

Abstract
Background:
Evidence for a role of human gut microbiota in multiple sclerosis (MS) risk is mounting, yet large variability is seen across studies. This owes, in part, to the lack of standardization of study protocols, sample collection methods and sequencing approaches.
Objective:
This study aims to address the effect of a household experimental design, sample collection and sequencing approaches in a gut microbiome study in MS subjects from a multi-city study population.
Methods:
We analyzed 128 MS patient and cohabiting healthy control pairs from the International Multiple Sclerosis Microbiome Study (iMSMS). A total of 1,005 snap frozen or desiccated Q-tip stool samples were collected and evaluated using 16S and shallow whole metagenome shotgun sequencing.
Results:
The intra-individual variance observed by different collection strategies was dramatically lower than inter-individual variance. Shallow shotgun highly correlated with 16S sequencing. Participant house and recruitment site accounted for the two largest sources of microbial variance, while higher microbial similarity was seen in household-matched participants as hypothesized. A significant proportion of the variance in dietary intake was also dominated by geographic distance.
Conclusion:
A household-pair study largely overcomes common inherent limitations and increases statistical power in population-based microbiome studies.
Keywords: multiple sclerosis, gut microbiome, 16S rRNA sequencing, shallow whole metagenome sequencing, diet
Introduction
Although genetic factors can account for a significant proportion of susceptibility to multiple sclerosis (MS), almost two thirds of monozygotic twins are discordant, suggesting a major role of the environment1. Among the environmental factors, low vitamin D, Epstein-Barr virus (EBV) infection, smoking and adiposity have been consistently reported2. Yet, additional environmental factors are expected to influence the onset and or perpetuation of MS. The gut microbiome has recently emerged as a potentially critical interface between environmental exposures and autoimmunity that may modulate both risk and phenotype across a number of diseases including MS3, 4. The composition of the gut microbiota was recently shown to differ between people with MS and healthy controls5–7. Furthermore, the observed differences appear to be of functional importance, as transplanting human MS microbiota into germ free mice results in exacerbation of symptoms in a murine model of the disease5.
Despite initial evidence for a role of gut microbiota in MS risk, few studies have been performed to date, and results are not always concordant. Several causes can be cited for the observed lack of replication. First, small sample size (ranging from a few dozen to a little over a hundred) and patient heterogeneity (disease sub-types, diet, medication use, etc). In addition, diverse experimental designs including the choice of a control population, sample collection strategies and sequencing techniques could also account for the observed heterogeneity.
The International MS Microbiome Study (iMSMS) is a global effort to collect biospecimens (blood and stool) and investigate how gut microbiota of individual patients impacts MS susceptibility, progression, and response to treatment. A large-scale study like this requires rigorous experimental design, assessment of existing technologies, and application of appropriate statistical methods for data interpretation.
Here, we present a pilot microbiome analysis of 128 household pairs (256 samples) of the iMSMS with a goal of addressing some of the challenges posed by large-scale microbiome research and validate the current experimental design. This analysis focused primarily on technical and computational work needed to accurately characterize microbiome structure with respect to the impact of study design, the environmental and dietary factors that most greatly influence variability in microbiome composition. Such knowledge is critical to ensure that appropriate control for these factors is achieved either during future analyses or by amendment to the experimental design for future sample capture.
Materials and methods
Recruitment
Participants were recruited for the study through multiple sclerosis clinics at UCSF (San Francisco, CA), Brigham and Women’s Hospital (Bostin, MA), Mount Sinai (New York, NY), the Anne Rowling Clinic (Edinburgh, UK), and FLENI (Buenos Aires, Argentina) and by self-referral through the iMSMS website (imsms.org). Each collaborating site obtained human subject research approval through their respective ethics review committees, following a master protocol established at UCSF (protocol no. 15–17061). Sample collection procedures were performed as specified by the protocol. All participants provided written informed consent and also signed a HIPAA Authorization that allows for use of their medical record for research purposes.
To be eligible, participants must carry a diagnosis of multiple sclerosis8; be of White (Hispanic or non-Hispanic) ethnicity (i.e. to match characteristic genetic risk profile of MS9); and be enrolled with a genetically unrelated household healthy control (HHC) that cohabitated for at least six months. MS and control subjects must have been free of other autoimmune disorders (excluding the patient’s MS), gastrointestinal infections, and other neurological disorders. Participants were excluded if they had been on oral antibiotics within the past three months, had received corticosteroids within the past 30 days, or were on a disease modifying therapy for less than three months. Participants were provided with a stool sample collection kit, and instructed to obtain two consecutive stool samples in the privacy of their own homes. Each stool sample time point included 3 collection vials - a Q-tip (Q, dry), a snap frozen vial (S, wet), and a vial filled with lysogeny broth (LB) and glycerol. Participants were instructed to freeze the samples for at least 12 hours, and ship them frozen. Samples were returned to each site via overnight shipping.
All data were collected and stored through secure REDCap questionnaires and a validated Block 2005 food frequency questionnaire (FFQ)10, which was set up through an external vendor (NutritionQuest). An analysis of nutritional intake was performed by NutritionQuest in a standardized fashion for all participants based on their responses to the FFQ.
Dietary analysis
37 items were summarized from the FFQ and grouped as antioxidants, average intake, B-vitamins, food group servings and minerals. Dietary dissimilarity was measured using Jaccard distance. The effect of confounders on the variation of diet and the effect of dietary items (covariates) on the variation of gut microbiome were accessed by PERMANOVA (Permutational multivariate analysis of variance)11. The test was performed by using the “adonis” function implemented in R package “vegan”12. The permuted P-value was obtained by running 999 permutations. Redundancy analysis (RDA) between microbial composition and dietary items was performed by using “rda” function implemented in R package “vegan”12.
Sample preparation for sequencing
Stool samples collected from each recruiting site were shipped in dry ice to the central processing lab at the University of California, San Francisco (UCSF) and immediately stored at −80° C. Bacterial DNA was obtained for each sample at UCSF and then shipped to the University of California San Diego, Institute for Genomic Medicine for sequencing. Q-tip samples (i.e. dry) and snap frozen (i.e. wet) samples were processed using the QIAamp PowerFecal DNA Kit (ref 12830–50). After lysis solution was added to bead beating tubes, dry samples were transferred by grinding the Q-tips into the bottom while snap frozen samples were chipped to an appropriate size for the kit. Sample processing was done on a QIAcube platform according to the protocols generated by the manufacturer. DNA sample quantity and purity were measured by NanoDrop spectrophotometry.
16S rRNA sequencing and data analysis
The V4 region of the bacteria 16S ribosomal RNA gene was amplified on an Illumina MiSeq platform using the Earth Microbiome Project protocol13. Amplicon reads were analyzed by QIIME14 to join the forward and reverse reads, trim short reads in length less than 250bp and assign filtered reads to OTUs using a closed-reference OTU picking protocol against Greengenes database (version 13.8) at 97% identity. Taxonomy was assigned to each read by accepting the Greengenes taxonomy string of the best matching Greengenes sequence. Samples were filtered to at least 10,000 sequences per sample, and OTUs were filtered to retain only OTUs present in at least 5% of samples, covering at least 500 total reads. OTU abundance for each individual was averaged from all corresponding Q-tip and snap frozen samples. 128 paired household MS and healthy controls were counted for downstream analyses.
Microbial diversity
The OTU abundance table generated in the previous step was rarefied to 10,000 sequences per sample. α-diversity was measured by Shannon15 and Chao116 indexes. Both weighted and unweighted UniFrac17 distances were computed between all samples, and principal coordinates analysis (PCoA) was applied to visualize the β-diversity. All these analyses were performed with QIIME. Bray-Curtis18 dissimilarities were calculated to compare gut microbiome among individuals in terms of geographic distance. Microbiome dissimilarity between each pair within the same recruitment site was compared with that between two individuals from different recruitment sites by controlling in same disease status. Since the MS and control subjects within household are often of different sex, the random comparisons between households utilized only heterosexual comparisons to control for the sex effect. Statistical significance was determined by ANOVA.
