

A chromosome-level giraffe genome assembly and gene-edited mice reveal molecular mechanisms underpinning its high stature.
Abstract
The suite of adaptations associated with the extreme stature of the giraffe has long interested biologists and physiologists. By generating a high-quality chromosome-level giraffe genome and a comprehensive comparison with other ruminant genomes, we identified a robust catalog of giraffe-specific mutations. These are primarily related to cardiovascular, bone growth, vision, hearing, and circadian functions. Among them, the giraffe FGFRL1 gene is an outlier with seven unique amino acid substitutions not found in any other ruminant. Gene-edited mice with the giraffe-type FGFRL1 show exceptional hypertension resistance and higher bone mineral density, both of which are tightly connected with giraffe adaptations to high stature. Our results facilitate a deeper understanding of the molecular mechanism underpinning distinct giraffe traits, and may provide insights into the study of hypertension in humans.
INTRODUCTION
Giraffes are immediately recognizable due to their exceptionally long necks and legs, making them the tallest terrestrial animals. Giraffes played a central role in different evolutionary schools of thought, including those of Lamarck and Darwin. Their unusual anatomy is thought to provide various selective advantages. In addition to allowing access to otherwise inaccessible food resources (1), their elevated head position provides an excellent vantage point for scanning the horizon and thus detecting predators or competitors, both of which are crucial for their survival (2). However, their exceptional anatomy is also accompanied by considerable physiological challenges. Most notably, the cardiovascular system has to tolerate twofold higher systemic blood pressure than most other mammals to supply the brain with blood (3). This elevated hydrostatic pressure has resulted in hypertrophy of their cardiac and arteriole walls (3) and adaptations of the circulation system that prevent sudden changes in blood pressure when a giraffe elevates or lowers its head (4). Giraffes also have neuromotor delays due to their long neural networks (5) and face difficulties in rising due to their long legs, which increases the danger associated with resting and drinking. They also require greatly enlarged and strengthened nuchal ligaments to support their long, heavy neck (6). Hence, the giraffe provides a unique case for studying co-adaptation or evolution in several different traits that are causally linked to an extreme body plan.
The okapi (Okapia johnstoni) is the only other extant member of the Giraffidae family and provides a useful point of genomic comparison. A study published in 2016 provided the first giraffe and okapi draft genomes and identified candidate genes and pathways involved in neck elongation and cardiovascular adaptations (7). However, these initial draft genomes were relatively fragmented, which can both introduce certain biases and limit the interpretation of some analyses (8). Furthermore, the comparative analyses carried out using these draft genomes were restricted to 17,210 genes, which were annotated by aligning with cattle (Bos taurus) reference transcripts, thus limiting the resolution power to explore genomic features unique to the giraffe, not least given the paucity of other ruminant genomes available at that time. Hence, the availability of a higher-quality giraffe genome assembly together with our recently published whole-genome dataset for ~50 ruminant species (9) opens up the possibility of identifying giraffe-specific mutations with a much higher accuracy and robustness. This, in turn, provides a better resource for inferring the true genomic changes that account for the unique body plan of the giraffe.
Here, we report an improved, chromosome-level genome assembly of a Rothschild’s giraffe (Giraffa camelopardalis rothschildi), the results of comparative analysis against the recently available ruminant genomes, and—crucially—functional validation of one key cardiovascular and skeletal gene in gene-edited mice. These results provide insights into the genetic basis of the giraffe anatomy and associated adaptations, with particular implications concerning the cardiovascular system, which may be helpful for treating human cardiovascular disease and hypertension.
RESULTS AND DISCUSSION
Genome assembly and comparative genomics
We sequenced the genome of a male Rothschild’s giraffe with a combination of single-molecule real-time sequencing (using an Oxford Nanopore platform), paired-end sequencing (with an Illumina HiSeq 2000 system), and Hi-C sequencing (figs. S1 and S2 and table S1). First, we used Nanopore data to generate initial contigs, and after polishing with Illumina reads, we obtained an assembly with contig N50 of 35.9 Mb (table S2). Next, Hi-C data were used to anchor the contigs into chromosomes, which yielded a final assembly of 2.44 Gb with ~97.95% of the bases successfully anchored to 15 chromosomes (2n = 30) (figs. S3 and S4 and table S3). A series of evaluations show that the genome assembly is of high quality (see the Supplementary Materials, figs. S2 to S6, and tables S4 to S11).
Chromosome evolution is related to genome size, gene family evolution, and speciation (10). The giraffe has many fewer chromosomes (2n = 30) than the putative ancestral karyotype of even-toed ungulates (2n = 58 to 60), suggesting the occurrence of multiple chromosome fusion events in its evolution (11). Using the genomes of cattle, goat, giraffe, and okapi, with sperm whale as outgroup, we reconstructed the ancestral karyotype of the Giraffidae and Bovidae families (2n = 60) (table S12), which corresponds to the ancestor of the Pecora suborder (11). The results indicate that just three fissions and three fusions occurred in the cattle lineage since the pecoran ancestor. Hence, most of the ancestral chromosome structure is retained in cattle, including the complement of 30 chromosomes. In contrast, a minimum of four fissions and 17 fusions occurred between the pecoran ancestor and the giraffe, resulting in a substantial decrease (to 15) in haploid chromosome number (Fig. 1). The functional significance—if any—of such prolific chromosome fusions in giraffes requires further research.
Fig. 1. Reconstruction of ancestral chromosomes of Giraffidae and Bovidae families with sperm whale as an outgroup.
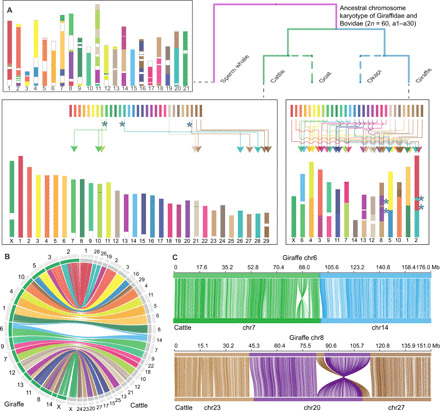
(A) The figure displays the distribution of ancestral chromosome segments in cattle and giraffe genomes, including interchromosome rearrangements and fission and fusion events in cattle and giraffe. Blue asterisks in the cattle chromosome diagram indicate chromosome fission events in cattle. Blue asterisks in the giraffe chromosome diagram indicate sites of chromosome rearrangements. (B) Circos plot showing syntenic relationships of chromosomes between giraffe (left) and cattle (right). Chromosomes are colored on the basis of the cattle homologies. (C) Two types of collinear relationship between giraffe and cattle. The top and bottom horizontal lines represent giraffe and cattle chromosomes, respectively, and the lines between them link the alignment blocks.
We next evaluated the adaptive divergence between giraffe and other mammals in coding regions, using both the branch and the branch-site models implemented in PAML (12). We detected 101 positively selected genes (PSGs) and 359 rapidly evolving genes (REGs) in the giraffe (P < 0.05 according to χ2 tests in both cases) (fig. S7 and tables S13 and S14) (13). This is a large increase compared to those found in the previous giraffe genome study, which identified 17 PSGs and 53 genes with adaptive divergence (high divergence compared with other mammals or unique substitutions) in giraffe (7). Notably, while 7 of the 17 PSGs from the previous study overlapped with our findings, the remaining 10 PSGs showed no positive selection signal in our analyses, which is primarily caused by the inclusion of many more ruminant branches as background. We show two examples of how the inclusion of a larger background panel or better genome quality refined our ability to identify giraffe-specific selection signals (fig. S8). Similarly, only 15 of the 53 previously identified adaptive divergence genes (7) were identified as PSGs or REGs in our analysis. Together, the improved genome assembly (better genome completeness, accuracy, and annotation) and higher number of accessible ruminant reference genomes allow us to substantially decrease both false positive and false negative signals of genes undergoing adaptive evolution in the giraffe. A Gene Ontology (GO) enrichment analysis showed that the 460 PSGs and REGs identified in the present study are primarily related to growth and development, nervous and visual systems, circadian rhythm, and blood pressure regulation (table S15). The KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway–based analysis suggested that the rapidly evolving pathways in giraffe compared to okapi are related to metabolic, circulatory, and immune systems (table S16).
