


Abstract
The DNA damage-responsive tumor suppressors p53 and HIPK2 are well established regulators of cell fate decision-making and regulate the cellular sensitivity to DNA-damaging drugs. Here, we identify Deleted in Azoospermia-associated protein 2 (DAZAP2), a small adaptor protein, as a novel regulator of HIPK2 and specifier of the DNA damage-induced p53 response. Knock-down or genetic deletion of DAZAP2 strongly potentiates cancer cell chemosensitivity both in cells and in vivo using a mouse tumour xenograft model. In unstressed cells, DAZAP2 stimulates HIPK2 polyubiquitination and degradation through interplay with the ubiquitin ligase SIAH1. Upon DNA damage, HIPK2 site-specifically phosphorylates DAZAP2, which terminates its HIPK2-degrading function and triggers its re-localization to the cell nucleus. Interestingly, nuclear DAZAP2 interacts with p53 and specifies target gene expression through modulating a defined subset of p53 target genes. Furthermore, our results suggest that DAZAP2 co-occupies p53 response elements to specify target gene expression. Collectively, our findings propose DAZAP2 as novel regulator of the DNA damage-induced p53 response that controls cancer cell chemosensitivity.
INTRODUCTION
The DNA damage response (DDR) lies at the heart of cancer etiology and poses an important barrier against genomic instability and cancer development (1–3). Furthermore, DNA-damaging chemotherapeutic drug treatments play a fundamental role in cancer therapy. Upon repairable damage, the DDR activates cellular signaling checkpoints, which block cell cycle progression and promote DNA repair (4,5). Irreparable DNA damage stimulates inactivation or elimination of damaged cells by induction of cellular senescence or cell death, respectively (6–8).
p53 is the most frequently mutated gene in human cancers and plays a fundamental role in tumor suppression (9,10). p53 is a master regulator of the DNA damage response and orchestrates DNA damage-induced cell fate by controlling distinct gene expression programs, which in turn regulate different cell fate options such as cell cycle arrest and DNA repair, cellular senescence and cell death (8,11–13). The molecular mechanisms by which the expression of specific p53 target gene sets is regulated and subsequent cell fate selection is specified still remains unclear. Unstressed cells keep p53 levels low and inactive through interaction with negative-regulatory ubiquitin ligases such as MDM2/HDM2 (14). Upon DNA damage, p53 is rapidly stabilized and activated by posttranslational modifications involving both phosphorylation of p53 itself and of its ubiquitin ligases, leading to the disruption of the p53–ubiquitin ligase complexes (14–16).
p53 is differentially phosphorylated in response to repairable versus irreparable DNA damage. This has been linked to the activity of the p53 Ser46 kinases, which are activated in an ATM-dependent manner upon irreparable DNA damage and trigger the cell death response through induction of apoptosis and ferroptosis (17,18). Currently the best understood p53 Ser46 kinase is the Homeodomain-interacting protein kinase 2 (HIPK2). HIPK2 is stabilized and activated by UV radiation, ionizing radiation and DNA-damaging chemotherapeutic drugs through the ATM and ATR pathway and plays a fundamental role in the DNA damage-induced cell death response (19–26). HIPK2 is largely regulated at the level of its protein stability through interaction with a set of ubiquitin ligases including SIAH1 and SIAH2, which control HIPK2 steady-state protein levels by catalyzing HIPK2 polyubiquitination and degradation by the proteasome (22,27,28). Also MDM2 can regulate HIPK2 protein levels, however, not the steady-state levels but its levels in response to cytostatic drug treatment (29). HIPK2 acts as a tumor suppressor and its depletion, inactivation or cancer-associated mutation inhibits cancer cell responsiveness to DNA-damaging chemotherapeutic drugs resulting in radio- and chemoresistance (30–34). Although HIPK2 is a well-established mediator of cancer cell radio- and chemosensitivity, only a few molecular players regulating HIPK2 function in chemotherapy have been identified to date.
Here, we identified the ubiquitously expressed 17 kDa adaptor protein Deleted in AZoospermia-Associated Protein 2 (DAZAP2; also termed proline-rich transcript in brain, PRTB) (35,36) as a novel regulator of HIPK2 and the p53 response. We provide evidence that DAZAP2 functions as a specifier of the p53 response regulating cancer cell chemosensitivity. Through interplay with the SIAH1 ligase DAZAP2 restricts HIPK2 steady-state protein levels in unstressed cells. In response to DNA damage, HIPK2 phosphorylates DAZAP2 at several Ser/Thr residues including Ser77, which inhibits its HIPK2-degrading function and targets it to the cell nucleus. Nuclear DAZAP2 binds to p53 and specifies p53 target gene expression by binding p53 response elements at the chromatin. Finally, we demonstrate that depletion or genetic deletion of DAZAP2 results in robust sensitization of cancer cells in cellulo and in vivo. Our findings propose DAZAP2 as a specifier of the p53 response and regulator of cancer cell chemosensitivity to DNA-damaging drugs.
MATERIALS AND METHODS
Cell culture and transfections
H1299, HCT116, HepG2, U2OS (all obtained from ATCC) were maintained in DMEM supplemented with 10% (v/v) FCS, 100 U/ml Penicillin, 100 μg/ml Streptomycin, 10 mM HEPES buffer and 2 mM l-glutamine at 37°C and 5% CO2. Transient transfections were performed using Lipofectamine 2000 (Life Technologies), Fugene® HD (Promega) or by standard CaPO4 precipitation. The Siah1/Siah2 double knock-out MEFs (37) were kindly provided by Andreas Möller and David Bowtell, the HCT116 p53–/– cell were kindly provided by Bert Vogelstein.
Chemotherapeutic drugs treatments
Cells were incubated with culture medium supplemented with Adriamycin (Sigma Aldrich), 5-Fluorouracil (Sigma Aldrich), Oxaliplatin (Biomol), MG-132 (Biomol) or z-VAD-FMK (BD Biosciences) at the specified concentrations for the indicated time points. 5-Fluorouracil (5-FU), MG-132 and z-VAD-FMK were dissolved in DMSO, Adriamycin and Oxaliplatin in water. Control treatments were done with the same amount of solvent. After incubation cells were harvested and processed as indicated.
Antibodies
GFP (#sc-8834), p53 (DO-1, #sc-126) p53 FL-393 (s#c-6243), Myc (9E10, #sc-40) and normal rabbit IgG (sc-2027) antibodies were purchased from Santa Cruz Biotechnologies. Flag (M2, #F3165) and α-tubulin (#T5168) antibodies were obtained from Sigma. ACTIN (C4, #69100) antibodies were purchased from MP Biomedicals and HA (3F10, #11867423001) antibodies from Roche. Ubiquitin (P4D1, #3936), and p53 phospho-Ser46 (#2521) antibodies were from Cell Signaling Technology. Cleaved PARP (Y34, #ab32561) was obtained from Abcam. The H4 (#07-108) antibodies were from Millipore. The affinity-purified HIPK2 antibody and the chicken SIAH1 antibody have been described previously (22). Rabbit polyclonal DAZAP2 antibodies were raised against the following keyhole limpet hemocyanine (KLH)-coupled peptide: H2N-MNSKGQYPTQPTYPVC-COOH. The rabbit sera were affinity-purified against the peptide prior to use. The phosphorylation specific DAZAP2 Ser77 antibody was raised against the following phospho-peptide: H2N-YLPMA-S(p)-VAVGPLC-COOH and the phospho-specific antibodies were subsequently affinity purified.
Expression constructs
The human Flag-HIPK2 constructs and GST-HIPK2 fusion constructs have been described previously (19,22,24,25,38,39). The human SIAH1 (22), HA-ubiquitin (22) and p53 (19) constructs were described previously. The DAZAP2 expression constructs, mCherry-HIPK2 and p53 deletion mutants were generated by standard PCR or gene synthesis (GeneArt, ThermoFisher) and standard cloning methods. Detailed cloning descriptions can be obtained from the authors on request. All PCR-generated constructs were verified by DNA sequencing.
RNA interference
siRNA duplexes were produced by Dharmacon Research (GE Healthcare, Dharmacon), Sigma Aldrich and Qiagen. The following siRNAs were used to specifically target human DAZAP2 (L-020985–00, Dharmacon), murine SIAH1b (L-044849–01, Dharmacon), murine DAZAP2 #1 (5′-AACAGCAAAGGTCAATATCCA) or murine DAZAP2 #2 (5′-AATAACTCAGCGCAAGGGAAA). For control experiments a luciferase duplex (5′-AACGUACGCGGAAUACUUCGA) was used. siRNA duplexes (final concentration 25–50 nM) were transfected using Lipofectamine RNAiMAX (Life Technologies) or HiPerFect (Qiagen) as specified by the manufacturer. HepG2 or H1299 cells were transduced with a retroviral control plasmid pGLenti-shLuciferase or pGLenti-shDAZAP2 to knock down DAZAP2 or pSUPER-shLuciferase or pSUPER-shSIAH1 to deplete SIAH1 by retroviral transduction after having produced the virus in 293T cells. The following shRNA sequences were used: human DAZAP2 (forward 5′-GATCCCCGCTCTATCGTCCGAGCTTTTTCAAGAGAAAAGCTCGGACGATAGAGCTTTTTGGAAA, reverse 5′-AGCTTTTCCAAAAAGCTCTATCGTCCGAGCTTTTCTCTTGAAAAAGCTCGGACGATAGAGCGGG), human SIAH1 (forward 5′-GATCCCCGATAGGAACACGCAAGCAATTCAAGAGATTGCTTGCGCGTGTTCCTATCTTTTTGGAAA, reverse 5′-AGCTTTTCCAAAAAGATAGGAACACGCAAGCAATCTCTTGAATTGCTTGCGTGTTCCTATCGGG), murine β-ACTIN (forward 5′-ACCCACACTGTGCCCATCTACGA, reverse 5′-CTTGCTGATCCACATCTGCTGGA).
RNA Sequencing and analysis of the RNA-seq data
Total RNA was collected using the TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's instructions. After RNA extraction, potential contaminating genomic DNA was removed with the RNA Clean & Concentrator Kit (Zymo Research) following the manufacturer's instructions. RNA quality was assessed by running 1 μl on a RNA ScreenTape on the TapeStation 2200 (Agilent) (Supplementary Table S3). Samples were submitted as biological triplicates to the German Cancer Research Center (dkfz) Genomics & Proteomics Core Facility. Transcriptome-sequencing libraries were prepared from total RNA extractions using the TruSEQ RNA sample preparation kit (Illumina) and sequenced on a HiSeq 2000 machine (Illumina) using 50-bp single reads. This resulted in each of the individual replicates having more than 45 million reads. Reads were then trimmed by removing stretches of bases having a quality score of <30 at the ends of the reads. Reads were mapped using Tophat 2.0.6 (40) against the hg19 assembly of the human genome. Differential expression was quantified using DESeq2 (41) and subjected to multiple testing corrections. Genes with a q-value <0.05 were considered to be differentially expressed. All files related to these experiments are available in the GEO database (GSE154146, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154146). FPKM values were computed using Cuffdiff 2.0 (42). Principal Component Analysis (PCA) plots, heatmaps and volcano plots were done in R using the FactoMineR or gplot packages.
