


Abstract
TFIIH is a crucial transcription and DNA repair factor consisting of the seven-subunit core. The core subunit p62 contains a pleckstrin homology domain (PH-D), which is essential for locating TFIIH at transcription initiation and DNA damage sites, and two BSD (BTF2-like transcription factors, synapse-associated proteins and DOS2-like proteins) domains. A recent cryo-electron microscopy (cryo-EM) structure of human TFIIH visualized most parts of core, except for the PH-D. Here, by nuclear magnetic resonance spectroscopy we have established the solution structure of human p62 PH-D connected to the BSD1 domain by a highly flexible linker, suggesting the flexibility of PH-D in TFIIH. Based on this dynamic character, the PH-D was modeled in the cryo-EM structure to obtain the whole human TFIIH core structure, which indicates that the PH-D moves around the surface of core with a specific but limited spatial distribution; these dynamic structures were refined by molecular dynamics (MD) simulations. Furthermore, we built models, also refined by MD simulations, of TFIIH in complex with five p62-binding partners, including transcription factors TFIIEα, p53 and DP1, and nucleotide excision repair factors XPC and UVSSA. The models explain why the PH-D is crucially targeted by these factors, which use their intrinsically disordered acidic regions for TFIIH recruitment.
INTRODUCTION
The general transcription factor TFIIH is a multifunctional protein complex involved in transcriptional initiation (1–3), activation (4–13) and elongation (14–16), the cell cycle (17–20) and nucleotide excision repair (21–24). TFIIH consists of 10 different subunits (∼500 kDa) and can be divided into two sub-complexes: the core TFIIH complex consisting of 7 subunits (XPB, XPD, p62, p52, p44, p34 and p8) and the Cdk-activating kinase (CAK) complex formed by 3 subunits (Cdk7, Cyclin H and MAT1). TFIIH possesses several enzymatic activities: for example, the ATPase/helicase activities of XPB and XPD, both of which are required for promoter melting at transcription initiation sites and damaged DNA opening at nucleotide excision repair sites; the ubiquitin ligase activity of p44 (in yeast), which is thought to mediate the transcriptional response to DNA damage; and the protein kinase activity of Cdk7, which is necessary for phosphorylation of the C-terminal domain of the largest subunit of RNA polymerase II, other transcription factors and nuclear receptors (25,26). In particular, mutations in XPB, XPD and p8 cause three autosomal recessive disorders: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy (27).
p62 is an essential subunit of TFIIH, not only for formation of the core TFIIH complex but also for recruiting TFIIH to appropriate functional sites. Human p62 has four structural domains: an N-terminal pleckstrin homology (PH) domain (PH-D) (residues 1–103); two BSD (BTF2-like transcription factors, synapse-associated proteins and DOS2-like proteins) domains, BSD1 (residues 109–147) and BSD2 (residues 189–225); and a C-terminal three-helix bundle (residues 454–548). The PH-D is responsible for recruitment of TFIIH through its specific interaction with intrinsically disordered acidic regions of various transcription factors and several nucleotide excision repair factors, such as TFIIEα (28,29), p53 (30,31), EKLF (32), DP1 (33), VP16 (34,35) and EBNA2 (36) during transcription, and XPC (37,38), UVSSA (39) and XPG (40) during nucleotide excision repair. Recent cryo-electron microscopy (cryo-EM) studies of human apo TFIIH (41) and yeast holo TFIIH in complex with the pre-initiation complex (PIC) and Mediator (42) have provided detailed pictures of the core TFIIH complex and how it is connected to the CAK complex. In the cryo-EM structure of human TFIIH, almost all parts of the seven subunits in the core can be visualized except for the N-terminal PH-D of p62.
To gain insight into the structure of the p62 PH-D in the core TFIIH complex, here, keeping in mind the fact that the BSD1 domain can be clearly identified in the cryo-EM structure, we have solved the solution structure of the tandem PH and BSD1 domains of human p62 (residues 1–158) by nuclear magnetic resonance (NMR) spectroscopy. These domains are connected by a short linker (residues 104–108) and do not interact with each other. The flexibility of the interdomain region has been evaluated by measuring 15N relaxation parameters and 1H exchange rates with solvent. Based on our dynamic NMR structures of the tandem PH and BSD1 domains coupled with the cryo-EM structure (43), we have built reasonable dynamic structural models of human TFIIH including the flexible p62 PH-D by superposing the BSD1 domains from the solution and cryo-EM structures; we have further refined these dynamic structures by using all-atom molecular dynamics (MD) simulations. In addition, we have modeled the structures, also refined by the MD simulations, of TFIIH in complex with five p62-binding partners that are responsible for recruiting TFIIH to sites of transcription initiation and DNA damage.
MATERIALS AND METHODS
Preparation of human p621–158
The 13C/15N-labeled fragment of human p621–158 (residues 1–158) was expressed as a hexa-histidine-tagged product in a pET15b vector (Merck Millipore) in Escherichia coli BL21 (DE3) Gold (Agilent Technologies). The cells were grown at 37°C in M9 minimal medium containing [15N]ammonium chloride and [13C]glucose. The product was expressed by induction with 1 mM isopropyl-β-d-thiogalactopyranoside. After 21 h of growth at 20°C, the cells were collected, resuspended in buffer A [20 mM Tris–HCl (pH 8.0), 10% glycerol, 1 M NaCl], lysed by sonication and then centrifuged. The supernatant was loaded onto a His-Tag Purification Resin (Roche) column equilibrated with buffer A, and the column was washed with buffer A containing 10 mM imidazole–HCl. The sample was eluted by 500 mM imidazole–HCl, peak fractions were pooled and the buffer was changed to thrombin cleavage buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 500 mM NaCl, 2.7 mM KCl, pH 7.3) by using an Amicon Ultra device (Merck Millipore). To remove the histidine tag, the sample was digested with thrombin for 63 h at 20°C. After concentration via an Amicon Ultra, the sample was applied to a Superdex75 (GE Healthcare) column equilibrated with 20 mM potassium phosphate (pH 6.8) and 500 mM NaCl.
NMR structure determination
For structure determination, we used 0.9–1.2 mM 13C/15N-labeled p621–158 in 20 mM potassium phosphate (pH 6.8) and 5 mM deuterated DTT prepared in either 90% H2O/10% D2O or 99.9% D2O. NMR experiments were performed at 20°C on Bruker AVANCE III HD 600-MHz and 950-MHz spectrometers, each equipped with a cryo-TCI probe. Backbone and side-chain resonances were assigned by using standard triple-resonance NMR experiments (44). Stereospecific assignments were obtained from a combination of HNHB, HN(CO)HB, HNCG, HN(CO)CG, and 13C-edited and 15N-edited NOESY-HSQC spectra. Distance restraints were obtained from 15N-edited NOESY-HSQC (τm = 150 ms) and 13C-edited NOESY-HSQC (τm = 50 and 100 ms) spectra. Side-chain torsion angles, χ1 and χ2, were obtained from a combination of HNHB, HN(CO)HB, HNCG, HN(CO)CG, and 13C-edited and 15N-edited NOESY-HSQC spectra. Hydrogen bond restraints were obtained by backbone amide H/D-exchange experiments. Spectra were processed by using NMRPipe (45) and analyzed by using NMRView (46).
Structure calculation
Interproton distance restraints derived from NOE (nuclear Overhauser effect) intensities were grouped into four distance ranges, 1.8–2.7 Å (1.8–2.9 Å for NOEs involving HN protons), 1.8–3.3 Å (1.8–3.5 Å for NOEs involving HN protons), 1.8–5.0 and 1.8–6.0 Å, corresponding to strong, medium, weak and very weak NOEs, respectively. The upper limit was corrected for constraints involving methyl groups, aromatic ring protons and non-stereospecifically assigned methylene protons. Dihedral angle restraints for φ and ψ were obtained from analysis of the backbone chemical shifts with TALOS+ (47). χ1 and χ2 angles were restrained ±30° for three side-chain rotamers. Structure calculations were performed by distance geometry and simulated annealing using the program Xplor-NIH (48,49). In total, 100 structures were calculated. All structures were then subjected to water refinement (50), in which the structures were immersed in a 7.0-Å layer of water molecules. After minimization with 120 steps, a heating stage from 100 to 500 K with 200 steps of MD for every 100-K increment, a refinement stage with 2500 steps at 500 K and a cooling stage from 500 to 25 K with 200 steps for every 25-K decrement were carried out. The refinement protocol was finished with 200 steps of minimization. Structural statistics for the 20 best structures are summarized in Table 1. Structures were analyzed and displayed by using PROCHECK-NMR (51), MOLMOL (52) and PyMol (http://www.pymol.org).
Table 1.