Variance of gut microbiome given by confounding factors
The PERMANOVA11 was used to assess the effect of host metadata categories (confounders): demography, lifestyle, diseases, medication and physiology, on the variation of microbiome abundance. The test was performed by using the “adonis” function with default method “bray” distance implemented in R package “vegan”12. The permuted P-value was obtained by running 999 permutations.
Power and sample-size estimation.
Effect size (corrected coefficient of determination ω2) and power calculation for the permutational multivariate analysis of variance test based on Bray-Curtis distances were performed using the R micropower package19. The power was estimated by bootstrapping the distance matrices 100 times with different sample sizes ranging from 2 to maximum samples in each group.
Shallow whole metagenome shotgun sequencing (WMGS) and data processing
1 ng of input DNA was used in a 1:10 miniaturized Kapa HyperPlus protocol. For samples with less than 1 ng DNA, a maximum volume of 3.5 μl input was used. Library concentration was determined with triplicate readings of the Kapa Illumina Library Quantification Kit; 20 fmol of sample libraries were pooled and size selected for fragments between 300 and 800 bp on the Sage Science PippinHT to exclude primer dimers. The pooled library was sequenced as a paired-end 150-cycle run on an Illumina HiSeq2500 v2 in Rapid Run mode at the UCSD IGM Genomics Center.
Raw Fastq files were processed using Atropos v1.1.1820 to remove adapters and filter reads with lower quality score than 15 and length less than 100 base pairs. Putative human genome contaminations were identified and removed by using Bowtie 2 v2.2.321 with the “--very-sensitive” option against the human reference genome GRCh37/hg19. Functional and taxonomic predictions of processed sequences were performed by SHOGUN in Qiita22, 23 with parameters “rep82_bowtie2” to align forward reads to representative bacterial genomes from the reference database RefSeq version number 82. 5% samples with the lowest shotgun sequencing library size were excluded from the analysis. Species classified were filtered by the criteria that present in at least 5% of samples. The correlation of phylum/genus abundance characterized by 16S rRNA and WMGS was measured by Pearson correlation.
Availability of data and materials
The datasets generated and analyzed during the current study are available in the EMBL-ENA repository (https://www.ebi.ac.uk) with accession number ERP115476.
Results
The iMSMS recruited 128 pairs of MS patients and their household healthy controls (HHC) between September 2015 and October 2016 from five sites (recruiting centers) located in San Francisco, Boston, New York, Buenos Aires and Edinburgh (Figure 1, Table 1, Datasets S1). In our paired household design, cases and controls (typically spouses) are of similar age, while as a consequence of the uneven sex distribution of MS, 71% of the MS participants were female, compared with 37.5% of controls (Table 1). The median disease duration of MS was 9.5 years (IQR: 4.75–17 years) and duration of cohabitation was 15 years (IQR: 8–27.5years). MS patients with disease courses of relapsing-remitting MS (RRMS), secondary progressive MS (SPMS) and primary progressive (PPMS) were included. As a natural consequence of the disease process, patients with progressive disease (SPMS and PPMS) had relatively higher Expanded Disability Status Scale (EDSS) scores, disease duration and cohabitation time with HHC than those with RRMS (Table 1, Figure S1). Given the small numbers of SP and PP patients, both groups were combined into progressive MS (PMS) for subsequent analyses.
Figure 1.

Study summary. (A) Geographic distribution of samples collected from five recruiting centers, and the percentile of each disease course among all samples. (B) Workflow of microbiome study in MS patients and household healthy controls (HHC).
Table 1.
Participants’ characteristics
| HHC | MS | RRMS | SPMS | PPMS | |
|---|---|---|---|---|---|
| Number | 128 | 128 | 99 (77%) | 12(9%) | 17(14%) |
| Age (y) | 47.5 (38–57.75) | 45(37–55) | 41(35–51.75) | 56.5(53.25–62) | 56(50–59) |
| Female (%) | 37.5% | 71.1% | 70% | 91.7% | 64.7% |
| EDSS | n/a | 2(0.75–4) | 1.5(0–3) | 6(4.75–6.5) | 4(3–6.5) |
| Disease duration (y) | n/a | 9.5(4.25–17) | 9(4–15) | 21.5(13.25–30.75) | 10(6–11) |
| Cohabitation (y) | 15 (8–27.5) | 15(8–27.5) | 12(6–21) | 32.5(28.5–39.75) | 27(18.5–33.25) |
Data are presented as median (interquartile range, IQR); y, year; n/a, data not available; EDSS, Expanded Disability Status Scale.
Patient demographics, disease status, medication, lifestyle and physiology data were summarized in Datasets S2. A summary of dietary questionnaires and the dietary intakes, including average intake, food group servings, antioxidants, minerals and vitamins et al. was provided in Datasets S3.
Inter-individual outweigh intra-individual differences in the microbiome
1005 stool samples were collected from the 256 participants by rubbing Q-tip (Q, dry) and snap freezing (S, wet) methods in two consecutive days (time point 1 and 2 when possible) (Figure 1B).
A total of 1206 microbial OTUs were identified from 867 samples that were qualified the sequencing depth (at least 10,000 reads per sample) by 16S rRNA sequencing (Datasets S4). The impact of the sample collection method on the microbial composition and diversity was first accessed. No significant difference of microbiome α-diversity (Shannon index) was observed between collection methods or across both time points (Figure 2A). A statistically significant but small difference in beta-diversity was observed by collection method (Q1 vs S1, Q2 vs S2) but not by time (Q1 vs Q2, S1 vs S2), however, the microbiome variability explained by sample collection was dramatically lower (Figure 2B, PERMANOVA R2= 0.0102). Indeed, the microbiome dissimilarity observed by collection method (Q-S) didn’t differ from the one observed by time (T1-T2) (Figure 2C). Samples collected by different methods over time (QT-ST) showed larger dissimilarity than each alone (Figure 2C); however, this intra-individual difference is almost negligible compared to the one measured across individuals (Figure 2D). The high intra-individual microbiome similarity opens the possibility of averaging microbiome abundance of samples from each individual and suggests that either collection methods, snap freezing or Q-tip, can be selected.
Figure 2.