Ingenuity pathway analysis
To assess the role of genes (n = 1404) differentially expressed in RNA-sequencing data from DAZAP2-depleted versus control cells and to identify upstream regulators of this gene set, Ingenuity Pathway Analysis (IPA) software (Qiagen) was used to. P-values were plotted to show the significance.
Subcellular fractionation
Subcellular fractionation was performed by modifying the protocol from Mendéz and Stillman (43). Briefly, cells were harvested, washed with phosphate-buffered saline (PBS) and resuspended in Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M Sucrose, 10% Glycerol, 1 mM DTT, Complete Protease Inhibitor Cocktail [Roche]). Triton X-100 was added to a final concentration of 0.1% and the cells were incubated on ice for 8 min. After removing a sample (= whole cell lysate), the nuclei were collected (1300 g, 5 min, 4°C). The supernatant was clarified by centrifugation (17 000 g, 5 min, 4°C) and transferred to a new microcentrifugation tube ( = cytoplasmic fraction). The collected nuclei were washed six times with Buffer A and then lysed in Buffer B (3 mM EDTA, 0.2 mM EGTA, 1 mM DTT, 1× Complete Protease Inhibitor Cocktail [Roche]) (= nuclear fraction). All collected fractions were sonicated before analyzing by immunoblotting.
Generation of DAZAP2 CRISPR/Cas9 KO cells
sgRNA target sites for DAZAP2 (ENSG00000183283) were identified using the ECRISP Design Tool (44). The following sgRNA sequences targeting DAZAP2 Exon 2 were selected: sgRNA 1 5′-CCTGGGAATCCAGTATACCC and sgRNA 2 5′-TATACCGATGCTCCACCTGC. Sequences for Control (CTRL) sgRNAs were taken from Wang et al. (45): CTRL sgRNA 1 5′-GTGTATCTCAGCACGCTAAC and CTRL sgRNA 2 5′-GAGATTCCGATGTAACGTAC. Single guide RNAs were cloned into plentiCRISPR v1 (Addgene #49535) as indicated by the supplier. Lentiviruses were produced in 293T cells and subsequently used for transduction of HCT116 cells followed by selection with puromycin (1 μg/ml). Monoclonal cell lines were obtained by limited dilution of the transduced and selected cell pools and analyzed for DAZAP2 expression by immunoblotting. Genomic DNA (gDNA) from clonal cell lines showing no DAZAP2 expression was extracted by resuspending harvested cells in lysis buffer (100 mM Tris–HCl pH 8.0, 200 mM NaCl, 5 mM EDTA, 0.2% (v/v) SDS) and adding Proteinase K (0.25 mg/ml), followed by alcohol precipitation. For validation of DAZAP2 KO clones, a 919 bp fragment from the DAZAP2 locus was amplified by PCR using the following primer: 5′-TGCTGTTACCCACGAAGTCTG and 5′-AGGCAGAAGAGAAGGCAAAATG. The PCR product was purified using the QIAquick PCR Purification Kit (Qiagen), sequenced or subcloned into the pC2.1 vector (Thermo Fisher Scientific) before sequencing. The following primer was used for sequencing: 5′-AGGAGCAGTGAGCAGTG. The obtained sequences were aligned against the DAZAP2 reference sequence to check for genetic disruption of the DAZAP2 locus.
Mouse tumor model
CRISPR/Cas9 DAZAP2 KO and CRISPR/Cas9 control HCT116 cells (106 cells each) were subcutaneously injected into the flanks of 10 weeks old NOD/SCID mice. When the tumors reached 100 mm3, the mice were treated with 5-FU. 5-FU was administered intraperitoneally and three injections were given per week (each injection 50 mg/kg) for a duration of 3 weeks. Control mice were injected with DMSO/PBS. DMSO and 5-FU were diluted in PBS before administration. Volume of the tumors was measured using a manual caliper at the end of the experiment.
RT-PCR analysis and RT-qPCR analysis
Total RNA was isolated using the RNAeasy kit (Qiagen). 2 μg total RNA was reverse transcribed using the cDNA Cycle Kit (Invitrogen) according to the manufacturer's instruction. 20% of the RT reaction was used as a template for PCR using the following primer pairs: human SIAH1 (forward 5′-CCTGTAAATGGCAAGGCTCTC, reverse 5′-CTAGTCTTTGACACCAGCATTG), human β-ACTIN (forward 5′-CCTCGCCTTTGCCGATCC, reverse 5′GGATCTTCATGAGGTAGTCAGTC), murine SIAH1B (forward 5′-ACTACCCCTTTGGCCTTTACACG, reverse 5′-CTAGCTGCTGCTGCTGCTGCTG), murine β-ACTIN (forward 5′-ACCCACACTGTGCCCATCTACGA, reverse 5′CTTGCTGATCCACATCTGCTGGA). PCR reactions were performed using the following conditions: 1 min 95°C, 1 min 58°C, 1 min 72°C, 25 cycles. Reactions were analyzed on agarose gels.
For quantitative RT-PCR, total RNA was extracted from cultured cells using TRIzol reagent (Invitrogen, Life Technologies GmbH) according to the manufacturer's instructions. Potential contaminating genomic DNA was removed with the RNA Clean & Concentrator Kit (Zymo Research). RNA concentration was measured on a Nanodrop 2000 and 2 μg total RNA were used for cDNA synthesis using High capacity RNA to cDNA kit (Applied Biosystems, Life Technologies) following the manufacturer's instructions. cDNA was diluted 1:200 in H2O and 2 μl used for each PCR reaction. Quantitative real-time PCR was performed using a real-time PCR system (Roche LightCycler 480 or Bio-Rad CFX384 Touch) and a real-time PCR master mix (iTaq Universal SYBR Green Supermix, Bio-Rad) with 0.5 μM gene-specific primers (primer sequences are shown in Supplementary Table S1) following the manufacturer's instructions. Non-template controls were included in each run. PCR conditions were as follows: step 1: 95°C for 5 min; step 2: 40 cycles of 95°C for 5 s followed by 60°C for 30 s and the generation of a melting curve. Quantitative data were normalized using GAPDH and B2M as endogenous reference genes and calculated using LightCycler® 480 (Roche) or CFX Maestro (Bio-Rad) software.
Chromatin Immunoprecipitation (ChIP)
Chromatin Immunoprecipitations were performed using the SimpleChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology) according to the manufacturer's instructions. 10 μg cross-linked chromatin were precipitated with 2 μg antibodies. p53 antibodies (1C12, #2524) and normal rabbit IgG (#2729) were obtained from Cell Signaling Technology. The DAZAP2 antibodies were self-generated and are described in the antibodies section. 1 μl of the precipitated DNA was used for quantitative PCR using the iTaq Universal SYBR Green Supermix (Bio-Rad) following the manufacturer's instructions using 0.5 μM gene-specific primers (primer sequences are shown in Supplementary Table S2). Non-template controls were included in each run. PCR conditions were as follows: step 1: 95°C for 5 min; step 2: 40 cycles of 95°C for 5 s followed by 60°C for 30 s and the generation of a melting curve. Cq values were determined using CFX Maestro (Bio-Rad) software and the fold change was calculated using Microsoft Excel (Version 2011).
GST-pulldown assays
GST fusion protein expression in Escherichia coli, protein purification and GST pulldown assays were performed as published previously (19,38). Briefly, GST fusion proteins were expressed in the E. coli BL21(DE3) pLysS strain and purified using glutathione Sepharose 4 Fast Flow beads (GE Healthcare). Bead-coupled GST fusion proteins were incubated with proteins generated by in vitro translation using the TNT Coupled Reticulocyte Lysate System (Promega Corporation). GST-pulldowns were performed in 1× PBS, 0.05% NP40, 1 mM PMSF, 1 mM Na3VO4 and analyzed by SDS-PAGE, Coomassie Brilliant Blue staining and autoradiography of the dried gels.
Immunofluorescence microscopy
Immunofluorescence analyses were performed essentially as described (24,25). Briefly, cells were seeded onto coverslips and transfected with the indicated constructs. Twenty-four hours after transfection, cells were fixed with 4% PFA in PBS for 25 min at RT. For direct fluorescence microscopy, DNA was stained with Hoechst (Sigma-Aldrich) 1:1000 (w/v) in PBS and cells were mounted on glass slides with Mowiol (Sigma-Aldrich). Images were taken using a confocal laser scanning microscope (FluoView1000, Olympus) with a ×60 oil objective using the sequential scanning mode. All images were collected and processed using the FluoView Software (Olympus).
Immunoprecipitation and immunoblotting
Immunoprecipitation analysis and immunoblotting was performed as published previously (19,22). Proteins were detected by Western Lightning Plus-ECL (Perkin Elmer) and Super Signal West Dura and Femto (Pierce) as described (19,22). Immunoprecipitation experiments were performed either in lysis buffer containing 20 mM Tris–HCl pH 7.4, 150 mM NaCl, 1% NP40, 0.1% SDS, 5 mM EDTA, 25 mM NaF or in 20 mM HEPES–KOH pH 7.9, 350 mM NaCl, 1 mM MgCl2, 20% glycerol, 1% NP40, 0.6 mM EDTA. Anti-Flag M2 affinity gel (Sigma) and rabbit IgG TrueBlot (Rockland Immunochemicals) were used according to the manufacturer's instructions. A normal rabbit IgG was used as a specificity control. Reactions were incubated for 2 h or overnight at 4°C on a rotating wheel and washed three times in lysis buffer. Samples were prepared for immunoblotting by adding 5× Laemmli buffer and boiled at 95°C for 5 min.
In vitro kinase assays
In vitro kinase assays were performed in principle as described previously (24,25). His-HIPK2 and GST-DAZAP2 were expressed in E. coli, Flag-HIPK2 in 293T cells. After protein purification His-HIPK2 or Flag-HIPK2 proteins were incubated with GST-DAZAP2 in 30 μl kinase buffer containing 40 μM cold ATP and 5 μCi [γ-32P] ATP. For the experiment shown in Figure 5D, exclusively 40 μM cold ATP was used. After incubation for 30 min at 30°C, the reaction was stopped by adding 5× Laemmli buffer. After separation by SDS–PAGE, gels were stained with Coomassie Brilliant Blue, dried and exposed to X-ray films. The experiment shown in Figure 5D was analyzed by immunoblotting as indicated.
Figure 5.

