

Abstract
Due to the therapeutic potential of targeting protein-protein interactions (PPIs) there is a need for easily executed assays to perform high throughput screening (HTS) of inhibitors. We have developed and optimized an innovative and robust fluorescence-based assay for detecting PPI inhibitors, called FluorIA (Fluorescence-based protein-protein Interaction Assay). Targeting the PPI of RAD52 with replication protein A (RPA) was used as an example, and the FluorIA protocol design, optimization and successful application to HTS of large chemical libraries are described. Here enhanced green fluorescent protein (EGFP)-tagged RAD52 detected the PPI using full-length RPA heterotrimer coated, black microtiter plates and loss in fluorescence intensity identified small molecule inhibitors (SMIs) that displaced the EGFP-tagged RAD52. The FluorIA design and protocol can be adapted and applied to detect PPIs for other protein systems. This should push forward efforts to develop targeted therapeutics against protein complexes in pathological processes.
Keywords: protein-protein interaction, fluorescent assay, small molecule inhibitor, high throughput screening
High throughput screening (HTS) is a key drug discovery tool 1, 2. With this advancement, the ambitions of scientific research shifted from the delivery of indiscriminate agents to drugs with more selectivity for the target. Current targets in cancer, for example, include abnormally expressed proteins, tumor microenvironment components, and alternative survival pathways adopted by tumor cells 3. In each of these categories of therapeutic approaches, targeting pathologic protein-protein interactions (PPIs) is as promising as targeting mutated genes. PPIs orchestrate the signaling of normal cellular proliferation, the very trait that defines malignancies when deregulated. Cancer cells sustain chronic proliferation by acquiring alternative signaling pathways defined by unique and specific PPIs 4. The first step in targeting PPIs is the HTS of chemical libraries to find initial hits for drug development. The successful design and optimization of PPI assays for HTS is currently the most significant barrier to overcome in the development of new therapies.
Historically modulation of PPI with small molecules was considered “undruggable” or the “high-hanging fruit” 5. However, over the past two decades interest in targeting PPIs has increased along with the accumulation of proteomic interaction network data. The large PPI interfaces that were once regarded as major challenges to target with small drug-like molecules are now desirable to bolster selectivity 6, 7. In addition, the community no longer thinks small molecule inhibitors (SMIs) must mimic the interacting partner of the targeted protein, and this realization led to a more comprehensive screening of available libraries 8–10. Mutational studies demonstrated that only some “hot spot” residues in the PPI contribute the energy for the SMIs to bind and block the targeted protein partner 6, 11. With this finding, the quest for targeting PPIs with SMIs became plausible, and the development of robust and cost-effective assays for HTS of SMI libraries is in demand. Successful HTS that target PPIs include nuclear magnetic resonance 12, differential scanning fluorimetry 13, fluorescence polarization/anisotropy 14, virtual screening with docking 15 and cell-free, enzyme-linked immunosorbent assay (ELISA) 16, 17. The fluorescent-based protein-protein interaction assay (FluorIA) described here is similar in some ways to ELISA in that a high protein binding plate is used to bind one of the protein partners. The FluorIA is completely different after this first step and replaces antibodies with a fluorescently-tagged protein binding partner to detect the PPI.
Here, we describe the design and optimization of FluorIA and use it in HTS. As an example, the FluorIA was used to target the PPI of two important DNA repair proteins: RAD52 and replication protein A (RPA). RAD52 is a multi-domain protein that mediates homologous recombination repair of DNA double-strand breaks, a role mainly played by BRCA2 in mammalian cells 18–21. Tumor cells that have lost BRCA2 function rely on RAD52 to survive the breaks encountered from replication stress or cytotoxic treatments 22–24, and this presents a therapeutic opportunity to target RAD52 activity in BRCA-deficient tumors 25–29. For homologous recombination, breaks are initially processed to produce single-stranded DNA tails that are then bound by RPA. The PPI of RAD52 with RPA is extensive (Fig. 1) and essential for RAD52 activity in the subsequent steps of repair 30–32. Only coarse domain maps are available for the PPI 31–33. A portion of the RPA:RAD52 PPI including the RPA32 wHLH domain and a peptide of RAD52 (residues 257–274) was characterized by solution NMR 34, 35. Plate and coworkers demonstrated by mutational analysis that the formation of RAD52 foci required the RPA-binding domain 36. Since most of the structure of the RPA:RAD52 PPI is unknown, the FluorIA was developed using full-length proteins and designed to screen for SMIs of the RPA:RAD52 PPI in an unbiased manner (Fig. 2). The application of this protocol to other PPIs will support quests to find therapeutic SMIs or modulators targeting cancer or other pathological conditions.
Fig. 1.
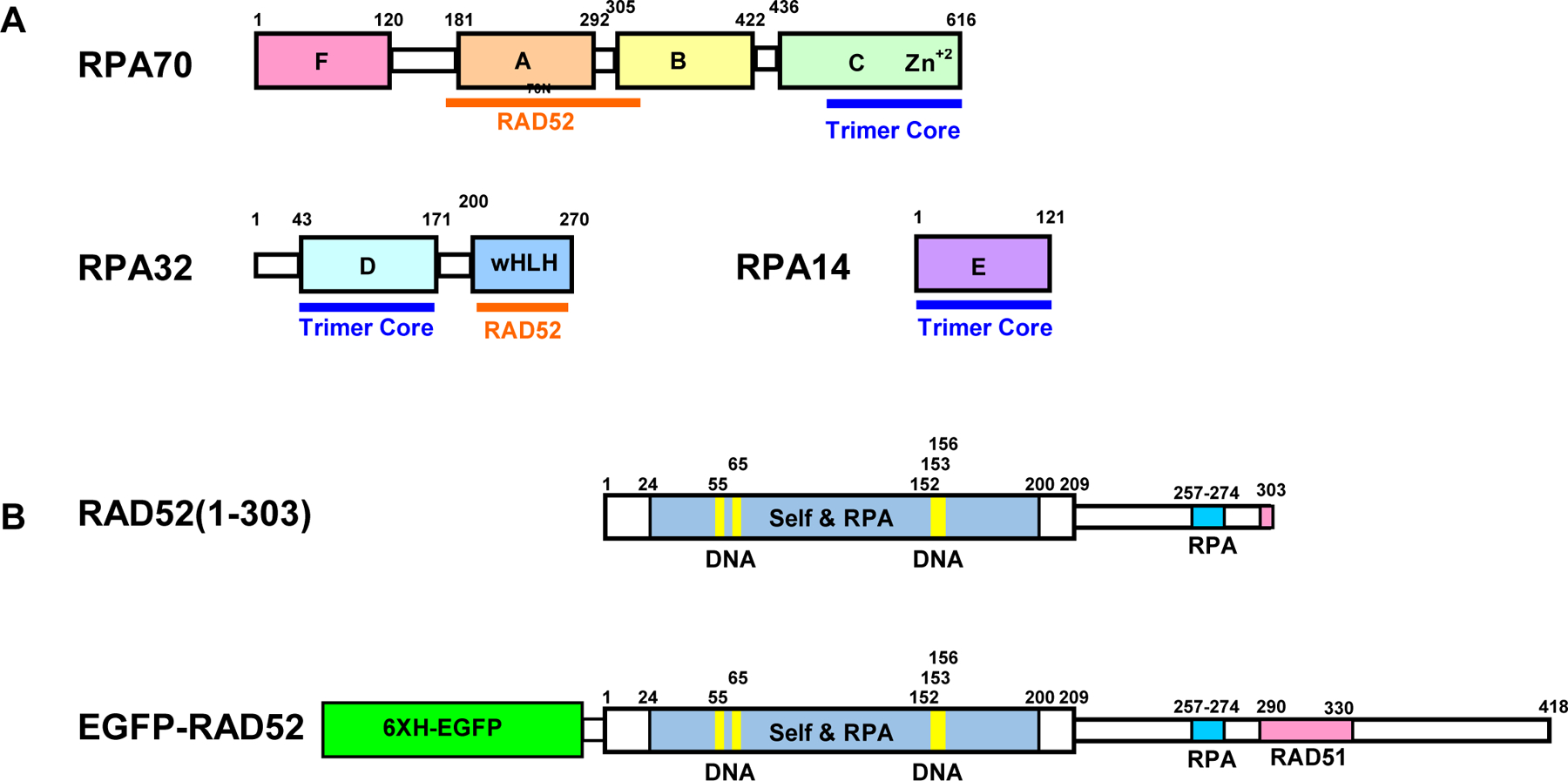
Domain maps of proteins used in this study. (A) RPA, (B), RAD52(1-303), (C) Full length RAD52 tagged with 6x His EGFP. Structured domains and flexible linkers indicated as thick and thin boxes, respectively.
