

Systematic comparison between two serial crystallography methods shows that both enable time-resolved measurements.
Abstract
For the two proteins myoglobin and fluoroacetate dehalogenase, we present a systematic comparison of crystallographic diffraction data collected by serial femtosecond (SFX) and serial synchrotron crystallography (SSX). To maximize comparability, we used the same batch of micron-sized crystals, the same sample delivery device, and the same data analysis software. Overall figures of merit indicate that the data of both radiation sources are of equivalent quality. For both proteins, reasonable data statistics can be obtained with approximately 5000 room-temperature diffraction images irrespective of the radiation source. The direct comparability of SSX and SFX data indicates that the quality of diffraction data obtained from these samples is linked to the properties of the crystals rather than to the radiation source. Therefore, for other systems with similar properties, time-resolved experiments can be conducted at the radiation source that best matches the desired time resolution.
INTRODUCTION
Structural biology has been highly successful in providing three-dimensional information about the architecture of biomolecules. Usually, these structures display equilibrium state conformations, important snapshots that provide insight into functional states of these biomolecules (1). The advent of x-ray free-electron laser (XFEL) sources promised the possibility to study ultrafast time scales down to the femtosecond domain to watch conformational changes and even chemical reactions (2). Addressing irreversible reactions at these ultrafast time scales and guaranteeing homogeneous reaction initiation required the use of micron-sized protein crystals. However, the extremely intense beam brightness of XFEL sources will destroy these protein microcrystals during a single exposure. This dilemma was circumvented by serial data collection schemes in which thousands of microcrystals are exploited, to each provide a single still diffraction pattern. Because of the diffraction-before-destruction approach, which allows the collection of a useful diffraction pattern by outrunning the onset of damage, it has been argued that serial femtosecond crystallography (SFX) data from XFELs are de facto radiation damage free (3–6).
Soon after the first protein crystal structures were solved from SFX data, the method was adapted for use at synchrotrons, giving rise to serial synchrotron crystallography (SSX) (7, 8). Most SSX experiments thus far involved static structures. However, since most enzymes have median turnover times in the hundreds of milliseconds range, synchrotrons represent a valid alternative for time-resolved experiments. Historically exploited via Laue diffraction on single crystals (9, 10), time-resolved experiments have recently been the subject of serial data collection approaches to study enzymes on time scales from milliseconds to many seconds (11–16). The results of those studies have sparked renewed and expanded interest in serial crystallography methods, and the implementation of time-resolved SSX at a number of synchrotron beamlines can be expected, in particular, as improving synchrotron technology will also bring faster time domains into reach.
Because of the large number of photons compressed into the x-ray laser pulses, SFX exposure times can be very short, down to the femtosecond domain (2, 17). As a consequence of the more limited photon flux at synchrotron sources, accordingly longer exposure times are needed in SSX, and consequently, radiation damage cannot be fully outrun. This is particularly severe for time-resolved experiments, because data need to be collected at room temperature, leading to much increased radiation sensitivity. However, while radiation damage is more pronounced at synchrotron sources, much of its deleterious effects can be mitigated by using low-dose exposure, which still leaves the electron density interpretable with little ambiguity (18, 19). The dose causing a 50% drop in the overall diffraction intensity for lysozyme at room-temperature SSX has been reported to be 380 kilograys (kGy) (20). While this represents the upper dose limit for serial crystallography experiments, which should not be exceeded at room temperature, the onset of site-specific damage could be observed at a much lower dose of approximately 80 kGy (20). Although this value is much lower than doses encountered during typical cryoexperiments, at modern microfocus beamlines, it is still possible to collect multiple frames from a single microcrystal at room temperature within this dose limit (19–21).
For a systematic comparison of SFX and SSX data, we analyzed two different proteins at both radiation sources. This study does not intend to discuss the scope of each type of instrument but to compare their performance in the overlapping micron-sized crystal regime. Smaller crystals are clearly beneficial for reaction initiation in time-resolved experiments, permitting a more homogeneous light activation and faster diffusion rates (22, 23). However, under practical considerations, crystals in the submicron domain display experimental challenges with respect to their generation and identification. Since nanocrystals are smaller than the wavelength of visible light, they escape convenient determination by canonical light microscopy, and alternative methods need to be sought. In addition, Ostwald ripening rather supports the growth of larger particles, which, in practice, favors the growth of crystals in the micron-size regime. At the same time, most enzymes have turnover numbers in the high millisecond time domain, and we have recently demonstrated that these can be effectively addressed both by optical excitation and by in situ mixing reaction initiation in micron-sized crystals (16, 24, 25). As indicated by the crystal sizes used in a number of recent experiments (11–16), practical simplicity suggests that there is a lot of use for crystals in the micron-size regime. This potential could be further substantiated if different dynamic aspects can be synergistically addressed at both synchrotrons and free-electron lasers without the need to change sample conditions. To minimize sources of potential discrepancies, we used the same batch of micron-sized crystals, the same sample delivery device, and the same data analysis software for each of the datasets.
RESULTS AND DISCUSSION
To evaluate potential differences in quality between microcrystal diffraction data obtained from XFEL (SFX) and those from synchrotron sources (SSX), we exploited two well-established protein systems: (i) the relatively radiation-tolerant fluoroacetate dehalogenase (FAcD) and (ii) the highly radiation-sensitive myoglobin (MB). FAcD is a homodimeric protein from the soil bacterium Rhodopseudomonas palustris and one of the very few enzymes that can cleave fluorine carbon bonds. FAcD shows half-of-the-sites reactivity and incorporates a covalent substrate-enzyme intermediate into its SN2 substitution mechanism (16, 26). MB is a monomeric heme protein involved in the oxygen exchange of mammalian muscle tissue. It was the first protein that had its three-dimensional structure determined and is known to be highly radiation sensitive because of the iron center of its heme chromophore (27, 28). Crystallization conditions for both systems were optimized to fall into a comparable low micron-size regime, a regime quite popular in multiple other protein XFEL and SSX studies (8, 29–33). For sample delivery, we made use of our previously described fixed target approach (18, 34–37). Briefly, approximately 25,000 protein microcrystals are mounted on a silicon support chip in random orientations. These chips are then raster-scanned in the x-ray beam by means of a fast and accurate closed-loop piezo translation stage system. This system allows the collection of data from up to four chips (i.e., approximately 100,000 microcrystals) per hour. The modular architecture of this system enables its use at different synchrotron and XFEL end stations (15, 18, 21, 25, 34–37). For the present comparison, data were collected at the European Molecular Biology Laboratory (EMBL) beamline P14 at the PETRA III synchrotron at Deutsches Elektronen-Synchrotron (DESY) and at SPring-8 Angstrom Compact free electron Laser (SACLA) at the RIKEN SPring-8 Center using comparable beam sizes, which are all smaller than the protein crystals. These installations represent third- and fourth-generation light sources, with a difference in peak brightness of 8 to 10 orders of magnitude (38). In our experiment, the pulse duration differed by nine orders of magnitude, but the total number of photons each crystal received differed by less than one order of magnitude. In addition to applying the same data collection hardware, we used protein crystals from the same crystallization batches in both experiments. The microcrystals of both proteins were isomorphous and of the same physical dimensions. This should reduce any crystal quality differences that may influence overall data quality parameters. Moreover, to reduce any further mismatches in the analysis, we processed the data with the same software package, CrystFEL 0.8.0 (39).