A pleiotropic role of FGFRL1 in hypertension resistance and bone growth
The giraffe fibroblast growth factor (FGF) receptor–like protein 1 (FGFRL1) gene has previously been identified as one of the most conspicuous targets of selection in the giraffe (7). FGFRL1 contains a cluster of seven nonsynonymous mutations in its key FGF binding domain when compared against sequences in other ruminants and outgroup mammals (fig. S9). Using our substantially expanded set of background genomes, we confirmed that these mutations are unique to the giraffe and that FGFRL1 contains more unique substitutions than any other giraffe gene (table S17). Mutations in FGFRL1 cause severe cardiovascular and skeletal defects in humans and mice (14, 15), and hence we follow Agaba et al. (7) in hypothesizing that FGFRL1 may be associated with the extreme cardiovascular and skeletal adaptations in the giraffe. To investigate the in vivo consequences of these substitutions, we introduced these seven mutations into the FGFRL1 gene of mice to obtain giraffe-type FGFRL1 mice, using CRISPR-Cas9 technology (fig. S10). In contrast to mice with targeted complete deletion of the gene (14), which die with multiple congenital malformations, giraffe-type FGFRL1 mice were viable and fertile.
The cardiovascular hemodynamic in the giraffe is characterized by exceptionally high blood pressure without related organ damage, in contrast to the typical detrimental effects of hypertension observed in other animals/humans (3). FGFRL1 is known to be involved in the cardiovascular system (14), and we hypothesized that some vascular adaptations in the giraffe may only be apparent in a hypertensive physiological setting. To test this, we induced high blood pressure in wild-type (WT) and mutant FGFRL1 mice. The mice were infused with angiotensin II (Ang II; 900 ng/kg per minute; fig. S11), which induces hypertension by vasoconstriction and sodium retention. Our giraffe-type FGFRL1 mice showed no signs of congenital heart defects (fig. S12) or any obvious alterations in heart rate compared to WT controls (fig. S13). Although the basal blood pressure was slightly higher in giraffe-type than in WT FGFRL1 mice, no significant difference was observed (fig. S13). After Ang II infusion for 28 days, the average systolic and diastolic blood pressure in WT controls were significantly increased to 158.97 ± 5.01 and 94.54 ± 8.60 mmHg (Fig. 2A), respectively, confirming that hypertension was successfully induced in them. Unexpectedly, the Ang II–induced hypertension was absent in giraffe-type FGFRL1 mice, which showed average systolic and diastolic pressures of 125.30 ± 5.97 and 83.43 ± 11.77 mmHg, respectively, after Ang II infusion for 28 days (Fig. 2A), not significantly different from giraffe-type FGFRL1 controls. Moreover, significantly less myocardial and renal fibrosis was observed in giraffe-type FGFRL1 mice, as manifested by significantly lower proportion of collagen fibers in their heart and kidney than in WT controls, which obviously resulted from the absence of Ang II–induced hypertension in giraffe-type FGFRL1 mice (Fig. 2B and fig. S14). In addition, the impaired heart function caused by hypertension in Ang II–treated WT mice was also significantly alleviated in giraffe-type FGFRL1 mice, as evidenced by improved left ventricular ejection fractions (LVEFs) and fractional shortening (LVFS) (fig. S15). Our findings collectively suggest that the giraffe-type FGFRL1 has little effect on cardiac development but can prevent Ang II–induced hypertension and thus avoid or at least alleviate a range of detrimental effects of hypertension. In addition, our molecular dynamics (MD) simulations suggested that the unique variants in giraffe-type FGFRL1 could affect its binding affinity with FGF ligands (fig. S16), potentially interfering with their cross-talk with renin-angiotensin-aldosterone system to modulate blood pressure and providing a possible mechanism by which giraffe-type FGFRL1 modulates blood pressure (16). Despite the differences in cardiovascular structure and physiology between mice and human and the possibility that other genes may have contributed to the observed systemic co-adaptation to hypertension, it is intriguing to speculate that FGFRL1 might hold promise as a therapeutic target for prevention or treatment of hypertension or cardiovascular diseases in humans. Nevertheless, we acknowledge that this perspective is tentative and awaits a thorough investigation of the mechanisms behind the observed cardiovascular effect of giraffe-type FGFRL1.
Fig. 2. Skeletal and cardiovascular characteristics of giraffe-type FGFRL1 mice.
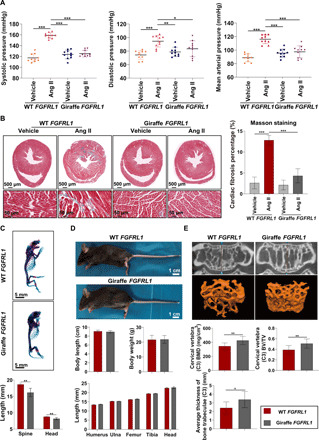
(A) Giraffe-type FGFRL1 mice showed significantly lower systolic, diastolic, and mean arterial pressures (mmHg) than WT FGFRL1 mice after Ang II infusion for 28 days. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA followed by Tukey’s post hoc test. (B) Giraffe-type FGFRL1 mice had significantly lower proportions of fibrotic areas in heart than WT FGFRL1 mice after 28 days of Ang II infusion. ***P < 0.001, one-way ANOVA followed by Tukey’s post hoc test. Error bars indicate SD. (C) Whole-mount skeletons of P0 mice showed hypoplasia of skeletal elements in giraffe-type FGFRL1 mice. **P < 0.01 by t test. (D) Adult giraffe-type FGFRL1 mice show no discernible body size and skeletal phenotype difference to WT mice. (E) Giraffe-type FGFRL1 mice showed significantly higher BMD, BV/TV, and average trabeculae thickness than WT mice. *P < 0.05, **P < 0.01 by t test. Photo credit: Jianbo Gao, The Fourth Military Medical University.
In addition to the observed cardiovascular effect, we noticed that postnatal day 0 (P0) giraffe-type FGFRL1 mice showed prenatal hypoplasia of skeletal elements, with a smaller body size, delayed craniofacial development, shortened axial/appendicular skeletons, and smaller vertebral lengths than the P0 WT mice (Fig. 2C and fig. S17). In contrast, adult giraffe-type FGFRL1 mice (24 to 26 g, 16 weeks) showed no discernible skeletal phenotype compared with WT mice or any significant deviation in body size and weight, limb length, or vertebral height (Fig. 2D and fig. S18). This suggests that mutations in this gene in itself are not sufficient for neck elongation in the giraffe, refuting a previous hypothesis (7), although again we must recognize the limitations of introducing a gene into a different genetic background. However, it also shows that giraffe-type FGFRL1-associated postnatal bone growth can compensate for the observed prenatal effects such that FGFRL1 may play an indirect role in the exceptional bone growth of giraffe, e.g., by accelerating bone formation to maintain bone mineral density (BMD), as in humans (17). Therefore, we next examined bone ultrastructure by micro computed tomography (microCT). Giraffe-type FGFRL1 mice achieved significantly higher BMD, bone volume/total volume (BV/TV) ratio, and average trabeculae thickness in vertebrae (C3) and distal femur (Fig. 2E and fig. S19). Skeletal growth rate tends to be inversely related to bone strength in animals (18), but despite having the highest skeletal growth rate among mammals, giraffes maintain normal BMD (19). In summary, we find indications for a pleiotropic adaptive effect of the highly unique giraffe-type FGFRL1 by not only significantly enhancing hypertension resistance but also achieving normal bone strength, despite the accelerated rate of bone growth in the giraffe.