Phosphorylation of DAZAP2 by HIPK2 regulates DAZAP2-mediated HIPK2 degradation and DAZAP2 localization. (A) FLAG-HIPK2 and kinase-dead FLAG-HIPK2 K221A were purified from 293T cells, recombinant GST-DAZAP2 was purified from E. coli. In vitro kinase assays were performed and DAZAP2 phosphorylation as well as HIPK2 autophosphorylation were examined by autoradiography. Protein levels were analyzed by Coomassie Brilliant Blue (CBB) staining. n = 2. (B) HIPK2 phosphorylates DAZAP2 on five Ser/Thr residues. In vitro kinase assays were performed using bacterially expressed His-HIPK2 and GST-DAZAP2 mutants and analyzed by SDS–PAGE and autoradiography. Protein levels were analyzed by CBB staining. n = 3. (C) Validation of the DAZAP2 phospho-Ser77 antibody. In vitro kinase assays were performed with His-HIPK2 and GST-DAZAP2. Non-phosphorylated GST-DAZAP2 and in vitro phosphorylated GST-DAZAP2 phosphorylation were examined by SDS–PAGE and immunoblotting. n = 3. (D) DAZAP2 is phosphorylated at Ser77 upon DNA damage in cells. Proteins were expressed in H1299 cells as indicated in the presence of the indicated compounds. Myc-DAZAP2 was precipitated from the lysates and analyzed by immunoblotting as indicated. n = 2. (E) Effect of DAZAP2 phosphorylation on its HIPK2-degrading capacity. H1299 cells were transfected with the indicated expression vectors and total protein lysates were analyzed by immunoblotting. n = 3. (F, G) DAZAP2 phosphorylation regulates its localization. The indicated EGFP-DAZAP2 fusion proteins were expressed in U2OS cells and their subcellular localization was assessed by confocal microscopy. DNA is stained by Hoechst (blue). n = 3. DAZAP23A: S77A, S85A, T86A; DAZAP25A: S62A, S69A, S77A, S85A, T86A; DAZAP25D: S62D, S69D, S77D, S85D, T86D. Representative experiments are shown.
In vitro and in cell ubiquitination assays
The in vitro and in cellulo ubiquitination assays were performed in principle as published previously (22,24). Briefly, for in vitro ubiquitination assays His-HIPK2, GST-DAZAP2 and GST-SIAH1 were expressed in E. coli BL21 and purified by standard protocols. Flag-HIPK2 was immunopurified from transfected 293T cells. The isolated proteins were incubated with UBE1 (100 ng, Boston Biochem), UBE2D2 (200 ng, Boston-Biochem) and HA-Ubiquitin (10 μg, Boston Biochem) as indicated in ubiquitination buffer (10 mM Tris–HCl pH 7.4, 5 mM MgCl2, 2 mM DTT). Reactions were incubated under constant shaking (800 rpm) at 30°C for 30 min. His-HIPK2 was subsequently purified from the reactions by Ni-NTA pulldown under denaturing conditions followed by immunoblotting with HA- and HIPK2-specific antibodies. Total protein levels were analyzed by Ponceau S Staining or Immunoblotting.
In cell ubiquitination was performed by transfecting H1299 as indicated. Cells were lysed in buffer A (6 M guanidinium–HCl, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris–HCl pH 8.0, 5 mM imidazole, 10 mM β-mercaptoethanol) and incubated with Ni2+-NTA sepharose (Qiagen) for 45 min at room temperature. Beads were washed with buffer B (8 M urea, 0.1 M Na2HPO4 /NaH2PO4, 0.01 M Tris–HCl pH 8.0, 10 mM β-mercaptoethanol) and C (8 M urea, 0.1 M Na2HPO4/NaH2PO4, 0.01 M Tris–HCl pH 6.8, 10 mM β-Mercaptoethanol), and bound proteins were eluted with buffer D (200 mM imidazole, 62.5 mM Tris–HCl pH 6.8, 2% glycerol, 0.72 M β-mercaptoethanol, 5% SDS, 0.005% (w/v) bromophenol blue) and analyzed by immunoblotting.
Apoptosis measurements
Trypsin-dissociated single cells were stained using the Annexin V-FITC Apoptosis Detection Kit (BioVision) according to the manufacturer's instructions. The stained samples were examined by flow cytometry (BD FACSCanto™ II, BD Biosciences) using the BD FACSDiva Software with excitation from a 488 nm laser and emission detection with a 530/30 filter for Annexin V-FITC. Ten thousand cells were analyzed per sample. Caspase 3/7 activation was assessed in live cells using the NucView® 488 Caspase-3 Assay Kit (Biotium) in accordance with the manufacturer's recommendations. Briefly, cells were incubated with culture medium supplemented with DMSO or 5-Fluorouracil and 3 μM NucView® 488 Caspase-3 substrate and incubated for the indicated time points. Images were acquired from three random fields with an Axio Vert.A1 microscope (Zeiss) using the ZEN 2 core imagine software (Zeiss) and analyzed with ImageJ software. The number of green fluorescent cells and the total cell number were counted, and the percentage of green fluorescent cells was calculated.
Statistical analysis
NucView® 488 Caspase-3, flow cytometry, real-time PCR, and ChIP-PCR were performed in triplicates and data are presented as the mean ± standard deviation. Data from the murine tumor model are shown as mean ± standard error of the mean. The statistical significance of differences between two groups was assessed by two-tailed unpaired t-test. A value of P < 0.05 was considered to be statistically significant. Statistical analysis was performed using Prism GraphPad version 8 (GraphPad Software). For testing the effect of two independent variables on gene expression, two-way ANOVA was performed using the AOV function in R.
RESULTS
DAZAP2 is a novel HIPK2 binding protein
To identify novel factors involved in DNA damage-induced cell fate regulation, we previously performed yeast two-hybrid screens using different domains of HIPK2 as bait (39). Among various HIPK2-interacting proteins previously reported by us (22,28,39), we also identified several cDNA clones coding for the 168 amino acid adaptor protein Deleted in azoospermia-associated protein 2 (DAZAP2) using the C-terminal domain of HIPK2 as a bait (Figure 1A). The interaction of DAZAP2 and HIPK2 was confirmed by in vitro pull-down assays using recombinant GST-DAZAP2 fusion protein and in vitro translated 35S-labeled HIPK2 (Figure 1B). In vitro interaction mapping revealed two DAZAP2-binding domains on the HIPK2 polypeptide located between amino acids 600–800 and 1019–1191 (Supplementary Figure S1A, B), indicating that the interaction of DAZAP2 and HIPK2 is established between defined domains.
Figure 1.

DAZAP2 binds to HIPK2 in vitro and in cells. (A) Schematic representation of the domain structure of HIPK2 and the HIPK2 truncation used as bait in the yeast two-hybrid screening that identified DAZAP2 as a binding partner of HIPK2. AID = autoinhibitory domain. (B) DAZAP2 associates with HIPK2 in vitro. GST pulldown assays were performed with bacterially expressed GST or GST-DAZAP2 and 35S-labeled HIPK2K221A and analyzed by SDS-PAGE, Coomassie Brilliant Blue (CBB) staining and autoradiography. n = 3. (C) Interaction of ectopically expressed DAZAP2 and HIPK2. FLAG-HIPK2 and Myc-DAZAP2 were expressed in H1299 cells as indicated in the presence of 20 μM MG-132. FLAG-HIPK2 was precipitated from the lysates and binding of Myc-DAZAP2 to FLAG-HIPK2 was analyzed by immunoblotting. n = 2. (D) Interaction of endogenous DAZAP2 and HIPK2. Lysates from HCT116 cells treated with MG-132 were subjected to immunoprecipitation with control or HIPK2 antibodies and analyzed by immunoblotting (n = 2). A non-specific band is marked with an asterisk. IgGL = Immunoglobulin G Light Chain. (E) DAZAP2 and HIPK2 colocalize. Co-localization of EGFP-DAZAP2 and mCherry-HIPK2 in U2OS cells was assessed by confocal microscopy (n = 2). DNA is stained by Hoechst (blue). Two representative cells showing nuclear (upper panel) or cytoplasmic (lower panel) HIPK2 and DAZAP2 expression are shown.
To further validate DAZAP2-HIPK2 complex formation, we co-expressed both proteins (in the presence of the proteasome inhibitor MG-132) and performed coimmunoprecipitation analysis. Indeed, DAZAP2 coimmunoprecipitated with HIPK2 (Figure 1C), demonstrating that the two proteins also interact in cells. Interaction mapping experiments revealed that HIPK2-DAZAP2 complex formation in cells requires HIPK2 amino acids 600–800, whereas the C-terminus of HIPK2 (aa 1019–1191) was not required for the interaction (Supplementary Figure S1A, C). These results suggest that the amino acid stretch 600–800 comprises the major DAZAP2-binding domain of HIPK2.
To determine whether endogenous HIPK2 and DAZAP2 form a protein complex, endogenous coimmunoprecipitation experiments were performed. Immunoblotting revealed that endogenous DAZAP2 coimmunoprecipitates with HIPK2, demonstrating that endogenous DAZAP2 and HIPK2 form a complex (Figure 1D).
Previous work demonstrated that HIPK2 can localize to the cytoplasm, the cell nucleus and nuclear bodies (19,20,28,33). DAZAP2 has been previously shown to localize to cytoplasmic stress granules, to the cell nucleus and to SC35 nuclear speckles (46–48). To investigate a potential colocalization of DAZAP2 and HIPK2, we analyzed their subcellular distribution using confocal microscopy. Both DAZAP2 and HIPK2 showed a nuclear (Figure 1E, upper panel) and cytoplasmic (Figure 1E, lower panel) subcellular distribution. Interestingly, DAZAP2 and HIPK2 efficiently colocalized at cytoplasmic speckles (likely stress granules) and at nuclear bodies (Figure 1E), suggesting that both proteins are capable to form protein complexes in the cytoplasm and the cell nucleus. Taken together, our results identify DAZAP2 as a novel HIPK2 binding protein.
DAZAP2 triggers SIAH1-dependent HIPK2 degradation
HIPK2 activity is regulated at the level of protein stability and by caspase-dependent cleavage (49,50). Interestingly, we observed that DAZAP2 depletion by RNA interference in human and in mouse cells resulted in increased HIPK2 steady-state protein levels suggesting that DAZAP2 may regulate HIPK2 protein stability (Figure 2A and Supplementary Figure S2A). Accordingly, ectopic expression of DAZAP2 dose-dependently reduced HIPK2 protein levels (Figure 2B). This effect was rescued in the presence of the proteasome inhibitor MG-132, whereas treatment with a pan-caspase inhibitor (Z-VAD-FMK) did not interfere with HIPK2 degradation stimulated by DAZAP2 expression (Figure 2C).
Figure 2.