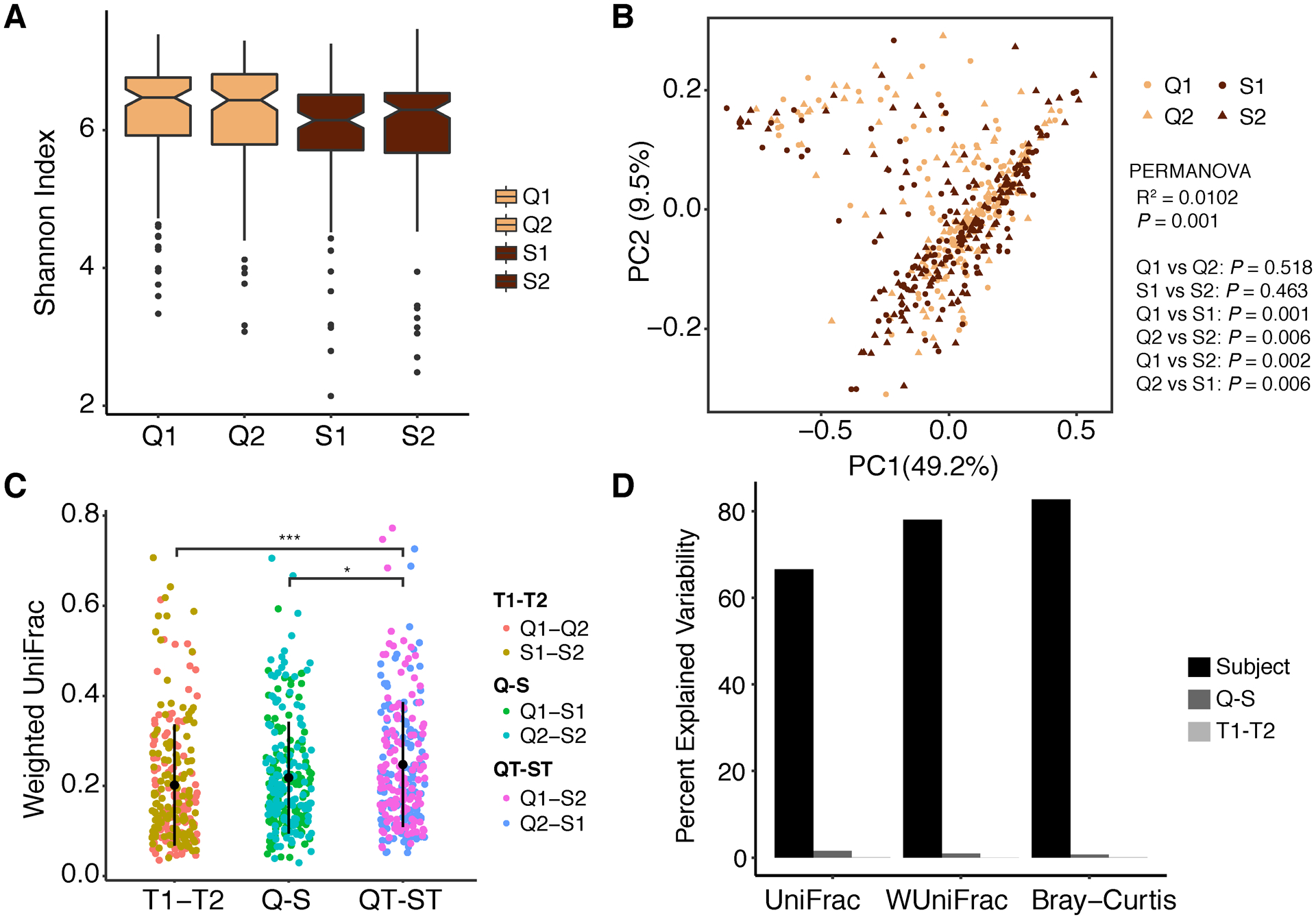
Intra-individual diversity. (A) Microbiome α-diversity measured by Shannon index was compared by stool collection method (Q, S) across consecutive two (1,2) days (ANOVA, not significant). (B) Principal Coordinates Analysis (PCoA) of weighted UniFrac distance compared by collection methods and times. Statistical significance was determined by PERMANOVA, multiple testing corrected P value. (C) Microbial dissimilarity (weighted UniFrac) compared between samples by controlling collection method, time, or cross methods and times (ANOVA, multiple testing corrected P value, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). (D) Percent explained variability (PERMANOVA R2) by each variable of participating subject, collection method and collection time. S, wet samples by snap freezing; Q, dry samples by Q-tip; T1, T2, sample collection time point 1 and continuous time point 2, respectively; S1, S2, S samples collected at time point 1 and 2, respectively; Q1, Q2, Q samples collected at time point 1 and 2, respectively.
Gut microbiome quantified by shallow WMGS sequencing
16S rRNA sequencing is widely used as the primary approach for bacterial classification and abundance estimation, but this approach which based on targeted sequencing of a short region (hypervariable region) of the 16S rRNA gene, is limited by its low resolution to resolve organisms on strain or species level24. In contrast, whole-genome (shotgun) metagenomic sequencing can provide higher resolution in identifying lower taxa but it is more costly, particularly for large-scale studies25. Shallow shotgun sequencing with as little as 0.5 million sequences per sample has been proposed as a cost-effective alternative to 16S rRNA sequencing22. We therefore applied shallow shotgun sequencing on 1005 stool samples.
Samples were sequenced at an average depth of 591,648 (±10,158) paired-end non-human genome reads, classified in 2146 species, among which 596 remained after filtering the less present and abundant species (Datasets S5). Even if the number of species is reduced after removing rare taxa (see Methods), this number is still dramatically higher than the species identified by 16S rRNA sequencing (Figure S2, Datasets S6). Thus, shallow WMGS offers a higher resolution of species classification, increasing the potential to discover new associations in a particular study.
A similar number of higher taxa (phyla, classes and orders) were identified by 16S and shallow shotgun sequencing. The microbe composition measured by the two methods is shown in Figure 3. Figure 3A shows the composition shift of phyla (up) and genera (down) among RRMS, PMS and HHC groups. 100% of phyla (n=10) identified by 16S rRNA sequencing were also identified by WMGS with similar structure of Proteobacteria, Verrucomicrobia and Actinobacteria, while detecting a decrease in Firmicutes and an increase in Bacteroidetes. As reported in other studies, these differences are likely due to the fact that more bacteroides species can be resolved using WMGS (Figure 3) and that the sequencing target genes and reference databases used for classification are different25. However, when specifically compared, a high correlation was detected between 16S rRNA and corresponding WMGS at both phylum and genus level (Figure 3B), confirming shallow shotgun sequencing is a cost-effective alternative of 16S to study the microbiome on large populations.
Figure 3.
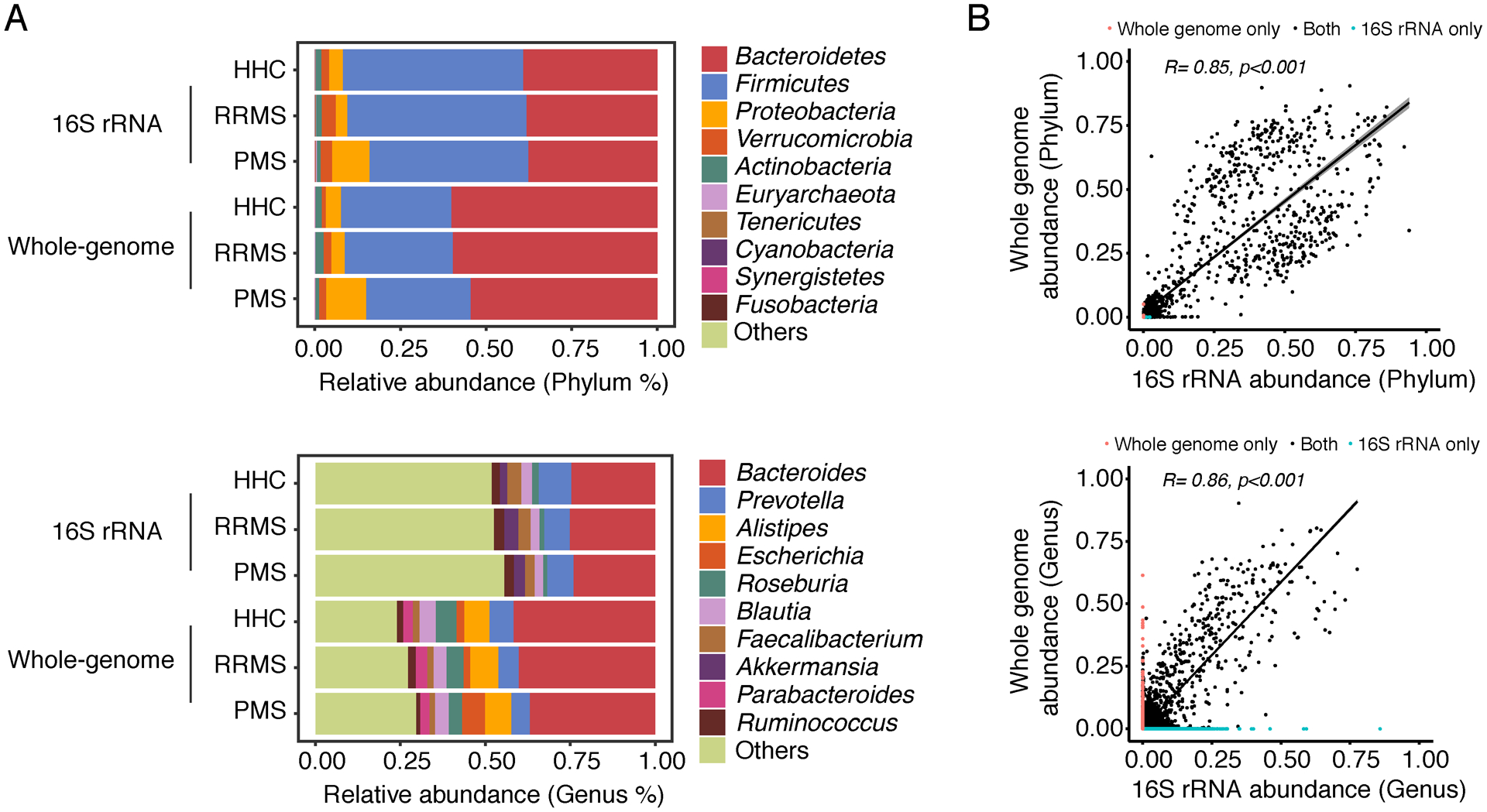
Microbial community compared between 16S rRNA and shallow whole-metagenome sequencing techniques (WMGS). (A) Average relative abundance of top 10 most abundant bacteria (phylum and genus level from top to bottom) in healthy control, RRMS and progressive MS. (B) Pearson’s correlation of bacteria (phylum and genus level from top to bottom) classified by 16S rRNA (x-axis) and shallow WMGS (y-axis).
