

Abstract
Hydrogen sulfide (H2S) is an environmental toxin and a heritage of ancient microbial metabolism, which has stimulated new interest following its discovery as a neuromodulator. While many physiological responses have been attributed to low H2S levels, higher levels inhibit complex IV in the electron transport chain. To prevent respiratory poisoning, a dedicated set of enzymes comprising the mitochondrial sulfide oxidation pathway exists to clear H2S. The committed step in this pathway is catalyzed by sulfide quinone oxidoreductase (SQOR), which couples sulfide oxidation to coenzyme Q10 reduction in the electron transport chain. The SQOR reaction prevents H2S accumulation and generates highly reactive persulfide species as products, which can be further oxidized or can modify cysteine residues in proteins via persulfidation. Here, we review the kinetic, and structural characteristics of human SQOR, and how its unconventional redox cofactor configuration and substrate promiscuity lead to sulfide clearance and potentially expand the signaling potential of H2S. This dual role of SQOR makes it a promising target for H2S-based therapeutics.
Keywords: protein structure, sulfide, flavin, redox chemistry, metabolism
Graphical Abstract
Regulating sulfide toxicity and signaling: Utilizing an active site cysteine trisulfide, human sulfide quinone oxidoreductase detoxifies hydrogen sulfide, a respiratory poison. Herein, we detail the remarkable enzymology of sulfide quinone oxidoreductase and its potential modulation of sulfide signaling.
Introduction
Hydrogen sulfide (H2S) is a volatile, flammable, and toxic gas, with a characteristic odor of rotten eggs that is detectable by the human nose at levels as low as 0.02–0.03 ppm.[1–2] The primary mechanism of H2S toxicity is via tight binding to heme a3 in cytochrome c oxidase, inhibiting mitochondrial respiration.[3] Owing to its toxicity, H2S has long been infamous as an occupational and environmental hazard,[1–2, 4] but its reputation underwent a paradigm shift in 1996 with the discovery of its neuromodulatory role.[5] Since its induction into the family of gaseous signaling molecules along with nitric oxide (NO) and carbon monoxide (CO), significant strides have been made in understanding the physiological effects of H2S, which span the cardiovascular, central nervous, and gastrointestinal systems.[6]
To elicit signaling responses, cellular H2S levels presumably increase sharply, before returning to low steady-state levels to prevent respiratory poisoning.[7] Alternatively, H2S-based signaling might originate from transient inhibition of mitochondrial bioenergetics, which is reversed by the action of SQOR.[8] Physiological effects in response to H2S typically display a bimodal response that is dependent on the concentration of H2S. High H2S levels have been implicated in peripheral and coronary artery diseases,[9] stimulation of the growth and plasticity of colon, liver, and breast cancers,[10–11] septic shock,[12] and inflammation,[13–14] while low H2S levels are implicated in hypertension,[15] acceleration of atherosclerosis,[16] and fibrotic disease in several organs.[17–18] Intriguingly, H2S has been reported to reversibly induce a state of suspended animation in mice.[19] The multitude of disease correlations have led to a growing interest in H2S-based therapeutics.[20–21]
While the physiological effects of H2S have been extensively studied, the mechanism by which these effects are elicited are still in the early stages of investigation. Unlike NO and CO,[22–23] H2S does not have a known second messenger that transduces its signal. A prevailing hypothesis for H2S-mediated signaling is that it involves protein persulfidation, which leads to the reversible modification of cysteine residues.[24–25] Under physiological conditions, H2S (pKa = 6.76 at 37 °C) exists predominantly as the nucleophilic sulfide anion (HS−),[26] which can react with oxidized cysteines such as S-glutathionyl, S-nitrosyl, or sulfenyl species forming the corresponding persulfide (Figure 1A).[27] Alternatively, reduced cysteines can be persulfidated by a low molecular weight persulfide (RSSH)[27] or in a reaction catalyzed by a sulfurtransferase (Figure 1B).[28] Not surprisingly, the number of persulfidated proteins in cells increases upon H2S treatment.[29] The association between protein persulfidation and the modulation of protein function in several biochemical processes has been reviewed previously.[27, 30–31] This review focuses on sulfide quinone oxidoreductase (SQOR), which catalyzes the first and committing step in the mitochondrial sulfide oxidation pathway, and could serve as the “off-switch” in H2S-based signaling or the “on-switch” in persulfide based signaling, or both. SQOR is a mitochondrial inner membrane-anchored flavoenzyme that is poised to play a critical role in H2S-based signaling while also serving as a guardian of the electron transfer chain against H2S poisoning.[8]
Figure 1. Mechanisms of protein persulfidation.
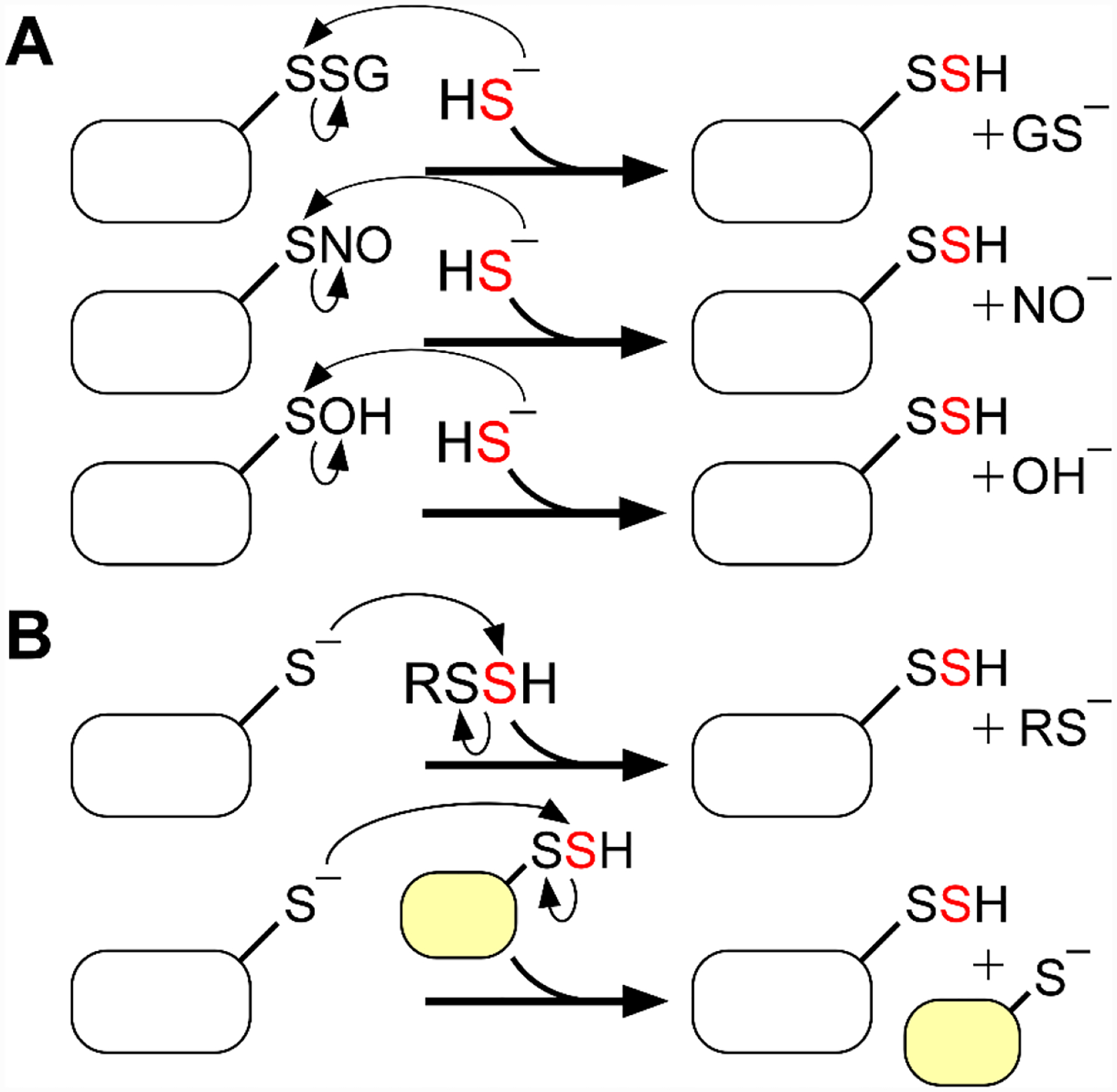
A, Persulfidation of oxidized cysteines on proteins. B, Persulfidation of reduced cysteines on proteins via reaction with a low molecular weight persulfide (RSSH) or a sulfurtransferase.
1. H2S biogenesis and catabolism
1.1. Enzymatic / microbial H2S production
Endogenous production of H2S can be catalyzed by at least three enzymes of which two are in the transsulfuration pathway, linking the methionine cycle to cysteine and glutathione (GSH) synthesis (Figure 2A).[32–33] Cystathionine β-synthase (CBS), the first enzyme in this pathway, produces H2S primarily via condensation of cysteine and homocysteine,[34] while cystathionine γ-lyase (CSE), the second enzyme in the pathway, produces H2S primarily via α, β-elimination of cysteine, forming pyruvate and ammonia in the process.[35] The relative contributions of CBS and CSE to H2S production can be estimated based on their tissue-specific expression levels, their reaction velocities, and the physiological concentrations of their substrates.[36–37] The third H2S-synthesizing enzyme, 3-mercaptopyruvate sulfurtransferase (MPST), is involved in cysteine catabolism and initially produces an enzyme-bound persulfide via the desulfuration of 3-mercaptopyruvate.[38] The final persulfide product can be a low molecular weight species (e.g. cysteine persulfide) or is protein bound (e.g. on thioredoxin), and can release H2S in the presence of a reductant, or transfer sulfane sulfur to an acceptor.[39] The two MPST isoforms in the cytoplasm (MPST1) and in the mitochondrion (MPST2), have nearly identical kinetic profiles, but differ in their tissue expression levels.[39] The steady-state H2S concentration in mammalian cells and tissues is estimated to range from 10–30 nM.[7, 40–41] An interesting exception is the aorta, in which significantly higher H2S levels are reported (~1.8 μM), potentially related to a role for H2S in vasodilation.[41]
Figure 2. H2S biosynthesis and oxidation pathways.
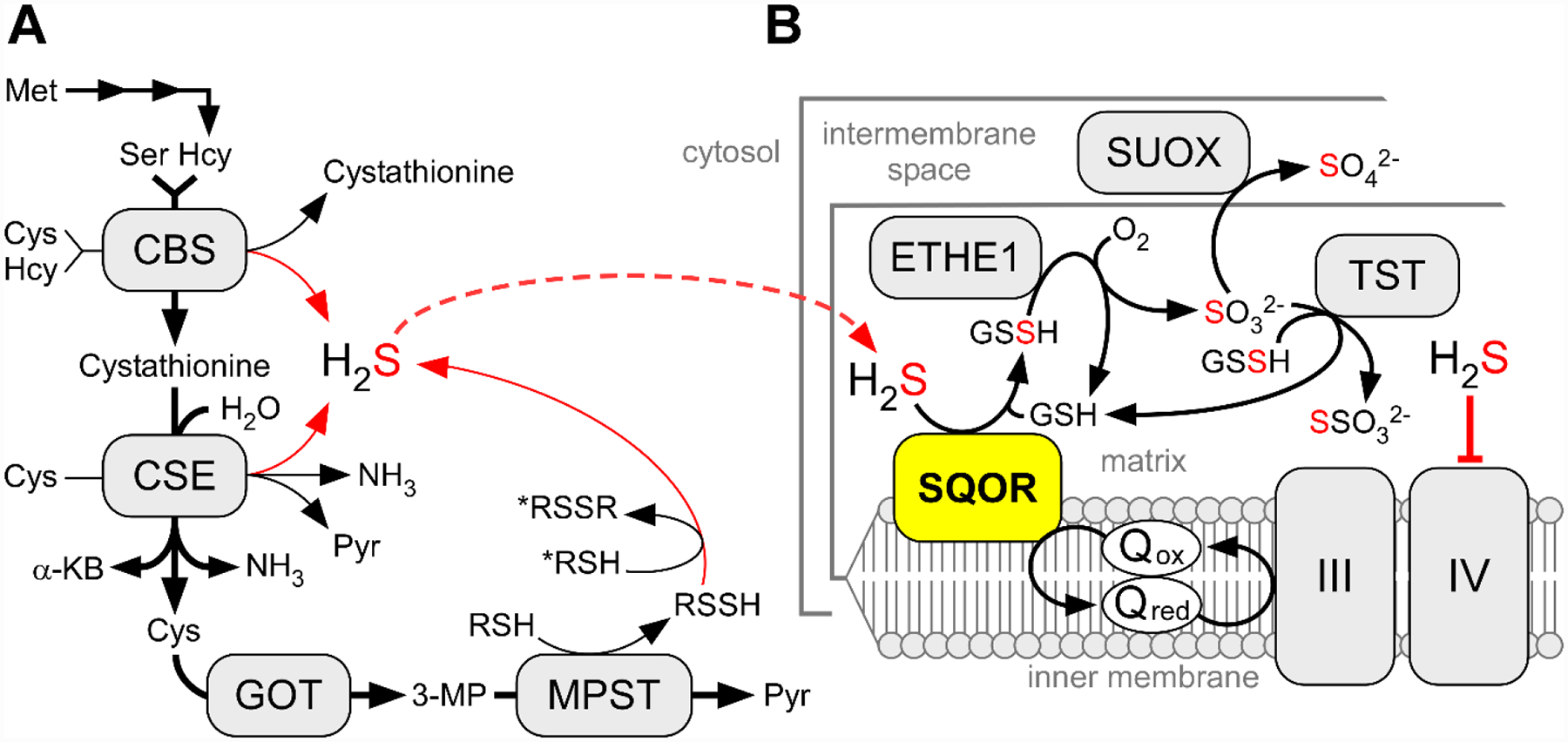
A, The canonical reactions catalyzed by CBS and CSE and MPST are depicted by bold black arrows while the H2S-synthesizing reactions are shown by thin black and red arrows. α-KB and Pyr denote α-ketobutyrate and pyruvate, respectively. GOT and 3-MP denote cysteine aminotransferase and its product, 3-mercaptopyruvate, respectively. To note, MPST has both cytoplasmic and mitochondrial isoforms. B, The mitochondrial sulfide oxidation pathway. TST, SUOX, III and IV denote rhodanese, sulfite oxidase, and respiratory complexes III and IV, respectively.
In contrast to most cells, colonocytes, which line the colonic lumen, are exposed to high H2S concentrations derived from microbial metabolism that are reported to range from 0.2–2.4 mM.[42–43] The gut microbial population is vast and is estimated to comprise 10–100 trillion organisms,[44] including sulfate-reducing bacteria.[45] Since H2S can freely diffuse across membranes,[46] it potentially exposes the colonic epithelium to toxic levels of this gas. Hence, to prevent respiratory poisoning, colonocytes must be adapted to withstand acute or steady exposure to high levels of H2S. The colonic epithelium is protected from invasive microbial growth by a two-layer barrier of mucus,[47] which is enriched with glycoproteins secreted by goblet cells.[48] However, it is largely unknown whether this protection extends to H2S entry. On the other hand, defective H2S clearance in colonocytes has been implicated in ulcerative colitis, which is marked by decreased butyrate oxidation[49–50] that, as discussed below, is an important source of energy for these cells.
1.2. Overview of the mitochondrial sulfide oxidation pathway
In mammals, the sulfide oxidation pathway resides in the mitochondrion and begins with SQOR (Figure 2B). The SQOR reaction couples H2S oxidation to coenzyme Q10 (CoQ) reduction.[51–54] Under physiological conditions, the primary sulfane sulfur acceptor for the SQOR reaction is GSH, generating glutathione persulfide (GSSH) as the product.[53–54] GSSH is then further oxidized by the iron-dependent persulfide dioxygenase, ETHE1, to produce sulfite and regenerate GSH.[55–56] GSSH is also a substrate for rhodanese (or TST), a sulfurtransferase that catalyzes the reaction of GSSH with sulfite to produce GSH and thiosulfate.[53, 57] Alternatively, sulfite is oxidized to sulfate by sulfite oxidase, which resides in the intermembrane space.[58] Electrons from the sulfide oxidation pathway enter the electron transfer chain at the level of Complex III (from SQOR) and cytochrome c/Complex IV (from sulfite oxidase).
The end products of the pathway, thiosulfate and sulfate, can be excreted. In serum, thiosulfate levels of ~0.6 μM[59] and significantly higher sulfate levels (~300 μM) have been reported.[60] Thiosulfate and sulfate concentrations are higher in urine at ~9 μM[61] and ~17 mM,[62] respectively. In sulfite oxidase deficiency thiosulfate levels increase dramatically and have been reported to be ~2 mM.[62] Thiosulfate levels in the blood and urine also increase after acute H2S exposure, thus serving as a biomarker of H2S poisoning.[63–64]
2. SQOR belongs to the flavin disulfide reductase superfamily
2.1. Flavin disulfide reductases
SQOR is a member of the diverse and extensive flavin disulfide reductase (FDR) superfamily.[65–66] FDRs often share a common mechanistic characteristic in which the flavin and the disulfide transfer electrons to one another via a transient covalent flavin C4a-S-Cys adduct. FDRs each have a tightly bound flavin cofactor, most often FAD, along with a cysteine-based center. In most instances, the flavin mediates electron transfer between thiol-disulfide exchange reactions at the cysteine-based center and organic redox molecules, including the pyridine nucleotides NADP(H) or NAD(H), quinones such as CoQ, or cytochrome c. A reduced cysteine-based center can transfer electrons to oxidized substrates including glutathione disulfide and lipoamide; alternatively, substrates including sulfide can drive electrons to an active site cysteine disulfide, which are then relayed to the flavin. A catalytic triad (Glu-His-Cys) that stabilizes the thiolate nucleophile is frequently, but not always, found in the active site of these enzymes.
The so-called classical Group 1 FDRs are typified by glutathione reductase, lipoyl dehydrogenase, and trypanothione reductase.[65–66] Group 2 also has the classical fold, but with an additional dithiol containing domain and include the high molecular weight form of thioredoxin reductase, mercuric reductase, and thioredoxin glutathione reductase. Group 1 and 2 FDRs are most often obligate homodimers because FAD, pyridine nucleotide, and cysteine domains are in one subunit, while the active site base including the catalytic triad is from another. In addition, both the binding and specificity of the non-pyridine nucleotide substrate are often contributed by both monomers.
Group 3 FDRs, including NADH peroxidase, do not contain a di-cysteine center, but instead, a single cysteine that undergoes 2-electron oxidation by H2O2 to sulfenic acid. The cysteine can form a charge transfer (CT) complex with the flavin, as seen with other enzymes in the FDR superfamily. NADH reduces the flavin, which transfers electrons to the sulfenic acid, eliminating H2O.[67] CoA disulfide reductase has a similar single cysteinyl center, which undergoes a thiol-disulfide exchange with CoA disulfide. This exchange releases one CoASH and concomitantly forms a covalent CoA disulfide with the active site cysteinyl residue. The Cys-S-S-CoA mixed disulfide is then reduced by NADPH via the flavin, releasing CoASH.[68]
Group 4 FDRs, which includes SQOR, is the most diverse both structurally and functionally. SQORs contain two redox active cysteines that are widely separated in the primary sequence but are spatially proximal.[69–73] A hallmark of this subgroup is that they can utilize diverse substrates. Many members, including SQOR and flavocytochrome c sulfide dehydrogenase (FCSD), do not use pyridine nucleotides, but instead, substitute CoQ (in SQOR), or cytochrome c (in FCSD) as an electron acceptor. In both of these cases, the enzymes oxidize sulfide. SQOR utilizes several of the chemical principles common to the FDR superfamily, as well as some unique sulfur-based chemistry during the catalytic cycle.
2.2. Classification of SQORs
SQORs are broadly distributed across all domains of life, owing to their pleiotropic roles spanning sulfide oxidation-driven energy production in bacteria and archaea to conveying sulfide tolerance to higher organisms. Following initial attempts to classify SQORs based on sequence data,[74–75] a clearer picture emerged as the first SQOR structures became available for the archaeal Acidianus ambivalens[70] and the bacterial Aquifex aeolicus[69] and Acidithiobacillus ferrooxidans[71] proteins. While the structures were similar overall, significant differences were seen in the FAD binding mode, the channel for active site access to sulfide, and the configuration of active site residues. Structure and function-based sequence fingerprints were used to classify SQORs into six groups.[76–77] Among these, the Type II SQORs encompass SQORs from all eukaryotes, including the human enzyme as well as SQOR from α-proteobacteria.
Mechanistically, Type II SQORs are more similar to FCSDs than to other SQOR types.[76] FCSDs are found exclusively in bacteria that live in sulfide-rich environments,[77] and couple H2S oxidation in the flavin-containing subunit to heme reduction in a separate cytochrome c subunit.[78] The FCSD reaction requires a cysteine disulfide, FAD, and a second equivalent of H2S as an acceptor, producing hydrodisulfide (HSSH). While FCSD and SQOR both contribute to energy production by driving sulfide-derived electrons into the respiratory chain, the SQOR reaction is more efficient since it enters earlier, at the level of complex III rather than IV, resulting in more protons being pumped and more ATP being generated. The purple sulfur bacterium Allochromatium vinosum, which possesses both FCSD and SQOR, preferentially uses the latter for sulfide oxidation, consistent with the higher energy yield from SQOR.[79–80]
3. Enzymology of human SQOR
3.1. Sulfide oxidation is coupled to CoQ reduction
The first structures of SQOR were reported a decade ago and were from bacteria.[69–71] The first structures of human SQOR were reported only recently,[72–73] and provided unexpected insights into the catalytic mechanism, as discussed later. Human SQOR is structurally similar to the Acidithiobacillus ferrooxidans SQOR (Figure 3A)[71] and to the flavoprotein subunit of FCSD (Figure 3B).[78] In each case, two tandem Rossmann folds harbor the FAD cofactor and two amphipathic helices at the C-terminus anchor the enzyme to the membrane. The redox-active cysteinyl residues in human SQOR, 379Cys and 201Cys, reside on the re face of FAD, and surprisingly, exist in a trisulfide configuration bridged by an additional sulfane sulfur atom (Figure 3C).[72–73] The si face of the FAD faces the CoQ binding pocket, which leads to the protein surface via a hydrophobic tunnel that presumably facilitates the movement of the hydrophobic CoQ between the active site and the lipid bilayer. Unlike the bacterial enzymes in which the active site appears to be protected from solvent, a large cavity on the surface of human SQOR leads to the active site and partially exposes 379Cys to the bulk solvent of the mitochondrial matrix (Figure 3C).
Figure 3. Structures of bacterial SQOR, FCSD, and human SQOR.
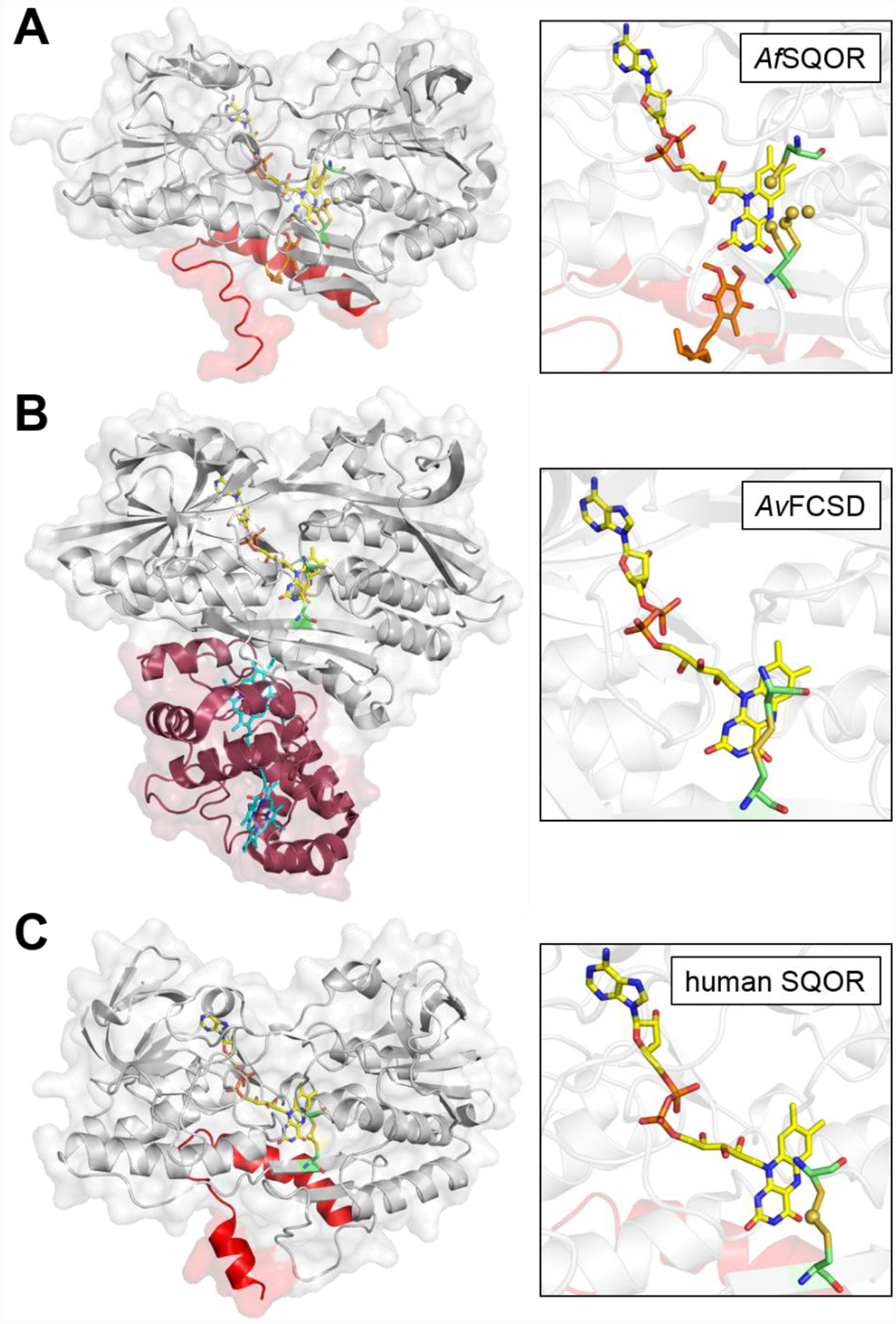
Cartoon and transparent surface overlay representations of A, Acidithiobacillus ferrooxidans SQOR (AfSQOR, PDB ID: 3T31); B, Allochromatium vinosum FCSD (AvFCSD, PDB ID: 1FCD); and C, human SQOR (PDB ID: 6OI5). The amphipathic helices of membrane-anchored AfSQOR and human SQOR are depicted in red, and the diheme cytochrome subunit of cytosolic AvFCSD is depicted in burgundy. Yellow sticks represent the FAD cofactor in each structure. The CoQ analog decylubiquinone in AfSQOR is shown in orange stick display and the dual heme cofactors in the cytochrome subunit of AvFCSD in cyan. An enlarged view of the active sites is shown in each panel on the right, with the redox active cysteines displayed in green sticks. Sulfane sulfur atoms in AfSQOR and bridging the active site cysteines in human SQOR, are shown as yellow spheres.
SQOR-catalyzed oxidation of H2S proceeds via two half reactions (Figure 4). The oxidative half reaction is initiated upon sulfide addition to the cysteine trisulfide, presumably by nucleophilic attack on the more exposed 379Cys. Of the resulting pair of persulfides,379Cys-SSH is surface exposed while 201Cys-SSH is engaged in an unusually intense charge transfer (CT) interaction with the FAD cofactor, exhibiting an absorbance maximum at 675 nm with an absorbance coefficient of ~8 mM–1 cm–1.[52, 54, 73, 81] The first half reaction is completed upon transfer of the sulfane sulfur from 379Cys-SSH to a small thiophilic acceptor, e.g. GSH, regenerating the cysteine trisulfide and leading to the two-electron reduction of FAD, presumably via the transient formation of a C4a adduct with the persulfide of 201Cys. The second half reaction then proceeds through electron transfer from FADH2 to CoQ, regenerating the resting enzyme. Reduced CoQ enters the electron transport chain at the level of complex III, making sulfide the first known inorganic substrate for human mitochondrial energy production.[82]
Figure 4. Proposed catalytic mechanism for human SQOR.
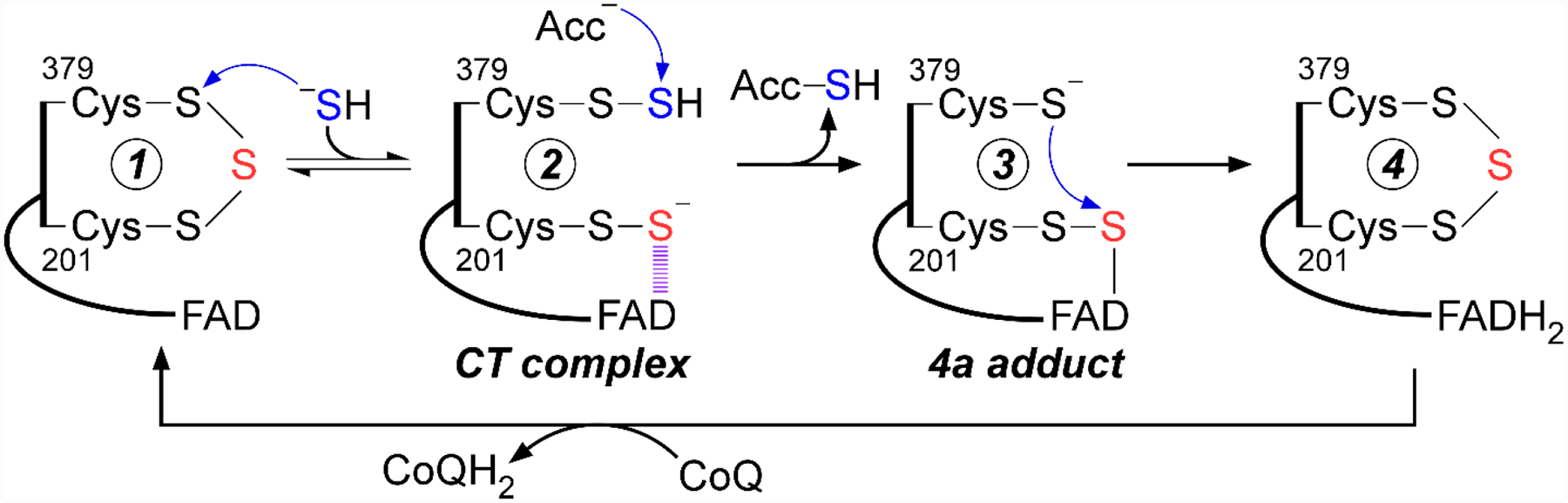
The bridging sulfur within the active site cysteine trisulfide is shown in red. The sulfur that undergoes oxidation and transfer to a thiophilic acceptor (Acc) is shown in blue.
3.2. A cysteine trisulfide is the redox-active motif in human SQOR
An unexpected surprise revealed by the crystal structures of human SQOR was that the redox active cysteines are ~4 Å apart, precluding formation of a direct disulfide bond in the absence of a large conformational change.[72–73] Instead, the two cysteines were linked via a bridging sulfur atom (Figure 3C), which was confirmed via sulfur anomalous diffraction analysis.[73] This configuration contrasts with that of other members of the FDR superfamily, in which the redox active cysteines exist as a disulfide in the oxidized state. The cysteine trisulfide configuration was initially ascribed as a purification artifact and was postulated to represent an inactive form of the enzyme.[72] A chemically unusual mechanism was proposed to explain how the trisulfide converted to a cysteine disulfide, involving Cβ-S bond cleavage in one of the cysteines and the mechanism by which the distance between the cysteines would be bridged was not addressed.[72] In contrast, studies in our laboratory led to the conclusion that the trisulfide represents the active form of SQOR, and it is reformed at the end of catalytic turnover.[73] To our knowledge, human SQOR is the first reported example of an enzyme in which the catalytically active redox cofactor has a cysteine trisulfide configuration. It is possible that the bacterial homologs have a similar configuration, although resting enzyme structures are not available. Instead, crystal structures have captured active site configurations with Cys-S-SSS-S-Cys[70, 83] and cyclooctasulfur-bound (Cys-S-S8) intermediates.[69] Cysteine trisulfides have been observed previously, e.g., as an artifact in recombinant human growth hormone.[84–87] They have also been proposed as intermediates in the catalytic cycle of Aquifex aeolicus SQOR, which generates polysulfide rather than a persulfide product,[69] and in the catalytic cycle of the bacterial dissimilatory sulfite reductase DsrC.[88]
The bridging sulfur in the human SQOR cysteine trisulfide is sensitive to cyanolysis, which was initially used to detect its presence biochemically.[73] Cyanolysis was shown to occur via nucleophilic addition of the cyanide to the trisulfide, forming a transient CT complex.[89] Subsequent decay of the CT complex leads to a disulfanyl-methanimido thioate intermediate, which was trapped in crystallo. The bridging sulfur is extracted from this intermediate in the presence of excess cyanide, preserving the oxidation state of the active site cysteines as a cyclized 201Cys-S-N=CHS-379Cys species in an inactive form of the enzyme.[89] Incubation of cyanide-treated SQOR with sulfide reforms the resting trisulfide, reactivating the enzyme. These analyses provided insights into how the trisulfide might be initially built in human SQOR, which is currently under investigation in our laboratory.
Minimally, two mechanisms can be considered for trisulfide formation. In the first, both cysteines are oxidized (e.g., to cysteine sulfenic acid), followed by the attack of a sulfide anion to form a persulfide (e.g., on the solvent accessible 379Cys (Figure 5A, 1 → 3). Then, attack of the 379Cys-SSH on the sulfenic acid moiety of 201Cys generates the trisulfide. In the second mechanism, both cysteines undergo persulfidation, an oxidative cysteine modification that has been detected in many proteins.[29] Low molecular weight persulfides (e.g. cysteine persulfide) could lead to the formation of a bis-persulfide intermediate (Figure 5B, 1 → 2) from which the trisulfide could be built. Low molecular weight persulfides are synthesized by all three H2S generating enzymes,[38–39, 90–91] and a potential role for these reactive sulfur species in signaling has been proposed.[27] Alternatively, generation of the trisulfide in SQOR could be catalyzed via sulfur transfer from a protein-bound persulfide. Candidate human sulfur transferases include rhodanese,[57] MPST,[38] and TSTD1.[28] We note that these two mechanisms could coexist. The cysteine trisulfide is more reactive than a cysteine disulfide both in terms of electrophilicity and leaving group potential. Computational modeling and molecular dynamics simulations estimate that the trisulfide configuration contributes an ~105-fold rate enhancement over a disulfide for the initial nucleophilic addition of sulfide,[89] accounting for much of the ~107-fold higher rate constant for sulfide addition to SQOR versus to a cysteine disulfide in solution.[92]
Figure 5. Possible mechanisms for in vivo building of the SQOR cysteine trisulfide.
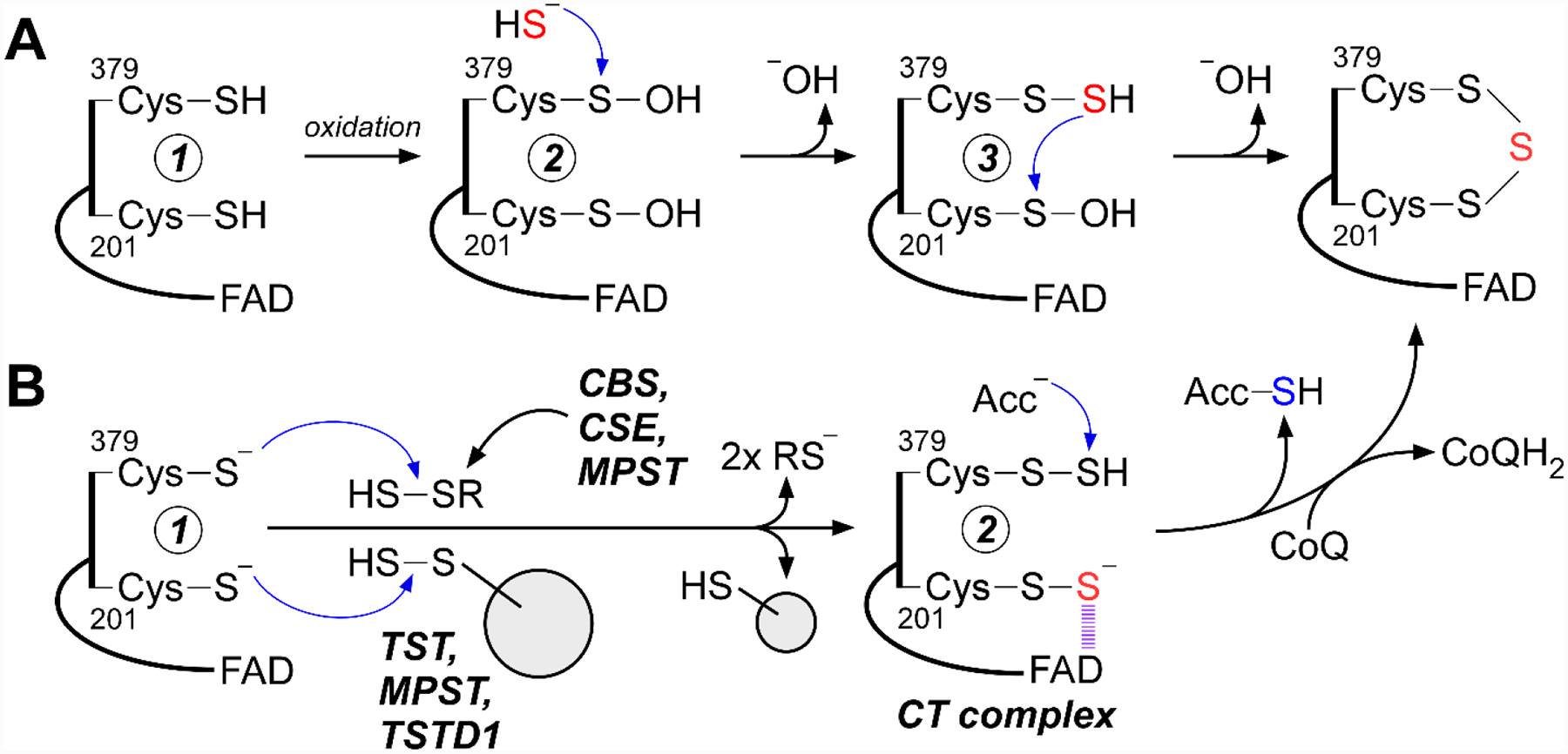
A, Oxidation of active site cysteines in newly synthesized SQOR (e.g., formation of cysteine sulfenic acid) (2) can promote cysteine persulfide formation (3) via nucleophilic attack of sulfide. Attack on the sulfenic acid by the persulfide generates the trisulfide. B, Persulfidation of SQOR at each cysteine by small molecule persulfides generated by the H2S synthesizing enzymes (CBS: cystathionine β-synthase; CSE: cystathionine γ-lyase; MPST: 3-mercaptopyruvate sulfurtransferase), or by an enzyme-catalyzed sulfur transfer via a sulfurtransferase (TST: rhodanese; MPST, or TSTD1), generates the CT complex (2). The latter leads to cysteine trisulfide formation following electron transfer to FAD and subsequent oxidation by CoQ.
3.3. Substrate promiscuity leads to dead-end complexes
Human SQOR exhibits remarkable substrate promiscuity, and in addition to sulfide, a number of nucleophiles can add to the resting trisulfide. Thus, in addition to sulfide (kon = 4 × 106 M−1s−1 at 4 °C),[54] other nucleophiles add to the trisulfide with rate constants that range over 105-fold under the same assay conditions, including sulfite (940 M−1s−1), GSH (0.38 M−1s−1), methanethiol (6.3 × 104 M−1s−1), and CoA.[73, 81, 93] The addition of alternative nucleophiles to resting SQOR leads to the corresponding 379Cys mixed disulfide and the 201Cys-SS− persulfide that forms an intense CT complex with FAD. Unlike the sulfide-induced CT complex, which decays quickly to yield FADH2,[52, 81] the alternative CT complexes represent dead-end complexes and decay slowly at rates that approximate the respective dissociation rate constants (koff) for the nucleophiles (Figure 6A).[93] Although these dead-end complexes could entrap SQOR in an unproductive state, their formation is suppressed to some extent by the membrane environment of SQOR, as revealed by a comparative kinetic analysis of the solubilized versus nanodisc-embedded protein.[93] Under certain pathological conditions, however, such as sulfite oxidase deficiency that is marked by elevated sulfite,[94] oxidative stress conditions that lead to GSH depletion,[95–96] or periodontitis marked by elevated methanethiol,[97–98] adventitious nucleophilic additions into the active site trisulfide in SQOR could become physiologically relevant and lead to impaired H2S clearance.
Figure 6. Promiscuous nucleophilic addition and active site persulfide formation.
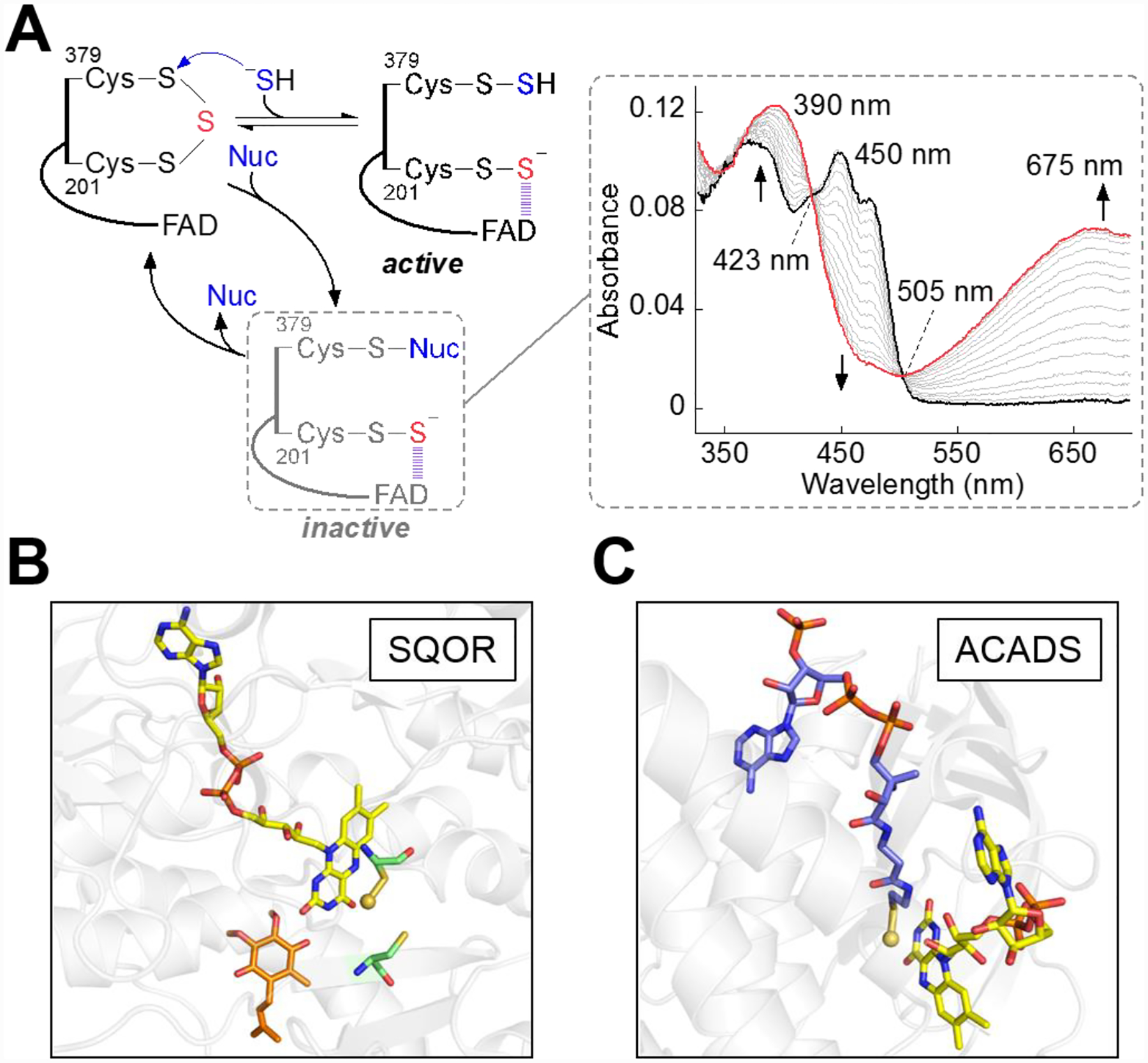
A, Alternative to sulfide, nucleophiles (Nuc) including GSH, sulfite, and methanethiol are capable of adding into the cysteine trisulfide to form a slowly decaying dead-end complex. The right panel (adapted from Ref. 93 Supplemental Figure 2) shows SQOR embedded in nanodiscs (20 μM, black trace) rapidly mixed with sulfite (500 μM) and monitored over a period of 14 s for the formation of an alternative CT complex (red trace), with an absorbance maximum at 675 nm. B, Active site of human SQOR co-crystallized with CoQ (orange sticks) and soaked with sulfite (PDB: 6OIC), which contained a stable 201Cys persulfide-to-FAD CT complex in crystallo. C, Active site of ACADS crystallized with bound CoA persulfide (PDB: 2VIG), shown as blue sticks. In panels B and C, the sulfane sulfur in 201Cys-SSH and in CoA-SSH persulfide are shown as yellow spheres.
The CT complex formed upon addition of sulfide or an alternative nucleophile has an unusually large absorbance coefficient (Figure 6A) relative to those of thiolate-to-FAD CT complexes observed in other FDRs,[99–103] including the structurally related FCSD.[104] The electron-rich persulfide-to-FAD interaction in SQOR that was predicted to form from the resting trisulfide has been captured crystallographically (Figure 6B),[73] revealing the structural basis for the unusual intensity of the CT complex. The CT complex in SQOR is reminiscent of the robust CT complex seen in short-chain acyl-CoA dehydrogenase (ACADS), which is formed between a tightly-bound CoA persulfide and FAD (Figure 6C).[105]
3.4. SQOR accommodates alternate sulfane sulfur acceptors
The substrate promiscuity of human SQOR also extends to the sulfur transfer step to a low molecular weight thiophilic acceptor, and potentially leads to a variety of reactive sulfur species as products (Figure 7). Identifying the physiologically relevant sulfane sulfur acceptor is germane to understanding the logic of how the sulfide oxidation pathway is organized. The two major proposals for the physiologically relevant acceptor are sulfite, which leads to thiosulfate formation[52] and GSH, which leads to GSSH formation.[53]
Figure 7. Catalytic promiscuity of the SQOR reaction.
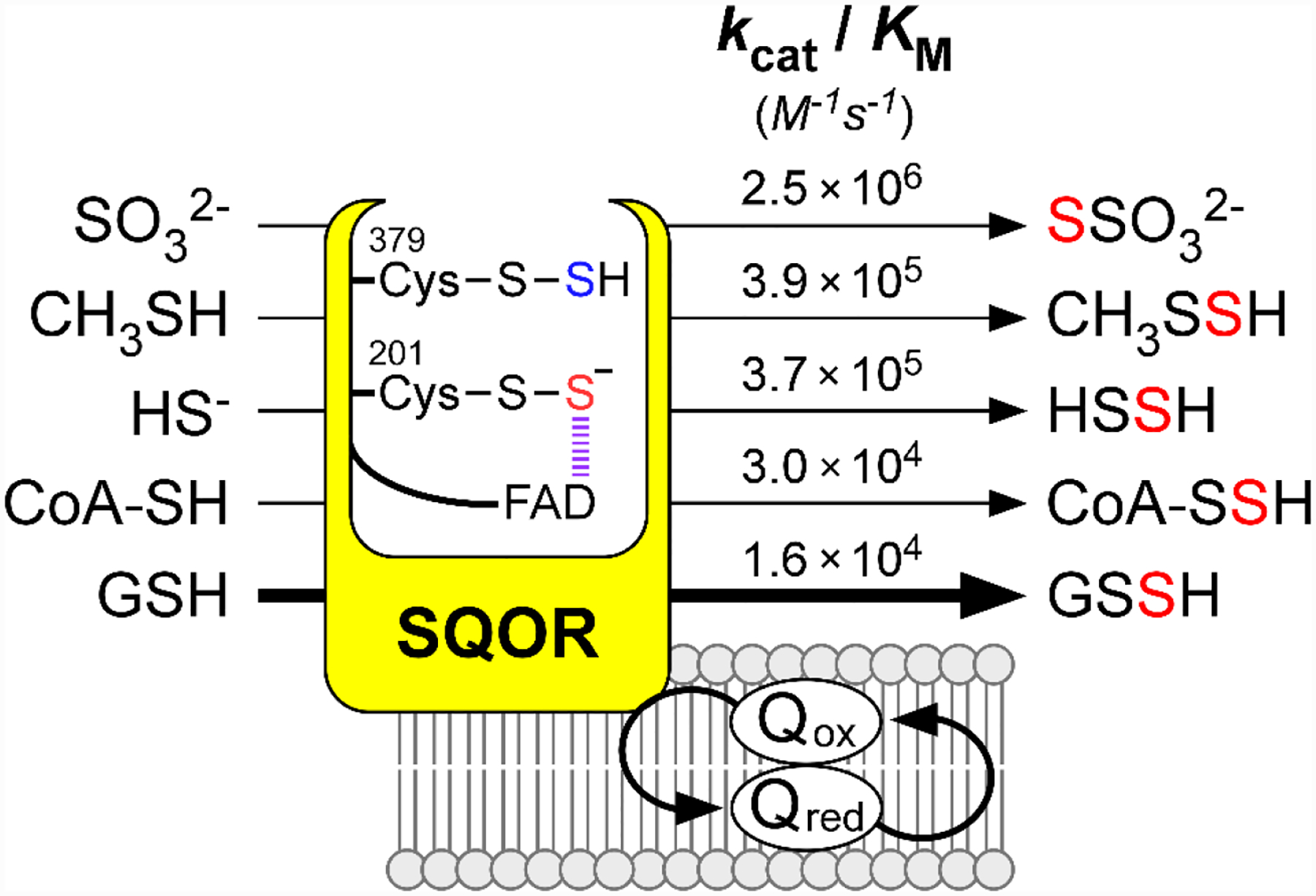
The catalytic efficiencies denoted for small thiophilic acceptors are derived from the most recent kinetic characterizations using SQOR embedded in nanodiscs (Refs. 54, 73, 93). In the CT complex intermediate, the sulfur derived from the active site cysteine trisulfide is shown in red, and the sulfur derived from H2S is shown in blue.
The proposal that sulfite is the primary physiological acceptor was largely based on in vitro kinetic data. The kcat/KM for sulfite is ~2 ☓ 106 M−1s−1,[52–54] and thiosulfate production was observed during sulfide oxidation in lugworm and rat liver mitochondria,[51] and in colon.[106] In this model, thiosulfate is postulated to be an intermediate in the production of sulfate,[107–108] which would require the kinetically unfavorable conversion of thiosulfate to GSSH via the rhodanese-catalyzed sulfur transfer reaction,[53] followed by oxidation of GSSH by ETHE1 to sulfite[55] for use by the SQOR catalytic cycle. However, this model is inconsistent with the very low intracellular concentration of sulfite, reported to range from <20 nM (free sulfite) to 0.47–4.6 μM (free + bound sulfite) in serum and plasma.[59, 109] These values are much lower than the KM for sulfite (260 ± 30 μM) that has been reported for the SQOR reaction.[54] Although higher sulfite levels in rat liver (9.2 μM) and heart (38 μM) were reported by the authors who proposed sulfite as a co-substrate for SQOR,[110] the lack of rigorous product identification raised questions about the validity of these values.[54] Since sulfite is inherently toxic[111–112] and is itself an oxidation product of sulfide, its use as a substrate for sulfide oxidation appears unlikely. Further, the buildup of toxic sulfite is averted by the activity of sulfite oxidase (kcat/KM (sulfite) = ~2.4 ☓ 106 M−1s−1).[58] However, under pathological conditions discussed above that lead to elevated sulfite, its use as an acceptor in the SQOR reaction might increase in significance.
In contrast to sulfite, GSH is highly abundant in cells (1–10 mM),[113–114] but is less efficient as an acceptor in the SQOR reaction (kcat/KM (GSH) = 1.1 ☓ 104 M−1s−1).[54] The KM(GSH) for the SQOR reaction was initially determined to be 22 ± 3 mM,[53] and later revised to 8 ± 1 mM after accounting for a non-enzymatic reduction of CoQ1 by GSH.[54] Incorporation of solubilized SQOR into nanodiscs slightly enhances the catalytic efficiencies of the reaction with either of the acceptors, with kcat/KM values of 2.5☓106 M−1 s−1 and 1.6 ☓ 104 M−1 s−1 for sulfite and GSH, respectively.[54] Kinetic simulations based on these values, and the intracellular concentrations of GSH and sulfite, predict that GSH is the dominant acceptor under physiological conditions.[53–54] The product, GSSH, is an efficient substrate for ETHE1 and is also the favored substrate for rhodanese, which converts GSSH to thiosulfate (Figure 2B). The pKa of GSSH is 5.45 (versus 8.94 for GSH) and it exists almost exclusively as the persulfide anion (GSS−) under physiological conditions.[115] This species is highly nucleophilic, which would prime it for facilitating cysteine persulfidation on proteins.
3.5. