

Abstract
BACKGROUND AND PURPOSE:
EPM1, caused by mutations in the CSTB gene, is the most common form of PME. The most incapacitating symptom of EPM1 is action-activated and stimulus-sensitive myoclonus. The clinical severity of the disease varies considerably among patients, but so far, no correlations have been observed between quantitative structural changes in the brain and clinical parameters such as duration of the disease, age at onset, or myoclonus severity. The aim of this study was to evaluate possible changes in CTH of patients with EPM1 compared with healthy controls and to correlate those changes with clinical parameters.
MATERIALS AND METHODS:
Fifty-three genetically verified patients with EPM1 and 70 healthy volunteers matched for age and sex underwent 1.5T MR imaging. T1-weighted 3D images were analyzed with CTH analysis to detect alterations. The patients were clinically evaluated for myoclonus severity by using the UMRS. Higher UMRS scores indicate more severe myoclonus.
RESULTS:
CTH analysis revealed significant thinning of the sensorimotor and visual and auditory cortices of patients with EPM1 compared with healthy controls. CTH was reduced with increasing age in both groups, but in patients, the changes were confined specifically to the aforementioned areas, while in controls, the changes were more diffuse. Duration of the disease and the severity of myoclonus correlated negatively with CTH.
CONCLUSIONS:
Cortical thinning in the sensorimotor areas in EPM1 correlated significantly with the degree of the severity of the myoclonus and is most likely related to the widespread stimulus sensitivity in EPM1.
PME are a heterogeneous group of neurologic disorders characterized by myoclonus, epilepsy, and progressive neurologic deterioration.1 The most common form of PME is EPM1 (Online Mendelian Inheritance in Man, 254800; http://www.omim.org), an autosomal recessively inherited disorder caused by mutations in the CSTB gene.2 EPM1 occurs worldwide, but its prevalence is increased in certain populations (eg, in Finland), where its incidence is about 1:20,000 births per year,3 and in the western Mediterranean countries.4 The symptoms at onset are action-activated and stimulus-sensitive myoclonic jerks or tonic-clonic epileptic seizures, or both. Patients exhibit spontaneous myoclonus, and they are also usually sensitive to exogenic stimuli, especially to photic stimulation, but also to light touch, sounds, passive movements, or initiation of motor activity. The age at onset is typically between 6 and 16 years. Cognitive decline is mild or absent.5
There are only a few previous imaging reports investigating EPM1.6–10 Loss of bulk in the pons and cerebellum6 and loss of GM volume in cortical motor areas and thalami have been reported.10 These structural findings may explain the occurrence of devastating myoclonic symptoms in EPM1.
The myoclonic jerks are often provoked by a variety of exogenic stimuli5; however, morphologic changes in the sensory cortical areas of the brain have not been reported, to our knowledge. In addition, there is considerable variation in the severity of the disease among patients and even within the same family,5 but to the best of our knowledge, no correlations between quantitative structural changes in the brain and clinical parameters such as the duration of the disease, age at onset, or myoclonus severity have been investigated so far.
CTH analysis is a novel method that has been proposed as being a more sophisticated alternative to volumetric and VBM methods to measure regional brain atrophy.11 This method has been shown to be reliable in detecting changes in cortical morphology both at the single subject and the group level, and it provides a direct quantitative measure of thickness of the cerebral cortex.12 It has shown promise as a sensitive imaging biomarker in mild cognitive impairment as well as in Alzheimer disease, autism, and temporal lobe epilepsy,13–15 but it has not been used in EPM1.
The aim of this study was to investigate possible regional alterations in the CTH of patients with EPM1 compared with healthy controls and to correlate those changes with clinical symptoms suggesting both motor and sensory cortical hyperexcitability.
Materials and Methods
Subjects
Fifty-three genetically verified patients with EPM1 homozygous for the dodecamer repeat expansion mutation in the CSTB gene (29 men and 24 women; mean age, 35 years; range, 18–64 years) participating in an ongoing clinical and molecular genetics study conducted by Kuopio Epilepsy Center, Kuopio University Hospital, jointly with the Folkhälsan Institute of Genetics and Neuroscience Center, University of Helsinki, were evaluated in the Kuopio University Hospital. Seventy healthy volunteers matched for age and sex (34 men and 36 women; mean age, 33 years; range, 18–59 years) provided control values for the cortical thickness analysis. The ethics committee of Kuopio University Hospital approved the study protocol, and written informed consent was obtained from all participants.