DAZAP2 enhances HIPK2 poly-ubiquitination and degradation through the ubiquitin ligase SIAH1. (A) DAZAP2 depletion increases HIPK2 protein levels. HepG2 cells were transduced with vectors encoding control (Luciferase) or DAZAP2 shRNAs. Total cell lysates were analyzed by immunoblotting using the indicated antibodies. A non-specific band is marked with an asterisk. n = 3. (B) Ectopic expression of DAZAP2 decreases HIPK2 levels. H1299 cells were transfected with the indicated expression vectors and total protein lysates were analyzed by immunoblotting. n = 2. (C) DAZAP2 potentiates proteasome-dependent HIPK2 degradation. H1299 cells were transfected with the indicated expression vectors and exposed to MG-132 or z-VAD-FMK as indicated. Total protein lysates were analyzed by immunoblotting. n = 3. (D) SIAH1 is essential for DAZAP2-mediated HIPK2 degradation. H1299 cells were transduced with the indicated shRNAs and transfected with the indicated expression vectors. Total protein lysates were analyzed by Immunoblotting. Depletion of SIAH1 was investigated by RT-PCR. n = 2. (E) DAZAP2 interacts with SIAH1. The proteins indicated were expressed in H1299 cells in the presence of MG-132. FLAG-SIAH1 was precipitated from the lysates and immunoprecipitates were analyzed by immunoblotting. n = 2. (F) DAZAP2 interacts with SIAH1 in vitro. Bacterially expressed GST or GST-SIAH1 were incubated with in vitro translated 35S-labeled DAZAP2. GST pulldown assays were performed and analyzed by SDS-PAGE, Coomassie Brilliant Blue (CBB) staining and autoradiography. n = 3. (G) DAZAP2 potentiates HIPK2-SIAH1 complex formation. The indicated proteins were expressed in H1299 cells in the presence of MG-132. FLAG-HIPK2 was precipitated from the lysates and immunoprecipitates were analyzed by immunoblotting. n = 3. (H) DAZAP2 stimulates SIAH1-dependent HIPK2 ubiquitination. Recombinant proteins were expressed in E. coli and purified by affinity chromatography. The isolated proteins were incubated with UBE1 (= E1 ubiquitin-activating enzyme), UBE2D2 ( = E2 ubiquitin-conjugating enzyme) and HA-Ubiquitin and in vitro ubiquitination assays were performed. Subsequently, His-HIPK2 was purified from the reactions using Ni-NTA-beads followed by immunoblotting with a Ubiquitin- and HIPK2-specific antibody. Total levels of GST-DAZAP2 and GST-SIAH1 were analyzed by Ponceau S staining and immunoblotting, respectively. n = 3. Representative experiments are shown.
Our previous work demonstrated that the E3 ubiquitin ligase SIAH1 is an important regulator of HIPK2 steady-state protein levels (22,24). Since DAZAP2 regulated HIPK2 steady-state levels but does not contain any known enzymatic activity, we speculated that DAZAP2 stimulates HIPK2 proteolysis by employing SIAH1. To test this hypothesis, we depleted SIAH1 in human cells, which resulted in a profound reduction in DAZAP2-mediated HIPK2 proteolysis (Figure 2D). Consistent with this result, we found that DAZAP2 cooperates with SIAH1 in mediating HIPK2 degradation (Supplementary Figure S2C). To confirm our findings, we used Siah1a/Siah2 double knock-out (DKO) mouse embryonic fibroblasts (MEFs) (37). Of note, in contrast to humans, mice harbor two functionally redundant genes coding for SIAH1, Siah1a and Siah1b (37). Ectopic expression of DAZAP2 readily stimulated HIPK2 degradation in the Siah1a/Siah2 DKO cells (Supplementary Figure S2B), suggesting that the remaining SIAH1b expression may mediate the degradation. Therefore, we co-depleted SIAH1b expression by RNA interference in Siah1a/Siah2 DKO MEFs. Indeed, DAZAP2 failed to stimulate HIPK2 degradation in SIAH1b co-depleted DKO MEFs (Supplementary Figure S2B), demonstrating an essential role of SIAH1 in DAZAP2-mediated HIPK2 degradation. Collectively, our results demonstrate that DAZAP2 mediates proteasome-dependent HIPK2 degradation in a SIAH1-dependent manner.
DAZAP2 interacts with SIAH1 and regulates HIPK2-SIAH1 complex formation
The functional link between DAZAP2 and SIAH1 prompted us to assess whether DAZAP2 may form a complex with SIAH1. Indeed, protein interaction analysis revealed that DAZAP2 coimmunoprecipitates with SIAH1 (Figure 2E) and vice versa (Supplementary Figure S2D). These results indicate that DAZAP2 and SIAH1 form a complex in cells. The interaction between SIAH1 and DAZAP2 was further confirmed by pull-down assays using GST-SIAH1 and in vitro translated 35S-labeled DAZAP2 (Figure 2F).
We next analyzed the effect of DAZAP2 on HIPK2-SIAH1 complex formation. We co-expressed HIPK2 and SIAH1 in the presence or absence of DAZAP2 and analyzed protein complex formation using coimmunoprecipitation analysis. Interestingly, in the presence of DAZAP2 the amount of SIAH1 coprecipitated with HIPK2 was clearly increased, indicating that DAZAP2 stimulates HIPK2-SIAH1 interaction (Figure 2G). Surprisingly, increased HIPK2-SIAH1 complex formation was not accompanied by elevated DAZAP2 coimmunoprecipitation suggesting that DAZAP2 does not act as a classical bridging factor between HIPK2 and SIAH1 but may act as a loading factor to potentiate the loading of SIAH1 on HIPK2 (Figure 2G).
To investigate whether DAZAP2-stimulated loading of SIAH1 on HIPK2 potentiates SIAH1-dependent HIPK2 polyubiquitination (22), we performed in vitro ubiquitination assays. All assay components were produced recombinantly in E. coli. Indeed, DAZAP2 potentiated SIAH1-mediated HIPK2 polyubiquitination in vitro (Figure 2H) and also in cellulo (Supplementary Figure S2E). Taken together, our results demonstrate that DAZAP2 potentiates HIPK2-SIAH1 complex formation and stimulates HIPK2 ubiquitination and subsequent proteolysis.
Depletion of DAZAP2 sensitizes cells to DNA-damaging drug treatments
Since DAZAP2 regulates HIPK2 steady-state levels and HIPK2 is a crucial regulator of cancer cell sensitivity to DNA-damaging chemotherapeutic drugs (8,32), we investigated whether DAZAP2 regulates cancer cell chemosensitivity by affecting cell death commitment. Therefore, we depleted DAZAP2 in colorectal cancer cells using RNA interference and treated the cells with the DNA-damaging chemotherapeutic drug 5-Fluorouracil (5-FU). Indeed, upon chemotherapeutic drug exposure, DAZAP2 depleted cells showed an increase in PARP cleavage, which is a marker of caspase-driven apoptotic cell death (Figure 3A), and in Annexin V-positivity, an additional apoptosis marker (Figure 3B). To corroborate the role of DAZAP2 in regulating cancer cell chemosensitivity, we used two other DNA-damaging chemotherapeutic drugs with a different mode of action. Strikingly, depletion of DAZAP2 also resulted in an increased sensitivity of the cells to Adriamycin/Doxorubicin or Oxaliplatin exposure (Figure 3C). These results indicate that loss of DAZAP2 potentiates cancer cell chemosensitivity and suggest that DAZAP2 acts as a chemoresistance factor compromising DNA damage-induced cell death.
Figure 3.

Loss of DAZAP2 sensitizes cancer cells to chemotherapeutic drug treatment. (A, B) DAZAP2 depletion sensitizes HCT116 cells to 5-FU treatment. HCT116 cells were transfected with DAZAP2-specific or control siRNAs and DNA damage was induced using 50 μM 5-FU. (A) Cells were harvested 24 h post-treatment and lysates were analyzed by immunoblotting. n = 3. (B) Cells were harvested 48 h post-treatment and co-stained with Annexin V and PI. Stained cells were analyzed by flow cytometry. Bar graph showing quantification of % Annexin V-positive cells + SD from three biological replicates. ** P-value ≤ 0.01, ns non-significant (un-paired t-test). n = 3. (C) DAZAP2 depletion sensitizes HCT116 cells to Adriamycin and Oxaliplatin. HCT116 cells were transfected with DAZAP2-specific or control siRNAs and DNA damage was induced for 24 h using 0.75 μg/ml Adriamycin or 50 μM Oxaliplatin. Cell lysates were analyzed by immunoblotting. n = 3. (D) DAZAP2 deletion chemosensitizes cells. HCT116 control CRISPR/Cas9 and DAZAP2 knockout CRISPR/Cas9 clonal cell lines were treated with 50 μM 5-FU for 24 h and total protein lysates were subjected to immunoblot analysis for the expression of the indicated proteins. n = 3. (E, F) DAZAP2 deletion increases Caspase-3/7 activity upon DNA damage. HCT116 CRISPR/Cas9 control and CRISPR/Cas9 DAZAP2 knockout clonal cell lines were treated with 50 μM 5-FU and Caspase-3/7 activity was assessed 24 h post-treatment by fluorescence microscopy. n = 3. (E) Representative image of cells with active Caspase-3/7 (green) are shown. (F) Bar graph represents mean and SD of % NucView-positive, green cells from three images. (G) DAZAP2 deletion sensitizes tumors to chemotherapeutic drug treatment in vivo. CRISPR/Cas9 DAZAP2 KO or control CRISPR/Cas9 HCT116 cells were subcutaneously injected into NOD/SCID mice. When the tumor size reached 100 mm3, mice were treated with DMSO or 50 mg/kg 5-FU for 21 days. Analysis of weight of tumor size after 21 days of treatment is shown. Data are displayed as sample mean ± standard error of the mean. * P-value ≤0.05, ns non-significant (unpaired t-test).
To exclude off-target effects of our RNAi approach we next used CRISPR/Cas9-mediated gene editing to generate DAZAP2 knock-out cells. DNA sequencing of the genomic DAZAP2 loci demonstrated that the generated cell clones showed frameshifts in the DAZAP2 coding sequence leading to a premature STOP codon in both alleles (Supplementary Figure S3A, B). Accordingly, no DAZAP2 protein expression was detectable in the DAZAP2 KO cells, while the WT control cells generated with control guide RNAs lacking any specific genomic targeting sequence, as expected, retained DAZAP2 expression at a similar level as the parental cells (Supplementary Figure S3C). Chemotherapeutic drug treatment of DAZAP2 WT and KO cells demonstrated increased chemosensitivity of the KO cells as revealed by increased PARP cleavage (Figure 3D). Furthermore, comparable results were obtained using a single cell-based life-cell cell death assay termed Nuc-View® assay, which visualizes cells with Caspase-3/-6 activity through accumulation of a green fluorochrome in the nucleus of the cells (Figure 3E, F). Collectively, our results show that both RNAi-mediated depletion as well as genetic deletion of DAZAP2 sensitizes cancer cells to chemotherapeutic drug treatment.
