


Abstract
We recently found that toxic PS-ASOs can cause P54nrb and PSF nucleolar mislocalization in an RNase H1-dependent manner. To better understand the underlying mechanisms of these observations, here we utilize different biochemical approaches to demonstrate that PS-ASO binding can alter the conformations of the bound proteins, as illustrated using recombinant RNase H1, P54nrb, PSF proteins and various isolated domains. While, in general, binding of PS-ASOs or ASO/RNA duplexes stabilizes the conformations of these proteins, PS-ASO binding may also cause the unfolding of RNase H1, including both the hybrid binding domain and the catalytic domain. The extent of conformational change correlates with the binding affinity of PS-ASOs to the proteins. Consequently, PS-ASO binding to RNase H1 induces the interaction of RNase H1 with P54nrb or PSF in a 2′-modification and sequence dependent manner, and toxic PS-ASOs tend to induce more interactions than non-toxic PS-ASOs. PS-ASO binding also enhances the interaction between P54nrb and PSF. However, the interaction between RNase H1 and P32 protein can be disrupted upon binding of PS-ASOs. Together, these results suggest that stronger binding of PS-ASOs can cause greater conformational changes of the bound proteins, subsequently affecting protein–protein interactions. These observations thus provide deeper understanding of the molecular basis of PS-ASO-induced protein mislocalization or degradation observed in cells and advance our understanding of why some PS-ASOs are cytotoxic.
INTRODUCTION
Antisense oligonucleotides (ASOs) as therapeutic agents have been explored for more than three decades and have now been successfully providing solutions for multiple previously untreatable diseases (1–3). A commonly used antisense mechanism utilizes the endoribonuclease RNase H1. RNase H1 can cleave the RNA strand in the RNA–DNA heteroduplex formed between target RNA and ASO, leading to a specific degradation of the target RNAs (4). To improve the pharmacological properties of ASOs, different chemical modifications of the backbone and 2′-ribose have been developed (5). The phosphorothioate (PS) backbone modification, with a sulfur atom replacing a non-bridging oxygen, increases the stability, distribution, and cell uptake of ASOs, as compared with phosphodiester (PO) ASOs. To further improve ASO drug potency and safety, PS-ASOs are designed as gapmers, with 8–10 deoxynucleotides in the central region (gap) to support RNase H1 activity, and 3–5 nucleotides (nts) at both ends (wings) of an ASO to enhance affinity for cognate sequences in target RNAs. Commonly used 2′ modifications include 2′-O-methy (OMe), 2′-O-methoxyethyl (MOE), constrained ethyl (cEt), locked nucleic acid (LNA) or 2′-fluoro (F) (6).
These various chemical modifications not only improve PS-ASO interactions with RNA, but also affect PS-ASO binding to proteins. Recent work showed that PS-ASO–protein interactions play essential roles in PS-ASO performance, affecting PS-ASO’s stability, tissue targeting, internalization, endosomal trafficking, subcellular localization, and activity (7,8). For example, SSB/Lupus La protein can enhance PS-ASO activity and cause PS-ASO nuclear retention; however, paraspeckle proteins, P54nrb and PSF, can inhibit ASO activity by competition with RNase H1 for binding to ASO/RNA heteroduplex (9). On the other hand, the interaction of PS-ASOs with proteins can affect the fate of proteins and alter their subcellular localization and stability, affecting the toxicity of PS-ASOs (7–8,10). Although off-target RNA cleavage may cause toxicity for certain PS-ASOs if the off-target genes are essential (11), we recently demonstrated that protein binding contributes to the toxicity of most toxic PS-ASOs (10). We found that toxic PS-ASOs tend to bind more proteins more tightly than non-toxic PS-ASOs, causing protein degradation or mislocalization, leading to apoptotic cell death (12,13). For example, toxic PS-ASOs cause paraspeckle protein P54nrb and PSF mislocalization to the nucleolus, protein degradation (especially by 2′ F-PS-ASO), and nucleolar stress (12,14). Importantly, the introduction of 2′-OMe at position 2 of the gap region (Gap2 OMe) reduces protein binding and dramatically mitigated toxicity (12). Such effects were observed for more than 90% toxic PS-ASOs tested, in different tissues or species, suggesting a common toxic mechanism mediated by PS-ASO–protein interactions. The mislocalization and degradation of these paraspeckle proteins induced by toxic PS-ASOs are RNase H1-dependent, as reduction of RNase H1 prevented paraspeckle protein mislocalization and degradation, and alleviated toxicity (12).
Human RNase H1 consists of three domains, the N-terminal domain (H1-NTD) or hybrid binding domain (HBD), Spacer domain and Catalytic domain (15). RNase H1 can localize to mitochondria, cytosol, and the nucleus, depending on the presence or absence of a mitochondria localization signal (MLS) located at the first 26 residues (16,17). The MLS deleted RNase H1 is generated by translation from a downstream start codon that is more efficiently utilized than the first start codon due to the presence of an upstream open reading frame (17). Till now, there is no structure solved for RNase H1 full-length protein; however, the structures of the catalytic domain and HBD have been solved (18). The presence of the HBD increases the affinity of RNase H1 for heteroduplex substrate and enhances cleavage specificity. Both NTD and catalytic domains have relatively high affinity to the RNA/DNA duplex (19). In this article, we define the spacer domain and catalytic domain as the entire C-terminal domain (H1-CTD).
The H1-NTD (27–73AA) has at least 25-fold weaker binding to double-stranded (ds)RNA (Kd = 4.9 μM) and dsDNA (Kd = 23 μM) than binding to RNA/DNA heteroduplex (Kd = ∼0.2 μM) (20). H1-NTD binds tightly to single strand (ss) PS-ASO, with the Kds, on average, approximately 12 and 15 nM for 2′ F-PS-ASO and 2′ MOE-PS-ASO, respectively (21). The catalytic domain also binds RNA/DNA heteroduplexes with a much higher affinity than dsRNA and dsDNA, with Kds at 0.6–0.7, 3.1 and 29 μM, respectively (14,22,23). Compared with the full-length protein, the catalytic domain itself has a decreased binding with RNA/PS-ASO substrate and therefore has a greater kcat for RNA/DNA duplexes (14,15).
RNase H1 can interact with different protein partners (14,24,25). We have demonstrated that P32 interacts with the NTD of RNase H1 and enhances the turnover rate by reducing the binding with the heteroduplex substrate (24). More recently, we found that toxic PS-ASOs can induce a unique interaction of RNase H1 with paraspeckle proteins P54nrb and PSF and cause nucleolar mislocalization of the PS-ASO and paraspeckle protein complex, as demonstrated using immunofluorescence staining and live-cell imaging assays by NanoBRET and NanoBiT (12,14). The spacer domain of RNase H1 was found to contribute to the interaction with P54nrb mediated by toxic PS-ASO in cells. Given the pivotal roles of RNase H1 and these paraspeckle proteins in PS-ASO activity and toxicity, it is important to understand the detailed molecular bases governing these toxic-PS-ASO-induced interactions.
In this study, we extended our research using different biophysical and biochemical approaches to understand in more detail whether and how PS-ASO-binding, different chemistry, and sequences affect protein conformation, protein thermostability and protein–protein interactions.
MATERIALS AND METHODS
Plasmids, reagents, antibodies and ASOs
Plasmids expressing GST-tagged RNase H1 full-length and different domains, P54nrb, and PSF were synthesized from GenScript and constructed on pGEX-6P1 vector. MBP-tagged RNase H1 was constructed on pMal-C5x vector. Detailed construction is described in Supplementary Table S1. PreScission protease (GE27-0843-01) was purchased from Sigma. Antibodies against P54nrb (sc-376865), PSF (sc-374502) and P32 (sc-48795) were purchased from Santa Cruz Biotechnologies. RNase H1 antibody (15606-1-AP) was purchased from ThermoFisher. Ku70 antibody (ab 83501) and Anti-GST antibody (ab19256) were from Abcam. Secondary anti-mouse or anti-rabbit antibodies were purchased from Bio-Rad. ASOs used in this study are listed in Supplementary Table S2.
Protein expression and purification
Plasmids were transformed into BL21 DE3 One-shot competent cells (Thermo Fisher, C606003). Overnight culture for protein expression was inoculated into Terrific medium (Thermo Fisher, A1374301) containing carbenicillin (100 μg/ml) and chloramphenicol (33 μg/ml), grown until the OD600 reached 0.6–0.8, and then induced with 0.5 mM IPTG overnight at room temperature. Cells were harvested and lysed with B-per lysate buffer (Thermo Fisher, 89822). The expressed proteins were enriched on glutathione resin 4B (Cytiva, 17075605) and washed seven times (30 ml/each) with buffer L [50 mM Tris (pH 7.5), 250 mM NaCl, 5% glycerol, and 1 mM DTT]. PreScission protease (20 units/ml resin) was used to release P54nrb, PSF, or RNase H1 domains from the resin. GST-tagged fusion proteins were eluted by 20 mM Glutathione in Buffer L and dialyzed into buffer L. MBP-RNase H1 was purified using amylose resin (New England Biolab, E8021L) and eluted by 10 mM maltose in Buffer L. All purified proteins were analyzed on Zenix C 300 and concentrated (concentrator, Fisher Scientific, 88528) and frozen at −80°C.
Fluorescence polarization (FP)
Different RNase H1 proteins were three times diluted serially with cold PBS buffer from 9 μM to 56 pM in 50 μl system, then incubated with 4 nM Alex647-ss-PS-ASO or PS-ASO/RNA duplex at RT for 30 min. The Fluorescence polarization results were read on Tecan plate reader (infinite M1000Pro) at λex = 635 nm, λem = 670 nm. The binding Kds were analyzed in Prism.
Electrophoretic mobility shift assay (EMSA)
Alex488-labeled PS-ASO was mixed with RNA (at 1:1.2 molar ratio), heated at 94°C for 2 min, then naturally cool down to RT to generate PS-ASO/RNA duplex. The duplex was used immediately or stored at −20°C. The single-strand PS-ASO or PS-ASO/RNA duplex was incubated with various amounts of RNase H1 domains on ice for 30 min in binding buffer (10 mM HEPES (pH 7.5), 0.5 mM EDTA, 100 mM NaCl, 5% glycerol, 1 mM TCEP). The protein–ASO complexes were resolved on 6% native polyacrylamide gels using 1× TBE buffer. Gels were scanned on STORM 860 WO Phosphor Screen and analyzed by ImageQantTL software.
RNase H1 cleavage assay
PS-ASO (558807) was annealed with complementary FITC labeled RNA in 1× PBS buffer. 0.3 μM duplex was incubated with different amount of RNase H1 protein in cleavage buffer (20 mM Tris–HCl (pH7.5), 50 mM NaCl, 10 mM MgCl2, 10 mM DTT), incubate at 37°C for 10 min. The cleavage reaction was stopped by adding loading dye. The reaction product was separated on 20% urea gel in 1× TBE buffer. Gels were scanned on STORM 860 WO Phosphor Screen and analyzed by ImageQantTL software.
