

Abstract
Oral microbiota has been implicated in pathogenesis of recurrent aphthous stomatitis (RAS), which is a common mucosal disorder with unclear etiology. This study has explored the association between oral microbiota disorder and RAS in high-risk young female population.
Forty-five young females were enrolled, including 24 RAS patients and 21 healthy individuals. Oral microbiome was analyzed by Illumina Miseq sequencing.
Oral microbiota associated with RAS was characterized by the lower alpha-diversity indices (Chao1 and ACE). Several infectious pathogens increased in RAS, such as genera Actinobacillus, Haemophilus, Prevotella and Vibrio. The PICRUSt analysis indicated that the oral microbiota might be related with the up-regulation of genes involving infectious and neurodegenerative diseases, environmental adaptation, the down-regulation of genes involving basal metabolism, such as carbohydrate, energy, and amino acid metabolism.
This study indicated that oral microbiota may play a significant role in RAS development.
Keywords: illumina miseq, oral microbiota, recurrent aphthous stomatitis, young females
1. Introduction
Recurrent aphthous stomatitis (RAS) is a common clinical condition producing painful ulcerations in the oral cavity.[1] Epidemiological studies have shown that the prevalence of RAS was approximately 20% in the general population, and it was especially higher (male, 48.3%; female, 57.2%) in professional-school students aged between 10 and 19 years.[2,3] RAS is characterized by chronic inflammation of the oral mucosa with multiple recurrent small, rounds, ovoid ulcers with circumscribed margins, erythematous haloes, and yellow or gray floors.[4] The potential trigger factors of RAS include genetic predisposition, immunologic disturbances, viral and bacterial infections, food allergies, systemic diseases, and stress; however, the etiology of RAS remains unknown.[5]
To date, it is widely accepted that genetic, immunological and environmental factors are involved with RAS.[6] Microbial infection and disturbance of the oral microbiota have been identified as the important environmental factors. Streptococcal species and Helicobacter pylori were the two main infectious bacteria that may contribute to the pathogenesis.[7,8] The infectious bacteria may act as either pathogens or as antigens that induce the production of antibodies, which can cross-react with oral mucosal keratinocytes.[9] However, a complex commensal microbiota exists in the oral cavity, which is characterized by the oral microbiota. Previous studies have reported the association between oral microbiota and RAS,[9–11] however, among various RAS populations, the profiles of the oral microbiota differed resulting from the intervening factors, such as diet, age, and host gene.[12–14] Therefore, the relationship between RAS and the oral microbiota in young adults may be different from the previous reported literature.
To weaken the intervening effects of other factors on the association between RAS and oral microbiota, forty-five females, aged 19–22 years, were enrolled in this study. Illumina MiSeq sequencing was used to reveal the oral microbiome differences between RAS and healthy students. The aim of this study was to ascertain the association between oral microbiota and RAS in young females.
2. Materials and methods
2.1. Study subjects
A cross-sectional collection of saliva samples was performed for female students from Zhejiang Chinese Medical University who were suffering from ulcer in the oral cavity and diagnosed with RAS. In addition, another group of female from the same university, who were matched by gender and age, were also enrolled for saliva sample collection as healthy controls (HCs). All participants were aged between 19 and 22 years old. Subjects who have received antibiotics or antibacterial mouth rinses or drugs within one month were excluded. Additional exclusion criteria included other types of oral mucosal lesions, smoking, and consumption of alcohol within one month. The study protocol was approved by the Ethics Committee of Zhejiang Chinese Medical University, and written informed consent was obtained from each participant.
The demographic data of enrolled subjects in the current study were summarized in Table 1. No difference in age and BMI index was observed between RAS and HCs. Additionally, information about ulcer numbers was also reordered. Twenty RAS females had a single ulcer, while four RAS females had multiple ulcers in the mouth. Compared with HCs, the level of C-reactive protein (CRP) was significantly increased in of RAS females (Table 1). All enrolled females have no other inflammation disorders, including chronic hypertension, diabetes mellitus and autoimmune disorders (Table 1).