Fig. 2.
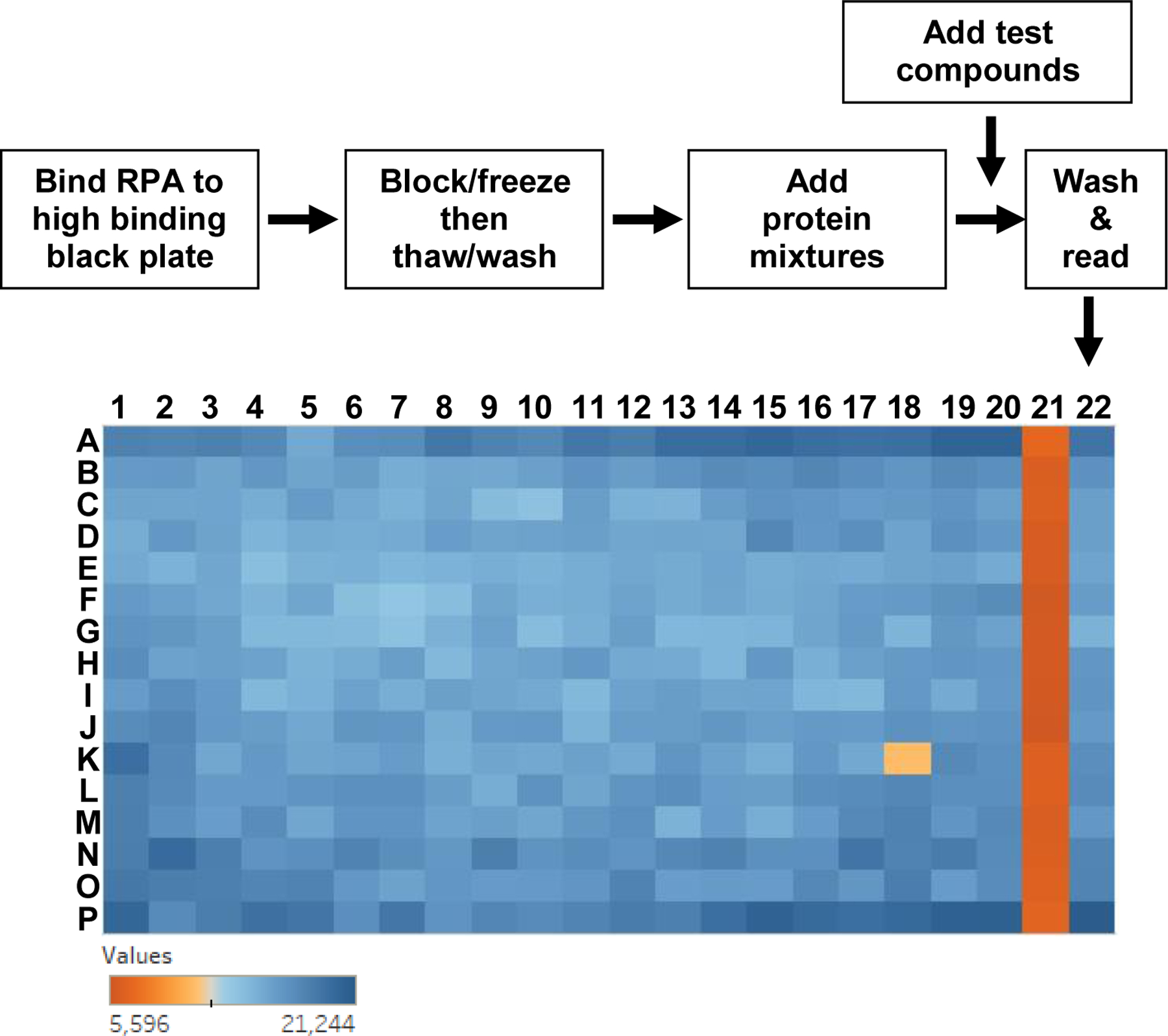
HTS FluorIA procedure diagram and representative data. In the flow diagram (top), protein mixtures included EGFP-RAD52, EGFP-RAD52 with a known inhibitor (RAD52(1-303) or salt) or EGFP alone. In the example 364 plate from HTS of the ChemBridge chemical library, Columns 1–20 are the screen of the chemical library with one test compound from the library added to each well. So for one plate, 320 SMIs were screened. Column 21 is inhibited with RAD52(1-303) control and column 22 is buffer control. For equation 2, column 21 is the PC and column 22 is the NC. Well K18 met the criteria to be a “hit”.
Results and Discussion
FluorIA design
Previously, we used an ELISA-based method that required antibodies and chemiluminescence for detection to analyze the domains of RPA interaction with RAD52 and vice versa 31. This method worked well for biochemical type assays but was too cumbersome, expensive, and used far too many wash steps to be practical for HTS. The FluorIA is a new approach that does not use antibodies, thus avoiding problems of non-specificity, and detects PPI simply with purified proteins with a minimal number of steps and low cost.
Special attention was given to the design of an enhanced green fluorescent protein (EGFP) -RAD52 fusion protein. EGFP was fused with a flexible linker to the N-terminus of RAD52 to avoid any interference with the RPA binding domains. In addition, the linker was designed to be soluble and disordered to avoid perturbing the structure of RAD52 and so that RAD52 would not quench EGFP. There were two features to the linker design. First, the eight C-terminal residues of EGFP in the crystal structure are disordered (PDBID) and so linkage to the EGFP C-terminus had this advantage. Second, the linker incorporated the disordered linker (GlyGlySer)4GlyGly between EGFP and RAD52. Supporting information contains the experimental details for the design of tagging RAD52 with EGFP to produce EGFP-RAD52 (Fig. S1).
Here, purified full-length RPA heterotrimer was directly bound to a black, high-binding microtiter plate (Fig. 2, flow diagram). After washing off excess RPA and blocking any exposed plastic with milk, the PPI was detected with RAD52 tagged with EGFP (Fig. 1C and 2). An increased relative fluorescent unit (RFU) was detected when EGFP-RAD52 was added to RPA (Fig. 2, blue color). A similar response was not detected with the EGFP tag alone indicating the interaction was between RPA and RAD52, not between RPA and the EGFP tag (Fig. 3A). For HTS screening, the test compounds are added after EGFP-RAD52, plates are incubated, washed and the fluorescence signal is read (Fig. 2). Several parameters were optimized before the FluorIA could be used for HTS and these are described below.
Fig. 3.
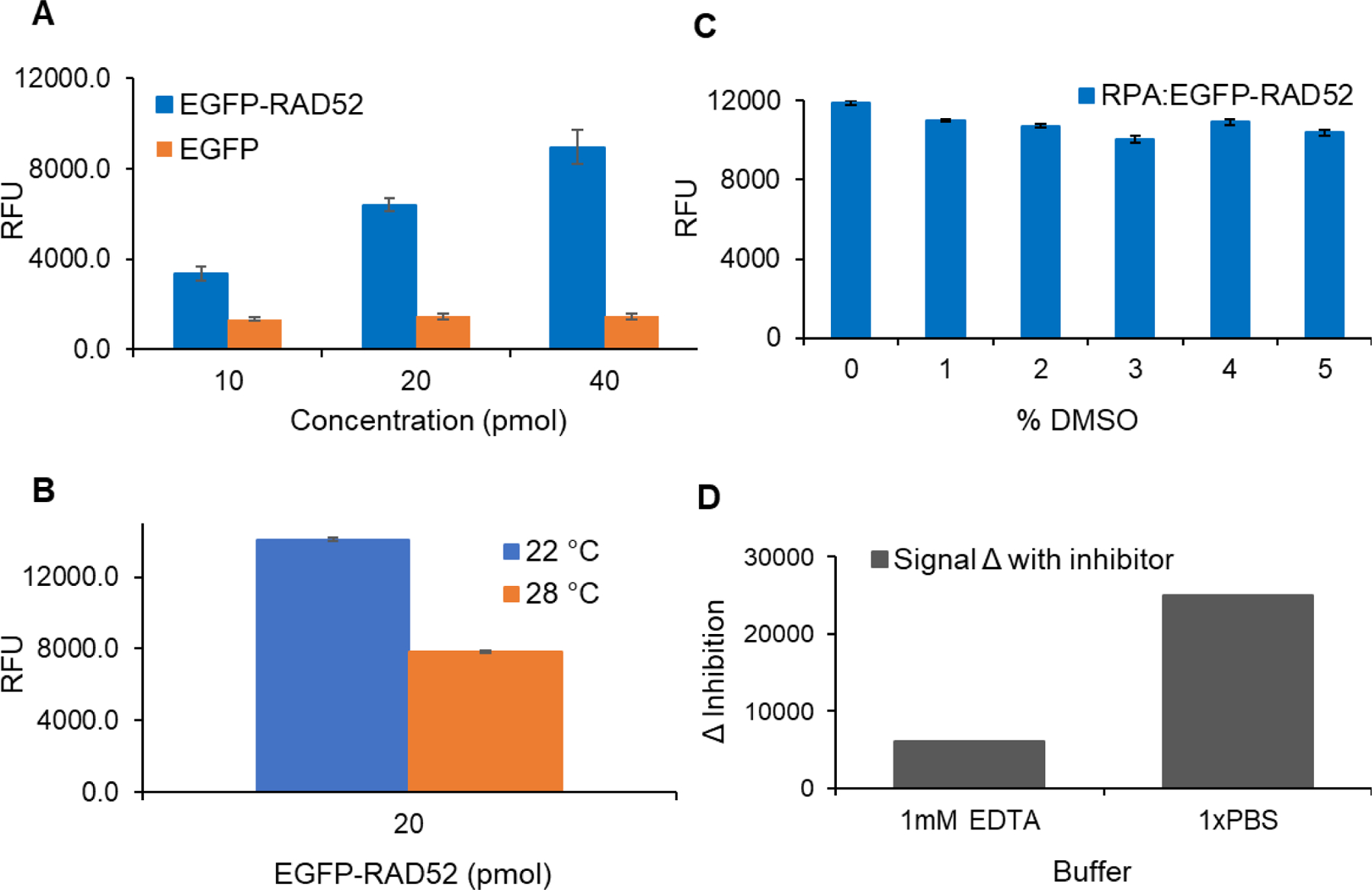
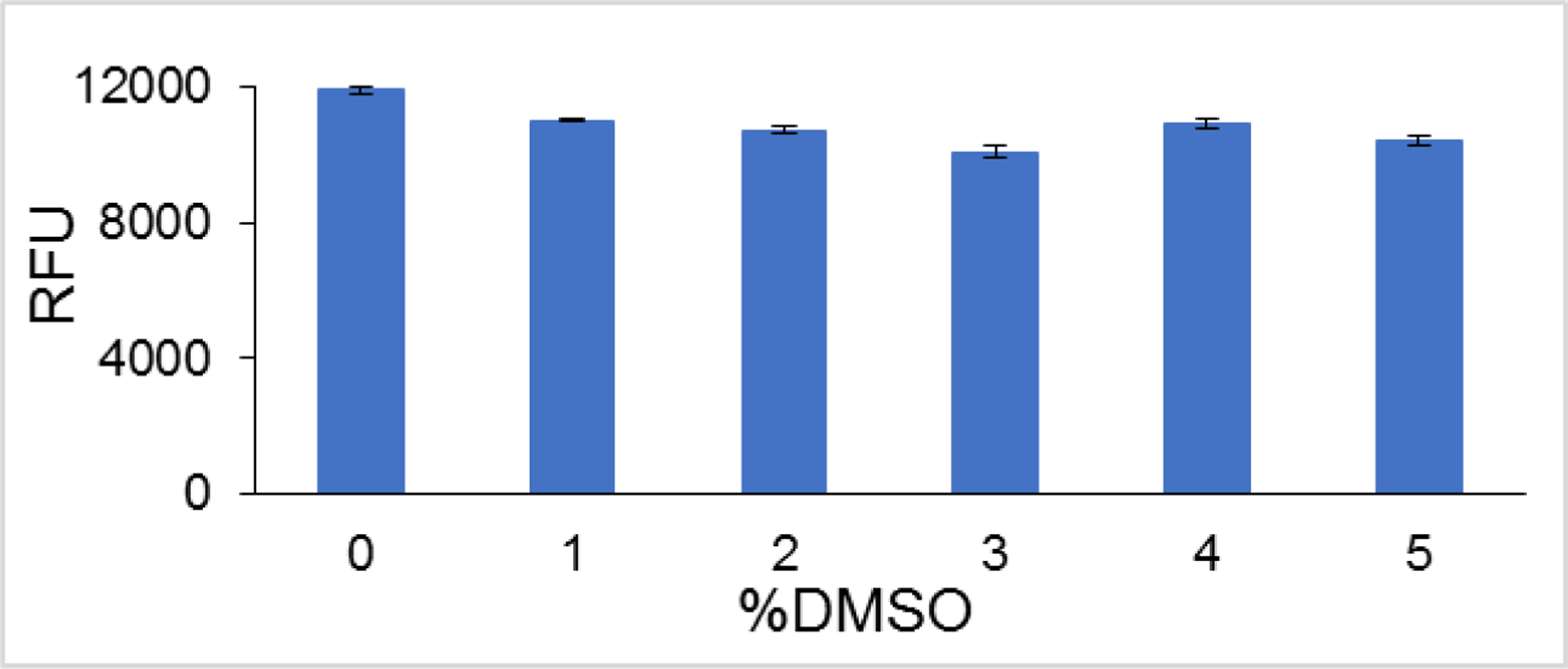
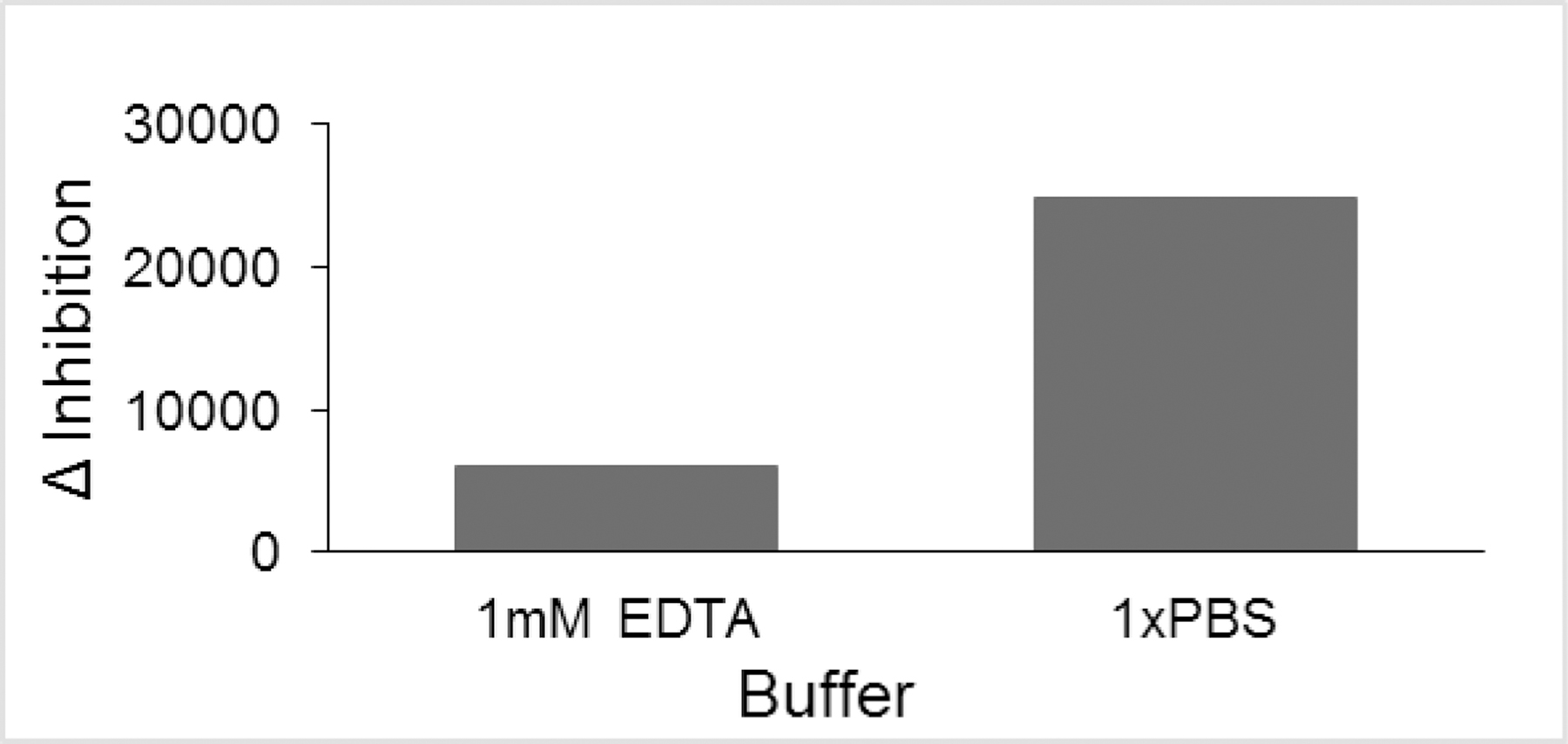
FluorIA optimization. A) Optimization of protein levels per well. The wells were coated with 10 pmol of RPA and the amount of EGFP-RAD52 or EGFP was varied. Z-factors were 0.39, 0.73 and 0.66 for 10, 20, and 40 pmol, respectively. Average values were graphed with standard deviation as error bars. B) FluorIA reaction carried at 22°C or 28°C to test the effect of temperature on proteins’ interaction readout. C) Assessing proteins’ activity in the presence of up to 5% DMSO in the FluorIA reactions. D) Optimal buffer choice. Relative fluorescence units (RFU) is the readout signal on the y-axis which positively correlate with the level of PPI.