Co-immunoprecipitation (IP)
GST-tagged proteins (2 μg) were bound on the glutathione resin and incubated with PS-ASO at 8 μM. After 2 h rotation, resin was washed with Buffer L for five times. Next, 4 μg P54nrb, PSF or 4 μg P54nrb/PSF complex in 50 μl Buffer L was added to the resin. After incubation for another 2 h, the complex was washed using Buffer L for five times. The resin was transferred to clean tubes, washed another five times. Then SDS loading buffer was added, and proteins were analyzed by western analyses.
For co-immunoprecipitation of RNase H1 and P32, cell lysates from HEK293 cells expressing Flag-tagged RNase H1 or Flag-tagged RNase H2 (24) were incubated without or with 2 μM ss-PS-ASO (116847) or PS-ASO/RNA heteroduplex formed with the same ASO and a 2′-OMe modified complementary RNA. Next, immunoprecipitation was performed using anti-Flag antibody. After seven times washing with wash buffer W100 [50 mM Tris–HCl (pH 7.5), 100 mM KCl, 5 mM ethylenediaminetetraacetic acid (EDTA), 0.1% NP-40, 0.05% sodium dodecyl sulphate (SDS)], isolated proteins were analyzed by western blot. For competition assay, immunoprecipitation was performed with anti-Flag beads using cell lysate prepared from HEK293 cells expressing Flag-tagged RNase H1. After seven times wash, beads bound proteins were eluted at room temperature for 20 min by competition using short, length matched PS-ASO/RNA duplex at different concentrations, and eluted proteins and proteins remained on beads were analyzed by western analysis.
Thermostability shift assay
The Protein Thermal Shift™ Starter Kit was purchased from Invitrogen (4462263), which includes the Dye Kit, similar to SYPRO orange dye, λex = 470 nm, λem = 570 nm, used ROX as reporter, and applied to monitor the temperature ramping induced denature/unfolding of different proteins. Upon temperature increases, protein unfolds and exposes its hydrophobic residues or surface; hence the fluorescent dye can bind the hydrophobic area and become unquenched. The midpoint of the melting curve was measured as melting temperature (Tm). The reaction was set up in 96-well PCR plates with 1–2 μg proteins with or without equal molar ss-PS-ASO or PS-ASO/RNA duplexes, and 2× dye in assay buffer (50 mM Tris–HCl pH 7.5, 100 mM NaCl, 0.5 mM EDTA and 1 mM DTT). Differential scanning fluorimetry was performed in a StepOnePlus real-time PCR Thermal Cycler and analyzed using Protein Thermal Shift software 1.3 (Invitrogen). Scans were recorded using the fluorescence Thermal Shift™ dye (ROX as reporter) between 25 and 99°C in 0.05°C increments with a Second equilibration time. Tm was determined from the first derivative of a plot of fluorescence intensity peak versus temperature (26). The standard error of Tm was calculated from four independent measurements using Protein Thermal Shift Software 1.3.
Limited proteolysis
Five μg proteins were first incubated without PS-ASO or with equal molar PS-ASO, or PS-ASO/RNA duplex on ice for 30 min. Chymotrypsin (VWR, PI90056) was three times diluted serially in buffer L, at 0.5 mg/ml as the start point. 2.5 μl diluted enzyme was mixed with 5 μg proteins complexed with or without PS-ASO or PS-ASO/RNA duplex in 12.5 μl system, incubated at RT for 20 min. The reaction was stopped by adding stop buffer (5.2 mM EDTA, 5.2 mM PMSF in 4× SDS buffer), boiled for 5 min. Proteins were separated by SDS-PAGE, and visualized using coomassie staining.
Affinity selection
Affinity selection using biotinylated PS-ASO 386652 was performed as described previously (27). Proteins were eluted using PS-ASOs 558807 or 549139, or PS-ASO/RNA duplexes formed with these ASOs and corresponding complementary RNAs modified with 2′-OMe. Eluted proteins were separated on 4 –12% SDS-PAGE gel, and proteins were detected by immunoblotting (28).
PS-ASO protein binding affinity measurement using nanoBRET assay
NanoBRET assay was performed to determine the binding affinities of PS-ASOs or ASO/RNA duplex to proteins, as described previously (21). Amino-terminal NLuc fusions to P54nrb and RNase H1 were created using the vector pFN31K Nluc CMV-neo (Promega) (28). Fusion proteins were expressed by transfecting the plasmids into 6 × 105 HEK 293 cells using effectene transfection reagent (Qiagen). Following a 24 h incubation, cells were removed from the plate by trypsinization, washed with PBS, then resuspended in 250 μl Pierce IP Lysis Buffer (Thermo Scientific). Lysates were incubated 30 min at 4°C while rotating, then debris pelleted by centrifugation at 15 000 rpm for 5 min. The fusion proteins were purified by adding 20 μl HisPur Ni-NTA Magnetic Beads (Thermo Scientific) and 10 mM imidazole then incubating for 2 h at 4°C. Beads were washed four times with wash buffer (1 × PBS, 10 mM imidazole, and 0.01% Tween-20). Fusion proteins were eluted in 100 μl 1 × PBS + 200 mM imidazole, followed by dilution with 200 μl IP buffer. BRET assays were performed in white 96-well plates. Alexa-linked ASOs at the indicated concentrations were incubated at room temperature for 15 min in 1 × binding buffer with 106 RLU/well of immunoprecipitated purified NLuc fusion protein or whole cell lysate. Following the incubation, NanoGlo substrate (Promega) was added at 0.1 μl/well. Readings were performed for 0.3 s using a Glomax Discover system using 450 nm/8 nm bandpass for the donor filter, and 600 nm long pass for the acceptor filter. BRET was calculated as the ratio of the emission at 600/450 nm (fluorescent excitation-emission/RLU). For competitive binding assays, the 5–10–5 Alexa-linked cEt PS-ASO was added at 10 nM and the unconjugated competing ASO added at the indicated concentrations in 50 μl water. 106 RLU/well of purified fusion protein or whole cell lysate was then added in 50 μl 2× binding buffer for a final volume of 100 μl. After incubation at room temperature for 15 min, substrate addition and BRET readings were carried out as detailed above. P54nrb 1× binding buffer: 100 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM EDTA, 0.1% NP40. RNase H1 1× binding buffer: 50 mM NaCl, 20 mM Tris–HCl pH 7.5, 1 mM MgCl2, 1 mM TCEP.
RESULTS
Purified RNase H1 protein interacts with both single-stranded ASO and ASO/RNA duplex with different affinities affected by 2′ modifications
The RNase H1 proteins, either full-length, H1-NTD or H1-CTD, as indicated in Supplementary Figure S1A, were expressed and purified from bacteria. The full-length RNase H1 protein (1–286 AA) is tagged with GST or MBP. The purified proteins were analyzed by coomassie blue staining, which showed >90% pure (Supplementary Figure S1B). On the other hand, the purified H1-NTD (27–73 AA) and H1-CTD (74–286 AA) were obtained through GST-tag, which was removed by PreScission cleavage, generating untagged H1-NTD and H1-CTD that migrate at the expected sizes in PAGE and with >95% purity (Supplementary Figure S1B). The full length and truncated domains of RNase H1 were confirmed by Western analysis or by Mass spectrometry (data not shown). The purified full-length MBP- or GST-tagged RNase H1 or untagged H1-CTD is active in cleaving the RNA substrate in a PS-ASO/RNA duplex, with a cleavage pattern similar to that generated using a full length His-tagged RNase H1 proteins (Supplementary Figure S1C).
To characterize the binding properties of purified RNase H1 to PS-ASOs with different chemistries, fluorescent polarization (FP) assays were performed using fluorophore-labeled 5–10–5 gapmer PS-ASOs containing 2′-MOE, cEt or F modifications (Supplementary Figure S2A, left panel). The results showed that, consistent with previous observations determined via NanoBRET assays with unpurified RNase H1 protein (14,21), the 2′-F PS-ASO bound more tightly to purified RNase H1 protein, either full-length or H1-CTD, H1-NTD, than the 2′-MOE PS-ASO. Similarly, FP was also performed for RNase H1 binding to a PS-ASO/RNA heteroduplex formed between a 2′ OMe-modified complementary oligoribonucleotide and the same PS-ASOs with different 2′ modifications (Supplementary Figure S2A, right panel). As expected, full-length RNase H1 can bind the heteroduplexes, and H1-NTD binds tighter than the H1-CTD, also consistent with previous observations (19,20). However, the effects of 2′ modifications of PS-ASOs on protein binding are much weaker for PS-ASO/RNA heteroduplexes than for ss-PS-ASO, which is not unexpected, as the difference of 2′-modifications can be significantly masked by the formation of PS-ASO/RNA duplex. In addition, full-length RNase H1 binds to ss-PS-ASOs and duplexes with similar affinity, and H1-CTD binds more tightly to ss-PS-ASO than to PS-ASO/RNA duplex. On the other hand, H1-NTD binds PS-ASO/RNA duplex slightly tighter than ss-PS-ASO, consistent with previous observations from NanoBRET assay (14).
To further characterize RNase H1 binding to PS-ASOs and PS-ASO/RNA heteroduplexes, EMSA assays were performed with the purified RNase H1 proteins complexed with ss 5–10–5 PS-MOE ASO or with a heteroduplex formed with the same PS-ASO and an uncleavable 2′-OMe modified complementary RNA. Consistent with the observations from the FP study that the H1-NTD binds tighter to a PS-ASO/RNA duplex than to ss-PS-ASO, the heteroduplex also caused a greater shift than ss-PS-ASO in EMSA (Supplementary Figure S2B, left panel). On the other hand, ss-PS-ASO caused a greater shift for both the H1-CTD and full-length RNase H1 protein than the PS-ASO/RNA duplex, as seen from the reduced levels of unbound free PS-ASOs or duplexes (Supplementary Figure S2B, middle and right panels), also consistent with FP results.
RNase H1 protein can bind ss-PS-ASO and PS-ASO/RNA duplexes with different binding modes
To determine the impact of binding of PS-ASOs and PS-ASO/RNA duplexes to RNase H1 on the protein conformation, thermoshift assays were performed. Protein thermoshift (PTS) is commonly used to detect protein conformational changes (29). Protein Thermal Shift™ Dye was used that can bind to the hydrophobic regions of the protein during the temperature ramping, which can monitor the protein conformation/hydrophobic surface changes from folded to unfolded stages. The midpoint of the protein unfolding transition is defined as the melting temperature (Tm). Tms correlate with the stability of the structure. Thus, increased Tm upon ligand binding suggests a condition that favors the stabilization of the conformation of the protein, whereas a reduced Tm suggests a condition that favors the destabilization/unfolding of the protein conformation (30).