Global data quality comparison
The global data quality parameters for both proteins indicate close equivalence of the SSX and SFX data over the whole resolution range (Table 1 and Fig. 1). For both proteins, signal-to-noise ratio (SNR), multiplicity, Rsplit, and overall completeness are almost identical for the SFX and SSX data (Fig. 2). Only minor resolution differences can be observed, as indicated by the resolution shell half-set correlation coefficients (CC1/2), Rsplit, and refinement Rfree values. While the MB dataset shows virtually no differences in B factors between the SSX and SFX dataset, the B factors are slightly lower for the SFX dataset of FAcD (Table 1). This may correlate with the marginally better data statistics in the high-resolution shell of FAcD as made evident by the SNR and Rsplit values. It is conceivable that the FAcD crystals would have diffracted to a slightly higher resolution in the XFEL beam relative to the synchrotron beam as indicated by the B factors. The current resolution cutoff was defined by the detector distance at the XFEL, allowing data collection to a resolution of 1.75 Å at the edge of the detector, which then was matched by setting the detector at the appropriate distance at the synchrotron. It is conceivable that a slightly higher resolution could have been obtained by a closer detector distance; however, the data quality falloff for the SFX and SSX data is highly similar, indicating only a marginal difference between both radiation sources.
Table 1. Data collection and refinement statistics.
Statistics for the highest-resolution shell are shown in parentheses.
| FAcD-SSX | FAcD-SFX | MB-SSX | MB-SFX | |
| Accession code | 7A42 | 7A43 | 7A44 | 7A45 |
| Resolution range | 33.08–1.75 (1.813–1.75) | 33.08–1.75 (1.813–1.75) | 31.47–1.75 (1.813–1.75) | 31.47–1.75 (1.813–1.75) |
| Space group | P21 | P21 | P212121 | P212121 |
| Unit cell | 41.9 79.9 84.8 90 103.4 90 | 41.9 79.9 84.8 90 103.4 90 | 37.9 47.9 83.5 90 90 90 | 37.9 47.9 83.5 90 90 90 |
| Unique reflections | 54827 (5456) | 54828 (5457) | 15863 (1492) | 15891 (1520) |
| Multiplicity | 139.52 (74.0) | 101.92 (66.0) | 114.79 (65.4) | 134.40 (76.2) |
| Completeness (%) | 100.00 (100.00) | 100.00 (100.00) | 99.53 (95.08) | 99.72 (97.19) |
| SNR | 4.18 (1.19) | 3.88 (2.06) | 5.54 (1.62) | 5.65 (1.61) |
| Wilson B factor | 18.73 | 13.83 | 22.21 | 24.83 |
| Rsplit (%) | 19.08 (85.41) | 21.70 (48.78) | 14.27 (77.14) | 13.78 (74.6) |
| CC1/2 (%) | 95.34 (52.52) | 93.10 (72.32) | 97.33 (50.03) | 97.35 (40.54) |
|
Reflections used in refinement |
54827 (4396) | 54828 (4873) | 15866 (1231) | 15332 (1280) |
| Reflections used for Rfree | 1852 (151) | 1838 (172) | 1522 (125) | 1529 (127) |
| Rwork (%) | 15.70 (28.89) | 16.70 (23.32) | 17.30 (27.70) | 16.83 (25.51) |
| Rfree (%) | 19.29 (30.64) | 19.95 (26.05) | 20.99 (32.08) | 20.85 (30.25) |
|
Number of non-hydrogen atoms |
5093 | 5051 | 1371 | 1384 |
| Macromolecules | 4791 | 4743 | 1247 | 1248 |
| Ligands | 2 | 2 | 50 | 50 |
| Solvent | 300 | 306 | 74 | 86 |
| Protein residues | 595 | 595 | 154 | 154 |
| RMS (bonds) | 0.009 | 0.012 | 0.016 | 0.016 |
| RMS (angles) | 1.08 | 1.25 | 1.32 | 1.34 |
| Ramachandran favored (%) | 97.80 | 97.97 | 98.03 | 98.03 |
| Ramachandran allowed (%) | 2.20 | 2.03 | 1.97 | 1.97 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 |
| Rotamer outliers (%) | 0.41 | 0.63 | 1.55 | 1.54 |
| Clashscore | 3.92 | 3.01 | 4.59 | 4.21 |
| Average B factor | 22.87 | 16.53 | 26.50 | 28.98 |
| Macromolecules | 22.28 | 15.87 | 25.97 | 28.32 |
| Ligands | 35.54 | 28.12 | 24.17 | 25.87 |
| Solvent | 32.22 | 26.72 | 37.11 | 40.36 |
Fig. 1. Global data quality parameters as a function of resolution.
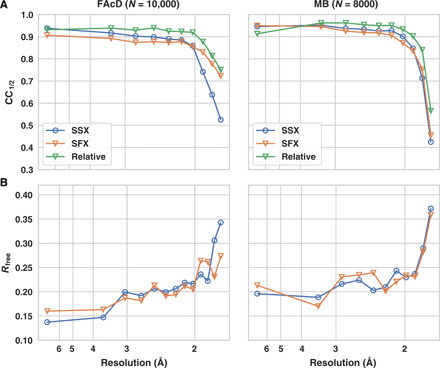
(A) Half-set correlation coefficient for SSX (blue, open circles) and SFX (orange, open triangles) for FAcD and MB datasets. The green curve displays the relative correlation coefficient between the SFX and SSX data. The datasets were limited to the same number of diffraction images (10,000 for FAcD and 8000 for MB), and the same resolution cutoff was applied to SSX and SFX data. (B) Corresponding refinement Rfree values.
Fig. 2. Global data quality parameters as a function of dataset size (number of crystals).
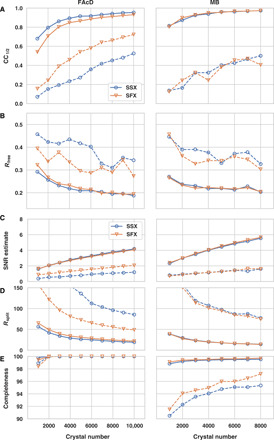
Global data quality parameters for SSX and XFEL datasets: (A) CC1/2 values, (B) Rfree values, (C) SNR estimate, (D) Rsplit values, and (E) completeness. SSX data are displayed as blue, open circles and SFX data as orange, open triangles. Overall values are shown as solid lines and highest-resolution shell values as dashed lines.