We next investigated how microbiome structure is impacted by an array of confounding factors.
The effect of co-habitation on gut microbiome variability
The abundance of 1206 OTUs (classified in 95 genera) by 16S rRNA sequencing, were averaged from snap freezing and Q-tip samples to individual data (Datasets S7), and analyzed for the inter-individual variability. Geography-specific microbial divergences were observed, as participants from different sites (recruiting centers) differed more than participants from the same site within the same disease status (Figure 4A) or disease course (Figure S3A). Furthermore, MS and healthy participants within the same house shared more gut microbiota than those from different houses at the same site (Figure 4B, Figure S3B). An even greater microbial dissimilarity was observed between MS and HHC from different sites in different houses (Figure 4B, Figure S3B).
Figure 4.
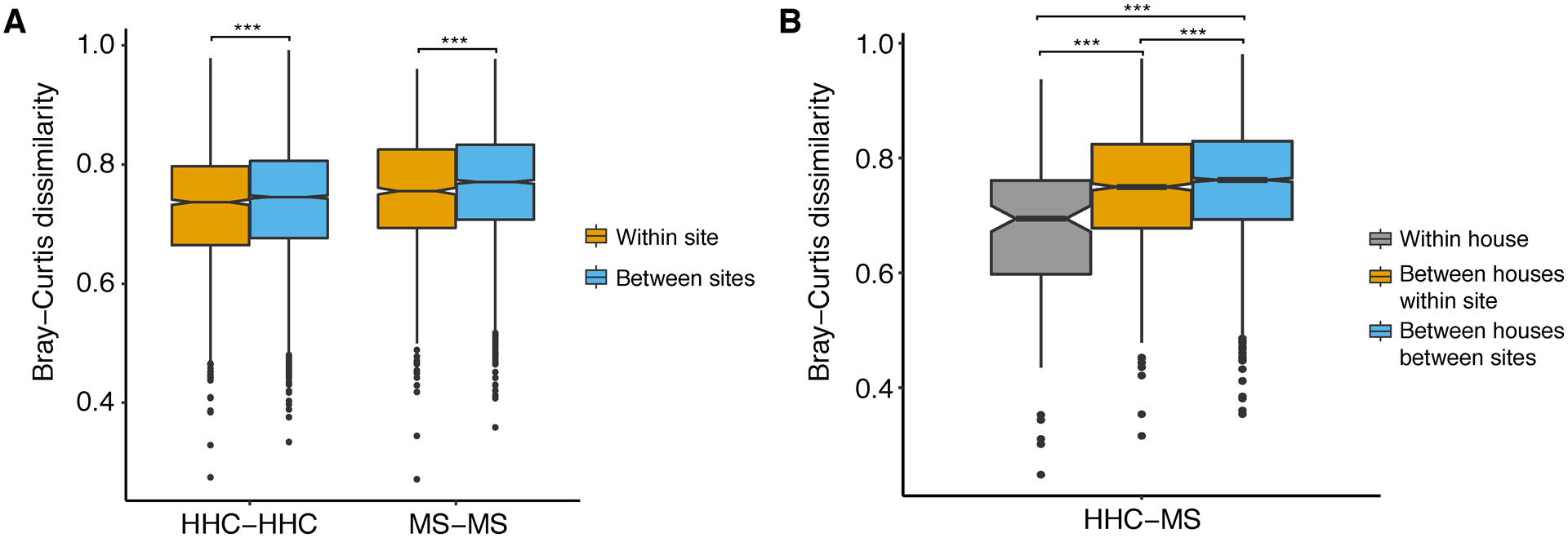
Microbial dissimilarity by geography. (A) Bray-Curtis dissimilarity of microbes measured between individuals in the same disease status within the same site (recruiting center) and in different sites. (B) Bray-Curtis dissimilarity of microbes measured between healthy control and MS within the same house, between different houses in the same time and between different houses in different sites. Random comparisons of healthy control and MS were female-male matched only to control sex effect. Statistical significance was determined by ANOVA (multiple testing corrected P value, ***P ≤ 0.001).
Our data suggest that the household case-control pair design indeed minimizes microbial differences by exposed environment, thus will potentially enhance the power to detect true differences between cases and controls collected from multi-cities.
Additional confounding factors affecting the gut microbiome
To understand how microbial composition differs between patients and household healthy controls, we compared the diversity of microbe communities characterized by 16S rRNA sequencing within (α-diversity) and between individuals (β-diversity).
Unsurprisingly, due to the small sample size, we found no significant difference in α-diversity between MS and HHC groups as measured by Shannon and Chao1 index (Figure 5A). β-diversity based sample clustering did not reveal major difference in disease status (Figure 5B, PEARMANOVA, P > 0.05), consistent with our earlier studies that no major global shift in bacterial community was observed between MS and control1, 5. A trend towards a decrease in microbiome diversity was observed in PMS compared to RRMS or HHC, but these differences were not significant (Figure S4A–B).
Figure 5.