Having demonstrated that DAZAP2 regulates cancer cell chemosensitivity in cellulo, we next tested whether DAZAP2 also controls cancer cell chemosensitivity in vivo. To this end we used a mouse xenograft tumor model. One million DAZAP2 KO and WT cells were injected subcutaneously into the opposing flanks of immune-compromised mice and tumor growth was allowed for a few days to obtain tumors of 100 mm3. Next, the animals were subjected to chemotherapy using 5-FU or DMSO as solvent control. Notably, tumor growth as well as chemotherapeutic treatment showed no generalized detrimental effects on the body weight of the animals (Supplementary Figure S3D). After nine cycles of chemotherapy the animals were sacrificed and the tumor volume was analyzed to determine the treatment response (Figure 3G). Tumors derived from DAZAP2 KO cells were significantly smaller upon 5-FU treatment than tumors derived from WT cells, indicating that DAZAP2 regulates cancer cell chemosensitivity in vivo. Collectively, our results indicate that DAZAP2 regulates cancer cell chemosensitivity.
A fraction of DAZAP2 is targeted to the cell nucleus and binds to p53 upon DNA damage
Next, we aimed to elucidate the underlying molecular mechanism how loss of DAZAP2 potentiates cancer cell chemosensitivity. Since DAZAP2 depletion increases HIPK2 steady-state levels and HIPK2 phosphorylates p53 at Ser46, which potently simulates the cell death response (18), we hypothesized that DAZAP2 depletion may impact on p53 Ser46 phosphorylation. However, DAZAP2 depletion showed no effect on p53 Ser46 phosphorylation and HIPK2 protein levels after chemotherapeutic drug treatment (Supplementary Figure S4A). Therefore, we postulated that, although DAZAP2 regulates HIPK2 steady-state protein levels in unstressed cells, HIPK2 may escape DAZAP2 regulation in response to DNA damage. Indeed, we will provide evidence for this conclusion later in this report.
To get insight into the molecular mechanism by which DAZAP2 regulates cancer cell chemosensitivity we investigated its subcellular distribution. Interestingly, subcellular fractionation experiments revealed that DAZAP2 alters its subcellular localization upon DNA damage and partially accumulates in the cell nucleus (Figure 4A). This finding prompted us to test whether DAZAP2 might interact with p53, which is known to accumulate in the cell nucleus upon DNA damage. Indeed, coimmunoprecipitation experiments demonstrated that endogenous DAZAP2 forms a complex with p53 in response to DNA damage, but not in the absence of genotoxic stress (Figure 4B).
Figure 4.

DAZAP2 interacts with p53. (A) A fraction of DAZAP2 accumulates in the nucleus upon DNA damage. HCT116 cells were treated with DMSO or 50 μM 5-FU for 24 h. Cells were fractionated into nuclear and cytoplasmic fractions, and equal total protein amounts of all fractions were analyzed by immunoblotting. WCL = whole cell lysate. Cyto = Cytoplasm. n = 3. (B) Interaction of endogenous DAZAP2 and p53. Lysates from untreated or Adriamycin-treated HepG2 cells in the presence of MG-132 were subjected to immunoprecipitation with control or p53 antibodies. Lysates and immunoprecipitates were analyzed by immunoblotting. n = 2. (C, D) In vitro interaction of DAZAP2 and p53. (C) GST pull-down assays were performed using in vitro translated 35S-labeled p53 and bacterially expressed GST-DAZAP2 or GST as a control. n = 3. (D) GST pull-down assays were performed using in vitro translated 35S-labeled DAZAP2 and bacterially expressed GST-p53 or GST as a control. n = 3. GST pulldown assays were analyzed by SDS-PAGE, Coomassie Brilliant Blue (CBB) staining and autoradiography. Representative experiments are shown.
To confirm that DAZAP2 binds to p53, we performed GST-pulldown assays. Therefore, we recombinantly expressed DAZAP2 in E. coli and purified GST-DAZAP2 via affinity purification. GST-pulldown assays were performed after incubation with 35S-labeled p53. Indeed, p53 specifically coprecipitated with GST-DAZAP2 (Figure 4C) and vice versa (Figure 4D), suggesting that DAZAP2 directly binds to p53. In order to map the DAZAP2-interaction domain on p53, we performed GST-pulldowns using various truncated GST-p53 proteins (Supplementary Figure S4B). The interaction mapping suggests that DAZAP2 interacts with the DNA-binding domain (DD) and the C-terminal domain of p53 (Supplementary Figure S4B). Taken together, our findings indicate that DAZAP2 accumulates upon DNA damage in the cell nucleus where it binds to p53.
HIPK2 site-specifically phosphorylates DAZAP2 in vitro and upon DNA damage
Post-translational protein modifications can regulate intracellular protein distribution. Since HIPK2 is a Ser/Thr protein kinase, we next performed in vitro kinase assays to determine if DAZAP2 is a substrate of HIPK2. Indeed, wild-type HIPK2 precipitated from 293T cells phosphorylated bacterially expressed GST-DAZAP2, while a kinase-dead HIPK2 point-mutant (K221A) failed to do so (Figure 5A). To show that HIPK2 directly phosphorylates DAZAP2, we repeated the in vitro kinase assays with bacterially expressed His-HIPK2 confirming that HIPK2 is able to phosphorylate DAZAP2 (Supplementary Figure S5A).
Next, we aimed to map the phosphorylation site(s). Previously identified HIPK2 substrates suggest that HIPK2 is a proline-directed Ser/Thr kinase (19,20,25,51). Intriguingly, the amino acid sequence of DAZAP2 lacks Ser/Thr-Pro motifs, indicating that HIPK2 can also phosphorylate substrates at none proline-flanked Ser/Thr residues. To experimentally map the phosphorylation site(s), various truncated GST-DAZAP2 fusion proteins were expressed in E. coli, affinity-purified and used in in vitro kinase assays. Our analyses enabled us to identify the central, proline-rich region of DAZAP2 as a critical region for HIPK2-mediated phosphorylation (Supplementary Figure S5B). Site-directed mutagenesis within the proline-rich region indicated that multiple Ser/Thr residues, Ser62, Ser68, Ser77 and Ser85/Thr86, are phosphorylated by HIPK2 in vitro (Figure 5B). Since the exchange of Ser77 to Ala resulted in a massive reduction in DAZAP2 phosphorylation (Figure 5B), our data identify Ser77 as the main phosphorylation site.
We raised a phospho-specific antibody against the phospho-Ser77 site of DAZAP2. The antibody was validated using non-phosphorylated DAZAP2 and DAZAP2 phosphorylated by HIPK2 in vitro. The affinity-purified DAZAP2 phospho-Ser77 antibody specifically recognized phosphorylated DAZAP2, but not the non-phosphorylated DAZAP2 protein, thus indicating its phospho-specificity (Figure 5C).
Next, we used this antibody to study whether DAZAP2 is phosphorylated at Ser77 in cells under conditions when HIPK2 gets activated. Thus, we co-expressed HIPK2 and DAZAP2, exposed the cells to DNA damage and subsequently immunoprecipitated DAZAP2 from the cell lysates. Immunoblotting using the pSer77 DAZAP2 antibody demonstrated phosphorylation of DAZAP2 upon Adriamycin treatment (Figure 5D), indicating that DAZAP2 is phosphorylated at Ser77 in response to DNA damage in cells.
Phosphorylation of DAZAP2 terminates its HIPK2-degrading function
To study the functional consequences of HIPK2-mediated DAZAP2 phosphorylation we generated phospho-mimetic (DAZAP25D) and phospho-deficient (DAZAP25A) DAZAP2 point-mutants and compared their effects on HIPK2 degradation to the wild-type protein. Intriguingly, DAZAP25D entirely lacked the capacity to trigger HIPK2 proteolysis when compared to wild-type DAZAP2 or to the corresponding phospho-deficient DAZAP25A point mutant (Figure 5E). Furthermore, protein-protein interaction analysis using coimmunoprecipitation experiments revealed a massive reduction in complex formation between phospho-mimetic DAZAP25D both with HIPK2 (Supplementary Figure S5C) and with SIAH1 (Supplementary Figure S5D). In contrast, phospho-deficient DAZAP25A and DAZAP2 wild-type showed a comparable, clear interaction with HIPK2 (Supplementary Figure S5C), excluding the possibility that the Ser/Thr residues mutated in the phospho-deficient and -mimetic versions are structurally essential for the interaction. These results suggest, as anticipated earlier in this study, that HIPK2 can escape the proteolysis-stimulating function of DAZAP2 upon DNA damage through phosphorylating DAZAP2. This is in line with our finding that depletion of DAZAP2 does not further increase HIPK2 protein levels and p53 Ser46 phosphorylation in response to DNA damage (Supplementary Figure S4A). Collectively, our findings indicate that upon DNA damage HIPK2 phosphorylates DAZAP2, which interferes with its HIPK2-degrading capacity.
Phosphorylation targets DAZAP2 to the cell nucleus
Next, we analyzed the subcellular distribution of wild-type DAZAP2, phospho-mimetic DAZAP25D and phospho-deficient DAZAP25A using confocal microscopy. Interestingly, we recognized that the phospho-mutant proteins massively differ in their subcellular distribution. While the phospho-deficient DAZAP25A almost exclusively localizes to the cytoplasm of the cells (Figure 5F), the phospho-mimetic DAZAP25D mutant predominantly localizes to the cell nucleus (Figure 5G). This latter distribution is reminiscent of the subcellular distribution of transcriptional regulators including p53.
DAZAP2 modulates the expression of a subset of p53 target genes upon DNA damage
So far, our results demonstrated that DAZAP2 accumulates in the cell nucleus and binds to p53 in response to chemotherapeutic drug-induced DNA damage. Since p53 is a transcription factor regulating well-defined target gene sets (52), we investigated the impact of DAZAP2 on p53 target gene expression. To this end, we performed transcriptome-wide RNA-seq analysis of DAZAP2-depleted and control cancer cells upon DNA-damaging chemotherapeutic drug treatment or DMSO as solvent control. Principal component analysis (PCA) revealed that the independent biological replicates used for our analysis readily clustered together (Supplementary Figure S6A). Samples from 5-FU treatment were strongly separated from controls, while DAZAP2 depletion showed a more moderate, but still highly consistent separation (Supplementary Figure S6). This suggests that 5-FU treatment resulted in substantial changes in gene expression, while only a small set of genes changed their expression upon DAZAP2 knockdown.
Since our previous findings indicated that DAZAP2 depletion potentiates chemosensitivity and cancer cell death, we identified the genes that were differentially regulated by DAZAP2 in response to 5-FU treatment. Overall, 1404 genes were found to be differentially regulated by DAZAP2 after 5-FU exposure (Figure 6A). Gene ontology (GO) analysis revealed that biological processes including DNA damage response, cell death and survival regulation are most affected upon DAZAP2 depletion (Figure 6B).
Figure 6.

DAZAP2 regulates a subset of p53 target genes and restricts cell death in a p53-mediated manner. (A) Results of RNA-sequencing analyses are visualized by a Volcano plot showing differentially expressed genes between DAZAP2-depleted and control HCT116 cells upon 5-FU treatment. Each dot represents one gene. Red dots indicate genes with higher expression in DAZAP2-depleted cells and a q-value ≤ 0.05, while blue dots represent genes with lower expression in DAZAP2-depleted cells and a q-value ≤ 0.05. Grey dots show the genes with an q-value ≥ 0.05. Highlighted in green is DAZAP2 expression. (B, C) Ingenuity pathway analysis of genes differentially expressed in RNA-seq data from DAZAP2-depleted versus control HCT116 cells upon 5-FU treatment. (B) Top predicted biological functions are ranked by -LOG(P-value). (C) Top predicted upstream regulators are ranked by -LOG(P-value). (D) Identification of DAZAP2-regulated p53 target genes. Venn diagram displaying the overlap between DAZAP2-regulated genes upon 5-FU treatment and p53 target genes(52). (E) Heatmap of the p53 target genes regulated by DAZAP2 upon genotoxic stress. RNA-Seq analysis was performed as described in (A). Relative expression of DAZAP2 is shown in the last row of the heatmap. R1–3 indicate the three biological replicates. (F) DAZAP2 depletion enhances the expression of a subset of cell death-associated p53 target genes upon 5-FU exposure. HCT116 or HCT116 p53−/− were transfected with DAZAP2-specific or control siRNAs and DNA damage was induced using 50 μM 5-FU. mRNA expression was analyzed by qRT-PCR 48 h post-treatment for the indicated genes. Gene expression was normalized to GAPDH/B2M expression and the fold change relative to the untreated cells was calculated. Data are shown as mean + SD from three replicates. *** P-value ≤0.001 (two-way ANOVA for left panel, un-paired t-test for right panel). (G, H) DAZAP2 depletion sensitizes HCT116 cells to cell death in a p53-regulated manner. HCT116 wt and p53−/− cells were transfected with DAZAP2-specific or control siRNAs and DNA damage was induced using 50 μM 5-FU. n = 3. (G) Total protein extracts were prepared 24 h post-treatment and analyzed by immunoblotting for the indicated proteins. A representative experiment is shown. (H) Cells were harvested 48 h post-treatment and co-stained with Annexin V and PI. Stained cells were analyzed by flow cytometry. Bar graph showing quantification of % Annexin V-positive cells + SD from three biological replicates. ** P-value ≤0.01, ns non-significant (un-paired t-test).
Notably, when we queried the upstream regulators of the DAZAP2-regulated genes, the top hit was p53 (Figure 6C). This result prompted us to compare the DAZAP2-regulated genes to a list of 343 validated, high-confidence p53 target genes (52). This comparison identified a subset of 53 p53 target genes to be modulated by DAZAP2 upon chemotherapeutic drug treatment (Figure 6D). Heat map visualization of the overlap indicated that 36 p53 target genes were upregulated in DAZAP2-depleted cells upon 5-FU treatment (Figure 6E). Interestingly, a substantial number of the p53 target genes upregulated upon DAZAP2 depletion are known regulators of cell death, which is in line with our finding that DAZAP2 regulates cancer cell chemosensitivity. Collectively, our RNA-seq analysis indicates that DAZAP2 specifies the p53 response by regulating a subset of p53 target genes upon DNA damage.
Next, we aimed at validating the identified DAZAP2-regulated p53 target genes using an independent approach. Since we found that DAZAP2 depletion sensitizes cells to chemotherapy-induced cell death, we focused on the cell death-associated DAZAP2-regulated p53 target genes. Reverse-transcription (RT) quantitative (q) PCR analysis confirmed that cell death-regulatory p53 target genes were significantly up-regulated in DAZAP2-depleted cells upon chemotherapeutic drug treatment (Figure 6F).
To discriminate whether p53 activation alone is sufficient for the observed changes in DAZAP2-regulated p53 target genes or if additional DNA damage-induced signals are involved, we used the MDM2 inhibitor Nutlin-3a to trigger p53 activation in absence of additional DNA damage signals. Interestingly, Nutlin-3a treatment did not increase p53 target gene activation upon DAZAP2 depletion (Supplementary Figure S6B) arguing that additional signals like DNA-damage induced DAZAP2 phosphorylation and subsequent nuclear accumulation are required for the observed changes in gene expression upon 5-FU exposure.
To test whether DAZAP2 regulates these cell death-linked target genes through its interplay with p53, we used isogenic HCT116 p53 wild-type and p53 KO cells and analyzed their response to drug-treatment upon DAZAP depletion (Figure 6F). qRT-PCR analysis showed that DAZAP2 depletion exclusively increases the expression of these target genes in the presence of p53, but not in p53-deficient cells (Figure 6F). Collectively, our results show that DAZAP2 specifies the p53 response by modulating a subset of p53 target genes.
DAZAP2 controls p53-dependent cell death upon chemotherapeutic drug treatment
Since DAZAP2 modulates the expression of cell death regulatory p53 target genes upon DNA damage, we investigated the role of p53 in chemo-sensitization of DAZAP2-depleted cancer cells. To this end, we used HCT116 p53 wild-type and p53 KO cells. While knock-down of DAZAP2 resulted in pronounced PARP cleavage in the presence of p53, PARP cleavage was largely absent in the p53 KO cells upon DAZAP2 depletion (Figure 6G). Furthermore, Annexin V staining and flow cytometry analysis also demonstrated that DAZAP2 depletion specifies the response towards cell death induction in p53-proficient cells (Figures 3A and 6H), but not in p53-deficient cells (Figure 6H). Collectively, these findings indicate that DAZAP2 specifies the p53-dependent cell death response through modulating a subset of p53 target genes.
DAZAP2 occupies p53 target gene promoters
To unravel the mechanism by which DAZAP2 regulates p53 target gene expression, we considered the following observations. (i) Since depletion of DAZAP2 shows no impact on total p53 protein levels (see Figure 6G), and (ii) since DAZAP2 regulates a select subset of p53 target genes, we concluded that DAZAP2 is not a general p53 regulator (for instance, by affecting p53 protein stability). Instead, DAZAP2 may specify p53 target gene expression through binding p53 target genes at promoter proximal response elements.
To test this hypothesis, we performed chromatin-immunoprecipitation (ChIP) analysis. First, we confirmed that the p53 binding elements of the DAZAP2-regulated p53 target genes were indeed occupied by p53 (Supplementary Figure S7). Next, we assessed whether DAZAP2 is recruited to p53 response elements by performing ChIP analysis using DAZAP2 antibodies. Indeed, our results demonstrated an association of DAZAP2 with the p53 binding elements of the DAZAP2-regulated target genes FAS, TNFRSF10B, TP53INP1, GPX1 and GADD45A (Figure 7A). These results suggest that DAZAP2 regulates p53 target gene expression through binding p53 at its response elements.
Figure 7.
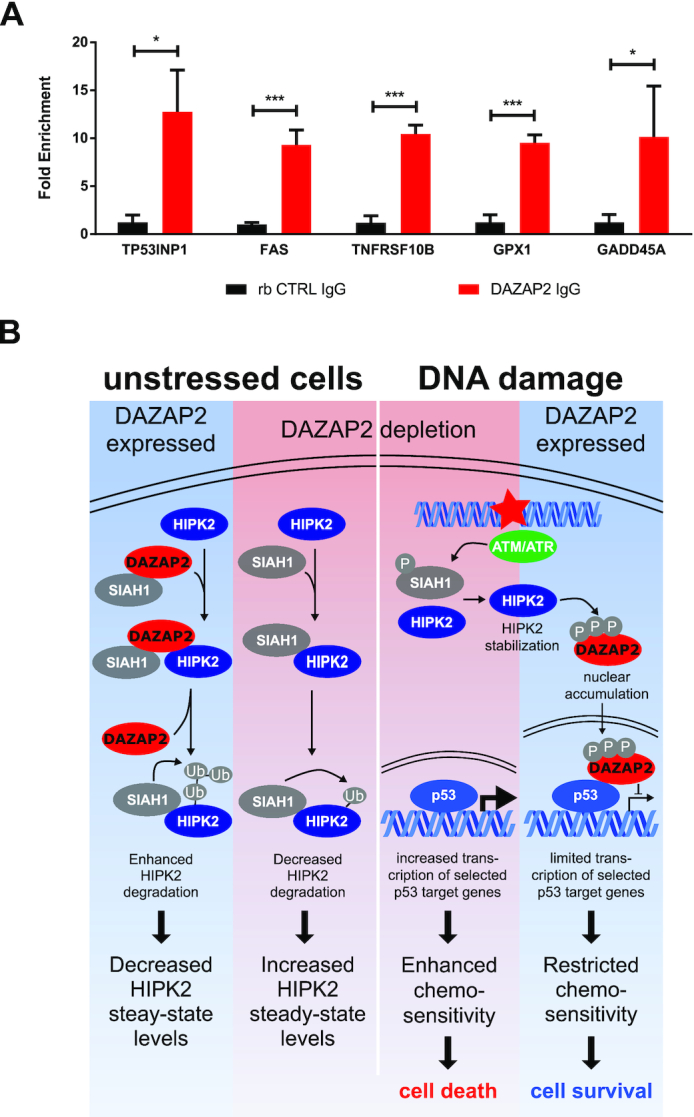
DAZAP2 co-occupies p53 response elements. (A) DAZAP2 binds to p53 response elements. ChIP analyses were performed with anti-DAZAP2 antibody in cross-linked lysates from HCT116 cells treated with 50 μM 5-FU for 24 h. A non-specific IgG was used as a negative binding control. Quantitative PCR was carried out on the immunoprecipitated DNA with primers specific for regions encompassing the p53 RE. Results are represented as fold enrichment + SD from three replicates. * P-value < 0.05, *** P-value < 0.001 (unpaired t-test). (B) Schematic model summarizing the regulation of HIPK2 and the p53 pathway by the adaptor protein DAZAP2 in undamaged cells and in response to DNA damage.
DISCUSSION
In this study we provide in vitro and in vivo evidence that the 17 kDa small adaptor protein DAZAP2 is a novel regulator of cancer cell sensitivity to DNA-damaging chemotherapeutic drugs. Mechanistically, we elucidate an intricate signaling network linking DAZAP2 to the p53 response via physical interaction with the DNA damage-activated kinase HIPK2 and the tumor suppressor p53 (Figure 7B). We demonstrate that in unstressed cells DAZAP2 associates with HIPK2 and the E3 ubiquitin ligase SIAH1 and stimulates SIAH1-catalysed HIPK2 ubiquitination and subsequent proteolysis by the proteasome, thus controlling HIPK2 steady-state protein levels. Our data suggest that DAZAP2 may act as a ‘loading factor’ that potentiates complex formation between SIAH1 and HIPK2. This interpretation is supported by our findings that, although DAZAP2 can directly form complexes both with HIPK2 and SIAH1, upon co-expression of DAZAP2, HIPK2 and SIAH1, DAZAP2 increases HIPK2-SIAH1 complex formation to the expense of its own binding to HIPK2. Interestingly, SIAH1 and DAZAP2 bind to each other and in addition share the same binding domain on HIPK2 (22). Thus, we hypothesize that DAZAP2 may function as a supportive SIAH1 landing platform, which gets released from HIPK2 upon HIPK2-SIAH1 binding.
In line with a previous observation, our data provide evidence that DAZAP2 can shuttle between the cytoplasm and the cell nucleus (46,47). Similarly, HIPK2 can reside both in the cell nucleus and in the cytoplasm (19,20,28,33). Previous studies have linked DAZAP2 to cytoplasmic stress granule (SG) formation, Wnt signaling as well as inflammatory signaling, and nuclear DAZAP2 to transcriptional regulation by SOX6 and TCF4/LEF (46,48,53–57). Our results indicate that DAZAP2 colocalizes with HIPK2 both in the nucleus and at cytoplasmic granules, which likely represent SGs. These findings suggest a potential link between the HIPK2-p53 cell fate regulatory signaling pathway and SGs, which are also known to regulate cellular stress responses and cell fate. A potential role of HIPK2 in SG biology remains to be elucidated in the future.
Our data demonstrate that DAZAP2 accumulates in the cell nucleus upon DNA damage (Figure 7B). Furthermore, our results provide evidence that phosphorylation targets DAZAP2 to the cell nucleus. We show that HIPK2 site-specifically phosphorylates DAZAP2 in vitro and in cellulo upon DNA damage at a set of Ser/Thr residues including Ser77. Remarkably, none of these phosphorylation sites is flanked by a Pro residue, demonstrating that HIPK2-mediated substrate phosphorylation is not restricted to proline-directed Ser/Thr residues as supposed previously. Site-specific phosphorylation of DAZAP2 negatively regulates its interaction with HIPK2 and SIAH1, providing an explanation why DAZAP2 selectively regulates HIPK2 steady-state protein levels in undamaged cells, and suggesting a mechanism by which HIPK2 escapes DAZAP2-regulated proteolysis upon DNA damage, which triggers HIPK2 stabilization and activation.
Upon DNA damage, DAZAP2 accumulates in the cell nucleus and binds to p53. Our in vitro analysis demonstrates that DAZAP2 binds p53, presumably by interacting with the DNA-binding domain and the C-terminal part of p53. Furthermore, RNA-seq analysis demonstrated that depletion of DAZAP2 regulates a subset of 53 p53 target genes in response to chemotherapeutic drug treatment. Mechanistically, our results obtained by ChIP analysis indicate that DAZAP2 impacts on p53 target genes expression at the level of the p53 response elements, which is in line with its physical interaction with p53.
Our experiments revealed that depletion of DAZAP2 potentiates chemotherapeutic drug-induced cell death through enhancing the expression of cell death-driving p53 target genes. This strongly supports the conclusion that the presence of DAZAP2 and its interaction with p53 reduces DNA damage-induced cell death by limiting the expression of cell death-mediating p53 target genes. Our findings indicate that upon DNA damage HIPK2 targets a fraction of DAZAP2 to the cell nucleus by a mechanism including site-specific phosphorylation at Ser77. Furthermore, nuclear DAZAP2 directly interacts with p53 and down-modulates apoptotic target gene expression. Actually, at first sight, such an involvement of HIPK2 in blunting the cell death response seems to be unexpected and counterintuitive, as HIPK2 is a well-established mediator of pro-apoptotic functions in response to irreparable DNA damage (8,18,58,59).
Although, due to technical limitations (low sensitivity) of the phospho-Ser77 DAZAP2 antibodies, we could not directly determine whether endogenous DAZAP2 is differentially phosphorylated in response to repairable versus irreparable DNA damage, the following hypothetic model would reconcile our findings on DAZAP2 with the current view of HIPK2. We hypothesize that DAZAP2 phosphorylation, its subsequent nuclear targeting and its p53-modulating effects restrict the cell death-inducing functions of HIPK2 in particular in response to mild, repairable DNA damage in order to favor cell survival. In line with this assumption, previous studies indicate that HIPK2 is also stabilized and activated upon mild, repairable DNA damage, but does not trigger the cell death response under such conditions (24,25). Thus, by specifically interfering with cell death-activating functions of HIPK2 upon mild DNA damage, DAZAP2 may provide an important contribution to the previously described cyto-protective role of HIPK2 in response to DNA damage (60). In summary, we propose that DAZAP2 fulfills an important function in guiding cell survival-promoting signaling events by HIPK2 upon mild, repairable DNA damage through blocking cell death-stimulating HIPK2 activities.
DAZAP2 knock-down or genetic deletion using CRISPR/Cas9-mediated gene editing results in a potent chemosensitization of cancer cells in cellulo and in vivo in a mouse xenograft tumor model. The profound chemosensitization achieved by DAZAP2 depletion was largely abrogated in p53-deficient cancer cells, indicating that the DAZAP2-p53 link identified in this study is of functional relevance for the outcome of DNA-damaging chemotherapeutic drug treatment. In line with our finding demonstrating that DAZAP2 depletion increases chemosensitivity, a recent report identified amplification of DAZAP2 by gene duplication, which presumably results in increased DAZAP2 expression, in Hodgkin lymphoma cells (61). Therefore, upregulation of DAZAP2 expression might mechanistically contribute to chemoresistance in cancer.
Collectively, our findings integrate the small adaptor protein DAZAP2 as a novel piece in the p53 puzzle and provide novel insight into the mechanism by which the p53 response and cell fate choice upon DNA damage gets specified.
DATA AVAILABILITY
The RNA-Seq data are available in the GEO database (GSE154146, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154146).
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Dr D. Sombroek, K. Scheuermann and D. Grieshober for help with experiments, and to Dr. A. Möller and Dr. D.D. Bowtell for kindly providing the Siah1/Siah2 double KO MEFs.
Contributor Information
Magdalena C Liebl, Institute of Toxicology, University Medical Center Mainz, Johannes Gutenberg University, Mainz, Germany.
Jutta Moehlenbrink, Institute of Toxicology, University Medical Center Mainz, Johannes Gutenberg University, Mainz, Germany.
Huong Becker, Institute of Toxicology, University Medical Center Mainz, Johannes Gutenberg University, Mainz, Germany.
Günter Raddatz, Division of Epigenetics, German Cancer Research Center (dkfz), Heidelberg, Germany.
Suhaib K Abdeen, The Concern Foundation Laboratories, The Lautenberg Center for Immunology and Cancer Research, Department of Immunology and Cancer Research-IMRIC, Hebrew University-Hadassah Medical School, Jerusalem, Israel.
Rami I Aqeilan, The Concern Foundation Laboratories, The Lautenberg Center for Immunology and Cancer Research, Department of Immunology and Cancer Research-IMRIC, Hebrew University-Hadassah Medical School, Jerusalem, Israel; Department of Cancer Biology and Genetics, Wexner Medical Center, The Ohio State University, Columbus, OH, USA.
Frank Lyko, Division of Epigenetics, German Cancer Research Center (dkfz), Heidelberg, Germany.
Thomas G Hofmann, Institute of Toxicology, University Medical Center Mainz, Johannes Gutenberg University, Mainz, Germany.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) [SFB 1036 (Project-ID 201348542), project 3 (to T.G.H.), SFB 1361 (Project-ID 393547839), project 19 (to T.G.H.)]. Funding for open access charge: Deutsche Forschungsgemeinschaft [SFB 1361].
Conflict of interest statement. None declared.