The interactions of H1-NTD protein with 5–10–5 gapmer PS-ASOs containing 2′-MOE, cEt or F modifications were tested using the PTS assay. The H1-NTD protein demonstrated clear Tm peaks in PTS (Figure 1A), with a Tm at approximately 60.7°C for the protein alone. However, binding of ss-PS-ASOs reduced the Tm of H1-NTD to 51.06, 45.69 and 45.03°C by the 2′-MOE, cEt and F-modified PS-ASOs, respectively (Figure 1B, C). Reduced Tm in the PST assay upon ligand binding has been observed previously. It suggests that the ligand prefers to bind the unfolded form of the protein in a disruptive way that destabilizes the protein structure (29). Interestingly, 2′-modification of the PS-ASOs influences the Tm change. The PS-F ASO caused a greater Tm reduction than the PS-MOE ASO (Figure 1A–C), suggesting that the PS-F ASO has a more disruptive effect on RNase H1 conformation than the PS-MOE ASO. We note that the PTS results are highly reproducible, as seen from the low standard deviations and comparable Tm values in different experiments (Figure 1C, H1-NTD data in left and right panels). In addition, the binding of 3–10–3 ss-PS-cEt ASOs with different sequences also reduced the Tm of H1-NTD (Supplementary Figure S2C). As a control, no significant Tm change was detected for H1-NTD when incubated with phosphodiester (PO) backbone ASOs (Supplementary Figure S2C), and PS-ASO itself did not show decent thermoshift signal (Figure 1A).
Figure 1.

PS-ASO binding can alter the stability of RNase H1 protein. (A) Thermostability assay was employed to determine the melting curve of RNase H1-NTD incubated without or with 5–10–5 gapmer PS-ASOs of the same sequence but modified with 2′-MOE (ASO 116847), 2′-cEt (ASO 582801) or 2′-Fluoro (ASO 404130). The top panel showed the real curves for the different groups; the bottom panel showed the derivatives of the melting curves. The vertical dashed lines indicate the Tms. (B) The protein melting temperature (Tm) of H1-NTD is determined using the protein thermostability assay (PTS). There are four repeats (red dots) for each experimental group; the green line represents the median; diamond shows the 95% lower and upper confidence limit. (C) Melting temperature (Tm, in °C) of RNase H1 domains complexed with ss-PS-ASOs or ASO/RNA duplexes, as determined by thermostability shift assay. The average values and standard deviations from four duplicates are shown.
The PTS assay was also performed for H1-NTD binding to heteroduplexes formed with complementary RNA and the PS-ASOs as described above. In contrast to the effects of binding to ss-PS-ASOs, H1-NTD binding to the heteroduplexes increased the Tm by 6–8°C (Figure 1C, right panel). Increased Tm was also observed with a PO-ASO/RNA heteroduplex (Supplementary Figure S2C), which has been shown to bind RNase H1 (20). These results suggest that binding of the ASO/RNA duplex tends to increase Tm of H1-NTD, likely by binding to H1-NTD in a more native conformation that stabilizes the structure.
Similar experiments were performed for the H1-CTD (Figure 1C, right panel). Although this domain did not show substantial Tm change upon binding to ss-PS-MOE ASO or to different PS-ASO/RNA duplexes, significant Tm reduction (>2°C) was observed upon binding to PS-cEt or PS-F ASO. These results suggest that ss-PS-ASOs tend to disrupt the conformation of H1-CTD, despite that the extent of the conformational change appears to be modest relative to the entire H1-CTD, which is >4-fold larger than H1-NTD. The purified full-length MBP-H1 and GST-H1 tagged proteins failed to response to PS-ASO binding in PTS assay (data not shown), most likely due to the stable tag and the presence of mitochondria targeting peptide which might stabilize the conformation of RNase H1, but mitochondria targeting peptide deleted RNase H1 protein responses well (see below). Despite this, the results from H1-NTD and H1-CTD clearly showed different binding modes between ss-PS-ASO and PS-ASO/RNA duplex to these domains.
The mitochondria localization signal peptide of RNase H1 may affect the folding and performance of the protein
It has been shown that endogenous RNase H1 can be translated from two start codons regulated by an upstream open reading frame, generating a full-length RNase H1 that contains the MLS peptide (1-26) and is transported to mitochondria, and an MLS-deleted version of RNase H1 (H1-delMit) that is localized to the nucleus (31). As we found previously that the sizes of RNase H1 in nuclear, cytosol, and mitochondria fractions are essentially the same (16), it is possible that the majority of cellular RNase H1 protein lacks the MLS peptide, thus showing different performance than the purified full-length protein.
To demonstrate this possibility, the H1-delMit was purified (Figure 2A). As expected, the H1-delMit has a similar size as the endogenous RNase H1 protein in different human cells and is obviously smaller than a his-tagged full-length RNase H1 (Figure 2B). This result confirmed that the H1-delMit is the most dominant form in mammalian cells, and the results of cleavage assay showed that the purified H1-delMit is active (Figure 2C). Given that the MLS deleted form of RNase H1 present in the cytosol and the nucleus is responsible for target RNA degradation induced by PS-ASOs, we characterized the conformational change of this protein upon binding to PS-ASOs.
Figure 2.

The MLS-deleted RNase H1 exhibits sensitivity to PS-ASO binding. (A) Coomassie blue staining of purified H1-delMit protein. (B) 30 μg whole cell lysate from different human cell lines, and 100 ng purified his-RNase H1 full-length and MLS deleted RNase H1 (H1-delMit), were separated on 4–12% SDS-PAGE using MOPs buffer. Western blot showed the endogenous RNase H1 in mammalian cells had similar size to MLS deleted RNase H1. (C) RNase H1 cleavage of PS-ASO/RNA duplex (ASO558807/FITC-GC-558807). The duplex was incubated without or with H1-delMit, and cleavage products were separated on a 20% PAGE gel, visualized with STORM 860 WO Phosphor Screen. (D) EMSA assay of H1-delMit protein complexed with 0.4 μM ss-PS-ASO or PS-ASO/RNA duplexes. The concentrations of the RNase H1 protein are shown above lanes. (E) Melting temperature (Tm, in °C) of H1-delMit complexed with ss-PS-ASOs or PS-ASO/RNA duplexes, as determined by thermostability shift assay. The average values and standard deviations from four duplicates are shown.
EMSA results showed that both ss-PS-ASOs and PS-ASO/RNA duplexes can bind to H1-delMit in two different complexes (Figure 2D). For ss-PS-ASO, the two complexes were formed at similar H1-delMit protein levels (protein:duplex ≥ 1:1). However, for PS-ASO/RNA duplexes, the smaller complex formed first at lower H1-delMit levels (protein:duplex = 1:1), and the larger complex formed at higher H1-delMit levels (protein:duplex > 2:1), accompanied with decreased level of the smaller complex. Compared to MBP-H1, which is difficult to migrate into the PAGE wells (Supplementary Figure S2B), the H1-delMit has much better folding and prevents oligomer formation, and can migrate into the wells of the native gel.
The H1-delMit protein also showed significant changes in the PTS assay. Similar to H1-NTD and H1-CTD, binding of ss-PS-ASOs to H1-delMit caused disruptive conformational changes, as evidenced by Tm reduction (Figure 2E). Consistently, 2′ modification of PS-ASOs also affects the conformational change, with PS-MOE ASO decreased the Tm by 4°C, and PS-cEt and PS-F ASOs decreased Tm by 5.5 and 6°C, respectively. PS-ASO/RNA duplexes didn’t significantly affect Tm of H1-delMit, consistent with H1-CTD (Figure 1C). These results suggest that the Tm changes of H1-NTD and H1-CTD domains can be reflected in the H1-DelMit protein, and that the failure of the tagged full length H1 protein to show Tm changes upon PS-ASO binding is likely due to the presence of MLS sequence and the tag peptide. Together, these results indicate that ss-PS-ASO interactions with H1-delMit, the endogenous form of RNase H1, caused disruptive conformational changes, but binding of the duplexes did not change the Tm of H1-delMit, likely due to the better fit with a site(s) that has evolved to bind RNA/DNA duplexes.
The binding affinity of PS-ASOs with H1-NTD correlates with the structural stability of the protein
As described above, the conformational stability of H1-NTD is decreased to different extents by binding of 5–10–5 ss-PS-ASOs containing different 2′ modifications, with the greatest reduction by the PS-F ASO and the least reduction by the PS-MOE ASO. Previously we have shown that 2′-modifications significantly affect the binding affinity of PS-ASOs to most proteins tested. Typically, PS-F ASO binds proteins more tightly than PS-cEt ASO, and PS-MOE ASO binds the least (21,32–33). These observations suggest that tighter binding of PS-ASO may cause stronger disruption of the conformation of H1-NTD.
To determine the impact of binding affinity of PS-ASOs to proteins on protein conformational change, we evaluated PS-ASOs with different 2′ modifications or PS-ASOs with varying numbers of 2′ modified nucleotides in the wings. The PTS study was performed for H1-NTD with a set of 3–10–3 PS-ASOs containing different modifications, as H1-NTD exhibits significant Tm change that is easier to monitor. Consistent with the 5–10–5 PS-ASOs, the 3–10–3 ss-PS-ASO also reduced Tm of H1-NTD, with PS-F ASO causing the greatest decrease, followed by the PS-cEt ASO and the PS-MOE ASO (Figure 3A), suggesting that 2′-modifications of PS-ASOs with higher binding affinity to proteins can cause stronger disruption of the H1-NTD conformation. To further confirm this observation, PS-MOE and PS-cEt ASOs with different wing lengths were tested, ranging from 2–10–2 to 5–10–5. The results showed that increasing the size of PS-ASO with MOE or cEt modified wing further reduced Tm (Figure 3B, C). The binding affinities of the PS-cEt ASOs to RNase H1 was measured using NanoBRET assay (Figure 3C). As expected, increasing the wing size increased the binding affinity to RNase H1, consistent with our previous observations (27,32). These results together indicate that tighter binding of PS-ASO to H1-NTD can cause a greater reduction in Tm, likely due to stronger disruption of the protein conformation.
Figure 3.