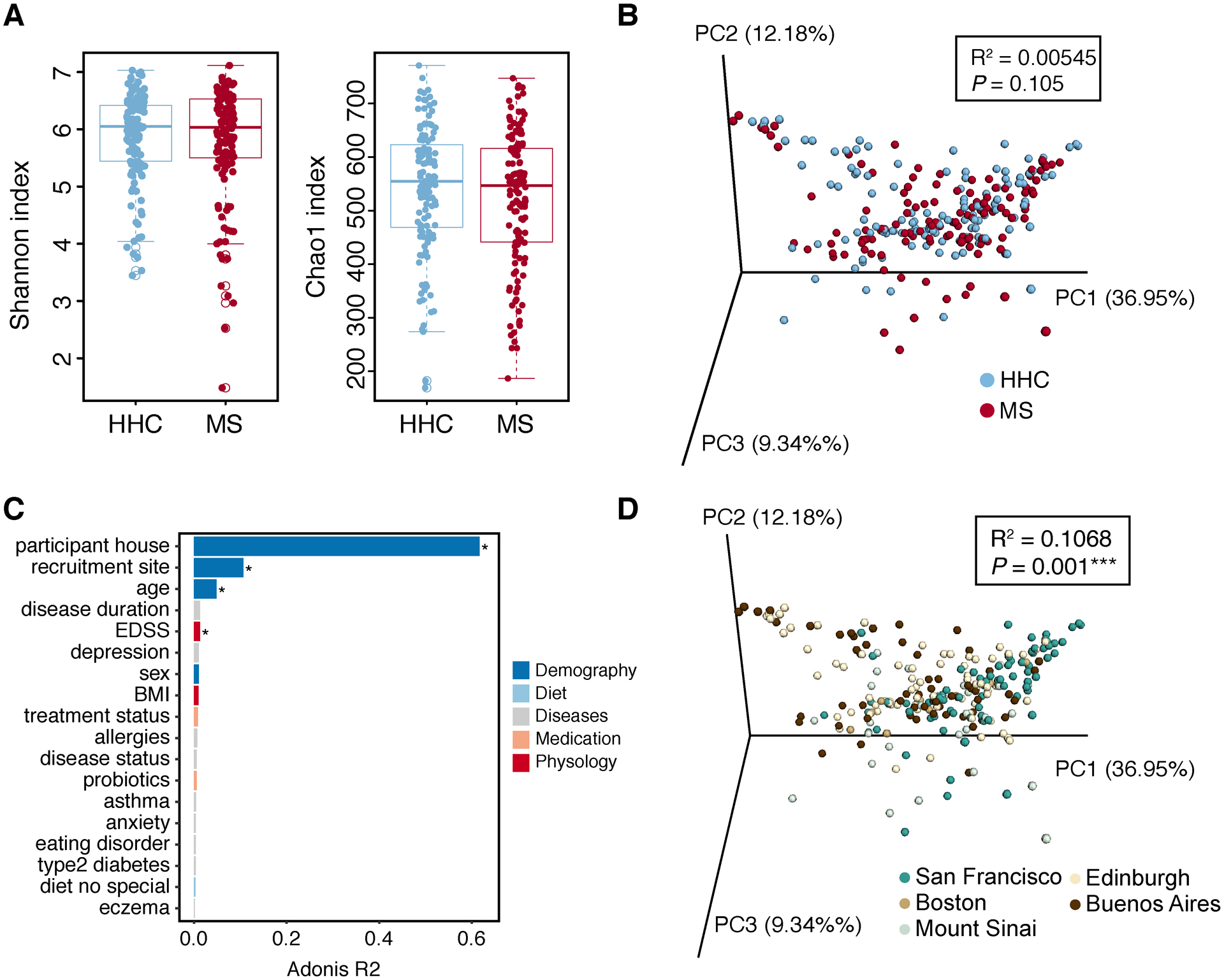
Microbial diversity. (A) Microbiome α-diversity measured by Shannon and Chao1 index was compared by disease status. Data are presented as mean ± SEM (ANOVA, not significant). (B) PCoA of weighted UniFrac community distance by disease status. (C) Bar plot showing the size effect (Adonis R2) of confounders associated with gut microbial variations (weighted UniFrac distance). Confounders showing significant impact on gut microbiome were labeled. EDSS, expanded disability status scale. (D) PCoA of weighted UniFrac community distance by recruitment sites. Statistical significance was determined by PERMANOVA (multiple testing corrected P value, * P ≤ 0.05, ***P ≤ 0.001).
As a large-scale study will introduce more confounding factors that shape the microbiome composition beyond disease status, we used PERMANOVA to test for effect size (quantitative differences between two or more groups) and power of metadata categories (i.e. confounders), including demography, lifestyle, disease, medication and physiology, on gut microbiome. Four confounders were identified to be significantly associated with gut microbial variation measured by inter-individual weighted uniFrac distances under a multiple testing corrected P value < 0.05 (Figure 5C). House location accounted for the largest effect size (Figure 5C, adonis R2=0.62), far more than any other metadata category. The geographical shape of the microbiome was in addition reflected by recruitment site, as also shown in several previous studies26–28. A PCoA of the microbiome beta-diversity showed the significant difference of samples from San Francisco, Edinburgh and Buenos Aires (Figure 5D, PERMANOVA, P < 0.05). Age also exerted significant effects (Figure 5C), consistent with previous studies showing that the gut microbiome can vary across the lifespan28, 29. The average difference of age in MS (age 47.7±12.9) and HHC (age 46.1±11.9) was small (3.85±5.92) in our data, thus age-associated variation can be also potentially reduced by the household design. Surprisingly, EDSS (a measure of disease severity) was significantly associated with microbiome diversity. We speculated this diversity could be partially explained by age, as a significantly positive correlation between EDSS and age was observed (Fig S4C). A smaller microbiota divergence was explained by sex, disease status, or medication use (Figure 5C), although the modest size and clinical heterogeneity of our sample limits interpretation of this observation. Similarly, MS comorbidities and lifestyle show a relatively small influence on gut microbiome.
To formally test whether a household design reduces the influence of geographic and environmental factors on microbiome diversity, we repeated the PERMANOVA test by constraining permutations within the house group (strata) as MS and healthy controls are nested in the same house. Remarkably, no significant influence was observed by recruitment site or age while a significant effect was observed for sex and EDSS (P value < 0.05 without multiple testing correction, Figure S4D). While encouraging, this is unsurprising as most MS patients are female and they naturally have higher disability than HHC (EDSS considered as 0). The human microbiome is influenced by sex despite a relatively weak effect across body sites30, 31. The microbiome differences given by confounders like sex will be adjusted in a lager cohort to resolve the true difference in microbiota between MS and control. In summary, the paired design indeed reduces the influencing of confounding factors, thereby potentially enhancing power to identify MS-associated microbiome.
Power and sample-size estimation
The power and sample size required to reveal a statistical difference between two or more groups depend on within-group distances (within-group sum of squares) and effect size (the difference between the between-group distance and within-group distance). To estimate the samples needed in a study, assumptions need to be made about desired effect size and variance within the data. However, to date no standardized effect size has been reported in any MS gut microbiome study. Here we applied micropower19, a simulation-based method for PERMANOVA-based beta diversity comparisons, to assess the effect size and statistical power of this pilot study with 128 pairs of MS and healthy controls, and expand the analyses to two previous published microbiome study in MS1, 5.
1206 OTUs were classified in the 256 subjects by at least 10,000 16S rRNA gene reads per subject. To estimate the minimum number of samples adequate to represent the total OTUs, we performed random sampling of different sample sizes and measured the observed OTUs in each recruitment site as we observed a strong geographic effect on gut microbiome. The number of observed OTUs increased proportionally to the sample size in all recruitment sites and approached a maximum at n=25 (Figure6A). This result suggests that at least 25 participants per group could capture the maximum composition of human gut microbiome. However, this sample size only provides approximately 60% power to detect differences between groups, even within each recruitment site, as measured by the corrected coefficient of determination ω2 (Figure 6B, San Francisco ω2=0.00068 with 64% power, Edinburgh ω2=0.0012 with 58% power, Buenos Aires ω2=0.0016 with 56% power). The modest power and effect size can be explained by the comparable within-group distance to between-group distance, and this varied from one recruitment site to another (Fig 6C). A sample size of only 37 participants from San Francisco provided 90% power to detect a difference at ω2 = 0.00068 while sample size in all other sites was much too low to reach a similar power. The highest effect size (ω2 = 0.0021) was detected in the “all sites” group which included samples from all recruitment sites (n=128). However, more samples (n=86) were required to reach 90% power to detect the difference in this combined sets compared to a unique site, underscoring sample and geographical heterogeneity.
Figure 6.

Sample size estimation. (A) The number of observed OTUs in each recruitment site by randomly sampling samples from total samples (100 permutations with replacement). (B) Statistical power of PERMANOVA testing on Bray-Curtis distance to detect the group-level effect in each recruitment site, based on bootstrap sampling. ω2, corrected coefficient of determination. (C) Within-group and between-group dissimilarity measured in each site.