Protein levels
A screening window coefficient (denoted Z-factor) was used to assess the quality of the FluorIA signal to see if it was sufficient for HTS 37.
| (1) |
Here, the assay value in the absence of inhibitor was the “sample” while the “control” was the value with the inhibited interaction. In some early pilot assays, EGFP alone was the control before a competitive inhibitor was found. If 1 > Z ≥ 0.5 then the separation band between the sample and control was large enough and the assay was considered excellent.
Z-factor analysis helped us select minimal protein levels that gave an optimal response (Fig. 3A). The best Z-factor value along with lower variation in replicates was obtained for 20 pmol of EGFP-RAD52 per well added to 10 pmol of RPA. Despite higher signal value with 40 pmol EGFP-RAD52, we used 20 pmol concentration for HTS as it gave a consistently good Z-score of ~0.7 in repeats and met the goal of lowering the cost of reagents. We also tested pilot assays with 25 μL/well or 75 μL/ well for each reaction. A 75 µL volume gave the best signal as it filled the small 384-well without overflowing and allowed RPA to bind the flat bottom as well as the walls of the well for the increased signal. As optimization progressed, we increased the amount of RPA bound to the well to 15 pmol to ensure sufficient RPA per well after multiple washing and handling procedures. Excellent Z-factors greater than 0.5 were obtained in the FluorIA mainly due to the low standard deviation of the screen and large difference between sample and control. Before performing HTS, several more parameters were studied to improve the robustness and ease of the FluorIA.
Temperature
The effect of temperature was studied by incubating two plates at our laboratory room temperature (22 °C) and at 28 °C (Fig. 3B). The results suggested that it was best to perform the screen at room temperature and to watch the thermostat for the room. Higher temperature fluctuations probably resulted in detachment of the bound RPA from the wells or bound EGFP-RAD52 from RPA, both resulting in the lower signal. Therefore, the FluorIA was conveniently conducted at room temperature.
SMI solvent effects
At this juncture, it was necessary to assess the resilience of the FluorIA readout signal with the added challenges of HTS. Typically, compounds in chemical libraries are dissolved in 100% DMSO, and that could be detrimental to the integrity of purified proteins, the main components of the FluorIA. We planned to limit the final concentration of DMSO to no more than 5% per reaction well in HTS. Different amounts of DMSO were added with EGFP-RAD52 before incubating in the RPA plates (Fig. 3C) to see if there were any effects. Fortunately, up to 5% DMSO had no significant effect on the FluorIA signal.
Buffer
Effects of the buffer system were explored. For most of the pilot optimization screens, an EDTA-containing buffer system was used. By exploring other buffer systems, we found that 1x PBS gave a larger change between positive and negative controls (Δ inhibition) contributing to the statistical quality of the assay (Fig. 3D).
Finding a known inhibitor
Further development of the assay required the use of a robust known inhibitor for the statistical analysis of each plate and hit identification. Previous work by our group showed that RAD52:RPA electrostatic interaction can be disrupted by increased salt concentration 31. Therefore, we diluted the EGFP-RAD52 with a range of 0–1 M KCl before adding it to the RPA plate. Consistent with our previous findings, inhibition was evident by a progressive decrease in fluorescence signal with increased salt concentration (Fig. 4A). However, it was evident that a high salt concentration was required to achieve the desired baseline inhibition. The possibility of salt interfering with the chemical libraries motivated us to search for an alternative inhibitor.
Fig. 4.
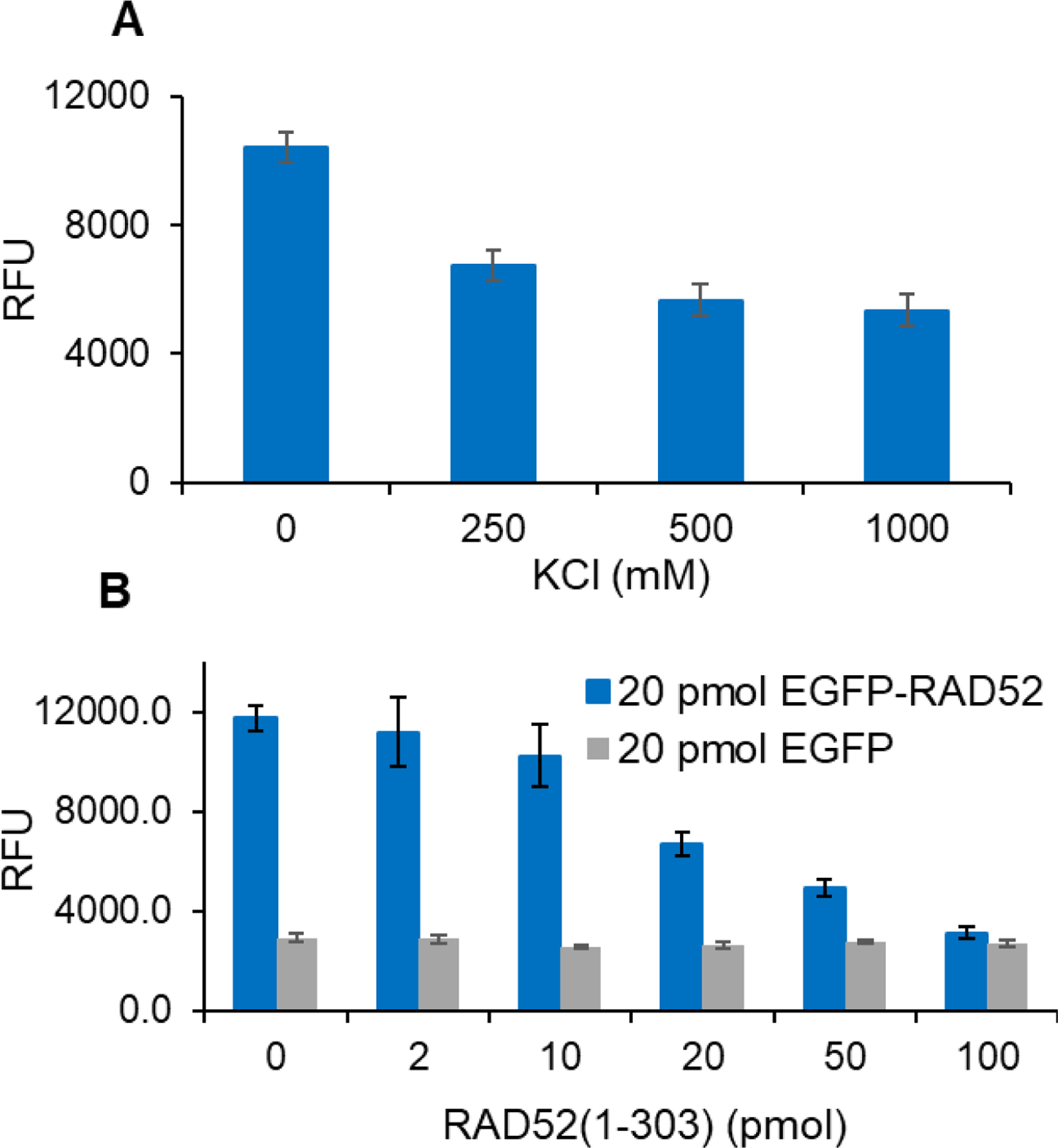
Finding suitable inhibitors to serve as positive controls (A) KCl or (B) RAD52(1-303).