Statistics for the 20 best structures of the PH and BSD1 domains of TFIIH p62
| Experimental restraints | |
|---|---|
| Total NOE | 3338 |
| Intra-residue | 465 |
| Sequential (i − j = 1) | 799 |
| Medium range (1 < i − j < 5) | 709 |
| Long range (i − j ≥ 5) | 1365 |
| Hydrogen bond | 48 × 2 |
| Number of dihedral restraints | |
| φ | 147 |
| ψ | 146 |
| χ1 | 75 |
| χ2 | 13 |
| Statistics for structure calculations | |
| Root-mean-square deviation (RMSD) from experimental restraintsa | |
| Distance (Å) | 0.038 ± 0.001 |
| Dihedral (°) | 0.384 ± 0.070 |
| RMSD from idealized covalent geometry | |
| Bonds (Å) | 0.00513 ± 0.00009 |
| Angles (°) | 0.642 ± 0.011 |
| Improper (°) | 0.649 ± 0.021 |
| Coordinate precision | |
| Average pairwise RMSD from the mean structure | |
| Backbone atoms (Å) | 0.46 ± 0.10,b 0.76 ± 0.34,c 0.45 ± 0.12d |
| Heavy atoms (Å) | 1.22 ± 0.18,b 1.49 ± 0.35,c 1.12 ± 0.19d |
| Ramachandran plot statistics | |
| Residues in most favored regions (%) | 84.0e |
| Residues in additional allowed regions (%) | 15.8e |
| Residues in generously allowed regions (%) | 0.2e |
| Residues in disallowed regions (%) | 0.0e |
| Clash score | 26 |
aNone of the structures exhibited distance violations >0.5 Å and dihedral angle violations >5°.
bThe value was calculated over residues 7–103.
cThe value was calculated over residues 109–147.
dThe value was calculated over residues 114–147.
eThe value was calculated over residues 5–154.
NMR relaxation analysis
The backbone 15N relaxation parameters were measured by using a 13C/15N-labeled sample (0.60 mM) at 20°C on Bruker AVANCE III HD 600-MHz and 950-MHz spectrometers equipped with a cryo-TCI probe. Relaxation durations of 24, 49, 64, 128, 256, 384, 512, 768, 1024, 1280, 1536, 1792 and 2048 ms for the longitudinal relaxation rate (R1) and 16.98, 33.95, 50.93, 67.90, 84.88, 101.86, 135.81, 203.71, 271.62, 339.52, 441.38 and 543.23 ms for the transverse relaxation rate (R2) were used. Heteronuclear 15N–{1H} NOE experiments were recorded in the presence and absence of proton saturation, which was achieved with a 5.0-s duration consisting of 120° 1H pulses applied every 5.0 ms. R1 and R2 were obtained by fitting peak intensities at a series of relaxation durations to an exponential decay curve using CurveFit (53). The uncertainties of the peak intensities were estimated by using duplicated data from the shortest relaxation delay. The uncertainties of the relaxation rates were determined by CurveFit using a Monte Carlo simulation. The steady-state 15N–{1H} NOE values were determined from peak intensity ratios obtained from spectra acquired with and without proton saturation. The uncertainties were determined from the standard deviation in background noise levels by using NMRView (46).
HETex-BEST-TROSY experiment
HETex-BEST-TROSY (54) experiments were performed at 20°C on a Bruker AVANCE III HD 950-MHz spectrometer equipped with a cryo-TCI probe. A series of spectra was collected with relaxation delays of 339, 709, 1080, 1450 and 2006 ms with and without water saturation. The water signal intensity was measured at each relaxation delay to determine the polarization of water using the 1H–15N BEST-TROSY pulse sequence with an additional small flip angle reading pulse (0.1 μs). The hydrogen exchange rate kex was determined by simple fitting using Equations (1) and (2) for the amide proton polarizations:
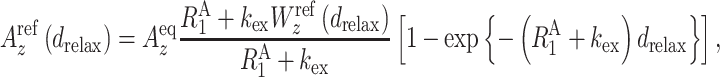 |
(1) |
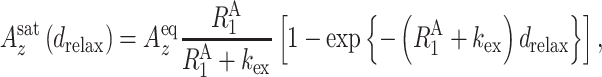 |
(2) |
where 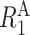 is the longitudinal relaxation rate of amide protons,
is the longitudinal relaxation rate of amide protons, 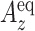 is the equilibrium amide proton polarization and
is the equilibrium amide proton polarization and 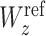 is the water polarization. All NMR spectra were processed by using the program NMRPipe (45) and analyzed by using the program NMRView (46). Least-squares fitting of the data was performed by using Gnuplot software (http://www.gnuplot.info).
is the water polarization. All NMR spectra were processed by using the program NMRPipe (45) and analyzed by using the program NMRView (46). Least-squares fitting of the data was performed by using Gnuplot software (http://www.gnuplot.info).
CLEANEX-PM experiment
CLEANEX-PM (phase-modulated CLEAN chemical exchange spectroscopy) (55) experiments were performed at 20°C on a Bruker AVANCE III HD 950-MHz spectrometer equipped with a cryo-TCI probe using a standard Bruker pulse program. A series of spectra was collected with mixing times for the CLEANEX spin lock of 7.5, 15, 30, 50 and 80 ms. The hydrogen exchange rate kex was determined by using established methods. All NMR spectra were processed by using the program NMRPipe (45) and analyzed by using the program NMRView (46). Least-squares fitting of the data was performed by using Gnuplot software (http://www.gnuplot.info).
Structural modeling of human TFIIH
Initially, 1000 structures of p621–158 were calculated as described in the ‘Structure calculation’ section, and then 399 structures were selected on the basis of violation criteria (distance violations <0.5 Å and dihedral angle violations <5°). These 399 structures were aligned to the BSD1 domain region (residues 107–147) of p62 in the cryo-EM structure of human apo TFIIH (PDB code 6NMI) by using the program PyMol (http://www.pymol.org). The BSD1 domain region (residues 107–147) of p62 in the cryo-EM structure was then replaced with the PH-D and BSD1 domain (residues 1–147) of p62 in the 399 structures of p621–158. Finally, undesirable models in which the PH-D of p62 clashed against other parts of TFIIH were removed, resulting in 275 model structures.
Docking structural modeling of human TFIIH and its p62-binding partners
To build structural models of TFIIH docking with its p62-binding partners, TFIIEα (29), p53 (31), DP1 (33), XPC (38) and UVSSA (39), five models in which the PH-D of p62 was located in free space were selected from the 275 structural models of TFIIH (Figure 5A). Structure calculation was done by the program Xplor-NIH (48,49) using the scripts ‘rigid_min.inp’ and ‘sa_cross_tor.inp’. We employed experimentally obtained intermolecular distance restraints in each complex of the p621–108 domain with TFIIEα (PDB code 2RNR), p53 (PDB code 2RUK), DP1 (PDB code 5GOW), XPC (PDB code 2RVB) and UVSSA (PDB code 5XV8).
Figure 5.
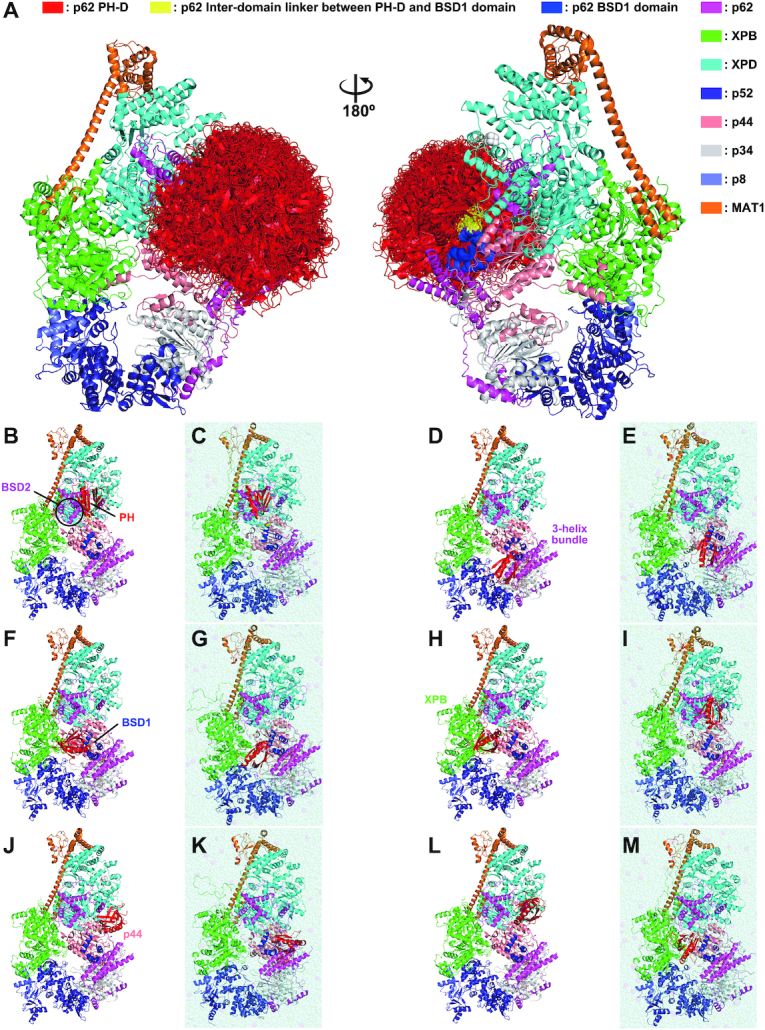
Structural model of human TFIIH. Structural model of human TFIIH built by a combination of the cryo-EM (PDB code 6NMI) and NMR (PDB code 7BUL) structures. The 275 models (A), single representative models (B, D, F, H, J, L) and their corresponding MD simulation models (C, E, G, I, K, M) are shown.