Clinical Assessment of Patients with EPM1
Patients' medical histories were collected from medical records and by interviewing patients and their relatives. Their score on the UMRS test panel was evaluated as part of the clinical examination. UMRS is a quantitative 74-item clinical rating instrument comprising 8 sections.16 In the present study, 3 sections were used to evaluate the severity of myoclonus: “Stimulus Sensitivity” (maximum score 17), “Myoclonus with Actio” (maximum score 160), and “Functional Test” (maximum score 28). These sections were video-recorded and evaluated by using a standard protocol.16 Higher UMRS scores are indicative of more severe myoclonus.
MR Imaging Acquisition Protocol and Data Analysis
MR imaging was performed on a clinical 1.5T scanner (Avanto; Siemens, Erlangen, Germany). The imaging protocol included T1- and T2-weighted spin-echo sequences, a fluid attenuated inversion recovery sequence, and T1-weighted 3D images (magnetization-prepared rapid acquisition of gradient echo: TR, 1980 ms; TE, 3.09 ms; flip angle, 15°; matrix, 256 × 256; 176 sagittal sections with section thickness varying between 1.0 and 1.2 mm depending on the size of the head; and in-section resolution of 1.0 × 1.0 mm). A neuroradiologist (P.K.) assessed all conventional images visually for focal abnormalities.
CTH Analysis
CTH analysis was conducted by using the pipelining method developed at the McConnell Brain Imaging Centre, Montreal Neurologic Institute, McGill University, Montreal, Canada (http://www2.bic.mni.mcgill.ca/). All subjects' T1-weighted 3D images were registered to standard space by using the International Consortium for Brain Mapping 152 template17 and corrected for nonuniformity artifacts.18 The images were then segmented into WM, GM, and CSF by using the intensity-normalized stereotaxic environment for classification of tissues (INSECT) algorithm,19 and the magnitude of the partial volume effect was estimated.20 The brains were then automatically divided into 2 separate hemispheres. By using the constrained Laplacian anatomic segmentation using proximity (CLASP) algorithm,21,22 the inner and outer surfaces of the cortex were extracted according to intersections between WM and GM (WM surface) as well as GM and CSF (GM surface), and the distance between the 2 surfaces was calculated at 40,962 nodes per hemisphere with the t-link metric.12 The thickness calculations were performed on each subject's native space and were then transformed back to standard space for the group analysis. The data were then smoothed with a 20-mm surface-based diffusion-smoothing kernel before the statistical analysis.23
The peak voxels of the significantly thinned cortical areas were defined by searching the highest t values and their coordinates in the images by using BrainView software (McConnell Brain Imaging Centre). The location of each peak voxel was then verified from an anatomic atlas.24
Statistical Analysis
Statistical analyses of CTH were performed according to the general linear model with Matlab R2007b (MathWorks, Natick, Massachusetts). Significant between-group differences were tested by using a t test with a threshold of P < .0001, corrected for multiple comparisons with the FDR method.25 Age, sex, and voxel size of the original image were used as nuisance variables. The correlations between clinical parameters and cortical thickness of patients with EPM1 were tested in every node by examining whether the variation in CTH correlated with the variation in clinical parameters. Sex and voxel size of the original image were used as nuisance variables.
Statistical analyses of demographic data were performed with the Statistical Package for the Social Sciences for Windows, Version 17 (SPSS, Chicago, Illinois).
Results
Demographic and clinical data of the patients are presented in Table 1. When assessed visually, no focal signal intensity abnormalities in the conventional images were found. Statistical differences in CTH between the patients with EPM1 and healthy controls at group level are presented in detail in Fig 1 and Table 2. The EPM1 group displayed widespread thinning of the cortex in both hemispheres. The effects were more extensive in the left hemisphere.