Previously, we demonstrated that the primary interaction sites for RPA on RAD52 are within residues 193–303 31. Also, RAD52(1-303), which has intact RPA binding domains but is missing the remaining 115 residues at the C-terminus, exhibited a slightly higher RPA binding activity compared to full-length RAD52 31. Interestingly, Plate and coworkers reported an enhanced repair activity for a yeast Rad52(1-307) construct compared to full-length Rad52 36. Accordingly, RAD52(1-303) inhibited the FluorIA by competing with EGFP-RAD52 for RPA binding. In a pilot screen, 20 pmol of EGFP-RAD52 was incubated with increasing concentrations of RAD52(1-303) in the RPA plate (Fig. 4B). At a molar ratio of 1:1, 50% inhibition was observed, and with increased concentration of RAD52(1-303), baseline inhibition was achieved. Therefore, 100 pmol of RAD52(1-303) was used as a known inhibitor (positive control) in the FluorIA.
Time and cost efficiency
The parameters defined above provide the essentials for an HTS assay, namely, sensitivity, reliability, and homogeneity. Subsequent work created a successful process for screening large libraries with minimum time and maximum cost efficiency. Assay steps need to be minimized and automated for simplicity and speed. The proper storage conditions for plates, plate-to-plate differences and the storage of assay plates over time were addressed. Once the storage condition in 30% glycerol at the blocking step was found to be optimal, three different RPA plates were tested after freezing overnight to assess plate-to-plate variability (Fig. 5A). As no significant difference was detected between plates, we conducted the freezing test for up to two weeks, and freezing did not lead to a significant change in signal output (Fig. 5B). Thus, making the RPA plates ahead of time and freezing them provided a convenient stopping place in the FluorIA protocol. Also, having the RPA incubation, washing, and blocking steps done ahead significantly mitigated the number of steps to be done on the day of HTS, reduced the risk of costly errors and allowed for screening of several thousand chemicals a day. The case was not the same with EGFP-RAD52. The EGFP signal diminished with storage time (Fig. 5C). EGFP-RAD52 had to be freshly purified the week of the screen. On an HTS day, it was critical to perform a test plate of all proteins and buffers to test activity and Z-factor before committing to a long day of screening. With the optimized FluorIA protocol described here, it was possible for two people to screen up to 10,000 compounds in a single day.
Fig. 5.
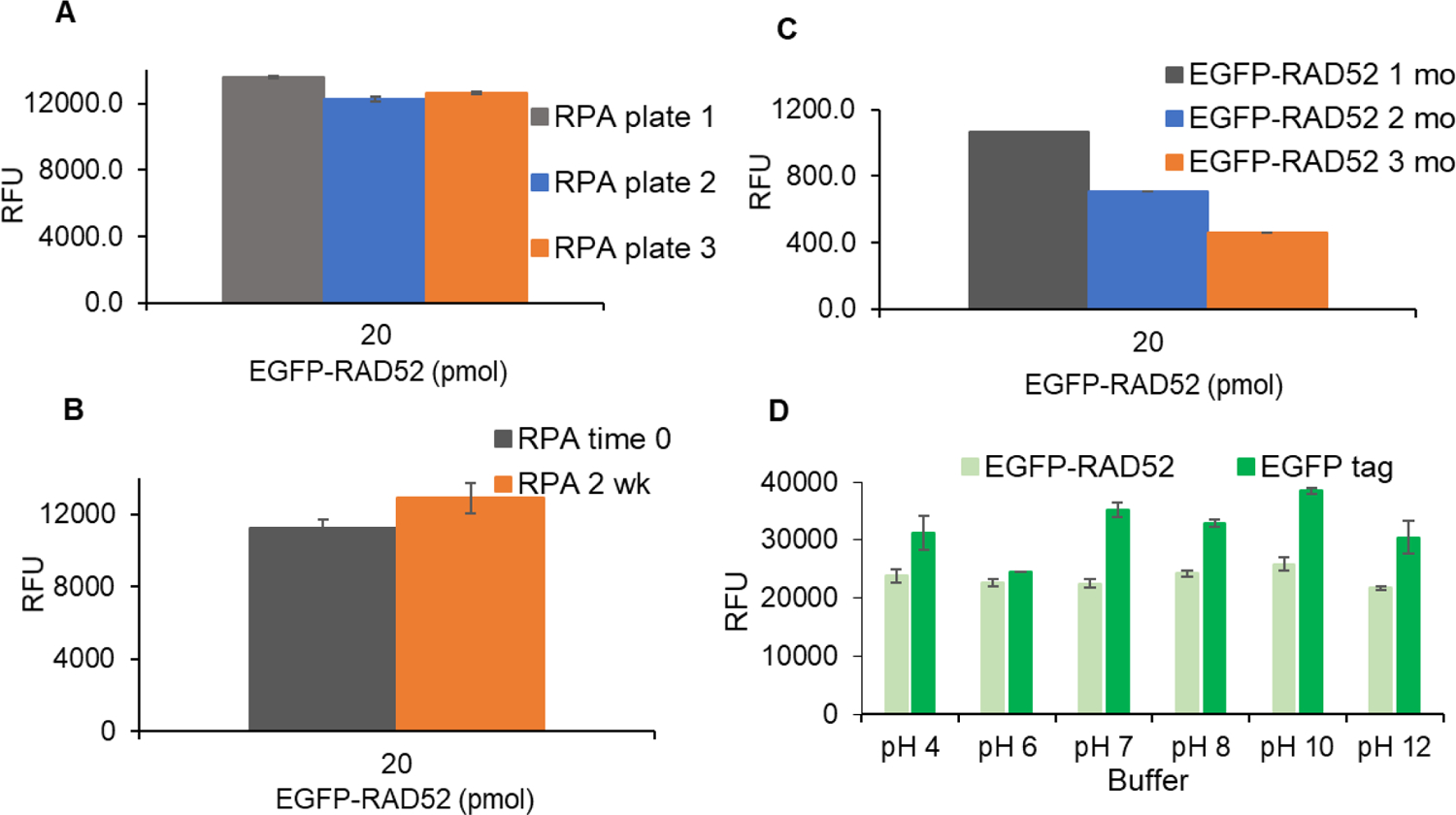
Reagent shelf life. (A) RPA plate variation with three different RPA assay plates prepared, sealed and frozen overnight. (B) Effect of short term freezing on RPA plates. (C) Effect of freezing on purified EGFP-RAD52 signal over three months (mo). (D) Effect of varying buffer’s pH on the EGFP signal. For part C and D, nonbinding plates were used.
pH
EGFP is known to have changes in fluorescence due to changes in pH with a reported pKa of 6.038. We looked at the emitted fluorescence of purified EGFP and EGFP-RAD52 in 1x PBS over a range of pH values (Fig. 5D). EGFP alone had the lowest emitted fluorescence at pH 6 and varied by 13% overall pH values examined. EGFP-RAD52 was more stable and did not vary as dramatically as EGFP alone. EGFP-RAD52 had only a 4% change in the pH range from 6 to 8 and a 6% change overall.
HTS results
First, a small screen of kinase inhibitors was performed. We did not expect a kinase inhibitor to inhibit the PPI. The kinase inhibitor produced no result and therefore no false positives. Then the1200 member Prestwick Chemical Library® was screened. Surprisingly, the Prestwick screen yielded three SMI hits and provided confidence in the screen. Then the 100,000 member ChemBridge library was screened. To conserve reagents and save time the ChemBridge was initially screened singularly and at 100 mM (Fig. 2). An additional eight hits were found and confirmed in triplicate. Thus in total, we have discovered eleven SMIs for the RPA:RAD52 complex that range in molecular weight from 253 to 543 daltons. We also used the FluorIA to test compounds from previous reports including the F79 aptamer and SMIs that target RAD52 ssDNA binding, as they might have some effect on RPA binding 27–29, 39 and they have no effect on the stability of the RPA:RAD52 complex. Thus our SMI hits that inhibit the RPA:RAD52 PPI appear to be unique reagents.
Conclusions
PPIs are important in biological processes as well as pathology. PPIs comprise key features that can be exploited in identifying and designing SMIs. It is now commonly believed that modulating PPI with small molecules is not only possible but advantageous in the quest of targeted therapeutics. Here the FluorIA was proven an effective assay in an HTS setting, and eleven unique SMI hits were discovered for future experiments. Here we demonstrated how the FluorIA could be used to find SMIs that disrupt PPI. FluorIA is applicable to any PPI of sufficient binding affinity where the binding activity can be maintained with purified components. Moreover, FluorIA application is not restricted to screening SMIs and can be applied in screening aptamers as well.
The FluorIA has several advantages to other assays to study PPI inhibitors. Linking a fluorescent protein to the protein-of-interest eliminates the need and expense of other potentially interfering detection methods such as antibodies or dyes. Since purified proteins are used the PPI is studied directly and any SMI found must bind these proteins. The assay can be used in a low throughput fashion or optimized, as we have, for HTS. Disadvantages include the use of protein adhered to the plastic surface of the plate which could be partially denaturing or inactivating. As long as the protein complex can be formed and optimized this is probably not an issue. Also, the proteins are purified and for some systems this is prohibitory.
