

Supplemental Digital Content is available in the text.
Keywords: actins, blood pressure, calcineurin, phosphorylation, vascular smooth muscle, vascular stiffness
Rationale:
BCL11B (B-cell leukemia 11b) is a transcription factor known as an essential regulator of T lymphocytes and neuronal development during embryogenesis. A genome-wide association study showed that a gene desert region downstream of BCL11B, known to function as a BCL11B enhancer, harbors single nucleotide polymorphisms associated with increased arterial stiffness. However, a role for BCL11B in the adult cardiovascular system is unknown.
Objective:
Based on these human findings, we sought to examine the relation between BCL11B and arterial function.
Methods and Results:
Here we report that BCL11B is expressed in the vascular smooth muscle where it regulates vascular stiffness. RNA sequencing of aortas from wild-type and Bcl11b null mice (BSMKO) identified the cGMP (cyclic guanosine monophosphate)-cGMP-dependent protein kinase G (PKG) as the most significant differentially regulated signaling pathway in BSMKO compared with wild-type mice. BSMKO aortas showed decreased levels of PKG1, increased levels of Ca++-calmodulin-dependent serine/threonine phosphatase calcineurin (PP2B) and decreased levels of their common phosphorylation target, phosphorylated vasodilator-stimulated phosphoprotein (pVASPS239), a regulator of cytoskelatal actin rearrangements. Decreased pVASPS239 in BSMKO aortas was associated with increased actin polymerization (filamentous/globular actin ratio). Functionally, aortic force, stress, wall tension, and stiffness, measured ex vivo in organ baths, were increased in BSMKO aortas, and BSMKO mice had increased pulse wave velocity, the in vivo index of arterial stiffness. Despite having no effect on blood pressure or microalbuminuria, increased arterial stiffness in BSMKO mice was associated with increased incidence of cerebral microbleeds compared with age-matched wild-type littermates.
Conclusions:
We have identified vascular smooth muscle BCL11B as a crucial regulator of aortic smooth muscle function and a potential therapeutic target for vascular stiffness.
Editorial, see p 769
Meet the First Author, see p 689
Cardiovascular disease (CVD) morbidity and mortality remain a dire public health burden, claiming more lives each year than all forms of cancer combined.1 Loss of compliance of large elastic arteries, also known as arterial stiffening, is a strong independent risk factor for CVD. Carotid-femoral pulse wave velocity (PWV), the gold standard clinical measure of aortic wall stiffness,2 predicts incident hypertension,3–5 heart failure, stroke and coronary artery disease2,6–9 and cognitive decline,10 independently of other CVD risk factors. Moreover, recent evidence suggests that increased pressure pulsatility due to large artery stiffness can lead to end-organ microcirculatory injury, potentially inciting structural and functional changes in organ systems, referred to as target-organ damage. Target-organ damage is highly associated with, and considered a precursor of, overt CVD.11 Therefore, understanding genetic determinants of arterial stiffness and molecular mechanisms thereof, may aid in the development of novel therapies to prevent target-organ damage and CVD.
A recent genome-wide association study of 9 discovery cohorts (20 634 participants; average age of cohort 34–75 years old) and 2 replication cohorts (N=5306) identified single nucleotide polymorphisms (SNPs) in a gene desert locus on chromosome 14 with highly significant association with PWV (P<5.6×10−11 for the top SNP, rs1381289C>T).12 Notably, the presence of each rs1381289 allele resulted in an estimated hazard ratio=1.05 for a first major coronary artery disease event and 1.10 for incident heart failure, suggesting that increased arterial stiffness associated with SNP variants in this locus may be causally linked to an increased risk of subsequently developing major CVD events.
The aortic stiffness locus spans 2 Mb between coding genes B-cell leukemia 11b (BCL11B) and vaccinia-related kinase 1 (VRK1). By performing a detailed analysis of this locus, we found an ≈1.9 kb sequence, located ≈850 kb downstream (3′) of BCL11B, known to function as an enhancer for BCL11B,13 but not VRK1. Enhancers are DNA regulatory elements that activate the expression of target genes independently of distance or orientation.14 In the present study, we sought to elucidate the mechanistic basis of the association of the chromosome 14 locus with aortic stiffness and a possible cause-effect relation between BCL11B and vascular function.
BCL11B, also known as CTIP2 (COUP-TF [chicken ovalbumin upstream promoter transcription factor] interacting protein-2), is a transcription factor15,16 best known for its critical role in T cells and innate lymphoid cells,17,18 as well as neuronal development during embryogenesis19,20; however, a role of BCL11B in the cardiovascular system has never been described. Here we report for the first time that BCL11B is expressed in the vascular smooth muscle (VSM) where it regulates vascular stiffness by increasing nonmuscle actin polymerization in VSM cells via the cyclic guanosine monophosphate (cGMP)/protein kinase G (PKG)/phosphorylated vasodilator-stimulated phosphoprotein (pVASPS239) signaling pathway. Notably, despite VSM Bcl11b deletion having profound effects on the aorta, contractile properties of resistance vessels, blood pressure, and microalbuminuria in tamoxifen-inducible VSM-specific Bcl11b null mice (BSMKO) remained comparable with wild-type (WT) mice. In contrast, we found that increased aortic stiffness in BSMKO was correlated with enhanced cerebral microbleeds compared with age-matched WT mice. Taken together, our study identifies VSM BCL11B as a novel and crucial regulator of VSM cytoskeletal assembly in large arteries affecting aortic wall stiffness and suggests that BCL11B or its downstream signaling targets are promising candidates for the translational development of therapies against arterial stiffness and related target organ microcirculatory damage.
Methods
Data Availability
A detailed description of materials and methods can be found in the Data Supplement. All data and supporting material are available upon request.
Results
BCL11B Is Expressed in VSM and Is Down-Regulated in Animal Models of Arterial Stiffness
A detailed analysis of the genetic locus on chromosome 14 with genome-wide association with elevated PWV12 revealed a highly conserved 550 bp sequence (>95% homology among species) within a BCL11B enhancer,13 in proximity of the highest significant SNP variant associated with increased PWV (rs1381289C>T), which has recently been shown to correlate with BCL11B expression.21 By using Basic Local Alignment Search Tool (BLAST), we identified this highly conserved 550 bp sequence in a locus named AI060616 (National Center for Biotechnology Information [NCBI] nomenclature) and amplified one specific polymerase chain reaction (PCR) band of expected 356 bp molecular size, confirmed by sequencing, in aortic extracts (Figure 1A).
Figure 1.
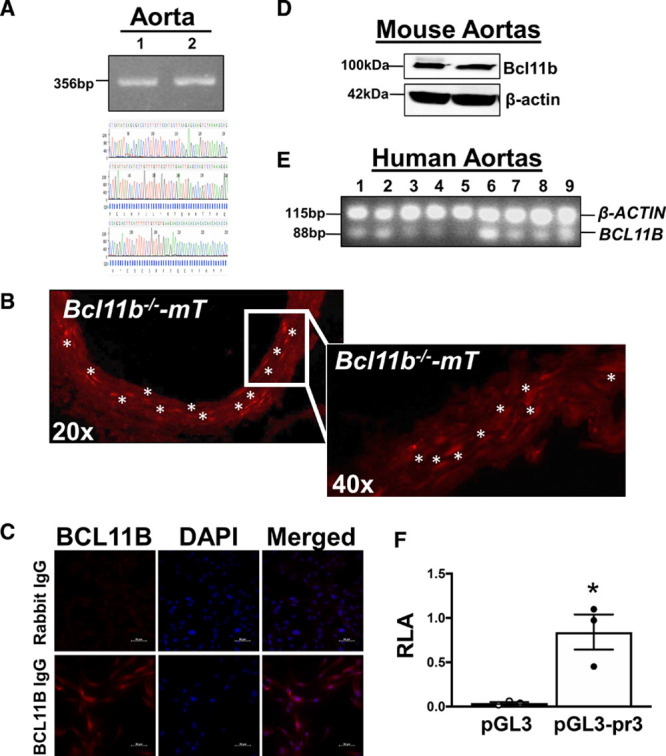
BCL11B (B-cell leukemia 11b) is expressed in vascular smooth muscle (VSM). A, A conserved region within the 3′-BCL11B genetic locus with SNPs highly associated with arterial stiffness is present in murine aortas and identified as AI060616 (National Center for Biotechnology Information [NCBI]). AI060616 356 bp nucleotide sequence was confirmed by sequencing polymerase chain reaction (PCR) products from aortic DNA obtained from 2 C57Bl/6J mice (1 and 2). Sequencing chromatogram tracing shown in lower panel. B, Representative fluorescent images of aortic sections, taken at 20× and 40× magnification, indicating Bcl11b’s localization in VSM. Red fluorescence indicates tomato (mT) expression in lieu of Bcl11b, upon tamoxifen-induced Bcl11b removal and mT induction in 3 ER-Cre-Bcl11bflox/flox-mTomato mice. Asterisks indicate clusters of VSM cells with high mT fluorescence intensity. C, Representative images (40× magnification) of human primary aortic smooth muscle cell immunostaining with anti-BCL11B; rabbit IgG serves as negative control for antibody specificity. DAPI indicates nuclei. n=3 replicates with 3 different cell lines, as described in materials and methods. D, Western blot on murine aortas confirmed Bcl11b protein expression, with a band of expected MW 100–120 kDa. β-actin serves as loading control. Each lane represents one mouse. E, Image of 1% agarose gel electrophoresis of qRT-PCR Taqman products indicates BCL11B mRNA expression in human aortas (n=9). Each lane represents one human subject. β-ACTIN used as endogenous housekeeping gene in multiplexed assay. F, Relative luciferase activity (RLA), expressed as a ratio with Renilla luciferase, in VSM cells transfected with luciferase reporter plasmids expressing an empty plasmid (pGL3) or Bcl11b promoter (pGL3-pr3). Results represent 3 replicate experiments. *P=2.0×10−2 by unpaired t test.
