table 2B and Supplemental Table 2C. HIF-2α levels were higher among patients with ECOG scores of 0, i.e. the patients with the best performance status, compared to those with ECOG ≥ 1. HIF-2α was also highly associated with T stage, but with an unexpected trend of decreasing levels with increasing tumor stage from stage T1-T4. Significantly, we noted a clear decrease in HIF-2α levels with increasing grade, tumor size, the presence of metastasis or necrosis, with greater variability within the same patient in smaller tumors. Thus, high HIF-2α levels were associated with lower tumor grade and T stage and were inversely correlated with metastasis.
HAF, like HIF-2α, decreased as T stage increased. HAF was inversely correlated with metastasis, tumor grade, tumor size, and lymphovascular invasion (LVI), (Table 1A and Supplemental Table 2C). Thus, HAF levels closely tracked with HIF-2α in that high HAF levels were associated with lower tumor grade and T stage and were inversely correlated with metastasis.
HIF-1α is primarily expressed within tumor stroma.
When examining the staining patterns for HIF-1α, we noted that unlike HIF-2α, which was mainly expressed in tumor cells, HIF-1α staining was almost exclusively observed in infiltrating non-tumor cells (tumor stroma: S1). Indeed, we observed weak HIF-1α intensity within ccRCC cells, whereas most patients showed intense HIF-1α staining within the tumor stroma (Supplemental Figures 1, 3). Thus, by setting low- and high-intensity detection thresholds, we were able differentially quantify tumor and stroma HIF-1α positivity (Supplemental Figure 3). When HIF-1α positivity using low (tumor) and high thresholds (stroma) were assessed, only 9 patients (2.4%) showed positivity for low threshold (tumor-specific) HIF-1α, which under-powered further statistical analysis, whereas all the original samples showed expression of high threshold (stroma-specific) HIF-1α. High threshold HIF-1α staining - HIF-1α (stroma) summaries in relation to tumor grade, stage and metastasis are shown in Table 1B with all measured parameters shown in Supplemental Table 2D. Similar to overall HIF-1α staining, HIF-1α (stroma), was significantly associated with higher grade and with increased heterogeneity in grade 4, larger tumors or tumors with LVI.
Table 1B:
Levels of HIF-1α (stroma) within Patients by Clinical Characteristics.
| Patient Characteristics | Within Patient HIF-1α (stroma) Values | |||
|---|---|---|---|---|
| Mean | SE | |||
| Median (IQR) | P-value | Median (IQR) | P-value | |
| All | 0.009 (0.004, 0.017) | 0.003 (0.001, 0.006) | ||
| Clinical T Stage | 0.13 | 0.17 | ||
| T1 | 0.010 (0.005, 0.018) | 0.003 (0.001, 0.005) | ||
| T2 | 0.006 (0.003, 0.016) | 0.002 (0.001, 0.007) | ||
| T3 | 0.009 (0.005, 0.016) | 0.003 (0.001, 0.007) | ||
| T4 | 0.032 (0.005, 0.082) | 0.011 (0.001, 0.035) | ||
| Clinical M Stage | 0.55 | 0.09 | ||
| No | 0.009 (0.004, 0.017) | 0.003 (0.001, 0.005) | ||
| Yes | 0.009 (0.002, 0.019) | 0.004 (0.001, 0.008) | ||
| Tumor Grade | 0.01 | <0.001 | ||
| 1 | 0.006 (0.004, 0.030) | 0.002 (0.001, 0.002) | ||
| 2 | 0.007 (0.003, 0.014) | 0.003 (0.001, 0.004) | ||
| 3 | 0.008 (0.004, 0.016) | 0.002 (0.001, 0.006) | ||
| 4 | 0.012 (0.007, 0.022) | 0.005 (0.003, 0.010) | ||
HIF-1α is associated with poor outcome whereas HIF-2α and HAF are associated with better outcome.
The univariable overall survival (OS) estimates by markers are shown in Table 1C, with full table with other clinical parameters shown in Supplemental Table 3. In this patient set, higher HIF-1α (stroma), and lower HIF-2α or HAF expression were significantly associated with decreased OS. Older age, ECOG scores >0, higher T stage, metastases, higher tumor grade, larger tumor size, LVI, or necrosis were each associated with decreased OS, as expected (Supplemental Table 3). Similar details for progression-free survival are shown in Supplemental Table 4. With no consistent cut-offs identified using recursive-partitioning analysis (RPA, Supplemental Tables 5, 6), HIF-1α, HIF-2α, and HIF-1α (stroma) are presented with divisions of 33rd and 67th percentiles. An increase of 0.10 in the expression of HIF-1α was associated with 28% increase in the risk of death (HR = 1.28 [1.04, 1.58]; p=0.02); whereas a corresponding increase in the expression of HIF-1α (stroma) was associated with 39% increase in risk of death (1.39 [1.06, 1.83]; p=0.02; Fig. 1A). By contrast, an increase of 0.10 in HIF-2α levels were associated with a 13% decrease in risk of death (0.87 [0.80, 0.93]; p<0.001; Fig. 1B). Similarly, each increase of 0.10 in HAF expression was associated with a 12% reduction in risk of death (HR=0.88 [95%CI: 0.80, 0.98]; p=0.02; Fig. 1C). We observed similar correlations of HIF-1α, HIF-2α and HAF with progression-free survival as with OS (Supplemental Figure 4). Thus, in the univariable setting, high HIF-1α or high HIF-1α (stroma) was significantly correlated with decreased OS, whereas high HAF or high HIF-2α was significantly correlated with increased OS.
Table 1C:
Univariable analysis of overall Survival by Patient Characteristics
| Patient Characteristic | Measurement | Overall Survival | ||
|---|---|---|---|---|
| Deaths/N | 5-Year (SD) | P-Value | ||
| All Patients | 195/380 | 66.7% (2.5%) | ||
| HAF Mean* | 0.02 | |||
| <0.14 | 133/229 | 61.0% (3.3%) | ||
| >=0.14 | 59/146 | 75.3% (3.6%) | ||
| HIF-2α Mean* | <0.001 | |||
| <0.102 | 60/117 | 60.5% (4.7%) | ||
| 0.102-<0.307 | 69/120 | 57.8% (4.6%) | ||
| >=0.307 | 51/119 | 79.6% (3.7%) | ||
| HIF-1α (total) Mean* | 0.02 | |||
| <0.0171 | 57/124 | 71.8% (4.1%) | ||
| 0.0171-<0.041 | 63/122 | 66.0% (4.4%) | ||
| >=0.041 | 70/124 | 61.1% (4.4%) | ||
| HIF-1α (Stroma) Mean* | 0.02 | |||
| <0.005 | 55/116 | 74.4% (4.1%) | ||
| 0.005-<0.014 | 65/135 | 66.6% (4.1%) | ||
| >=0.014 | 71/120 | 57.8% (4.6%) | ||
These variables are continuous measures. The p-value is based on the continuous variable, and the groupings are used for presentation of time estimates only.