MD simulations of human TFIIH and its complexes with p62-binding partners
The initial structure used for MD simulations was prepared from the whole apo human TFIIH core complex, in which the NMR structure was used for the PH-D and BSD1 domain (residues 1–147) of p62 and the cryo-EM structure (PDB code 6NMI) was used for the other parts of TFIIH. The 275 model structures described above were classified in terms of the position of the PH-D, and 6 representative model structures (Figure 5B, D, F, H, J and L) were subjected to MD simulations. First, in a typical structure (Figure 5L), the missing regions and side chains in the cryo-EM structure, as well as the region joining the NMR and cryo-EM structures of p62 (residues 145–147), were built by MODELLER (56). The long loops at the termini (residues 1–31 and 731–782 of XPB, residues 1–14 of p44, residues 294–308 of p34, residues 69–71 of p8 and residues 213–309 of MAT1) were not modeled. Next, using the model structure in Figure 5L, the other five whole TFIIH structures (Figure 5B, D, F, H and J) and five TFIIH complexes (Figure 6) were built: the whole TFIIH structures were aligned to the model structure by using a helix of the three-helix bundle of p62 (residues 487–511), and the PH-D and BSD1 domain (residues 1–143) of each structure were replaced with the same domains of the model structure.
Figure 6.

MD simulation models of the complexes of human TFIIH with p62-binding partners. (A) Overall view. Extended view of the area indicated by the square in (A): (B) complex of TFIIH and TFIIEα; (C) complex of TFIIH and p53; (D) complex of TFIIH and DP1; (E) complex of TFIIH and XPC; and (F) complex of TFIIH and UVSSA.
The systems for MD simulations of the model TFIIH structures were prepared by using the psfgen, solvate and autoionize plugins implemented in VMD (57). The protonation states of His, Asp, Glu and Lys were determined by using pKa values calculated with PROPKA (58,59) implemented in PDB2PQR (60) under the pH 7 condition. Missing hydrogen atoms were inserted in accordance with topology in the CHARMM36m force field (61–63). The N- and C-termini were set to NH3+ and COO−, respectively. The coordinate bonds among anionic cysteine or histidine residues and zinc ions were incorporated, as defined in the CHARMM36m force field, and the oxidized iron–sulfur cluster and the coordinate bonds of anionic cysteine residues with the iron–sulfur cluster were also incorporated by using the topologies and parameters determined by Chang and Kim (64). Here, both phosphorylated serine and threonine in the p62-binding partner p53 were set to the dianionic state. The MD unit cell was set to a rectangular cell, and the cell size was determined such that the distance between the edge of the cell and the centered model structure was at least 15 Å. Using the TIP3P water model (65), the cell was filled with water molecules, and counterions (Na+) and 150 mM NaCl were added. The final systems consisted of ∼504 000 atoms.
The all-atom MD simulations were performed by using the MD program package GROMACS version 2019.6 (66–68) with the CHARMM36m force field under periodic boundary conditions. The topology and parameter files were converted into the GROMACS format by using ParmEd (https://github.com/ParmEd/ParmEd). Electrostatic interactions were handled by the smooth particle mesh Ewald method (69), and the van der Waals interactions were truncated by the switching function with a range of 10–12 Å. Bond lengths involving hydrogen atoms were constrained by the P-LINKS algorithm (70). In all simulations, position constraints at the Cα atoms with a 60 kJ mol−2 nm−2 force constant were applied to the TFIIH structure, except for the PH-D, the interdomain linker and the N-terminal part of the BSD1 domain (residues 1–114) of p62, the modeled loops that were originally missing in the cryo-EM structure, and the p62-binding partners. The temperature and pressure were 298 K and 1 atm, respectively. Before the production run, energy minimization by the steepest descent method and three equilibration runs were performed sequentially: the first 125-ps run with NVT ensemble and a 1-fs timestep, and the second and third 100-ps runs with NPT ensemble and a 2-fs timestep. The thermostat in the first and third equilibration runs and the production run was the Nosé–Hoover scheme (71,72), and that in the second equilibration run was the Berendsen algorithm (73). The barostat in the second equilibration run was the Berendsen algorithm (73), and that in the third equilibration run and the production run was the Parrinello–Rahman approach (74,75). After the equilibration runs, a 100-ns production run was carried out with NPT ensemble and a 2-fs timestep. These MD simulations were performed for each initial model structure of the six whole TFIIH models and the five complex models with p62-binding partners (1.1 μs in aggregation of the production runs).
The root-mean-square fluctuation (RMSF) of Cα atoms, the solvent-accessible surface area (SASA) of the amide hydrogen and the protein secondary structures were calculated by using each 100-ns trajectory. Snapshots were saved every 1 ns (100 snapshots in a trajectory). As an estimate of the RMSF, an averaged structure was obtained by structural alignment of the snapshots using Cα atoms in the PH-D except for the N-terminus (residues 9–100). The RMSF values of the six apo TFIIH simulations or the five complex simulations were then averaged. The SASA of the amide hydrogen was calculated by the GROMACS module. The average SASA of each residue was obtained from each trajectory, and then the averaged SASA was calculated from the simulations for each apo TFIIH or each complex. Protein secondary structures were determined by using the DSSP program (76) implemented in CPPTRJ (77). The population of snapshots forming the helix or 310 helix structure was obtained for each trajectory, and its averaged value was calculated.
NMR chemical shift perturbation experiments
Samples of p62-binding partners, including TFIIEα (residues 378–439) (29), Ser46- and Thr55-phosphorylated p53 (residues 41–62) (31), DP1 (residues 392–410) (33), XPC (residues 124–142) (38) and UVSSA (residues 399–419) (39), were prepared as previously described. Each of the unlabeled samples was added to 0.1 mM 13C/15N-labeled p621–158 at a molar ratio of 1:1 except for DP1 (molar ratio 1:4) in 20 mM potassium phosphate (pH 6.8), 100 mM sodium chloride, 5 mM deuterated DTT and 10% D2O. 1H–15N HSQC spectra were acquired before and after the addition of unlabeled samples at 20°C on a Bruker AVANCE III HD 600-MHz spectrometer equipped with a cryo-TCI probe. Spectra were processed by NMRPipe (45) and analyzed by NMRView (46).
RESULTS AND DISCUSSION
Invisible PH-D of p62 in the cryo-EM structure of human TFIIH
Understanding the structure and dynamics of the PH-D of p62 in TFIIH is indispensable for elucidating the mechanism of recruitment of human TFIIH. At present, high-resolution cryo-EM structures of human and yeast TFIIH are available. The structure of human TFIIH has been resolved for an apo form (41,43), while that of yeast TFIIH has been resolved for a holo form in the transcription PIC (42). The corresponding structures of each subunit and the overall architecture of the core TFIIH are relatively similar, except for the corresponding PH-Ds of the yeast homolog Tfb1 (Saccharomyces cerevisiae) and human p62: the Tfb1 PH-D is clearly visible, whereas the human p62 PH-D is invisible (Supplementary Figure S1). Recently, an interesting structural study of human PIC has been reported in which all available cryo-EM data have been integrated to build a structural model of PIC in which the PH-D of p62 is visible (78).
Here, we initially compared the amino acid sequence of human p62 with that of its well-studied homolog Tfb1 (Figure 1A and Supplementary Figure S2). The sequence alignment identified a marked difference in the region between the PH and BSD1 domains. In human p62, the PH-D is immediately followed by the BSD1 domain; in yeast Tfb1, in contrast, a linker of ∼50 residues is present between these domains. This substantial sequence dissimilarity is reflected in their cryo-EM structures (Figure 1B). In both p62 and Tfb1, the BSD1 and BSD2 domains are aligned at nearly the same position with a beads-on-a-string-like topology, and contribute to stabilizing the core architecture by interacting with other four subunits: XPD, p52, p44 and p34 for p62; and Rad3, Tfb2, Ssl1 and Tfb4 for Tfb1. The PH-D of Tfb1 is also visualized in the yeast holo TFIIH structure (Figure 1B, right). Its PH-D lies apart from the BSD1 domain owing to its long linker, and interacts with another general transcription factor, Tfa1/TFIIEα (not shown in Figure 1B). In contrast, the PH-D of p62 is disordered and invisible in the human apo TFIIH structure (Figure 1B, left), suggesting that, despite the short interdomain linker, this domain is surface exposed and behaves flexibly in the absence of proteins other than TFIIH. The possible state of p62 in human holo TFIIH is discussed in detail below.
Figure 1.
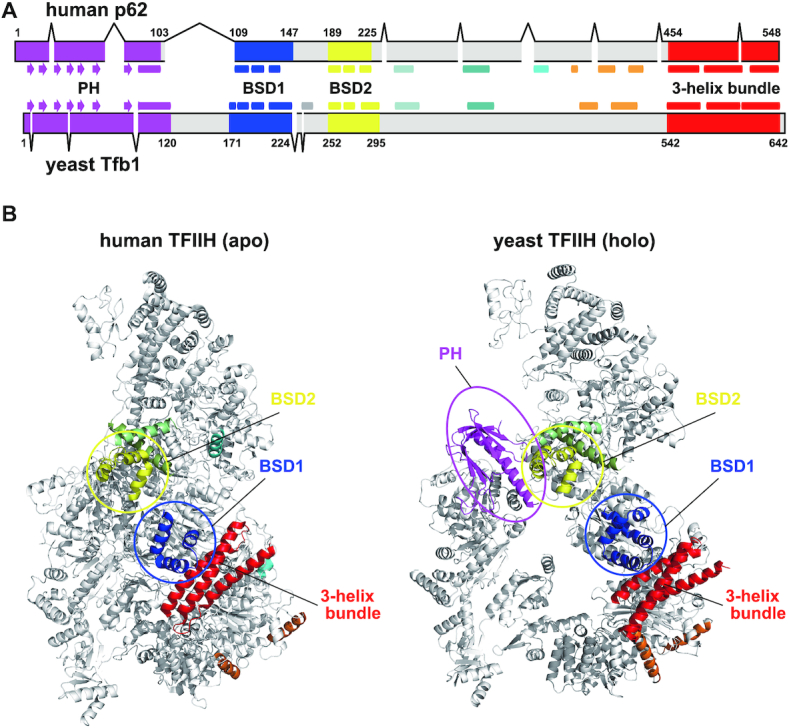
Structure comparison between human and yeast TFIIH p62/Tfb1. (A) Domain organization of TFIIH p62/Tfb1. The figure is based on sequence alignment of the two proteins (Supplementary Figure S2). Arrow, β-strand; rectangle, α-helix. (B) Ribbon representation of cryo-EM TFIIH structures. Left, human TFIIH, apo form (PDB code 6NMI); right, yeast TFIIH, holo form (PDB code 5OQM). For clarity, only the p62/Tfb1 domain is colored.