Adaptive changes in other cardiovascular genes
Previous anatomical and physiological analyses suggest that multiple giraffe organs are involved in associated adaptations of the cardiovascular system, including hypertrophy of the left ventricle and interventricular walls (3), thickening of blood vessels of the lower extremities, and low glomerular filtration rates (20). Our results revealed that several pathways involving tissues influenced by high blood pressure, such as blood vessels, heart, and kidney, were significantly diverged between giraffe and other ruminants (table S16). The “platelet activation” pathway plays an important role in hypertension-associated thrombosis (Fig. 3A) (21). Three REGs (COL1A2, LYN, and PLCB1) and a number of genes with giraffe-specific amino acid variations are involved in the two major platelet activation, shape change, and platelet aggregation paths. A further set of PSGs and REGs that participate in phosphatidylinositol metabolism (PIP4K2A, ISYNA1, MTMR3, CDS1, and INPP1) may also be involved in the regulation of platelet activation (22). Another giraffe-divergent pathway is the “adrenergic signaling pathway in cardiomyocytes,” which is related to cardiac contraction and possibly the morphological remodeling of the giraffe heart (Fig. 3B) (23). Highly divergent genes in this pathway are mainly involved in ion transport (SCN7A, SLC9A1, ATP1A4, and CACNA2D4), which is important for myocardial function (24). We also found strong signals of adaptation in two major adrenergic receptors (ADRA1A and ADRA2B), as previously reported (7). Although ADRA2B is mainly expressed in the nervous system, both of these receptors are strongly related to blood pressure regulation (25, 26). Last, we detected strong giraffe-specific divergence in genes related to the “proximal tubule bicarbonate reclamation” and “endocrine and other factor-regulated calcium reabsorption” pathways. Changes in these pathways may reduce the pressure gradient across membranes in the giraffe kidney and protect it from hypertensive damage (Fig. 3C) (20). The REG AQP1 encodes a water-transporting protein in cell membranes of kidney proximal tubules and is involved in kidney development and injury responses (27). Two REGs (PLCB1 and ATP1A4) that are reportedly involved in hypertension or related organ damage participate in more than one of the mentioned pathways (28, 29), in accordance with expectations of co-adaptation of the blood vessel–heart–kidney axis in giraffe. In addition to genes in the mentioned pathways, we also detected other PSGs and REGs that may help to avoid hypertensive damage, including ANGPTL1, which is associated with the integrity of vascular endothelium (30), and TGFB1, which is strongly implicated in multiorgan fibrosis associated with hypertension (31). The finding of multiple genes involved in several phenotypic traits that share evolutionary constraints due to the extreme stature of the giraffe suggests that pleiotropy may play an important role in evolving such an extreme body plan.
Fig. 3. Genes and pathways related to high blood pressure adaptations in giraffe blood vessels, heart, and kidney.
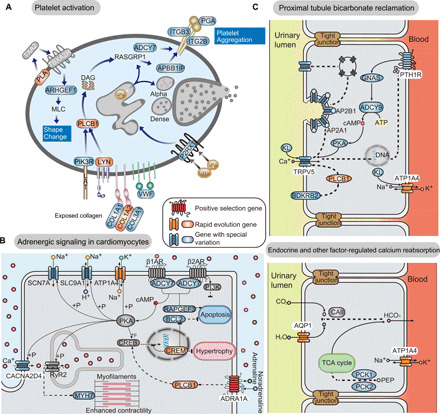
(A) Modifications of genes in the platelet activation pathway may help to prevent damage to giraffe blood vessels. (B) Genes in the adrenergic signaling in cardiomyocytes that show high divergence in giraffe. (C) The proximal tubule bicarbonate reclamation (top) and endocrine and other factor-regulated calcium reabsorption (bottom) pathways may help to prevent kidney damage.
Co-adaptations in sensory systems
For herbivorous ungulates subject to predation, vigilance is crucial for survival and has two components: gathering information and instigating muscular action after signal transduction through the nervous system (32). Giraffes are thought to have a distinctive retinal cone topography that provides the best visual acuity in the Artiodactyla, which, together with the elevated head, enhances the capacity for horizon scanning (33). Accordingly, we found not only a number of PSGs and REGs that contribute to eye development and vision but also a number of genes that are related to Usher syndrome in humans (CDH23, PCDH15, USH2A, NINL, and UBR3), which affects vision, hearing, and balance (34), suggesting a related suite of sensory co-adaptations in giraffe (Fig. 4A). Similar to all other ruminants, we found only two opsin genes; thus, we could not verify that giraffes see color, at least not trichromatic color as has been hypothesized before (35). We found indications that the sense of smell in the giraffe may be degenerated. Compared to okapi, giraffe has lost at least 53 olfactory-related genes, including 50 encoding olfactory receptors, two encoding vomeronasal receptors, and one encoding an odorant binding protein (table S18). Further analysis shows that most of these olfactory receptors are spatially clustered and were lost because of a segmental deletion (Fig. 4B and figs. S20 and S21). Moreover, the contracted gene families in giraffe were also enriched in olfactory receptor activity (fig. S22 and tables S19 and S20). This may be an evolutionary consequence of enhanced vision, consistent with the hypothesized trade-off in sensory acuity found in many taxa (36) and/or with reduction in competition for food with other browsers.
Fig. 4. Adaptations for vigilance.
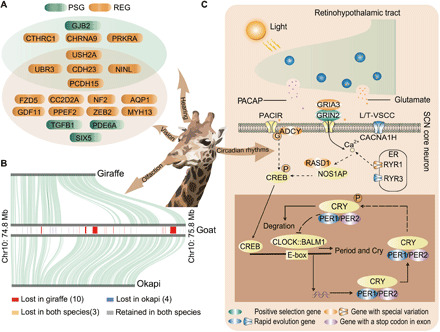
(A) PSGs and REGs associated with giraffes’ visual, auditory, and balance systems. (B) Giraffes have lost several olfactory receptors (for example, on chromosome 10 of goat) compared to okapi. The location of genes on goat chromosome is shown in the rectangle, and the collinear relationship of giraffe-goat and okapi-goat is shown in the top and bottom panels, respectively. (C) Genetic changes involved in light-mediated regulation of the molecular clock in giraffe suprachiasmatic nucleus (SCN) neurons.
Moreover, the extreme morphology of the giraffe increases its vulnerability when asleep by increasing the time required to become upright. Expectedly, therefore, given their needs for vigilance and high food intakes, giraffe sleep durations are among the lowest recorded (37). Concordantly, we found evidence of rapid evolution of PER1 in giraffe, a period family gene critical for the maintenance of circadian rhythm (38) and the emergence of a stop codon in the first exon of PER2 (Fig. 4C and fig. S23), possibly altering the transcript of this important circadian rhythm gene. HCRT, which plays a role in the regulation of sleep and arousal (39), also shows accelerated evolution in giraffe. Together, there is evidence that adaptive modifications of circadian rhythm and sleep arousal systems in giraffe have promoted short and fragmented sleep patterns. Overall, the comparative genomic analysis highlights that the unique stature of the giraffe has led to a series of necessary behavioral co-adaptations.
Conclusion
Because of its exceptional anatomy and suite of evolutionary adaptations, the giraffe has always been an outstanding case of animal evolution and physiology. Our high-quality genome assembly provides more precise comparative insights into the genetic basis of a broad spectrum of its biological features. We identified genetic changes underlying co-adaptations to the singular stature, notably related to the adaptation of high blood pressure, bone growth, and vigilance, all of which are traits that are intricately associated with the unique stature and biology of the giraffe. The strongest selective signal was found in FGFRL1, a gene for which our gene edition experiments suggest pleiotropic effects related to cardiovascular challenges and postnatal bone formation. Specifically, giraffe-specific FGFRL1 can prevent or alleviate Ang II–induced hypertension and associated organ damages and may therefore play a key role in the hypertension resistance of giraffes. Our results potentially facilitate the formulation of new strategies for treating or preventing cardiovascular diseases in humans.
In addition to these results, we found evidence of evolution in genes related to sensory perception and behavior. The sensory environment at 5-m elevation differs fundamentally from that experienced by all other pecorans and all other terrestrial mammals (40). It was therefore interesting to note that the giraffe has lost many olfactory-receptor genes while showing positive selection in other genes related to vision, a sensory trade-off otherwise observed in arboreal species (36).
Overall, these results show that pleiotropy is a plausible mechanism for contributing to the suite of co-adaptations necessary in the evolution of the giraffe’s towering stature. However, because of the complexity of cardiovascular and sensory systems, more research on the functional consequences of giraffe-specific genetic variants is needed.