Experimental procedures
Protein expression and purification
Protein expression and purification.
Several proteins were purified for the FluorIA (Fig. 6). The plasmid for full-length RPA heterotrimer in pET29a vector, a gift from Dr. Marc Wold, was transformed into Rosetta2(DE3) Escherichia coli with chloramphenicol and kanamycin selection. A single colony was inoculated into 5 mL starter culture and grown with selection for 6–8 hours at 37 °C. Large cultures were made by inoculating 1 mL starter culture into 2 L sterilized LB with antibiotics in 4 L flasks and incubating at room temperature or 37 °C overnight without shaking. The following day, the cultures were incubated at 37 °C with shaking and induced with 0.5 mM isopropyl-β-D-l-thiogalactopyranoside (IPTG) at an A600 of 0.6–0.8. After 3 hours, cells were collected by centrifugation at 8,500 xg for 20 min. Pellets were divided into 5 gram portions and stored frozen at −20 °C.
Fig. 6.
SDS-PAGE gel of the purified proteins.
Each RPA cell pellet was thawed and resuspended in 25 mL of HI-0 buffer [30 mM HEPES pH 7.8, 0.25% inositol, 0.25 mM EDTA, and 1mM dithiothreitol] with 500 mM sodium thiocyanate (NaSCN) plus 250 μL of protease inhibitor cocktail (PIC, Sigma, Cat. #P8849) before lysis using an Emulsflex-C3. Lysate was clarified by centrifugation at 45,000 xg for 30–45 min. The NaSCN content was reduced by either dilution or dialysis against HI-0 buffer and then filtered through a 0.45 μm filter. All chromatography steps employed HI-0 with varying salts. First, the lysate was applied to a HiTrap Blue HP column (5 mL, GE Healthcare # 17-0412-01) and washed for 5 CVs with 800 mM KCl followed by 5 CVs with 0.5 M NaSCN and then eluted with 1.5 M NaSCN into 1 mL fractions. The NaSCN content was reduced by either dilution or dialysis against HI-0 buffer and then applied to a hydroxyapatite column (3 mL, BioRAD # 157–0040) and eluted in a 25 CV gradient of 0–75% 160 mM NaPO4. The column was cleaned by washing with 2 M KPO4 in HI-0 after each use. RPA fractions were pooled and diluted with 4 volumes HI-0 before polishing with a monoQ anion exchange column (1 mL, GE Healthcare # 17-5166-01). RPA was eluted in a 25 CV gradient of 0–100% 1 M KCl. Throughout the purification procedures described above, fractions were examined by 10% polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie stain (Fig. 6, lane 5). A NanoDrop1000 was used to measure concentrations using a molecular weight of 110 kDa and ɛ280=87.2 M−1cm−1. The yield from 1 L of culture was 1 to 1.5 mg purified RPA. Purified RPA was stored at −20 °C in 500 mM KCl in HI-0 with 30% glycerol.
The RAD52(1-303) in pET28b plasmid was a gift from Dr. Min Park. It has a 6xHis tag on the C-terminus that is not cleavable. Transformation was done into Rosetta2(DE3). Expression and growth were the same as described for RPA with the exception of allowing large cultures to cool in an ice bath for 30 min before inducing with IPTG. Cultures were then transferred to a pre-chilled shaker at 18 °C for 15–18 hours. Pellets were processed in the same manner described above.
A thawed RAD52(1-303) cell pellet was resuspended in 25 mL of buffer A [50 mM Tris-HCl pH 7.8, 300 mM KCl, 2 mM β-mercaptoethanol (β-ME), 10 mM imidazole, and 10% glycerol] with 250 μL of PIC and lysed. Clarified and filtered lysate was then loaded onto a HisTrap FF column (5 mL, GE Healthcare). The protein eluted with a 25 CV gradient to 1 M imidazole in buffer A. Eluted fractions were pooled and dialyzed against heparin buffer (50 mM Tris-HCl pH 7.5, 200 mM KCl, 2 mM β-ME, 0.5 mM EDTA, and 10% glycerol). The dialyzed protein was loaded onto a HiTrap Heparin HP column (5 mL, GE Healthcare) and eluted with a 25 CV gradient to 1 M KCl in heparin buffer (Fig. 6, lane 4). The recovered protein was dialyzed overnight into a storage buffer (20 mM HEPES pH 6, 400 mM NaCl, 100 mM KCl, 1 mM EDTA, 2 mM β-ME, and 10% glycerol). A NanoDrop1000 was used to measure concentrations using a molecular weight of 34.6 kDa and ɛ280=20.4 M−1cm−1. The yield from 1 L of culture was 0.75 to 1.0 mg purified RAD52(1-303). The protein was stored frozen with 30% glycerol.
Full-length RAD52 tagged with 6X-His enhanced green fluorescent protein (EGFP-RAD52), in pET28a vector was ordered from GenScript with codons optimized for E. coli expression and the overall design is described in the supplement. BL21(DE3) cells were transformed and selected with kanamycin. Expression, growth, and pellet storage was as described for RAD52(1-303). It is noteworthy that overnight growth at 18 °C was mandatory for protein expression.
Thawed EGFP-RAD52 cell pellets were resuspended in 25 mL buffer B (50 mM BICINE pH 9, 300 mM KCl, 200 mM NaCl, 10 mM imidazole, 10% glycerol, and 2 mM β-ME) with 250 μL of PIC and lysed. The clarified lysate was loaded onto a HisTrap column and eluted in a 25 CV gradient to 1 M imidazole in buffer B. Keeping the lysate and buffers ice-cold throughout was necessary to prevent the loss of the EGFP tag from the protein. A NanoDrop1000 was used to measure concentrations using a molecular weight of 76 kDa and ɛ280=63.0 M−1cm−1. The protein product was verified by SDS-PAGE gel (Fig. 6, lane 2). The yield from 1 L of culture was 0.5 to 0.75 mg purified EGFP-RAD52. EGFP-RAD52 must be made fresh the week of HTS.
A plasmid of 6xHis-tagged EGFP in pET28a vector was ordered from GenScript with E. coli optimized codons and transformed into BL21(DE3) with kanamycin selection. Expression, growth, and pellet storage was as described for RAD52(1-303). EGFP cells were resuspended in 25 mL buffer C (50 mM BICINE; pH 9, 20 mM imidazole, 200 mM NaCl, and 2 mM β-ME) with 50 μL of PIC and lysed. The lysate was loaded onto HisTrap column and eluted with 25 CV gradient up to 500 mM imidazole in buffer C. The tag was removed by thrombin digestion to give pure EGFP (Fig. 6, lane 3).
FluorIA procedure
The first step of the FluorIA procedure was to prepare RPA plates (Fig. 2 top left). Purified RPA, diluted to 0.2 µM with milli-Q water, was dispensed by Multidrop Combi (ThermoScientific) to uniformly fill each well of a 384-well, high-binding, “MaxiSorp”, black plastic, flat bottom, opaque microtiter plate (Thermo Fisher Scientific, #460518) to 75 µL volume. Centrifugation at 500 xg for a few seconds prevented air bubble formation and evenly coated the well. Plates were incubated on an orbital shaker for 1 hour at room temperature; then excess protein solution was decanted by flicking plates upside down several times and repeatedly striking against a pad of paper toweling to remove residual liquid. Unbound RPA was removed by two washes, 85 µL per well, using wash buffer (1X PBS with 0.2% Tween-20) with the elimination of residual liquid after each washing step as described above. Freezing-blocking buffer (5% milk in 1X PBS with 30% glycerol) was then dispensed at 85 µL per well and plates briefly centrifuged. Following a 10 min room temperature incubation on an orbital shaker, each plate was sealed with an aluminum adhesive sheet (Thermo Scientific, Cat. # AB-0626) and stored in the −20 °C freezer until use. Typically, we made 60 to 80 RPA plates per day.