To explore the predicted flexible behavior of the PH-D of p62 in human apo TFIIH, we applied solution NMR because it is a powerful tool for investigating both the dynamics and structure of proteins in solution. Although NMR is not advantageous for investigating large proteins like TFIIH, the PH-D of p62 is likely to be free in apo TFIIH; we considered therefore that we would not need to target the whole TFIIH protein for our purpose. In the cryo-EM structure, the p62 subunit is visualized from Ala107 onward, just after the PH-D and before the BSD1 domain (Figure 1B, left). Considering this, we focused on the region including both the PH and BSD1 domains (residues 1–158, hereafter designated p621–158) (Figure 2A), and analyzed the secondary structure of p621–158 by NMR.
Figure 2.
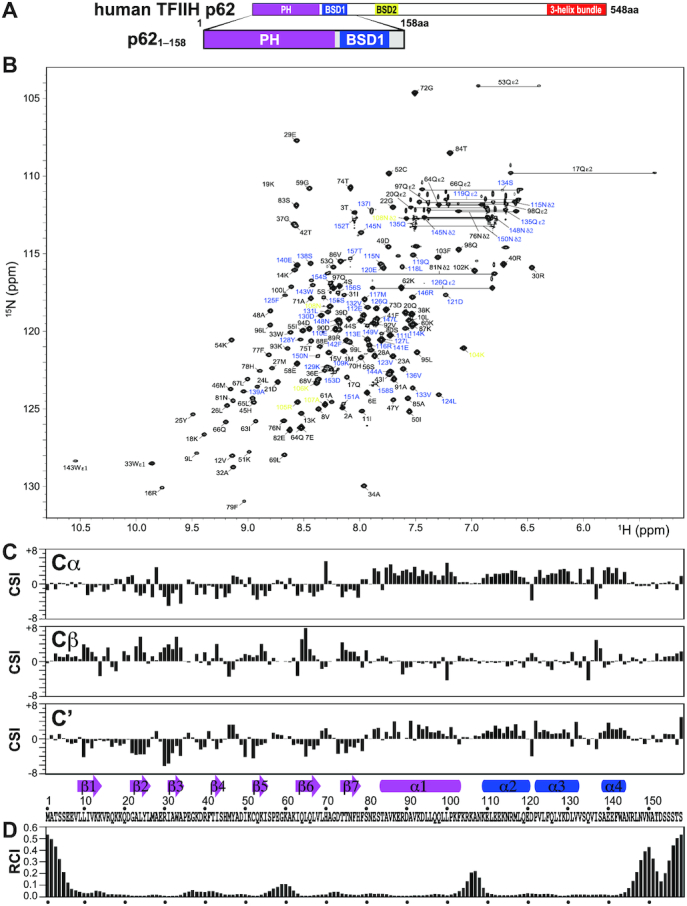
NMR signal assignment and secondary structure of the PH and BSD1 domains of human TFIIH p62. (A) p621–158 construct used in the NMR experiments. (B) 1H–15N-HSQC spectrum. Residues 1–103, 104–108 and 109–158 are labeled in black, yellow and blue, respectively. (C) Chemical shift index (CSI) of 13Cα, 13Cβ and 13C′, and identified secondary structure elements. Arrow: β-strand; rectangle; α-helix. The secondary structure identification was performed using the CSI 2.0 (http://csi.wishartlab.com) web server (79). (D) Random coil index (RCI), calculated by using the difference between observed and reference random coil shifts of 13Cα, 13Cβ, 13C′, 15N and 1H. RCI should be proportional to the flexibility of the protein backbone (80).
The p621–158 construct exhibited well-dispersed signals on a 1H–15N-HSQC spectrum (Figure 2B). Almost all backbone and side-chain signals were assigned without using any deuterated samples. The chemical shift data (CSI) (79) of the backbone atoms indicated that there were seven β-strands and one α-helix in the PH-D and three α-helices in the BSD1 domain, consistent with the structures of their isolated forms (40). No secondary structure elements were found in the interdomain linker region between the α1 and α2 helices (Figure 2C). Based on the chemical shift data, it is predicted (RCI) (80) that the interdomain linker region is flexible (Figure 2D).
Structure of the tandem PH and BSD1 domains of human p62
Next, we determined the tertiary structure of p621–158 by using NMR (Figure 3). In total, 3338 NOE-derived distance restraints, 96 hydrogen bond restraints and 381 dihedral angle restraints were collected and used to define the structure (Figure 3A–D and Table 1). Although the RMSD of the backbone atoms for residues 7–147 in the 20 structures was as high as 8.81 ± 2.55 Å, each domain structure was well determined with RMSD values of 0.46 ± 0.10 Å for residues 7–103 (PH-D) and 0.76 ± 0.34 Å for residues 109–147 (BSD1), suggesting that the interdomain linker (104–108) has high mobility (Table 1). In the PH-D, the seven β-strands (β1–β7) form a β-sandwich, in which strands β1–β4 and β5–β7 form the first and second antiparallel β-sheets, respectively. These two β-sheets are linked by a contact between strands β1 and β7, and by a 310 helix connecting strands β4 and β5 (Figure 3E). The following α-helix (α1) is inserted between the two β-sheets and extensively interacts with residues in both, thereby stabilizing the β-sandwich. Pro101 is likely to participate in the α-helix because Phe103 stabilizes the helix by inserting into the hydrophobic core. The structure of the PH-D in p621–158 is essentially identical to that in the isolated form (40). Likewise, the structure of the BSD1 domain in p621–158 is essentially the same as that in the isolated form, which has been determined by another research group (PDB code 2DII) although the study has not been published.
Figure 3.
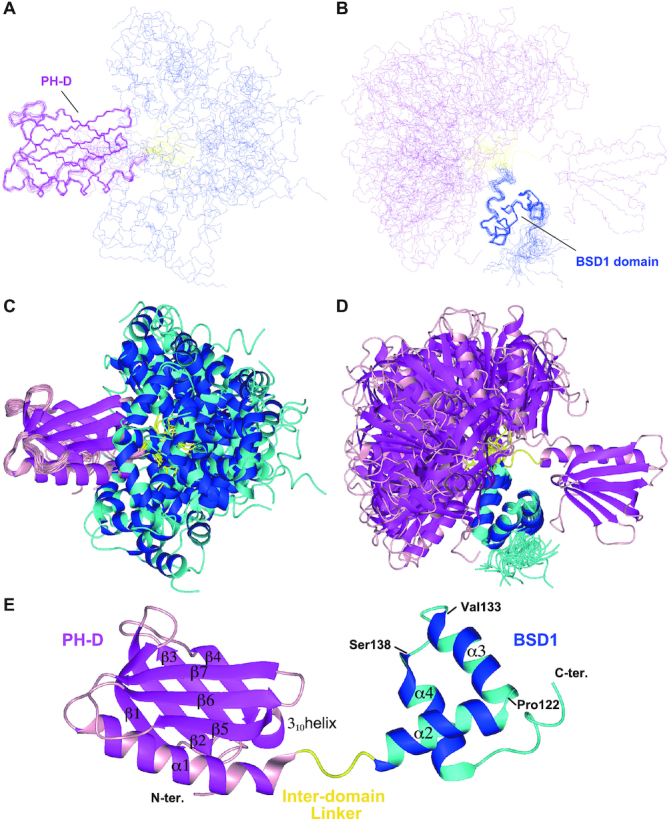
Solution structure of the PH and BSD1 domains of human TFIIH p62. Overlay of the backbone structures (A, B) and ribbon representation (C, D) of the 20 best structures, superimposed on the PH (A, C) and BSD1 (B, D) domains. (E) Ribbon representation of a single structure. The PH-D is shown in magenta/pink, the BSD1 domain in blue/cyan and the interdomain linker in yellow.
Here, we describe the structure in a little more detail. The first helix (α2) of the BSD1 domain bends nearly at a right angle to the second helix (α3) at Pro122 (Figure 3E). After the turn (Val133–Ser138), the third helix (α4) runs antiparallel to helix α3 and perpendicularly to helix α2. These three helices interact with one another, rendering the BSD1 domain stable and compact. Lys114, Met117 and Leu118 in α2, Val123, Leu124, Phe125, Leu127, Tyr128, Leu131 and Val132 in α3, and Ala139, Phe142, Trp143, Arg146 and Leu147 in α4 form the hydrophobic core. The turn between helices α3 and α4, where Val136 and Ile137 closely interact with Leu131, looks rigid. Ile137 also contacts Val132 and Phe142, participating in the formation of the hydrophobic core.
Dynamics of the tandem PH and BSD1 domains of human p62
To investigate the suggested high mobility of the interdomain linker quantitatively, we measured the 15N relaxation rates R1 and R2 (Figure 4A–C and Supplementary Figure S3) and 15N–{1H} NOE for p621–158 by NMR (Figure 4D and Supplementary Figure S3). These relaxation data define how much each residue in the interdomain linker contributes to the mobility. Lys104 and Arg105 clearly displayed higher mobility as compared with the residues before the linker. Lys104 and Arg105 exhibited, respectively, R2/R1 values of 24.1 and 27.5, and 15N–{1H} NOE values of 0.556 and 0.502. These values were close to those of 21.7 and 25.6 (R2/R1), and 0.562 and 0.605 (15N–{1H} NOE) observed for Glu110 and Leu111, the N-terminal residues in the first helix (α2) of the BSD1 domain. The following four residues, Lys106, Ala107, Asn108 and Lys109, displayed even higher mobility with R2/R1 values of 17.9, 12.9, 9.5 and 11.6, and 15N–{1H} NOE values of 0.451, 0.319, 0.297 and 0.431, respectively.
Figure 4.
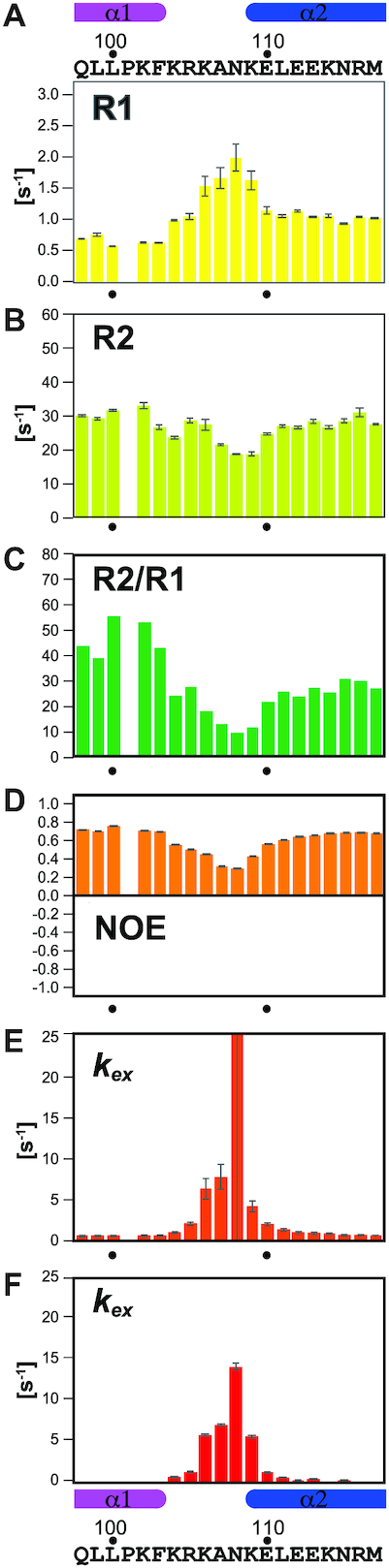
Dynamics of the linker between the PH and BSD1 domains of human TFIIH p62. (A) 15N longitudinal relaxation rate (R1). (B) Transverse relaxation rate (R2). (C) Ratio of R2 to R1. (D) 15N–{1H} NOE. Water–amide proton exchange rate (kex) measured by HETex-BEST-TROSY (54) (E) and CLEANEX-PM (55) (F).
The high mobility of the interdomain linker residues was confirmed in both the HETex-BEST-TROSY experiment, where the amide proton solvent exchange rates were measured as 1.05 ± 0.11 s−1 (Lys104), 2.08 ± 0.20 s−1 (Arg105), 6.32 ± 1.22 s−1 (Lys106), 7.73 ± 1.51 s−1 (Ala107), >10 s−1 (Asn108), 4.21 ± 0.64 s−1 (Lys109), 2.05 ± 0.18 s−1 (Glu110) and 1.40 ± 0.14 s−1 (Leu111) (Figure 4E and Supplementary Figure S3), and the CLEANEX-PM experiment, in which the rates were 0.52 ± 0.05 s−1 (Lys104), 1.17 ± 0.08 s−1 (Arg105), 5.63 ± 0.09 s−1 (Lys106), 6.86 ± 0.15 s−1 (Ala107), 13.9 ± 0.40 s−1 (Asn108), 5.42 ± 0.18 s−1 (Lys109), 1.10 ± 0.06 s−1 (Glu110) and 0.45 ± 0.05 s−1 (Leu111) (Figure 4F and Supplementary Figure S3). In all experiments, Asn108 displayed the highest mobility among the interdomain linker residues. Thus, this highly mobile interdomain linker causes the dynamic behavior of the PH and BSD1 domains in solution.
Structural model of human TFIIH
Our results enabled us to complete a structural model of the whole human apo TFIIH core complex by first combining the cryo-EM structure with our NMR structure of p621–158 and then considering the dynamics of p62. To build the structural model, initially 399 NMR structures of p621–158 were chosen from the total 1000 calculated structures on the basis of violation criteria. The structure of the BSD1 domain in p621–158 is essentially identical to that of the isolated form (PDB code 2DII), which was used to build p62 in the cryo-EM structure of human apo TFIIH, enabling the BSD1 domain of p621–158 to be placed at the same position in the cryo-EM structure. Hence, the BSD1 domain in the cryo-EM structure could be replaced with the PH and BSD1 domains of our 399 NMR structures of p621–158. Ultimately, we selected 275 models in which the PH-D of p62 did not clash against other parts of TFIIH. Of note, nearly 70% (275/399) of the models had no steric clashes.
The structural models allow us to visualize the dynamic behavior of the PH-D of p62 in apo TFIIH (Figure 5A). The PH-D can move close to both the BSD2 domain (Figure 5B) and the three-helix bundle (Figure 5D), as well as the BSD1 domain (Figure 5F). In addition to its own p62 subunit, it can move close to the C-terminal RecA-like (RecA2) domain of XPB (Figure 5H) and the von Willebrand factor A domain of p44 (Figure 5J). This spatially proximity is in accordance with the results of a chemical cross-linking/mass spectrometry study that detected cross-links between Lys38 of p62 and Lys549 of XPB, and between Lys38 of p62 and Lys126 of p44 (81). Notably, in the majority of our models, the PH-D occupies free space (Figure 5L).
To refine the docking structural models, we performed all-atom MD simulations of the apo whole TFIIH structure. The MD simulations for six representative structural models (Figure 5B, D, F, H, J and L) were carried out in solution, and the snapshot structures obtained at 100 ns are illustrated in Figure 5C, E, G, I, K and M. In the MD simulations, the PH-D, the interdomain linker and the N-terminal part of BSD1 domain of p62 (residues 1–114) were freely moving, whereas the Cα atoms in the other part were restrained to the cryo-EM structure (see the ‘Materials and Methods’ section). The structures of the PH-D itself were retained (Supplementary Figure S4A), but their positions and orientations changed from the initial structure. In MD simulations of the model structures in Figure 5B, D and F, the PH-D fluctuated around its initial position because it interacted with the BSD2 domain (Figure 5C), the three-helix bundle (Figure 5E) or both the BSD1 domain and the three-helix bundle (Figure 5G) of p62. In the MD simulations of the model structures in Figure 5H, J and L, the PH-D moved from its initial position largely because of the weak interactions that held it in its initial place (Figure 5I, K and M).
This dynamic behavior of the PH-D originates from the high mobility of the interdomain linker (residues 104–108) between the PH-D and BSD1 domain. To estimate the interdomain linker mobility, the RMSF of Cα atoms was calculated from the MD trajectories (Supplementary Figure S5A). The fluctuation in the PH-D except for the N-terminus was up to ∼2 Å. In contrast, the RMSF of the interdomain linker region was markedly higher, indicating that the conformation of the linker is flexible. We found that the flexibility of the interdomain linker region is related to the SASA of the amide hydrogen in backbones (Supplementary Figure S6A). The amide hydrogens in the linker region are exposed to solvent. The dynamic behavior of the PH-D and the mobility of the interdomain linker region observed in MD simulations are in good agreement with our NMR measurements.
Thus, the PH-D of p62 is able to move dynamically around the surface of the core TFIIH complex, transiently interacting with some regions; however, the spatial distribution is limited to some extent owing to the short interdomain linker. As mentioned above, the structural model of human PIC has been reported (78). In that model, the PH-D of p62 is unambiguously positioned between XPB and XPD (Supplementary Figure S7A, left). In our structural model of apo TFIIH (Supplementary Figure S7A, right), it is impossible for the PH-D to sit in the same position because it is too far away from the BSD1 domain. Structural changes of either the PH-D or BSD1 domain, or both, would be required to achieve that positioning. Notably, the C-terminal part of the α1 helix of the PH-D (residues 101–103) and the N-terminal part of the α2 helix of the BSD1 domain (residues 109–111) were unfolded in the published model of holo TFIIH (78) (Supplementary Figure S7B, left; compare with our apo TFIIH model in Figure S7B, right). During formation of the PIC at a transcription start site, the PH and BSD1 domains extensively interact with another transcription factor, TFIIEα (28,29,78). It is not clear whether the partial helix unfolding of the PH-D and BSD1 domain is a result of binding to TFIIEα. Further studies are needed to explore the possibility.
Complex structural models of human TFIIH and p62-binding partners
We have previously determined the solution structures of the PH-D of p62 in complex with five binding partners: the acidic domain of TFIIEα (29); the transactivation domains of the tumor suppressor p53 (31) and the cell cycle regulatory transcription factor DP1 (33); and the acidic regions of the nucleotide excision repair factors XPC (38) and UVSSA (39). All of these proteins bind specifically to the PH-D of p62 via an intrinsically disordered acidic region. Upon binding, this region forms an extended string-like conformation and interacts with a basic surface of the PH-D via extensive electrostatic contacts. Simultaneously, it inserts a phenylalanine or tryptophan residue surrounded by acidic residues into a pocket in the PH-D, enhancing binding specificity. In those studies, we used the isolated form of the PH-D, consisting of residues 1–108 (p621–108). The current study now demonstrates that this construct design is appropriate because Asn108 is located at the C-terminus of the interdomain linker. Nevertheless, it has been suggested that some binding partners interact with several residues in the C-terminal region of the p621–108 construct (29,38,39). Here, based on the complete structural model of the human apo TFIIH core complex (Figure 5), we have explored whether the p62-binding partners would be able to bind to the PH-D of p62 in TFIIH in the same manner as they bind to p621–108.
To build structural models of TFIIH docked to its p62-binding partners, we selected five structures with sufficient variety from the 275 structural models of TFIIH (Figure 5A). In almost all calculated models, TFIIEα, p53, DP1, XPC and UVSSA could bind to the PH-D of p62 in TFIIH without steric clashes except for the TFIIEα–TFIIH complex, in which the C-terminus of the acidic domain of TFIIEα collided with the N-terminus of the BSD1 domain of p62 (data not shown).
To refine the docking models of the TFIIH with its p62-binding partners, MD simulations were performed for the complex structures. The interactions between the PH-D and the p62-binding partner were maintained for all complexes, and none of the partners dissociated during the simulations (Figure 6). The relative positions and orientations of the PH-D and the binding partner changed with respect to the whole TFIIH, owing to the flexibility of the linker region of p62. As observed for apo TFIIH, the RMSF (Supplementary Figure S5B) and SASA of the amide hydrogens (Supplementary Figure S6B) indicated that the linker region is flexible, even in complex with binding partners.
It should be noted that TFIIEα differs from the other p62-binding partners in possessing some structure in the binding site, although the N-terminal intrinsically disordered acidic region is mainly responsible for binding to the PH-D (29); however, our MD simulation indicates that a stable complex forms between the TFIIEα acidic domain and the p62 PH-D in human TFIIH without any helix unfolding of the PH-D or BSD1 domain (Supplementary Figure S4B). The present complex model of the TFIIEα acidic domain bound to p62 PH-D is likely to correspond to the initiation step in formation of the final PIC. As mentioned above, yeast Tfb1 PH-D is followed by a long (∼50-residue) linker, with the result that the PH-D sits at a different location in PIC as compared with the position of the human p62 PH-D. Even if the p62 PH-D could be located at the same position as the Tfb1 PH-D, it seems unlikely that TFIIEα could bind to the p62 PH-D due to steric hindrance from the structured part of the acidic domain; both the structure in the binding site of TFIIEα and the length of the linker of p62 are likely to determine their positions in PIC.
These results verify the importance of an intrinsically disordered region in recognition of the PH-D of p62 in TFIIH. Namely, the PH-D in TFIIH is sufficiently free to allow access to intrinsically disordered partners, but not completely free to allow access by a structured protein because of the short interdomain linker of p62, which restricts the range of motion of the PH-D in TFIIH as mentioned above. Although the dynamic behavior of the PH-D is essential for TFIIH recruitment, a completely free state would allow approach by not only true partners but also many other irrelevant proteins, resulting in inefficient recruitment. It is likely that this proper spatial restriction serves to enhance target selectivity.
It has recently been reported that monoubiquitination of Lys414 of UVSSA facilitates transfer of TFIIH to RNA polymerase II stalled at DNA lesions during transcription-coupled nucleotide excision repair (82). Our MD simulation shows that Lys414, located at the C-terminus of the p62-binding site, remains accessible even though UVSSA is bound to the PH-D of p62 in TFIIH.
The refined complex models also suggest some electrostatic interactions between acidic residues around the C-terminus of the binding sites of partners and lysine residues in the interdomain linker of p62: namely, Asp436 of TFIIEα and Lys104 of p62; Glu435 of TFIIEα and Lys106 of p62 (Supplementary Figure S8A); Glu62 of p53 and Lys106 of p62 (Supplementary Figure S8B); Glu138 of XPC and Lys104 of p62 (Supplementary Figure S8D); and Glu413 of UVSSA and Lys104 of p62 (Supplementary Figure S8E). However, there were no electrostatic interactions in the DP1–TFIIH complex, even though DP1 has four consecutive acidic residues at the C-terminus (Supplementary Figure S8C).
Thus, we performed NMR chemical shift perturbation experiments for 15N-labeled p621–158 with the five binding partners and identified significant chemical shift changes of these lysine residues in the interdomain linker of p62 when adding all binding partners except for DP1 (Supplementary Figure S9). The NMR titration experiments were well correlated with the complex models observed in MD simulations. In contrast, Arg105 of p62 displayed a large chemical shift change, but the MD models indicated that this basic residue does not participate in direct binding to the partners except for TFIIEα, where Glu435 or Asp436 might interact with Arg105 of p62; the large chemical shift change of Arg105 in other cases is likely to be caused by interactions of the neighboring lysine residues, Lys104 and/or Lys106, with binding partners (Supplementary Figure S9).
CONCLUSION
Among the 10 subunits of TFIIH, p62 is relatively unique. In addition to its involvement in both formation of the core architecture and recruitment of TFIIH, the cryo-EM structure revealed that an unstructured and extended segment of p62 (residues 266–287) is inserted into the DNA-binding cavity of XPD, suggesting that p62 may function in regulating the helicase activity of XPD (42,43,78). Again, the dynamical behavior of p62 is crucial for TFIIH function. In the present study, we have revealed the dynamic structural model of human apo TFIIH core by using NMR and MD simulations based the cryo-EM structure, and provided the first structural view of the dynamic behavior of the PH-D of p62 in human apo TFIIH, revealing a different range of motion from the PH-D in the previously modeled human holo TFIIH (78) and yeast holo TFIIH (42) structures. We have also presented models of TFIIH complexed with five p62-binding partners refined by MD simulations. These will be useful for discussing interactions between the full-length proteins. For example, a previous study presented a model of a cryo-EM structure of the full-length p53–RNA polymerase II complex docked in the PIC; however, the N-terminal transactivation domain of p53 was invisible owing to its structural flexibility (83). We believe that the complementary approach (NMR, MD and cryo-EM) demonstrated here will help us to better understand the dynamics of such key proteins in large, multi-subunit protein complexes.
DATA AVAILABILITY
Coordinates for the TFIIH p621–158 have been deposited in the RCSB Protein Data Bank (PDB) under ID code 7BUL, and chemical shifts, 15N relaxation parameters (T1 = 1/R1, T2 = 1/R2 and heteronuclear NOE) and hydrogen exchange rates (kex) have been deposited in the BioMagResBank (BMRB) under accession number 36340.
Supplementary Material
ACKNOWLEDGEMENTS
This research used the computational resources of TSUBAME3.0 provided by the Tokyo Institute of Technology through BINDS (project ID: JP20am0101009).
Contributor Information
Masahiko Okuda, Graduate School of Medical Life Science, Yokohama City University, 1-7-29 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan.
Toru Ekimoto, Graduate School of Medical Life Science, Yokohama City University, 1-7-29 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan.
Jun-ichi Kurita, Graduate School of Medical Life Science, Yokohama City University, 1-7-29 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan.
Mitsunori Ikeguchi, Graduate School of Medical Life Science, Yokohama City University, 1-7-29 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan; RIKEN Medical Sciences Innovation Hub Program, 1-7-22 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan.
Yoshifumi Nishimura, Graduate School of Medical Life Science, Yokohama City University, 1-7-29 Suehiro-cho, Tsurumi-ku, Yokohama 230-0045, Japan; Graduate School of Integrated Sciences for Life, Hiroshima University, 1-4-4 Kagamiyama, Higashi-Hiroshima 739-8258, Japan.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ministry of Education, Culture, Sports, Science and Technology [07022019 to Y.N., hp200129 and 18H05426 to M.I.]; Japan Agency for Medical Research and Development [JP18am0101033 (support number 0004) to Y.N. and JP20am0101009 (support number 2515) to M.I.]; Japan Society for the Promotion of Science [ 16K07277 to M.O.]; RIKEN Dynamic Structural Biology Project [to M.I.]. Funding for open access charge: Japan Agency for Medical Research and Development [JP18am0101033 (support number 0004) to Y.N.]:
Conflict of interest statement. None declared.
REFERENCES
- 1. Conaway R.C., Conaway J.W.. An RNA polymerase II transcription factor has an associated DNA-dependent ATPase (dATPase) activity strongly stimulated by the TATA region of promoters. Proc. Natl Acad. Sci. U.S.A. 1989; 86:7356–7360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Feaver W.J., Gileadi O., Kornberg R.D.. Purification and characterization of yeast RNA polymerase II transcription factor b. J. Biol. Chem. 1991; 266:19000–19005. [PubMed] [Google Scholar]
- 3. Gerard M., Fischer L., Moncollin V., Chipoulet J.M., Chambon P., Egly J.M.. Purification and interaction properties of the human RNA polymerase B(II) general transcription factor BTF2. J. Biol. Chem. 1991; 266:20940–20945. [PubMed] [Google Scholar]
- 4. Xiao H., Pearson A., Coulombe B., Truant R., Zhang S., Regier J.L., Triezenberg S.J., Reinberg D., Flores O., Ingles C.J.et al.. Binding of basal transcription factor TFIIH to the acidic activation domains of VP16 and p53. Mol. Cell. Biol. 1994; 14:7013–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tong X., Drapkin R., Reinberg D., Kieff E.. The 62- and 80-kDa subunits of transcription factor IIH mediate the interaction with Epstein–Barr virus nuclear protein 2. Proc. Natl Acad. Sci. U.S.A. 1995; 92:3259–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Qadri I., Conaway J.W., Conaway R.C., Schaack J., Siddiqui A.. Hepatitis B virus transactivator protein, HBx, associates with the components of TFIIH and stimulates the DNA helicase activity of TFIIH. Proc. Natl Acad. Sci. U.S.A. 1996; 93:10578–10583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu H., Fisher R.P., Bailey P., Levine A.J.. The CDK7–cycH–p36 complex of transcription factor IIH phosphorylates p53, enhancing its sequence-specific DNA binding activity in vitro. Mol. Cell. Biol. 1997; 17:5923–5934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rochette-Egly C., Adam S., Rossignol M., Egly J.M., Chambon P.. Stimulation of RAR alpha activation function AF-1 through binding to the general transcription factor TFIIH and phosphorylation by CDK7. Cell. 1997; 90:97–107. [DOI] [PubMed] [Google Scholar]
- 9. Chen D., Riedl T., Washbrook E., Pace P.E., Coombes R.C., Egly J.M., Ali S.. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol. Cell. 2000; 6:127–137. [PubMed] [Google Scholar]
- 10. Lee D.K., Duan H.O., Chang C.. From androgen receptor to the general transcription factor TFIIH. Identification of cdk activating kinase (CAK) as an androgen receptor NH2-terminal associated coactivator. J. Biol. Chem. 2000; 275:9308–9313. [DOI] [PubMed] [Google Scholar]
- 11. Liu J., He L., Collins I., Ge H., Libutti D., Li J., Egly J.M., Levens D.. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol. Cell. 2000; 5:331–341. [DOI] [PubMed] [Google Scholar]
- 12. Compe E., Drané P., Laurent C., Diderich K., Braun C., Hoeijmakers J.H., Egly J.M.. Dysregulation of the peroxisome proliferator-activated receptor target genes by XPD mutations. Mol. Cell. Biol. 2005; 25:6065–6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Compe E., Malerba M., Soler L., Marescaux J., Borrelli E., Egly J.M.. Neurological defects in trichothiodystrophy reveal a coactivator function of TFIIH. Nat. Neurosci. 2007; 10:1414–1422. [DOI] [PubMed] [Google Scholar]
- 14. Yankulov K.Y., Pandes M., McCracken S., Bouchard D., Bentley D.L.. TFIIH functions in regulating transcriptional elongation by RNA polymerase II in Xenopus oocytes. Mol. Cell. Biol. 1996; 16:3291–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Assfalg R., Lebedev A., Gonzalez O.G., Schelling A., Koch S., Iben S.. TFIIH is an elongation factor of RNA polymerase I. Nucleic Acids Res. 2012; 40:650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Narita T., Narita K., Takedachi A., Saijo M., Tanaka K.. Regulation of transcription elongation by the XPG–TFIIH complex is implicated in Cockayne syndrome. Mol. Cell. Biol. 2015; 35:3178–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roy R., Adamczewski J.P., Seroz T., Vermeulen W., Tassan J.P., Schaeffer L., Nigg E.A., Hoeijmakers J.H., Egly J.M.. The MO15 cell cycle kinase is associated with the TFIIH transcription–DNA repair factor. Cell. 1994; 79:1093–1101. [DOI] [PubMed] [Google Scholar]
- 18. Fesquet D., Morin N., Doree M., Devault A.. Is Cdk7/cyclin H/MAT1 the genuine cdk activating kinase in cycling Xenopus egg extracts?. Oncogene. 1997; 15:1303–1307. [DOI] [PubMed] [Google Scholar]
- 19. Larochelle S., Pandur J., Fisher R.P., Salz H.K., Suter B.. Cdk7 is essential for mitosis and for in vivo Cdk-activating kinase activity. Genes Dev. 1998; 12:370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu L., Chen P., Hwang J.J., Barsky L.W., Weinberg K.I., Jong A., Starnes V.A.. RNA antisense abrogation of MAT1 induces G1 phase arrest and triggers apoptosis in aortic smooth muscle cells. J. Biol. Chem. 1999; 274:5564–5572. [DOI] [PubMed] [Google Scholar]
- 21. Schaeffer L., Roy R., Humbert S., Moncollin V., Vermeulen W., Hoeijmakers J.H., Chambon P., Egly J.M.. DNA repair helicase: a component of BTF2 (TFIIH) basic transcription factor. Science. 1993; 260:58–63. [DOI] [PubMed] [Google Scholar]
- 22. Drapkin R., Reardon J.T., Ansari A., Huang J.C., Zawel L., Ahn K., Sancar A., Reinberg D.. Dual role of TFIIH in DNA excision repair and in transcription by RNA polymerase II. Nature. 1994; 368:769–772. [DOI] [PubMed] [Google Scholar]
- 23. Guzder S.N., Qiu H., Sommers C.H., Sung P., Prakash L., Prakash S.. DNA repair gene RAD3 of S. cerevisiae is essential for transcription by RNA polymerase II. Nature. 1994; 367:91–94. [DOI] [PubMed] [Google Scholar]
- 24. Guzder S.N., Sung P., Bailly V., Prakash L., Prakash S.. RAD25 is a DNA helicase required for DNA repair and RNA polymerase II transcription. Nature. 1994; 369:578–581. [DOI] [PubMed] [Google Scholar]
- 25. Egly J.M., Coin F.. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst.). 2011; 10:714–721. [DOI] [PubMed] [Google Scholar]
- 26. Compe E., Egly J.M.. TFIIH: when transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012; 13:343–354. [DOI] [PubMed] [Google Scholar]
- 27. Lehmann A.R. DNA repair-deficient diseases, xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. Biochimie. 2003; 85:1101–1111. [DOI] [PubMed] [Google Scholar]
- 28. Di Lello P., Jenkins M.M.L., Mas C., Langlois C., Malitskaya E., Fradet-Turcotte A., Archambault J., Legault P., Omichinski J.G.. p53 and TFIIEalpha share a common binding site on the Tfb1/p62 subunit of TFIIH. Proc. Natl Acad. Sci. U.S.A. 2008; 105:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Okuda M., Tanaka A., Satoh M., Mizuta S., Takazawa M., Ohkuma Y., Nishimura Y.. Structural insight into the TFIIE–TFIIH interaction: TFIIE and p53 share the binding region on TFIIH. EMBO J. 2008; 27:1161–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Lello P., Jenkins L.M., Jones T.N., Nguyen B.D., Hara T., Yamaguchi H., Dikeakos J.D., Appella E., Legault P., Omichinski J.G.. Structure of the Tfb1/p53 complex: insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell. 2006; 22:731–740. [DOI] [PubMed] [Google Scholar]
- 31. Okuda M., Nishimura Y.. Extended string binding mode of the phosphorylated transactivation domain of tumor suppressor p53. J. Am. Chem. Soc. 2014; 136:14143–14152. [DOI] [PubMed] [Google Scholar]
- 32. Mas C., Lussier-Price M., Soni S., Morse T., Arseneault G., Di Lello P., Lafrance-Vanasse J., Bieker J.J., Omichinski J.G.. Structural and functional characterization of an atypical activation domain in erythroid Kruppel-like factor (EKLF). Proc. Natl Acad. Sci. U.S.A. 2011; 108:10484–10489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Okuda M., Araki K., Ohtani K., Nishimura Y.. The interaction mode of the acidic region of the cell cycle transcription factor DP1 with TFIIH. J. Mol. Biol. 2016; 428:4993–5006. [DOI] [PubMed] [Google Scholar]
- 34. Di Lello P., Nguyen B.D., Jones T.N., Potempa K, Kobor M.S., Legault P., Omichinski J.G.. NMR structure of the amino-terminal domain from the Tfb1 subunit of TFIIH and characterization of its phosphoinositide and VP16 binding sites. Biochemistry. 2005; 44:7678–7686. [DOI] [PubMed] [Google Scholar]
- 35. Langlois C., Mas C., Di Lello P., Jenkins L.M., Legault P., Omichinski J.G.. NMR structure of the complex between the Tfb1 subunit of TFIIH and the activation domain of VP16: structural similarities between VP16 and p53. J. Am. Chem. Soc. 2008; 130:10596–10604. [DOI] [PubMed] [Google Scholar]
- 36. Chabot P.R., Raiola L., Lussier-Price M., Morse T., Arseneault G., Archambault J., Omichinski J.G.. Structural and functional characterization of a complex between the acidic transactivation domain of EBNA2 and the Tfb1/p62 subunit of TFIIH. PLoS Pathog. 2014; 10:e1004042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lafrance-Vanasse J., Arseneault G., Cappadocia L., Legault P., Omichinski J.G.. Structural and functional evidence that Rad4 competes with Rad2 for binding to the Tfb1 subunit of TFIIH in NER. Nucleic Acids Res. 2013; 41:2736–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Okuda M., Kinoshita M., Kakumu E., Sugasawa K., Nishimura Y.. Structural insight into the mechanism of TFIIH recognition by the acidic string of the nucleotide excision repair factor XPC. Structure. 2015; 23:1827–1837. [DOI] [PubMed] [Google Scholar]