MATERIALS AND METHODS
Genome sequencing and assembly
Procedures applied in sample collection and animal experiments were reviewed and approved by the Institutional Ethics Committee of the Northwestern Polytechnical University and Fourth Military Medical University. Fresh blood samples of a male Rothschild’s giraffe used for genome sequencing were acquired during a routine physical examination at the Guangzhou Zoo in China. High-quality genomic DNA was extracted using a Qiagen DNA purification kit, then used to construct libraries, and sequenced with Illumina HiSeq and Oxford Nanopore GridIOn platforms. Data (199.64 and 140.56 Gb) were obtained, after filtering, from these platforms. In addition, lymphocytes collected from the same blood sample were used for Hi-C library construction, and 138.71-Gb data were obtained using the Illumina HiSeq X Ten platform.
Contigs were assembled by Wtdbg software (v1.2.8) (41), and the assembled contig-level genomes were polished by Racon (v1.2.1) (42) and Pilon (v1.22) (43). Last, the contigs were anchored into chromosomes by Hi-C sequencing reads through the Juicer (version 1.5) (44) and 3D-DNA (version 180922) (45) software workflow. To further improve the chromosome-scale assembly, it was subjected to manual review and refinement using Juicebox Assembly Tools (https://github.com/theaidenlab/juicebox). Last, genome quality was estimated with BUSCO (version 3.0.2) (46), whole-genome synteny with cattle (UMD3.1) genome, and k-mer analysis and by mapping back the initial reads to the assembly.
According to the good genome synteny with cattle genome (Fig. 1B), we assigned the chromosome numbers of our assembly as indicated by previous research (11). Our assembly agrees with the giraffe karyotype revealed before: 13 biarmed autosomal pairs and an acrocentric autosomal pair plus the sex chromosomes (47). Then, we mapped both the Nanopore reads and Illumina reads used for the assembly back onto it. More than 98% of the Nanopore raw reads could be mapped to the assembly properly with an average depth of 54×, and 99.99% of genome has a reads depth more than 50×, with chromosome X excluded (fig. S5 and table S4). Furthermore, 97.14% of the Illumina reads could be mapped to the genome properly with an average depth of 79× (fig. S5 and table S5). Last, the assembly also recovered 96.15% of the expected single-copy orthologous genes according to BUSCO analysis (table S6), the highest coverage yet for the reported Giraffidae genomes (table S7).
Annotation of repetitive sequences and protein-coding genes
Tandem repeats were predicted by Tandem Repeats Finder software (v4.04) (48). RepeatMasker (open-4.0.7) (49), RepeatModeler (v1.0.8) (49), and RepeatProteinMask (v1.0.8) were used together to predict transposable elements. Gene structures were determined by combining ab initio and homology methods. For ab initio annotation, we used Augustus (v3.2.1) (50) and GENSCAN (v1.0) (51) to analyze the repeat-masked genome. For homolog-based annotation, protein sequences of cattle (B. taurus; ensemble 87 release), sheep (Ovis aries; ensemble 87 release), and human (Homo sapiens; ensemble 87 release) genomes were aligned to giraffe sequences using BLAST software (v2.3.0) (52) and GeneWise (v2.4.1) (53). Then, results from the three methods were integrated by EVidenceModeler software (v1.1.1) (54). To annotate the gene functions, the integrated gene set was aligned against public databases, including KEGG, Swiss-Prot, TrEMBL, COG, and NR with BLAST (v2.3.0) (52), and merged with annotations by InterProScan (v4.8) (55) software. The integrity of annotation was estimated by comparison with reference genome annotations and BUSCO (version 3.0.2) (46). On the basis of homology and ab initio gene prediction, we annotated 21,580 protein-coding genes in the genome (fig. S6 and tables S8 to S11), with 96.81% completeness according to BUSCO analysis, suggesting that our annotation also has high quality (table S6).
Taxonomic identification
The complete mitochondrial cytochrome b (Cytb) gene (1140 base pairs) was used to investigate the phylogenetic status of our sample. In addition, previously published cytb sequences of 160 giraffes and outgroup (okapi and pronghorn) were retrieved from the National Center for Biotechnology Information (NCBI) according to the accession number provided by a research before (56). These sequences were aligned with our data using ClustalW in MEGA7 (57) with default parameters and subsequently adjusted manually to maximize positional homology. Last, the remaining sequences were used to infer the phylogenetic tree using IQ-TREE (58) under parameters “-nt AUTO -m MFP -bb 1000 -bnni -o Pronghorn.” As a result, the specimen used for genome sequencing was clustered together with the giraffe subspecies Rothschild’s giraffe (G. camelopardalis rothschildi) with high support (ultra fast bootstrap value = 93).
Chromosome evolution
We reconstructed the ancestral chromosome karyotype of Giraffidae and Bovidae families using the genomes of cattle, goat, giraffe, okapi, and sperm whale (as outgroup). With giraffe as the reference genome, we carried out pairwise alignments with other species as target using LASTZ (v1.1) with parameters “T=2 C=2 H=2000 Y=3400 L=6000 K=2200 --format=axt.” Then, axtChain, chainMergeSort, chainPreNet, and ChainNet were used to generate “chain” and “net” files as input for DESCHRAMBLER (59). Last, we identified 1502 conserved segments by DESCHRAMBLER at a 300-kb resolution and reconstructed 30 predicted ancestral chromosomes (2n = 60) with a total length of ~2.25 Gb.
Identification of PSGs and REGs
To minimize effects of annotation, pseudogenes, and genome quality, we used conserved genome synteny methodology to establish a high-confidence orthologous gene set that included four nonruminants (human, dog, horse, and pig) and six ruminants (pronghorn, giraffe, okapi, forest musk deer, reindeer, and cattle). Using the goat genome sequence (ARS1) as a reference, we performed synteny alignment for these ten species with Last (version 894) (60) and generated pairwise whole-genome alignments with Multiz (version 11.2) (61) using the default parameters. A total of 13,776 genes were extracted from the synteny alignments. We used the Codeml program in the PAML package (version 4.8) (12) to estimate the lineage-specific evolutionary rate for each branch with the phylogenetic extracted from a ruminant study before (9). First, the branch-site model was used for detecting PSGs. The giraffe lineage was specified as the foreground branch, and a likelihood ratio test (LRT) was conducted to examine whether the branch-site model containing positively selected codons (omega > 1) fits better than the null model, which only includes neutral selection or negative selection (omega ≤ 1). The P values for model comparison were computed based on chi-square statistics. Besides, the potential positive selection of codon sites was assessed by their posterior probabilities calculated with the Bayes empirical Bayes (BEB) method. The genes with an LRT P < 0.05 and with the sites with a posterior probability of positive selection over 0.95 from the BEB method were treated as PSGs. Then, the branch model that was used for detecting REGs used the same orthologous genes as above. We tested whether the foreground branch (the giraffe lineage) exhibited a significantly higher omega (regardless of whether it is greater than 1) than the background branch (the other lineages) using the LRT test. The genes with an LRT P < 0.05 were treated as REGs in giraffe. The combined set of PSGs and REGs was subjected to KEGG and GO enrichment analysis (P < 0.05) with the online tool Metascape (v1.0) (62).
Gene family expansion and contraction
We used 12 species to construct gene families, including human, horse, dog, pig, killer whale, camel, pronghorn, giraffe, okapi, white-lipped deer, forest musk deer, and cattle. Proteins with premature stop codons, nontriplet codon lengths, and fewer than 30 amino acids were removed. Last, we used OrthoMCL (v2.0.9) (63) for protein clustering with a dataset of 256,596 protein sequences. Family expansion or contraction analysis was performed by CAFE (v3.1) (64), and the phylogenetic tree was extracted from the ruminant study before (9). Gene expansion and contraction results for each branch of the phylogenetic tree were estimated, and enrichment analysis about the gene families expanded or contracted in giraffe was performed with KOBAS (v3.0) (65).