Table 1.
Demographic data of HCs and RAS.
| Healthy females (n = 21) | RAS females (n = 24) | |
| Age | 19–22 | 19–22 |
| Ulcer numbers | NA | Single: 20 Multiple: 4 |
| BMI | 21.98 ± 0.85 | 22.48 ± 1.20 |
| CRP (mg/L) | 0.48 ± 0.14 | 1.98 ± 0.73 |
| Medical history | ||
| Chronic hypertension | NA | NA |
| Diabetes mellitus | NA | NA |
| Autoimmune disease | NA | NA |
2.2. Sample collection and DNA extraction
For sampling, the saliva of RAS females was collected on the third day after ulcer appearance. Unstimulated whole saliva was collected from healthy females. A minimum of 2 ml saliva was collected from each subject and stored at −80°C until DNA extraction.
Before DNA extraction, the frozen saliva samples underwent a freeze-drying process for removing moisture. The concentrated samples were used to extract genomic DNA using the QIAamp DNA Micro Kit (Qiagen, Hilden, Germany) according to the manufacturer's protocols. The DNA concentration was determined using NanoDrop (Thermo Scientific) and DNA was visualized using agarose gel electrophoresis.
2.3. 16S rRNA gene amplification and sequencing
The hyper-variable V3-V4 regions of the bacterial 16S rRNA gene sequences were amplified from the diluted DNA using bacterial primers 338F (5’- ACTCCTACGGGAGGCAGCA-3’) and 806R (5’-GGACTACHVGGGTWTCTAA T-3’), with the reverse primer containing a 6-bp barcode used to tag each sample. PCR amplification was performed in a 30 μl mixture containing 1.0 μl of forward primer (10 μM), 1.0 μl of reverse primer (10 μM), 5.0 μl of dNTP (100 mM), 2 μl of DNA template (20 ng/μl), 5.0 μl of 5×reaction buffer, 5.0 μl of 5×GC buffer, 9.0 μl of ddH2O and 2.0 μl of Q5 High-Fidelity DNA Polymerase (NEB). The reactions were hot-started at 98°C for 2 min, followed by 25 cycles of 98°C for 15 s, 55°C for 30 s, and 72°C for 30 s, with a final extension step at 72°C for 5 min.
Amplicons were extracted from 1.5% agarose gel and purified using the QIAquick Gel Extraction kit (Qiagen, Valencia, CA, USA). Purified amplicons were pooled in equimolar quantities and pair-end sequenced (2 x 250) on an Illumina MiSeq platform (Personal Biotechnology Co., Ltd, Shanghai) according to the standard protocols. Libraries were constructed according to the TruSeqTM DNA Sample Prep Kit protocol (Illumina, USA).
2.4. Processing of sequencing data
After sequencing, raw FASTQ files were demultiplexed and quality-filtered using Quantitative Insights Into Microbial Ecology (QIIME, version 1.17) under the following criteria: (i) reads were truncated at any site receiving an average quality score of <20 bp over a 50-bp sliding window, and truncated reads shorter than 50 bp were discarded; (ii) exact barcode matching, two nucleotide mismatch in primer matching, and reads containing ambiguous characters were removed; (iii) only sequences that overlapped for more than 20 bp were merged according to their overlap sequence, reads that could not be merged were discarded.
The high-quality sequences were clustered into the 16S rRNA Operational Taxonomic Units (OTUs) with 97% similarity cutoff using UPARSE (version 7.1). Chimera detection and removal were assessed using the GOLD reference databased and UCHIME.[15] The taxonomic affiliation of each OTU was analyzed using Ribosomal Database Project (RDP) classifier against the SILVA 16S rRNA database.[16]
2.5. Sequence analysis
Bacterial diversity was determined by sampling based analysis of OTUs, and shown by Chao1, ACE, Shannon and Simpson indices that were calculated by using R program package “vegan”. Principal coordinates analysis (PCoA) based on OTUs distribution was conducted by the R package to visualize interaction among bacterial communities.