To generalize the difference of gut microbiome in MS, we calculated the effect sizes in two previous MS microbiome studies1, 5. Similar within-group and between-group distances, but a smaller effect size (ω2 = 0.00012) were observed in Cekanaviciute et al5 (n=71 cases and 71 controls) study compared to those from the UCSF site of this study (n=40 pairs, both recruited in San Francisco) (Figure S5). This reduction of the effect size required more samples (n=59) to provide 90% power to detect MS associated differences compared to the 37 UCSF pairs required in the present study (Figure 6). These results suggest the environment-controlled paired design reduces confounding effects and increases the MS-associated effect size. Although lower dissimilarity and higher effect size (ω2 = 0.0057) were found in Berer et al1 (a discordant twin study, n=34), the small sample size limited the power to detect global microbiome difference (Figure S5).
Diet and gut microbiome
Giving the significant role of diet in shaping the gut microbiome32, 33, we hypothesized that studying its interaction with the gut microbiome might shed light on how diet influences the immune system in MS6, 34. We first explored whether diet differs by disease course, household, recruitment site and other confounders. Furthermore, we assessed which types of dietary intake were associated with a change of microbial composition.
A high proportion of participants (93.75%, n=240) completed online food frequency questionnaires in which 38 specific dietary components were quantitated (Datasets S4). The average intake structure, measured by the percentage of nutrient intake, was similar between MS and HHC (Figure S6A). In fact, when visualized in a PCA, samples were not clustered by disease status based on diet (Figure S6B), and the difference between the diets of HHC and MS subjects was not statistically significant. Based on these results, we then tested to what extent other confounders were involved in the divergence of diet. The two largest sources of variance in dietary intake were associated with participant house and recruitment site (Figure 7A). This finding is in agreement with the diversity of gut microbiome by geography we observed previously, as it is known microbial communities are significantly shaped by dietary habits.
Figure 7.
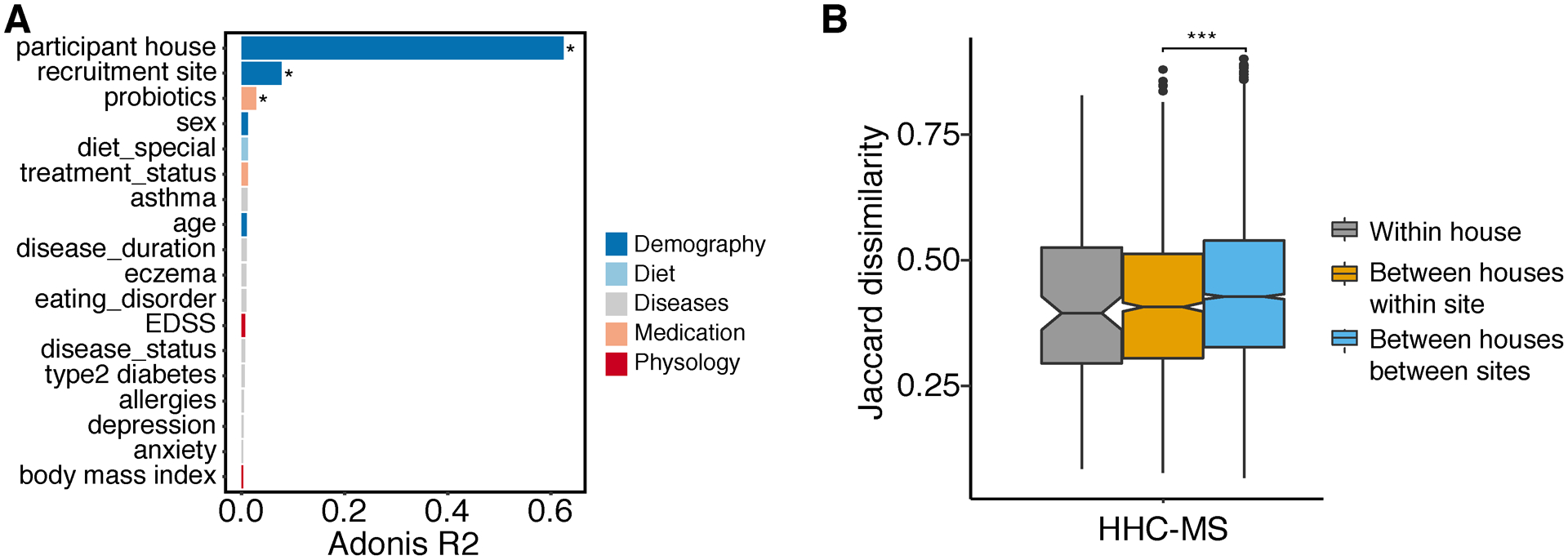
Dietary dissimilarity. (A) Bar plot showing the size effect (Adonis R2) of confounders associated with dietary variations (Jaccard dissimilarity). Confounders showing significant impact on gut microbiome were labeled (PERMANOVA, multiple testing corrected P value, *P ≤ 0.05). (B) Jaccard dissimilarity of dietary intakes measured between healthy control and MS within the same house, between different houses in the same time and between different houses in different sites. Random comparisons of healthy control and MS were female-male matched only to control sex effect. Statistical significance was determined by ANOVA (multiple testing corrected P value, ***P ≤ 0.001).
The observation that gut microbiomes are most similar between household individuals led to the hypothesis that the similarity was related to shared environment and diet. However, despite observing a trend, we were not able to identify a significantly similar diet within household pairs, even those within the same city (Figure 7B). We speculate this might be due to the relatively small sample size. We did, however, observe a significantly lower diet dissimilarity across pairs from the same city compared to pairs randomly assembled from different cities (Figure 7B, P < 0.001), which could reflect distinct dietary habits across cities (some of them in different continents).
A deeper analysis of the relationship between gut microbiome and diet led us to identify that sweets (as a % of total calories), whole grains, and “good oils” in foods explain the largest variation of microbiome composition (Figure S7A). Redundancy analysis (RDA), used to evaluate the relationship between explanatory variables diet and response variables microbial community, also revealed that sweets, whole grains, “good oils” were highly correlated with microbiome composition, explaining 42.3% and 24.1% of the variance even the samples were not clustered by disease status (Figure S7B). We found several relationships between nutrients and bacteria (e.g. sweets and Bacteroides, Bifidobacterium, “good oils” and Akkermansia, Blautia, Ruminococcus, whole grains and Roseburia), as illustrated by their close proximity in the same quadrant of Figure S6B (angles between lines of response variables and lines of explanatory variables represent a two-dimensional approximation of correlations). Similar relationships were previously reported: Bacteroides was enriched in mice taking saccharin35 and non-digestible carbohydrates from whole grain have been shown to increase the abundance of Roseburia33. Further elucidating how diet shapes the gut microbiome and how diet is related to disease course require will to study much larger sample sizes, which the iMSMS is set out to do.
Discussion
The precise mechanism by which the gut microbiome may be involved in the pathogenesis of MS still remains unclear. To address that question, the iMSMS consortium is recruiting patients and controls in the US, Europe and South America. However, a large study faces numerous challenges in design, not the least of which are developing the appropriate experimental protocols, computational methods to model associations between the gut microbiome and disease phenotypes such as progression or treatment. Although no disease associated microbiota were studied in this pilot analysis, it provided invaluable information allowing us to identify the main sources of expected microbiome variance (technical, biological, and related to the experimental design) observed.
Prior to DNA extraction and sequencing on large-scale samples, sample collection and storage methods, e.g. room temperature storage or immediately fresh freezing, should also be considered as they have been shown to impact microbial community analyses36, 37. This study assessed the impact of two technical covariates, sample storage methods and sequencing strategies, on the structure and diversity of bacterial community. Gut microbes characterized in Q-tip (dry) and snap frozen (wet) samples showed higher correlation of structure and no difference in alpha-diversity, implying either method can be used for sample collection. Notably, the intra-individual dissimilarity as measured in experimental dry and wet samples is dramatically lower than the inter-individual microbial diversity as measured among participants stratified by sampling sites or even by house.