In addition to the integration statistics, the general quality of the refined structures needs to be assessed. Globally, this can be achieved by comparing the free R values (Rfree) of the SSX and SFX structures (Table 1). The absolute percentage differences between the overall Rfree values for FAcD and MB are negligible, while for the high-resolution shell, absolute Rfree value differences of ~4.6 and ~1.8% can be observed, respectively. On a per-residue basis, model differences between the SFX and SSX structures can conveniently be assessed via the root mean square deviation (RMSD) and B factor values. The all-atom RMSD of the SFX and SSX structures for FAcD is 0.12 Å, compared to a coordinate error of 0.20 Å. For MB, the all-atom RMSD of the SFX and SSX structures is 0.13 Å, compared to a coordinate error of 0.23 Å. By collecting room-temperature data from the same batch of micron-sized crystals, using the same sample delivery device and the same software for data analysis, we have attempted to maximize the comparability of data collection at XFEL and synchrotron sources, leaving only the photon flux and energy as the distinctive parameters in this experiment. This experimental similarity is reflected in highly comparable data statistics for the SFX and SSX data for both protein systems. The data quality indicators obtained (such as CC1/2, Rfree, and RMSD) are nearly identical for data from both radiation sources. However, thus far, this statement only holds true for crystals of suitable size (i.e., on the order of a few micrometers), which enable data collection at synchrotrons. XFELs can make use of much higher flux densities, thereby accommodating much smaller crystals, down to the nanometer scale, which currently is outside the range of synchrotrons (40). In summary, by normalizing all experimental parameters, this comparison demonstrates that in (static) serial x-ray crystallography, data quality is predominantly a crystal-dependent property. Our observations reveal that neither obtainable resolution nor CC1/2 or Rfree values strongly depend on the radiation source. As a consequence, the diffraction properties of protein microcrystals can be explored at synchrotron sources to test their suitability for XFEL experiments. This would relieve XFEL beamlines of the high burden of screening time and increase opportunities for testing crystals, as beamtime at synchrotrons is much more readily available. This not only includes static data collection but, of course, also extends to time-resolved experiments, where crystals can be used for experiments at synchrotron sources for observing somewhat slower time domains, which, for current generation synchrotrons at monochromatic wavelengths, encompasses time points longer than the mid-microsecond regime.
Scaling with dataset size
Subsequently, we correlated the quality of the datasets with the number of crystals used for structure determination, randomly picking subsets of the total acquired datasets. Analysis of correlation coefficients, Rfree values, SNR, Rsplit, and completeness again shows a high degree of similarity between SSX and SFX data. Although the SFX data were recorded with a bandwidth approximately two orders of magnitude larger than available at monochromatic synchrotron beams, the number of diffraction patterns needed to converge is similar for both datasets. While true Laue serial diffraction data require fewer images for convergence, this criterion is apparently not met by the increased bandwidth of XFEL beams compared to monochromatic synchrotron beams (41).
In agreement with our previous observations, the datasets of both proteins show almost full completeness, which is more than 95% in the highest-resolution shell, even for a low number (<1000) of diffraction patterns (42). This indicates that, for serial data collection, completeness should be the first data quality metric to be assessed before turning to other quality indicators such as CC1/2 or Rsplit. We argue that if the data are not fully complete, then other data quality indicators are probably meaningless. For both FAcD and MB, the overall quality indicators CC1/2 and Rfree start to converge to reasonable levels at approximately 5000 diffraction images by standard assessments (Fig. 2). While there are only minor improvements for the overall data quality indicators, the high-resolution shell quality indicators still undergo substantial improvements if more than 5000 diffraction patterns are included. The FAcD and MB crystals used in this analysis were of monoclinic and orthorhombic symmetry, respectively—representing the two most commonly encountered crystal systems in the Protein Data Bank (PDB). This suggests that, for the vast majority of cases, reasonable data statistics should be achievable with a similar number of diffraction images. For triclinic symmetry (representing less than 5% of the structures in the PDB), however, a higher number of diffraction patterns would be required.
This corroborates our previous findings that protein structures and even protein small-molecule complexes obtained by SFX and SSX can be solved from ~5000 diffraction patterns—or fewer if high symmetry permits—yielding reasonable electron density (42, 43). However, merging and refinement statistics, as well as the quality of electron density, clearly improve if further diffraction patterns are added. This can be especially important in time-resolved analyses to reveal low occupancy details or to distinguish subtle electron density differences from the noise.
Thus, as a rule of thumb, the present data suggest obtaining approximately 5000 diffraction patterns for each structure, as, at this point, the overall dataset quality has mostly converged to reasonable quality indicators. Provided that micron-sized crystals are used, SFX and SSX appear to be equivalent alternatives not only regarding the quality of the data but also with respect to the required number of diffraction patterns. Exciting developments in nanofocus x-ray beamlines combined with the increased brilliance of next-generation synchrotron sources may narrow the gap of useful crystal sizes in the future, thereby also enabling the use of nanocrystals at synchrotrons (44, 45). Since microsecond time resolution can also be obtained at current generation synchrotron sources, time resolutions faster than this regime are a practical long time range limit for SFX experiments, although longer time delays are technically feasible via, e.g., the hit-and-return (HARE) method (15). XFELs can therefore be focused on ultrafast time-resolution experiments, the time domain on which XFELs are essential.
Radiation damage
For traditional crystallographic experiments at synchrotrons, radiation damage is a key limiting factor in obtaining high-resolution structures even at cryogenic temperatures. Damage is caused by the interaction of the x-ray photons with the electrons in the protein crystals and the energy they deposit in the sample, either caused by the photoelectric effect or inelastic scattering (Compton scattering) of x-rays, leading to primary, secondary, and tertiary damage (46, 47). The traditional Garman limit for cryotemperature structures is 30 MGy (48). The situation is worse at room temperature, typically encountered during serial crystallography experiments. Radiation damage manifests either globally as a loss of diffraction intensity or specifically via chemical modification of protein residues, i.e., breaking of disulfide bonds (46, 49). Recently, dose limits for room-temperature SSX were inferred from lysozyme crystals, indicating a half-diffraction dose of 380 kGy and site-specific damage starting to appear at approximately 80 kGy (20). Notably, the authors implied that the onset of site-specific damage may occur at much lower doses for radiation-sensitive proteins. This agrees well with previous findings that the dose limit at room temperature is highly sample dependent, varying over an order of magnitude for 15 different structures (50). In comparing synchrotron and XFEL sources radiation, one needs to further distinguish between direct and indirect damage. While direct damage occurs via the primary x-ray absorption at an atom in the protein, indirect damage occurs via x-ray absorption in the bulk solvent (46). The latter leads to the formation of reactive oxygen species on the nanosecond-millisecond time scale that can diffuse through the crystal (51). Thus, the advantages of XFELs with respect to radiation damage originate in their short pulse lengths, which outrun those damage mechanisms caused by the diffusion of radicals encountered during longer exposure times at synchrotrons (52, 53). Because of the short pulse length (10 fs) and high intensity of the SACLA beam, established dose criteria cannot be applied to SFX data in an unmodified manner. For reference, we therefore used an updated version of RADDOSE (RADDOSE-XFEL), which now also allows to correctly calculate XFEL and SSX doses (54). We estimated the average dose in the exposed region, indicating that the 80-kGy dose limit was exceeded by a factor of ~2 for the SSX data (Table 2).