Identification of rapidly evolving KEGG pathways (66)
For each KEGG pathway with more than 20 genes, we counted numbers of nonsynonymous and synonymous mutations between giraffe and its most recent common ancestor (Nh and Sh, respectively). We also counted numbers of nonsynonymous mutations and synonymous mutations between okapi and the MCRA (Nt and St, respectively). We formulated a null hypothesis that the probabilities of nonsynonymous mutations compare to synonymous mutations in giraffe and okapi are similar and then applied a one-sided binomial test to identify rapidly evolving pathways with significantly more nonsynonymous mutations than expected. The binomial test included three parameters for each KEGG pathway: the number of “successes” (Nh), the number of trials (Nh + Sh), and the hypothetical probability of “success” is given by , where All is the genome-wide value. Last, the rapidly KEGG pathways were identified using a threshold of P < 0.05 (one-sided binomial test).
Identification of unique amino acid substitutions in giraffe
Through analysis of conserved genome synteny with goat, we obtained a highly confident set of orthologous genes of mammals (including mouse, human, cat, cheetah, dog, rhinoceros, horse, camel, pig, dolphin, killer whale, and sperm whale) and 51 ruminant species. Domain regions of the encoded proteins were predicted with Pfam (67). Then, we scanned the domain regions in the syntenic alignments and identified the giraffe-specific amino acid substitutions compared to all other species. The substitutions that were not fixed in all published giraffe genomes were further filtered. Last, we identified 414 giraffe genes that have unique substitutions in domain regions, of which 33 genes have more than three unique substitutions (table S17).
Identification of gene loss
The Illumina short reads of giraffe and okapi were mapped onto the cattle genome (UMD3.1). For every gene, the read depth was counted with SAMtools (68) along the coding sequence (CDS). For the CDS region, if more than 50% of the sites had not been mapped with reads in the giraffe, but if more than 50% sites had been mapped with more than 10 × reads in okapi, the gene was assumed to be specifically lost in giraffe. It turned out that giraffe uniquely lost 83 genes compared to okapi. To avoid sequencing problem in one research and validate the result, we repeated the same analysis with previously published genomic short reads of another giraffe (9), and it showed that giraffe uniquely lost 78 genes with 63 genes overlapping the result before. However, okapi lost only 13 genes uniquely under the same analysis. We noticed that 53 of the 63 genes lost in giraffe were related to the sense of smell and that they located spatially clustered on chromosomes 10 and 15 on cattle genome (table S18). Furthermore, to validate the result on genome level and to avoid the influence of using cattle genome, we checked the synteny alignment between giraffe-goat (ARS1) and okapi-goat (ARS1), and it verified again that giraffe lost more olfactory-receptor genes on chromosomes 10 and 15 of goat (Fig. 4B and fig. S20). We further checked the deletions on giraffe’s chromosome 7 (chromosome 10 of goat) with the long Nanopore reads mapped back to the giraffe genome and goat genome; the deletion region can be finely span in giraffe (fig. S21).
Homology modeling of protein structures
Because the 3D structure of the complexes of FGFR1 (a major FGF receptor and was thought to be involved in FGFRL1 signaling) and FGF23 (the ligand) has been revealed (69), we built an in silico 3D structure model of the giraffe FGFRL1 (from IG-II to IG-III domain) by homolog modeling and docked the model with FGF23 to assess possible effects of the mutations on the proteins’ interaction. The 3D structure model of mtFGFRL1 (seven sites in giraffe type) and WT FGFRL1 (seven sites changed to common type) was separately generated with homolog modeling methods by PROMALS3D (70) with several FGFR structures as templates [Protein Data Bank (PDB) nos. 1E0O, 1EV2, 1II4, 1IIL, 1NUN, 1RY7, 2FDB, 3GRW, 3OJ2, 3OJV, 4J23, and 5W59]. The FGF23 structure was obtained from PDB no. 5w21. The structure of FGFRL1 and FGF23 complexes was produced by the Rosetta (71) protein docking program, using the docking conformation of FGFR1 and FGF23 in PDB no. 5w21 as the initial docking pose.
MD simulation and binding free energy calculation
MD simulations were performed by using the Amber (version 18) software (72) in combination with the ff14SB (version 1.0) force field (73). Protein systems were solvated in the TIP3P water model with an edge distance of 12 Å, and systems were neutralized (pH 7) by adding suitable counterions (Na+ or Cl−). Before performing MD simulations, each system was minimized by means of the steepest descent and conjugate gradient methods through 2000 steps. NPT (constant number of atoms, pressure, and temperature) simulations were then carried out to heat the system from 0 to 300 K using Langevin dynamics for temperature control and the SHAKE algorithm on hydrogen atom constraint. MD simulations were run for 100 ns with the time step set to 2 fs. Last, determination of the relative binding free energy was performed using the molecular mechanics generalized Born surface area method in the Amber package (version 18) (72).
Generation of giraffe-type FGFRL1 mice
To elucidate the giraffe-type FGFRL1 gene’s role in skeletogenesis and the cardiovascular system, the seven unique substitutions in giraffe-type FGFRL1 were introduced into the FGFRL1 gene in mice (giraffe-type FGFRL1 mice) by CRISPR-Cas9–mediated genome editing as follows. First, single-guide RNA (sgRNA) expression constructs were prepared, based on the pUC57-sgRNA expression vector (no. 51132; Addgene), using oligonucleotide sequences listed in table S21. Next, the sgRNA expression plasmids were linearized and prepared as templates for in vitro transcription using a MEGAshortscript kit (Ambion, AM1354). The sgRNA was purified using a MEGAclear kit (Ambion, AM1908). Fertilized eggs were injected with a mixture of Cas9 protein, sgRNAs, and homologous DNA template. Genomic DNA was then extracted from the tails of 7-day-old mice (“new pups”) using phenol-chloroform and recovered by alcohol precipitation to detect the mutations. Polymerase chain reaction primers for targeting sites are listed in table S22. Last, mice with expected mutations were mated with WT mice to get enough heterozygous mutant mice, and then homozygous mutant mice were produced by crossing and prepared for consequence experiments.
Whole-mount skeletal staining of P0 WT FGFRL1 and giraffe-type FGFRL1 mice
Neonates (P0) were subjected to whole-mount skeletal staining. Briefly, both P0 WT FGFRL1 (n = 5) and giraffe-type FGFRL1 (n = 5) mice were fixed in 90% ethanol for 12 hours at 4°C. Next, specimens were transferred into acetone for 12 hours at room temperature and then into a cartilage staining solution containing 0.03% Alcian blue (w/v; Sigma-Aldrich, USA), 80% ethanol, and 20% acetic acid overnight. The samples were washed with 20% acetic acid, and the ossified tissues were stained in a solution with 0.005% Alizarin red (w/v) overnight at 4°C. The specimens were transferred into 1% KOH (w/v) until the muscle tissue was transparent and then saved with 50% glycerol solution containing 1% KOH (w/v). Whole skeleton images were then obtained with an M205 FA stereoscopic microscope (Leica, Germany), and ImageJ software (version 1.46; National Institutes of Health, USA) was used to obtain the following measurements: head length (distance from the frontal tip of the maxilla to the caudal tip of the occipital bone in lateral view), spine length (distance from the annular vertebrae to the tail root), and length of limbs (distance between the two tips of limbs). The measurements were repeated three times for each sample, and average values were obtained.
X-ray imaging of skeletons of adult giraffe-type FGFRL1 mice
Adult (16 weeks, 24 to 26 g) WT FGFRL1 mice (n = 8) and giraffe-type FGFRL1 mice (n = 8) were randomly selected and anesthetized by intraperitoneal 1% (w/v) sodium pentobarbital solution (40 mg/kg). The body weight and length (from nose to tail root) of each mouse was measured. Then, x-ray images of the head, lumbar vertebra, and limbs of both sets of mice (n = 3) were acquired using a SkyScan 1276 high-resolution in vivo x-ray microtomography (Bruker, Germany). Digital images were obtained under identical imaging conditions using the same acquisition parameters, and ImageJ software was used to obtain the following measurements: head length (as defined above), height of the L1 lumbar vertebra (distance between the upper and lower endplates of the vertebral body), and length of limbs (as defined above). The measurements were repeated three times for each sample, and average values were obtained.