Figure 1: High HIF-1α expression in tumor stroma is associated with worse outcome, whereas high tumor HAF or HIF-2α expression is associated with better outcome.
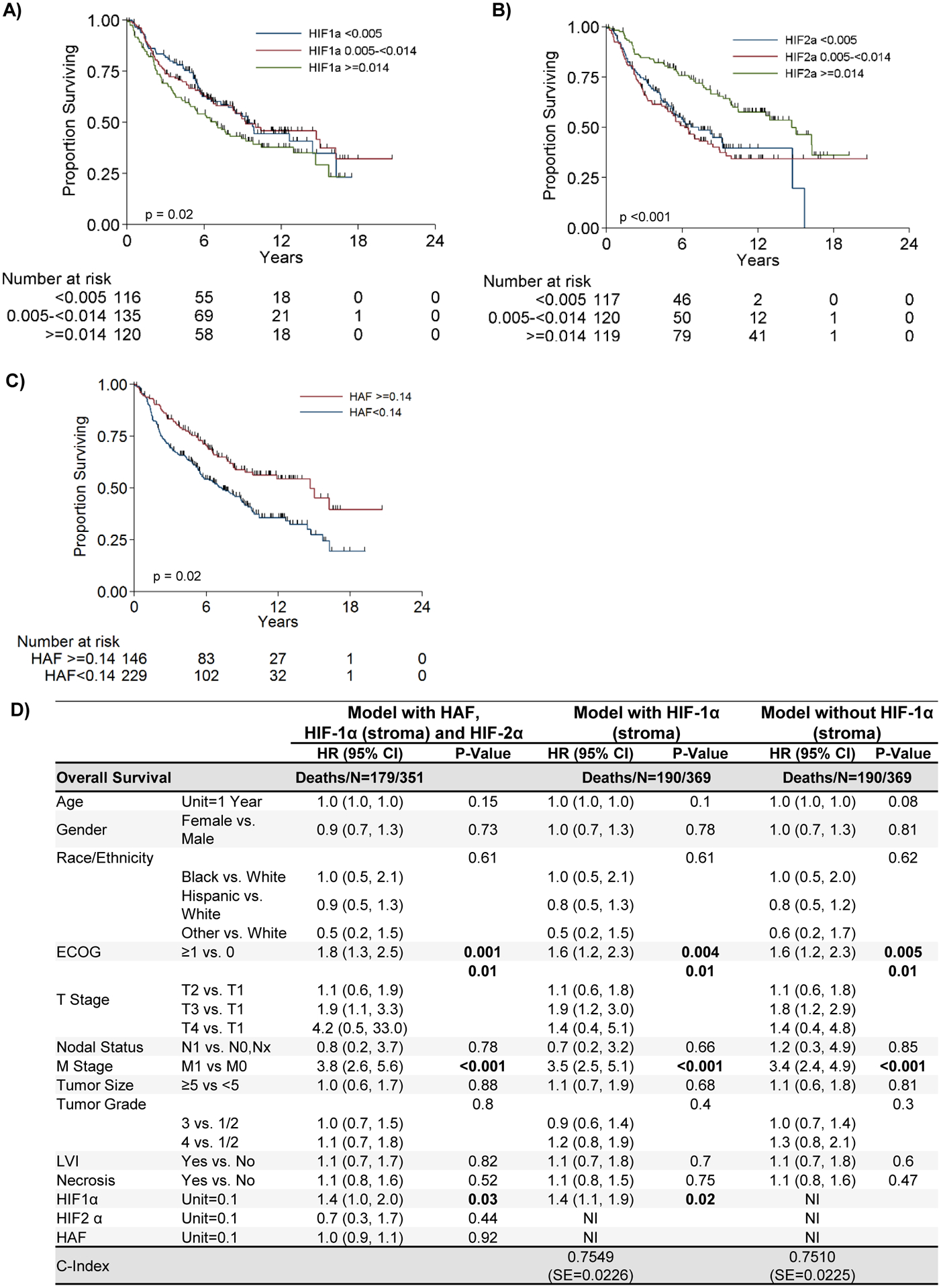
Kaplan Meier plots showing associations of HIF-1α (stroma), (A), HIF-2α, (B), and HAF, (C) with overall survival. D) Table showing multivariable overall survival analysis by patient and tumor characteristics. NI=Not Included.
HIF-1α (stroma) significantly predicts for decreased OS in the multivariable setting.
Figure 1D presents the multivariable OS model with clinically relevant variables together with levels of the HIFs and HAF to determine whether these markers could contribute additional information once the common clinical measures were accounted for. Similar details for progression-free survival are shown in Supplemental Table 7. After accounting for all the variables in the model, higher ECOG, T and M stages, and HIF-1α (stroma) were all associated with a higher risk of death. Neither HAF nor HIF-2α levels contributed additional information about OS after accounting for the standard clinical predictors. Most importantly, HIF-1α (stroma) remained significantly associated with OS, with a 40% increased risk of death for each increase of 0.10 in expression (1.4 [1.1, 1.9]; p=0.02). A likelihood ratio test between the two multivariable models (either with or without HIF-1α [stroma]) indicates that the model which includes HIF-1α (stroma) is significantly better at predicting survival (p=0.04). Thus, HIF-1α (stroma) is a significant predictor of poor overall survival in the multivariable setting.
HAF, HIF-1α and HIF-2α levels in ccRCC versus uninvolved kidney in T stage 1 disease.
To determine the relative levels of HAF and the HIFs in tumor versus paired uninvolved normal kidney tissue, we used large sections of additional tissue (containing both tumor and uninvolved kidney) obtained from 12 patients with early stage tumors (T stage 1 comprising grades 2–3) quantitated using Aperio (Supplemental Figures 5–8). HIF-1α and HIF-2α were expressed only at low levels in uninvolved kidney, although HIF-1α levels were slightly higher within the glomerulus compared to the tubules (Fig. 2A, Supplemental Figure 6). HIF-2α levels were significantly elevated in ccRCC (compared to paired uninvolved tissue (Fig. 2B, Supplemental Figure 7), whereas HIF-1α levels were not significantly different in ccRCC compared to paired uninvolved tissue. Intriguingly, we observed that HIF-1α was expressed within both tumor and stromal cells within these T Stage 1 tumors (Supplemental Figure 6). HAF was highly expressed within tubular epithelial cells and was decreased in paired ccRCC tissue (Fig. 2A–B and Supplemental Figure 8). Thus, HIF-2α is increased, HAF is decreased, and HIF-1α (tumor and stroma) is not significantly different in ccRCC compared to uninvolved normal kidney in T stage 1 ccRCC tissue.
Figure 2: Levels of HAF and the HIFs during ccRCC progression.

A) Representative images of large sections of paired uninvolved and T stage 1 tumor sections stained for HAF, HIF-2α and HIF-1α. Scale bars = 100μM. B-D) Quantitation of staining percent nuclear positivity of large tissue sections from T stage 1 tumors containing both tumor and uninvolved tissue (B), multi-grade tumor microarrays (C), and large tissue sections (D), based on an arbitrary threshold for HAF, HIF-2α and HIF-1α as determined by Aperio digital imaging software. Each dot on the plots indicates a single patient with patient numbers indicated in brackets. Average values ± SEM are shown with p-values indicated for (B), students’ paired T-tests, or (C,D) Mann Whitney U- tests. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05. NS: not significant.