We next sought to determine whether BCL11B, a downstream target of this genomic locus, is present in the vasculature. By using double mutant mice expressing a red fluorescent protein (mTomato) upon removal of Bcl11b after tamoxifen administration (ER-Cre-Bcl11bflox/flox-mTomato mice), we were able to indirectly visualize Bcl11b’s localization in aortic sections and, specifically, in the tunica media (Figure 1B). These findings were confirmed by immunostaining human aortic smooth muscle cells with an antibody specific to BCL11B (Figure 1C), Western blotting of murine aortas (Figure 1D), and performing qRT-PCR of human aortas (Figure 1E). In addition to the aorta, Bcl11b was visualized in VSM of arteries and arterioles in heart, lung, and kidney of tamoxifen-treated ER-Cre-Bcl11bflox/flox-mTomato mice (Figure I in the Data Supplement). Last, a pGL3 luciferase reporter construct containing Bcl11b promoter region Chr12: 108004359-108003144 (GRCm38.p6), demonstrated that VSM cells are transcriptionally competent to sustain Bcl11b expression compared with an empty (control) plasmid (Figure 1F).
To determine whether Bcl11b expression in the vasculature was linked to arterial function, we measured Bcl11b in aortas of high-fat, high-sucrose (HFHS)-fed obese mice, a model of arterial stiffness we previously described.22,23 We found that Bcl11b mRNA and protein levels were significantly decreased in aortas of HFHS- compared with normal diet (ND)-fed mice (Figure 2A and 2B, quantitation values on graphs). To further elucidate a functional role of vascular Bcl11b in vivo, we measured PWV in mice with an inducible global Bcl11b knockout (ER-Cre-Bcl11bflox/flox treated with tamoxifen). We found that PWV was significantly increased in 10-month-old Bcl11b null mice compared with WT littermate controls (area under the curve: 260.3±4.7 m/s×mm Hg in WT versus 285.6±2.8 m/s×mm Hg in Bcl11b−/−, P=6.0×10−3; over a range of mean arterial pressures, Figure 2C). Notably, PWV in Bcl11b−/− mice increased to a similar extent as HFHS-fed obese mice (shadowed box in Figure 2C corresponding to PWV values in HFHS-fed mice, in a similar mean arterial pressure range, adapted from our previous publication22). Taken together, our novel findings demonstrate that BCL11B is present in the VSM of the aortic wall, and that aortic BCL11B downregulation may increase aortic stiffness.
Figure 2.
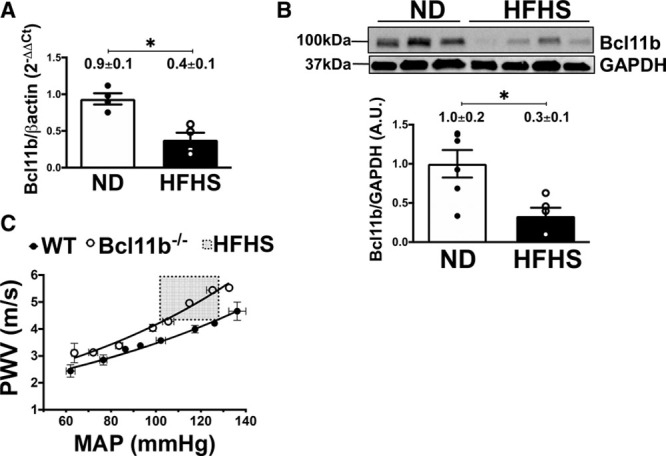
BCL11B (B-cell leukemia 11b) downregulation is associated with increased arterial stiffness. A, Bcl11b mRNA levels measured by quantitative real-time polymerase chain reaction (PCR) (2−ΔΔCt) are decreased in aortas of high fat, high-sucrose (HFHS)-fed obese mice, a model of arterial stiffness, compared with normal diet (ND)-fed mice. n=4 mice in each group; *P=3.0×10−2 by Mann-Whitney nonparametric test. B, Representative Western Blot of Bcl11b protein expression in aortic homogenates of ND- and HFHS-fed mice. Each lane represents one mouse. GAPDH serves as loading control. Bar graph summarizes protein band quantitation (ratio of Bcl11b over GAPDH band intensities), each dot represents one mouse. n=6 mice in each group; *P=1.0×10−2 by unpaired t test. C, Pulse wave velocity (PWV, m/s), the in vivo index of arterial stiffness, measured over a range of mean arterial pressures (MAP, mm Hg), is increased in 10-month old mice lacking Bcl11b compared with wild-type (WT) littermates. n=4 mice in each group; area under the curve (AUC): 260.3±4.7 m/s×mm Hg in WT vs 285.6±2.8 m/s×mm Hg in Bcl11b−/−; *P=6.0×10−3 by unpaired t test for AUC. Shadowed box indicates PWV values (4–6 m/s) for HFHS-fed mice in comparable ranges of MAP (100–130 mm Hg), adapted from reference.22
VSM BCL11B Regulates Aortic Tone and Stiffness
Based on our novel observation that BCL11B is expressed in VSM and its decreased aortic levels are associated with increased arterial stiffness, we generated mice with tamoxifen-inducible Bcl11b deletion in VSM (BSMKO; Figure II in the Data Supplement). Bcl11b deletion in VSM did not affect gross aortic morphology, as indicated by comparable aortic media thickness (54.5±1.0 μm in WT, n=5 versus 55.1±1.1 μm in BSMKO, n=7; P=6.9×10−1) and diameters (unloaded dimensions: 0.74±0.02 mm in WT, n=5 versus 0.75±0.01 mm in BSMKO, n=7, P=8.1×10−1; and Figure III in the Data Supplement for in vivo measurements) between WT and BSMKO mice. However, force, wall tension, and stress, generated by BSMKO aortic rings (n=7) in organ baths were significantly increased compared with WT (n=5; Figure 3A–3D). Likewise, aortic stiffness measured by PWV in vivo or derived from ex vivo elastic modulus with the Moens-Kortweg equation, was significantly increased in BSMKO mice (n=17) 2 months after VSM Bcl11b removal, compared with WT littermate controls (n=14; Figure 3E and 3F). Interestingly, aortic KCl- and phenylephrine-induced stress were not significantly affected by Bcl11b deletion (Figure 3G and 3H) indicating that Bcl11b regulates VSM force generation but not via KCl- and phenylephrine-stimulated pathways.
Figure 3.
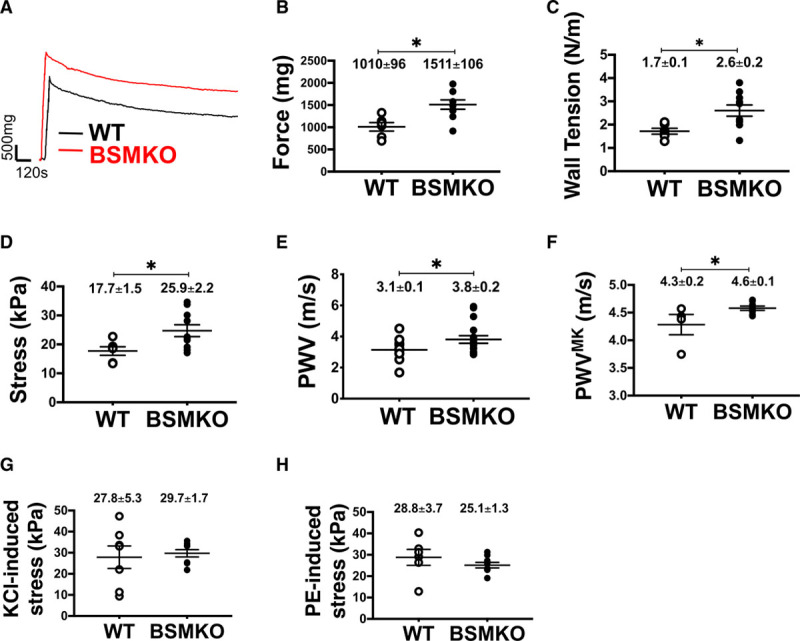
Vascular smooth muscle (VSM) bcl11b (B-cell leukemia 11b) deletion increases contractile force of aortic rings and arterial stiffness. A, Representative force tracings recorded ex vivo in organ baths in aortic rings from wild-type (WT; n=5) and tamoxifen-inducible VSM-specific Bcl11b null mice (BSMKO; n=7). Scale on graph. Scatter plots indicate individual values for (B) force (mg); *P=6.0×10−3; (C) wall tension (N/m), *P=2.0×10−2; (D) stress (kPa), *P=3.0×10−2; (F) stiffness, expressed as pulse wave velocity (PWV) calculated via the Moens-Kortweg equation (PWVMK), *P=4.0×10−2; (G) KCl-, P=7.5×10−1; and (H) phenylephrine (PE)-induced stress (kPa), P=2.9×10−1. Each dot represents an aortic ring from one mouse; mean±SEM on graphs. E, Pulse wave velocity (m/s) measured in vivo by Doppler echocardiography in WT (n=14) and BSMKO (n=17) mice. *P=4.0×10−2 by unpaired t test. Details in Methods.