REFERENCES
- 1. Halazonetis T.D., Gorgoulis V.G., Bartek J.. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319:1352–1355. [DOI] [PubMed] [Google Scholar]
- 2. Kastan M.B., Bartek J.. Cell-cycle checkpoints and cancer. Nature. 2004; 432:316–323. [DOI] [PubMed] [Google Scholar]
- 3. Carbone M., Arron S.T., Beutler B., Bononi A., Cavenee W., Cleaver J.E., Croce C.M., D’Andrea A., Foulkes W.D., Gaudino G.et al.. Tumour predisposition and cancer syndromes as models to study gene-environment interactions. Nat. Rev. Cancer. 2020; 20:533–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bartek J., Lukas J.. DNA damage checkpoints: from initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007; 19:238–245. [DOI] [PubMed] [Google Scholar]
- 5. Jackson S.P., Bartek J.. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. d’Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nat. Rev. Cancer. 2008; 8:512–522. [DOI] [PubMed] [Google Scholar]
- 7. Zhivotovsky B., Kroemer G.. Apoptosis and genomic instability. Nat. Rev. Mol. Cell Biol. 2004; 5:752–762. [DOI] [PubMed] [Google Scholar]
- 8. Matt S., Hofmann T.G.. The DNA damage-induced cell death response: a roadmap to kill cancer cells. Cell. Mol. Life Sci. 2016; 73:2829–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hollstein M., Sidransky D., Vogelstein B., Harris C.C.. p53 mutations in human cancers. Science. 1991; 253:49–53. [DOI] [PubMed] [Google Scholar]
- 10. Charni M., Aloni-Grinstein R., Molchadsky A., Rotter V.. p53 on the crossroad between regeneration and cancer. Cell Death Differ. 2017; 24:8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003; 10:431–442. [DOI] [PubMed] [Google Scholar]
- 12. Appella E., Anderson C.W.. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. 2001; 268:2764–2772. [DOI] [PubMed] [Google Scholar]
- 13. Vousden K.H., Prives C.. Blinded by the light: the growing complexity of p53. Cell. 2009; 137:413–431. [DOI] [PubMed] [Google Scholar]
- 14. Brooks C.L., Gu W.. p53 ubiquitination: Mdm2 and beyond. Mol. Cell. 2006; 21:307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boehme K.A., Blattner C.. Regulation of p53 - insights into a complex process. Crit. Rev. Biochem. Mol. Biol. 2009; 44:367–392. [DOI] [PubMed] [Google Scholar]
- 16. Lee J.T., Gu W.. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 2010; 17:86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oda K., Arakawa H., Tanaka T., Matsuda K., Tanikawa C., Mori T., Nishimori H., Tamai K., Tokino T., Nakamura Y.et al.. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000; 102:849–862. [DOI] [PubMed] [Google Scholar]
- 18. Liebl M.C., Hofmann T.G.. Cell fate regulation upon DNA damage: p53 serine 46 kinases pave the cell death road. Bioessays. 2019; 41:e1900127. [DOI] [PubMed] [Google Scholar]
- 19. Hofmann T.G., Moller A., Sirma H., Zentgraf H., Taya Y., Droge W., Will H., Schmitz M.L.. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat. Cell Biol. 2002; 4:1–10. [DOI] [PubMed] [Google Scholar]
- 20. D’Orazi G., Cecchinelli B., Bruno T., Manni I., Higashimoto Y., Saito S., Gostissa M., Coen S., Marchetti A., Del Sal G.et al.. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat. Cell Biol. 2002; 4:11–19. [DOI] [PubMed] [Google Scholar]
- 21. Dauth I., Kruger J., Hofmann T.G.. Homeodomain-interacting protein kinase 2 is the ionizing radiation-activated p53 serine 46 kinase and is regulated by ATM. Cancer Res. 2007; 67:2274–2279. [DOI] [PubMed] [Google Scholar]
- 22. Winter M., Sombroek D., Dauth I., Moehlenbrink J., Scheuermann K., Crone J., Hofmann T.G.. Control of HIPK2 stability by ubiquitin ligase Siah-1 and checkpoint kinases ATM and ATR. Nat. Cell Biol. 2008; 10:812–824. [DOI] [PubMed] [Google Scholar]
- 23. Puca R., Nardinocchi L., D’Orazi G.. Regulation of vascular endothelial growth factor expression by homeodomain-interacting protein kinase-2. J. Exp. Clin. Cancer Res. 2008; 27:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bitomsky N., Conrad E., Moritz C., Polonio-Vallon T., Sombroek D., Schultheiss K., Glas C., Greiner V., Herbel C., Mantovani F.et al.. Autophosphorylation and Pin1 binding coordinate DNA damage-induced HIPK2 activation and cell death. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E4203–E4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Conrad E., Polonio-Vallon T., Meister M., Matt S., Bitomsky N., Herbel C., Liebl M., Greiner V., Kriznik B., Schumacher S.et al.. HIPK2 restricts SIRT1 activity upon severe DNA damage by a phosphorylation-controlled mechanism. Cell Death Differ. 2016; 23:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. He Y., Roos W.P., Wu Q., Hofmann T.G., Kaina B.. The SIAH1-HIPK2-p53ser46 damage response pathway is involved in temozolomide-induced glioblastoma cell death. Mol. Cancer Res. 2019; 17:1129–1141. [DOI] [PubMed] [Google Scholar]
- 27. Calzado M.A., de la Vega L., Moller A., Bowtell D.D., Schmitz M.L.. An inducible autoregulatory loop between HIPK2 and Siah2 at the apex of the hypoxic response. Nat. Cell Biol. 2009; 11:85–91. [DOI] [PubMed] [Google Scholar]
- 28. Crone J., Glas C., Schultheiss K., Moehlenbrink J., Krieghoff-Henning E., Hofmann T.G.. Zyxin is a critical regulator of the apoptotic HIPK2-p53 signaling axis. Cancer Res. 2011; 71:2350–2359. [DOI] [PubMed] [Google Scholar]
- 29. Rinaldo C., Prodosmo A., Mancini F., Iacovelli S., Sacchi A., Moretti F., Soddu S.. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol. Cell. 2007; 25:739–750. [DOI] [PubMed] [Google Scholar]
- 30. Wei G., Ku S., Ma G.K., Saito S., Tang A.A., Zhang J., Mao J.H., Appella E., Balmain A., Huang E.J.. HIPK2 represses beta-catenin-mediated transcription, epidermal stem cell expansion, and skin tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:13040–13045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moehlenbrink J., Bitomsky N., Hofmann T.G.. Hypoxia suppresses chemotherapeutic drug-induced p53 Serine 46 phosphorylation by triggering HIPK2 degradation. Cancer Lett. 2010; 292:119–124. [DOI] [PubMed] [Google Scholar]
- 32. Hofmann T.G., Glas C., Bitomsky N.. HIPK2: A tumour suppressor that controls DNA damage-induced cell fate and cytokinesis. Bioessays. 2013; 35:55–64. [DOI] [PubMed] [Google Scholar]
- 33. Polonio-Vallon T., Kirkpatrick J., Krijgsveld J., Hofmann T.G.. Src kinase modulates the apoptotic p53 pathway by altering HIPK2 localization. Cell Cycle. 2014; 13:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sung K.S., Kim S.J., Cho S.W., Park Y.J., Tae K., Choi C.Y.. Functional impairment of the HIPK2 small ubiquitin-like modifier (SUMO)-interacting motif in acute myeloid leukemia. Am. J. Cancer Res. 2019; 9:94–107. [PMC free article] [PubMed] [Google Scholar]
- 35. Yang W., Mansour S.L.. Expression and genetic analysis of prtb, a gene that encodes a highly conserved proline-rich protein expressed in the brain. Dev. Dyn. 1999; 215:108–116. [DOI] [PubMed] [Google Scholar]
- 36. Tsui S., Dai T., Roettger S., Schempp W., Salido E.C., Yen P.H.. Identification of two novel proteins that interact with germ-cell-specific RNA-binding proteins DAZ and DAZL1. Genomics. 2000; 65:266–273. [DOI] [PubMed] [Google Scholar]
- 37. Frew I.J., Dickins R.A., Cuddihy A.R., Del Rosario M., Reinhard C., O’Connell M.J., Bowtell D.D. Normal p53 function in primary cells deficient for siah genes. Mol. Cell. Biol. 2002; 22:8155–8164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hofmann T.G., Stollberg N., Schmitz M.L., Will H.. HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells. Cancer Res. 2003; 63:8271–8277. [PubMed] [Google Scholar]
- 39. Hofmann T.G., Jaffray E., Stollberg N., Hay R.T., Will H.. Regulation of homeodomain-interacting protein kinase 2 (HIPK2) effector function through dynamic small ubiquitin-related modifier-1 (SUMO-1) modification. J. Biol. Chem. 2005; 280:29224–29232. [DOI] [PubMed] [Google Scholar]
- 40. Kim D., Pertea G., Trapnell C., Pimentel H., Kelley R., Salzberg S.L.. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013; 14:R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Love M.I., Huber W., Anders S.. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014; 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L.. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotech. 2013; 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mendez J., Stillman B.. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol. Cell Biol. 2000; 20:8602–8612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heigwer F., Kerr G., Boutros M.. E-CRISP: fast CRISPR target site identification. Nat. Methods. 2014; 11:122–123. [DOI] [PubMed] [Google Scholar]
- 45. Wang T., Wei J.J., Sabatini D.M., Lander E.S.. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014; 343:80–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lukas J., Mazna P., Valenta T., Doubravska L., Pospichalova V., Vojtechova M., Fafilek B., Ivanek R., Plachy J., Novak J.et al.. Dazap2 modulates transcription driven by the Wnt effector TCF-4. Nucleic Acids Res. 2009; 37:3007–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matsunami M., Yoshioka T., Minoura T., Okano Y., Muto Y.. Evolutionary features and intracellular behavior of the PRTB protein. Biochem. Genetics. 2011; 49:458–473. [DOI] [PubMed] [Google Scholar]
- 48. Popova A., Kzhyshkowska J., Nurgazieva D., Goerdt S., Gratchev A.. Smurf2 regulates IL17RB by proteasomal degradation of its novel binding partner DAZAP2. Immunobiology. 2012; 217:321–328. [DOI] [PubMed] [Google Scholar]
- 49. Sombroek D., Hofmann T.G.. How cells switch HIPK2 on and off. Cell Death Differ. 2009; 16:187–194. [DOI] [PubMed] [Google Scholar]
- 50. Gresko E., Roscic A., Ritterhoff S., Vichalkovski A., Del Sal G., Schmitz M.L.. Autoregulatory control of the p53 response by caspase-mediated processing of HIPK2. EMBO J. 2006; 25:1883–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhang Q., Yoshimatsu Y., Hildebrand J., Frisch S.M., Goodman R.H.. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell. 2003; 115:177–186. [DOI] [PubMed] [Google Scholar]
- 52. Fischer M. Census and evaluation of p53 target genes. Oncogene. 2017; 36:3943–3956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kim J.E., Ryu I., Kim W.J., Song O.K., Ryu J., Kwon M.Y., Kim J.H., Jang S.K.. Proline-rich transcript in brain protein induces stress granule formation. Mol. Cell Biol. 2008; 28:803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zepp J.A., Wu L., Qian W., Ouyang W., Aronica M., Erzurum S., Li X.. TRAF4-SMURF2-mediated DAZAP2 degradation is critical for IL-25 signaling and allergic airway inflammation. J. Immunol. 2015; 194:2826–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cohen-Barak O., Yi Z., Hagiwara N., Monzen K., Komuro I., Brilliant M.H.. Sox6 regulation of cardiac myocyte development. Nucleic. Acids. Res. 2003; 31:5941–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yang L., Li X., Qin X., Wang Q., Zhou K., Li H., Zhang X., Wang Q., Li W.. Deleted in azoospermia-associated protein 2 regulates innate immunity by stimulating Hippo signaling in crab. J. Biol. Chem. 2019; 294:14704–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Winkel A., Stricker S., Tylzanowski P., Seiffart V., Mundlos S., Gross G., Hoffmann A.. Wnt-ligand-dependent interaction of TAK1 (TGF-beta-activated kinase-1) with the receptor tyrosine kinase Ror2 modulates canonical Wnt-signalling. Cell. Signal. 2008; 20:2134–2144. [DOI] [PubMed] [Google Scholar]
- 58. D’Orazi G., Rinaldo C., Soddu S.. Updates on HIPK2: a resourceful oncosuppressor for clearing cancer. J. Exp. Clin. Cancer Res. 2012; 31:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saul V.V., Schmitz M.L.. Posttranslational modifications regulate HIPK2, a driver of proliferative diseases. J. Mol. Med. 2013; 91:1051–1058. [DOI] [PubMed] [Google Scholar]
- 60. Torrente L., Sanchez C., Moreno R., Chowdhry S., Cabello P., Isono K., Koseki H., Honda T., Hayes J.D., Dinkova-Kostova A.T.et al.. Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene. 2017; 36:6204–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Nagel S., MacLeod R.A.F., Pommerenke C., Meyer C., Kaufmann M., Drexler H.G.. NKL homeobox gene NKX2-2 is aberrantly expressed in Hodgkin lymphoma. Oncotarget. 2018; 9:37480–37496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-Seq data are available in the GEO database (GSE154146, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154146).