Validation of HAF/HIF relationship with ccRCC grade.
To validate the relationships of HAF/HIFs to tumor grade, we analyzed a TMA comprising uninvolved, low grade and high grade ccRCC tissue. HAF levels were significantly decreased in low grade ccRCC compared to uninvolved kidney but was not significantly different between high grade and low grade ccRCC (Fig. 2C). HIF-2α, was significantly increased in low grade ccRCC compared to uninvolved kidney but was then significantly decreased in high grade compared to low grade ccRCC (Fig. 2C). HIF-1α levels were almost exclusively stromal (referred to as HIF-1α [stroma]) and was significantly increased in low grade ccRCC compared to uninvolved kidney, but was not significantly different between high grade and low grade ccRCC (Fig. 2C). In a second cohort, we observed a significant decrease in HAF levels in high grade compared to low grade tumors, and a significant increase in HIF-1α (stroma) in high grade compared to low grade tumor, whereas HIF-2α levels were not significantly different but trended lower in high grade tumors. (Fig. 2D, Supplemental Figure 9). Taken together, a validation cohort from 49 additional patients confirm an association of decreased HAF and HIF-2α levels with increasing tumor grade, and an association of increased HIF-1α (stroma) with tumor grade.
HIF-1α and HIF-2α are expressed in ccRCC tumor cells whereas only HIF-1α is expressed in immune infiltrating cells.
To address the issue of HIF-1α localization, we enlisted GU pathologists (NK, DS) to examine additional surgical specimens obtained from Huntsman Cancer Institute. These were comprised of low-grade (10 cases), high-grade (13 cases), and uninvolved normal tissue obtained from separate blocks and distal from the tumor (15 cases). Within tumor cell nuclei, HIF-1α and HIF-2α staining was very heterogeneous and were primarily localized to tumor margins and bordering regions of necrosis. HIF-1α expression was also detected in approximately 5% of immune cells in both uninvolved and ccRCC tissue, whereas HIF-2α was not detectable in immune cells (median expression <1% of immune cells (Fig. 3B–C). These immune cells were heterogeneously distributed throughout the tumor, particularly within local regions of inflammation, but did not have any specific localization to tumor margins or necrotic regions. Although we observed a significant increase in the percentage of immune cells within high grade ccRCC tissue compared to uninvolved kidney, the percentage of immune cells that were HIF-1α positive was not significantly different (Fig. 3A, C). Representative images show consistent HIF-1α staining in immune infiltrating cells but rarely in tumor cells, and frequent HIF-2α staining in tumor cells, but never in immune infiltrating cells (Fig. 3D–E). Thus, we confirm expression of only HIF-1α in immune infiltrating cells.
Figure 3: HIF-1α is primarily expressed in tumor-associated macrophages (TAMs).
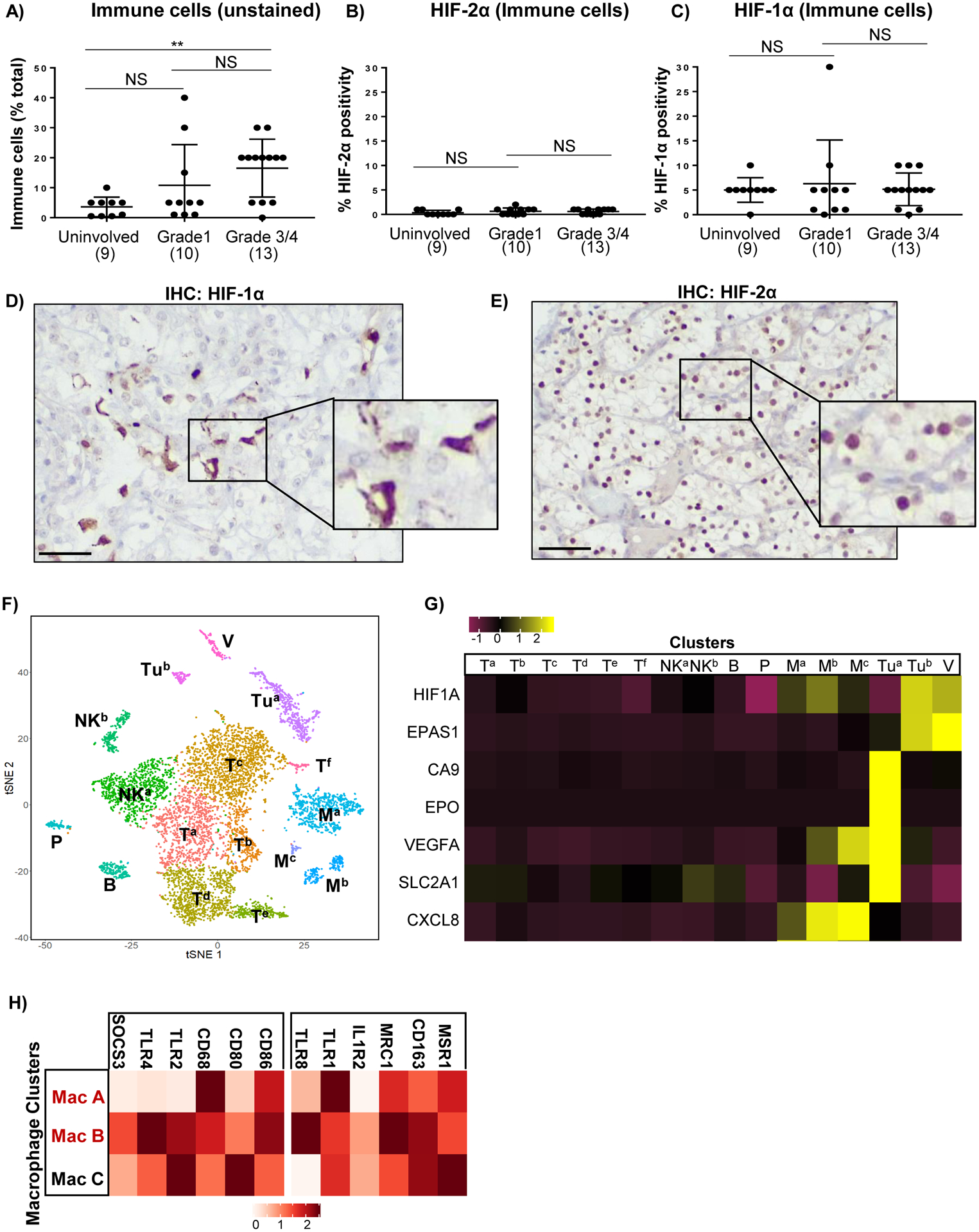
A) Percent of immune cells as a percentage of total cells in tumors as determined by pathologists’ analysis of hematoxylin and eosin (H&E) stained tissue sections. B,C) Percent of immune cells that stained positive for HIF-2α (B) or HIF-1α (C) as determined by pathologists’ analysis of HIF-1/2α stained sections with reference to H&E stained sections. Each dot on the plots indicates a single patient with patient numbers indicated in brackets. D-E) IHC showing HIF-1α (D) and HIF-2α (E) localization in matched high grade ccRCC. Scale bars = 50μM. F) T-SNE plots showing clustering of indicated cell types from single cell sequencing of ccRCC tissue from 3 patients. V = vasculature; Tu = tumor cells; T = T-cells, NK = NK cells, P = plasma cells, B = B-cells; M = macrophages. G) Heatmap showing average transcript expression levels for HIF-1α (HIF1A), HIF2-α (EPAS1) and a panel of HIF regulated genes for the indicated cell type clusters. H) Heatmap showing average expression levels of transcripts associated with M1 or M2 polarization in the three macrophage clusters.