Bcl11b deletion in VSM did not significantly affect blood pressure or heart rate, measured by radiotelemetry in 4- or 15-month old, conscious mice over 8 consecutive days, compared with age-matched WT littermates (Table I in the Data Supplement). Aging comparably increased systolic and mean arterial pressures in both groups (Table I in the Data Supplement).
pVASPs239 Is Down-Regulated in Aortas of BSMKO Mice
We used RNA sequencing to identify molecular mechanisms that may contribute to increased VSM tone and stiffness in BSMKO aortas. Four hundred fifty-eight and 329 genes were differentially up- and down-regulated, respectively, in aortic mRNA extracts of BSMKO (n=5) compared with WT (n=5) mice (Figure 4A and 4B and Figure IV in the Data Supplement, in which the heatmap illustrates individual levels of expression for replicate mice for each of the top 40 differentially expressed genes; P=2.7×10−7 to 3.4×10−4). Pathway enrichment analysis (Database for Annotation, Visualization and Integrated Discovery [DAVID]) on differentially expressed genes revealed that cGMP-PKG signaling pathway was the most significantly down-regulated pathway in BSMKO compared with WT aortas (FDR; q=9.4×10−3; Figure 4C and Figures V and VI in the Data Supplement for a list of differentially regulated pathways as well as individual levels of expression for each gene within the cGMP-PKG pathway). We further validated RNA sequencing findings by analyzing levels of GC (guanylate cyclase) and cGMP-dependent PKG1 (PKG, isoform 1), the enzymes directly upstream and downstream of cGMP. We found that GC catalytic subunit isoforms (α1, α2, β1) mRNA (Figure 4D) and PKG1 protein levels (Figure 4E, quantitation in graph) were significantly down-regulated in VSM cells isolated from BSMKO aortas compared with WT. Similarly, downregulating Bcl11b with a validated siRNA, decreased PKG1 expression in VSM cells (Figure 4F, quantitation in graph).
Figure 4.
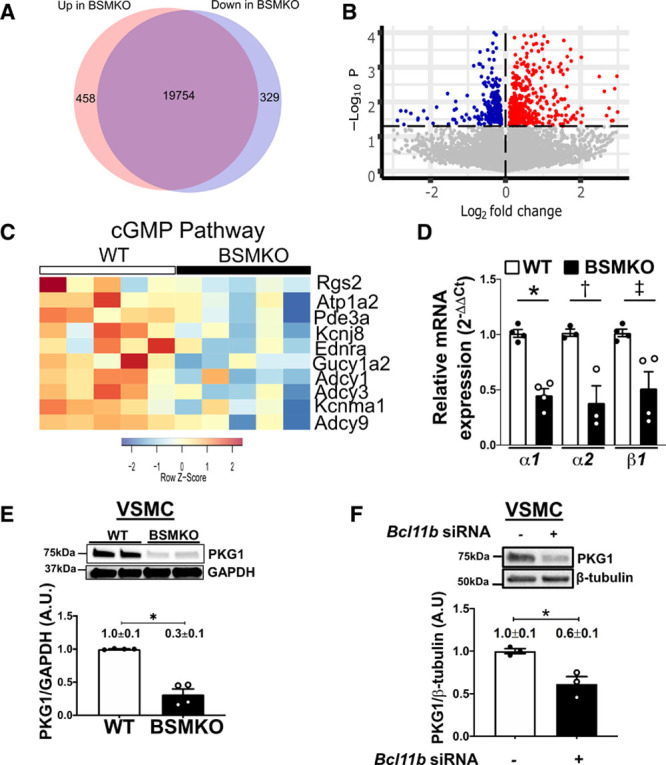
cGMP (cyclic guanosine monophosphate)-protein kinase G (PKG)-signaling pathway is down-regulated in aortas with vascular smooth muscle (VSM) bcl11b (B-cell leukemia 11b) deletion. A, Venn diagram of genes differentially up- and down-regulated in aortas of tamoxifen-inducible VSM-specific Bcl11b null mice (BSMKO; n=5) compared with wild-type (WT; n=5) mice assessed by RNA sequencing. B, Volcano plot of differentially up- (red) and down-(blue) regulated genes in aortas of BSMKO (n=5) compared with WT (n=5) mice. The horizontal line indicates a threshold of P=0.05. For a list of the top 40 differentially regulated genes see Figure IV in the Data Supplement. C, List of genes within the cGMP-PKG signaling pathway, the most significantly regulated signaling pathway in BSMKO aortas after Database for Annotation, Visualization and Integrated Discovery (DAVID) network analysis of differentially expressed genes (FDR, q=0.0094). For quantitation of individual genes see Figure VI in the Data Supplement. D, Quantitative RT-polymerase chain reaction (PCR) for guanylyl cyclase isoforms Gucy1a1, Gucy1a2, and Gucy1b1 in mRNA extracts from WT and BSMKO VSM cells; n=4 replicate experiments. Data expressed as fold change vs WT. *P=2.8×10−2; †P=1.6×10−2; ‡P=2.8×10−2 by Mann-Whitney nonparametric test. E, Representative Western Blot for PKG1 (PKG, isoform 1) protein levels in VSM cells from WT and BSMKO mice. GAPDH used as loading control. Quantitation of 4 replicate experiments in graph. *P=3.0×10−2 by Mann-Whitney nonparametric test. F, Representative Western Blot for PKG1 in mouse VSM cells treated with a scrambled siRNA (control) or a validated Bcl11b siRNA. Quantitation of 3 replicate experiments on 3 different mouse VSM cell lines in graph. β-tubulin used as loading control. *P=4.0×10−2 by unpaired t test.
As VASP (vasodilator-stimulated phosphoprotein) is a PKG1 phosphorylation target in VSM, we examined whether VASP phosphorylation was affected by Bcl11b deletion in VSM. Specifically, we analyzed VASP phosphorylation at serine 239, since we previously showed that VASP phosphorylation at this residue inversely correlates with arterial stiffness.23 Levels of pVASPS239 were dramatically decreased in BSMKO VSM cells compared with WT cells, cultured with or without fetal bovine serum (FBS) (Figure 5A; quantitation in graph), and in BSMKO aortas compared with WT controls (Figure 5B; quantitation in graph), while total VASP remained unchanged. Similar findings were obtained in aortas of HFHS-fed obese mice (ie, with decreased aortic Bcl11b, Figure 2B), compared with ND-fed mice (Figure 5C; quantitation in graph). Our findings of decreased pVASPS239 were corroborated in males (n=4) and females (n=4) of a second animal model in which constitutive Bcl11b removal in VSM was achieved with a Sm22α (transgelin) promoter-driven Cre recombinase transgene (Figure VIIA in the Data Supplement). Last, no statistically significant differences in pVASPS239 expression were observed in tamoxifen- (n=3) compared with vehicle (oil)-treated Bcl11bfl/fl mice (n=3), excluding the possibility that tamoxifen administration per se may have decreased pVASPS239 levels in the aorta (Figure VIIB in the Data Supplement).
Figure 5.
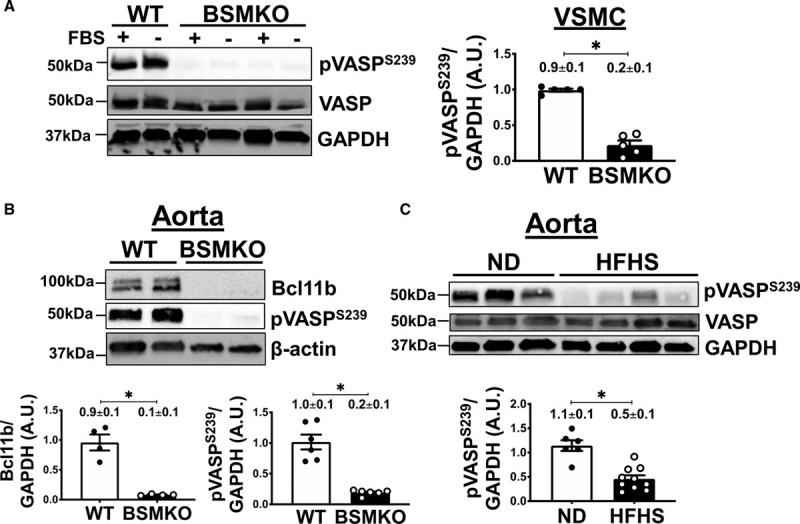
Vascular smooth muscle (VSM) bcl11b (B-cell leukemia 11b) deletion is associated with pVASPS239 (phosphorylated vasodilator-stimulated phosphoprotein) downregulation in aortas. A, VASP (vasodilator-stimulated phosphoprotein) phosphorylation at serine 239 (pVASPS239) was significantly decreased in tamoxifen-inducible VSM-specific Bcl11b null mice (BSMKO) VSM cells, cultured with or without fetal bovine serum (FBS), compared with wild-type (WT) cells. Total VASP remained unchanged. GAPDH serves as loading control. n=5 replicate experiments. *P=8.0×10−3 by Mann-Whitney nonparametric test. B, Representative Western Blot images demonstrating pVASPS239 in aortas of BSMKO mice (n=6) compared with WT littermate controls (n=6). β-actin serves as loading control. Each lane represents one mouse. *P=5.3×10−5 by unpaired t test. C, pVASPS239 was significantly decreased in aortas of high fat, high sucrose (HFHS)-fed mice (n=10) compared with ND-fed controls (n=6; same aortic samples as in Figure 2B). Total VASP was similar in the two groups. Each lane represents one mouse. Band intensity quantitation summarized in graphs. *P=9.6×10−5 by unpaired t test.