On a screening day, the desired number of RPA plates were thawed for 1 hour at room temperature, then the aluminum seal was removed and the freezing-blocking buffer decanted. The plates were washed once and decanted. Two protein mixtures were prepared on the day of the screen: mixture-A is 1.33 µM RAD52(1-303) with 2.67 µM EGFP-RAD52 in 1xPBS-5% milk, and mixture-B is 2.67 µM EGFP-RAD52 alone in 1xPBS-5% milk. In the optimized protocol, 75 µL of each protein mixture was used per well. Mixture-A was dispensed into one column per plate to serve as a positive control (known inhibitor, Fig. 2, column 21) while mixture-B was dispensed into all wells that receive a test compound from the chemical library or buffer only control. A column was reserved for 1xPBS-5%-milk as a buffer blank (Fig. 2, column 22). At this point, the RPA plates were ready to receive compounds from a chemical library. A Biomek F/X liquid handler (Beckman Coulter LifeSciences) automated the transfer of test compounds from a chemical library source plate into the wells of an RPA plate. After test compound addition, RPA plates were centrifuged at 500 xg for a few seconds. The plates were covered with aluminum foil and placed on an orbital shaker for 1 hour at room temperature. Then three washes were performed to remove unbound chemicals, and excess protein before the relative fluorescence (RFU) was measured using a POLARstar OPTIMA plate reader (BMG LABTECH) on emptied plates at an excitation/emission (for EGFP) of 485/520 nm at 2500 gain setting. Plates must be read decanted as reading them with liquid present increased variation in the data.
Screening Libraries of Compounds:
All screening procedures were done at the UNMC Fred & Pamela Cancer Center Molecular Biology/HTS Core Facility. Three libraries were screened: the 355 member SelleckChem Kinase Inhibitor Library, the 1200 member Prestwick Chemical Library, and the 100,000 member ChemBridge library. The SelleckChem Kinase Inhibitor and the Prestwick screens were done at two concentrations of the chemicals, 10 and 100 μM each in duplicate. For the large ChemBridge library, the screen was performed at a single concentration, 100 μM. Then, to eliminate false hits, plates that showed potential hits were repeated. Potential hits were further verified in triplicate at 10 and 100 μM.
Statistical analysis
Candidate SMI hits were identified as follows: On each screening plate, the average value of a designated column containing no potential inhibitors and buffer only was the negative control (NC). The average value of a column containing the known inhibitor RAD52(1-303) was the positive control (PC). Then
| (2) |
A well with SMI was considered a hit if its value was lower than Xhit. We decided to accept hits if the value was lower than 50% of the NC-PC value (or n=2). The rationale for this was that the RPA:RAD52 PPI under study might involve two separate binding surfaces and a hit might bind to only half of the uncharacterized surface, diminishing the binding by half and so the value of n was 2 in this example. The arbitrary value of 50% could also take into account the possible lower binding affinity of the SMI relative to the PPI. Typically, the hits were also three standard deviations below the NC. For the example data in Figure 2, the value of NC was 17,053; PC was 6,116 and Xhit was 11,584. Well K18 (Fig. 2, light orange) was a hit with a value of 10,941 that is less than Xhit.
Supplementary Material
Figure 7.
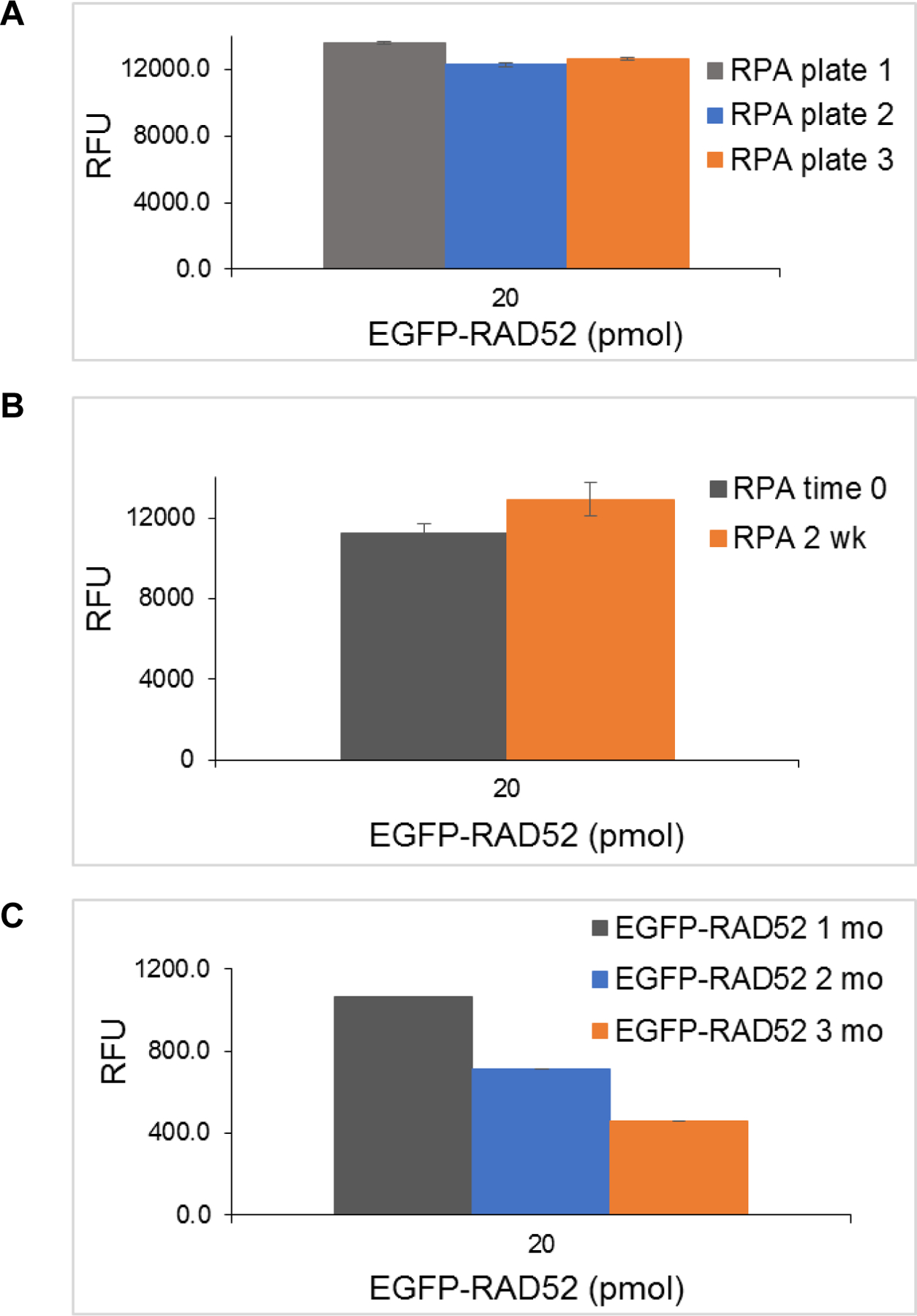
Figure 8.
Acknowledgements:
We thank William Lutz, Jacob Remsza, Admir Kellezi, Jeff Lovelace and Amy Wells for technical assistance. We also thank UNMC Fred & Pamela Cancer Center Molecular Biology/HTS Core Facility. The research was supported by the Fred & Pamela Buffett Cancer Center Support Grant (P30CA036727) pilot project funding, the Nebraska Research Initiative and the Nebraska Department of Health and Human Services. MAM also acknowledges the U.S. Department of Education GAANN (P200A120231) and Nebraska NASA EPSCoR Space Grant for student fellowships.