The specific characterization of oral microbiota was analyzed by linear discriminant analysis (LDA) effect size (LEfSe) method (http://huttenhower.sph.harvard.edu/lefse/).[17] LEfSe performs the Kruskall-Wallis test to determine the features with significantly different abundances between RAS and HCs and LDA is used to assess the effect size of each feature.
Microbial function was inferred using PICRUST that predict the molecular functions of each sample based on the 16S rRNA maker gene sequences. These predictions were pre-calculated for genes in databases including KEGG and COGs. Briefly, the PICRUST that consisted of two steps: gene content inference and metagenome inference was performed as described previously.[18]
Multiple group differences in diversity indices and predicted metabolic function were analyzed via Mann-Whitney non-parametric test in SPSS software 16.0.
3. Results
3.1. Structure and composition of the oral microbiome in RAS
Alpha and beta diversity was calculated to assess overall differences in microbial community structure of saliva between RAS and HCs. As shown in Figure 1, the two alpha-diversity indices (Chao1 and ACE) of RAS were significantly lower than HCs (p < 0.05), however, the other two alpha-diversity indices (Shannon and Simpson) were not statistically different. The above results indicated that lower diversity of oral microbiome was accompanied with RAS in young females.
Figure 1.
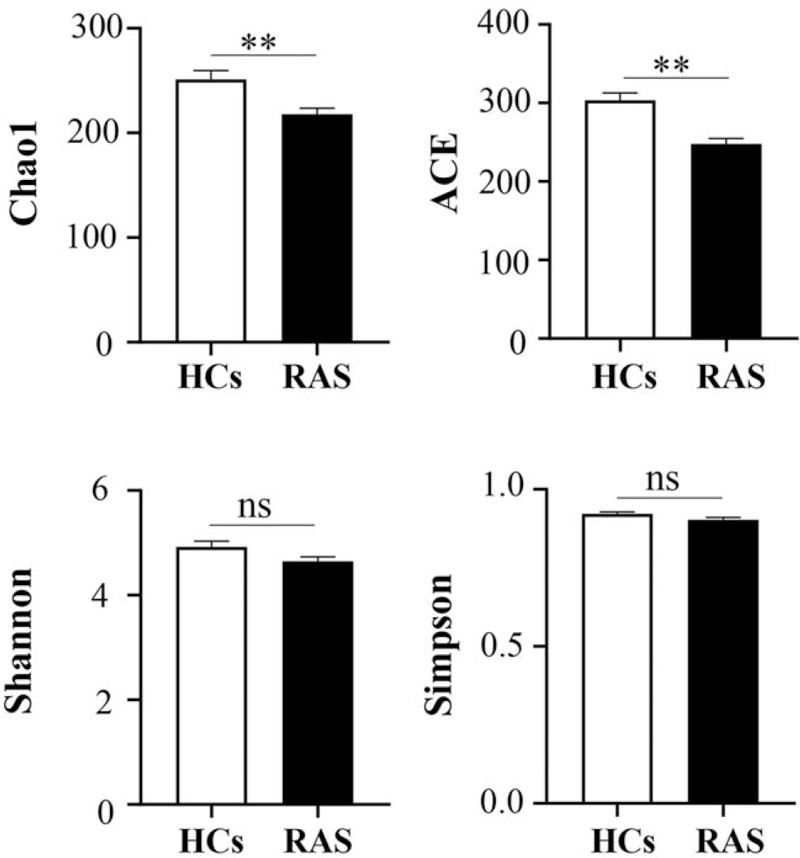
Alpha-diversity indices of oral microbiome between HCs and RAS.