Table 2. Results from RADDOSE-XFEL.
|
Average diffraction weighted dose (MGy) |
RADDOSE- XFEL average dose–exposed region (MGy) |
RADDOSE-3D style average dose–exposed region (MGy) |
|
| MB-SSX | 0.252 | – | 0.184 |
| MB-SFX | – | 0.200 | – |
| FAcD-SSX | 0.229 | – | 0.167 |
| FAcD-SFX | – | 0.163 | – |
No global structural differences were observed in either FAcD or MB. To address the site-specific differences between the SFX and SSX data, we used a resolution shell scaling tool to calculate isomorphous difference densities (55). The difference density maps obtained this way do not display the absolute damage but rather indicate relative differences between the two datasets (Fig. 3). For FAcD, the difference density peaks are evenly distributed over the whole structure, indicating no preferred site of radiation damage. By contrast for MB, damage is less homogeneously dispersed throughout the structure; rather, local differences are primarily concentrated around certain areas such as the radiation-sensitive heme center. This agrees with canonical radiation damage mechanisms induced by the reduction of the iron center (Fig. 3) (28). While the SSX data are clearly not radiation damage free, the serial data collection approach apparently allows to collect data that show only minimal differences to SFX data, which outrun the most deleterious radiation damage effects, even for very radiation damage–sensitive proteins such as MB. While the present SSX data were collected at doses of approximately 170 kGy, with new rapid acquisition detectors (Eiger), it is possible to collect SSX data with <2-ms exposure time, thus at much lower doses (<5 kGy), further mitigating the differences between the SFX and SSX data (20).
Fig. 3. Isomorphous difference maps for FAcD and MB, respectively.
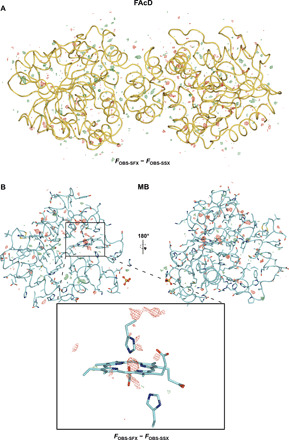
Resolution shell–scaled Fobs-SFX − Fobs-SSX difference maps of FAcD (A) and MB (B). Difference map peaks are homogeneously distributed over FAcD, while difference map peaks are more concentrated around the radiation-sensitive heme center in MB. Proteins are shown as cartoon representations; all maps are shown at ±3 σ.
Data obtained via serial crystallography approaches at synchrotron and XFEL sources can be of equivalent quality. For example, for the samples tested in this study, data quality is a crystal-dependent property and, in first approximation, does not depend on the radiation source.
We conclude that, as a rule of thumb, ~5000 diffraction patterns are required to obtain data statistics and electron density maps of reasonable quality; higher numbers may be required to tease out lowly populated electron density states. Furthermore, our findings suggest that it is possible to tailor SSX data collection to largely avoid the effects of radiation damage, thereby preserving the clean biological interpretability of the results. Because of the overlapping data quality, the choice of the radiation source should primarily be guided by the required time resolution for time-resolved experiments. This study was limited to crystals in the micrometer-size regime and only carried out at two respective radiation sources. Further research with a larger sample pool is required to establish whether our conclusions hold true for a broader spectrum of crystal sizes and SFX data obtained at even brighter radiation sources. However, since other light sources provide similar peak brightness and exposure times, we anticipate that similar results will also be achievable at further instruments, enabling mutually supportive experiments without requiring a change in sample conditions. Experiments with substantially brighter synchrotrons and XFELs, different (more complex) samples, sample delivery systems, timing regimes, detectors, experimental strategies, data analysis methods, etc. are likely to alter such comparisons in the future.
MATERIALS AND METHODS
Protein expression, purification, and crystallization
Recombinant FAcD was purified from Escherichia coli BL21(DE3) as described (26). FAcD was extracted from E. coli cell-free lysate using Ni chromatography with subsequent cleavage of the His6-tag using tobacco etch virus protease. Size exclusion chromatography was completed using 50 mM tris-H2SO4 (pH 8.5) and 150 mM NaCl and buffer-exchanged to remove the NaCl as a final step in purification. FAcD crystals were grown in crystallization buffer [18 to 20% (w/v) polyethylene glycol (PEG) 3350, 200 mM CaCl2, and 100 mM tris-HCl (pH 8.5)]. From these crystals, a microseed stock was generated at a 4 to 8% higher PEG3350 concentration, using a seed bead kit from Hampton Research (HR2-320). Microcrystals were produced using batch crystallization; 100 to 200 μl of seed stock and an equal volume of 0.5 mM FAcD solution were mixed. Within 24 to 72 hours, crystals grew to approximately 20 × 20 × 10 μm3 in size, with a very homogeneous size distribution as determined by light microscopy. CO-bound sperm whale MB was prepared and crystallized as described previously (18, 35). For microcrystallization, 180 to 200 μl of crystallization buffer [50 to 60 mg/ml of MB in 10 mM tris-HCl (pH 9.0) and 2.5 to 2.6 M ammonium sulfate] was added to 3 ml of CO gas–filled Monoject blood collection tubes (Covidien, Mansfield, MA, USA) at 1 atm. To promote nucleation, the solution was seeded with 10 μl of a 1:100 diluted seed solution [crushed MB crystals in 10 mM tris-HCl (pH 9.0) and 3.2 M ammonium sulfate]. To control the size distribution of the final crystals, 180 μl of buffer [10 mM tris-HCl (pH 9.0) and 3.2 mM ammonium sulfate presaturated with CO gas] was added to each tube after the first crystals appeared. MB crystals displayed a larger size distribution and varied from approximately 50 × 50 × 25 to approximately 10 × 10 × 5 μm3 in size. However, most of the crystals had a size of approximately 30 × 30 × 15 μm3. The MB crystals’ size distribution was determined manually when located inside the features of the chip using a Hirox RH-2000 microscope, part of user facilities on site at SACLA (RIKEN, Japan).
SSX data collection
Single-crystalline silicon chips provide a suitable scaffold material for fixed target crystallography. Chip fabrication and sample loading process are described in detail in (35–37). The HARE chips hold randomly oriented crystals in precisely defined, bottomless wells (also called features). Within a humid environment, 100 to 200 μl of a suspension of crystals was loaded onto a single chip by applying vacuum suction of 0.4 mbar for approximately 30 s as described previously. The loaded chips were then covered with 2.5 μm of mylar foil to prevent evaporation, and data were collected immediately after chip loading. SSX diffraction data were collected at room temperature (294 K) at EMBL beamline P14 at DESY, Hamburg, using our previously described fixed-target setup (18, 34). Crystals were not rotated during an exposure; still images were recorded at a wavelength of 0.976 Å (12.703 keV) and an exposure time of 37 ms using an Eiger 16M detector (Dectris, Switzerland). The synchrotron beam had a Gaussian profile with a flux of 4 × 1012 photon/s and full width at half maximum (FWHM) dimensions of 5 × 10 μm2, with a rectangular collimation of 15 × 30 μm2. SSX diffraction data were processed using CrystFEL 0.8.0 (39).