HIF-1α is primarily expressed in ccRCC tumor-associated macrophages (TAMs).
To more definitively identify the HIF-1α expressing cell-types, we performed single-cell sequencing mRNA-sequencing (scRNA-Seq) on three grade III/IV ccRCC cases with paired uninvolved kidney. Quality control and data filtering performed produced a total of 7569 ccRCC and 2464 uninvolved cells for analysis. Clustering was performed using the Seurat package with cell identities assigned manually using previously described cell-specific markers in renal cancer (Fig. 3F, Supplemental Figure 10A). Tumor clusters were identified by NDUFA4L2 expression, which has been characterized as a robust expression marker for ccRCC with high expression localized to two clusters in our scRNAseq data (Tua, Tub; Supplemental Figure 10B) (29,30). In comparison, CAIX, a commonly used protein maker of ccRCC, was not localized to any specific cluster (apart from a few cells in Tua [Supplemental Figure 10B]), reducing its utility as a tumor-specific marker in this study. Analysis of average gene expression within each cluster showed that HIF1A and EPAS1 transcript were strongly expressed within a minority subset of tumor cells (Tub), and within the vasculature, but this was not associated with increased transcription of HIF target genes. However, classical HIF target genes (for example, CA9, EPO, VEGFA and GLUT1) were highly expressed in the majority subset of tumor cells (Tua), where HIF1A and EPAS1 transcript were poorly expressed, consistent with HIF-α post-transcriptional protein stabilization by hypoxia or pVHL loss (Fig. 3G, Supplemental Figure 10C). A cluster heatmap with a more exhaustive list of validated HIF target genes is shown in Supplemental Figure 11. HIF1A (but not EPAS1) transcript was also elevated in the macrophage Ma and Mb clusters, and this was co-incident with the activation of HIF targets, VEGFA or CXCL8 (Fig. 3G, Supplemental Figure 10C), suggesting a transcriptional mechanism of HIF activation in these macrophages By contrast, the Mc cluster also showed upregulation of HIF targets, but had low HIF1A transcript levels, suggesting that this cluster may activate HIF through the canonical hypoxia-mediated pathway. All three macrophage clusters expressed a spectrum of M1 and M2 activation markers and did not clearly correspond to any specific macrophage activation subtype (Fig. 3H). RNA in situ hybridization (RNA-ISH) confirmed expression of HIF1A transcript in CD163 expressing macrophages (a marker of alternatively activated, M2 polarized macrophages), whereas expression of the HIF target, SLC2A1 (also known as glucose transporter 1, GLUT1) was mainly localized in NDUFA4L2 expressing ccRCC tumor cells, in the absence of HIF1A transcript expression (Fig. 4A). Manual quantitation showed significantly higher levels of HIF1A mRNA molecules in close proximity to CD163 than to NDUFA4L2, and significantly increased levels of GLUT1 mRNA molecules in close proximity to NDUFA4L2 rather than to CD163 (Fig. 4B). Besides tumor cells, cells of the vasculature and macrophages, no other cell type showed clear upregulation of HIF1A or HIF target genes.
Figure 4: Characterization of HIF-1α expressing TAMs.
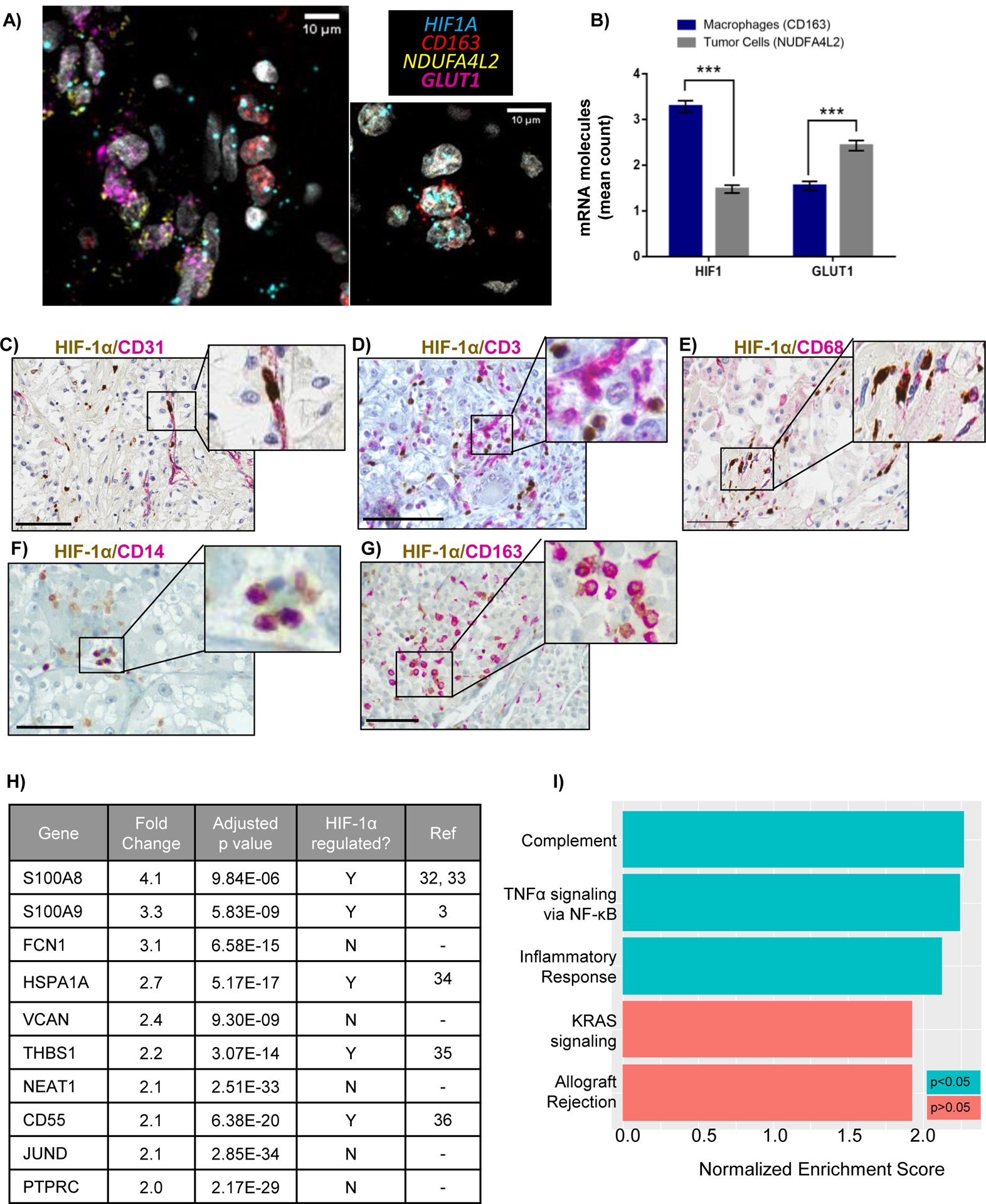
A) RNA in-situ hybridization (RNA-ISH) showing proximity of HIF1A and CD163 in TAMs, and HIF target, GLUT1 (SLC2A1), with ccRCC tumor cell -specific transcript, NDUFA4L2. Scale bars = 10μM. B) Quantitation of average number of HIF1A or GLUT1 mRNA molecules within a 5μM radius of NDUFA4L2 and CD163 molecules. At least 200 NDUFA4L2 and CD163 molecules were assessed across all patients. *** p < 0.0001. C-G) Double staining of HIF-1α with indicated markers. Scale bar = 100μM. H) Top 10 upregulated genes in HIF1A+ macrophages from differential gene expression analysis of HIF1A+ TAMs versus HIF1A− TAMs I) The most positively enriched pathways identified from gene set enrichment analysis on upregulated genes (using a 10% FDR cut-off) in HIF1A+ TAMs.