Abbreviations:
- EGFP
enhanced green fluorescent protein
- ELISA
enzyme-linked immunosorbent assay
- FluorIA
fluorescence-based protein-protein Interaction Assay
- HTS
high throughput screening
- PPI
protein-protein interactions
- RFU
relative fluorescent unit
- RPA
replication protein A
- SMI
small molecule inhibitor
References
- 1.Fox S, Farr-Jones S, Sopchak L, Boggs A, Nicely HW, Khoury R, and Biros M, High-throughput screening: update on practices and success. J Biomol Screen, 2006. 11(7): p. 864–9. [DOI] [PubMed] [Google Scholar]
- 2.Inglese J, Johnson RL, Simeonov A, Xia M, Zheng W, Austin CP, and Auld DS, High-throughput screening assays for the identification of chemical probes. Nat Chem Biol, 2007. 3(8): p. 466–79. [DOI] [PubMed] [Google Scholar]
- 3.Polyak K and Garber J, Targeting the missing links for cancer therapy. Nat Med, 2011. 17(3): p. 283–4. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D and Weinberg RA, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646–74. [DOI] [PubMed] [Google Scholar]
- 5.Wells JA and McClendon CL, Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature, 2007. 450(7172): p. 1001–9. [DOI] [PubMed] [Google Scholar]
- 6.Laraia L, McKenzie G, Spring DR, Venkitaraman AR, and Huggins DJ, Overcoming Chemical, Biological, and Computational Challenges in the Development of Inhibitors Targeting Protein-Protein Interactions. Chem Biol, 2015. 22(6): p. 689–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scott DE, Bayly AR, Abell C, and Skidmore J, Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov, 2016. 15(8): p. 533–50. [DOI] [PubMed] [Google Scholar]
- 8.Mayer JP and Dimarchi RD, Drugging the undruggable. Chem Biol, 2005. 12(8): p. 860–1. [DOI] [PubMed] [Google Scholar]
- 9.Murray JK and Gellman SH, Targeting protein-protein interactions: lessons from p53/MDM2. Biopolymers, 2007. 88(5): p. 657–86. [DOI] [PubMed] [Google Scholar]
- 10.Villoutreix BO, Kuenemann MA, Poyet JL, Bruzzoni-Giovanelli H, Labbe C, Lagorce D, Sperandio O, and Miteva MA, Drug-Like Protein-Protein Interaction Modulators: Challenges and Opportunities for Drug Discovery and Chemical Biology. Mol Inform, 2014. 33(6–7): p. 414–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arkin MR, Tang Y, and Wells JA, Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol, 2014. 21(9): p. 1102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yu JL, Chen TT, Zhou C, Lian FL, Tang XL, Wen Y, Shen JK, Xu YC, Xiong B, and Zhang NX, NMR-based platform for fragment-based lead discovery used in screening BRD4-targeted compounds. Acta Pharmacol Sin, 2016. 37(7): p. 984–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wan KF, Wang S, Brown CJ, Yu VC, Entzeroth M, Lane DP, and Lee MA, Differential scanning fluorimetry as secondary screening platform for small molecule inhibitors of Bcl-XL. Cell Cycle, 2009. 8(23): p. 3943–52. [DOI] [PubMed] [Google Scholar]
- 14.Du Y, Fluorescence polarization assay to quantify protein-protein interactions in an HTS format. Methods Mol Biol, 2015. 1278: p. 529–44. [DOI] [PubMed] [Google Scholar]
- 15.Szelag M, Czerwoniec A, Wesoly J, and Bluyssen HA, Identification of STAT1 and STAT3 specific inhibitors using comparative virtual screening and docking validation. PLoS One, 2015. 10(2): p. e0116688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yasui T, Yamamoto T, Sakai N, Asano K, Takai T, Yoshitomi Y, Davis M, Takagi T, Sakamoto K, Sogabe S, Kamada Y, Lane W, Snell G, Iwata M, Goto M, Inooka H, Sakamoto JI, Nakada Y, and Imaeda Y, Discovery of a novel B-cell lymphoma 6 (BCL6)-corepressor interaction inhibitor by utilizing structure-based drug design. Bioorg Med Chem, 2017. 25(17): p. 4876–4886. [DOI] [PubMed] [Google Scholar]
- 17.Sameshima T, Yamamoto T, Sano O, Sogabe S, Igaki S, Sakamoto K, Ida K, Gotou M, Imaeda Y, Sakamoto J, and Miyahisa I, Discovery of an Irreversible and Cell-Active BCL6 Inhibitor Selectively Targeting Cys53 Located at the Protein-Protein Interaction Interface. Biochemistry, 2018. [DOI] [PubMed]
- 18.Moynahan ME, Pierce AJ, and Jasin M, BRCA2 is required for homology-directed repair of chromosomal breaks. Mol Cell, 2001. 7(2): p. 263–72. [DOI] [PubMed] [Google Scholar]
- 19.Sung P, Mediating repair. Nat Struct Mol Biol, 2005. 12(3): p. 213–4. [DOI] [PubMed] [Google Scholar]
- 20.Jensen RB, Carreira A, and Kowalczykowski SC, Purified human BRCA2 stimulates RAD51-mediated recombination. Nature, 2010. 467(7316): p. 678–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu J, Doty T, Gibson B, and Heyer WD, Human BRCA2 protein promotes RAD51 filament formation on RPA-covered single-stranded DNA. Nat Struct Mol Biol, 2010. 17(10): p. 1260–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu J and Heyer WD, Who’s who in human recombination: BRCA2 and RAD52. Proc Natl Acad Sci U S A, 2011. 108(2): p. 441–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaheen M, Allen C, Nickoloff JA, and Hromas R, Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood, 2011. 117(23): p. 6074–82. [DOI] [PubMed] [Google Scholar]
- 24.Hanamshet K, Mazina OM, and Mazin AV, Reappearance from Obscurity: Mammalian Rad52 in Homologous Recombination. Genes (Basel), 2016. 7(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng Z, Scott SP, Bussen W, Sharma GG, Guo G, Pandita TK, and Powell SN, Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc Natl Acad Sci U S A, 2011. 108(2): p. 686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lok BH, Carley AC, Tchang B, and Powell SN, RAD52 inactivation is synthetically lethal with deficiencies in BRCA1 and PALB2 in addition to BRCA2 through RAD51-mediated homologous recombination. Oncogene, 2013. 32(30): p. 3552–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cramer-Morales K, Nieborowska-Skorska M, Scheibner K, Padget M, Irvine DA, Sliwinski T, Haas K, Lee J, Geng H, Roy D, Slupianek A, Rassool FV, Wasik MA, Childers W, Copland M, Muschen M, Civin CI, and Skorski T, Personalized synthetic lethality induced by targeting RAD52 in leukemias identified by gene mutation and expression profile. Blood, 2013. 122(7): p. 1293–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandramouly G, McDevitt S, Sullivan K, Kent T, Luz A, Glickman JF, Andrake M, Skorski T, and Pomerantz RT, Small-Molecule Disruption of RAD52 Rings as a Mechanism for Precision Medicine in BRCA-Deficient Cancers. Chem Biol, 2015. 22(11): p. 1491–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang F, Goyal N, Sullivan K, Hanamshet K, Patel M, Mazina OM, Wang CX, An WF, Spoonamore J, Metkar S, Emmitte KA, Cocklin S, Skorski T, and Mazin AV, Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res, 2016. 44(9): p. 4189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sugiyama T, New JH, and Kowalczykowski SC, DNA annealing by RAD52 protein is stimulated by specific interaction with the complex of replication protein A and single-stranded DNA. Proc Natl Acad Sci U S A, 1998. 95(11): p. 6049–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jackson D, Dhar K, Wahl JK, Wold MS, and Borgstahl GE, Analysis of the human replication protein A:Rad52 complex: evidence for crosstalk between RPA32, RPA70, Rad52 and DNA. J Mol Biol, 2002. 321(1): p. 133–48. [DOI] [PubMed] [Google Scholar]
- 32.Deng X, Prakash A, Dhar K, Baia GS, Kolar C, Oakley GG, and Borgstahl GE, Human replication protein A-Rad52-single-stranded DNA complex: stoichiometry and evidence for strand transfer regulation by phosphorylation. Biochemistry, 2009. 48(28): p. 6633–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park MS, Ludwig DL, Stigger E, and Lee SH, Physical interaction between human RAD52 and RPA is required for homologous recombination in mammalian cells. J Biol Chem, 1996. 271(31): p. 18996–9000. [DOI] [PubMed] [Google Scholar]
- 34.Mer G, Bochkarev A, Gupta R, Bochkareva E, Frappier L, Ingles CJ, Edwards AM, and Chazin WJ, Structural basis for the recognition of DNA repair proteins UNG2, XPA, and RAD52 by replication factor RPA. Cell, 2000. 103(3): p. 449–56. [DOI] [PubMed] [Google Scholar]
- 35.Mer G, Edwards AM, and Chazin WJ, 1H, 15N and 13C resonance assignments for the C-terminal protein interaction region of the 32 kDa subunit of human replication protein A. J Biomol NMR, 2000. 17(2): p. 179–80. [DOI] [PubMed] [Google Scholar]
- 36.Plate I, Hallwyl SC, Shi I, Krejci L, Muller C, Albertsen L, Sung P, and Mortensen UH, Interaction with RPA is necessary for Rad52 repair center formation and for its mediator activity. J Biol Chem, 2008. 283(43): p. 29077–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang JH, Chung TD, and Oldenburg KR, A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen, 1999. 4(2): p. 67–73. [DOI] [PubMed] [Google Scholar]
- 38.Campbell TN and Choy FY, Large-scale colony screening and insert orientation determination using PCR. Biotechniques, 2001. 30(1): p. 32–4. [DOI] [PubMed] [Google Scholar]
- 39.Sullivan K, Cramer-Morales K, McElroy DL, Ostrov DA, Haas K, Childers W, Hromas R, and Skorski T, Identification of a Small Molecule Inhibitor of RAD52 by Structure-Based Selection. PLoS One, 2016. 11(1): p. e0147230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.