

Transition-state molecular switches enable hyper responsive detection of SARS-CoV-2 in native clinical samples.
Abstract
Despite the importance of nucleic acid testing in managing the COVID-19 pandemic, current detection approaches remain limited due to their high complexity and extensive processing. Here, we describe a molecular nanotechnology that enables direct and sensitive detection of viral RNA targets in native clinical samples. The technology, termed catalytic amplification by transition-state molecular switch (CATCH), leverages DNA-enzyme hybrid complexes to form a molecular switch. By ratiometric tuning of its constituents, the multicomponent molecular switch is prepared in a hyperresponsive state—the transition state—that can be readily activated upon the binding of sparse RNA targets to turn on substantial enzymatic activity. CATCH thus achieves superior performance (~8 RNA copies/μl), direct fluorescence detection that bypasses all steps of PCR (<1 hour at room temperature), and versatile implementation (high-throughput 96-well format and portable microfluidic assay). When applied for clinical COVID-19 diagnostics, CATCH demonstrated direct and accurate detection in minimally processed patient swab samples.
INTRODUCTION
The rapid global spread of coronavirus disease 2019 (COVID-19) has stretched the limits of healthcare resources (1). Person-to-person transmissions from infected individuals with no or mild symptoms have been widely reported (2, 3). Aggressive testing for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causal pathogen of COVID-19 (4), is important in controlling the disease spread and devising safety measures. To date, quantitative reverse transcription polymerase chain reaction (RT-qPCR) remains the primary assay for detecting SARS-CoV-2 (5). Albeit its sensitive performance, the technology requires extensive sample preparation (e.g., RNA extraction), exquisite primer design, specialized instrument, and trained personnel (6). These limitations not only result in a long assay turnaround time but also hinder its large-scale implementation and adaptation in a rapidly evolving pandemic. These shortcomings are particularly apparent when challenged under the severe pressure of COVID-19; a global shortage of reagents and the emergence of new mutations and false negatives pose critical challenges for RT-qPCR–based detection (7–9). Accurate, rapid, and easy-to-use molecular diagnostic tests for SARS-CoV-2 are crucially needed across the globe (10).
Molecular nanotechnology offers unparalleled precision and programmability to construct a variety of self-assembled functional nanostructures (11–14). These nanostructures can be designed as versatile, multifunction machines, which can not only recognize external stimuli but also respond and actuate various activities (15, 16). We have previously developed a molecular nanotechnology platform for rapid detection of nucleic acids (17). Instead of relying on the traditional approach of target amplification (as in conventional RT-qPCR), the technology detects through target hybridization. It leverages enzyme-DNA hybrid nanocomplexes as molecular switches; upon the direct binding of specific nucleic acids (even RNA targets), the nanocomplexes dissociate to activate strong enzymatic activity. The technology is highly programmable; new assays can be readily developed by modifying the highly configurable nanocomplexes, without needing complex design of PCR primers and dedicated fluorescent probes (e.g., TaqMan probes). Because of this unique sensing mechanism and high programmability, we thus envision that the technology could enable direct detection of SARS-CoV-2, bypassing many steps and challenges of PCR detection (e.g., reverse transcription and thermal cycling). Nevertheless, given that a substantial proportion of COVID-19 patients are reported to have a very low viral load (18), our previously developed assay, with a limit of detection (LOD) of ~10 amol, would have a limited sensitivity to diagnose a broad spectrum of COVID-19 patients.
To bridge this gap in detection sensitivity, motivated by the multicomponent nature of individual nanocomplexes, we reason that they can be tuned to establish highly responsive molecular switches. Specifically, the nanocomplex switches are self-assembled from multiple molecular constituents—Taq polymerase and distinct DNA strands—which exist in a dynamic equilibrium and exert different effects on overall switch characteristics. Through ratiometric tuning of these molecular constituents, we found that the most responsive state is a metastable state, where even trace amounts of target nucleic acids can readily activate the molecular switches to induce strong enzymatic activity. Leveraging molecular switches in this hyperresponsive state, which we call the transition state, we develop a highly sensitive and direct nucleic acid detection assay for SARS-CoV-2. The technology, termed catalytic amplification by transition-state molecular switch (CATCH), benefits from dual catalytic amplification: Its transition-state molecular switches are readily activated upon the direct binding of even sparse amounts of viral RNA targets to liberate substantial enzymatic activity; this switch activation further recruits additional enzymatic cascades to transduce strong signal output.
Harnessing its hyperresponsiveness, CATCH achieves superior performance. It enables sensitive and specific detection of RNA targets, against a complex biological background, and reports a LOD of ~8 copies of target per microliter, which is >10,000-fold more sensitive than our previous platform. The detection is also direct and rapid; the entire assay can be completed in <1 hour at room temperature and can be applied to a variety of sample types (e.g., purified RNA and complex clinical samples), bypassing all steps of conventional RT-qPCR (i.e., RNA extraction, reverse transcription, and thermal cycling amplification). CATCH enables versatile assay implementation. To support different diagnostic needs, the assay can be implemented in a 96-well format for high-throughput analysis and as a miniaturized microfluidic cartridge for portable smartphone-based measurement. When applied for clinical detection of SARS-CoV-2, CATCH demonstrated accurate and sensitive detection in both extracted RNA samples and inactivated patient swabs.
RESULTS
CATCH platform
The working principle of the CATCH assay is illustrated in Fig. 1A. Clinical samples containing SARS-CoV-2 viral RNA targets are mixed with a DNA-enzyme molecular switch for direct and sensitive detection. The hybrid switch consists of an inhibitory DNA complex—comprising an inhibitor strand and an enhancer strand—that binds and inactivates Taq DNA polymerase (19). We design the inhibitory DNA complex to be complementary to various SARS-CoV-2 RNA targets (fig. S1A); only in the presence of specific target RNA, the enhancer hybridizes with the target and the inhibitor is displaced, thereby releasing and activating the polymerase. The inhibitor strand is a stem-loop structure, which consists of a conserved region (loop) and a variable region (stem). We found that while the inhibitor strand alone can weakly decrease the polymerase activity, simultaneous addition of the enhancer strand strongly inhibits the polymerase activity (fig. S1B). This is likely due to the improved stabilization of the stem-loop conformation as a result of the hybridization of the enhancer strand to the stem of the inhibitor strand, resulting in an enhancement of its inhibitory effect (19, 20). Motivated by the strong toggling effect by the enhancer strand and the multicomponent dynamic equilibrium (i.e., intraswitch and interswitch), we reason that by adjusting the ratio of molecular constituents of individual switches, we can tune the molecular switches to achieve different states of target responsiveness: closed, transition, and open (Fig. 1A, right). In the closed state, most of the molecular switches are fully inactivated, through polymerase binding with excess inhibitory complexes; turning on the polymerase activity thus requires a large amount of RNA targets. In the open state, most of the molecular switches are fully activated; turning on additional polymerase activity amidst a high initial background results in a low net signal. In the transition state, different forms of molecular switches (i.e., inactivated, intermediate, and activated) (fig. S1C) exist in a delicate dynamic equilibrium; a small amount of target molecules can readily shift the equilibrium to favor the formation of more activated switches, thereby triggering a large increase in overall polymerase activity.
Fig. 1. Catalytic amplification by transition-state molecular switch.

(A) Schematic representation of the CATCH assay. The CATCH assay leverages the specific binding of nucleic acid targets (SARS-CoV-2 viral RNA) to activate molecular switches. Each molecular switch consists of a Taq DNA polymerase and an inhibitory DNA complex, comprising an inhibitor strand and an enhancer strand, that binds and inactivates the polymerase. As the viral RNA target hybridizes with the enhancer strand, it destabilizes the inhibitory complex and releases the active polymerase (left). By adjusting the ratio of molecular constituents in individual switches, we prepare molecular switches in different states of target responsiveness: closed, transition, and open (right). In the closed state, switches are fully inactivated, due to excess inhibitory complexes, and cannot be readily activated by sparse RNA targets. In the open state, switches are fully activated and largely unresponsive to targets due to a high initial background. In the transition state, different forms of switches exist in a delicate equilibrium—that a small amount of RNA targets can readily shift this equilibrium to favor the formation of more activated switches. The transition-state switches thus demonstrate maximal responsiveness (i.e., the largest change in polymerase activity within the shortest time span). (B) Signal generation. To enhance the detection signal, the CATCH assay recruits additional enzymatic cascades to transduce and amplify the target-induced polymerase activity as a fluorescence readout (see Fig. 3A for more details). As compared to that using the closed- or open-state molecular switches, the CATCH assay (transition state) generates strong signals from clinical samples with a low viral load. (C) Different assay formats. The CATCH assay can be performed in a 96-well format for high-throughput applications (top) or a miniaturized microfluidic device for portable, smartphone-based detection (bottom).
By ratiometric tuning of various switch components, we found the transition-state switches to be hyperresponsive to RNA targets and leveraged this state to develop the CATCH assay for rapid, sensitive detection of SARS-CoV-2. To further enhance the detection signal, we measured the changes in polymerase activity through additional enzymatic amplification (Fig. 1B). Specifically, we used the target-induced polymerase activity to incorporate biotin-modified deoxynucleotide triphosphates (dNTPs) into immobilized hairpin oligonucleotides and this incorporation to recruit streptavidin-conjugated enzymes [horseradish peroxidase (HRP)] for the development of chemifluorescence signal. As compared to assays using the closed- or open-state molecular switches, the CATCH assay (transition state) generates strong signals from mildly positive patients with a low viral load. The CATCH assay could be versatilely implemented to accommodate different diagnostic needs (Fig. 1C). For example, the signaling oligonucleotides can be immobilized onto a 96-well plate for high-throughput applications; this assay configuration closely resembles conventional enzyme-linked immunosorbent assay (ELISA) in terms of assay workflow and readout, enabling its easy adaptation in clinical laboratories with standard instrumentation. The CATCH assay can also be implemented on a miniaturized microfluidic device (figs. S2 and S3). Furthermore, chemifluorescence signals can be readily detected through a portable, smartphone-based fluorescence detector with comparable performance (fig. S4).
Transition-state molecular switches
To develop the CATCH assay for detecting SARS-CoV-2, we designed molecular switches as specific probes against the viral RNA. We chose regions of the spike (S) gene (21) and the nucleocapsid (N) gene (22) of the virus as specific targets and constructed distinct molecular switches based on these sequences (Fig. 2A and table S1). To identify the transition state of the molecular switches, we first evaluated the effect of the inhibitory complex on Taq polymerase. Specifically, to a fixed concentration of polymerase, we titrated an increasing concentration of the inhibitory complex (i.e., varying the complex concentration but keeping the ratio of enhancer:inhibitor to 1:1). We observed that polymerase activity was markedly inhibited when incubated with >20 nM of inhibitory complex (Fig. 2B). Plotting the first derivative of the inhibition curve, we categorized the molecular switches in three groups, namely, open, responsive, and closed (Fig. 2B, inset, and fig. S5A). Open-state molecular switches were made with a low concentration of inhibitory complex (<20 nM). In the responsive range, switches were prepared with a moderate concentration of inhibitory complex and remained responsive to changes in the inhibitory complex concentration (i.e., at the vertex, where switches were the most responsive, switches were made with 36 nM of inhibitory complex). Closed-state molecular switches were made with a high concentration of inhibitory complex (>60 nM). When we perturbed the system through a reduction in the amount of enhancer strand (i.e., reducing the ratio of enhancer:inhibitor), molecular switches in the responsive state demonstrated large changes in their polymerase activity (fig. S5B).
Fig. 2. Hyperresponsive molecular switches for SARS-CoV-2 detection.
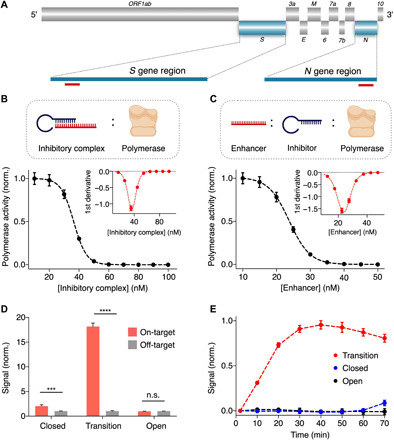
(A) Map of the SARS-CoV-2 genome. Molecular switches are designed to recognize the spike (S) gene and the nucleocapsid (N) gene. Enhancer strands of respective molecular switches are represented by red rectangles. Not drawn to scale. (B) Switch responsiveness to inhibitory complex. To a fixed concentration of polymerase, we added a varying concentration of inhibitory complex (with the enhancer:inhibitor ratio kept at 1:1) and measured the resultant polymerase activity. Inset shows the first derivative plot for visualization of switch responsiveness to inhibitory complex. Switch composition at the vertex was deemed the most responsive and selected for further optimization. (C) Determination of the transition state. We further varied the concentration of the enhancer strand with fixed inhibitor strand concentration and measured the resultant polymerase activity. Inset shows the first derivative plot for visualization of switch responsiveness to enhancer strand. We defined the transition state as the vertex composition. (D) Performance of the molecular switches. The closed-, open-, and transition-state molecular switches were incubated with on-target and off-target sequences. The transition-state molecular switches exhibited significant polymerase activation with target binding while maintaining a low background. (E) Activation kinetics. The transition-state molecular switches achieved fast activation upon incubation with SARS-CoV-2 S gene target. Different molecular switches in (D) and (E) were prepared at the following representative compositions (inhibitor and enhancer strand, respectively): open state, 1 and 1 nM; transition state, 36 and 24 nM; closed state, 100 and 100 nM. All measurements were performed in triplicate, and the data in (B) to (E) are presented as mean ± SD (****P < 0.0001 and ***P < 0.005; n.s., not significant, Student’s t test).
To establish the transition state, we further tuned the responsive-state molecular switches by titrating the amount of enhancer strand (i.e., through which target hybridizes and activates the switch) while keeping constant the amount of inhibitor strand (Fig. 2C). Through this optimization, we defined the transition state as the vertex on the first derivative inhibition plot (Fig. 2C, inset). This identified transition state demonstrated further improvement in its responsiveness, producing the largest increase in polymerase activity, while the open- and closed-state switches failed to produce any distinguishable signal (fig. S5C). We further evaluated the performance of the transition-state molecular switches. The ratiometric-tuned switches not only demonstrated significant polymerase activity upon incubating with complementary on-target RNA sequences but also maintained a low background activity when treated with off-target sequences (Fig. 2D). For both the S-gene (Fig. 2E) and N-gene molecular switches (fig. S5D), the transition-state switches achieved much faster activation kinetics. As compared to switches prepared in the other states, the transition-state switches enabled rapid polymerase activation. Different target concentrations could be distinguished within 30 min of incubation at room temperature (fig. S5E).
Signal generation and amplification
Next, we devised a signaling mechanism to enzymatically amplify and measure the switch-induced polymerase activity. Specifically, we designed two signaling oligonucleotide structures to leverage different types of polymerase activity (i.e., elongation versus exonuclease activity) and recruit additional enzymatic cascades (i.e., HRP) for signal amplification (Fig. 3A). We immobilized the oligonucleotide structures on a 96-well ELISA plate through protein scaffold (fig. S6). In the elongation-based strategy, the active polymerase incorporates biotin-modified dNTPs to the growing chains of the self-primed hairpin oligonucleotides (3′-end). Fluorescence signal is then generated after the addition of streptavidin-conjugated HRP and chemifluorescence substrate. In the exonuclease-based strategy, we constructed a dumbbell-shaped signaling oligonucleotide with biotin modifications at its 5′-end. Active polymerase extends the 3′-end of the oligonucleotide and, upon reaching the self-hybridized 5′-end, cleaves the biotin-modified nucleotides; when reacted with streptavidin-conjugated HRP, this removal of biotin groups reduces the amount of fluorescence signal. We evaluated the two strategies by treating both oligonucleotide structures with an equal amount of active polymerase and measured the resultant changes in fluorescence signals. The elongation-based strategy showed a significantly higher signal as compared with the exonuclease-based strategy (Fig. 3B, left). We thus incorporated the elongation approach for CATCH signaling. In comparison to measurements based on sole polymerase activity, the additional HRP recruitment significantly enhanced the signal output (Fig. 3B, right) and expanded the detection dynamic range (fig. S7A).
Fig. 3. Signal enhancement through multienzyme cascades.
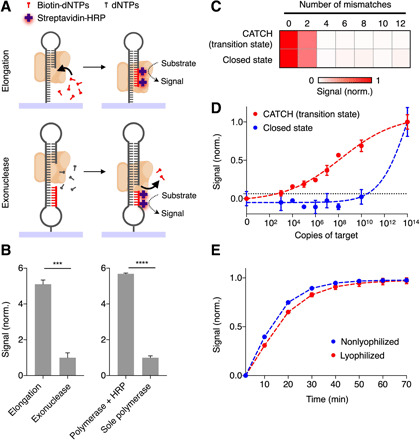
(A) Schematic of two signaling oligonucleotide structures for the measurement of elongation and exonuclease activity of polymerase, respectively. In the elongation-based strategy, active polymerase incorporates biotin-modified dNTPs to the growing 3′-ends of self-primed hairpin oligonucleotides. After the addition of streptavidin-conjugated HRP and substrate, fluorescence signal can be read out. In the exonuclease-based strategy, active polymerase cleaves biotin-modified nucleotides upon reaching the self-hybridized 5′-ends, thereby reducing the amount of HRP incorporation and the resultant fluorescence signal. (B) Elongation-based signal enhancement. When treated with an equal amount of polymerase, the elongation-based strategy showed a higher signal as compared to that by the exonuclease-based strategy (left). The recruitment of an additional enzymatic cascade (HRP) enhanced the signal significantly as compared to measurements based with sole polymerase activity (right). (C) Specificity of the CATCH assay. The CATCH assay, which uses transition-state molecular switches, showed uncompromised specificity against target mismatches, as compared to that by the closed-state molecular switches. (D) Sensitivity of the CATCH assay. The detection limits (dotted lines) were defined as 3× SD of the no-target controls and determined by titrating known quantities of target in a total volume of 50 μl and measuring their corresponding fluorescence signal. (E) Lyophilization of the CATCH assay. Assay reagents [molecular switches and biotin-modified dNTPs (biotin-dNTPs)] were lyophilized to facilitate portable applications. The lyophilization preserved the switch performance. All measurements were performed in triplicate, and the data are presented as mean ± SD in (B), (D), and (E) and as mean in (C) (****P < 0.0001 and ***P < 0.005, Student’s t test).
Motivated by the signaling performance, we developed the CATCH assay workflow to use transition-state molecular switches for responsive target recognition and elongation-based multienzyme cascade for signal enhancement. Specifically, we mixed RNA targets with transition-state switches and directly incubated the reaction with immobilized oligonucleotides (30 min at room temperature) for signal transduction and enhancement. As compared to a similar assay using closed-state molecular switches (i.e., fully inactivated molecular switches and HRP-based signal enhancement), the CATCH assay demonstrated comparable specificity against target mismatches, even when the mismatches were introduced against the most sensitive segment of the switches (Fig. 3C and table S1). The transition-state switches showed superior performance. In a titration experiment, where target samples were serially diluted and incubated with different-state molecular switches, the CATCH assay achieved >107-fold improvement in its LOD (LOD of ~8 copies of target per microliter) as compared to the closed-state molecular switches (Fig. 3D and fig. S7B). To facilitate portable clinical application, we lyophilized the assay reagents (i.e., molecular switches and biotin-modified dNTPs) within the microfluidic device. The lyophilization not only preserved the assay performance but also conferred excellent long-term stability (Fig. 3E and fig. S7, C and D).
Assessment of CATCH assay in cellular lysates
To address the need for extensive sample preparation in conventional qPCR (i.e., RNA extraction), we next determined whether the CATCH assay could be developed to bypass this crucial and limiting step. Using specific molecular switches designed for SARS-CoV-2 RNA targets (i.e., S-gene and N-gene switches), which demonstrated specific detection and minimal activity against sequences of other closely related human coronaviruses [SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV)] as well as other viruses causing diseases with similar symptoms (dengue virus and influenza A subtype H1N1 virus) (Fig. 4A), we evaluated the performance of the CATCH assay in different cellular lysates.
Fig. 4. Specificity of CATCH in detecting SARS-CoV-2 in cellular lysates.

Specificity of the CATCH assay in detecting S and N gene targets of SARS-CoV-2. Assay specificity was evaluated against sequences of other closely related human coronaviruses (SARS-CoV and MERS-CoV) and other viruses causing diseases with similar symptoms (dengue virus and influenza A subtype H1N1 virus). Synthetic targets were spiked in (A) pure buffer or (B and C) cell lysates. Lysates were prepared through (B) thermal incubation at different temperature and duration or (C) chemical lysis using different combinations of detergents. The molecular switches maintained specific detection for SARS-CoV-2 targets and showed minimal cross-reactivity with off-target viral sequences, across all lysis conditions. All measurements were performed in triplicate, and the data are presented as mean ± SD.
Specifically, we explored two modes of direct lysis, namely, thermal (Fig. 4B) and chemical lysis (Fig. 4C), and used the lysates for direct CATCH detection. For thermal lysis, we investigated the effects of different temperature and heating duration on the lysis efficiency; three different temperature conditions—56°C for 30 min, 70°C for 5 min, and 90°C for 5 min—were selected on the basis of published studies (23, 24). For chemical lysis, to optimize the treatment conditions, we first evaluated polymerase activity in the presence of single detergents (fig. S8). Polymerase activity was found to be highly inhibited in the presence of SDS and gradually inhibited with increasing concentration of saponin. Other tested detergents (e.g., Triton X-100) showed negligible effects on the polymerase activity. Using this information, we next optimized detergent combinations for rapid cell lysis (fig. S9A) while maintaining good polymerase activity (fig. S9, B and C). We determined that an optimal ratio of 1:10 between SDS and Triton X-100 could both preserve polymerase activity and lyse cells within 5 min.
With these selected thermal and chemical lysis protocols, we first validated the ability of these methods to release and preserve endogenous RNA targets [i.e., glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and β-actin] in human lung epithelial cells. We demonstrated that for both endogenous targets tested, when assayed via RT-qPCR, all lysates generated similar cycle threshold (Ct) values as compared with the gold-standard extracted RNA samples (fig. S10). We further evaluated the compatibility of the lysis protocols with the developed CATCH assay. Using synthetic targets spiked into thermal lysates (Fig. 4B) and chemical lysates (Fig. 4C), we incubated the lysate mixtures with molecular switches for CATCH detection. Across all lysis conditions, the CATCH assay not only maintained strong and specific detection for SARS-CoV-2 but also showed minimal cross-reactivity with off-target viruses.
SARS-CoV-2 detection in clinical swab samples
To test the clinical utility of the CATCH platform for SARS-CoV-2 detection, we conducted a feasibility study with patient samples. We aimed at addressing the following questions: (i) if the CATCH assay can be applied directly to detect extracted RNA of nasopharyngeal swab samples (i.e., bypassing RT-qPCR), (ii) if the CATCH platform can be used for direct detection of swab lysates (i.e., bypassing RNA extraction), and (iii) the accuracy of CATCH in COVID-19 diagnosis.
We first tested swab-extracted RNA samples (n = 49) using the CATCH assay. RNA samples were extracted through commercial columns and incubated directly with the CATCH mixture for 30 min at room temperature. Of the 49 extracted RNA samples, 24 were determined by gold-standard RT-qPCR assay as positive for COVID-19 infection and 25 as negative. The positive and negative diagnostic prediction of CATCH relative to the clinical RT-qPCR outcome were 100 and 92%, respectively (Fig. 5A). We further tested our assay in heat-treated swab samples (n = 24), thereby omitting the RNA extraction steps. Of the 24 patient swab samples obtained, 9 were positive for COVID-19 infection, as determined by RT-qPCR assay, and 15 were negative for COVID-19 infection. The CATCH assay correctly identified 9 of 9 (100%) positive samples and 14 of 15 (93.34%) negative samples (Fig. 5B).
Fig. 5. Clinical validation of CATCH for COVID-19 diagnosis.

The CATCH assay was performed on (A) extracted RNA of nasopharyngeal swab samples (positive, n = 24; negative, n = 25) and (B) heat-inactivated swab lysates (positive, n = 9; negative, n = 15). (C) Correlation of CATCH assay with clinical RT-qPCR Ct values. The CATCH assay demonstrated a good agreement with the clinical results (R = 0.8261). (D) Receiver operating characteristic (ROC) curves of the CATCH platform. The CATCH analysis showed a high accuracy for SARS-CoV-2 detection (AUC = 0.9803 for combined samples; AUC = 0.9833 for extracted patient RNA; AUC = 0.9704 for heat-inactivated swab samples). All measurements were performed in triplicate, and the data are presented as mean ± SD in (A) to (C). a.u., arbitrary unit; AUC, area under curve.
To evaluate the clinical performance, across all tested clinical samples, we correlated the CATCH assay with the matched RT-qPCR Ct values (Fig. 5C). The CATCH assay demonstrated a good agreement with the clinical results (R = 0.8261) and could sensitively detect samples with a low viral load (Ct > 35). Compared with the RT-qPCR–based clinical diagnoses, the CATCH platform demonstrated a high accuracy for SARS-CoV-2 detection [Fig. 5D; area under the curve (AUC) = 0.9803 for combined samples; AUC = 0.9833 for extracted patient RNA; AUC = 0.9704 for heat-inactivated swab samples]. CATCH’s ability to diagnose COVID-19 without the need for RNA extraction and RT-qPCR could thus facilitate faster, simpler, and cheaper diagnostic tests.
DISCUSSION
The COVID-19 pandemic has caused an unprecedented public health crisis around the world. Among current testing protocols, nucleic acid detection, particularly RT-qPCR, remains the gold standard. Nevertheless, the approach is almost exclusively performed in large, centralized clinical laboratories due to its extensive processing, high complexity, and need for trained personnel; reliance on RT-qPCR has thus placed much pressure on public health systems (1, 10), leading to a significant global supply shortage and delayed diagnoses. For prompt detection and efficient management, rapid and accurate diagnostic assays are urgently needed (25, 26). Building on our previous molecular nanotechnology platform (17), we developed the CATCH assay as an alternative nucleic acid detection method to complement the current gold standard. Specifically, the CATCH assay demonstrates distinct advantages, through its unique assay mechanism and facile clinical adaptation, to address multiple challenges of COVID-19 diagnostics.
From the assay perspective, CATCH leverages DNA-enzyme hybrid complexes as hyperresponsive molecular switches. By tuning their molecular composition, the multicomponent molecular switches are prepared in a hyperresponsive state—the transition state—that can be readily activated upon the direct hybridization of even sparse RNA targets to turn on substantial enzymatic activity. CATCH thus achieves an enhanced response that that is not only bigger in magnitude but also faster in kinetics. Yet, CATCH retains all key advantages inherent to molecular switching: (i) It is highly specific and activates only when complementary targets bind to the switches, (ii) it can be readily integrated with other enzyme cascades (e.g., HRP) for further signal enhancement, and (iii) it enables programmable design and rapid new assay prototyping. In comparison to our previously developed molecular nanotechnology, CATCH demonstrated significant improvements in analytical performance; it achieved an LOD of ~8 RNA copies/μl (>10,000-fold more sensitive than our previous platform), could be completed in <1 hour at room temperature, and could be applied directly to a variety of sample types (e.g., swab lysates). Its superior performance enables CATCH to accurately detect SARS-CoV-2 even in patient samples with a low viral load.
For clinical adaptation, CATCH detects through target hybridization, instead of conventional target amplification (as in RT-qPCR). This enables the technology to bypass essentially all critical steps of RT-qPCR (i.e., RNA extraction, reverse transcription, and thermal cycling amplification). CATCH supports versatile assay implementation to accommodate the different diagnostic needs of COVID-19. In its 96-well format, the assay configuration closely resembles conventional ELISA in terms of assay workflow and readout, and can be readily adapted for high-throughput analysis, using existing infrastructure of clinical laboratories (e.g., plate reader and trained personnel). In its portable format, CATCH is implemented through a miniaturized microfluidic cartridge, where assay reagents are lyophilized within the device for user-friendly application and smartphone-based detection (27–29). For different clinical applications, the CATCH assay threshold should be adjusted with respect to the proposed application. This threshold setting presents a tradeoff between assay sensitivity versus specificity. For example, considering the potential application of CATCH as a preliminary screening test, we prioritized assay sensitivity when setting the current detection threshold (100% sensitivity, minimal false negatives, and maximal false positives); even at this assay threshold, we determined a low incidence of false positives (<8%), which is within the range reported of existing assays (0 to 16.7%) (30–32). The cause of false results in nucleic acid tests could be assay-associated or PCR-based misclassification, both of which have been reported (31, 33).
The technology has the potential to be expanded further. For COVID-19 diagnostics, in view of the rapidly evolving pandemic, we envision the integration of multiple CATCH switches, designed to recognize different genetic loci of SARS-CoV-2, to not only enhance the detection coverage of the infection but also enable subtype differentiation and mutation identification (9). With its robust performance in minimally processed clinical lysates, CATCH could be readily expanded to investigate other more accessible sample types (e.g., saliva and sputum) (34, 35). To further improve user-friendliness, the microfluidic CATCH platform could be integrated with automated liquid handling systems (e.g., computer-programmed fluidics and pumps for compact liquid handling) (36, 37). These sample expansion and system automation could facilitate new clinical opportunities for repeat testing and self-testing. Last, beyond the current COVID-19 pandemic, CATCH can be further developed to discover and measure new biomarker signatures. The platform could be applied across a spectrum of diseases (e.g., infectious diseases, cancers, and neurodegenerative diseases) to facilitate sensitive detection of nucleic acid targets and composite signatures (38). Further technical improvements, such as multiplexed microfluidic compartmentalization (39–41), could enable microarray-type assay implementation for highly parallel biomarker discovery and large-scale clinical validation.
METHODS
Molecular switch design and preparation
All oligonucleotide sequences can be found in table S1 and were purchased from Integrated DNA Technologies (IDT). Genome sequences of SARS-CoV-2 (NC_045512), SARS-CoV (FJ882957), MERS (NC_019843), dengue virus (NC_001477), and influenza A subtype H1N1 virus [strain A/California/07/2009(H1N1), NC_026431–NC_026438] were obtained from National Center for Biotechnology Information (NCBI) RefSeq. Multiple sequence alignment was performed using the UGENE suite of tools (42). To prepare molecular switches, we mixed inhibitor and enhancer oligonucleotides (table S1; IDT) in a reaction buffer made up of 50 mM NaCl, 1.5 mM MgCl2, and 50 mM tris-HCl (pH 8.5). The mixture was incubated at 95°C for 5 min and slowly cooled at 0.1°C/s until the reaction reached 25°C to form the inhibitory complex. Taq DNA polymerase (Promega) was then added to form the complete molecular switch.
Transition-state characterization
To identify various states of the molecular switch, we varied the ratio of its constituents, first with the inhibitor and enhancer strand at 1:1 ratio and then at varying ratios of these two components. The resultant polymerase activity was measured through 5′ exonuclease degradation of fluorescent signaling probe. Briefly, equimolar amounts of fluorescent probe, template, and primer (IDT) were mixed with dNTPs (Thermo Fisher Scientific) in the reaction buffer. The mixture was incubated at 95°C for 5 min and slowly cooled to 25°C at 0.1°C/s. Molecular switches were then added to the probe mixture and incubated at 25°C, while fluorescence readings were taken. On the basis of the observed changes in polymerase activity, we defined the different states of molecular switches: The open state is where the inhibitory complex is lacking (<20 nM), the closed state is where the inhibitory complex is in excess (>60 nM), and the transition state is the most responsive state (i.e., the vertex of the first derivative of the inhibition curve, where a small change in the switch composition would result in the largest change in polymerase activity). To characterize the responsiveness of the different switch states to nucleic acid targets, we prepared switches at the following representative composition and incubated the switches with target oligonucleotides: open state, 1 nM of inhibitor strand and 1 nM of enhancer strand; closed state, 100 nM of inhibitor strand and 100 nM of enhancer strand; and the transition state, 36 nM of inhibitor strand and 24 nM of enhancer strand. All experiments were also performed with scrambled oligonucleotides to determine background off-target signal.
Immobilization of signaling oligonucleotides
Signaling oligonucleotides were immobilized on an ELISA plate as illustrated in fig. S6. Briefly, bovine serum albumin [BSA; 5% (w/v), Sigma-Aldrich] was adsorbed onto an ELISA plate (Thermo Fisher Scientific) as protein scaffold and activated by incubating with sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (sulfo-SMCC; 0.5 mg/ml; Pierce) for 30 min at room temperature. Plates were then washed with phosphate-buffered saline (Thermo Fisher Scientific) with 0.05% (v/v) Tween 20 (Sigma-Aldrich) (PBST). Separately, thiol-modified signaling oligonucleotides (table S1; IDT) were activated by incubating with tris(2-carboxyethyl)phosphine (TCEP) reducing gel (Pierce) to reduce the disulfide bonds for 1 hour at room temperature. The reaction was then filtered, and the gel was washed several times to recover the activated oligonucleotides. The activated oligonucleotides were then added to the prepared BSA-coated plate and incubated for 2 hours at room temperature. After washing with PBST, the plate was blocked with 2% BSA for 1 hour at room temperature. The plate was then washed with PBST and the reaction buffer before sample application.
Amplification of polymerase activity by HRP
Two forms of signaling oligonucleotides were used and evaluated to amplify and transduce different types of polymerase activity, namely, exonuclease- and elongation-based activity. In both approaches, control wells containing no polymerase were run concurrently to provide the baseline signal. For the exonuclease-based strategy, we immobilized dumbbell DNA signaling structures on the plate and measured the polymerase activity (5′ exonuclease activity) through the catalytic removal of biotin-modified nucleotides from the immobilized dumbbells. Briefly, we mixed sample targets with transition-state molecular switches and directly incubated the reaction with immobilized oligonucleotides, in the presence of dNTPs (Thermo Fisher Scientific), for 30 min at room temperature. Following washing steps with PBST and incubation with streptavidin-conjugated HRP (Thermo Fisher Scientific), we applied QuantaRed chemifluorescence substrate (Thermo Fisher Scientific) and measured the fluorescence intensity (Tecan) to evaluate the removal of biotin-modified nucleotides.
For the elongation-based strategy, polymerase activity was measured through the incorporation of biotin-modified nucleotides to self-priming, hairpin DNA signaling structures immobilized on the plate. Sample and molecular switches were added to the signaling structures and incubated in the presence of biotin-modified dNTP mixture (TriLink BioTechnologies). Following incubation for 30 min at room temperature and washing with PBST, we incubated streptavidin-conjugated HRP (Thermo Fisher Scientific). After washing, we applied QuantaRed chemifluorescence substrate (Thermo Fisher Scientific) and measured the fluorescence intensity (Tecan) to evaluate the addition of biotin-modified nucleotides.
CATCH assay (plate format)
Transition-state molecular switches were prepared as previously described. Sample containing target was mixed with the prepared molecular switches to a final volume of 50 μl. The mixture was added to the self-priming DNA signaling structures, immobilized on the plate, in the presence of biotin-modified dNTP mixture. The reaction mixture was incubated for 30 min at room temperature. Following washing steps with PBST and incubation with streptavidin-conjugated HRP (Thermo Fisher Scientific), we applied QuantaRed chemifluorescence substrate (Thermo Fisher Scientific) and measured the fluorescence intensity (Tecan). For each sample, sample-matched positive (containing polymerase without inhibitory complex) and negative (scrambled molecular switch) controls were run concurrently for data normalization.
Device fabrication
A prototype microfluidic device was fabricated through standard soft lithography as previously described (29). Briefly, 50-μm-thick cast molds were patterned with SU-8 photoresist and silicon wafers using a cleanroom mask aligner (SUSS MicroTec) and developed after ultraviolet (UV) exposure. Polydimethylsiloxane (PDMS; Dow Corning) and cross-linker were mixed at a ratio of 10:1 and casted on the SU-8 mold. The polymer was first cured at 75°C for 30 min. Then, multiple nylon screws and hex nuts (RS Components) were positioned on the PDMS film over their respective channels and embedded in the PDMS, before a final curing step.
Device preparation
To immobilize the signaling oligonucleotides on the device, we treated the device’s glass surface with (3-aminopropyl)triethoxysilane (APTES; 2% v/v, Sigma) in 95% ethanol for 1 hour at room temperature. The chambers were then flushed with ethanol to remove excess APTES and dried. Separately, thiol-modified signaling oligonucleotides were activated as previously described. The activated oligonucleotides were then flowed in and incubated for 2 hours at room temperature. After flushing with PBST to remove excess oligonucleotides, the chambers were blocked with 2% BSA for 1 hour at room temperature. The chambers were then washed with PBST and the reaction buffer. To prepare the device for operation, we lyophilized the assay reagents within the device. The reagent mixture, containing inhibitor strand, enhancer strand, polymerase, and biotin-modified dNTP mixture, was flowed into the device and lyophilized overnight (Labconco).
CATCH assay (microfluidic chip format)
Operation steps of the microfluidic device are illustrated in fig. S3. In a typical assay, 5 μl of sample was introduced to each of the three inlets for measurement of sample, sample-matched positive, and negative controls. Positive pressure was applied to flow the samples to the respective detection chambers. The solution was incubated within the device for 30 min at room temperature. After flushing with PBST, 5 μl of streptavidin-conjugated HRP was introduced and incubated for 5 min at room temperature. The unbound streptavidin conjugates were then removed, and 5 μl of QuantaRed chemifluorescence substrate (Thermo Fisher Scientific) was added. The resultant fluorescence intensity was measured with a smartphone-based optical sensor.
Smartphone-based optical sensor
To enable smartphone analysis of the microfluidic CATCH assay, we developed a sensor that comprised a light-emitting diode (LED) source, an optical filter, and a magnification lens within a three-dimensional (3D)–printed optical cage as previously described (29). The optical cage was fabricated from a UV-curable resin (HTM 140) using a desktop 3D printer (Aureus). The central wavelengths of the LED light source (Chaoziran S&T) and optical filter (Thorlabs) were 500 and 600 nm, respectively. The magnification lens (Thorlabs) was placed before the smartphone camera to improve the image quality. The assembled system measured 45 mm (width) by 45 mm (length) by 50 mm (height) in dimension and was equipped with two sliding slots for quick attachment to smartphones (Apple). Sensor performance was evaluated against a commercial microplate reader (Tecan) for different fluorescent dyes and intensities.
Data normalization
where Inorm is the normalized fluorescence intensity; Itarget is the fluorescence intensity of the sample incubated with molecular switches against the target; Icontrol is the fluorescence intensity of the sample-matched negative control, incubated with scrambled control molecular switches; and Ipol is the fluorescence intensity of the sample-matched positive control, incubated with active polymerase.
Evaluation of CATCH performance
To evaluate the specificity of the transition state compared to that of the closed state, molecular switches were mixed with targets with varying number of mismatches at positions that would most drastically affect the signal produced by the molecular switches (17) (table S1). The resultant polymerase activity was measured using the assay on the plate as previously described. To characterize the sensitivity of the assay, we prepared serial 10-fold dilutions of the target and mixed the target samples with molecular switches in distinct states (e.g., transition versus closed states) to evaluate changes in polymerase activity. To investigate the incubation time required to recover the functionality of lyophilized switches, we reconstituted the lyophilized reagents with the reaction buffer and incubated the mixture for less than 1, 5, 10, and 30 min before mixing with target and transferring to the functionalized plate for signaling. To evaluate the performance of lyophilized switches, we mixed lyophilized and nonlyophilized switches with target, and the resultant polymerase activity was measured through 5′ exonuclease degradation of fluorescent signaling probe as previously described.
Cell culture and lysis
Human lung epithelial cell line (PC9) was obtained from the American Type Culture Collection (ATCC) and grown in RPMI 1640 medium (HyClone) supplemented with 10% fetal bovine serum (HyClone) and 1% penicillin-streptomycin (Gibco) in a humidified 37°C incubator with 5% CO2. The cell line was tested and free of mycoplasma contamination (MycoAlert Mycoplasma Detection Kit, Lonza, LT07-418). To evaluate the performance of the assay in biological samples, we prepared cell lysates through different protocols and spiked in synthetic target oligonucleotides, before testing the samples with molecular switches. Ribonuclease inhibitor was added to all lysate mixtures. Specifically, we lysed cell pellets through heating or incubating with detergent buffer. For heat treatment, cell pellets were resuspended in the reaction buffer and heated at 56°C for 30 min, 70°C for 5 min, or 90°C for 5 min (23, 24). For chemical lysis, we prepared lysis buffers, by mixing the reaction buffer with varying amounts of single or a mixture of detergents: Triton X-100, SDS, Saponin, Tween 20, Igepal CA-630, and NP-40 (Sigma-Aldrich). To optimize the chemical lysis composition and incubation duration, we evaluated various lysis conditions for their ability to rapidly lyse cells while maintaining good polymerase activity. To assess cell lysis efficiency, cells were incubated with the lysis buffers and the resultant cell numbers were counted using Countess II Automated Cell Counter (Thermo Fisher Scientific). Polymerase activity was measured through 5′ exonuclease degradation of fluorescent signaling probe, as described above.
RNA extraction and detection
RNA extraction was performed with a commercially available kit (RNeasy Mini, Qiagen) per the manufacturer’s protocol. Extracted RNA was quantified with a NanoDrop spectrophotometer (Thermo Fisher Scientific). To detect specific RNA targets through gold-standard RT-qPCR analysis, extracted RNA was first reverse-transcribed to generate first-strand complementary DNA (MultiScribe Reverse Transcriptase, Thermo Fisher Scientific). For PCR analysis, to detect housekeeping genes (i.e., GAPDH and β-actin), we used TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) and primer sets (TaqMan gene expression assays, Thermo Fisher Scientific) as recommended by the manufacturer. Amplification conditions consisted of 1 cycle of 95°C for 2 min and 45 cycles of 95°C for 1 s and 60°C for 20 s. All thermal cycling was performed on a QuantStudio 5 real-time PCR system (Applied Biosystems).
Clinical measurements
The study was approved by the National Healthcare Group Domain Specific Review Board (reference: 2020/00106 and 2020/00120) and the National University of Singapore (NUS) Institutional Review Board (IRB; H-19-001). All individuals were recruited according to IRB-approved protocols after obtaining informed consent. SARS-CoV-2–positive clinical samples were handled according to the Singapore Ministry of Health Biosafety Branch and the NUS Institutional Biosafety Committee regulations in the Biosafety Level 3 (BSL-3) or Biosafety Level 2+ (BSL-2+) laboratories where appropriate. A total of 73 samples consisting of extracted RNA and heat-inactivated swabs were evaluated in this study. To determine the diagnostic performance of the CATCH assay, extracted RNA samples (positive, n = 24; negative, n = 25) were used directly on the CATCH assay, while swab lysates (positive, n = 9; negative, n = 15) were prepared through heating at 70°C for 30 min, before measurement by the CATCH assay. SARS-CoV-2 clinical diagnoses were generated by commercial RT-qPCR assay (Fortitude Kit, MiRXES). Amplification conditions consisted of 1 cycle of 48°C for 15 min, 1 cycle of 95°C for 150 s, and 42 cycles of 95°C for 10 s and 59°C for 42 s. Ct value of <40 was determined as positive as per Centers for Disease Control and Prevention’s (CDC) guidelines (6). All measurements on clinical samples were performed in an anonymized and blinded fashion and finalized before comparison with clinical Ct value.
Statistical analyses
Unless otherwise stated, all measurements were performed in biological triplicate, and the data are presented as mean ± SD. For intersample comparisons, multiple pairs of samples were each tested via Student’s t test, and the resulting P values were adjusted for multiple hypothesis testing using Bonferroni correction. An adjusted P < 0.05 was determined as significant. Receiver operating characteristic (ROC) curves were generated from patient profiling data and constructed by plotting sensitivity versus (1 − specificity), and the values of AUC were computed using the trapezoidal rule. The clinical reports were used as classifiers (true positives and true negatives). Detection sensitivity, specificity, and accuracy were calculated using standard formulas. Statistical analyses were performed using GraphPad Prism (version 7.0c).
Supplementary Material
Acknowledgments
We thank Y. J. Tan and J. M. Hong for their biosafety input; R. Jureen, B. Yan, and C. K. Lee for facilitating clinical sample transfer; and P. M. Thong and X. Mao for assistance with clinical sample collection and access to BSL-2+ facilities. Funding: This work was supported, in part, by funding from National University of Singapore (NUS), NUS Research Scholarship, National Medical Research Council, Ministry of Education, Institute for Health Innovation&Technology, IMCB Independent Fellowship, NUS Early Career Research Award, National University Health System Seed Grant, and National Centre for Infectious Diseases Grant. N.R.Y.H. received funding from the National Medical Research Council (Open Fund Young Investigator Research Grant, MOH-000234). Author contributions: N.R.S., A.N., Y.L., and H.S. designed the research. N.R.S., A.N., Y.L., N.R.Y.H., H.Z., and Y.C. performed the research. Q.H.M. and Y.W. performed BSL-3 experiments that allowed clinical sample testing. D.L.L.B., K.L.C., D.C., P.A.T., and C.W.M.O. provided deidentified clinical samples and data. N.R.S., A.N., Y.L., N.R.Y.H., H.Z., Y.C., and H.S. analyzed the data and wrote the paper. All authors contributed to the manuscript. Competing interests: N.R.S., A.N., Y.L., and H.S. are inventors on a pending patent application related to this work filed by the National University of Singapore. N.R.Y.H. and H.S. are inventors on a patent related to this work filed by the National University of Singapore (no. WO/2020/009660, published on 9 January 2020). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe5940/DC1
REFERENCES AND NOTES
- 1.Huang H., Fan C., Li M., Nie H.-L., Wang F.-B., Wang H., Wang R., Xia J., Zheng X., Zuo X., Huang J., COVID-19: A call for physical scientists and engineers. ACS Nano 14, 3747–3754 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Bai Y., Yao L., Wei T., Tian F., Jin D. Y., Chen L., Wang M., Presumed asymptomatic carrier transmission of COVID-19. JAMA 323, 1406–1407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rothe C., Schunk M., Sothmann P., Bretzel G., Froeschl G., Wallrauch C., Zimmer T., Thiel V., Janke C., Guggemos W., Seilmaier M., Drosten C., Vollmar P., Zwirglmaier K., Zange S., Wölfel R., Hoelscher M., Transmission of 2019-nCoV infection from an asymptomatic contact in Germany. N. Engl. J. Med. 382, 970–971 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gorbalenya A. E., Baker S. C., Baric R. S., de Groot R. J., Drosten C., Gulyaeva A. A., Haagmans B. L., Lauber C., Leontovich A. M., Neuman B. W., Penzar D., Perlman S., Poon L. L. M., Samborskiy D. V., Sidorov I. A., Sola I., Ziebuhr J., Coronaviridae study group of the International Committee on taxonomy of viruses, The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 5, 536–544 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter L. J., Garner L. V., Smoot J. W., Li Y., Zhou Q., Saveson C. J., Sasso J. M., Gregg A. C., Soares D. J., Beskid T. R., Jervey S. R., Liu C., Assay techniques and test development for COVID-19 diagnosis. ACS Cent. Sci. 6, 591–605 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Centers for Disease Control and Prevention, CDC 2019-Novel Coronavirus (2019-nCoV) Real-Time RT-PCR Diagnostic Panel (Centers for Disease Control and Prevention, 2020); https://www.fda.gov/media/134922/download.
- 7.Bruce E. A., Huang M. L., Perchetti G. A., Tighe S., Laaguiby P., Hoffman J. J., Gerrard D. L., Nalla A. K., Wei Y., Greninger A. L., Diehl S. A., Shirley D. J., Leonard D. G. B., Huston C. D., Kirkpatrick B. D., Dragon J. A., Crothers J. W., Jerome K. R., Botten J. W., Direct RT-qPCR detection of SARS-CoV-2 RNA from patient nasopharyngeal swabs without an RNA extraction step. PLOS Biol. 18, e3000896 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y., Yao L., Li J., Chen L., Song Y., Cai Z., Yang C., Stability issues of RT-PCR testing of SARS-CoV-2 for hospitalized patients clinically diagnosed with COVID-19. J. Med. Virol. 92, 903–908 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toyoshima Y., Nemoto K., Matsumoto S., Nakamura Y., Kiyotani K., SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 65, 1075–1082 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weissleder R., Lee H., Ko J., Pittet M. J., COVID-19 diagnostics in context. Sci. Transl. Med. 12, eabc1931 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Li J., Green A. A., Yan H., Fan C., Engineering nucleic acid structures for programmable molecular circuitry and intracellular biocomputation. Nat. Chem. 9, 1056–1067 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y., Cu Y. T., Luo D., Multiplexed detection of pathogen DNA with DNA-based fluorescence nanobarcodes. Nat. Biotechnol. 23, 885–889 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones M. R., Seeman N. C., Mirkin C. A., Nanomaterials. Programmable materials and the nature of the DNA bond. Science 347, 1260901 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Sundah N. R., Ho N. R. Y., Lim G. S., Natalia A., Ding X., Liu Y., Seet J. E., Chan C. W., Loh T. P., Shao H., Barcoded DNA nanostructures for the multiplexed profiling of subcellular protein distribution. Nat. Biomed. Eng. 3, 684–694 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Wilner O. I., Weizmann Y., Gill R., Lioubashevski O., Freeman R., Willner I., Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 4, 249–254 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Song P., Shen J., Ye D., Dong B., Wang F., Pei H., Wang J., Shi J., Wang L., Xue W., Huang Y., Huang G., Zuo X., Fan C., Programming bulk enzyme heterojunctions for biosensor development with tetrahedral DNA framework. Nat. Commun. 11, 838 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho N. R. Y., Lim G. S., Sundah N. R., Lim D., Loh T. P., Shao H., Visual and modular detection of pathogen nucleic acids with enzyme-DNA molecular complexes. Nat. Commun. 9, 3238 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pan Y., Zhang D., Yang P., Poon L. L. M., Wang Q., Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 20, 411–412 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dang C., Jayasena S. D., Oligonucleotide inhibitors of Taq DNA polymerase facilitate detection of low copy number targets by PCR. J. Mol. Biol. 264, 268–278 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Hasegawa H., Savory N., Abe K., Ikebukuro K., Methods for improving aptamer binding affinity. Molecules 21, 421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yan C., Cui J., Huang L., Du B., Chen L., Xue G., Li S., Zhang W., Zhao L., Sun Y., Yao H., Li N., Zhao H., Feng Y., Liu S., Zhang Q., Liu D., Yuan J., Rapid and visual detection of 2019 novel coronavirus (SARS-CoV-2) by a reverse transcription loop-mediated isothermal amplification assay. Clin. Microbiol. Infect. 26, 773–779 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Broughton J. P., Deng X., Yu G., Fasching C. L., Servellita V., Singh J., Miao X., Streithorst J. A., Granados A., Sotomayor-Gonzalez A., Zorn K., Gopez A., Hsu E., Gu W., Miller S., Pan C. Y., Guevara H., Wadford D. A., Chen J. S., Chiu C. Y., CRISPR-Cas12-based detection of SARS-CoV-2. Nat. Biotechnol. 38, 870–874 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chin A. W. H., Chu J. T. S., Perera M. R. A., Hui K. P. Y., Yen H. L., Chan M. C. W., Peiris M., Poon L. L. M., Stability of SARS-CoV-2 in different environmental conditions. Lancet Microbe 1, e10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ladha A., Joung J., Abudayyeh O., Gootenberg J., Zhang F., A 5-min RNA preparation method for COVID-19 detection with RT-qPCR. medRxiv, (2020). [Google Scholar]
- 25.Ong C. W. M., Migliori G. B., Raviglione M., MacGregor-Skinner G., Sotgiu G., Alffenaar J. W., Tiberi S., Adlhoch C., Alonzi T., Archuleta S., Brusin S., Cambau E., Capobianchi M. R., Castilletti C., Centis R., Cirillo D. M., D’Ambrosio L., Delogu G., Esposito S. M. R., Figueroa J., Friedland J. S., Heng B. H. C., Ippolito G., Jankovic M., Kim H. Y., Klintz S. R., Ködmön C., Lalle E., Leo Y. S., Leung C. C., Märtson A. G., Melazzini M., Fard S. N., Penttinen P., Petrone L., Petruccioli E., Pontali E., Saderi L., Santin M., Spanevello A., van Crevel R., van der Werf M. J., Visca D., Viveiros M., Zellweger J. P., Zumla A., Goletti D., Epidemic and pandemic viral infections: Impact on tuberculosis and the lung. A consensus by the World Association for Infectious Diseases and Immunological Disorders (WAidid), Global Tuberculosis Network (GTN) and members# of ESCMID Study Group for Mycobacterial Infections (ESGMYC). Eur. Respir. J. 56, 2001727 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ong C. W. M., Goletti D., Impact of the global COVID-19 outbreak on the management of other communicable diseases. Int. J. Tuberc. Lung Dis. 24, 547–548 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Yelleswarapu V., Buser J. R., Haber M., Baron J., Inapuri E., Issadore D., Mobile platform for rapid sub-picogram-per-milliliter, multiplexed, digital droplet detection of proteins. Proc. Natl. Acad. Sci. U.S.A. 116, 4489–4495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu H., Xia A., Wang D., Zhang Y., Deng S., Lu W., Luo J., Zhong Q., Zhang F., Zhou L., Zhang W., Wang Y., Yang C., Chang K., Fu W., Cui J., Gan M., Luo D., Chen M., An ultraportable and versatile point-of-care DNA testing platform. Sci. Adv. 6, eaaz7445 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu X., Zhao H., Natalia A., Lim C. Z. J., Ho N. R. Y., Ong C. J., Teo M. C. C., So J. B. Y., Shao H., Exosome-templated nanoplasmonics for multiparametric molecular profiling. Sci. Adv. 6, eaba2556 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Surkova E., Nikolayevskyy V., Drobniewski F., False-positive COVID-19 results: Hidden problems and costs. Lancet Respir. Med. 8, 1167–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cohen A. N., Kessel B., Milgroom M. G., Diagnosing COVID-19 infection: The danger of over-reliance on positive test results. medRxiv, (2020). [Google Scholar]
- 32.Cohen A. N., Kessel B., False positives in reverse transcription PCR testing for SARS-CoV-2. medRxiv, (2020). [Google Scholar]
- 33.Vogels C. B. F., Brito A. F., Wyllie A. L., Fauver J. R., Ott I. M., Kalinich C. C., Petrone M. E., Casanovas-Massana A., Muenker M. C., Moore A. J., Klein J., Lu P., Lu-Culligan A., Jiang X., Kim D. J., Kudo E., Mao T., Moriyama M., Oh J. E., Park A., Silva J., Song E., Takahashi T., Taura M., Tokuyama M., Venkataraman A., Weizman O. E., Wong P., Yang Y., Cheemarla N. R., White E. B., Lapidus S., Earnest R., Geng B., Vijayakumar P., Odio C., Fournier J., Bermejo S., Farhadian S., Dela Cruz C. S., Iwasaki A., Ko A. I., Landry M. L., Foxman E. F., Grubaugh N. D., Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 5, 1299–1305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garg N., Boyle D., Randall A., Teng A., Pablo J., Liang X., Camerini D., Lee A. P., Rapid immunodiagnostics of multiple viral infections in an acoustic microstreaming device with serum and saliva samples. Lab Chip 19, 1524–1533 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jeong J. H., Kim K. H., Jeong S. H., Park J. W., Lee S. M., Seo Y. H., Comparison of sputum and nasopharyngeal swabs for detection of respiratory viruses. J. Med. Virol. 86, 2122–2127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shaffer S. M., Joshi R. P., Chambers B. S., Sterken D., Biaesch A. G., Gabrieli D. J., Li Y., Feemster K. A., Hensley S. E., Issadore D., Raj A., Multiplexed detection of viral infections using rapid in situ RNA analysis on a chip. Lab Chip 15, 3170–3182 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeh E. C., Fu C. C., Hu L., Thakur R., Feng J., Lee L. P., Self-powered integrated microfluidic point-of-care low-cost enabling (SIMPLE) chip. Sci. Adv. 3, e1501645 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lim C. Z. J., Zhang Y., Chen Y., Zhao H., Stephenson M. C., Ho N. R. Y., Chen Y., Chung J., Reilhac A., Loh T. P., Chen C. L. H., Shao H., Subtyping of circulating exosome-bound amyloid β reflects brain plaque deposition. Nat. Commun. 10, 1144 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duncombe T. A., Tentori A. M., Herr A. E., Microfluidics: Reframing biological enquiry. Nat. Rev. Mol. Cell Biol. 16, 554–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tokeshi M., Minagawa T., Uchiyama K., Hibara A., Sato K., Hisamoto H., Kitamori T., Continuous-flow chemical processing on a microchip by combining microunit operations and a multiphase flow network. Anal. Chem. 74, 1565–1571 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Shao H., Chung J., Lee K., Balaj L., Min C., Carter B. S., Hochberg F. H., Breakefield X. O., Lee H., Weissleder R., Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 6, 6999 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okonechnikov K., Golosova O., Fursov M.; UGENE team , Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 28, 1166–1167 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe5940/DC1