The data suggest that increased HIF1A transcript may translate to increased HIF-1α protein within macrophages but not within tumor cells, where activation of HIF target genes is likely to be primarily HIF-2α-dependent. Intriguingly, HIF-1α protein was detected in both the nucleus and cytoplasm in macrophages, unlike in tumor cells, where HIF-1α, when expressed, was primarily localized within the nucleus (Supplemental Figure 3). Double staining experiments with the endothelial cell marker CD31 confirmed that while many HIF-1α expressing cells resided within blood vessels, the endothelial cells were not themselves HIF-1α positive (Fig. 4C). Similarly, double staining with CD3 indicated that although HIF-1α expressing cells were within proximity with T cells, T cells themselves did not express HIF-1α (Fig. 4D). However, HIF-1α co-localized with the monocyte/macrophage markers CD68 and CD14 and M2 marker CD163 (Fig. 4E–G). Despite expression of the classically activated, M1 macrophage marker CD86 via scRNA-Seq, we could not detect more than a few CD86 positive cells in ccRCC tissue (10 cases examined), and none co-localized with HIF-1α (not shown). Co-staining of HIF-1α with other markers such as B-cell markers CD19, CD20 and CD21, and dendritic cell marker S100 were also attempted but only a few positive cells were observed and again, none co-localized with HIF-1α (not shown). Taken together, our scRNA-Seq, RNA-ISH and IHC data suggest that the highest HIF-1α expressing cells are TAMs, primarily those that also expressed the M2 marker, CD163.
To further characterize the HIF1A expressing macrophages, we performed differential gene expression analysis on HIF1A+ (HIF1A expression > 0) and HIF1A− (HIF1A ≤ 0) macrophages, followed by gene set enrichment analysis (GSEA) (31). We identified 361 upregulated and 19 downregulated genes in HIF1A+ compared to HIF1A− macrophages. Of the top 10 upregulated genes, 5 were previously known to be regulated by HIF-1α (Fig. 4H) (32–36). GSEA identified complement, TNF-α signaling via NF-κB, and inflammatory response as positively enriched pathways (p<0.05) in HIF1A+ macrophages (Fig. 4I). Intriguingly, the complement pathway has previously been linked to ccRCC progression, whereas TNF-α has been implicated in ccRCC metastasis and treatment resistance, while HIF-1 has long been implicated in both inflammation and tumorigenesis (37–39).
To investigate the mechanism(s) by which the tumor microenvironment results in HIF-1α upregulation within infiltrating TAMs, but loss of HIF-1α expression in the tumor, we used the THP-1 monocytic cell line and a panel of human RCC cell lines. We found that both HIF1A transcript and protein was strongly induced only in hypoxic M2-polarized macrophages (Fig. 5A–C) (40). When we investigated the impact of hypoxia on RCC cells, we found that while cells with wild-type VHL showed robust HIF-1α and HIF-2α accumulation under hypoxic conditions, pVHL deficient cells showed constitutively low levels of HIF-1α in both normoxia and hypoxia, supporting the previously reported notion of HIF-1α loss in ccRCC (Fig. 5D–F) (28). By contrast, HIF-2α levels remained relatively constant in pVHL mutant cells irrespective of oxygen tension, whereas HAF levels generally decreased in hypoxia similar to our previous observations (Fig. 5F–G) (41). Thus, the data suggest that the combination of hypoxia and M2 polarizing conditions within the tumor microenvironment promotes the upregulation of HIF1A transcript and protein in ccRCC TAMs, whereas intrinsic factors associated with pVHL loss inhibits the production of HIF-1α protein in tumor cells regardless of oxygen tension.
Figure 5: HIF-1α is upregulated in hypoxic M2-polarized macrophages and is associated with resistance to antiangiogenic therapy.