Current high throughput sequencing techniques, known as 16S rRNA and whole metagenome shotgun sequencing (WMGS), are widely applied to characterize the microbial community. 16S rRNA amplicon sequencing is more cost-effective and suited for large-scale studies, but offers limited taxonomical (especially species) and functional resolution24, while WMGS increases the identification of species or strains and provides additional functional information but is not always affordable for large population studies38, 39. The shallow shotgun metagenomics sequencing with as few as 0.5 million sequences per sample was proposed as an alternative of 16S rRNA sequencing on large studies22. Several studies reported a difference on microbial community detected by 16S rRNA and WMGS as observed in our study, but also showed an overall similar microbial diversity and composition40, 41. We found a high correlation of microbiome structure identified between 16S rRNA sequencing and shallow WMGS at both the phylum and genus level, suggesting that either of these two technologies could be sufficient for microbial characterization, but a higher resolution of species classification and functional profiling by WMGS will provide a higher resolution picture of bacteria-host interactions associated with the disease42.
Although larger sample sizes generally increase statistical power, recruitment of subjects from multiple sites also introduces more cofounders such as geography, demography, host lifestyle, diet, and medication use that could shape the gut microbiome26, 27. A better understanding of how such confounders contribute to microbial variation will help inform appropriate statistical approaches for data analyses. As individuals living in the same house share similar gut, skin and oral microbiome, a household case-control design has been suggested as optimal for disease-associated microbiome studies43–46. Not surprisingly, household case-control comparisons shared a more similar microbiome than between houses. The largest variation of microbiome was driven by house location confirming that the paired design of the iMSMS will help reduce the influence of geographic and environmental factors, thereby potentially enhancing power to identify MS-associated microbiome. While this paired design may lead to underestimation of MS-associated microbes, it plays a critical role in controlling false positive observations introduced by numerous confounding variables in non-household designs. Specifically, our study design, effectively minimizes confounders such as recruitment site, sex and diet all of which are major drivers of microbiome differences found in smaller, uncontrolled studies.
This MS microbiome study accessed for first time effect sizes on gut microbiome differences due to the disease, taking into account recruitment site and also estimated the sample size required to achieve adequate power. The global effect depends on the heterogeneity of participants (e.g. geography, medication use and genetic background), and thus either environment-controlled (household pairs) or genetic-controlled (twin pairs) studies will increase the effect size. Given that colonization by some gut bacteria is a heritable trait47, genetic variants (either within or outside of the HLA region), might affect the statistical power to detect disease-associated communities. The effect of gene–environment interaction on shaping gut microbiome needs to be investigated in a larger cohort.
Western diet, generally recognized as hypercaloric and high in saturated fat and sugar, has been associated with a higher prevalence of MS48. Studies have investigated the role of dietary intervention in experimental autoimmune encephalomyelitis (EAE), an animal model of MS, revealing that specific dietary regimens may have either proinflammatory or anti-inflammatory effects. The ‘Swank diet’, a low saturated fat diet (no more than 10–15 g/day), has been widely used by MS patients since 1950s and shown, possibly, to modestly reduce MS disease activity and disability progression in follow-up studies49–51. Complementary and alternative medicines (CAM), usually with vitamins, minerals and essential fatty acids, also have been taken by MS patients in conjunction with conventional treatments but with limited research evaluating their safety and effectiveness52, 53. We did not observe a significant difference of dietary structure between MS and household healthy controls in this relatively small cohort study, but a correlation of diet and gut microbiome was detected, which will be further explored in the larger study.
The study of gut microbiome in MS is undoubtedly promising and potentially revolutionary, both for patient care and drug discovery. Developing methods that make best use of the high-quality data produced in large-scale studies to identify unambiguous associations is the key for meaningful new discoveries in this field. By considering the challenges summarized above, appropriate design and suitable statistical models should be possible.
Supplementary Material
Acknowledgements
We would like to thank all members and participants of iMSMS consortium contributing to this study. X.Z. performed all computational analyses. S.S. led recruitment at UCSF and coordinated all centers; R.B., J.L. and S.J.C. prepared the samples for sequencing; X.J., A.-K.P., P.B., P.C., I.K.S., Z.X., H.W., T.C., S.C., P.C., D.O., T.T.T., J.C., L.N., M.F., R.H., J.G., A.B., J.R.O., T.W., J.C., B.A.C.C. and S.L.H. helped with patient recruitment; R.K. led microbiome sequencing and data submission. S.E.B. and S.L.H. conceived and designed the study. S.E.B. supervised the study. X.Z and S.E.B wrote the manuscript. All authors read, revised and approved the final manuscript.
Funding
This study was funded by the Valhalla Charitable Foundation, and by a grant by the National Multiple sclerosis Society (CA 1072-A-7 to S.E.B).
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflict of interest.
Contributor Information
The iMSMS Consortium:
Xiaoyuan Zhou, Sneha Singh, Ryan Baumann, Patrick Barba, James Landefeld, Patrizia Casaccia, Ilana Katz Sand, Zongqi Xia, Howard Weiner, Tanuja Chitnis, Siddharthan Chandran, Peter Connick, David Otaegui, Tamara Castillo-Triviño, Stacy J. Caillier, Adam Santaniello, Gail Ackermann, Greg Humphrey, Laura Negrotto, Mauricio Farez, Reinhard Hohlfeld, Anne-Katrin Pröbstel, Xiaoming Jia, Jennifer Graves, Amit Bar-or, Jorge R. Oksenberg, Jeffery Gelfand, Michael R. Wilson, Elizabeth Crabtree, Scott S Zamvil, Jorge Correale, Bruce A.C. Cree, Stephen L. Hauser, Rob Knight, and Sergio E. Baranzini
References
- 1.Berer K, Gerdes LA, Cekanaviciute E, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A 2017; 114: 10719–10724. DOI: 10.1073/pnas.1711233114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alfredsson L and Olsson T. Lifestyle and Environmental Factors in Multiple Sclerosis. Cold Spring Harbor perspectives in medicine 2019; 9. DOI: 10.1101/cshperspect.a028944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ochoa-Reparaz J, Kirby TO and Kasper LH. The Gut Microbiome and Multiple Sclerosis. Cold Spring Harbor perspectives in medicine 2018; 8. DOI: 10.1101/cshperspect.a029017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Luca F and Shoenfeld Y. The microbiome in autoimmune diseases. Clinical and experimental immunology 2019; 195: 74–85. DOI: 10.1111/cei.13158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cekanaviciute E, Yoo BB, Runia TF, et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc Natl Acad Sci U S A 2017; 114: 10713–10718. DOI: 10.1073/pnas.1711235114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jangi S, Gandhi R, Cox LM, et al. Alterations of the human gut microbiome in multiple sclerosis. Nature communications 2016; 7: 12015. DOI: 10.1038/ncomms12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, Chia N, Kalari KR, et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Scientific reports 2016; 6: 28484. DOI: 10.1038/srep28484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDonald WI, Compston A, Edan G, et al. Recommended diagnostic criteria for multiple sclerosis: guidelines from the International Panel on the diagnosis of multiple sclerosis. Annals of neurology 2001; 50: 121–127. [DOI] [PubMed] [Google Scholar]
- 9.Baranzini SE and Oksenberg JR. The Genetics of Multiple Sclerosis: From 0 to 200 in 50 Years. Trends in genetics : TIG 2017; 33: 960–970. DOI: 10.1016/j.tig.2017.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Block G Block Adult Questionnaire. Retrieved 2007-04-30 from the NutritionQuest Questionnaires and Screener Page:http://www.nutritionquest.com/products/questionnaires_screeners.htm. 2005.