SFX data collection
SFX diffraction data of FAcD and MB were collected at room temperature (294 K) at SACLA at SPring-8, Japan, using the same setup as described above. The uncollimated FWHM size of the beam was 4.7 × 3.4 μm2 with an energy of 10.2 keV, a pulse energy of 0.4 mJ, and an energy distribution of 0.5% (FWHM). The crystals were exposed for 10 fs, with each pulse carrying 2 × 1011 photons. SFX diffraction data were processed using CrystFEL 0.8.0 (39).
Molecular replacement and refinement
The structures were determined by molecular replacement in Phaser (56) using PDB IDs 5K3D and 5JOM as search models for FAcD and MB, respectively. Structure refinement was completed by iterative cycles of refinement in phenix.refine (57) and manual model building in COOT (58, 59).
Dose and radiation damage
Doses for each dataset were calculated using the software RADDOSE-3D (v. 4.0) and its subprogram RADDOSE-XFEL, respectively (54). Cuboid crystal forms with dimensions of 20 × 20 × 10 μm3 for MB and 20 × 20 × 15 μm3 for FAcD were applied. For the SSX data, a Gaussian beam profile with a flux of 4 × 1012 photon/s and a FWHM beam of 5 × 10 μm2, with a rectangular collimation of 15 × 30 μm2, was considered at an energy of 12.703 keV for an exposure time of 37 ms and a rotation wedge of 0°. RADDOSE-3D was run with a resolution of 5 pixels/μm and default angular resolution for a wedge size of 0°. For the XFEL data, a Gaussian beam profile, with an uncollimated FWHM size of 4.7 × 3.4 μm2, an energy of 10.2 keV, a pulse energy of 0.4 mJ, and an energy distribution of 0.5% (FWHM) as well as an exposure time of 10 fs were applied. RADDOSE-XFEL was run with a resolution of 0.5 pixels/μm and simulated 1,000,000 photons. Results from three runs were averaged and are summarized in Table 2. The average dose over the exposed region of the crystal is the correct metric to be used for SSX data, while the average diffraction weighted dose is generally an overestimation in this case. Similarly, RADDOSE-3D style average dose (1 MGy) does not apply for SFX data (20). Isomorphous difference maps were calculated in analogy to a previously published approach scaling the datasets to their resolution shells (55). The SFX datasets were scaled to the reference SSX datasets by dividing the limiting resolution range into 20 equal volume bins containing similar numbers of reflections. The reflection amplitudes in each bin were multiplied by a constant scale factor, calculated to set the average amplitude in each bin equal to that of the reference. The code for shell scaling is part of the Vagabond software suite. The resulting scaled datasets were plugged into sftools (CCP4 software suite), and difference structure factors were calculated Fobs-SFX − Fobs-SSX for both FAcD and MB, respectively (60).
Data analysis
Molecular images were generated in PyMOL (Schrödinger LLC).
Acknowledgments
The SSX data were collected at beamlines P14 operated by EMBL Hamburg at the PETRA III storage ring (DESY, Hamburg, Germany). We thank A. R. Pearson (University of Hamburg), S. Horrell (Diamond Light Source), M. Agthe, and T. R. Schneider (EMBL Hamburg) for assistance in using the beamline and for continuous support and helpful discussions in the implementation and improvement of SSX instruments at EMBL Hamburg. We acknowledge T. White for most helpful discussions on data processing. The XFEL experiments were performed at the BL3 of SACLA with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (proposal nos. 2016A8036 and 2016B8052). We thank the beamline scientists for excellent support. Funding: Support was provided by the Max Planck Society and by the Cluster of Excellence “The Hamburg Centre for Ultrafast Imaging” of the Deutsche Forschungsgemeinschaft (DFG)–EXC 1074–project ID 194651731 (to R.J.D.M.), the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007–2013) under REA grant no. 623994 (to H.M.M.-W.), the Joachim Herz Foundation (Biomedical Physics of Infection) (to E.C.S. and R.J.D.M.), grant RGPIN-2015-04877 from the Natural Sciences and Engineering Research Council of Canada, the Canada Research Chairs Program (to E.F.P.), and the Canada Excellence Research Chairs Program (to O.P.E.). O.P.E. holds the Anne and Max Tanenbaum Chair in Neuroscience at the University of Toronto, and O.P.E. and R.J.D.M. are CIFAR fellows. Author contributions: E.C.S. and P.M. initiated the project. P.M., T.M., and A.K. prepared the protein and crystals. E.C.S., P.M., D.v.S., H.M.M.-W., A.M., H.S., H.M.G., D.A.S., D.A., R.L.O., F.T., A.K., J.E.B., T.M., W.-L.O., O.P.E., E.F.P., and R.J.D.M. collected the data. E.C.S., P.M., R.B., H.M.G., B.T.E., and A.M. processed the data. E.C.S., P.M., R.B., D.v.S., and G.B. analyzed the data. E.C.S., P.M., and R.B. wrote the manuscript. All authors discussed and corrected the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper. Additional data related to this paper may be requested from the authors. The data are available via the RCSB PDB under the accession codes 7A42, 7A43, 7A44, and 7A45.