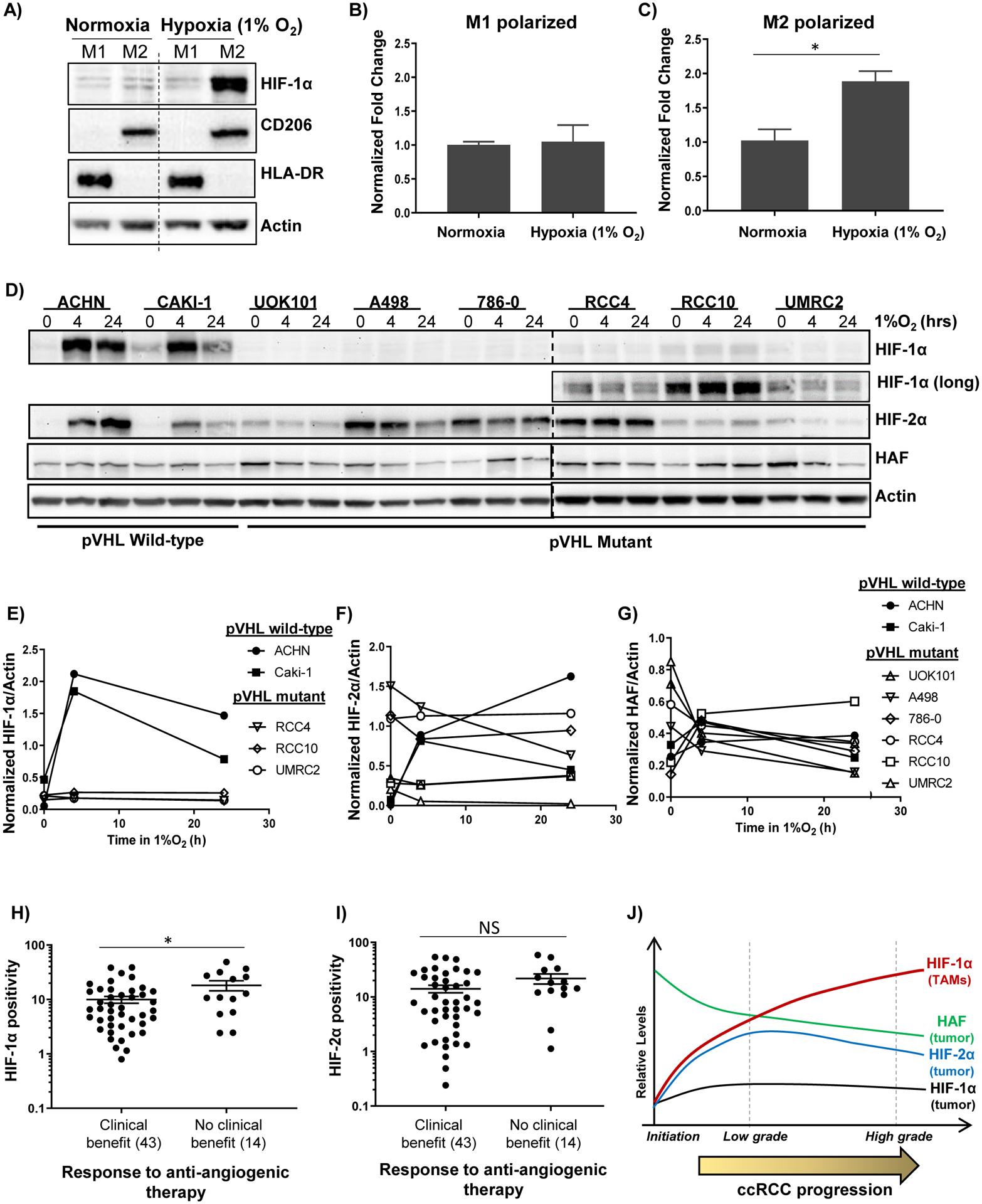
A) Western blots showing effect of exposure of PMA-differentiated THP-1 macrophages to M1 or M2-polarizing cytokines in normoxia or hypoxia. B-C) Taqman-qRT-PCR showing effects of hypoxia on HIF1A normalized to B2M in M1 or M2 polarized macrophages ± SEM. D-G) Western blots showing impact of indicated durations of hypoxia on a panel of RCC cells (D), with quantitation in E-G). H-I) HIF-1α (H) and HIF-2α (I) levels in ccRCC tissue and association with response to angiogenic therapy. *p < 0.05 using unpaired t-tests for log-transformed values. (J) Revised role of HAF and the HIFs in ccRCC: HIF-1α positive TAMs increase with ccRCC progression in the context of HIF-1α loss in tumor cells and is an independent predictor of poor outcome. By contrast, HAF and HIF-2α levels decrease with tumor stage and predict for better outcome.
To explore the clinical utility of HIF-1α, we measured baseline HIF-1α levels in primary tumors from 57 patients with advanced/metastatic RCC who were subsequently treated with antiangiogenic therapy and assessed the relationship between HIF-1α levels and response (Supplemental Figure 12A). As observed previously in prior datasets, HIF-1α staining was primarily stromal (Supplemental Figure 12B–C). Importantly, HIF-1α (stroma) levels were significantly higher (p = 0.03) in patients who derived no clinical benefit (CB) from antiangiogenic therapy (median PFS - 3 months; median OS - 18.5 months) compared to those that derived CB (median PFS - 15 months; median OS - 42 months) (Fig. 5H). By contrast, no significant associations were observed between HIF-2α and CB (Fig. 5I). Patient with stable disease, partial response or complete response following therapy were considered to have derived CB, whereas those with progressive disease were considered to have derived no CB. As expected, patients deriving no CB had significantly increased risk of progression or death compared to patients who derived CB (Supplemental Figure 12D–E). Thus, HIF-1α (stroma) is significantly associated with resistance to antiangiogenic therapy.
Discussion
Here, we have undertaken a comprehensive study to delineate the relationship between the HIFs and HAF during ccRCC progression. Although HIF-1α had been proposed as a tumor suppressor in ccRCC, our study has revealed an unexpected role of HIF-1α in TAMs as an independent indicator of poor outcome. We show that the exposure to a combination of hypoxia and M2 polarizing cytokines, conditions frequently present within the tumor microenvironment, induce HIF1A transcription and HIF-1α protein expression in TAMs. Our study highlights the potential contribution of TAM HIF-1α to ccRCC aggressiveness in the context of HIF-1α loss within tumor cells, suggests its utility as a prognostic marker, and potential therapeutic target. Surprisingly, HIF-2α, which is widely considered to be a tumor driver, was significantly decreased with tumor grade, and associated with better patient prognosis, similar to its co-activator, HAF (Table 1A, Fig. 2C–D) (13,28). However, we note that these findings do not bely the contribution of HIF-2α to the ccRCC phenotype, and its potential as a therapeutic target, as has been previously outlined (21–24).
HIF-2α is widely recognized as the major driving factor and a promising therapeutic target in ccRCC (21–24). Intriguingly, a recent meta-analysis found that although HIF-2α was negatively associated with overall survival in a variety of solid tumor types, HIF-2α was a positive prognostic biomarker for metastasis-free survival in kidney cancer (42). Another study showed a significant negative correlation of cytoplasmic HIF-2α with overall survival, but a significant positive correlation of nuclear HIF-2α with increased overall survival, which is in agreement with our findings (43). Only HIF-2α nuclear staining was considered in the analyses performed throughout our study due to the paucity of cytoplasmic HIF-2α detected in our tissue sections.
Although HIF-1α has previously been associated with classically activated, pro-inflammatory M1 macrophage polarization, we found that HIF-1α primarily co-localized with the M2 marker, CD163, at both the RNA and protein levels, whereas M1 markers such as CD86 were poorly expressed in the ccRCC cases we examined. No differences were observed between HIF-1α protein levels within TAMs compared to macrophages derived from uninvolved kidney suggesting that increased stromal HIF-1α in high-grade ccRCC may be due to increased numbers of immune cells rather than increased HIF-1α expression (Fig. 3A). It is also possible that due to the proximity of the uninvolved kidney to the tumor site, macrophages identified within uninvolved kidney may also consist of TAMs that have migrated away from the tumor site. We note that although HIF-2α has been linked to M2 polarization, we could not detect macrophage-specific HIF-2α expression in any of the ccRCC tissues examined (44).