- 11.McArdle BH and Anderson MJ. Fitting Multivariate Models to Community Data: A Comment on Distance-Based Redundancy Analysis. Ecology 2001; 82: 290–297. DOI: 10.1890/0012-9658(2001)082[0290:FMMTCD]2.0.CO;2. [DOI] [Google Scholar]
- 12.Zapala MA and Schork NJ. Multivariate regression analysis of distance matrices for testing associations between gene expression patterns and related variables. Proc Natl Acad Sci U S A 2006; 103: 19430–19435. DOI: 10.1073/pnas.0609333103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. The ISME journal 2012; 6: 1621–1624. DOI: 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7: 335–336. DOI: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shannon CE. The mathematical theory of communication. 1963. MD Comput 1997; 14: 306–317. [PubMed] [Google Scholar]
- 16.Chao A Non-parametric estimation of the number of classes in a population. Scand J Stat 1984; 11: 265–270. [Google Scholar]
- 17.Lozupone C and Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005; 71: 8228–8235. DOI: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bray JR CJ. An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 1957; 27: 325–349. DOI: 10.2307/1942268. [DOI] [Google Scholar]
- 19.Kelly BJ, Gross R, Bittinger K, et al. Power and sample-size estimation for microbiome studies using pairwise distances and PERMANOVA. Bioinformatics 2015; 31: 2461–2468. DOI: 10.1093/bioinformatics/btv183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Didion JP, Martin M and Collins FS. Atropos: specific, sensitive, and speedy trimming of sequencing reads. PeerJ 2017; 5: e3720. DOI: 10.7717/peerj.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langmead B, Trapnell C, Pop M, et al. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 2009; 10: R25. DOI: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hillmann B, Al-Ghalith GA, Shields-Cutler RR, et al. Evaluating the Information Content of Shallow Shotgun Metagenomics. mSystems 2018; 3. DOI: 10.1128/mSystems.00069-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gonzalez A, Navas-Molina JA, Kosciolek T, et al. Qiita: rapid, web-enabled microbiome meta-analysis. Nat Methods 2018; 15: 796–798. DOI: 10.1038/s41592-018-0141-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janda JM and Abbott SL. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: pluses, perils, and pitfalls. J Clin Microbiol 2007; 45: 2761–2764. DOI: 10.1128/JCM.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jovel J, Patterson J, Wang W, et al. Characterization of the Gut Microbiome Using 16S or Shotgun Metagenomics. Frontiers in microbiology 2016; 7: 459. DOI: 10.3389/fmicb.2016.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta VK, Paul S and Dutta C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Frontiers in microbiology 2017; 8: 1162. DOI: 10.3389/fmicb.2017.01162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothschild D, Weissbrod O, Barkan E, et al. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018; 555: 210–215. DOI: 10.1038/nature25973. [DOI] [PubMed] [Google Scholar]
- 28.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature 2012; 486: 222–227. DOI: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odamaki T, Kato K, Sugahara H, et al. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. Bmc Microbiol 2016; 16: 90. DOI: 10.1186/s12866-016-0708-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Costello EK, Lauber CL, Hamady M, et al. Bacterial community variation in human body habitats across space and time. Science (New York, NY) 2009; 326: 1694–1697. DOI: 10.1126/science.1177486.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haro C, Rangel-Zuniga OA, Alcala-Diaz JF, et al. Intestinal Microbiota Is Influenced by Gender and Body Mass Index. Plos One 2016; 11: e0154090. DOI: 10.1371/journal.pone.0154090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gentile CL and Weir TL. The gut microbiota at the intersection of diet and human health. Science (New York, NY) 2018; 362: 776–780. DOI: 10.1126/science.aau5812. [DOI] [PubMed] [Google Scholar]
- 33.Singh RK, Chang HW, Yan D, et al. Influence of diet on the gut microbiome and implications for human health. J Transl Med 2017; 15: 73. DOI: 10.1186/s12967-017-1175-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saresella M, Mendozzi L, Rossi V, et al. Immunological and Clinical Effect of Diet Modulation of the Gut Microbiome in Multiple Sclerosis Patients: A Pilot Study. Front Immunol 2017; 8: 1391. DOI: 10.3389/fimmu.2017.01391.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suez J, Korem T, Zilberman-Schapira G, et al. Non-caloric artificial sweeteners and the microbiome: findings and challenges. Gut Microbes 2015; 6: 149–155. DOI: 10.1080/19490976.2015.1017700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dominianni C, Wu J, Hayes RB, et al. Comparison of methods for fecal microbiome biospecimen collection. BMC Microbiol 2014; 14: 103. DOI: 10.1186/1471-2180-14-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sinha R, Chen J, Amir A, et al. Collecting Fecal Samples for Microbiome Analyses in Epidemiology Studies. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 2016; 25: 407–416. DOI: 10.1158/1055-9965.EPI-15-0951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitra S, Forster-Fromme K, Damms-Machado A, et al. Analysis of the intestinal microbiota using SOLiD 16S rRNA gene sequencing and SOLiD shotgun sequencing. Bmc Genomics 2013; 14 Suppl 5: S16. DOI: 10.1186/1471-2164-14-S5-S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ranjan R, Rani A, Metwally A, et al. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochemical and biophysical research communications 2016; 469: 967–977. DOI: 10.1016/j.bbrc.2015.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fierer N, Leff JW, Adams BJ, et al. Cross-biome metagenomic analyses of soil microbial communities and their functional attributes. Proc Natl Acad Sci U S A 2012; 109: 21390–21395. DOI: 10.1073/pnas.1215210110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Poretsky R, Rodriguez RL, Luo C, et al. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS One 2014; 9: e93827. DOI: 10.1371/journal.pone.0093827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Franzosa EA, Hsu T, Sirota-Madi A, et al. Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat Rev Microbiol 2015; 13: 360–372. DOI: 10.1038/nrmicro3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goodrich JK, Di Rienzi SC, Poole AC, et al. Conducting a microbiome study. Cell 2014; 158: 250–262. DOI: 10.1016/j.cell.2014.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abeles SR, Jones MB, Santiago-Rodriguez TM, et al. Microbial diversity in individuals and their household contacts following typical antibiotic courses. Microbiome 2016; 4: 39. DOI: 10.1186/s40168-016-0187-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lax S, Smith DP, Hampton-Marcell J, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science (New York, NY) 2014; 345: 1048–1052. DOI: 10.1126/science.1254529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song SJ, Lauber C, Costello EK, et al. Cohabiting family members share microbiota with one another and with their dogs. eLife 2013; 2: e00458. DOI: 10.7554/eLife.00458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurilshikov A, Wijmenga C, Fu J, et al. Host Genetics and Gut Microbiome: Challenges and Perspectives. Trends in immunology 2017; 38: 633–647. DOI: 10.1016/j.it.2017.06.003. [DOI] [PubMed] [Google Scholar]
- 48.Alter M, Yamoor M and Harshe M. Multiple sclerosis and nutrition. Archives of neurology 1974; 31: 267–272. [DOI] [PubMed] [Google Scholar]
- 49.Swank RL. Treatment of multiple sclerosis with low-fat diet. AMA Arch Neurol Psychiatry 1953; 69: 91–103. [DOI] [PubMed] [Google Scholar]
- 50.Swank RL and Dugan BB. Effect of low saturated fat diet in early and late cases of multiple sclerosis. Lancet 1990; 336: 37–39. [DOI] [PubMed] [Google Scholar]
- 51.Swank RL and Goodwin J. Review of MS patient survival on a Swank low saturated fat diet. Nutrition 2003; 19: 161–162. [DOI] [PubMed] [Google Scholar]
- 52.Leong EM, Semple SJ, Angley M, et al. Complementary and alternative medicines and dietary interventions in multiple sclerosis: what is being used in South Australia and why? Complement Ther Med 2009; 17: 216–223. DOI: 10.1016/j.ctim.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 53.Yadav V, Shinto L and Bourdette D. Complementary and alternative medicine for the treatment of multiple sclerosis. Expert Rev Clin Immunol 2010; 6: 381–395. DOI: 10.1586/eci.10.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated and analyzed during the current study are available in the EMBL-ENA repository (https://www.ebi.ac.uk) with accession number ERP115476.