REFERENCES AND NOTES
- 1.Muench S. P., Antonyuk S. V., Hasnain S. S., The expanding toolkit for structural biology: Synchrotrons. X-ray lasers and cryoEM. IUCrJ 6, 167–177 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chapman H. N., X-ray free-electron lasers for the structure and dynamics of macromolecules. Annu. Rev. Biochem. 88, 35–58 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Neutze R., Wouts R., van der Spoel D., Weckert E., Hajdu J., Potential for biomolecular imaging with femtosecond x-ray pulses. Nature 406, 752–757 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Schlichting I., Serial femtosecond crystallography: The first five years. IUCrJ 2, 246–255 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schlichting I., Miao J., Emerging opportunities in structural biology with x-ray free-electron lasers. Curr. Opin. Struct. Biol. 22, 613–626 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirata K., Shinzawa-Itoh K., Yano N., Takemura S., Kato K., Hatanaka M., Muramoto K., Kawahara T., Tsukihara T., Yamashita E., Tono K., Ueno G., Hikima T., Murakami H., Inubushi Y., Yabashi M., Ishikawa T., Yamamoto M., Ogura T., Sugimoto H., Shen J.-R., Yoshikawa S., Ago H., Determination of damage-free crystal structure of an x-ray–sensitive protein using an XFEL. Nat. Methods 11, 734–736 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Gati C., Bourenkov G., Klinge M., Rehders D., Stellato F., Oberthür D., Yefanov O., Sommer B. P., Mogk S., Duszenko M., Betzel C., Schneider T. R., Chapman H. N., Redecke L., Serial crystallography on in vivo grown microcrystals using synchrotron radiation. IUCrJ 1, 87–94 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stellato F., Oberthür D., Liang M., Bean R., Gati C., Yefanov O., Barty A., Burkhardt A., Fischer P., Galli L., Kirian R. A., Meyer J., Panneerselvam S., Yoon C. H., Chervinskii F., Speller E., White T. A., Betzel C., Meents A., Chapman H. N., Room-temperature macromolecular serial crystallography using synchrotron radiation. IUCrJ 1, 204–212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moffat K., Time-resolved macromolecular crystallography. Annu. Rev. Biophys. Chem. 18, 309–332 (1989). [DOI] [PubMed] [Google Scholar]
- 10.Hajdu J., Acharya K. R., Stuart D. I., McLaughlin P. J., Barford D., Oikonomakos N. G., Klein H., Johnson L. N., Catalysis in the crystal: Synchrotron radiation studies with glycogen phosphorylase b. EMBO J. 6, 539–546 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weinert T., Olieric N., Cheng R., Brünle S., James D., Ozerov D., Gashi D., Vera L., Marsh M., Jaeger K., Dworkowski F., Panepucci E., Basu S., Skopintsev P., Doré A. S., Geng T., Cooke R. M., Liang M., Prota A. E., Panneels V., Nogly P., Ermler U., Schertler G., Hennig M., Steinmetz M. O., Wang M., Standfuss J., Serial millisecond crystallography for routine room-temperature structure determination at synchrotrons. Nat. Commun. 8, 542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin-Garcia J. M., Conrad C. E., Nelson G., Stander N., Zatsepin N. A., Zook J., Zhu L., Geiger J., Chun E., Kissick D., Hilgart M. C., Ogata C., Ishchenko A., Nagaratnam N., Roy-Chowdhury S., Coe J., Subramanian G., Schaffer A., James D., Ketwala G., Venugopalan N., Xu S., Corcoran S., Ferguson D., Weierstall U., Spence J. C. H., Cherezov V., Fromme P., Fischetti R. F., Liu W., Serial millisecond crystallography of membrane and soluble protein microcrystals using synchrotron radiation. IUCrJ 4, 439–454 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beyerlein K. R., Dierksmeyer D., Mariani V., Kuhn M., Sarrou I., Ottaviano A., Awel S., Knoska J., Fuglerud S., Jönsson O., Stern S., Wiedorn M. O., Yefanov O., Adriano L., Bean R., Burkhardt A., Fischer P., Heymann M., Horke D. A., Jungnickel K. E. J., Kovaleva E., Lorbeer O., Metz M., Meyer J., Morgan A., Pande K., Panneerselvam S., Seuring C., Tolstikova A., Lieske J., Aplin S., Roessle M., White T. A., Chapman H. N., Meents A., Oberthuer D., Mix-and-diffuse serial synchrotron crystallography. IUCrJ 4, 769–777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinert T., Skopintsev P., James D., Dworkowski F., Panepucci E., Kekilli D., Furrer A., Brünle S., Mous S., Ozerov D., Nogly P., Wang M., Standfuss J., Proton uptake mechanism in bacteriorhodopsin captured by serial synchrotron crystallography. Science 365, 61–65 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Schulz E. C., Mehrabi P., Müller-Werkmeister H. M., Tellkamp F., Jha A., Stuart W., Persch E., De Gasparo R., Diederich F., Pai E. F., Dwayne Miller R. J., The hit-and-return system enables efficient time-resolved serial synchrotron crystallography. Nat. Methods 15, 901–904 (2018). [DOI] [PubMed] [Google Scholar]
- 16.Mehrabi P., Schulz E. C., Dsouza R., Müller-Werkmeister H. M., Tellkamp F., Dwayne Miller R. J., Pai E. F., Time-resolved crystallography reveals allosteric communication aligned with molecular breathing. Science 365, 1167–1170 (2019). [DOI] [PubMed] [Google Scholar]
- 17.Inoue I., Tamasaku K., Osaka T., Inubushi Y., Yabashi M., Determination of x-ray pulse duration via intensity correlation measurements of x-ray fluorescence. J. Synchrotron Radiat. 26, 2050–2054 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Owen R. L., Axford D., Sherrell D. A., Kuo A., Ernst O. P., Schulz E. C., Dwayne Miller R. J., Mueller-Werkmeister H. M., Low-dose fixed-target serial synchrotron crystallography. Acta Crystallogr. Sect. D Struct. Biol. 73, 373–378 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Appleby M. V., Axford D., Beale J., Moreno-Chicano T., Sherrell D. A., Strange R. W., Hough M. A., Owen R. L., Resolving polymorphs and radiation-driven effects in microcrystals using fixed-target serial synchrotron crystallography. Acta Crystallogr. Sect. D Struct. Biol. 75, 151–159 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de la Mora E., Coquelle N., Bury C. S., Rosenthal M., Holton J. M., Carmichael I., Garman E. F., Burghammer M., Colletier J.-P., Weik M., Radiation damage and dose limits in serial synchrotron crystallography at cryo- and room temperatures. Proc. Natl. Acad. Sci. U.S.A. 117, 4142–4151 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebrahim A., Moreno-Chicano T., Appleby M. V., Chaplin A. K., Beale J. H., Sherrell D. A., Duyvesteyn H. M. E., Owada S., Tono K., Sugimoto H., Strange R. W., Worrall J. A. R., Axford D., Owen R. L., Hough M. A., Dose-resolved serial synchrotron and XFEL structures of radiation-sensitive metalloproteins. IUCrJ 6, 543–551 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schmidt M., Mix and inject: Reaction initiation by diffusion for time-resolved macromolecular crystallography. Adv. Condens. Matter Phys. 2013, 167276 (2013). [Google Scholar]