Microstructural analysis of skeletons by microCT scanning
After x-ray imaging, the adult WT FGFRL1 mice (n = 10) and giraffe-type FGFRL1 mice (n = 10) were sacrificed by an intraperitoneal pentobarbital (Sigma-Aldrich, USA) overdose. The skeleton of each mouse was harvested and fixed by 4.0% formalin. The formalin-fixed femurs and cervical vertebrae were scanned, reconstructed, and analyzed using a GE-LSP industrial microCT system (GE Healthcare, Chicago, IL, USA) with the following parameters: 80 kV, 80 μA, and 3.0-s exposure time per projection. The BMD, average trabeculae thickness, and BV/TV of the distal femur (n = 6) and C3 vertebra (n = 10) were measured. In addition, the maximum transverse diameter, average thickness of cortical bone, and both inner and outer perimeters of their femurs (at mid-diaphysis) were measured.
Ang II–induced hypertension in mice
Hypertension was induced using Ang II (Sigma-Aldrich, USA) delivered using Alzet-1004 osmotic mini-pumps (Cupertino, CA). Briefly, WT FGFRL1 and giraffe-type FGFRL1 mice (16 weeks old, 24 to 26 g) were anesthetized with isoflurane (1% at 1.5 liters/min oxygen). A 1-cm incision was then made on the back, and an osmotic mini-pump containing Ang II (n = 10) or an equivalent volume of vehicle (saline, n = 10) was embedded. Ang II (900 ng/kg per minute) was infused at a rate of 10 μl/hour for 28 days. At the end of the infusion, the systolic, diastolic, and mean arterial blood pressures were measured using a tail-cuff sphygmomanometer. In addition, cardiac function was evaluated by echocardiography, and hypertension-related cardiac remodeling was examined histologically.
Blood pressure measurement
Blood pressure was measured using a BP2010A intelligent noninvasive sphygmomanometer for mice (Softron, Japan), which was calibrated and validated before recording. The reliability of tail-cuff determination of mouse blood pressure was independently validated by radiotelemetry before making critical assessment in mice. Before measurement, mice were acclimated to a restraint box and tail-cuff inflation in a quiet area with designated temperature (22° ± 2°C) for 5 days. On the day of testing, mice typically remained relatively calm and still in the restrainer after acclimation period. The tail-cuff was positioned at the base of the tail and a heating pad, supplied as an accessory for the tail-cuff sphygmomanometer, and was preheated to 35°C. Blood pressure recordings were acquired after the mice had prewarmed for 10 min. Briefly, the cuff was inflated to 250 mmHg and deflated over 20 s. Ten inflation and deflation cycles were included for each recording. The first three cycles were regarded as “acclimation” cycles and not included in the analysis. The highest and lowest values in the remaining seven cycles were discarded, and the remaining five readings were averaged for a single session value in further analysis. Changes in tail volume were detected by the pressurized receptor when the blood returned to the tail during cuff deflation. Measurements of the mice were obtained for 3 consecutive days before the Ang II or control treatment to obtain their baseline blood pressure.
Echocardiographic measurements of cardiac function in mice
Transthoracic echocardiography was performed to evaluate cardiac function using a Vevo 2100 instrument (VisualSonics, Canada) equipped with a 18- to 38-MHz MS-400 imaging sensor. Briefly, the mice were anesthetized with 1% isoflurane via an anesthetic gas machine and maintained in a supine position with limbs fixed, and body temperature was kept stable through a heat pad, while respiration and heart beats were continuously monitored. M-mode images were analyzed to obtain estimates of LVEF, LVFS, left ventricular posterior wall thickness at end diastole, and left ventricular internal diameter at end diastole. For this analysis, a dedicated software (Vevo 2100 version 1.4, VisualSonics, Canada) was used.
Cardiac and renal histological observation
At the end of Ang II infusion, the heart (n = 6) and kidney (n = 8) of each vehicle- and Ang II–treated mouse were harvested and fixed with 4.0% formalin (Sigma-Aldrich, USA). Histological sections, 5 μm thick, were prepared following standard fixation, clearing, dehydration, waxing, and paraffin-embedding procedures. Representative histological slides were used for histological staining, as follows.
Hematoxylin and eosin staining
The heart and kidney sections were processed by routine dewaxing in xylene followed by hydration with an ethanol concentration gradient. Thereafter, nuclei and cytoplasm in the sections were stained by hematoxylin and eosin (G1004; Servicebio, China), respectively. The sections were then dehydrated, cleared, and mounted. Staining was observed, and images were captured using a BX53+R6 light microscope (Olympus, Tokyo, Japan).
Masson trichrome staining
Heart and kidney tissues were subjected to Masson trichrome staining using a kit and protocols provided by the manufacturer (Sigma-Aldrich, USA). Heart and kidney fibrosis were then measured in terms of the proportion of collagen using ImageJ software. Three randomly selected regions of identical size in each heart or kidney slice were examined, and the average values obtained from them were recorded.
Sirius red staining
The heart sections were stained by incubation in a 0.1% (w/v) solution of Sirius red (G1018; Servicebio, China) in saturated aqueous picric acid for 1 hour. The slides were then washed, dehydrated, and mounted. This treatment stains collagen and noncollagen components red and orange, respectively. Heart fibrosis was measured in terms of the proportion of red-colored collagen using ImageJ software. Three randomly selected regions of identical size in each heart slice were examined, and the average values obtained from them were recorded.
Statistical analysis
Measurements of continuous variables were expressed as means ± SD. All statistical analysis was performed with SPSS software (version 19.0; Chicago, USA). Independent Student’s t tests were used to compare baseline values of WT FGFRL1 and giraffe FGFRL1 mice groups. One-way analysis of variance (ANOVA) was used to compare mean values. If there was a significant overall difference among groups, then Tukey’s post hoc test was used for multiple comparisons between groups. A value of P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank D. Wu for providing the giraffe sample. Funding: This study is supported by the Talents Team Construction Fund of Northwestern Polytechnical University (NWPU) to Q.Q. and W.W., the National Program for Support of Top-notch Young Professionals to Q.Q., the Research Funds for Interdisciplinary Subject, NWPU (19SH030408) to Q.Q., the 1000 Talent Project of Shaanxi Province to Q.Q. and W.W., the National Natural Science Foundation of China (81972052, 81672148, and 81802143) to J.H., and the Independent Research Fund Denmark (8049-00098B) to R.H. Author contributions: Q.Q., J.H., W.W., and R.H. designed this project and research aspects. Zhipeng Li, H.S., G.L., and Q.L. performed sample collection, and D.C. performed sequencing library construction. C.L., L.C., Yuan Yuan, Y.Z., T.Q., M.H., B.Z., Chenglong Zhu, C.Z., and K.W. performed data analysis including genome assembly, annotation, gene family, gene loss, and chromosome evolution. J.G., L.M., and X.C. conducted the experiments for mice. Zihe Li and Y.X. built 3D modeling of proteins. L.Z., Zeshan Lin, Yuan Yin, and W.X. contributed to figure designing. C.L., Q.Q., and J.H. wrote the manuscript. W.W., R.H., and M.T.P.G. performed manuscript amending. Competing interests: J.H., Q.Q., J.G., C.L., and W.W. are inventors on a patent application related to this work filed by the Fourth Military Medical University and the Northwestern Polytechnical University (no. 2020110969712, filed on 14 October 2020). The authors declare no other competing interests. Data and materials availability: All sequencing data and assembled genome have been deposited on the NCBI database with accession ID PRJNA627604. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe9459/DC1
REFERENCES AND NOTES