When we compared the transcriptomes of HIF1A+ TAMs with that of HIF1A− TAMs, we found that the most highly enriched gene sets were those associated with the complement pathway, TNF-α signaling via NF-κB and the inflammatory response (Fig. 4I). However, it is both possible and likely that other HIF-1α-driven pathways may contribute to the pro-tumorigenic roles of TAMs, as described previously (45). Indeed, a known limitation of scRNA-Seq technology is the relatively low mRNA capture efficiency and sequencing depth compared to bulk mRNA sequencing. Additionally, the proportions of tumor and stromal populations characterized through scRNA-Seq may vary from the proportions found in situ due to differences in efficiency of tissue disaggregation.
While our study is designed to address limitations of previous investigations, there are limitations of our own work that should be considered. First, the primary evaluation of the prognostic significance of HAF and the HIFs was performed using TMAs. Related to this, intratumor heterogeneity (ITH) occurs in ccRCC, and prognostic biomarkers may be influenced by tumor sampling or interrater variability. Additionally, there is potential for technical batch effects associated with the tissue distribution over multiple paraffin blocks (Supplemental Table 1). We addressed these issues by the inclusion of additional TMAs and large tissue sections from different institutions (the validation cohort), which confirmed the association of low HIF-2α or HAF with high tumor grade, and high HIF-1α with high tumor grade (Fig. 3B–C). Thus, despite these limitations, our data suggest that mean HIF-1α levels contribute additional value using fairly straightforward quantitative measurement techniques. Second, although our scRNA-Seq studies have revealed potential downstream targets of HIF-1α in TAMs, lack of sequencing depth has precluded a more granular analysis of the HIF-1α TAM transcriptome, and certainly those of other non-neoplastic cells within the tumor mass. Finally, we also note the use of OS instead of cancer-specific survival due to lack of data specifically confirming cancer as cause of death.
Our studies build on our current understanding of the role of the HIFs in ccRCC (Fig. 5J). We show that HIF-2α but not HIF-1α is increased in ccRCC tumor cells compared to paired uninvolved kidney. HAF is decreased during ccRCC progression, and both HAF and HIF-2α are associated with lower tumor grade/stage and better outcome. Most notably, HIF-1α expressing TAMs increase with tumor progression and is an independent negative prognostic indicator, suggesting that HIF-1α inhibition such as by small molecule inhibitors may be a promising strategy for targeting the tumor-promoting properties of TAMs in ccRCC (Fig. 5I) (46,47). We also demonstrate the potential utility of HIF-1α as a predictor of response to antiangiogenic therapy. Although future prospective studies will be required to confirm the prognostic role of HIF-1α and its predictive value, our work has established TAM HIF-1α as a potential contributor to ccRCC progression in the context of HIF-1α loss in tumor cells, and identified downstream targets of TAM HIF-1α that warrant further investigation.
Supplementary Material
Significance Statement.
Here, we highlight a novel prognostic role for HIF-1α in tumor associated macrophages (TAMs) in kidney cancer, in the context of its tumor suppressor role within kidney cancer tumor cells. Further, we identify an unexpected association of HIF-2α and its activating co-factor, HAF, with improved patient outcome and reduced incidence of metastasis. Taken together, these findings have important implications for the development of HIF-2α inhibitors currently underway for the treatment of kidney cancer and support the evaluation of TAM HIF-1α as a therapeutic target in kidney cancer.
Acknowledgements
Supported by NIH/NCI grants CA181106 and CA217905 to MYK, and NIH/NCI R01CA224917 to THH. Research in this publication utilized the High Throughput Genomics and Bioinformatics Resource and the Cancer Biostatistics Resource funded by NIH/NCI grant P30CA042014 awarded to the Huntsman Cancer Institute at the University of Utah, and the Biostatistics Resource Group at the U.T. M.D. Anderson Cancer Center funded by NIH/NCI P30CA016672. We acknowledge financial support by the Huntsman Cancer Foundation and the Experimental Therapeutics and Cell Response and Regulation Programs at HCI. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. This manuscript is dedicated to the memory of Adam A. Olalde.
Footnotes
Conflict of interest: MYK is founder and has equity in Kuda Therapeutics Inc. All other authors declare no conflict of interest.
References
- 1.Lara PN, Jonasch E. Kidney cancer : principles and practice. Heidelberg: Springer-Verlag; 2012. 309 p. [Google Scholar]
- 2.Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994;7(1):85–90 doi 10.1038/ng0594-85. [DOI] [PubMed] [Google Scholar]
- 3.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A 1995;92(12):5510–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer 2011;11(6):393–410 doi http://www.nature.com/nrc/journal/v11/n6/suppinfo/nrc3064_S1.html. [DOI] [PubMed] [Google Scholar]
- 5.Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med 2014;211(5):781–90 doi 10.1084/jem.20131916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruf M, Moch H, Schraml P. PD-L1 expression is regulated by hypoxia inducible factor in clear cell renal cell carcinoma. Int J Cancer 2016;139(2):396–403 doi 10.1002/ijc.30077. [DOI] [PubMed] [Google Scholar]
- 7.Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol 2003;23(24):9361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kapitsinou PP, Liu Q, Unger TL, Rha J, Davidoff O, Keith B, et al. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood 2010;116(16):3039–48 doi blood-2010-02-270322 [pii] 10.1182/blood-2010-02-270322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.LaGory EL, Giaccia AJ. The ever-expanding role of HIF in tumour and stromal biology. Nat Cell Biol 2016;18(4):356–65 doi 10.1038/ncb3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koh MY, Powis G. Passing the baton: the HIF switch. Trends Biochem Sci 2012;37(9):364–72 doi S0968–0004(12)00079–5 [pii] 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koh MY, Darnay BG, Powis G. Hypoxia-associated factor, a novel E3-ubiquitin ligase, binds and ubiquitinates hypoxia-inducible factor 1alpha, leading to its oxygen-independent degradation. Mol Cell Biol 2008;28(23):7081–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koh MY, Lemos R Jr., Liu X, Powis G. The Hypoxia-Associated Factor Switches Cells from HIF-1{alpha}- to HIF-2{alpha}-Dependent Signaling Promoting Stem Cell Characteristics, Aggressive Tumor Growth and Invasion. Cancer Res 2011;71(11):4015–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koh MY, Nguyen V, Lemos R, Darnay BG, Kiriakova G, Abdelmelek M, et al. Hypoxia-Induced SUMOylation of E3 Ligase HAF Determines Specific Activation of HIF2 in Clear-Cell Renal Cell Carcinoma. Cancer Research 2015;75(2):316–29 doi 10.1158/0008-5472.can-13-2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, et al. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 2005;25(13):5675–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen C, Beroukhim R, Schumacher S, Zhou J, Chang M, Signoretti S, et al. Genetic and Functional Studies Implicate HIF1α as a 14q Kidney Cancer Suppressor Gene. Cancer Discovery 2011. doi 10.1158/2159-8290.cd-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, et al. HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 2002;1(5):459–68. [DOI] [PubMed] [Google Scholar]