- 23.Grünbein M. L., Stricker M., Kovacs G. N., Kloos M., Doak R. B., Shoeman R. L., Reinstein J., Lecler S., Haacke S., Schlichting I., Illumination guidelines for ultrafast pump–probe experiments by serial femtosecond crystallography. Nat. Methods 17, 681–684 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Bar-Even A., Noor E., Savir Y., Liebermeister W., Davidi D., Tawfik D. S., Milo R., The moderately efficient enzyme: Evolutionary and physicochemical trends shaping enzyme parameters. Biochemistry 50, 4402–4410 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Mehrabi P., Schulz E. C., Agthe M., Horrell S., Bourenkov G., von Stetten D., Leimkohl J.-P., Schikora H., Schneider T. R., Pearson A. R., Tellkamp F., Miller R. J. D., Liquid application method for time-resolved analyses by serial synchrotron crystallography. Nat. Methods 16, 979–982 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Kim T. H., Mehrabi P., Ren Z., Sljoka A., Ing C., Bezginov A., Ye L., Pomès R., Prosser R. S., Pai E. F., The role of dimer asymmetry and protomer dynamics in enzyme catalysis. Science 355, eaag2355 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Kendrew J. C., Bodo G., Dintzis H. M., Parrish R. G., Wyckoff H., Phillips D. C., A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature 181, 662–666 (1958). [DOI] [PubMed] [Google Scholar]
- 28.C. C. F. Blake, D. C. Phillips, Effects of x-irradiation on single crystals of myoglobin, in Proceedings of the Symposium on the Biological Effects of Ionizing Radiation at the Molecular Level (Int. Atomic Energy Agency, 1962), pp. 183–191. [Google Scholar]
- 29.Lučič M., Svistunenko D. A., Wilson M. T., Chaplin A. K., Davy B., Ebrahim A., Axford D., Tosha T., Sugimoto H., Owada S., Dworkowski F. S. N., Tews I., Owen R. L., Hough M. A., Worrall J. A. R., Serial femtosecond zero dose crystallography captures a water-free distal heme site in a dye-decolourising peroxidase to reveal a catalytic role for an arginine in FeIV=O formation. Angew. Chemie 59, 21656 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wiedorn M. O., Oberthür D., Bean R., Schubert R., Werner N., Abbey B., Aepfelbacher M., Adriano L., Allahgholi A., Al-Qudami N., Andreasson J., Aplin S., Awel S., Ayyer K., Bajt S., Barák I., Bari S., Bielecki J., Botha S., Boukhelef D., Brehm W., Brockhauser S., Cheviakov I., Coleman M. A., Cruz-Mazo F., Danilevski C., Darmanin C., Bruce Doak R., Domaracky M., Dörner K., Du Y., Fangohr H., Fleckenstein H., Frank M., Fromme P., Gañán-Calvo A. M., Gevorkov Y., Giewekemeyer K., Ginn H. M., Graafsma H., Graceffa R., Greiffenberg D., Gumprecht L., Göttlicher P., Hajdu J., Hauf S., Heymann M., Holmes S., Horke D. A., Hunter M. S., Imlau S., Kaukher A., Kim Y., Klyuev A., Knoška J., Kobe B., Kuhn M., Kupitz C., Küpper J., Lahey-Rudolph J. M., Laurus T., Cong K. L., Letrun R., Lourdu Xavier P., Maia L., Maia F. R. N. C., Mariani V., Messerschmidt M., Metz M., Mezza D., Michelat T., Mills G., Monteiro D. C. F., Morgan A., Mühlig K., Munke A., Münnich A., Nette J., Nugent K. A., Nuguid T., Orville A. M., Pandey S., Pena G., Villanueva-Perez P., Poehlsen J., Previtali G., Redecke L., Riekehr W. M., Rohde H., Round A., Safenreiter T., Sarrou I., Sato T., Schmidt M., Schmitt B., Schönherr R., Schulz J., Sellberg J. A., Marvin Seibert M., Seuring C., Shelby M. L., Shoeman R. L., Sikorski M., Silenzi A., Stan C. A., Shi X., Stern S., Sztuk-Dambietz J., Szuba J., Tolstikova A., Trebbin M., Trunk U., Vagovic P., Ve T., Weinhausen B., White T. A., Wrona K., Xu C., Yefanov O., Zatsepin N., Zhang J., Perbandt M., Mancuso A. P., Betzel C., Chapman H., Barty A., Megahertz serial crystallography. Nat. Commun. 9, 4025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grünbein M. L., Bielecki J., Gorel A., Stricker M., Bean R., Cammarata M., Dörner K., Fröhlich L., Hartmann E., Hauf S., Hilpert M., Kim Y., Kloos M., Letrun R., Messerschmidt M., Mills G., Kovacs G. N., Ramilli M., Roome C. M., Sato T., Scholz M., Sliwa M., Sztuk-Dambietz J., Weik M., Weinhausen B., Al-Qudami N., Boukhelef D., Brockhauser S., Ehsan W., Emons M., Esenov S., Fangohr H., Kaukher A., Kluyver T., Lederer M., Maia L., Manetti M., Michelat T., Münnich A., Pallas F., Palmer G., Previtali G., Raab N., Silenzi A., Szuba J., Venkatesan S., Wrona K., Zhu J., Bruce Doak R., Shoeman R. L., Foucar L., Colletier J.-P., Mancuso A. P., Barends T. R. M., Stan C. A., Schlichting I., Megahertz data collection from protein microcrystals at an x-ray free-electron laser. Nat. Commun. 9, 3487 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugahara M., Mizohata E., Nango E., Suzuki M., Tanaka T., Masuda T., Tanaka R., Shimamura T., Tanaka Y., Suno C., Ihara K., Pan D., Kakinouchi K., Sugiyama S., Murata M., Inoue T., Tono K., Song C., Park J., Kameshima T., Hatsui T., Joti Y., Yabashi M., Iwata S., Grease matrix as a versatile carrier of proteins for serial crystallography. Nat. Methods 12, 61–63 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Gisriel C., Coe J., Letrun R., Yefanov O. M., Luna-Chavez C., Stander N. E., Lisova S., Mariani V., Kuhn M., Aplin S., Grant T. D., Dörner K., Sato T., Echelmeier A., Villarreal J. C., Hunter M. S., Wiedorn M. O., Knoska J., Mazalova V., Roy-Chowdhury S., Yang J.-H., Jones A., Bean R., Bielecki J., Kim Y., Mills G., Weinhausen B., Meza J. D., Al-Qudami N., Bajt S., Brehm G., Botha S., Boukhelef D., Brockhauser S., Bruce B. D., Coleman M. A., Danilevski C., Discianno E., Dobson Z., Fangohr H., Martin-Garcia J. M., Gevorkov Y., Hauf S., Hosseinizadeh A., Januschek F., Ketawala G. K., Kupitz C., Maia L., Manetti M., Messerschmidt M., Michelat T., Mondal J., Ourmazd A., Previtali G., Sarrou I., Schön S., Schwander P., Shelby M. L., Silenzi A., Sztuk-Dambietz J., Szuba J., Turcato M., White T. A., Wrona K., Xu C., Abdellatif M. H., Zook J. D., Spence J. C. H., Chapman H. N., Barty A., Kirian R. A., Frank M., Ros A., Schmidt M., Fromme R., Mancuso A. P., Fromme P., Zatsepin N. A., Membrane protein megahertz crystallography at the European XFEL. Nat. Commun. 10, 5021 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sherrell D. A., Foster A. J., Hudson L., Nutter B., O’Hea J., Nelson S., Paré-Labrosse O., Oghbaey S., Dwayne Miller R. J., Owen R. L., A modular and compact portable mini-endstation for high-precision, high-speed fixed target serial crystallography at FEL and synchrotron sources. J. Synchrotron Radiat. 