In contrast to the aorta, pVASPS239 levels in mesenteric arteries isolated from the mesenteric plexus (Figure VIIIA in the Data Supplement) were similar between WT (n=6) and BSMKO (n=6) mice (Figure VIIIB in the Data Supplement). Moreover, no statistically significant differences were observed in the contractile responses to phenylephrine or vasodilation responses to papaverine of mesenteric arteries from WT (n=8) and BSMKO (n=8; Figure VIIIC and VIIID in the Data Supplement), consistent with comparable blood pressures between WT and BSMKO mice (Table I in the Data Supplement). Taken together, these data suggest that the BCL11B—pVASPS239 axis is important for the regulation of large artery stiffness but is dispensable for vasoconstriction of resistance vessels.
In addition to protein kinases PKG and PKA (cAMP-dependent protein kinase),24 VASP phosphorylation in VSM cells is finely regulated by protein phosphatases (PP1, PP2A, PP2B [Ca2+/calmodulin-dependent serine-threonine phosphatase calcineurin], and PP2C)25 and Rho kinases (ROCK1 and ROCK2). Of interest, PP2B has been shown to directly interact with BCL11B to regulate gene expression in T cells.26 Therefore, we examined whether PP2B may also contribute to decreased pVASPS239 levels in BSMKO VSM cells. We found that protein levels of PP2B and NFAT2, a major PP2B phosphatase target, were significantly upregulated in BSMKO VSM cells compared with WT (Figure 6A). On the contrary, expression of ROCK1, a Rho-dependent kinase involved in cytoskeletal rearrangements and an upstream VASP regulator,27 was not significantly affected by Bcl11b deletion (Figure VIIC in the Data Supplement). Furthermore, overnight treatment with 2 PP2B inhibitors, cyclosporine A (1–10 μmol/L) or the more specific PP2B inhibitor CAIP (calcineurin autoinhibitory peptide, 10–100 μmol/L), restored pVASPS239 towards control levels in BSMKO VSM cells in a dose-dependent manner (Figure 6B and 6C; quantitation in graph), indicating that decreased pVASPS239 in BSMKO VSM is dependent, at least in part, on increased PP2B activity. Last, overexpressing Bcl11b in aortic media rings by transient transfection for 3 days was sufficient to significantly restore PKG, pVASPS239 and decrease PP2B to control levels (Figure 6D).
Figure 6.
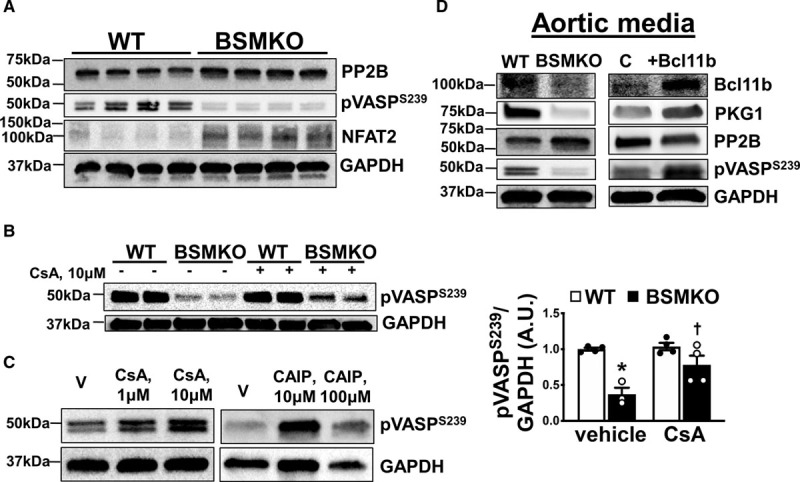
Calcineurin (PP2B) regulates VASPS239 phosphorylation after vascular smooth muscle (VSM) bcl11b (B-cell leukemia 11b) deletion. A, Representative Western blot images demonstrating increased PP2B expression, increased NFAT2 and impaired VASP (vasodilator-stimulated phosphoprotein) phosphorylation at serine 239 (pVASPS239) in tamoxifen-inducible VSM-specific Bcl11b null mice (BSMKO) VSM cells compared with wild-type (WT) controls. GAPDH serves as loading control. Each lane represents a cell preparation from one mouse for a total of 4 replicates. B, Treatment of BSMKO VSM cells with 1 or 10 μmol/L cyclosporine A (CsA), a calcineurin inhibitor, or (C) a specific calcineurin autoinhibitory peptide (CAIP; 10 or 100 μmol/L) reversed pVASPS239 in BSMO cells towards WT levels, in a dose-dependent manner. V, vehicle control. Quantitation of band intensities in graph (n=3 replicate experiments). *P=1.5×10−2 vs WT/vehicle; †P=3.0×10−2 vs BSMKO/vehicle by 1-way ANOVA with Tukey multiple comparisons test. D, Western blots for Bcl11b, PKG1 (PKG, isoform 1), PP2B and pVASPS239 in WT and BSMKO aortic media (adventitia removed) without (left) or with (right) transient transfection with vehicle (lipofectamine C, control) or 20 μg Bcl11b plasmid. GAPDH serves as loading control.
Actin Polymerization Is Dependent on VASP Phosphorylation in VSM Bcl11b Deleted Aortas
VASP is an important regulator of nonmuscle actin polymerization-dependent VSM tone28 whereas phosphorylation of VASP at serine 239 (pVASPS239) inhibits actin polymerization in VSM cells,29 thereby regulating cytoskeletal actin assembly. We found increased filamentous to globular actin ratio, indicative of increased actin polymerization, in BSMKO aortas compared with WT (Figure 7A). Moreover, α-actinin, a major VASP-interacting protein during actin polymerization, was significantly upregulated in BSMKO (n=4) compared with WT (n=4) aortas (Figure 7B, quantitation in graph). Notably, preincubation with the specific PP2B inhibitor CAIP (10 μmol/L, 1 hour, 37 °C), decreased both filamentous/globular actin ratio (Figure 7C) and aortic stiffness (from 245.7±4.3 kPa in BSMKO, n=4 to 184.0±17.2 kPa in BSMKO/CAIP, n=4; P=3.0×10−2; Figure 7D) in BSMKO aortic rings. Taken together, our data indicate that lack of VSM BCL11B increases aortic stiffness, at least in part, because of decreased pVASPS239 levels and associated increased actin polymerization.
Figure 7.
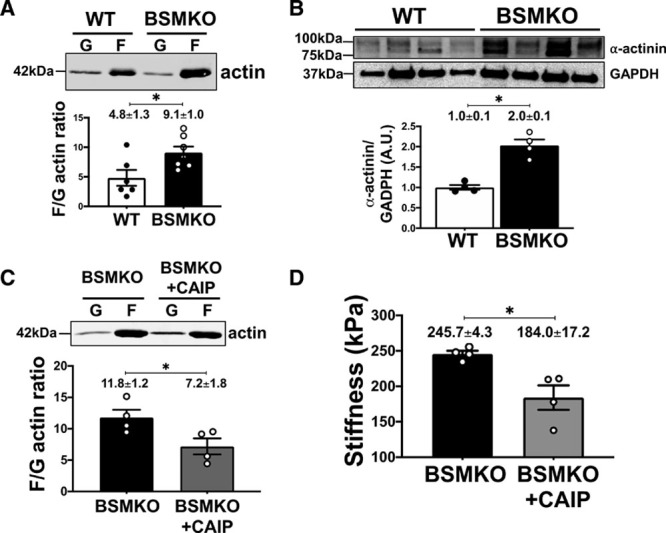
Vascular smooth muscle (VSM) bcl11b (B-cell leukemia 11b) regulates cytoskeletal actin polymerization and pVASPS239 (phosphorylated vasodilator-stimulated phosphoprotein). A, Representative Western blot images of filamentous (F) and globular (G) actin in wild-type (WT; n=6) and tamoxifen-inducible VSM-specific Bcl11b null mice (BSMKO; n=7) aortas. (F/G) actin ratio quantitation in graph. *P=2.0×10−2 by unpaired t test. B, Representative Western blot images of α-actinin in WT (n=4) and BSMKO (n=4) aortas. Each lane represents one mouse. GAPDH used as loading control. Quantitation in graph. *P=3.0×10−2 by Mann-Whitney nonparametric test. C, Representative Western blot images of F and G actin in BSMKO aortas treated with vehicle (n=4) or CAIP (calcineurin autoinhibitory peptide; 10 μmol/L; n=4). F/G actin ratio quantitation in graph. *P=5.0×10−2 by Mann-Whitney nonparametric test. D, Treatment of BSMKO aortas with CAIP (10 μmol/L) decreased stiffness, measured ex vivo on aortic rings in organ bath (n=4). *P=3.0×10−2 by Mann-Whitney nonparametric test.
VSM Bcl11b Deletion Increased the Incidence of Cerebral Microbleeds
Emerging evidence strongly correlates measures of arterial stiffness (PWV and increased pulse pressure) with microcirculatory end-organ damage, including kidney disease30,31 and cognitive impairment.32–34 We sought to determine whether increased aortic stiffness in BSMKO mice is associated with increased indexes of end-organ microcirculatory injury (microalbuminuria, cerebral microbleeds and retinal vessel density). We found that the urinary albumin to creatinine ratio was not significantly affected by VSM Bcl11b deletion compared with WT littermates (23.2±5.6 μg/mg in WT, n=12 and 12.0±5.3 μg/mg in BSMKO, n=8; P=1.9×10−1; Figure IXA in the Data Supplement). In contrast, a significantly higher number of cerebral microbleeds was identified by magnetic resonance imaging mainly in the thalamus of BSMKO compared with WT mice (1.0±0.4 in WT, n=5 versus 6.5±1.7 in BSMKO, n=6; P=1.0×10−2; Figure 8A). Histological staining of brain sections with Prussian blue confirmed an increased number of cerebral microbleeds in BSMKO compared with WT mice (5.7±1.0×103 μm2 in WT, n=12 versus 11.2±2.2×103 μm2 in BSMKO, n=12; P=3.0×10−2; Figure 8B). Interestingly, cerebral microbleeds in BSMKO mice were comparable to HFHS-fed mice, in which we similarly observed significant increases in cerebral microbleeds compared with ND-fed control mice (7.0±1.4×103 μm2 in ND, n=6 versus 12.6±2.2×103 μm2 in HFHS, n=6; P=5.0×10−2; Figure 8C). No statistically significant differences were observed between aged WT and BSMKO (24-months old; 13.8±4.2×103 μm2 in WT, n=4 versus 12.0±8.0×103 μm2 in BSMKO, n=4; P=3.9×10−1), indicating that VSM Bcl11b deletion accelerated the development of aging-associated cerebral microbleeds, which then plateaued to levels comparable to aged WT. Independently of the presence of cerebral microbleeds, no statistically significant difference in cognitive function was detected between 5-month or 24-month old WT and BSMKO mice as assessed by a novel object recognition test (Figure IXB in the Data Supplement).