Table 1:
Basic clinical characteristics and demographics of 53 patients with EPM1 and 70 controlsa)
| Patients with EPM1 | Controls | |
|---|---|---|
| Age (yr) | 35 ± 11.5 | 33 ± 10.7 |
| Sex (M/F) | 29/24 | 34/36 |
| Handedness (R/L/B/unknown) | 35/6/1/11 | 62/4/3/1 |
| Age at EPM1 onset (yr) | 10.7 ± 2.9 (6–25) | |
| Duration of the disease (yr) | 24.2 ± 10.4 (4–44) | |
| UMRS: stimulus sensitivity | 1.92 ± 1.9 (0–8) | |
| UMRS: myoclonus with action | 48.5 ± 28.4 (2–120) | |
| UMRS: functional tests | 10.35 ± 7 (0–28) |
Note:—R indicates right; L, left; B, bilateral.
a Data are presented as means unless otherwise indicated.
Fig 1.
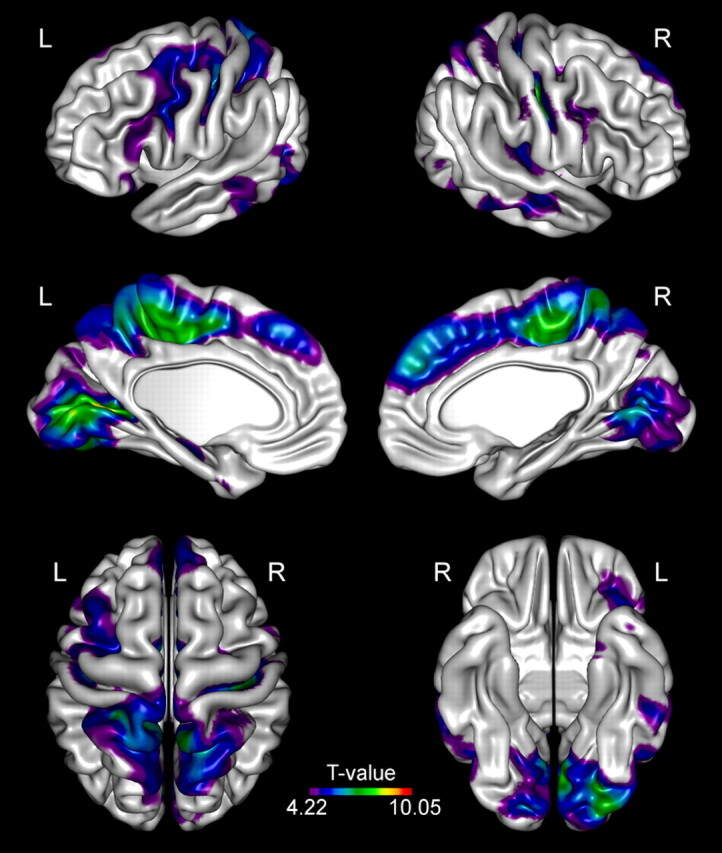
Group analysis of CTH. Brain regions demonstrate significantly (P < .0001, FDR corrected) reduced CTH in patients with EPM1 compared with healthy controls.
Table 2:
Areas of significant difference in CTH between the EPM1 group and controls
| Brain Region | Hemisphere | Maximum T Value | MNI Coordinates, (x, y, z)a |
|---|---|---|---|
| Central sulcus | Right | 8.42 | 42, −16, 36 |
| Left | 6.69 | −39, −19, 46 | |
| Precentral sulcus | Left | 6.05 | −40, 7, 27 |
| Paracentral lobule | Right | 8.11 | 5, −27, 50 |
| Postcentral gyrus | Right | 7.32 | 6, −49, 65 |
| Cingulate sulcus | Left | 8.06 | −10, −19, 46 |
| Postcentral sulcus | Left | 6.79 | −24, −40, 60 |
| Superior frontal gyrus | Right | 6.48 | 7, 45, 29 |
| Calcarine sulcus | Left | 8.99 | −20, −70, 6 |
| Lingual gyrus | Right | 6.52 | 5, −68, 3 |
| Left | 8.20 | −3, −76, 5 | |
| Superior temporal sulcus | Right | 5.32 | 54, −16, −14 |
| Transverse temporal gyrus | Right | 5.33 | 43, −28, 10 |
| Middle temporal gyrus | Left | 5.98 | −48, −66, 4 |
| Inferior occipital gyrus | Left | 7.39 | −27, −89, −17 |
Note:—MNI indicates Montreal Neurological Institute.
MNI coordinates are based on a standard brain template defined by using multiple MR images of healthy controls.
Group Analysis of CTH
The most significant alterations in the sensorimotor cortex were detected in the paracentral lobule bilaterally and in the depth of the central sulcus bilaterally. There was also thinning of the bilateral premotor and supplementary motor cortices and Broca area.
The most pronounced changes in the visual cortical areas were detected bilaterally in the primary visual cortices (pericalcarine cortex). There was also thinning of the associative visual areas in the occipital lobes and the parietal associative cortex, as well as bilaterally in the temporal association cortex.
Changes in the auditory areas were detected in the left primary auditory cortex. The parietal association cortex (precuneus) was bilaterally involved.
CTH Correlations to Clinical Parameters
Figure 2 illustrates the correlation between the mean CTH and age in both patients with EPM1 and controls. CTH was reduced with increasing age in both groups, but in patients, the alterations were confined to more limited areas of the sensorimotor, primary, and associative visual and primary auditory cortices (P < .01) (Fig 2A), whereas in controls, the age-related thinning was more diffuse throughout the hemispheres (P < .01) (Fig 2B). Furthermore, the cortex in the affected areas in patients with EPM1 was thinner than that in controls.
Fig 2.

Brain regions with significant (P < .01, FDR-corrected) negative correlations between age and CTH in patients with EPM1 (A) and controls (B). Scatterplots indicate correlations between age and mean CTH in individual subjects. The individual mean thicknesses have been calculated on the basis of the regions surviving the statistical threshold.
In a univariate analysis, there was a significant negative correlation between the duration of the disease and CTH in the same regions as the age effects in EPM1 (P < .01). When age was included in the analysis as a nuisance variable, the correlations did not survive the FDR correction.
There was also a significant negative correlation between the “Myoclonus with Action” score and CTH, especially in the pars opercularis and supramarginal gyrus bilaterally, but also in the superior and inferior parietal gyri (parietal association cortex), visual areas, and the premotor and prefrontal cortices bilaterally (P < .05) (Fig 3). When illness duration was included as a nuisance variable, the correlations did not survive the FDR correction.
Fig 3.
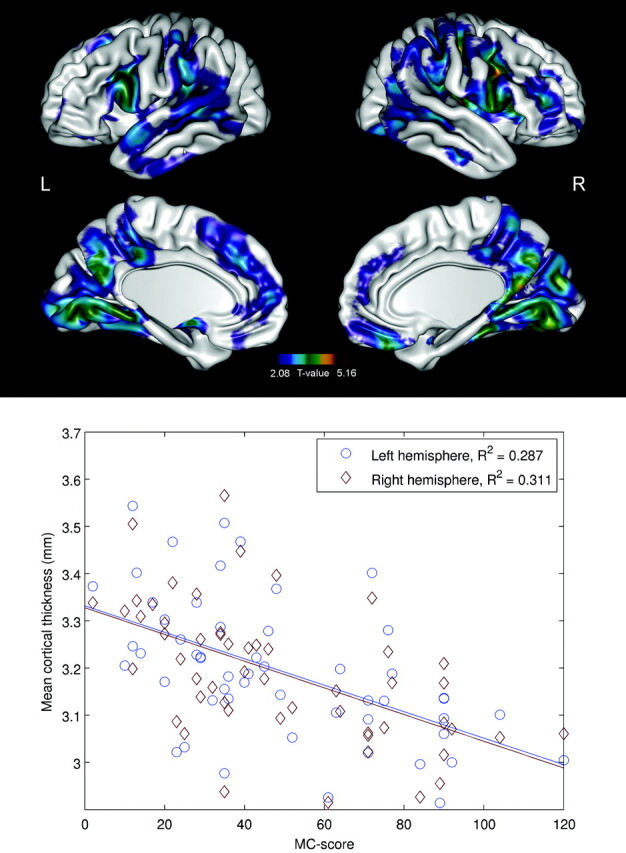
Brain regions with significant (P < .05, FDR-corrected) negative correlations between the “Myoclonus with Action” score and CTH in patients with EPM1. Scatterplots indicate correlations between the “Myoclonus with Action” score and mean CTH in individual subjects. The individual mean thicknesses have been calculated on the basis of the regions surviving the statistical threshold.
The correlation analysis between CTH and the age at onset did not reveal any significant results.
Discussion
The aim of the present MR imaging study was to evaluate possible regional alterations in CTH between patients with EPM1 and healthy controls in a larger patient population by using CTH as a detailed quantitative measure. Furthermore, the correlations between the cortical changes and several clinical parameters such as the “Myoclonus with Action” score were explored to study whether the imaging biomarkers would be sensitive and specific for the disease and could possibly further elucidate the complex clinical picture of EPM1. In agreement with previous VBM findings,10 significant and progressive thinning of the cortical motor areas of patients with EPM1 was found in comparison with those of the healthy controls. This type of thinning of the motor cortex has not been reported previously, to our knowledge, and it may be related to the most disabling symptoms in EPM1 (ie, myoclonus). In previous MEG studies, the origin of cortical myoclonus has been localized to the sensorimotor cortex.26,27
As a novel finding, we discovered extensive changes in the cortical sensory areas, the visual areas, and the auditory areas as well. Cortical thinning in the sensory areas extended well beyond the primary and secondary somatosensory, visual, and auditory cortices. For instance, the parietal association cortex was extensively affected. This area is involved in important cortical functions, (eg, visual localization of objects; elaboration of motor programs necessary to reach these objects; spatial perception; spatial memory in collaboration with the visual association cortex; and auditory functions, especially in the supramarginal gyrus in conjunction with the auditory association cortex).28 The areas of regionally reduced cortical thickness were in parallel with the severity of the action myoclonic symptoms of EPM1. Alterations in the visual and auditory cortices thus seem to be congruent with the stimulus-sensitive nature of the symptoms. The myoclonus in EPM1 can be triggered by different kinds of sensory stimuli, such as photic stimuli, sudden or loud noises, or touching (eg, during a neurologic examination). These stimuli can provoke myoclonic jerks that can be severe enough to cause the subject to fall down. They may also generalize into a series of myoclonic seizures or even evoke status myoclonicus (continuous myoclonic jerks involving a semiloss of consciousness).5
The cellular mechanisms causing the cortical thinning in EPM1 remain unclear because the pathophysiology of the disease is still poorly understood. The limited histopathologic data of EPM1 have revealed a loss of cerebellar Purkinje and granular cells and degeneration and loss of neurons in the cerebral cortex, striatum, thalamus, and brain stem nuclei.29–32
Transcranial magnetic stimulation33,34 and MEG35 studies have pointed to impaired sensorimotor cortical inhibition in EPM1. An absence of activation of the SII has been reported in patients who exhibited more severe motor symptoms,36 and the authors speculated that deficient activation of the SII could account for the disturbed sensorimotor integration, contributing to impaired movement coordination. The same phenomenon may be reflected in our finding that the “Myoclonus with Action” score correlated negatively with CTH in the sensory areas, including the SII area. Patients with more severe myoclonus had thinner sensorimotor cortical areas than patients with milder symptoms.
On the basis of the results of the present study, one cannot be absolutely certain that the process of cortical thinning in EPM1 is progressive, though the duration of the disease correlated negatively with CTH in univariate analysis. Because all the clinical parameters we have evaluated seem to correlate with each other as well as with CTH, it is difficult to totally separate their effects. Age correlated negatively with CTH in both patients and controls, but in patients, the findings were more limited. Thus the progression of cortical thinning cannot be explained by age alone. As can be seen in the scatterplots in Fig 2, the affected cortical areas seemed to be thinner in patients with EPM1 than in controls, even in young individuals. The onset of EPM1 is in early childhood or late adolescence, the peak being around 12–13 years37; before the onset of symptoms, the disease process has probably already had an effect on the brain. This might explain the more limited age-related thinning of the cortex in patients with EPM1.
In the control group, the effects of aging were consistent with previous reports of age affecting the prefrontal and parietal cortices negatively, as well as the GM around the central sulcus, and the relative sparing of the temporal and parahippocampal cortices.38,39 Other investigations have also found that the occipital lobes, particularly the visual cortex, are negatively affected by age.38,40 In a study of normal aging with 883 participants, the results were fairly similar to those in our study.41 In the present study, the pericalcarine cortex was relatively spared in healthy controls, but most interesting, it was more extensively affected in patients with EPM1. The mean age of the control group in our study was 33 years, which is substantially younger than that in previous reports, and this might explain some of the differences in the results.
There was some variation (1–1.2 mm) in the section thickness of the MR images, which is a limitation in the present study. However, from 123 scans, in only 16 did the section thickness differ from 1 mm, and section thickness was also included as a nuisance variable in the statistical analyses. Therefore, it should not affect the results significantly. CTH analysis concentrates on the cerebral cortex and cannot be applied to deep GM, the cerebellum, or WM, which can be considered as another limitation of this study. In the future, it may be possible to gather more information on the subcortical pathways by using diffusion tensor imaging.
Conclusions
CTH analysis revealed extensive changes in the sensorimotor, visual, and auditory cortical areas of patients with EPM1. Regional thinning of those areas seemed to increase with the duration of the disease and correlated significantly with the degree of the complex clinical motor symptoms. Alterations in the visual and auditory cortices seem to be congruent with the stimulus-sensitive nature of the symptoms. With these results, we hope to advance our understanding of the functional foundation underpinning EPM1. Future MR imaging and neurophysiologic studies will be needed to investigate possible abnormalities in subcortical structures such as the WM tracts to gain more insight into the pathophysiology and causative mechanisms of EPM1.
Acknowledgments
We wish to thank all the participants for their cooperation in the present study.
ABBREVIATIONS:
- CSTB
cystatin B gene
- CTH
cortical thickness
- EPM1
Unverricht-Lundborg disease
- FDR
false discovery rate
- GM
gray matter
- MEG
magnetoencephalography
- PME
progressive myoclonus epilepsies
- SII
secondary sensory cortex
- UMRS
Unified Myoclonus Rating Scale
- VBM
voxel-based morphometry
Footnotes
Disclosures: Hilkka Soininen—UNRELATED: Board Membership: AC Immune, Comments: member of data monitoring committee, Consultancy: Orion, Grants/Grants Pending: Academy of Finland,* European Union.* Reetta Kälviäinen—UNRELATED: Grants/Grants Pending: EuroEPINOMICS: European Science Foundation, Payment for Lectures (including service on Speakers Bureaus): Eisai, Pfizer, GlaxoSmithKline, UCB Pharma SA, Orion, Sanofi Aventis. * Money paid to institution.
This work was supported by the Academy of Finland/the NEURO Research Programme, the Vaajasalo Foundation, UCB Pharma SA, the Finnish Cultural Society, EVO grant 5772751 from the Kuopio University Hospital and Strategic Funding for UEFBRAIN consortium from University of Eastern Finland.
References
- 1. Shahwan A, Farrell M, Delanty N. Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol 2005;4:239–48 [DOI] [PubMed] [Google Scholar]
- 2. Pennacchio LA, Lehesjoki AE, Stone NE, et al. Mutations in the gene encoding cystatin B in progressive myoclonus epilepsy (EPM1). Science 1996;271:1731–34 [DOI] [PubMed] [Google Scholar]
- 3. Norio R, Koskiniemi M. Progressive myoclonus epilepsy: genetic and nosological aspects with special reference to 107 Finnish patients. Clin Genet 1979;15:382–98 [DOI] [PubMed] [Google Scholar]
- 4. Genton P, Malafosse A, Moulard B, et al. Progressive myoclonus epilepsies. In: Roger J, Bureau M, Dravet C, et al. eds. Epileptic Syndromes in Infancy, Childhood and Adolescence. 4th ed. Montrouge, France; John Libbey Eurotext Ltd; 2005:441–65 [Google Scholar]
- 5. Kälviäinen R, Khyuppenen J, Koskenkorva P, et al. Clinical picture of EPM1-Unverricht-Lundborg disease. Epilepsia 2008;49:549–56 [DOI] [PubMed] [Google Scholar]
- 6. Mascalchi M, Michelucci R, Cosottini M, et al. Brainstem involvement in Unverricht-Lundborg disease (EPM1): an MRI and 1H MRS study. Neurology 2002;58:1686–89 [DOI] [PubMed] [Google Scholar]
- 7. Parmeggiani A, Lehesjoki AE, Carelli V, et al. Familial Unverricht-Lundborg disease: a clinical, neurophysiologic, and genetic study. Epilepsia 1997;38:637–41 [DOI] [PubMed] [Google Scholar]
- 8. Santoshkumar B, Turnbull J, Minassian BA. Unverricht-Lundborg progressive myoclonus epilepsy in Oman. Pediatr Neurol 2008;38:252–55 [DOI] [PubMed] [Google Scholar]
- 9. Chew NK, Mir P, Edwards MJ, et al. The natural history of Unverricht-Lundborg disease: a report of eight genetically proven cases. Mov Disord 2008;23:107–13 [DOI] [PubMed] [Google Scholar]
- 10. Koskenkorva P, Khyuppenen J, Niskanen E, et al. Motor cortex and thalamic atrophy in Unverricht-Lundborg disease: voxel-based morphometric study. Neurology 2009;73:606–11 [DOI] [PubMed] [Google Scholar]
- 11. Lerch JP, Pruessner JC, Zijdenbos A, et al. Focal decline of cortical thickness in Alzheimer's disease identified by computational neuroanatomy. Cereb Cortex 2005;15:995–1001 [DOI] [PubMed] [Google Scholar]
- 12. Lerch JP, Evans AC. Cortical thickness analysis examined through power analysis and a population simulation. Neuroimage 2005;24:163–73 [DOI] [PubMed] [Google Scholar]
- 13. Julkunen V, Niskanen E, Muehlboeck S, et al. Cortical thickness analysis to detect progressive mild cognitive impairment: a reference to Alzheimer's disease. Dement Geriatr Cogn Disord 2009;28:404–12 [DOI] [PubMed] [Google Scholar]
- 14. Hyde KL, Samson F, Evans AC, et al. Neuroanatomical differences in brain areas implicated in perceptual and other core features of autism revealed by cortical thickness analysis and voxel-based morphometry. Hum Brain Mapp 2010;31:556–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bernhardt BC, Worsley KJ, Besson P, et al. Mapping limbic network organization in temporal lobe epilepsy using morphometric correlations: insights on the relation between mesiotemporal connectivity and cortical atrophy. Neuroimage 2008;42:515–24 [DOI] [PubMed] [Google Scholar]
- 16. Frucht SJ, Leurgans SE, Hallett M, et al. The Unified Myoclonus Rating Scale. Adv Neurol 2002;89:361–76 [PubMed] [Google Scholar]
- 17. Mazziotta J, Toga A, Evans A, et al. A probabilistic atlas and reference system for the human brain: International Consortium for Brain Mapping (ICBM). Philos Trans R Soc Lond B Biol Sci 2001;356:1293–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sled JG, Zijdenbos AP, Evans AC. A nonparametric method for automatic correction of intensity nonuniformity in MRI data. IEEE Trans Med Imaging 1998;17:87–97 [DOI] [PubMed] [Google Scholar]
- 19. Zijdenbos AP, Forghani R, Evans AC. Automatic “pipeline” analysis of 3-D MRI data for clinical trials: application to multiple sclerosis. IEEE Trans Med Imaging 2002;21:1280–91 [DOI] [PubMed] [Google Scholar]
- 20. Tohka J, Zijdenbos A, Evans A. Fast and robust parameter estimation for statistical partial volume models in brain MRI. Neuroimage 2004;23:84–97 [DOI] [PubMed] [Google Scholar]
- 21. Kim JS, Singh V, Lee JK, et al. Automated 3-D extraction and evaluation of the inner and outer cortical surfaces using a Laplacian map and partial volume effect classification. Neuroimage 2005;27:210–21 [DOI] [PubMed] [Google Scholar]
- 22. MacDonald D, Kabani N, Avis D, et al. Automated 3-D extraction of inner and outer surfaces of cerebral cortex from MRI. Neuroimage 2000;12:340–56 [DOI] [PubMed] [Google Scholar]
- 23. Chung M, Taylor J. Diffusion smoothing on brain surface via finite element method. Proc IEEE Int Symp Biomed Imaging 2004;1:432–35 [Google Scholar]
- 24. Duvernoy HM. The Human Brain: Surface, Three-Dimensional Sectional Anatomy with MRI, and Blood Supply. New York: Springer-Verlag Wien; 1999 [Google Scholar]
- 25. Genovese CR, Lazar NA, Nichols T. Thresholding of statistical maps in functional neuroimaging using the false discovery rate. Neuroimage 2002;15:870–78 [DOI] [PubMed] [Google Scholar]
- 26. Mima T, Nagamine T, Ikeda A, et al. Pathogenesis of cortical myoclonus studied by magnetoencephalography. Ann Neurol 1998;43:598–607 [DOI] [PubMed] [Google Scholar]
- 27. Mima T, Nagamine T, Nishitani N, et al. Cortical myoclonus: sensorimotor hyperexcitability. Neurology 1998;50:933–42 [DOI] [PubMed] [Google Scholar]
- 28. Mesulam MM. From sensation to cognition. Brain 1998;121(pt 6):1013–52 [DOI] [PubMed] [Google Scholar]
- 29. Koskiniemi M, Donner M, Majuri H, et al. Progressive myoclonus epilepsy: a clinical and histopathological study. Acta Neurol Scand 1974;50:307–32 [PubMed] [Google Scholar]
- 30. Haltia M, Kristensson K, Sourander P. Neuropathological studies in three Scandinavian cases of progressive myoclonus epilepsy. Acta Neurol Scand 1969;45:63–77 [DOI] [PubMed] [Google Scholar]
- 31. Eldridge R, Iivanainen M, Stern R, et al. “Baltic” myoclonus epilepsy: hereditary disorder of childhood made worse by phenytoin. Lancet 1983;2:838–42 [DOI] [PubMed] [Google Scholar]
- 32. Cohen NR, Hammans SR, Macpherson J, et al. New neuropathological findings in Unverricht-Lundborg disease: neuronal intranuclear and cytoplasmic inclusions. Acta Neuropathol 2011;121:421–27 [DOI] [PubMed] [Google Scholar]
- 33. Danner N, Julkunen P, Khyuppenen J, et al. Altered cortical inhibition in Unverricht-Lundborg type progressive myoclonus epilepsy (EPM1). Epilepsy Res 2009;85:81–88 [DOI] [PubMed] [Google Scholar]
- 34. Canafoglia L, Ciano C, Panzica F, et al. Sensorimotor cortex excitability in Unverricht-Lundborg disease and Lafora body disease. Neurology 2004;63:2309–15 [DOI] [PubMed] [Google Scholar]
- 35. Silen T, Forss N, Jensen O, et al. Abnormal reactivity of the approximately 20-Hz motor cortex rhythm in Unverricht-Lundborg type progressive myoclonus epilepsy. Neuroimage 2000;12:707–12 [DOI] [PubMed] [Google Scholar]
- 36. Forss N, Silen T, Karjalainen T. Lack of activation of human secondary somatosensory cortex in Unverricht-Lundborg type of progressive myoclonus epilepsy. Ann Neurol 2001;49:90–97 [PubMed] [Google Scholar]
- 37. Genton P. Unverricht-Lundborg disease (EPM1). Epilepsia 2010;51(suppl 1):37–39 [DOI] [PubMed] [Google Scholar]
- 38. Salat DH, Buckner RL, Snyder AZ, et al. Thinning of the cerebral cortex in aging. Cereb Cortex 2004;14:721–30 [DOI] [PubMed] [Google Scholar]
- 39. Thambisetty M, Wan J, Carass A, et al. Longitudinal changes in cortical thickness associated with normal aging. Neuroimage 2010;52:1215–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gaetz W, Roberts TP, Singh KD, et al. Functional and structural correlates of the aging brain: relating visual cortex (V1) gamma band responses to age-related structural change. Hum Brain Mapp 2011. July 18. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fjell AM, Westlye LT, Amlien I, et al. High consistency of regional cortical thinning in aging across multiple samples. Cereb Cortex 2009;19:2001–12 [DOI] [PMC free article] [PubMed] [Google Scholar]