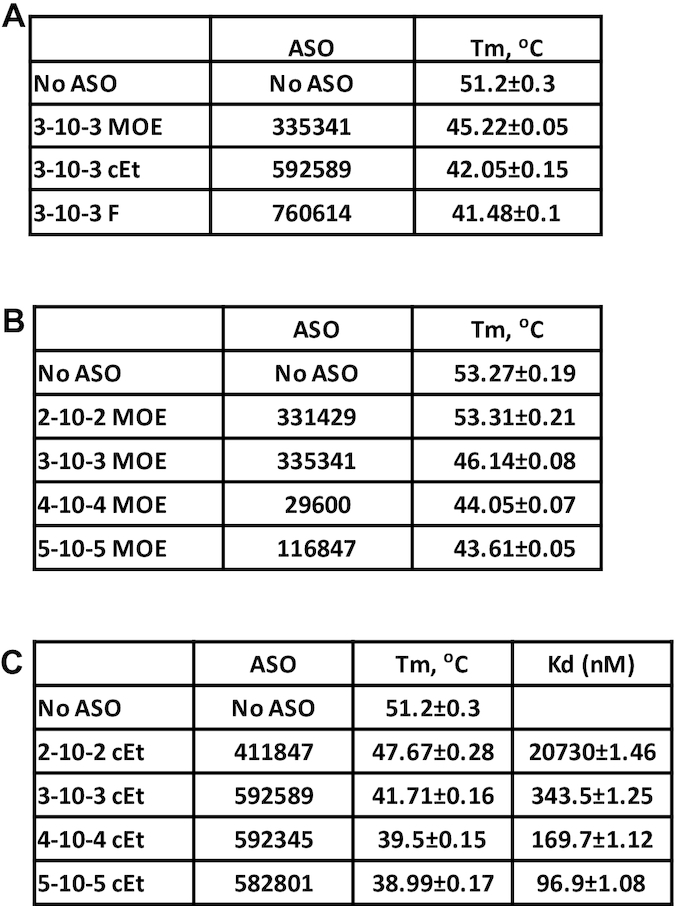
Altering the length of PS-ASOs affects the stability of RNase H1. (A) Thermostability assay was performed to determine the Tm change of H1-NTD upon binding of 3–10–3 gapmer ASOs of the same sequence but with different 2′-modifications. Buffer (Tris-EDTA: 50mM Tris, 100 mM NaCl and 0.5 mM EDTA) is different from Figure 1C, which caused different Tm value. (B, C) H1-NTD stability change upon binding to 2′-MOE (B) or 2′-cEt (C) modified PS-ASOs with different lengths, as determined using PTS assay. Protein binding affinity (Kd, nM) was determined using NanoBRET assay for RNase H1 protein and 2′-cEt modified PS-ASOs of different lengths. The binding Kd (nM) was calculated using Prism. The average values and standard deviations from four duplicates are shown.
ss-PS-ASO binding causes disruptive conformational change of RNase H1 protein
The observations of reduced Tm of H1-NTD, H1-CTD and H1-delMit upon ss-PS-ASO binding suggest that the protein is most likely unfolded. To evaluate this possibility, a limited proteolysis assay was applied to partially degrade the H1-NTD protein with chymotrypsin, in the absence or presence of PS-ASOs or PS-ASO/RNA duplexes. We reasoned that if ligand-binding altered the conformation of the protein, a different cleavage pattern or cleavage efficiency might be observed. Due to the small size (∼5 kDa) of H1-NTD, no distinct cleavage bands were observed by coomassie staining of the PAGE gel (Figure 4A). However, the presence of ss-PS-cEt or PS-F ASO caused more degradation of H1-NTD by chymotrypsin (Figure 4A, upper panel), relative to no ASO or PS-MOE ASO-containing samples, as shown by comparing the levels of the top band which is the intact protein. In addition, a protein smear representing cleaved products of H1-NTD was observed only in the presence of ss-PS-ASOs (Figure 4A, lower panel). These results indicate that binding of ss-PS-ASOs enhanced the degradation of H1-NTD by chymotrypsin, most likely by causing H1-NTD more accessible to the enzyme. However, the presence of PS-ASO/RNA duplex did not alter H1-NTD degradation, suggesting no substantial disruptive conformational change. These observations are consistent with the PTS results, which showed that ss-PS-ASO binding reduced Tm likely due to disruptive binding, whereas PS-ASO/RNA duplex binding increased Tm of H1-NTD most likely due to binding with a native conformation that further stabilizes the folding of H1-NTD.
Figure 4.

Limited proteinase digestion of RNase H1 domains. (A) Purified H1-NTD protein was incubated with ss-PS-ASOs of different 2′-modifications or with PS-ASO/RNA duplexes (Figure 1C, right panel), and subjected to digestion with 2.5 μl 0.5 mg/ml of chymotrypsin (Chym), as described in Materials and Methods. The digested proteins were separated on 10–12% SDS-PAGE in MOPs buffer (A, B), followed by coomassie blue staining. The arrows indicate increased digestion upon binding to ss-PS-ASOs. The signal intensity of the top band (marked by open arrow) was quantified and shown below the lanes. Lower panel, a stronger signal intensity. (B) Coomassie blue staining of H1-CTD protein incubated with PS-ASOs or ASO/RNA duplexes, as in panel A, followed by digestion with different concentrations of chymotrypsin. The arrows indicate enhanced digestion by ss-PS-ASOs compared with no ASO. The bands denoted by the open arrow were quantified using ImageJ and normalized to that in no ASO control sample, which were digested by the 0.17 mg/ml chymotrypsin. (C) Purified H1-delMit protein was incubated with ss-PS-ASOs of different 2′ modifications or with PS-ASO/RNA duplexes, and subjected to digestion with 0.5 mg/ml of chymotrypsin (Chym). The digested proteins were separated on 10–12% SDS-PAGE in Tris-glycine buffer, followed by coomassie blue staining. The red arrows indicate increased digestion upon binding to ss-PS-ASOs. The protected fragments by the duplex are marked with an open white arrow. The protein bands denoted by open arrows were quantified using ImageJ and normalized to that in no ASO control sample. The relative levels of the peptides are listed on the top for the band marked with a yellow arrow; at the bottom for the band marked with a white arrow.
To further confirm these observations, H1-CTD was also treated with chymotrypsin in the absence or presence of ss-PS-ASO or PS-ASO/RNA duplex. The results showed that, similar to H1-NTD, ss-PS-ASO binding substantially enhanced degradation of the H1-CTD protein, with PS-F ASO triggered greater degradation (1% protein left) than the PS-cEt (59%) and PS-MOE ASOs (66%), as compared to the level of the band marked with an open arrow in no ASO control sample (Figure 4B). This trend is also consistent with the PTS data, further indicating that ss-PS-ASO binding altered H1-CTD conformation in a disruptive manner, and that 2′-modification of PS-ASOs influenced the degree of conformational change. However, binding to PS-ASO/RNA heteroduplex did not substantially affect the degradation of the H1-CTD by chymotrypsin, and no substantial difference was observed for duplexes formed with PS-ASOs containing different 2′ modifications, consistent with what was observed with H1-NTD.
Next, chymotrypsin digestion was performed with H1-delMit protein, with or without different ss-PS-ASOs or ASO/RNA duplexes (Figure 4C). For a major degradation product (marked with a yellow arrow), binding of ss-PS-F ASO, and to a lesser extend of PS-cEt ASO, caused slightly more H1-delMit protein degradation by chymotrypsin, as compared with PS-MOE ASO, which showed similar degradation as no ASO control. However, similar pattern with stronger difference was observed for an additional protein band, as marked with a white arrow, of which the levels are 51%, 25% and 14% upon binding of ss-PS-MOE, cEt and F ASOs, respectively, relative to that in no ASO control sample. On the other hand, the binding of different PS-ASO/RNA duplexes to H1-delMit caused a similar pattern of chymotrypsin digestion, and all duplexes showed reduced cleavage of two degradation intermediates and accumulated four-fold of the peptides relative to that in control sample, suggesting that these duplexes have similar stabilization effects upon binding to H1-delMit. Together, these results suggest that ss-PS-ASO binding to H1-NTD, H1-CTD, or H1-delMit can cause disruptive conformational change, of which the extent is affected by the 2′ modifications of the PS-ASOs. Modifications with tighter protein binding can cause greater conformational changes. However, the binding of PS-ASO/RNA duplexes most likely occurs in a more native conformation of the protein. Moreover, PS-ASO/RNA duplexes binding stabilized and prevented degradation of the fragments of H1-delMit, which was not observed for H1-NTD and H1-CTD. It is possible that H1-delMit may have interactions between the two domains and form different conformations compared with the two individual domains, thus leading to more protections from chymotrypsin digestion.
Binding of both ss-PS-ASOs and PS-ASO/RNA duplexes to P54nrb stabilizes the conformation of the protein and protects from chymotrypsin digestion
To further characterize the effects of PS-ASO binding on protein conformation, P54nrb and PSF proteins were evaluated. These paraspeckle proteins are involved in multiple cellular activities such as splicing, editing, gene expression regulation, and DNA repair (34). Previously we have shown that these proteins affect the activity and toxicity of PS-ASOs (9,12). Toxic, and not non-toxic PS-ASOs, can cause nucleolar mislocalization of P54nrb and PSF upon transfection in an RNase H1-dependent manner (14). The GST-tagged full-length P54nrb and PSF were purified from bacteria, and the GST tag was removed by PreScission cleavage, resulting in >95% pure proteins (Supplementary Figure S1B). PSF has two bands after PreScission cleavage. N-terminal sequencing results showed that the smaller band starts at amino acid 114 of PSF due to enzyme cleavage.
PTS assay was performed with the purified P54nrb protein. The binding of PS-ASO/RNA duplexes formed with complementary RNA and 5–10–5 PS-ASOs containing different 2′-modifications increased Tm of P54nrb to similar degree (∼7°C) (Figure 5A). As a control, a PO-ASO/RNA duplex did not substantially increase Tm, consistent with the fact that P54nrb does not tightly bind PO-ASO/RNA duplex (Supplementary Figure S3). These results suggest that the binding of the PS-ASO-containing duplexes stabilizes the conformation of the protein, and that the 2′-modifications of the PS-ASOs in a duplex may not be well distinguished by P54nrb, similar to what was observed for H1-NTD.
Figure 5.

PS-ASO binding can affect the stability of P54nrb protein and cause conformational change. (A) Thermostability assay was performed to determine the Tm change of P54nrb upon binding of 5–10–5 gapmer ASOs of the same sequence but with different 2′-modifications or with ASO/RNA duplexes formed with a complementary RNA and these PS-ASOs. (B, C) P54nrb protein stability change upon binding to 3–10–3 PS-ASOs with different 2′-modifications (B), or to PS-cEt ASOs with different lengths (C), as determined using PTS assay. Protein binding affinity was determined using NanoBRET assay for P54nrb protein and 2′-cEt modified PS-ASOs of different lengths. The binding Kds (nM) were calculated using Prism. The average values and standard deviations from four duplicates are shown in related panels A, B and C. (D) Coomassie blue staining of P54nrb protein incubated with PS-ASOs or ASO/RNA duplexes, as in panel A, followed by digestion with different concentrations of chymotrypsin. The arrows indicate enhanced digestion by ss-PS-ASOs compared with PS-ASO/RNA duplex. The protein bands marked by the open arrow were quantified and the levels in percentage relative to that in PS-MOE ASO sample are listed.
However, contrary to what was observed with RNase H1 protein, the binding of 5–10–5 ss-PS-ASOs significantly increased the Tm of P54nrb (Figure 5A), suggesting that ss-PS-ASO binding stabilized the conformation of the protein. Importantly, 2′-modification of ss-PS-ASOs substantially affected the Tm of P54nrb. The PS-MOE ASO caused the highest increase in Tm (∼10°C), whereas the PS-F ASO caused the least increase (∼5°C). A similar observation was also made with 3–10–3 PS-ASOs (Figure 5B). Previously we have found that PS-F ASOs have a higher binding affinity to P54nrb (21). Thus, the degree of Tm increase by these different PS-ASOs does not seem to depend solely on the binding affinity. Rather, it is possible that PS-ASOs with different 2′-modifications may have different effects on the conformation of the P54nrb protein, with PS-MOE ASO being the least disruptive, whereas PS-F ASO the most disruptive relative to the native conformation, although all these PS-ASOs could stabilize the overall conformation of this protein.