- 17.Schietke RE, Hackenbeck T, Tran M, Gunther R, Klanke B, Warnecke CL, et al. Renal tubular HIF-2alpha expression requires VHL inactivation and causes fibrosis and cysts. PLoS One 2012;7(1):e31034 doi 10.1371/journal.pone.0031034 PONE-D-11–21123 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dowd AA, Ibrahim FI, Mohammed MM. Renal cell carcinoma as a cause of iron deficiency anemia. African Journal of Urology 2014;20(1):25–7 doi 10.1016/j.afju.2013.11.001. [DOI] [Google Scholar]
- 19.Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 2008;14(6):435–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi H, Chun Y-S, Kim T-Y, Park J-W. HIF-2α Enhances β-Catenin/TCF-Driven Transcription by Interacting with β-Catenin. Cancer Research 2010;70(24):10101–11 doi 10.1158/0008-5472.can-10-0505. [DOI] [PubMed] [Google Scholar]
- 21.Chen W, Hill H, Christie A, Kim MS, Holloman E, Pavia-Jimenez A, et al. Targeting Renal Cell Carcinoma with a HIF-2 antagonist. Nature 2016. doi 10.1038/nature19796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cho H, Du X, Rizzi JP, Liberzon E, Chakraborty AA, Gao W, et al. On-Target Efficacy of a HIF2alpha Antagonist in Preclinical Kidney Cancer Models. Nature 2016. doi 10.1038/nature19795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martinez-Saez O, Gajate Borau P, Alonso-Gordoa T, Molina-Cerrillo J, Grande E. Targeting HIF-2 alpha in clear cell renal cell carcinoma: A promising therapeutic strategy. Crit Rev Oncol Hematol 2017;111:117–23 doi 10.1016/j.critrevonc.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 24.Ricketts CJ, Crooks DR, Linehan WM. Targeting HIF2alpha in Clear-Cell Renal Cell Carcinoma. Cancer Cell 2016;30(4):515–7 doi 10.1016/j.ccell.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 25.Gudas LJ, Fu L, Minton DR, Mongan NP, Nanus DM. The role of HIF1alpha in renal cell carcinoma tumorigenesis. J Mol Med (Berl) 2014;92(8):825–36 doi 10.1007/s00109-014-1180-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minardi D, Lucarini G, Santoni M, Mazzucchelli R, Burattini L, Conti A, et al. Survival in patients with clear cell renal cell carcinoma is predicted by HIF-1alpha expression. Anticancer Res 2015;35(1):433–8. [PubMed] [Google Scholar]
- 27.Kondo K, Kim WY, Lechpammer M, Kaelin WG, Jr. Inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 2003;1(3):E83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, et al. Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov 2011;1(3):222–35 doi 2159–8290.CD-11–0098 [pii] 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Young MD, Mitchell TJ, Vieira Braga FA, Tran MGB, Stewart BJ, Ferdinand JR, et al. Single-cell transcriptomes from human kidneys reveal the cellular identity of renal tumors. Science 2018;361(6402):594–9 doi 10.1126/science.aat1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang L, Peng Z, Wang K, Qi Y, Yang Y, Zhang Y, et al. NDUFA4L2 is associated with clear cell renal cell carcinoma malignancy and is regulated by ELK1. PeerJ 2017;5:e4065 doi 10.7717/peerj.4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 2015;1(6):417–25 doi 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahn GO, Seita J, Hong BJ, Kim YE, Bok S, Lee CJ, et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc Natl Acad Sci U S A 2014;111(7):2698–703 doi 10.1073/pnas.1320243111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grebhardt S, Veltkamp C, Strobel P, Mayer D. Hypoxia and HIF-1 increase S100A8 and S100A9 expression in prostate cancer. Int J Cancer 2012;131(12):2785–94 doi 10.1002/ijc.27591. [DOI] [PubMed] [Google Scholar]
- 34.Tsuchida S, Arai Y, Takahashi KA, Kishida T, Terauchi R, Honjo K, et al. HIF-1alpha-induced HSP70 regulates anabolic responses in articular chondrocytes under hypoxic conditions. J Orthop Res 2014;32(8):975–80 doi 10.1002/jor.22623. [DOI] [PubMed] [Google Scholar]
- 35.Daubon T, Léon C, Clarke K, Andrique L, Salabert L, Darbo E, et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nature Communications 2019;10(1):1146 doi 10.1038/s41467-019-08480-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pandya PH, Fisher AJ, Mickler EA, Temm CJ, Lipking KP, Gracon A, et al. Hypoxia-Inducible Factor-1alpha Regulates CD55 in Airway Epithelium. Am J Respir Cell Mol Biol 2016;55(6):889–98 doi 10.1165/rcmb.2015-0237OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roumenina LT, Daugan MV, Noe R, Petitprez F, Vano YA, Sanchez-Salas R, et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol Res 2019. doi 10.1158/2326-6066.CIR-18-0891. [DOI] [PubMed] [Google Scholar]
- 38.Mikami S, Mizuno R, Kosaka T, Saya H, Oya M, Okada Y. Expression of TNF-α and CD44 is implicated in poor prognosis, cancer cell invasion, metastasis and resistance to the sunitinib treatment in clear cell renal cell carcinomas. International Journal of Cancer 2015;136(7):1504–14 doi 10.1002/ijc.29137. [DOI] [PubMed] [Google Scholar]
- 39.Balamurugan K HIF-1 at the crossroads of hypoxia, inflammation, and cancer. International journal of cancer 2016;138(5):1058–66 doi 10.1002/ijc.29519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer 2015;15:577 doi 10.1186/s12885-015-1546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Green YS, Sargis T, Reichert EC, Rudasi E, Fuja D, Jonasch E, et al. Hypoxia-Associated Factor (HAF) Mediates Neurofibromin Ubiquitination and Degradation Leading to Ras-ERK Pathway Activation in Hypoxia. Mol Cancer Res 2019. doi 10.1158/1541-7786.MCR-18-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreno Roig E, Yaromina A, Houben R, Groot AJ, Dubois L, Vooijs M. Prognostic Role of Hypoxia-Inducible Factor-2alpha Tumor Cell Expression in Cancer Patients: A Meta-Analysis. Front Oncol 2018;8:224 doi 10.3389/fonc.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kroeger N, Seligson DB, Signoretti S, Yu H, Magyar CE, Huang J, et al. Poor prognosis and advanced clinicopathological features of clear cell renal cell carcinoma (ccRCC) are associated with cytoplasmic subcellular localisation of Hypoxia inducible factor-2alpha. Eur J Cancer 2014;50(8):1531–40 doi 10.1016/j.ejca.2014.01.031. [DOI] [PubMed] [Google Scholar]
- 44.Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev 2010;24(5):491–501 doi 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chittezhath M, Dhillon MK, Lim JY, Laoui D, Shalova IN, Teo YL, et al. Molecular profiling reveals a tumor-promoting phenotype of monocytes and macrophages in human cancer progression. Immunity 2014;41(5):815–29 doi 10.1016/j.immuni.2014.09.014. [DOI] [PubMed] [Google Scholar]
- 46.Xia Y, Choi H-K, Lee K. Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. European Journal of Medicinal Chemistry 2012;49:24–40 doi 10.1016/j.ejmech.2012.01.033. [DOI] [PubMed] [Google Scholar]
- 47.Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, et al. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther 2008;7(1):90–100. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.