22, 1372–1378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oghbaey S., Sarracini A., Ginn H. M., Pare-Labrosse O., Kuo A., Marx A., Epp S. W., Sherrell D. A., Eger B. T., Zhong Y., Loch R., Mariani V., Alonso-Mori R., Nelson S., Lemke H. T., Owen R. L., Pearson A. R., Stuart D. I., Ernst O. P., Mueller-Werkmeister H. M., D. Miller R. J., Fixed target combined with spectral mapping: Approaching 100% hit rates for serial crystallography. Acta Crystallogr. Sect. D Struct. Biol. 72, 944–955 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mueller C., Marx A., Epp S. W., Zhong Y., Kuo A., Balo A. R., Soman J., Schotte F., Lemke H. T., Owen R. L., Pai E. F., Pearson A. R., Olson J. S., Anfinrud P. A., Ernst O. P., D. Miller R. J., Fixed target matrix for femtosecond time-resolved and in situ serial micro-crystallography. Struct. Dyn. 2, 054302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mehrabi P., Müller-Werkmeister H. M., Leimkohl J.-P., Schikora H., Ninkovic J., Krivokuca S., Andriček L., Epp S. W., Sherrell D., Owen R. L., Pearson A. R., Tellkamp F., Schulz E. C., Dwayne Miller R. J., The HARE chip for efficient time-resolved serial synchrotron crystallography. J. Synchrotron Radiat. 27, 360–370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weckert E., The potential of future light sources to explore the structure and function of matter. IUCrJ 2, 230–245 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.White T. A., Kirian R. A., Martin A. V., Aquila A., Nass K., Barty A., Chapman H. N., CrystFEL: A software suite for snapshot serial crystallography. J. Appl. Cryst. 45, 335–341 (2012). [Google Scholar]
- 40.Chapman H. N., Fromme P., Barty A., White T. A., Kirian R. A., Aquila A., Hunter M. S., Schulz J., De Ponte D. P., Weierstall U., Bruce Doak R., Maia F. R. N. C., Martin A. V., Schlichting I., Lomb L., Coppola N., Shoeman R. L., Epp S. W., Hartmann R., Rolles D., Rudenko A., Foucar L., Kimmel N., Weidenspointner G., Holl P., Liang M., Barthelmess M., Caleman C., Boutet S., Bogan M. J., Krzywinski J., Bostedt C., Bajt S., Gumprecht L., Rudek B., Erk B., Schmidt C., Hömke A., Reich C., Pietschner D., Strüder L., Hauser G., Gorke H., Ullrich J., Herrmann S., Schaller G., Schopper F., Soltau H., Kühnel K.-U., Messerschmidt M., Bozek J. D., Hau-Riege S. P., Frank M., Hampton C. Y., Sierra R. G., Starodub D., Williams G. J., Hajdu J., Timneanu N., M. Seibert M., Andreasson J., Rocker A., Jönsson O., Svenda M., Stern S., Nass K., Andritschke R., Schröter C.-D., Krasniqi F., Bott M., Schmidt K. E., Wang X., Grotjohann I., Holton J. M., Barends T. R. M., Neutze R., Marchesini S., Fromme R., Schorb S., Rupp D., Adolph M., Gorkhover T., Andersson I., Hirsemann H., Potdevin G., Graafsma H., Nilsson B., Spence J. C. H., Femtosecond x-ray protein nanocrystallography. Nature 470, 73–77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meents A., Wiedorn M. O., Srajer V., Henning R., Sarrou I., Bergtholdt J., Barthelmess M., Reinke P. Y. A., Dierksmeyer D., Tolstikova A., Schaible S., Messerschmidt M., Ogata C. M., Kissick D. J., Taft M. H., Manstein D. J., Lieske J., Oberthuer D., Fischetti R. F., Chapman H. N., Pink-beam serial crystallography. Nat. Commun. 8, 1281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moreno-Chicano T., Ebrahim A., Axford D., Appleby M. V., Beale J. H., Chaplin A. K., Duyvesteyn H. M. E., Ghiladi R. A., Owada S., Sherrell D. A., Strange R. W., Sugimoto H., Tono K., Worrall J. A. R., Owen R. L., Hough M. A., High-throughput structures of protein-ligand complexes at room temperature using serial femtosecond crystallography. IUCrJ 6, 1074–1085 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulz E. C., Kaub J., Busse F., Mehrabi P., Müller-Werkmeister H. M., Pai E. F., Robertson W. D., Miller R. J. D., Protein crystals IR laser ablated from aqueous solution at high speed retain their diffractive properties: Applications in high-speed serial crystallography. J. Appl. Cryst. 50, 1773–1781 (2017). [Google Scholar]
- 44.Beale E. V., Warren A. J., Trincão J., Beilsten-Edmands J., Crawshaw A. D., Sutton G., Stuart D., Evans G., Scanning electron microscopy as a method for sample visualization in protein x-ray crystallography. IUCrJ 7, 500–508 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laundy D., Sawhney K., Nistea I., Alcock S. G., Pape I., Sutter J., Alianelli L., Evans G., Development of a multi-lane x-ray mirror providing variable beam sizes. Rev. Sci. Instrum. 87, 051802 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Garman E. F., Radiation damage in macromolecular crystallography: What is it and why should we care? Acta Crystallogr. D Biol. Crystallogr. 66, 339–351 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Henderson R., The potential and limitations of neutrons, electrons and x-rays for atomic-resolution microscopy of unstained biological molecules. Q. Rev. Biophys. 28, 171–193 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Owen R. L., Rudiño-Piñera E., Garman E. F., Experimental determination of the radiation dose limit for cryocooled protein crystals. Proc. Natl. Acad. Sci. U.S.A. 103, 4912–4917 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holton J. M., A beginner’s guide to radiation damage. J. Synchrotron Radiat. 16, 133–142 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Leal R. M. F., Bourenkov G., Russi S., Popov A. N., A survey of global radiation damage to 15 different protein crystal types at room temperature: A new decay model. J. Synchrotron Radiat. 20, 14–22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Le Caer S., Water radiolysis: Influence of oxide surfaces on H2 production under ionizing radiation. Water 3, 235–253 (2011). [Google Scholar]
- 52.Tenboer J., Basu S., Zatsepin N., Pande K., Milathianaki D., Frank M., Hunter M., Boutet S., Williams G. J., Koglin J. E., Oberthuer D., Heymann M., Kupitz C., Conrad C., Coe J., Roy-Chowdhury S., Weierstall U., James D., Wang D., Grant T., Barty A., Yefanov O., Scales J., Gati C., Seuring C., Srajer V., Henning R., Schwander P., Fromme R., Ourmazd A., Moffat K., Van Thor J. J., Spence J. C. H., Fromme P., Chapman H. N., Schmidt M., Time-resolved serial crystallography captures high-resolution intermediates of photoactive yellow protein. Science 346, 1242–1246 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nass K., Radiation damage in protein crystallography at x-ray free-electron lasers. Acta Crystallogr. Sect. D Struct. Biol. D75, 211–218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dickerson J. L., McCubbin P. T. N., Garman E. F., RADDOSE-XFEL: Femtosecond time-resolved dose estimates for macromolecular x-ray free-electron laser experiments. J. Appl. Cryst. 53, 549–560 (2020). [Google Scholar]
- 55.Ginn H. M., Stuart D. I., Recovery of data from perfectly twinned virus crystals revisited. Sect. D Struct. Biol. 72, 817–822 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L.-W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H., PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G. W., Coy A. M., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]