The beta-diversity difference using the unweighted UniFrac distance was shown by PCoA analysis (Fig. 2). On the PCoA plot, oral microbial communities separated in RAS and HC samples, suggesting a unique oral microbiota in RAS.
Figure 2.
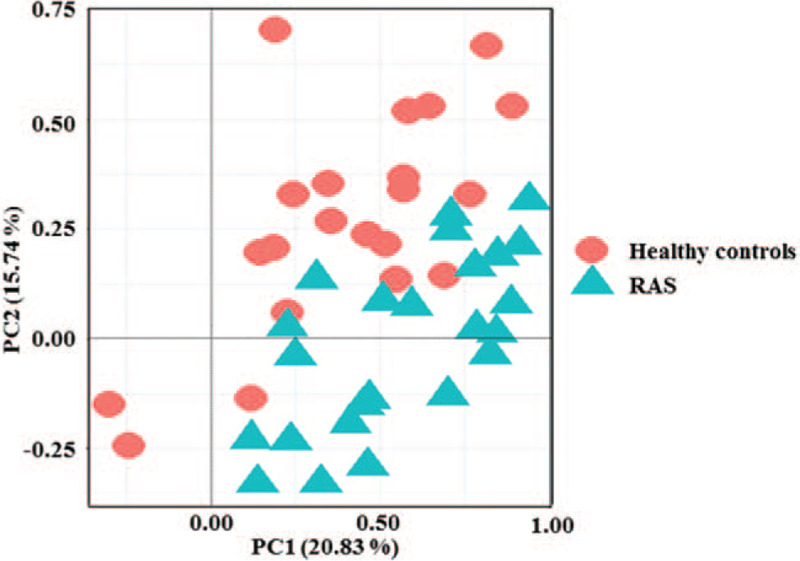
Unweighted PCoA score plots of HCs and RAS based on the oral microbial composition.
3.2. RAS-associated microbiota changes in saliva
LEfSe method was used to display the greatest differences of microbial structure between HCs and RAS. Seventeen different microbial phylotypes with LDA score >2 and p value < 0.05 were displayed in Figure 3. Compared with HCs, 10 phylotypes increased while 7 phylotypes decreased in RAS.
Figure 3.
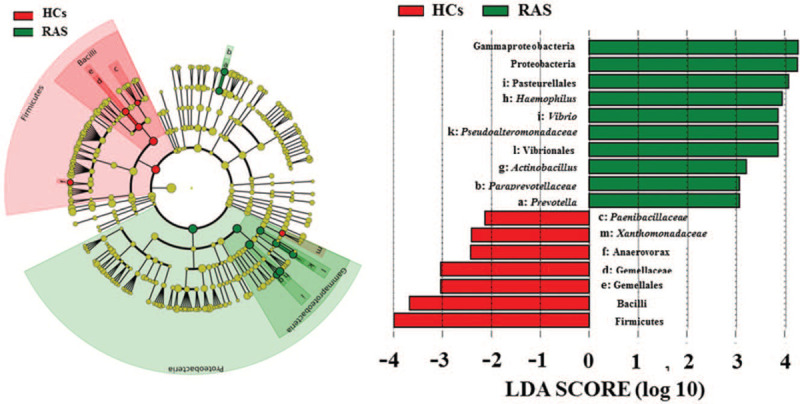
LEfSe identified the most different oral microbiota between HCs and RAS. Red and green represent HCs and RAS, respectively.
At the phylum level, Proteobacteria was significantly more abundant in the oral microbiota of the RAS than that in the HCs (Fig. 3). The class Gammaproteobacteria and its derivates (Order Pasteurellales, genus Actinobacillus and Haemophilus, and order Vibrionales, Family Pseudoalteromonadaceae, genus Vibrio) contributed to the significant increase of the phylum Proteobacteria in RAS. In addition, Family Paraprevotellaceae and its genus Prevotella were also significantly more enriched in the oral microbiota of RAS.
Conversely, Firmicutes were significantly reduced in the oral microbiota of RAS (Fig. 3). Compared with the HCs, the class Bacilli and its derivates (Order Gemellales, Family Gemellaceae and Order Bacillales, Family Paenibacillaceae) decreased in the RAS. Meanwhile, the decreased phylotypes of RAS also included the genus Anaerovorax and Xanthomonadaceae.
3.3. Predicted functional profile of oral microbiota
Phylogeny and function of oral microbial community were connected. Hence, the PICRUSt approach was used to analyze the KEGG pathways compositions in bacterial population based on 16S rRNA maker gene sequences. 12 second-level KEGG pathways differed in abundance between RAS and HCs (Fig. 4, p < 0.05).
Figure 4.
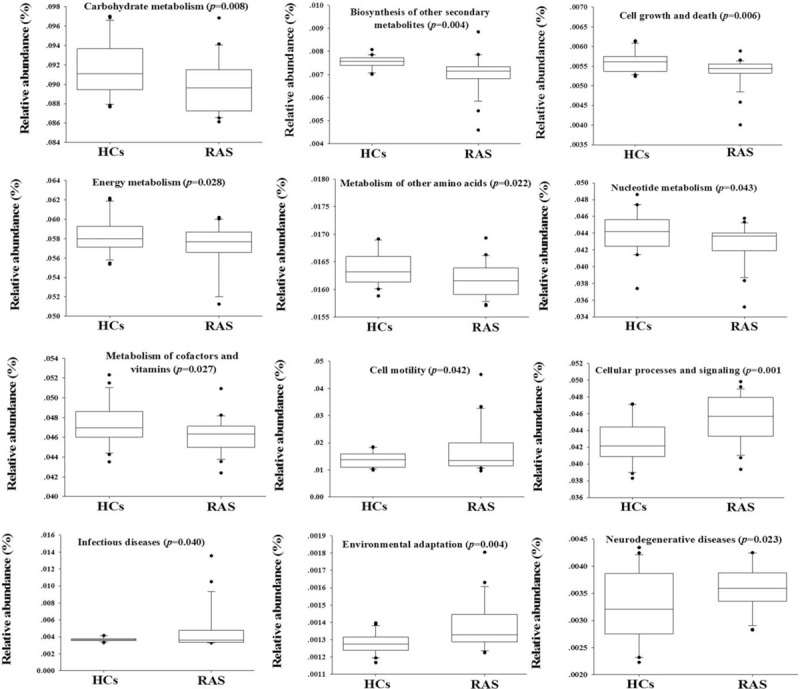
Significantly predicated functional profiling of PICRUSt analysis based on oral microbial composition.
Firstly, the pathways of infectious diseases and neurodegenerative diseases significantly increased in oral microbiota of RAS (Fig. 4). Secondly, the PICRUSt analyses also indicated that oral microbiota in RAS had the significantly more abundant genes involving in cell motility, cellular processes and signaling, as well as environment adaptation. Thirdly, oral microbiota of RAS had the significantly lower basal metabolism, such as carbohydrate metabolism, biosynthesis of other secondary metabolites, cell growth and death, energy metabolism, amino acid metabolism, and nucleotide metabolism.
4. Discussion
It has been well known that microbial factor is a causative agent of RAS.[19] Therefore, imbalances of oral microbiota are frequently accompanied with RAS.[9–11] The present study also characterized the oral microbiome of RAS. The study cohort was selected in young females aged from 19 to 22, since the onset of RAS seems to peak in young females according to epidemiologic studies.[20]
Using Illumina Miseq sequencing, the present study showed a significantly decreased alpha diversity in the oral microbiota of RAS. The previous literature also studied alpha diversity of oral microbiota in RAS, but there is no obvious difference in alpha diversity between healthy controls and RAS in study population with the mean age of 44.[11] It has been widely accepted that the lower alpha diversity represented more severe imbalances of microbial community.[21,22] Therefore, the young adults suffered a sharper imbalance of oral microbiota than the middle-aged adults with RAS.
In young females, several associations between RAS and oral microbiota was consistent with the former studies. It was well known that Streptococcal species and Helicobacter pylori were the main RAS-related pathogenic bacteria. Due to the limit of 16S rRNA Miseq sequencing, Helicobacter pylori was not identified in this study. In addition, Streptococcal species were not found being the significantly altered microbial phylotypes. Indeed, Streptococcal species and Helicobacter pylori have been proposed to provoke development of RAS,[7,23] but there is no evidence to prove that they are related with RAS in oral microbiome.[2,11] The reason might be that Streptococcal species were initial factors perturbing the global oral microbiota at the early stage of inflammation in RAS but their proportion were lower to normal levels after oral microbiota imbalances. In addition, the genus Haemophilus and Prevotella, which were increased in RAS, were also reported to be positive with RAS in the previous literature.[11] It has been well-known that genus Haemophilus and Prevotella were two infectious pathogen through intervening immunity, energy metabolism and amino acid metabolism.[24,25]
Furthermore, there are some alterations of oral microbiota specifically being found in this study. In oral cavity of young females with RAS, the family Pseudoalteromonadaceae which was known as the infectious bacteria also significantly increased. The family Pseudoalteromonadaceae was always considered as the exogenous invasion in the oral cavity, and its genus Vibrio was a common type of infectious bacterium in human oral cavity.[26] In addition, the increased genus Actinobacillus of RAS has been strongly implicated in the development of periodontitis.[27,28] The above specific findings may be the underlying reasons leading to the high risk of RAS in young adults.
Phylogeny and functions of gut microbiota are connected.[18] The altered oral microbiota could determine the potential functional of oral microbiota in the samples. RAS is a system disease with pathogenesis of infection, immunity and metabolism disorders.[29] The study inferred that oral microbiota could contribute to RAS pathogenesis due to the different function pathways of oral microbiota in the infectious diseases and basal metabolisms. The appearance of RAS could change the micro-environment of the oral mucosal with inflammation and immune activity.[30] The above stress would have adverse effects on oral microbiota and decline general metabolic functions (such as amino acid metabolism, carbohydrate metabolism and nucleotide metabolism) of oral microbiota in RAS. This is consistent with effects of microbiota on other diseases.[31,32] Meanwhile, the adverse environment in oral mucosal of RAS could induce some microbiota increasing, which could perform functional pathway (environmental adaptation) in decreasing negative effects.
5. Conclusions
In conclusion, the present study explored association between oral microbiota and RAS in young females for the first time. Some specific oral microbiota significantly altered in young females suffered from RAS, such as Haemophilus, Prevotella, Vibrio and Actinobacillus. The predicted function pathways using PICRUSt also verified the correlation between oral microbiota and RAS. Thus, our study may facilitate understanding the role of oral microbiota in RAS development in young females. However, this study also had several limitations. Firstly, the sample population come from a small group of people, and both sample source and size need to be expanded to verify our results. Secondly, there is little basic clinical information of sample population, and more clinical information needs to be collected to exclude their influence on oral microbiota.
Author contributions
Conceptualization: Tiejuan Shao, Yongsheng Fan, Zhixing He.
Data curation: Zhengyang Zhu, Zhixing He, Tiejuan Shao.
Formal analysis: Guanqun Xie.
Funding acquisition: Yongsheng Fan.
Methodology: Zhengyang Zhu, Guanqun Xie.
Software: Zhixing He.
Supervision: Guanqun Xie.
Validation: Guanqun Xie.
Writing - original draft: Zhengyang Zhu, Tiejuan Shao.
Writing-review & editing: Zhengyang Zhu, Tiejuan Shao, Yongsheng Fan, Zhixing He.
Footnotes
Abbreviations: BMI = Body Mass index, CRP = C-reactive protein, HCs = healthy controls, KEGG = kyoto encyclopedia of genes and genomes, LDA = linear discriminant analysis, OTUs = Operational Taxonomic Units, PCoA = principal coordinates analysis, QIIME = quantitative insights into microbial ecology, RAS = recurrent aphthous stomatitis.
How to cite this article: Zhu Z, He Z, Xie G, Fan Y, Shao T. Altered oral microbiota composition associated with recurrent aphthous stomatitis in young females. Medicine. 2021;100:10(e24742).
Funding: The National Basic Research Program of China (973 Program, No.2014CB543001) financially supported this study.
Availability of data and materials: Raw sequence data of this study has been deposited to the NCBI Sequence Read Archive with accession No. PRJNA413647
Ethics approval and consent to participate: All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional, national research committee and with the 1964 Helsinki declaration and its later amendments. Informed consent was obtained from all individual participants included in the study.
Consent for publication: Not applicable
Competing interests: The authors declare no conflicts of interest.
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
- [1].Edgar NR, Saleh D, Miller RA. Recurrent Aphthous Stomatitis: A Review. The Journal of clinical and aesthetic dermatology 2017;10:26–36. [PMC free article] [PubMed] [Google Scholar]
- [2].Gallo Cde B, Mimura MA, Sugaya NN. Psychological stress and recurrent aphthous stomatitis. Clinics (Sao Paulo) 2009;64:645–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chavan M, Jain H, Diwan N, et al. Recurrent aphthous stomatitis: a review. Journal of oral pathology & medicine: official publication of the International Association of Oral Pathologists and the American Academy of Oral Pathology 2012;41:577–83. [DOI] [PubMed] [Google Scholar]
- [4].Jurge S, Kuffer R, Scully C, et al. Mucosal disease series. Number VI. Recurrent aphthous stomatitis. Oral diseases 2006;12:1–21. [DOI] [PubMed] [Google Scholar]
- [5].Slebioda Z, Szponar E, Kowalska A. Etiopathogenesis of recurrent aphthous stomatitis and the role of immunologic aspects: literature review. Arch Immunol Ther Exp (Warsz) 2014;62:205–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Akintoye SO, Greenberg MS. Recurrent aphthous stomatitis. Dent Clin North Am 2014;58:281–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Di Pierro F, Adami T, Rapacioli G, et al. Clinical evaluation of the oral probiotic Streptococcus salivarius K12 in the prevention of recurrent pharyngitis and/or tonsillitis caused by Streptococcus pyogenes in adults. Expert Opin Biol Th 2013;13:339–43. [DOI] [PubMed] [Google Scholar]
- [8].Gulseren D, Karaduman A, Kutsal D, et al. The relationship between recurrent aphthous stomatitis, and periodontal disease and Helicobacter Pylori infection. Clin Oral Invest 2016;20:2055–60. [DOI] [PubMed] [Google Scholar]
- [9].Bankvall M, Sjoberg F, Gale G, et al. The oral microbiota of patients with recurrent aphthous stomatitis. J Oral Microbiol 2014; 6:25739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hijazi K, Lowe T, Meharg C, et al. Mucosal Microbiome in Patients with Recurrent Aphthous Stomatitis. J Dent Res 2015;94:87s–94s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim YJ, Choi YS, Baek KJ, et al. Mucosal and salivary microbiota associated with recurrent aphthous stomatitis. Bmc Microbiol 2016;16 Suppl 1:57. doi: 10.1186/s12866-016-0673-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Adler CJ, Dobney K, Weyrich LS, et al. Sequencing ancient calcified dental plaque shows changes in oral microbiota with dietary shifts of the Neolithic and Industrial revolutions. Nat Genet 2013;45:450–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mason MR, Nagaraja HN, Camerlengo T, et al. Deep Sequencing Identifies Ethnicity-Specific Bacterial Signatures in the Oral Microbiome. Plos One 2013;8:e77287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shoemark DK, Allen SJ. The Microbiome and Disease: Reviewing the Links between the Oral Microbiome, Aging, and Alzheimer's Disease. J Alzheimers Dis 2015;43:725–38. [DOI] [PubMed] [Google Scholar]
- [15].Edgar RC, Haas BJ, Clemente JC, et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011;27:2194–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Quast C, Pruesse E, Yilmaz P, et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 2013;41(D1):D590–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Segata N, Izard J, Waldron L, et al. Metagenomic biomarker discovery and explanation. Genome Biol 2011;12:R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Langille MGI, Zaneveld J, Caporaso JG, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013;31:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Akintoye SO, Greenberg MS. Recurrent aphthous stomatitis. Dental clinics of North America 2014;58:281–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ship JA, Chavez EM, Doerr PA, et al. Recurrent aphthous stomatitis. Quintessence Int 2000;31:95–112. [PubMed] [Google Scholar]
- [21].Ott SJ, Musfeldt M, Wenderoth DF, et al. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004;53:685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Choi EB, Hong SW, Kim DK, et al. Decreased diversity of nasal microbiota and their secreted extracellular vesicles in patients with chronic rhinosinusitis based on a metagenomic analysis. Allergy 2014;69:517–26. [DOI] [PubMed] [Google Scholar]
- [23].Tas DA, Yakar T, Sakalli H, et al. Impact of Helicobacter pylori on the clinical course of recurrent aphthous stomatitis. Journal of Oral Pathology & Medicine 2013;42:89–94. [DOI] [PubMed] [Google Scholar]
- [24].Macedo N, Rovira A, Torremorell M. Haemophilus parasuis: infection, immunity and enrofloxacin. Vet Res 2015;46:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Baddal B, Muzzi A, Censini S, et al. Dual RNA-seq of Nontypeable Haemophilus influenzae and Host Cell Transcriptomes Reveals Novel Insights into Host-Pathogen Cross Talk (vol 6, e01765, 2015). Mbio 2016;7:e00373–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Soonthornchai W, Rungrassamee W, Karoonuthaisiri N, et al. Expression of immune-related genes in the digestive organ of shrimp, Penaeus monodon, after an oral infection by Vibrio harveyi. Dev Comp Immunol 2010;34:19–28. [DOI] [PubMed] [Google Scholar]
- [27].Slots J, Listgarten MA. Bacteroides gingivalis, Bacteroides intermedius and Actinobacillus actinomycetemcomitans in human periodontal diseases. Journal of clinical periodontology 1988;15:85–93. [DOI] [PubMed] [Google Scholar]
- [28].Johansson A, Buhlin K, Sorsa T, et al. Systemic Aggregatibacter actinomycetemcomitans Leukotoxin-Neutralizing Antibodies in Periodontitis. Journal of periodontology 2017;88:122–9. [DOI] [PubMed] [Google Scholar]
- [29].Scully C, Porter S. Oral mucosal disease: Recurrent aphthous stomatitis. Brit J Oral Max Surg 2008;46:198–206. [DOI] [PubMed] [Google Scholar]
- [30].Al-Samadi A, Kouri VP, Salem A, et al. IL-17C and its receptor IL-17RA/IL-17RE identify human oral epithelial cell as an inflammatory cell in recurrent aphthous ulcer. Journal of Oral Pathology & Medicine 2014;43:117–24. [DOI] [PubMed] [Google Scholar]
- [31].Davenport M, Poles J, Leung JM, et al. Metabolic Alterations to the Mucosal Microbiota in Inflammatory Bowel Disease. Inflamm Bowel Dis 2014;20:723–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kirst ME, Li EC, Alfant B, et al. Dysbiosis and Alterations in Predicted Functions of the Subgingival Microbiome in Chronic Periodontitis. Appl Environ Microb 2015;81:783–93. [DOI] [PMC free article] [PubMed] [Google Scholar]