- 1.Cameron E. Z., du Toit J. T., Winning by a neck: Tall giraffes avoid competing with shorter browsers. Am. Nat. 169, 130–135 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Coimbra J. P., Hart N. S., Collin S. P., Manger P. R., Scene from above: Retinal ganglion cell topography and spatial resolving power in the giraffe (Giraffa camelopardalis). J. Comp. Neurol. 521, 2042–2057 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Mitchell G., Skinner J. D., An allometric analysis of the giraffe cardiovascular system. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 154, 523–529 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Brondum E., Hasenkam J. M., Secher N. H., Bertelsen M. F., Grondahl C., Petersen K. K., Buhl R., Aalkjaer C., Baandrup U., Nygaard H., Smerup M., Stegmann F., Sloth E., Ostergaard K. H., Nissen P., Runge M., Pitsillides K., Wang T., Jugular venous pooling during lowering of the head affects blood pressure of the anesthetized giraffe. Am. J. Physiol. Regul. Integr. Comp. Physiol. 297, R1058–R1065 (2009). [DOI] [PubMed] [Google Scholar]
- 5.More H. L., O’Connor S. M., Brondum E., Wang T., Bertelsen M. F., Grondahl C., Kastberg K., Horlyck A., Funder J., Donelan J. M., Sensorimotor responsiveness and resolution in the giraffe. J. Exp. Biol. 216, 1003–1011 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Endo H., Yamagiwa D., Fujisawa M., Kimura J., Kurohmaru M., Hayashi Y., Modified neck muscular system of the giraffe (Giraffa camelopardalis). Ann. Anat. 179, 481–485 (1997). [DOI] [PubMed] [Google Scholar]
- 7.Agaba M., Ishengoma E., Miller W. C., McGrath B. C., Hudson C. N., Bedoya Reina O. C., Ratan A., Burhans R., Chikhi R., Medvedev P., Praul C. A., Wu-Cavener L., Wood B., Robertson H., Penfold L., Cavener D. R., Giraffe genome sequence reveals clues to its unique morphology and physiology. Nat. Commun. 7, 11519 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mallick S., Gnerre S., Muller P., Reich D., The difficulty of avoiding false positives in genome scans for natural selection. Genome Res. 19, 922–933 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L., Qiu Q., Jiang Y., Wang K., Lin Z., Li Z., Bibi F., Yang Y., Wang J., Nie W., Su W., Liu G., Li Q., Fu W., Pan X., Liu C., Yang J., Zhang C., Yin Y., Wang Y., Zhao Y., Zhang C., Wang Z., Qin Y., Liu W., Wang B., Ren Y., Zhang R., Zeng Y., da Fonseca R. R., Wei B., Li R., Wan W., Zhao R., Zhu W., Wang Y., Duan S., Gao Y., Zhang Y. E., Chen C., Hvilsom C., Epps C. W., Chemnick L. G., Dong Y., Mirarab S., Siegismund H. R., Ryder O. A., Gilbert M. T. P., Lewin H. A., Zhang G., Heller R., Wang W., Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 364, eaav6202 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Eichler E. E., Sankoff D., Structural dynamics of eukaryotic chromosome evolution. Science 301, 793–797 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Cernohorska H., Kubickova S., Kopecna O., Kulemzina A. I., Perelman P. L., Elder F. F. B., Robinson T. J., Graphodatsky A. S., Rubes J., Molecular cytogenetic insights to the phylogenetic affinities of the giraffe (Giraffa camelopardalis) and pronghorn (Antilocapra americana). Chromosome Res. 21, 447–460 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Yang Z., PAML: A program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Gharib W. H., Robinson-Rechavi M., The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC. Mol. Biol. Evol. 30, 1675–1686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catela C., Bilbao-Cortes D., Slonimsky E., Kratsios P., Rosenthal N., Te Welscher P., Multiple congenital malformations of Wolf-Hirschhorn syndrome are recapitulated in Fgfrl1 null mice. Dis. Model. Mech. 2, 283–294 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engbers H., van der Smagt J. J., van’t Slot R., Vermeesch J. R., Hochstenbach R., Poot M., Wolf-Hirschhorn syndrome facial dysmorphic features in a patient with a terminal 4p16.3 deletion telomeric to the WHSCR and WHSCR 2 regions. Eur. J. Hum. Genet. 17, 129–132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pan X., Shao Y., Wu F., Wang Y., Xiong R., Zheng J., Tian H., Wang B., Wang Y., Zhang Y., Han Z., Qu A., Xu H., Lu A., Yang T., Li X., Xu A., Du J., Lin Z., FGF21 prevents angiotensin II-induced hypertension and vascular dysfunction by activation of ACE2/angiotensin-(1-7) axis in mice. Cell Metab. 27, 1323–1337.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Niu T., Liu N., Zhao M., Xie G., Zhang L., Li J., Pei Y. F., Shen H., Fu X., He H., Lu S., Chen X. D., Tan L. J., Yang T. L., Guo Y., Leo P. J., Duncan E. L., Shen J., Guo Y. F., Nicholson G. C., Prince R. L., Eisman J. A., Jones G., Sambrook P. N., Hu X., Das P. M., Tian Q., Zhu X. Z., Papasian C. J., Brown M. A., Uitterlinden A. G., Wang Y. P., Xiang S., Deng H. W., Identification of a novel FGFRL1 MicroRNA target site polymorphism for bone mineral density in meta-analyses of genome-wide association studies. Hum. Mol. Genet. 24, 4710–4727 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rawlinson S. C. F., Murray D. H., Mosley J. R., Wright C. D. P., Bredl J. C., Saxon L. K., Loveridge N., Leterrier C., Constantin P., Farquharson C., Pitsillides A. A., Genetic selection for fast growth generates bone architecture characterised by enhanced periosteal expansion and limited consolidation of the cortices but a diminution in the early responses to mechanical loading. Bone 45, 357–366 (2009). [DOI] [PubMed] [Google Scholar]
- 19.van Schalkwyk O. L., Skinner J. D., Mitchell G., A comparison of the bone density and morphology of giraffe (Giraffa camelopardalis) and buffalo (Syncerus caffer) skeletons. J. Zool. 264, 307–315 (1999). [Google Scholar]
- 20.Damkjaer M., Wang T., Brondum E., Ostergaard K. H., Baandrup U., Horlyck A., Hasenkam J. M., Smerup M., Funder J., Marcussen N., Danielsen C. C., Bertelsen M. F., Grondahl C., Pedersen M., Agger P., Candy G., Aalkjaer C., Bie P., The giraffe kidney tolerates high arterial blood pressure by high renal interstitial pressure and low glomerular filtration rate. Acta Physiol. (Oxf.) 214, 497–510 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Maxwell M. J., Dopheide S. M., Turner S. J., Jackson S. P., Shear induces a unique series of morphological changes in translocating platelets: Effects of morphology on translocation dynamics. Arterioscler. Thromb. Vasc. Biol. 26, 663–669 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Jackson S. F., Schoenwaelder S. M., Type I phosphoinositide 3-kinases: Potential antithrombotic targets? Cell. Mol. Life Sci. 63, 1085–1090 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xu Z., Wu J., Xin J., Feng Y., Hu G., Shen J., Li M., Zhang Y., Xiao H., Wang L., β3-adrenergic receptor activation induces TGFβ1 expression in cardiomyocytes via the PKG/JNK/c-Jun pathway. Biochem. Biophys. Res. Commun. 503, 146–151 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Zhang B., Li M., Wang L., Li C., Lou Y., Liu J., Liu Y., Wang Z., Wen S., The association between the polymorphisms in a sodium channel gene SCN7A and essential hypertension: A case-control study in the Northern Han Chinese. Ann. Hum. Genet. 79, 28–36 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Grassi G., Padmanabhan S., Menni C., Seravalle G., Lee W. K., Bombelli M., Brambilla G., Quarti-Trevano F., Giannattasio C., Cesana G., Dominiczak A., Mancia G., Association between ADRA1A gene and the metabolic syndrome: Candidate genes and functional counterpart in the PAMELA population. J. Hypertens. 29, 1121–1127 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Muszkat M., Kurnik D., Solus J., Sofowora G. G., Xie H.-G., Jiang L., McMunn C., Ihrie P., Harris J. R., Dawson E. P., Williams S. M., Wood A. J. J., Stein C. M., Variation in the alpha2B-adrenergic receptor gene (ADRA2B) and its relationship to vascular response in vivo. Pharmacogenet. Genomics 15, 407–414 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Hara-Chikuma M., Verkman A. S., Aquaporin-1 facilitates epithelial cell migration in kidney proximal tubule. J. Am. Soc. Nephrol. 17, 39–45 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Gordienko D., Povstyan O., Sukhanova K., Raphaël M., Harhun M., Dyskina Y., Lehen’kyi V., Jama A., Lu Z.-L., Skryma R., Prevarskaya N., Impaired P2X signalling pathways in renal microvascular myocytes in genetic hypertension. Cardiovasc. Res. 105, 131–142 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Tian G., Dang C., Lu Z., The change and significance of the Na+-K+-ATPase alpha-subunit in ouabain-hypertensive rats. Hypertens. Res. 24, 729–734 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Ehrlich K. C., Lacey M., Ehrlich M., Tissue-specific epigenetics of atherosclerosis-related ANGPT and ANGPTL genes. Epigenomics 11, 169–186 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H., Cai H., Deng J., Tu X., Sun Y., Huang Z., Ding Z., Dong L., Chen J., Zang Y., Zhang J., TGF-β-mediated upregulation of Sox9 in fibroblast promotes renal fibrosis. Biochim. Biophys. Acta. Mol. Basis Dis. 1864, 520–532 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Williams E. M., Giraffe stature and neck elongation: Vigilance as an evolutionary mechanism. Biology (Basel) 5, 35 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Veilleux C. C., Kirk E. C., Visual acuity in mammals: Effects of eye size and ecology. Brain Behav. Evol. 83, 43–53 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Mathur P., Yang J., Usher syndrome: Hearing loss, retinal degeneration and associated abnormalities. Biochim. Biophys. Acta 1852, 406–420 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Backhaus D., Experimentelle Prüfung des Farbsehvermögens einer Massai-Giraffe (Giraffa camelopardalis tippelskirchi Matschie 1898). Ethology 16, 468–477 (1959). [Google Scholar]