- 39. Okuda M., Nakazawa Y., Guo C., Ogi T., Nishimura Y.. Common TFIIH recruitment mechanism in global genome and transcription-coupled repair subpathways. Nucleic Acids Res. 2017; 45:13043–13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gervais V., Lamour V., Jawhari A., Frindel F., Wasielewski E., Dubaele S., Egly J.M., Thierry J.C., Kieffer B., Poterszman A.. TFIIH contains a PH domain involved in DNA nucleotide excision repair. Nat. Struct. Mol. Biol. 2004; 11:616–622. [DOI] [PubMed] [Google Scholar]
- 41. Greber B.J., Nguyen T.H.D., Fang J., Afonine P.V., Adams P.D., Nogales E.. The cryo-electron microscopy structure of human transcription factor IIH. Nature. 2017; 549:414–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schilbach S., Hantsche M., Tegunov D., Dienemann C., Wigge C., Urlaub H., Cramer P.. Structures of transcription pre-initiation complex with TFIIH and Mediator. Nature. 2017; 551:204–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greber B.J., Toso D.B., Fang J., Nogales E.. The complete structure of the human TFIIH core complex. eLife. 2019; 8:e44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cavanagh J., Fairbroher W.J., Palmer A.G. III, Skelton N.J.. Protein NMR Spectroscopy. 1996; San Diego, CA: Academic Press. [Google Scholar]
- 45. Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A.. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995; 6:277–293. [DOI] [PubMed] [Google Scholar]
- 46. Johnson B.A., Blevins R.A.. NMRView: a computer program for the visualization and analysis of NMR data. J. Biomol. NMR. 1994; 4:603–614. [DOI] [PubMed] [Google Scholar]
- 47. Cornilescu G., Delaglio F., Bax A.. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J. Biomol. NMR. 1999; 13:289–302. [DOI] [PubMed] [Google Scholar]
- 48. Brünger A.T. X-PLOR Version 3.1: A System for X-Ray Crystallography and NMR. 1993; New Haven, CT: Yale University Press. [Google Scholar]
- 49. Schwieters C.D., Kuszewski J.J., Tjandra N., Clore G.M.. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003; 160:65–73. [DOI] [PubMed] [Google Scholar]
- 50. Linge J.P., Williams M.A., Spronk C.A., Bonvin A.M., Nilges M.. Refinement of protein structures in explicit solvent. Proteins. 2003; 50:496–506. [DOI] [PubMed] [Google Scholar]
- 51. Laskowski R.A., Rullmann J.A.C., MacArthur M.W., Kaptein R., Thornton J.M.. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR. 1996; 8:477–486. [DOI] [PubMed] [Google Scholar]
- 52. Koradi R., Billeter M., Wüthrich K.. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 1996; 14:51–55. [DOI] [PubMed] [Google Scholar]
- 53. Mandel A.M., Akke M., Palmer A.G. III. Backbone dynamics of Escherichia coli ribonuclease HI: correlations with structure and function in an active enzyme. J. Mol. Biol. 1995; 246:144–163. [DOI] [PubMed] [Google Scholar]
- 54. Rennella E., Solyom Z., Brutscher B.. Measuring hydrogen exchange in proteins by selective water saturation in 1H–15N SOFAST/BEST-type experiments: advantages and limitations. J. Biomol. NMR. 2014; 60:99–107. [DOI] [PubMed] [Google Scholar]
- 55. Hwang T.L., van Zijl P.C., Mori S.. Accurate quantitation of water–amide proton exchange rates using the phase-modulated CLEAN chemical EXchange (CLEANEX-PM) approach with a Fast-HSQC (FHSQC) detection scheme. J. Biomol. NMR. 1998; 11:221–226. [DOI] [PubMed] [Google Scholar]
- 56. Sali A., Blundell T.L.. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 1993; 234:779–815. [DOI] [PubMed] [Google Scholar]
- 57. Humphrey A., Dalke A., Schulten K.. VMD: visual molecular dynamics. J. Mol. Graph. 1996; 14:33–38. [DOI] [PubMed] [Google Scholar]
- 58. Chresten R.S., Olsson H.M.M., Rostkowski M., Jensen H.J.. Improved treatment of ligands and coupling effects in empirical calculation and rationalization of pKa values. J. Chem. Theory Comput. 2011; 7:2284–2295. [DOI] [PubMed] [Google Scholar]
- 59. Mats H.M.O., Sondergaard R.C., Rotkowski M., Jensen H.J.. PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011; 7:525–537. [DOI] [PubMed] [Google Scholar]
- 60. Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A.. PDB2PAR: an automated pipeline for the setup, execution, and analysis of Poisson–Boltzmann electrostatics calculations. Nucleic Acids Res. 2004; 32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot L.B., Grubmükker H., Mackerell D.A.. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods. 2017; 14:71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. MacKerell D.A., Bashford D., Bellott M., Dunbrack L.R., Evanseck D.J., Field J.M., Fischer S., Gao J., Guo H., Ha S.et al.. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 1998; 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 63. MacKerell D.A., Feig M., Brooks L.C.. Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 2004; 126:698–699. [DOI] [PubMed] [Google Scholar]
- 64. Chang H.C., Kim K.. Density functional theory calculation of bonding and charge parameters for molecular dynamics studies on [FeFe] hydrogenases. J. Chem. Theory Comput. 2009; 5:1137–1145. [DOI] [PubMed] [Google Scholar]
- 65. Jorgensen L.W., Chandrasekhar J., Madura D.J., Impey M.R., Klein L.M.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 66. Abraham J.M., Murtola T., Schuls R., Páll S., Smith C.J., Hess B., Lindahl E.. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015; 1:19–25. [Google Scholar]
- 67. Páll S., Abraham J.M., Kutzner C., Hess B., Lindahl E.. Markidis S., Laure E.. Tackling exascale software challenges in molecular dynamics simulations with GROMACS. Solving Software Challenges for Exascale. 2015; 8759:Cham, Switzerland: Springer; 3–27. [Google Scholar]
- 68. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts R.M., Smith C.J., Kasson M.P., van der Spoel D.et al.. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013; 29:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Essmann U., Perera L., Berkowitz L.M., Darden T., Lee H., Pedersen G.L.. A smooth particle mesh Ewald method. J. Chem. Phys. 1995; 103:8577–8593. [Google Scholar]
- 70. Hess B. P-LINKS: a parallel linear constraint solver for molecular simulation. J. Chem. Theory Comput. 2008; 4:116–122. [DOI] [PubMed] [Google Scholar]
- 71. Nosé S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984; 52:255–268. [Google Scholar]
- 72. Hoover G.W. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 1985; 31:1695–1697. [DOI] [PubMed] [Google Scholar]
- 73. Berendsen C.J.H., Postma M.P.J., van Gunsteren F.W., DiNola A., Haak R.J.. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984; 81:3684–3690. [Google Scholar]
- 74. Parrinello M., Rahman A.. Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 1981; 52:7182–7190. [Google Scholar]
- 75. Nosé S., Klein L.M.. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983; 50:1055–1076. [Google Scholar]
- 76. Kabsch W., Sander C.. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983; 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 77. Roe R.D., Cheatham E.T. III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013; 9:3084–3095. [DOI] [PubMed] [Google Scholar]
- 78. Yan C., Dodd T., He Y., Tainer J.A., Tsutakawa S.E., Ivanov I.. Transcription preinitiation complex structure and dynamics provide insight into genetic diseases. Nat. Struct. Mol. Biol. 2019; 26:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hafsa N.E., Wishart D.S.. CSI 2.0: a significantly improved version of the chemical shift index. J. Biomol. NMR. 2014; 60:131–146. [DOI] [PubMed] [Google Scholar]
- 80. Berjanskii M.V., Wishart D.S.. A simple method to predict protein flexibility using secondary chemical shifts. J. Am. Chem. Soc. 2005; 127:14970–14971. [DOI] [PubMed] [Google Scholar]
- 81. Luo J., Cimermancic P., Viswanath S., Ebmeier C.C., Kim B., Dehecq M., Raman V., Greenberg C.H., Pellarin R., Sali A.et al.. Architecture of the human and yeast general transcription and DNA repair factor TFIIH. Mol. Cell. 2015; 59:794–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nakazawa Y., Hara Y., Oka Y., Komine O., van den Heuvel D., Guo C., Daigaku Y., Isono M., He Y., Shimada M.et al.. Ubiquitination of DNA damage-stalled RNAPII promotes transcription-coupled repair. Cell. 2020; 180:1228–1244. [DOI] [PubMed] [Google Scholar]
- 83. Singh S.K., Qiao Z., Song L., Jani V., Rice W., Eng E., Coleman R.A., Liu W.L.. Structural visualization of the p53/RNA polymerase II assembly. Genes Dev. 2016; 30:2527–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates for the TFIIH p621–158 have been deposited in the RCSB Protein Data Bank (PDB) under ID code 7BUL, and chemical shifts, 15N relaxation parameters (T1 = 1/R1, T2 = 1/R2 and heteronuclear NOE) and hydrogen exchange rates (kex) have been deposited in the BioMagResBank (BMRB) under accession number 36340.