Figure 8.
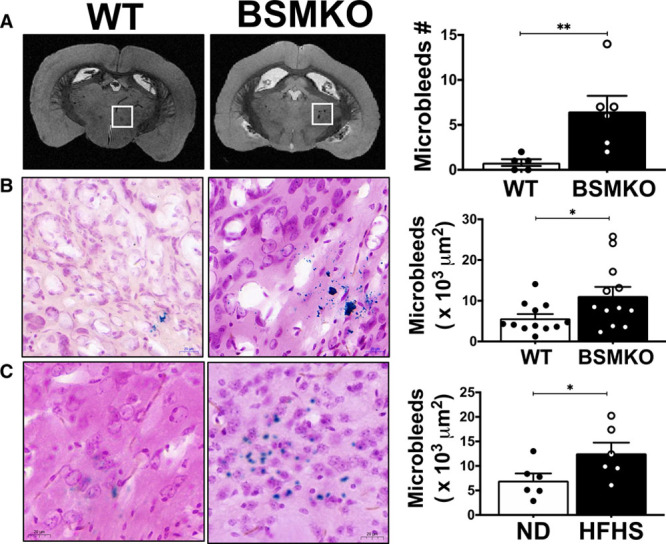
Cerebral microbleeds in wild-type (WT) vs tamoxifen-inducible VSM-specific Bcl11b null mice (BSMKO). A, Representative magnetic resonance imaging (MRI) of whole brains from WT (n=5) and BSMKO mice (n=6). A microbleed-rich region is highlighted with the white box. Quantitation in graph. **P=1.0×10−2 by unpaired t test. B, Representative histological staining of WT (n=12) and BSMKO (n=12) brain sections (40× magnification); areas stained in blue are indicative of cerebral microbleeds. Quantitation in graph (µm2). Each dot represents the average of at least 5 sections, corresponding to a cumulative thickness of 150 µm, for each mouse. *P=3.0×10−2 by unpaired t test. C, Representative histological staining of cerebral microbleeds in normal diet (ND)- (n=6) and high fat, high sucrose (HFHS)-fed (n=6) mice. Quantitation in graph. *P=5.0×10−2 by unpaired t test.
Consistent with cerebral microvascular damage, total and average vessel length and branching, measured in isolectin B4-stained retinal flat-mounts, trended to increase in BSMKO compared with WT as well as in HFHS-fed compared with ND-fed mice (Figure IXC in the Data Supplement), suggesting a stimulation of neovessel growth in a damaged retinal microvasculature.
Discussion
Arterial stiffness, or loss of elastic compliance of large arteries, is a strong, independent risk factor for CVD.2,6 Elevated PWV, the gold standard measure of aortic wall stiffness, strongly associates with adverse cardiovascular outcomes.6–8 Moreover, mounting evidence correlates measures of arterial stiffness and pressure pulsatility (PWV, pulse pressure) to kidney disease30,31 and cognitive impairment/dementia.32,34 However, genetic and molecular cues of aortic wall stiffening are not fully understood hampering the discovery of therapeutic targets that can slow or reverse arterial stiffening thereby decreasing target organ damage and the risk of developing overt CVD.
Arterial stiffness trait loci are moderately heritable,35 but little is known about genetic determinants of arterial stiffness. A recent genome-wide association study demonstrated that SNPs in the vicinity of the BCL11B genetic locus are associated with increased arterial stiffness and subsequent risk of developing CVD.12 Interestingly, the most significant SNP variant (rs1381289 C>T) in this aortic stiffness locus falls in a highly conserved sequence within a BCL11B enhancer. We postulated that SNP variants in the 3′-BCL11B locus may alter the BCL11B gene enhancer function and may play a causal role in the pathogenesis of arterial stiffness by regulating BCL11B expression. A recent report showed that rs1381289 C>T genotype in the 3′-BCL11B locus inversely correlates with BCL11B mRNA expression in aortic rings from transplant donors,21 despite the authors not detecting any BCL11B protein expression in the same aortic rings. These observations prompted us to further examine whether BCL11B is present in the vasculature and whether there is a cause-effect relation between BCL11B and vascular function.
Our novel findings demonstrate that BCL11B, previously known solely for its role in T lymphocyte36 and neuronal19 lineage commitment, is expressed in the VSM of the aortic wall. Importantly, here we report for the first time that VSM BCL11B is a crucial regulator of VSM structural components and aortic stiffness, as corroborated by the following findings: (1) in vivo Bcl11b deletion in VSM (BSMKO mice) resulted in increased nonmuscle actin polymerization (filamentous/globular actin ratio); (2) mice lacking Bcl11b globally or specifically in VSM (BSMKO) have increased PWV, the in vivo index of arterial stiffness, compared with WT littermates; (3) aortic rings from BSMKO mice have increased force, stress, wall tension, and stiffness compared with WT; and (4) Bcl11b is down-regulated in aortas of high fat, high sucrose-fed obese mice, a mouse model of arterial stiffness that we previously described.22 Although a major BCL11B interacting protein, COUP-TFII, has been previously shown to partake in atria and blood vessel development during embryogenesis,37 to the best of our knowledge, this is the first report of a functional role of BCL11B in the adult cardiovascular system.
Furthermore, we uncovered a pivotal role of VSM BCL11B in the regulation of VSM cytoskeletal filaments, which form a coordinated system to efficiently transduce contractile forces to the extracellular matrix and among adjacent VSM cells, thereby sustaining aortic wall mechanics and compliance. In response to pressure or mechanical stretch, thin filament dynamic assembly, namely nonmuscle actin polymerization, become a major determinant of VSM contraction and basal tone,38,39 independently of myosin light chain 2 phosphorylation and actino-myosin cross-bridges cycles.40,41 Cytoskeletal actin polymerization can sustain VSM force generation and cytoskeletal stiffness to maintain vessel diameter in response to wall tension or stretch,42 as it may occur in the aorta exposed to cyclic strain induced by cardiac contraction, particularly in the proximal regions. Therefore, our finding of increased filamentous/globular actin ratio, indicative of increased actin polymerization, is consistent with increased force and wall tension in BSMKO aortas and increased PWV in BSMKO mice compared with WT controls.
At the molecular level, we found that the cGMP/PKG/pVASPS239 signaling pathway was dramatically decreased in BSMKO aortas and VSM cells, while total VASP remained unchanged. VASP has emerged as an important mediator of actin polymerization-dependent VSM force generation. Specifically, VASP interacts with α-actinin, vinculin, zyxin, and other components of thin filament assembly at focal adhesion and dense bodies, which are important sites of VSM contractile filament attachment, cell-cell interactions and cell adhesion to the extracellular matrix, thereby contributing to VSM tone and stiffness independently of myosin light chain 2 phosphorylation.28,43–45 The drug cytochalasin D, commonly used to block actin polymerization in a variety of cell types, is known to interfere with VASP localization to nascent F-actin filaments46 underscoring the pivotal role of VASP in cytoskeletal actin rearrangement. Moreover, VASP overexpression has been shown to induce F-actin assembly47; in contrast, VASP phosphorylation at serine 239 is sufficient to inhibit actin filament polymerization.29 Interestingly, we found that partially restoring decreased pVASPS239 with a calcineurin inhibitor was able to reverse the increased actin polymerization and aortic stiffness in BSMKO aortic rings towards normal levels. Overall, our results suggest that cyclosporinA or specific PP2B inhibitors as well as compounds that inhibit actin filament assembly may become potential pharmacological candidates for arterial stiffness.
Interestingly, despite having profound effects on the aorta, lack of VSM Bcl11b did not affect pVASPS239 levels and contractile properties of resistance arteries, nor blood pressure in BSMKO mice suggesting a dispensable role of BCL11B in the regulation of VSM tone in resistance vessels. This intriguing finding on a differential role of BCL11B in VSM of large versus small arteries could be explained by the fact that smooth muscle along the adult vascular tree is not homogeneous but rather a mosaic of phenotypically and functionally distinct smooth muscle cell types.48 Elegant lineage mapping studies have demonstrated that smooth muscle of proximal regions of the aorta, which are the most susceptible to vascular stiffening, namely the outflow tract, innominate, carotid and subclavian arteries, but not the smooth muscle of resistance vessels, developmentally originates from the cranial neural crest.49 Considering that BCL11B is highly expressed and required for neuronal development during embryogenesis,19,20 BCL11B may be a regulator of VSM-specific gene programs of neural crest-derived proximal, but not distal, vascular regions. Further studies are warranted to fully elucidate the role of BCL11B in the development of the cardiovascular system during embryogenesis.