To determine how increasing the binding affinity of PS-ASOs to proteins with the same 2′-modification affects the conformation of P54nrb, PS-cEt ASOs of different wing lengths were evaluated. PTS results showed that greater Tm increase was achieved with longer PS-cEt ASOs (Figure 5C), which is consistent with increased binding affinity, as determined using NanoBRET assay (Figure 5C), suggesting that increasing the binding affinity of PS-ASOs with the same 2′ modification can further stabilize the conformation of the proteins.
Since binding of either ss-PS-ASOs or duplexes increased Tm of P54nrb, suggesting a stabilized conformation of P54nrb, we next evaluated this hypothesis using partial digestion with chymotrypsin. Without PS-ASO, P54nrb was almost completely degraded by the enzyme under the experimental conditions (Figure 5D). However, binding of either ss-PS-ASO or PS-ASO/RNA duplex protected the protein from being degraded, as clearly seen at low concentration of chymotrypsin. For ss-PS-ASOs, PS-MOE ASO provided the greatest protection and PS-F the least protection, as clearly seen based on the quantification of the level of the top band, relative to that in the PS-MOE ASO sample. However, the duplexes formed with PS-ASOs of different 2′ modifications provided similar protection. These observations are consistent with the Tm changes of P54nrb determined in the PTS assay, with 2′-modifications showing obvious influence for ss-PS-ASOs, and not for duplexes. These results together suggest that the binding of PS-ASO or PS-ASO/RNA duplex stabilized the P54nrb protein structure. The different protective effects of ss-PS-ASOs modified with different 2′-modifications imply that either these PS-ASOs bind to different sites in the protein or binding of the PS-ASOs caused different degrees of conformational change.
PS-ASO-binding can induce the interactions of RNase H1 with P54nrb and PSF proteins
Altered conformation of proteins upon PS-ASO binding may affect the interaction of the proteins with their partners. Previously we have found that toxic PS-ASOs could cause RNase H1-dependent mislocalization of P54nrb or PSF to the nucleolus (12,35), and toxic PS-ASOs result in the formation of a complex(es) of RNase H1, PSF, P54nrb and other proteins that accumulate in the nucleolus in live cells using the NanoBiT assay (14). To determine whether those interactions detected in cells are directly mediated by RNase H1 and P54nrb or bridged by other proteins or RNA factors, co-immunoprecipitation was performed using purified full-length RNase H1 as a bait. The RNase H1-coated beads were incubated without or with a toxic, 3–10–3 PS-cEt ASO (ASO 558807) to allow binding of PS-ASO to RNase H1, and subsequently incubated with P54nrb, PSF, or pre-complexed P54nrb/PSF proteins (Figure 6A). Co-IPed proteins were analyzed by western analysis (Figure 6B). In the absence of PS-ASOs, no P54nrb, PSF, or P54nrb/PSF complex was co-isolated with either MBP- or GST-tagged full-length RNase H1 proteins, suggesting that RNase H1 does not normally interact with these proteins. However, both P54nrb and PSF could be co-precipitated with RNase H1 preincubated with PS-ASOs, confirming that PS-ASOs can induce direct interactions between RNase H1 and these paraspeckle proteins.
Figure 6.

PS-ASO binding to RNase H1 can induce the interaction of RNase H1 with P54nrb. (A) Schematic presentation of the experimental procedures of co-isolation of P54nrb, PSF, or pre-complexed P54nrb/PSF with RNase H1. Full-length or domains of RNase H1 was bound to the beads, incubated with or without PS-ASO to allow binding of PS-ASO with RNase H1. After the removal of unbound PS-ASOs, P54nrb/PSF complex was incubated with beads and co-precipitated. (B) Western analyses of P54nrb and PSF co-isolated with MBP-H1 or GST-H1. The bait protein RNase H1 was also probed. (C) Western analysis of P54nrb co-isolated with different domains or full-length RNase H1, in the absence or presence of toxic (T, ASO 558807) or gap2 OMe modified safe (S, ASO 936053) PS-ASOs. Ponceau staining of bait proteins is shown in lower panels (C, D, E, F and H). (D) Western analysis of PSF co-isolated with different domains or full-length RNase H1, in the absence or presence of toxic (T) or safe (S) PS-ASOs. Note that the lower band of PSF is a truncated form of the protein. (E) Western analysis of P54nrb co-isolated with H1-delMit, in the absence or presence of toxic (T, ASO 558807) or safe (S, ASO 936053) PS-ASOs. (F) Western analysis of P54 co-isolated with H1-delMit upon binding of different toxic PS-ASOs and their non-toxic gap 2′-OMe modified PS-ASO counterparts, as in panel F. Two pairs of different ASOs were analyzed in the same experiment, but with different exposure times for the Western blot in the right panel. (G) The paired Tms of toxic and non-toxic gap 2′-OMe PS-ASOs in panel F determined in PTS. (H) Western analysis of P54nrb co-isolated with H1-delMit, in the absence or presence of different 3–10–3 cEt toxic (red) or safe PS-ASOs (listed in panel J). (I) The average level of coisolated P54nrb mediated by toxic versus safe PS-ASOs. (J) The Tms of toxic and safe PS-ASOs are determined in PTS. The average values and standard deviations from 4 duplicates are shown in related panels.
The spacer domain of RNase H1 has been shown to be involved in the PS-ASO-induced interaction with P54nrb in cells (14). Consistently, when H1-NTD or H1-CTD domains were used as bait in the Co-IP assay, H1-CTD, which contains both spacer domain and catalytic domain, could robustly interact with purified P54nrb and PSF in the presence of either a toxic or a non-toxic PS-ASO (Figure 6C and D), similar to the full-length RNase H1 proteins (Figure 6C and D). The H1-NTD domain also caused weaker but detectable interactions with P54nrb in the presence of PS-ASOs (data not shown and see below). These results indicate that the H1-CTD domain could provide major and direct interaction with P54nrb and PSF in the presence of PS-ASOs, without the need of other cellular protein or RNA factors.
Toxic PS-ASOs tend to induce more interactions between RNase H1 and P54nrb proteins
Previously we have demonstrated that toxic PS-ASOs tend to have tighter protein binding than non-toxic PS-ASOs, and introducing 2′ OMe at position 2 in the DNA gap region (Gap2 OMe) reduced protein binding and mitigated toxicity (12) (14). Consistently, in the co-IP assay, we observed that a gap2 OMe-modified PS-ASO (936053) induced less interactions between RNase H1 and P54nrb than the parental toxic PS-ASO (558807) (Figure 6C). These observations raise a possibility that toxic PS-ASOs may induce greater conformational change of RNase H1 than non-toxic PS-ASOs, leading to enhanced interactions with P54nrb, likely due to tighter binding affinity.
To evaluate this possibility, H1-delMit protein was used as bait in the co-IP assay, as this protein represents the form of endogenous RNase H1 that contains both HBD and CTD domains and behaves well in EMSA assay. Again, no H1-delMit interaction with P54nrb was observed in the absence of PS-ASOs (Figure 6E). Similar to H1-CTD and full-length proteins as in Figure 6C, binding of the parental toxic PS-ASO to H1-delMit robustly induced the interaction between H1-delMit and P54nrb, as compared with the gap2 OMe modified PS-ASO, which showed reduced toxicity and protein binding as demonstrated previously (13). To determine whether this observation is specific to this pair of PS-ASOs, co-IP and PTS were applied to investigate the P54nrb interaction with H1-delMit using additional four pairs of parental toxic and gap2 OMe-modified PS-ASOs (Figure 6F, G). Although the parental and gap2 OMe modified PS-ASOs did not show substantial differences in Tm changes of H1-delMit and P54nrb proteins (Figure 6G, Supplementary Figure S4A), three gap2 OMe modified PS-ASOs induced less interactions between H1-delMit and P54nrb, compared to their corresponding parental PS-ASOs, indicating that the reduced interaction between H1-delMit with P54nrb by Gap2 OMe modification is not specific to a particular toxic PS-ASO sequence.
However, for one toxic PS-ASO sequence (546061), the gap2 OMe PS-ASO (1132959) appeared to induce more interactions between RNase H1 and P54nrb, suggesting that in cells, the interactions between RNase H1 and P54nrb and RNase H1-dependent P54nrb nucleolar mislocalization induced by certain toxic PS-ASOs may be more complicated and may involve additional protein (and/or RNA) factors. To evaluate this possibility, affinity selection was performed to isolate PS-ASO binding proteins from HeLa cell lysate to mimic the cellular environment. Silver staining results showed that the gap2 OMe ASO (1132959) had slightly reduced binding to several protein bands as compared with the parental PS-ASO (546061) (Supplementary Figure S4B), and western analysis (Supplementary Figure S4C) showed that the gap2 OMe ASO has reduced binding to PSF and P54nrb but slightly increased binding to RNase H1. This may explain why this gap2 OMe PS-ASO induced more interactions between RNase H1 and P54nrb in the co-IP assay (Figure 6F). The reduced binding to PSF and P54nrb by gap2 OMe modification was also observed for two additional PS-ASOs, consistent with our previous observations (13). Together, these observations suggest that gap2 OMe modification that reduces PS-ASO toxicity tends to reduce protein binding and PS-ASO-induced interactions between RNase H1 and P54nrb. However, the RNase H1–P54nrb interactions induced by certain toxic PS-ASOs may be affected by other proteins in cells, contributing to RNase H1-dependent mislocalization of P54nrb protein and PS-ASO toxicity.
To further determine if toxic PS-ASOs tend to induce more interactions between RNase H1 and P54nrb, different toxic and non-toxic PS-ASO sequences were evaluated. Co-IP was applied to investigate the P54nrb binding levels to H1-delMit mediated by four toxic and four non-toxic PS-ASOs with different sequences (Figure 6H). In general, toxic PS-ASOs induced more P54nrb interaction with RNase H1 than non-toxic PS-ASOs. Compared with the toxic PS-ASOs, on average, the non-toxic PS-ASOs induce ∼24% of P54nrb binding to RNase H1 relative to toxic PS-ASOs (Figure 6I). PTS was also performed to determine the Tm changes of H1-delMit and P54nrb by the binding of these PS-ASOs (Figure 6J). Toxic ASOs caused greater Tm reduction of H1-delMit than the safe PS-ASOs (>2°C), indicating greater conformational disruptions that may facilitate more P54nrb binding to RNase H1. On the other hand, in the PTS assay, toxic PS-ASOs did not show significant difference in Tm changes of P54nrb compared with safe PS-ASOs, though a slightly greater Tm increase (<1°C) could be observed with toxic PS-ASOs than with safe PS-ASOs, and such trend still holds when more PS-ASO sequences were evaluated (Supplementary Figure S4A). Together, these results suggest that toxic PS-ASOs tend to induce more RNase H1-P54nrb interactions, likely due to tighter protein binding than non-toxic PS-ASOs.
2′ Modifications of ss-PS-ASOs that affect protein binding affinity can affect the interaction of RNase H1 with paraspeckle proteins
To further confirm that PS-ASO binding affinity to proteins affects protein–protein interactions, we next evaluated the effects of 2′-modification on PS-ASO-induced interactions between RNase H1 and P54nrb or PSF. 5–10–5 PS-ASOs with 2′-MOE, cEt, or F modifications were tested either as single-strands or as duplexes formed with complementary RNA. Although H1-NTD has weaker PS-ASO-induced interactions with P54nrb or PSF compared to H1-CTD, the H1-NTD exhibits greater Tm change upon PS-ASO binding in PTS assay. Thus, co-IP with H1-NTD was first performed as described above, and the results showed that ss-PS-F ASO induced the strongest, and ss-PS-MOE ASO induced the weakest, interaction of H1-NTD with P54nrb (Figure 7A). However, pre-binding of H1-NTD with PS-ASO/RNA or PO-ASO/RNA heteroduplexes caused no to very modest interaction between P54nrb and H1-NTD. A similar trend was also observed for H1-NTD interaction with PSF, though in this case, PS-cEt and PS-F ASOs induced similarly more interactions of H1-NTD and PSF than the PS-MOE ASO did (Figure 7B), and no or little interaction was induced by PS-ASO/RNA or PO-ASO/RNA heteroduplexes.
Figure 7.

PS-ASO induced interactions of RNase H1 with P54nrb or with PSF are affected by 2′-modifications of PS-ASOs. Co-precipitation was performed as in Figure 6, using GST-H1-NTD (A and B) or GST-H1-CTD (C and D) or GST-H1-delMit (E and F) as baits, in the absence or presence of different PS-ASOs of different 2′-modifications, or different ASO/RNA duplexes (ASOs are as in Figure 1C, right panel). The beads were incubated with either P54nrb (A, C and E), or with PSF (B, D and F), and isolated proteins were detected by western analyses. Ponceau staining of the bait proteins is shown.
Next, we evaluated the effects of 2′ modifications of PS-ASOs on PS-ASO-induced H1-CTD and P54nrb interactions. Consistent with H1-NTD, 2′-modification of PS-ASOs also affected the interactions of H1-CTD with P54nrb or with PSF, and ss-PS-ASOs induced stronger interaction than ASO/RNA duplexes (Figure 7C, D). However, the ss-PS-cEt ASO induced slightly stronger interactions between H1-CTD and these paraspeckle proteins, compared with the ss-PS-F ASO, although PS-MOE ASO still triggered the weakest interaction. On the other hand, the PS-F ASO/RNA duplex caused the strongest interaction between RNase H1 domains and these paraspeckle proteins.
The H1-delMit interaction with P54nrb (Figure 7E) or with PSF (Figure 7F) triggered by PS-ASOs is also affected by 2′ modifications, with PS-F and PS-cEt ASOs induced greater interaction than PS-MOE ASO. PS-ASO/RNA duplexes induced no or modest interaction of H1-delMit with P54nrb or PSF, and no H1-delMit interaction with these proteins was observed without PS-ASO or with the PO-ASO/RNA duplex. The results are similar to what was observed for individual H1 domains. The weak effect of PS-ASO/RNA duplex to induce RNase H1-P54nrb interaction is consistent with our previous observations that P54nrb can compete with RNase H1 for binding to PS-ASO/RNA duplexes (9).
Together, these results indicate that 2′-modification of PS-ASOs with higher protein binding affinity tends to induce more interactions between RNase H1 and paraspeckle proteins, and that ss-PS-ASOs may play a major role in mediating these protein–protein interactions in cells as compared with PS-ASO/RNA duplexes.
PS-ASOs can enhance the interaction between P54nrb and PSF proteins
P54nrb and PSF have been shown to form a complex in cells under normal conditions (34,36,37). Since PS-ASO binding also affects the conformation of P54nrb and PSF proteins as demonstrated in partial digestion assay (see above); next, we evaluated if PS-ASO binding alters the interaction between these two purified proteins. Co-IP was performed using GST-P54nrb as a bait (Figure 8A). The results showed that consistent with previous reports, PSF could be co-precipitated with P54nrb in the absence of PS-ASO (Figure 8B). However, upon PS-ASO binding to P54nrb, substantially more PSF was co-precipitated with P54nrb, indicating that PS-ASO can further enhance the interactions between P54nrb and PSF. On the other hand, slightly more PSF was co-precipitated with P54nrb upon binding of the toxic PS-ASO, as compared with the non-toxic PS-ASO. This is consistent with the observations that P54nrb and PSF tend to bind more tightly to toxic PS-ASOs than to non-toxic PS-ASOs, as shown previously (12) and also demonstrated here by affinity selection using biotinylated PS-ASO (Figure 8C). Western results showed that the toxic PS-ASO (ASO 558807) bound more PSF and P54nrb proteins than the non-toxic PS-ASO (ASO 549139), and that these proteins bound less PS-ASO/RNA heteroduplexes than ss-PS-ASOs. As a control, P32, an RNase H1-interacting protein, does not bind either the ss-PS-ASO or PS-ASO/RNA heteroduplex. Together, these results suggest that PS-ASO binding to proteins can not only induce new interactions like RNase H1-P54nrb interactions, but also enhance the interactions of existing protein partners.
Figure 8.

PS-ASO binding to P54nrb enhances the interaction between P54nrb and PSF, but does not induce the interaction between P54nrb and H1-CTD. (A) Schematic presentation of the experimental procedures of co-isolation of PSF with P54nrb. GST-P54nrb was bound to the beads, incubated without or with toxic (558807) or safe (936053) PS-ASO to allow binding of the PS-ASOs to P54nrb. After the removal of unbound PS-ASOs, PSF was incubated with beads and co-precipitated. (B) Western analysis of PSF protein co-isolated with P54nrb. P54nrb was probed. The signal intensity of PSF was quantified, normalized to P54nrb, and the ratios of PSF/P54nrb signals were compared with that in no-ASO control isolation and plotted. (C) Western analysis of proteins co-isolated from HeLa cell lysate through affinity selection using biotinylated PS-ASO 902459 then competitively eluted with PS-ASO or ASO/RNA duplex. P32 and Ku70 were probed as negative and positive controls, respectively. (D) Schematic presentation of potential modes of ASO-induced interaction between RNase H1 and P54nrb. (E) Western analysis of co-isolated P54nrb protein with H1-CTD. The H1-CTD was bound to beads, incubated with ss-PS-ASO (116847), followed by incubation of complementary RNA (XL279) or a control RNA (XL1052). After washing, the beads were incubated with P54nrb, and co-isolated P54nrb was analyzed by western analysis. Ponceau staining of the membrane was shown below the lanes. (F) Western analysis of H1-CTD co-isolated using GST-P54nrb as bait, similar to panel A. After PS-ASO binding to GST-P54nrb, H1-CTD was incubated with the beads, and co-isolated proteins were analyzed by western for RNase H1 or P54nrb. (G) Western analysis of H1-delMit co-isolated using GST-P54nrb as bait, similar to panel A. After PS-ASO binding to GST-P54nrb, H1-delMit was incubated with the beads, and co-isolated proteins were analyzed by western for RNase H1. Ponceau staining of the membrane was shown below the lanes.
PS-ASO-induced interactions between RNase H1 and P54nrb are due to PS-ASO binding to RNase H1
Since PS-ASO is able to bind both RNase H1 and P54nrb, affecting protein–protein interactions, we next examine how PS-ASO binding induces interaction of RNase H1 with P54nrb. The PS-ASO induced interactions between these two proteins may occur through direct protein–protein interaction; however, it is also possible that RNase H1 may not bind entire PS-ASO, and some unbound portion of the PS-ASO may be stretched out and available for P54nrb binding, thus serving as a bridge between RNase H1 and P54nrb proteins (Figure 8D). We reasoned that for this latter case, incubation with a complementary RNA to hybridize with the unbound portion of the PS-ASO should reduce the co-isolation of P54nrb with RNase H1, since PS-ASO/RNA duplexes do not induce substantial interactions between RNase H1 and P54nrb. Co-IP was thus performed using H1-CTD as a bait. The beads-bound H1-CTD was incubated with ss-PS-ASO, and subsequently with either a complementary uncleavable RNA, or a control RNA, and finally with purified P54nrb. Co-isolated P54nrb was analyzed by western analysis, which showed that the presence of complementary RNA did not affect the interaction of P54nrb with H1-CTD induced by PS-ASO (Figure 8E). These results suggest that the interaction between P54nrb and RNase H1 is at least not sorely bridged by a long stretch of ss-PS-ASO since decent binding of P54nrb to PS-ASO requires 11–13 PS-nucleotides (9,38), and such a long stretch ASO has the potential to hybridize with the complementary RNA to inhibit P54nrb binding.
On the other hand, since PS-ASOs can also bind P54nrb protein and cause conformational change, leading to enhanced interaction with PSF, it is possible that PS-ASO-binding to P54nrb may induce the interactions with RNase H1. To determine whether this is the case, beads-bound GST-P54nrb protein were pre-incubated with toxic or non-toxic PS-ASOs, followed by incubation with H1-CTD or H1-delMit. Co-precipitated proteins were analyzed by western analysis (Figure 8F, G). Neither H1-CTD nor H1-delMit was found to interact with P54nrb when PS-ASOs bound to P54nrb (Figure 8F, G), confirming that the PS-ASOs mediated interactions between RNase H1 and P54nrb are triggered by PS-ASO binding to RNase H1, which may cause RNase H1 conformational change, leading to gained interaction with P54nrb.
PS-ASO binding to RNase H1 can disrupt the interaction of RNase H1 and P32 in cells
RNase H1 has been shown to interact through HBD with P32 in cells, a protein that can affect RNase H1 activity (24). The altered conformation of RNase H1 upon PS-ASO binding may also affect the interaction of RNase H1 with this protein. To evaluate this possibility, Flag-tagged full-length RNase H1 was expressed in HEK293 cells, and the lysate was incubated without or with a 5–10–5 PS-MOE ASO, or with a PS-ASO/RNA heteroduplex formed with a 2′-OMe modified complementary RNA. Immunoprecipitation was then performed using anti-Flag beads, and coprecipitated protein was analyzed by Western analyses (Figure 9A). In the absence of PS-ASO or heteroduplexes, P32 was clearly co-isolated with Flag-RNase H1, and not with a control protein Flag-RNase H2, consistent with our previous findings (24). However, in the presence of either ss-PS-ASOs or with PS-ASO/RNA heteroduplex, RNase H1 failed to co-isolate P32, suggesting that RNase H1 binding to either ss-PS-ASO or PS-ASO/RNA duplex inhibited RNase H1 interaction with P32. To further confirm this observation, immunoprecipitation was performed using Flag-RNase H1 to isolate P32, followed by incubation of the beads with PS-ASO/RNA heteroduplex. P32 was substantially eluted from the beads by the PS-ASO/RNA heteroduplex (Figure 9B). The altered RNase H1-P32 interaction was not due to the binding of the PS-ASO or the duplex to P32, since P32 does not meaningfully bind PS-ASO or PS-ASO/RNA duplex (Figure 8C). Together, these results indicate that RNase H1 binding to PS-ASO or duplex may also disrupt the interaction of RNase H1 with certain partner proteins, likely due to conformational change of RNase H1.
Figure 9.

Western analysis of P32 co-isolated with RNase H1 in the absence or presence of ss-PS-ASO or ASO/RNA duplex. (A) Co-immunoprecipitation was performed with anti-Flag beads using lysates of HEK293 cells expressing Flag-RNase H1 or Flag-RNase H2 as described in materials and methods section. Co-isolated proteins were separated by SDS-PAGE and subjected to western blotting. (B) Western analysis of P32 protein co-isolated with Flag-RNase H1, then eluted with ASO/RNA duplex at different concentrations. Eluted and beads-bound proteins were separated by SDS-PAGE, and probed by western analysis. The upper panel indicates the experimental procedures.
DISCUSSION
Interactions of PS-ASOs with proteins can significantly affect the activity of PS-ASOs by, for example, affecting delivery, subcellular trafficking, distribution, and recruitment of RNase H1. On the other hand, PS-ASO–protein interactions also affect the fate of proteins bound, altering protein localization, stability, and interactions with other proteins, thus contributing to the toxicity of PS-ASOs. PS-ASOs can localize to and induce the formation of different cellular structures that contain different proteins. Though it has been well established that both sequence and chemistry can affect PS-ASO–protein interactions, the detailed mechanism(s) underlying the altered protein localization, stability and interactions with other proteins have not been well characterized. Here, we show for the first time that PS-ASO binding can alter the conformation of proteins in a chemistry and sequence dependent manner, which can alter the interactions with other proteins.
Using RNase H1, P54nrb and PSF as model proteins, we characterized the effects of PS-ASO or PS-ASO/RNA duplex binding on the conformation of these proteins. For RNase H1, ss-PS-ASO binding disrupted the conformation of the protein. Upon ss-PS-ASO binding, H1-NTD and H1-CTD all showed reduced Tm in PTS assay and more degradation by the proteinase, indicating that ss-PS-ASO binding may cause unfolding of the protein. The H1-delMit, the dominant form of endogenous RNase H1, also showed Tm reduction and disrupted conformation upon binding to ss-PS-ASOs, consistent with individual domains. The unfolding of RNase H1 protein upon ss-PS-ASO binding may expose more surface area capable of interacting with paraspeckle proteins.
On the contrary, the PS-ASO/RNA duplex binding to RNase H1 displays a different conformational change than ss-PS-ASO. The heteroduplex increased the Tm of H1-NTD, suggesting a more stable conformation of H1-NTD, which is supported by the observation that duplex binding did not alter the degradation pattern or level in the partial digestion assay. It is possible that, as a natural substrate, heteroduplex binding might strengthen the protein folding, facilitating the enzyme activity. Similarly, PS-ASO/RNA duplex binding to H1-CTD or H1-delMit also did not cause significant Tm change nor affected the proteinase digestion. Moreover, PS-ASO/RNA duplex binding to H1-delMit protected two intermediates in the proteinase digestion study (Figure 4C), further suggesting that PS-ASO/RNA duplex may bind to the protein in a non-disruptive manner.
PS-ASO binding also altered the conformation of P54nrb but differently from what was observed for RNase H1. Both PS-ASOs and PS-ASO/RNA duplexes caused protective effects on the conformation of P54nrb, as shown with an increased Tm in PTS assay and inhibited degradation by partial chymotrypsin digestion. However, 2′ modification in ss-PS-ASOs, and not in PS-ASO/RNA duplexes, significantly affected the Tm change and chymotrypsin digestion of P54nrb upon binding to ss-PS-ASO, and not PS-ASO/RNA duplex. These results suggest that ss-PS-ASO and duplexes can stabilize the protein folding, though it is possible that the conformational change induced by ss-PS-ASO may be different from that by PS-ASO/RNA duplex. The ss-PS-ASOs and PS-ASO/RNA duplexes might bind to different domains or different pockets of P54nrb. Previous publications reported that the N-terminus of P54nrb contributed to the interaction with ss-DNA or RNA, and the C-terminus with the coiled-coiled domain bound the DNA/RNA duplex, which might explain the different effects of the interaction of ss-PS-ASO and ASO/RNA duplex with P54nrb (39,40).
It is known that 2′-modifications of PS-ASOs affect protein binding affinity. 2′-F modified PS-ASOs bind tighter to most PS-ASO binding proteins than 2′-cEt modified PS-ASOs, which bind tighter than 2′-MOE modified PS-ASOs. Similarly, 2′-modifications also affect the conformational change of proteins bound. The binding affinity between PS-ASO and proteins appear to be correlated with the extent of the conformational change of the proteins, since longer PS-ASOs bound tighter to proteins (27,32) and caused stronger Tm change of proteins bound. Moreover, PS-ASOs with higher affinity 2′-modifications also caused greater conformational change. Compared with 2′-MOE PS-ASO, the 2′-F PS-ASO caused greater Tm reduction of RNase H1 protein in PTS assay, and more protein degradation in partial digestion assay. These results suggest that relative to 2′-MOE PS-ASO, tighter binding of the 2′-F PS-ASO may cause greater disruption of the conformation of RNase H1, resulting more protein partner binding or toxicity in cells.
However, the effects are more complex for P54nrb protein. Though both 2′-F and 2′-MOE PS-ASOs increased Tm of P54nrb as shown in PTS assay, weaker increase was found for 2′-F PS-ASO than for 2′-MOE PS-ASO. It is possible that PS-MOE ASOs bind to the protein in a more native conformation and further enhanced the folding, leading to increased stability. However, binding of 2′-F PS-ASO may cause a different or greater local conformational change of P54nrb, leading to the unfolding of different areas of the protein, resulting in less Tm increase in PTS assay, and less protective effects of the entire protein in partial digestion assay. On the other hand, it is also possible that 2′-F PS-ASO may bind to different positions of P54nrb, leading to different conformational changes compared with PS-MOE ASOs. Nevertheless, the different conformational changes of P54nrb by binding to 2′-F PS-ASOs and to 2′-MOE PS-ASOs may explain why in cells 2′-F PS-ASOs tend to cause rapid degradation of P54nrb (and PSF) proteins (33).
Although 2′ modifications of ss-PS-ASOs showed significant influence on protein conformation, the effects of 2′-modifications on protein conformational change are dramatically reduced when the PS-ASOs were duplexed with complementary RNA, as shown by PTS and partial proteinase digestion assays for both RNase H1 and P54nrb proteins. This is not unexpected, since the influence of 2′-modifications can be masked when PS-ASOs hybridize with complementary RNA to generate a duplex, which has much defined helical structures than ss-PS-ASOs. Given that 2′ modifications significantly affect PS-ASO toxicity, these observations also imply that toxic ASOs are most likely recognized by proteins in the form of ss-PS-ASO, and not heteroduplex.
The altered protein conformation may change the surface properties of the proteins, thus affecting interactions with other partners. Indeed, PS-ASO can induce the interactions between RNase H1 and paraspeckle proteins P54nrb/PSF. Such induced interaction between these two proteins is due to PS-ASO binding to RNase H1, rather than PS-ASO binding to P54nrb, since P54nrb could be co-isolated with PS-ASO-bound full-length or MLS-deleted RNase H1 or H1-CTD, and not vice versa. In addition, consistent with the observations that PS-ASOs with tighter protein binding affinity (e.g. 2′-F PS-ASO) caused greater protein conformational changes, the 2′-F PS-ASO also induced greater interaction between RNase H1 and paraspeckle proteins compared with 2′-MOE PS-ASOs. Similarly, toxic PS-ASOs tend to induce greater interaction between H1-delMit and P54nrb than non-toxic PS-ASOs, as shown with different parental toxic PS-ASO and gap2 OMe modified PS-ASO pairs, as well as different toxic and non-toxic PS-ASO sequences. This is consistent with previous observations that toxic PS-ASOs tend to bind more proteins more tightly than non-toxic PS-ASOs, and to induce the interaction of paraspeckle proteins with endogenous RNase H1. These observations provide an explanation of why RNase H1 is required for toxic PS-ASO-induced nucleolar mislocalization of P54nrb and PSF proteins in cells (12,14,40). It is possible that toxic PS-ASO binding to RNase H1 causes a significant conformational change of this protein, generating new surface properties (domains) that gain strong interaction with P54nrb and PSF, mediating relocalization of these paraspeckle proteins to the nucleolus (14,35).
However, as certain toxic PS-ASOs may not induce strong direct interaction between purified RNase H1 and P54nrb proteins in test tubes, and almost all toxic PS-ASOs can cause RNase H1-dependent paraspeckle protein mislocalization to the nucleolus (13), it is possible that for certain toxic PS-ASOs, additional cellular proteins may also mediate RNase H1-dependent paraspeckle protein mislocalization to the nucleolus. In a cellular system, other proteins could affect the degree of RNase H1 or P54nrb/PSF binding to PS-ASOs, due to the competition for binding to PS-ASOs. In addition, PS-ASO binding to RNase H1 may also induce interactions with additional cellular proteins. All these altered protein–protein interactions upon PS-ASO binding may act together to affect the activity, duration of action or toxicity of PS-ASOs.
On the other hand, the binding of PS-ASOs or PS-ASO/RNA duplexes to RNase H1 disrupted the interaction between RNase H1 and P32 in cell lysate, indicating that the altered conformation of RNase H1 can have different effects on the interactions with other proteins, likely due to the fact that RNase H1 contact P32 and P54/PSF through different domains. Indeed, it has been shown that P32 interacts with HBD, whereas P54nrb contacts the spacer domains of RNase H1 (14,24).
As compared with ss-PS-ASOs, PS-ASO/RNA duplexes induced no or mild interactions between RNase H1 and these paraspeckle proteins. This is consistent with the observations that PS-ASO/RNA duplex binding stabilized the RNase H1 protein, whereas ss-PS-ASO binding causes a more disruptive conformational change, which may expose certain RNase H1 domains to the surface and enables the interaction with P54nrb/PSF. Once again, these results suggest that ss-PS-ASO can bind proteins more tightly and maybe in different binding modes compared with PS-ASO/RNA duplex, causing more altered protein–protein interactions. Together, our observations can explain the molecular basis of PS-ASO induced protein mislocalization and altered protein–protein interactions in cells, and suggest that ss-PS-ASO is the major contributor of such effects, which are largely influenced by the chemical modifications of PS-ASOs. These findings will thus facilitate the design of PS-ASOs with improved performance by modulating PS-ASO–protein interactions through medicinal chemistry efforts.
Supplementary Material
ACKNOWLEDGEMENTS
We wish to thank Punit Seth and Michael Migawa for stimulating discussions, Karla Bernardo and Cheryl L. De Hoyos for technical assistance, Hans Gaus for help in fluorescence polarization assay.
Contributor Information
Lingdi Zhang, Core Antisense Research, Ionis Pharmaceuticals, Inc., 2855 Gazelle Court, Carlsbad, CA 92010, USA.
Timothy A Vickers, Core Antisense Research, Ionis Pharmaceuticals, Inc., 2855 Gazelle Court, Carlsbad, CA 92010, USA.
Hong Sun, Antisense Drug discovery, Ionis Pharmaceuticals, Inc. Carlsbad, CA 92010, USA.
Xue-hai Liang, Core Antisense Research, Ionis Pharmaceuticals, Inc., 2855 Gazelle Court, Carlsbad, CA 92010, USA.
Stanley T Crooke, Core Antisense Research, Ionis Pharmaceuticals, Inc., 2855 Gazelle Court, Carlsbad, CA 92010, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Ionis Pharmaceuticals. Funding for open access charge: IONIS Pharmaceuticals, Inc.
Conflict of interest statement. None declared.
REFERENCES
- 1. Crooke S.T., Vickers T.A., Lima W.F., Wu H.-J.. Crooke S.T. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; 2nd ednBoca Raton, FL: CRC Press; 3–46. [Google Scholar]
- 2. Crooke S.T., Witztum J.L., Bennett C.F., Baker B.F.. RNA-targeted therapeutics. Cell Metab. 2018; 27:714–739. [DOI] [PubMed] [Google Scholar]
- 3. Bennett C.F., Krainer A.R., Cleveland D.W.. Antisense oligonucleotide therapies for neurodegenerative diseases. Annu. Rev. Neurosci. 2019; 42:385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lima W., Wu H., Crooke S.T.. Crooke S.T. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; 2nd ednBoca Raton, FL: CRC Press; 47–74. [Google Scholar]
- 5. Swayze E.E., Bhat B.. Crooke S.T. Antisense Drug Technology - Principles, Strategies, and Applications. 2008; 2nd ednBoca Raton, FL: CRC Press; 143–182. [Google Scholar]
- 6. Kurreck J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003; 270:1628–1644. [DOI] [PubMed] [Google Scholar]
- 7. Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H.. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:230–237. [DOI] [PubMed] [Google Scholar]
- 8. Crooke S.T., Vickers T.A., Liang X.H.. Phosphorothioate modified oligonucleotide-protein interactions. Nucleic Acids Res. 2020; 48:5235–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liang X.H., Sun H., Shen W., Crooke S.T.. Identification and characterization of intracellular proteins that bind oligonucleotides with phosphorothioate linkages. Nucleic Acids Res. 2015; 43:2927–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Crooke S.T., Seth P.P., Vickers T.A., Liang X.-H.. The interaction of phosphorothioate-containing RNA targeted drugs with proteins is a critical determinant of the therapeutic effects of these agents. J. Am. Chem. Soc. 2020; 142:14754–14771. [DOI] [PubMed] [Google Scholar]
- 11. Burel S.A., Hart C.E., Cauntay P., Hsiao J., Machemer T., Katz M., Watt A., Bui H.H., Younis H., Sabripour M.et al.. Hepatotoxicity of high affinity gapmer antisense oligonucleotides is mediated by RNase H1 dependent promiscuous reduction of very long pre-mRNA transcripts. Nucleic Acids Res. 2015; 44:2093–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shen W., De Hoyos C.L., Migawa M.T., Vickers T.A., Sun H., Low A., Bell T.A. 3rd, Rahdar M., Mukhopadhyay S., Hart C.E.et al.. Chemical modification of PS-ASO therapeutics reduces cellular protein-binding and improves the therapeutic index. Nat. Biotechnol. 2019; 37:640–650. [DOI] [PubMed] [Google Scholar]
- 13. Migawa M.T., Shen W., Wan W.B., Vasquez G., Oestergaard M.E., Low A., De Hoyos C.L., Gupta R., Murray S., Tanowitz M.et al.. Site-specific replacement of phosphorothioate with alkyl phosphonate linkages enhances the therapeutic profile of gapmer ASOs by modulating interactions with cellular proteins. Nucleic Acids Res. 2019; 47:5465–5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vickers T.A., Rahdar M., Prakash T.P., Crooke S.T.. Kinetic and subcellular analysis of PS-ASO/protein interactions with P54nrb and RNase H1. Nucleic Acids Res. 2019; 47:10865–10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wu H., Lima W.F., Crooke S.T.. Investigating the structure of human RNase H1 by site-directed mutagenesis. J. Biol. Chem. 2001; 276:23547–23553. [DOI] [PubMed] [Google Scholar]
- 16. Liang X.H., Sun H., Nichols J.G., Crooke S.T.. RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol. Ther. 2017; 25:2075–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzuki Y., Holmes J.B., Cerritelli S.M., Sakhuja K., Minczuk M., Holt I.J., Crouch R.J.. An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol. Cell. Biol. 2010; 30:5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hyjek M., Figiel M., Nowotny M.. RNases H: Structure and mechanism. DNA Repair (Amst.). 2019; 84:102672. [DOI] [PubMed] [Google Scholar]
- 19. Lima W.F., Wu H., Nichols J.G., Prakash T.P., Ravikumar V., Crooke S.T.. Human RNase H1 uses one tryptophan and two lysines to position the enzyme at the 3′-DNA/5′-RNA terminus of the heteroduplex substrate. J. Biol. Chem. 2003; 278:49860–49867. [DOI] [PubMed] [Google Scholar]
- 20. Nowotny M., Cerritelli S.M., Ghirlando R., Gaidamakov S.A., Crouch R.J., Yang W.. Specific recognition of RNA/DNA hybrid and enhancement of human RNase H1 activity by HBD. EMBO J. 2008; 27:1172–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vickers T.A., Crooke S.T.. Development of a Quantitative BRET affinity assay for nucleic acid-protein interactions. PLoS One. 2016; 11:e0161930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lima W.F., Crooke S.T.. Binding affinity and specificity of Escherichia coli RNase H1: impact on the kinetics of catalysis of antisense oligonucleotide-RNA hybrids. Biochemistry. 1997; 36:390–398. [DOI] [PubMed] [Google Scholar]
- 23. Nowotny M., Gaidamakov S.A., Ghirlando R., Cerritelli S.M., Crouch R.J., Yang W.. Structure of human RNase H1 complexed with an RNA/DNA hybrid: insight into HIV reverse transcription. Mol. Cell. 2007; 28:264–276. [DOI] [PubMed] [Google Scholar]
- 24. Wu H., Sun H., Liang X., Lima W.F., Crooke S.T.. Human RNase H1 is associated with protein P32 and is involved in mitochondrial pre-rRNA processing. PLoS One. 2013; 8:e71006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen H.D., Yadav T., Giri S., Saez B., Graubert T.A., Zou L.. Functions of replication protein A as a sensor of R loops and a regulator of RNaseH1. Mol. Cell. 2017; 65:832–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Matulis D., Kranz J.K., Salemme F.R., Todd M.J.. Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using thermofluor. Biochemistry. 2005; 44:5258–5266. [DOI] [PubMed] [Google Scholar]
- 27. Liang X.H., Shen W., Sun H., Prakash T.P., Crooke S.T.. TCP1 complex proteins interact with phosphorothioate oligonucleotides and can co-localize in oligonucleotide-induced nuclear bodies in mammalian cells. Nucleic Acids Res. 2014; 42:7819–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vickers T.A., Rahdar M., Prakash T.P., Crooke S.T.. Kinetic and subcellular analysis of PS-ASO/protein interactions with P54nrb and RNase H1. Nucleic Acids Res. 2019; 47:10865–10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ericsson U.B., Hallberg B.M., Detitta G.T., Dekker N., Nordlund P.. Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 2006; 357:289–298. [DOI] [PubMed] [Google Scholar]
- 30. Seabrook S.A., Newman J.. High-throughput thermal scanning for protein stability: making a good technique more robust. ACS Combinator. Science. 2013; 15:387–392. [DOI] [PubMed] [Google Scholar]
- 31. Suzuki Y., Holmes J.B., Cerritelli S.M., Sakhuja K., Minczuk M., Holt I.J., Crouch R.J.. An upstream open reading frame and the context of the two AUG codons affect the abundance of mitochondrial and nuclear RNase H1. Mol. Cell. Biol. 2010; 30:5123–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liang X.H., Shen W., Sun H., Kinberger G.A., Prakash T.P., Nichols J.G., Crooke S.T.. Hsp90 protein interacts with phosphorothioate oligonucleotides containing hydrophobic 2′-modifications and enhances antisense activity. Nucleic Acids Res. 2016; 44:3892–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shen W., Liang X.H., Sun H., Crooke S.T.. 2′-Fluoro-modified phosphorothioate oligonucleotide can cause rapid degradation of P54nrb and PSF. Nucleic Acids Res. 2015; 43:4569–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li S., Kuhne W.W., Kulharya A., Hudson F.Z., Ha K., Cao Z., Dynan W.S.. Involvement of p54(nrb), a PSF partner protein, in DNA double-strand break repair and radioresistance. Nucleic Acids Res. 2009; 37:6746–6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shen W., Sun H., De Hoyos C.L., Bailey J.K., Liang X.H., Crooke S.T.. Dynamic nucleoplasmic and nucleolar localization of mammalian RNase H1 in response to RNAP I transcriptional R-loops. Nucleic Acids Res. 2017; 45:10672–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fox A.H. P54nrb forms a heterodimer with PSP1 that localizes to paraspeckles in an RNA-dependent manner. Mol. Biol. Cell. 2005; 16:5304–5315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shav-Tal Y., Zipori D.. PSF and p54(nrb)/NonO–multi-functional nuclear proteins. FEBS Lett. 2002; 531:109–114. [DOI] [PubMed] [Google Scholar]
- 38. Shen W., Liang X.H., Crooke S.T.. Phosphorothioate oligonucleotides can displace NEAT1 RNA and form nuclear paraspeckle-like structures. Nucleic Acids Res. 2014; 42:8648–8662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yang Y.S., Hanke J.H., Carayannopoulos L., Craft C.M., Capra J.D., Tucker P.W.. NonO, a non-POU-domain-containing, octamer-binding protein, is the mammalian homolog of Drosophila nonAdiss. Mol. Cell. Biol. 1993; 13:5593–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hu S.-B., Xiang J.-F., Li X., Xu Y., Xue W., Huang M., Wong C.C., Sagum C.A., Bedford M.T., Yang L.et al.. Protein arginine methyltransferase CARM1 attenuates the paraspeckle-mediated nuclear retention of mRNAs containing IRAlus. Genes Dev. 2015; 29:630–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.