- 36.Nummela S., Pihlstrom H., Puolamaki K., Fortelius M., Hemila S., Reuter T., Exploring the mammalian sensory space: Co-operations and trade-offs among senses. J. Comp. Physiol. A Neuroethol. Sens. Neural Behav. Physiol. 199, 1077–1092 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Siegel J. M., Clues to the functions of mammalian sleep. Nature 437, 1264–1271 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu A. C., Welsh D. K., Ko C. H., Tran H. G., Zhang E. E., Priest A. A., Buhr E. D., Singer O., Meeker K., Verma I. M., Doyle F. J. III, Takahashi J. S., Kay S. A., Intercellular coupling confers robustness against mutations in the SCN circadian clock network. Cell 129, 605–616 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiyashchenko L. I., Mileykovskiy B. Y., Maidment N., Lam H. A., Wu M. F., John J., Peever J., Siegel J. M., Release of hypocretin (orexin) during waking and sleep states. J. Neurosci. 22, 5282–5286 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marten K., Marler P., Sound transmission and its significance for animal vocalization: I. Temperate habitats. Behav. Ecol. Sociobiol. 2, 271–290 (1977). [Google Scholar]
- 41.Ruan J., Li H., Fast and accurate long-read assembly with wtdbg2. Nat. Methods 17, 155–158 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vaser R., Sovic I., Nagarajan N., Sikic M., Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C. A., Zeng Q. D., Wortman J., Young S. K., Earl A. M., Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLOS ONE 9, e112963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durand N. C., Shamim M. S., Machol I., Rao S. S., Huntley M. H., Lander E. S., Aiden E. L., Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dudchenko O., Batra S. S., Omer A. D., Nyquist S. K., Hoeger M., Durand N. C., Shamim M. S., Machol I., Lander E. S., Aiden A. P., Aiden E. L., De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simao F. A., Waterhouse R. M., Ioannidis P., Kriventseva E. V., Zdobnov E. M., BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Huang L., Nesterenko A., Nie W., Wang J., Su W., Graphodatsky A. S., Yang F., Karyotype evolution of giraffes (Giraffa camelopardalis) revealed by cross-species chromosome painting with Chinese muntjac (Muntiacus reevesi) and human (Homo sapiens) paints. Cytogenet. Genome Res. 122, 132–138 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Benson G., Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bedell J. A., Korf I., Gish W., MaskerAid: A performance enhancement to RepeatMasker. Bioinformatics 16, 1040–1041 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Stanke M., Waack S., Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 19, ii215–ii225 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Burge C., Karlin S., Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268, 78–94 (1997). [DOI] [PubMed] [Google Scholar]
- 52.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J., Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 53.Birney E., Clamp M., Durbin R., GeneWise and genomewise. Genome Res. 14, 988–995 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haas B. J., Salzberg S. L., Zhu W., Pertea M., Allen J. E., Orvis J., White O., Buell C. R., Wortman J. R., Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, R7 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zdobnov E. M., Apweiler R., InterProScan—An integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001). [DOI] [PubMed] [Google Scholar]
- 56.Fennessy J., Bidon T., Reuss F., Kumar V., Elkan P., Nilsson M. A., Vamberger M., Fritz U., Janke A., Multi-locus analyses reveal four giraffe species instead of one. Curr. Biol. 26, 2543–2549 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Kumar S., Stecher G., Tamura K., MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Minh B. Q., Schmidt H. A., Chernomor O., Schrempf D., Woodhams M. D., von Haeseler A., Lanfear R., IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J., Farre M., Auvil L., Capitanu B., Larkin D. M., Ma J., Lewin H. A., Reconstruction and evolutionary history of eutherian chromosomes. Proc. Natl. Acad. Sci. U.S.A. 114, E5379–E5388 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kielbasa S. M., Wan R., Sato K., Horton P., Frith M. C., Adaptive seeds tame genomic sequence comparison. Genome Res. 21, 487–493 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Blanchette M., Kent W. J., Riemer C., Elnitski L., Smit A. F., Roskin K. M., Baertsch R., Rosenbloom K., Clawson H., Green E. D., Haussler D., Miller W., Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14, 708–715 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhou Y., Zhou B., Pache L., Chang M., Khodabakhshi A. H., Tanaseichuk O., Benner C., Chanda S. K., Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 10, 1523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li L., Stoeckert C. J. Jr., Roos D. S., OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.De Bie T., Cristianini N., Demuth J. P., Hahn M. W., CAFE: A computational tool for the study of gene family evolution. Bioinformatics 22, 1269–1271 (2006). [DOI] [PubMed] [Google Scholar]
- 65.Xie C., Mao X., Huang J., Ding Y., Wu J., Dong S., Kong L., Gao G., Li C. Y., Wei L., KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 39, W316–W322 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang K., Shen Y., Yang Y., Gan X., Liu G., Hu K., Li Y., Gao Z., Zhu L., Yan G., He L., Shan X., Yang L., Lu S., Zeng H., Pan X., Liu C., Yuan Y., Feng C., Xu W., Zhu C., Xiao W., Dong Y., Wang W., Qiu Q., He S., Morphology and genome of a snailfish from the Mariana Trench provide insights into deep-sea adaptation. Nat. Ecol. Evol. 3, 823–833 (2019). [DOI] [PubMed] [Google Scholar]
- 67.El-Gebali S., Mistry J., Bateman A., Eddy S. R., Luciani A., Potter S. C., Qureshi M., Richardson L. J., Salazar G. A., Smart A., Sonnhammer E. L. L., Hirsh L., Paladin L., Piovesan D., Tosatto S. C. E., Finn R. D., The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome S., The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen G., Liu Y., Goetz R., Fu L., Jayaraman S., Hu M.-C., Moe O. W., Liang G., Li X., Mohammadi M., α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 553, 461–466 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pei J., Tang M., Grishin N. V., PROMALS3D web server for accurate multiple protein sequence and structure alignments. Nucleic Acids Res. 36, W30–W34 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Song Y., DiMaio F., Wang R. Y.-R., Kim D., Miles C., Brunette T., Thompson J., Baker D., High-resolution comparative modeling with RosettaCM. Structure 21, 1735–1742 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Case D. A., Cheatham T. E. III, Darden T., Gohlke H., Luo R., Merz K. M. Jr., Onufriev A., Simmerling C., Wang B., Woods R. J., The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Maier J. A., Martinez C., Kasavajhala K., Wickstrom L., Hauser K. E., Simmerling C., ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Farre M., Li Q., Darolti I., Zhou Y., Damas J., Proskuryakova A. A., Kulemzina A. I., Chemnick L. G., Kim J., Ryder O. A., Ma J., Graphodatsky A. S., Zhang G., Larkin D. M., Lewin H. A., An integrated chromosome-scale genome assembly of the Masai giraffe (Giraffa camelopardalis tippelskirchi). Gigascience 8, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Letunic I., Bork P., 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H., Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 32, 2103–2110 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/12/eabe9459/DC1