Arterial stiffness markedly increases the risk of adverse cardiovascular outcomes6,7,50 including heart failure,51,52 coronary artery disease,9 and hypertension.3,4 In addition, emerging evidence correlates measures of arterial stiffness (PWV and pulse pressure) with cognitive decline32,34 and kidney disease.30,31 Elastic compliance of large arteries is paramount to dampen the pulsatility of cardiac contraction and to ensure a steady blood supply to peripheral organs while limiting the pulsatile energy that penetrates into the microcirculation. However, as the aortic wall stiffens with age and obesity,53 this buffering effect is lost, thereby increasing the amount of pressure and flow pulsatility transmitted to the downstream microcirculation, where it triggers microvascular remodeling, impaired reactivity, and abnormal flow autoregulation.54,55 Of clinical importance, this microcirculatory injury may lead to structural and functional changes generally referred to as target organ damage, particularly in high flow/low resistance organs, such as brain and kidneys. An interesting finding of our study is that mice lacking Bcl11b in the VSM, with elevated arterial stiffness, have a significant increase in cerebral microbleeds compared with age-matched WT controls. Thalamus and hippocampus were the brain regions mostly affected - a finding consistent with epidemiological studies reporting that white matter hyperintensities and consequent cognitive decline and dementia associated with arterial stiffness are generally observed in deep central regions of the brain, which are particularly sensitive to pulsatile stress transmitted via branches of the carotid arteries penetrating the deep parenchyma.56,57 Although further studies are warranted to assess the effect of VSM Bcl11b deletion on the cerebral microvasculature, lack of Bcl11b in VSM did not affect blood pressure, or microalbuminuria in BSMKO mice compared with age-matched WT controls, consistent with the cerebral microcirculation being particularly sensitive to excessive pressure pulsatility triggered by arterial stiffness in the absence of VSM Bcl11b compared with renal or other resistive vessel beds.
In conclusion, our study has uncovered a novel and crucial role for VSM BCL11B in aortic structural and functional integrity and strongly supports BCL11B or its downstream regulated pathways as potential therapeutic targets against arterial stiffness (illustrated in the graphic summary). Although, the cause-effect relationship as well as the temporal progression from arterial stiffness to target organ damage, including hypertension, remains to be fully unraveled, as we previously pointed out,58 targeting arterial stiffness could represent a novel clinical approach to prevent the progression into target organ damage leading to cardiovascular complications and cognitive decline. Further studies in human populations from different ethnic groups are warranted to establish whether the genotype at the 3′-BCL11B locus may be used as diagnostic biomarker to identify individuals at increased risk of developing arterial stiffness and other vascular diseases.
Author Contributions
J.A.C. Valisno, J. May, L. Venegas, E. Budbazar, J.B. Goodman, C.J. Nicholson, K. Singh performed experiments, contributed to study design, data interpretation and reviewed the article; E.Y. Helm analyzed RNA sequencing data sets; DA provided Bcl11bflox/flox mouse strain used in the study; R.A. Cohen, D. Avram, G.F. Mitchell, K.G. Morgan provided critical comments to the study and the article; F. Seta contributed to the study design, coordinated the study, designed and performed experiments, analyzed and interpreted the data and wrote the article.
Acknowledgments
We would like to thank the Boston University Medical Campus Analytical Instrumentation, Cellular Imaging and Magnetic Resonance Imaging (MRI) Cores for the expert technical support.
Sources of Funding
This work has been possible thanks to the financial support of National Institutes of Health grants HL136311 to F. Seta; HL105287 to R.A. Cohen; AG053274, AG050599, and HL136224 to K.G. Morgan; AI067846 to D. Avram; National Heart, Lung, and Blood Institute’s Framingham Heart Study Contracts No. N01-HC-25195 and HHSN268201500001I and grants HL070100, HL080124, HL107385, and HL126136 to G.F. Mitchell; NIH T32 training grant HL007224 to J. May and J.B. Goodman; pilot grants 1UL1TR001430 and 5P30DK046200 to F. Seta and by the Evans Center for Interdisciplinary Biomedical Research Arterial Stiffness ARC at Boston University (http://www.bumc.bu.edu/evanscenteribr/).
Disclosures
G.F. Mitchell is the owner of Cardiovascular Engineering Inc, a manufacturer of biomedical devices for arterial stiffness measurements.
Supplemental Materials
Expanded Materials & Methods
Major Resources Table
Supplemental Figures I–XI
Online Table I
References 59–71
Supplementary Material
Nonstandard Abbreviations and Acronyms
- BCL11B
- B-cell leukemia 11b
- BSMKO
- tamoxifen-inducible VSM-specific Bcl11b null mice
- CAIP
- calcineurin autoinhibitory peptide
- CTIP2
- COUP-TF interacting protein-2
- CVD
- cardiovascular disease
- GC
- guanylate cyclase
- HFHS
- high-fat, high-sucrose
- ND
- normal diet
- PKA
- cAMP-dependent protein kinase
- PKG1
- PKG, isoform 1
- PP2B
- Ca2+/calmodulin-dependent serine-threonine phosphatase calcineurin
- pVASP
- phosphorylated vasodilator-stimulated phosphoprotein
- PWV
- pulse wave velocity
- SNPs
- single nucleotide polymorphisms
- VASP
- vasodilator-stimulated phosphoprotein
- VRK1
- vaccinia-related kinase 1
- VSM
- vascular smooth muscle
- WT
- wild-type
The Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.120.316666.
For Sources of Funding and Disclosures, see page 767.
Contributor Information
Jeff Arni C. Valisno, Email: jvalisno@bu.edu.
Joel May, Email: joel.b.may@gmail.com.
Kuldeep Singh, Email: kuldeepmsb@gmail.com.
Eric Y. Helm, Email: Eric.Helm@medicine.ufl.edu.
Lisia Venegas, Email: lisven82@gmail.com.
Enkhjargal Budbazar, Email: budbazar@bu.edu.
Jena B. Goodman, Email: goodmanj@bu.edu.
Christopher J. Nicholson, Email: CJNICHOLSON@mgh.harvard.edu.
Dorina Avram, Email: dorina.avram@moffitt.org.
Richard A. Cohen, Email: racohen@bu.edu.
Gary F. Mitchell, Email: garyfmitchell@gmail.com.
Kathleen G. Morgan, Email: kmorgan@bu.edu.
Novelty and Significance
What Is Known?
Arterial stiffness is a strong, independent risk factor for cardiovascular disease; however, genetic and molecular determinants of arterial stiffness are not fully understood, hampering the discovery of potential therapeutic targets to prevent cardiovascular disease.
Single nucleotide polymorphisms in a genetic locus on chromosome 14, upstream of the gene BCL11B (B-cell leukemia 11b), have been shown to be significantly associated with increased pulse wave velocity, the gold standard measure of aortic wall stiffness (P<5.6×10−11 for the top single nucleotide polymorphism, rs1381289C>T).
What New Information Does This Article Contribute?
We identified BCL11B in the vascular smooth muscle (VSM) as a novel and crucial regulator of vascular stiffness.
VSM BCL11B contributes to maintaining aortic compliance via the cGMP (cyclic guanosine monophosphate)/protein kinase G (PKG)/pVASPS239 (phosphorylated vasodilator-stimulated phosphoprotein) signaling pathway, whose impairment, in absence of Bcl11b, leads to increased nonmuscle actin polymerization in VSM cells and increased VSM stiffness.
Increased aortic stiffness is associated with increased cerebral microbleeds, underscoring the pivotal role of large elastic arteries in preventing microvascular damage and associated target organ damage.
Single nucleotide polymorphisms in a genetic locus on chromosome 14, known to function as an enhancer for the transcription factor BCL11B, are significantly associated with increased pulse wave velocity, the gold standard measure of aortic wall stiffness. Here we report, for the first time, that BCL11B, previously known uniquely for its role in T cell and neuronal lineage commitment, is present in VSM where it regulates vascular stiffness. Specifically, BCL11B modulates VSM cytoskeletal actin polymerization via the cGMP/PKG/pVASPS239 signaling pathway thereby regulating aortic stiffness. Notably, increased arterial stiffness in mice lacking VSM Bcl11b as well as in obese mice, a mouse model of arterial stiffness we previously described, is associated with increased incidence of cerebral microbleeds, suggesting a crucial role of large elastic arteries in preventing microvascular damage in downstream organs. Interestingly, despite profound effects on the aorta, BCL11B has a dispensable role in VSM contraction in resistance arteries. The present study strongly supports BCL11B or its downstream-regulated pathways as potential therapeutic targets against arterial stiffness and related target organ damage, to prevent overt cardiovascular disease, which remains a major cause of morbidity and mortality worldwide.
References
- 1.Virani SS, Alonso A, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Delling FN, et al. Heart disease and stroke statistics—2020 update: a report from the American Heart Association. Circulation. 2020;141:e139–e596 [DOI] [PubMed] [Google Scholar]
- 2.Willum-Hansen T, Staessen JA, Torp-Pedersen C, Rasmussen S, Thijs L, Ibsen H, Jeppesen J. Prognostic value of aortic pulse wave velocity as index of arterial stiffness in the general population. Circulation. 2006;113:664–670. doi: 10.1161/CIRCULATIONAHA.105.579342 [DOI] [PubMed] [Google Scholar]
- 3.Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. JAMA. 2012;308:875–881. doi: 10.1001/2012.jama.10503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Najjar SS, Scuteri A, Shetty V, Wright JG, Muller DC, Fleg JL, Spurgeon HP, Ferrucci L, Lakatta EG. Pulse wave velocity is an independent predictor of the longitudinal increase in systolic blood pressure and of incident hypertension in the Baltimore Longitudinal Study of Aging. J Am Coll Cardiol. 2008;51:1377–1383. doi: 10.1016/j.jacc.2007.10.065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.AlGhatrif M, Strait JB, Morrell CH, Canepa M, Wright J, Elango P, Scuteri A, Najjar SS, Ferrucci L, Lakatta EG. Longitudinal trajectories of arterial stiffness and the role of blood pressure: the Baltimore Longitudinal Study of Aging. Hypertension. 2013;62:934–941. doi: 10.1161/HYPERTENSIONAHA.113.01445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitchell GF, Hwang SJ, Vasan RS, Larson MG, Pencina MJ, Hamburg NM, Vita JA, Levy D, Benjamin EJ. Arterial stiffness and cardiovascular events: the Framingham Heart Study. Circulation. 2010;121:505–511. doi: 10.1161/CIRCULATIONAHA.109.886655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sutton-Tyrrell K, Najjar SS, Boudreau RM, Venkitachalam L, Kupelian V, Simonsick EM, Havlik R, Lakatta EG, Spurgeon H, Kritchevsky S, et al. ; Health ABC Study. Elevated aortic pulse wave velocity, a marker of arterial stiffness, predicts cardiovascular events in well-functioning older adults. Circulation. 2005;111:3384–3390. doi: 10.1161/CIRCULATIONAHA.104.483628 [DOI] [PubMed] [Google Scholar]
- 8.Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55:1318–1327. doi: 10.1016/j.jacc.2009.10.061 [DOI] [PubMed] [Google Scholar]
- 9.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, et al. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation. 2006;113:657–663. doi: 10.1161/CIRCULATIONAHA.105.555235 [DOI] [PubMed] [Google Scholar]
- 10.Ding J, Mitchell GF, Bots ML, Sigurdsson S, Harris TB, Garcia M, Eiriksdottir G, van Buchem MA, Gudnason V, Launer LJ. Carotid arterial stiffness and risk of incident cerebral microbleeds in older people: the Age, Gene/Environment Susceptibility (AGES)-Reykjavik study. Arterioscler Thromb Vasc Biol. 2015;35:1889–1895. doi: 10.1161/ATVBAHA.115.305451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, Clement DL, Coca A, de Simone G, Dominiczak A, et al. ; ESC Scientific Document Group. 2018 ESC/ESH guidelines for the management of arterial hypertension. Eur Heart J. 2018;39:3021–3104. doi: 10.1093/eurheartj/ehy339 [DOI] [PubMed] [Google Scholar]
- 12.Mitchell GF, Verwoert GC, Tarasov KV, Isaacs A, Smith AV, Yasmin, Rietzschel ER, Tanaka T, Liu Y, Parsa A, et al. Common genetic variation in the 3’-BCL11B gene desert is associated with carotid-femoral pulse wave velocity and excess cardiovascular disease risk: the AortaGen Consortium. Circ Cardiovasc Genet. 2012;5:81–90. doi: 10.1161/CIRCGENETICS.111.959817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li L, Zhang JA, Dose M, Kueh HY, Mosadeghi R, Gounari F, Rothenberg EV. A far downstream enhancer for murine Bcl11b controls its T-cell specific expression. Blood. 2013;122:902–911. doi: 10.1182/blood-2012-08-447839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annu Rev Genet. 2012;46:1–19. doi: 10.1146/annurev-genet-110711-155459 [DOI] [PubMed] [Google Scholar]
- 15.Avram D, Fields A, Senawong T, Topark-Ngarm A, Leid M. COUP-TF (chicken ovalbumin upstream promoter transcription factor)-interacting protein 1 (CTIP1) is a sequence-specific DNA binding protein. Biochem J. 2002;368:555–563. doi: 10.1042/BJ20020496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Avram D, Fields A, Pretty On Top K, Nevrivy DJ, Ishmael JE, Leid M. Isolation of a novel family of C(2)H(2) zinc finger proteins implicated in transcriptional repression mediated by chicken ovalbumin upstream promoter transcription factor (COUP-TF) orphan nuclear receptors. J Biol Chem. 2000;275:10315–10322. doi: 10.1074/jbc.275.14.10315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Avram D, Califano D. The multifaceted roles of Bcl11b in thymic and peripheral T cells: impact on immune diseases. J Immunol. 2014;193:2059–2065. doi: 10.4049/jimmunol.1400930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Califano D, Cho JJ, Uddin MN, Lorentsen KJ, Yang Q, Bhandoola A, Li H, Avram D. Transcription factor Bcl11b controls identity and function of mature Type 2 innate lymphoid cells. Immunity. 2015;43:354–368. doi: 10.1016/j.immuni.2015.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Srinivasan K, Leone DP, Bateson RK, Dobreva G, Kohwi Y, Kohwi-Shigematsu T, Grosschedl R, McConnell SK. A network of genetic repression and derepression specifies projection fates in the developing neocortex. Proc Natl Acad Sci USA. 2012;109:19071–19078. doi: 10.1073/pnas.1216793109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simon R, Brylka H, Schwegler H, Venkataramanappa S, Andratschke J, Wiegreffe C, Liu P, Fuchs E, Jenkins NA, Copeland NG, et al. A dual function of Bcl11b/Ctip2 in hippocampal neurogenesis. EMBO J. 2012;31:2922–2936. doi: 10.1038/emboj.2012.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maskari RA, Hardege I, Cleary S, Figg N, Li Y, Siew K, Khir A, Yu Y, Liu P, Wilkinson I, et al. Functional characterization of common BCL11B gene desert variants suggests a lymphocyte-mediated association of BCL11B with aortic stiffness. Eur J Hum Genet. 2018;26:1648–1657. doi: 10.1038/s41431-018-0226-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weisbrod RM, Shiang T, Al Sayah L, Fry JL, Bajpai S, Reinhart-King CA, Lob HE, Santhanam L, Mitchell G, Cohen RA, et al. Arterial stiffening precedes systolic hypertension in diet-induced obesity. Hypertension. 2013;62:1105–1110. doi: 10.1161/HYPERTENSIONAHA.113.01744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fry JL, Al Sayah L, Weisbrod RM, Van Roy I, Weng X, Cohen RA, Bachschmid MM, Seta F. Vascular smooth muscle sirtuin-1 protects against diet-induced aortic stiffness. Hypertension. 2016;68:775–784. doi: 10.1161/HYPERTENSIONAHA.116.07622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Butt E, Abel K, Krieger M, Palm D, Hoppe V, Hoppe J, Walter U. cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J Biol Chem. 1994;269:14509–14517 [PubMed] [Google Scholar]
- 25.Abel K, Mieskes G, Walter U. Dephosphorylation of the focal adhesion protein VASP in vitro and in intact human platelets. FEBS Lett. 1995;370:184–188. doi: 10.1016/0014-5793(95)00817-s [DOI] [PubMed] [Google Scholar]
- 26.Tosello V, Saccomani V, Yu J, Bordin F, Amadori A, Piovan E. Calcineurin complex isolated from T-cell acute lymphoblastic leukemia (T-ALL) cells identifies new signaling pathways including mTOR/AKT/S6K whose inhibition synergize with calcineurin inhibition to promote T-ALL cell death. Oncotarget. 2016;7:45715–45729. doi: 10.18632/oncotarget.9933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Döppler H, Storz P. Regulation of VASP by phosphorylation. Cell Adh Migr. 2013;7:492–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim HR, Graceffa P, Ferron F, Gallant C, Boczkowska M, Dominguez R, Morgan KG. Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator-stimulated phosphoprotein. Am J Physiol Cell Physiol. 2010;298:C559–C571. doi: 10.1152/ajpcell.00431.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benz PM, Blume C, Seifert S, Wilhelm S, Waschke J, Schuh K, Gertler F, Münzel T, Renné T. Differential VASP phosphorylation controls remodeling of the actin cytoskeleton. J Cell Sci. 2009;122:3954–3965. doi: 10.1242/jcs.044537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodard T, Sigurdsson S, Gotal JD, Torjesen AA, Inker LA, Aspelund T, Eiriksdottir G, Gudnason V, Harris TB, Launer LJ, et al. Mediation analysis of aortic stiffness and renal microvascular function. J Am Soc Nephrol. 2015;26:1181–1187. doi: 10.1681/ASN.2014050450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moody WE, Edwards NC, Chue CD, Ferro CJ, Townend JN. Arterial disease in chronic kidney disease. Heart. 2013;99:365–372. doi: 10.1136/heartjnl-2012-302818 [DOI] [PubMed] [Google Scholar]
- 32.Ding J, Mitchell GF, Bots ML, Sigurdsson S, Harris TB, Garcia M, Eiriksdottir G, van Buchem MA, Gudnason V, Launer LJ. Carotid arterial stiffness and risk of incident cerebral microbleeds in older people: the Age, Gene/Environment Susceptibility (AGES)-Reykjavik study. Arterioscler Thromb Vasc Biol. 2015;35:1889–1895. doi: 10.1161/ATVBAHA.115.305451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meyer ML, Palta P, Tanaka H, Deal JA, Wright J, Jack C, Knopman D, Griswold M, Mosley TH, Heiss GH. Association of arterial stiffness and pressure amplification with mild cognitive impairment and dementia: The atherosclerosis risk in communities study-neurocognitive study (ARIC-NCS). J Alzheimers Dis. 2017;57:195–204. doi: 10.3233/JAD-161041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cooper LL, Woodard T, Sigurdsson S, van Buchem MA, Torjesen AA, Inker LA, Aspelund T, Eiriksdottir G, Harris TB, Gudnason V, et al. Cerebrovascular damage mediates relations between aortic stiffness and memory. Hypertension. 2016;67:176–182. doi: 10.1161/HYPERTENSIONAHA.115.06398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitchell GF, DeStefano AL, Larson MG, Benjamin EJ, Chen MH, Vasan RS, Vita JA, Levy D. Heritability and a genome-wide linkage scan for arterial stiffness, wave reflection, and mean arterial pressure: the Framingham Heart Study. Circulation. 2005;112:194–199. doi: 10.1161/CIRCULATIONAHA.104.530675 [DOI] [PubMed] [Google Scholar]
- 36.Wakabayashi Y, Watanabe H, Inoue J, Takeda N, Sakata J, Mishima Y, Hitomi J, Yamamoto T, Utsuyama M, Niwa O, et al. Bcl11b is required for differentiation and survival of alphabeta T lymphocytes. Nat Immunol. 2003;4:533–539. doi: 10.1038/ni927 [DOI] [PubMed] [Google Scholar]
- 37.Wu SP, Cheng CM, Lanz RB, Wang T, Respress JL, Ather S, Chen W, Tsai SJ, Wehrens XH, Tsai MJ, et al. Atrial identity is determined by a COUP-TFII regulatory network. Dev Cell. 2013;25:417–426. doi: 10.1016/j.devcel.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Davis GE, Hill MA, Meininger GA. Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol. 2001;280:H1427–H1433. doi: 10.1152/ajpheart.2001.280.4.H1427 [DOI] [PubMed] [Google Scholar]
- 39.Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161 [DOI] [PubMed] [Google Scholar]
- 40.Chatterjee M, Murphy RA. Calcium-dependent stress maintenance without myosin phosphorylation in skinned smooth muscle. Science. 1983;221:464–466. doi: 10.1126/science.6867722 [DOI] [PubMed] [Google Scholar]
- 41.Dillon PF, Aksoy MO, Driska SP, Murphy RA. Myosin phosphorylation and the cross-bridge cycle in arterial smooth muscle. Science. 1981;211:495–497. doi: 10.1126/science.6893872 [DOI] [PubMed] [Google Scholar]
- 42.Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16:72–76. doi: 10.1096/cj.01-0104hyp [DOI] [PubMed] [Google Scholar]
- 43.Krause M, Dent EW, Bear JE, Loureiro JJ, Gertler FB. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 2003;19:541–564. doi: 10.1146/annurev.cellbio.19.050103.103356 [DOI] [PubMed] [Google Scholar]
- 44.Reinhard M, Giehl K, Abel K, Haffner C, Jarchau T, Hoppe V, Jockusch BM, Walter U. The proline-rich focal adhesion and microfilament protein VASP is a ligand for profilins. EMBO J. 1995;14:1583–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reinhard M, Jouvenal K, Tripier D, Walter U. Identification, purification, and characterization of a zyxin-related protein that binds the focal adhesion and microfilament protein VASP (vasodilator-stimulated phosphoprotein). Proc Natl Acad Sci U S A. 1995;92:7956–7960. doi: 10.1073/pnas.92.17.7956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bear JE, Svitkina TM, Krause M, Schafer DA, Loureiro JJ, Strasser GA, Maly IV, Chaga OY, Cooper JA, Borisy GG, et al. Antagonism between Ena/VASP proteins and actin filament capping regulates fibroblast motility. Cell. 2002;109:509–521. doi: 10.1016/s0092-8674(02)00731-6 [DOI] [PubMed] [Google Scholar]
- 47.Grosse R, Copeland JW, Newsome TP, Way M, Treisman R. A role for VASP in RhoA-Diaphanous signalling to actin dynamics and SRF activity. EMBO J. 2003;22:3050–3061. doi: 10.1093/emboj/cdg287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069 [DOI] [PubMed] [Google Scholar]
- 49.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616 [DOI] [PubMed] [Google Scholar]
- 50.Vlachopoulos C, Aznaouridis K, O’Rourke MF, Safar ME, Baou K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with central haemodynamics: a systematic review and meta-analysis. Eur Heart J. 2010;31:1865–1871. doi: 10.1093/eurheartj/ehq024 [DOI] [PubMed] [Google Scholar]
- 51.Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Cheng S, Aragam J, Levy D, Benjamin EJ, Vasan RS, et al. Relations of central hemodynamics and aortic stiffness with left ventricular structure and function: the Framingham Heart Study. J Am Heart Assoc. 2016;5:e002693. doi: 10.1161/JAHA.115.002693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bell V, McCabe EL, Larson MG, Rong J, Merz AA, Osypiuk E, Lehman BT, Stantchev P, Aragam J, Benjamin EJ, et al. Relations between aortic stiffness and left ventricular mechanical function in the community. J Am Heart Assoc. 2017;6:e004903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sutton-Tyrrell K, Newman A, Simonsick EM, Havlik R, Pahor M, Lakatta E, Spurgeon H, Vaitkevicius P. Aortic stiffness is associated with visceral adiposity in older adults enrolled in the study of health, aging, and body composition. Hypertension. 2001;38:429–433. doi: 10.1161/01.hyp.38.3.429 [DOI] [PubMed] [Google Scholar]
- 54.Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol (1985). 2008;105:1652–1660. doi: 10.1152/japplphysiol.90549.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cooper LL, Palmisano JN, Benjamin EJ, Larson MG, Vasan RS, Mitchell GF, Hamburg NM. Microvascular function contributes to the relation between aortic stiffness and cardiovascular events. Circ Cardiovasc Imaging. 2016;9:e004979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van den Bergh R. Centrifugal elements in the vascular pattern of the deep intracerebral blood supply. Angiology. 1969;20:88–94. doi: 10.1177/000331976902000205 [DOI] [PubMed] [Google Scholar]
- 57.Mitchell GF, van Buchem MA, Sigurdsson S, Gotal JD, Jonsdottir MK, Kjartansson Ó, Garcia M, Aspelund T, Harris TB, Gudnason V, et al. Arterial stiffness, pressure and flow pulsatility and brain structure and function: the Age, Gene/Environment Susceptibility–Reykjavik study. Brain. 2011;134:3398–3407. doi: 10.1093/brain/awr253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oh YS, Berkowitz DE, Cohen RA, Figueroa CA, Harrison DG, Humphrey JD, Larson DF, Leopold JA, Mecham RP, Ruiz-Opazo N, et al. A special report on the NHLBI initiative to study cellular and molecular mechanisms of arterial stiffness and its association with hypertension. Circ Res. 2017;121:1216–1218. doi: 10.1161/CIRCRESAHA.117.311703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li P, Burke S, Wang J, Chen X, Ortiz M, Lee SC, Lu D, Campos L, Goulding D, Ng BL, et al. Reprogramming of T cells to natural killer-like cells upon Bcl11b deletion. Science. 2010;329:85–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fry JL, Shiraishi Y, Turcotte R, Yu X, Gao YZ, Akiki R, Bachschmid M, Zhang Y, Morgan KG, Cohen RA, et al. Vascular smooth muscle sirtuin-1 protects against aortic dissection during angiotensin II-induced hypertension. J Am Heart Assoc. 2015;4:e002384. doi: 10.1161/JAHA.115.002384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nicholson CJ, Seta F, Lee S, Morgan KG. MicroRNA-203 mimics age-related aortic smooth muscle dysfunction of cytoskeletal pathways. J Cell Mol Med. 2017;21:81–95. doi: 10.1111/jcmm.12940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qin Z, Hou X, Weisbrod RM, Seta F, Cohen RA, Tong X. Nox2 mediates high fat high sucrose diet-induced nitric oxide dysfunction and inflammation in aortic smooth muscle cells. J Mol Cell Cardiol. 2014;72:56–63. doi: 10.1016/j.yjmcc.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Rourke M. Arterial stiffness, systolic blood pressure, and logical treatment of arterial hypertension. Hypertension. 1990;15:339–347. doi: 10.1161/01.hyp.15.4.339 [DOI] [PubMed] [Google Scholar]
- 64.Saphirstein RJ, Gao YZ, Lin QQ, Morgan KG. Cortical actin regulation modulates vascular contractility and compliance in veins. J Physiol. 2015;593:3929–3941. doi: 10.1113/JP270845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao YZ, Saphirstein RJ, Yamin R, Suki B, Morgan KG. Aging impairs smooth muscle-mediated regulation of aortic stiffness: a defect in shock absorption function? Am J Physiol Heart Circ Physiol. 2014;307:H1252–H1261. doi: 10.1152/ajpheart.00392.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lorentsen KJ, Cho JJ, Luo X, Zuniga AN, Urban JF, Zhou L, Gharaibeh R, Jobin C, Kladde MP, Avram D. Bcl11b is essential for licensing Th2 differentiation during helminth infection and allergic asthma. Nat Commun. 2018;9:1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim HR, Gallant C, Leavis PC, Gunst SJ, Morgan KG. Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. 2008;295:C768–C778. doi: 10.1152/ajpcell.00174.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, Launer LJ, Van Buchem MA, Breteler MM; Microbleed Study Group. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009;8:165–174. doi: 10.1016/S1474-4422(09)70013-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zudaire E, Gambardella L, Kurcz C, Vermeren S. A computational tool for quantitative analysis of vascular networks. PLoS One. 2011;6:e27385. doi: 10.1371/journal.pone.0027385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, Freret T. Object recognition test in mice. Nat Protoc. 2013;8:2531–2537. doi: 10.1038/nprot.2013.155 [DOI] [PubMed] [Google Scholar]
- 71.Pérez-Escudero A, Vicente-Page J, Hinz RC, Arganda S, de Polavieja GG. idTracker: tracking individuals in a group by automatic identification of unmarked animals. Nat Methods. 2014;11:743–748. doi: 10.1038/nmeth.2994 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A detailed description of materials and methods can be found in the Data Supplement. All data and supporting material are available upon request.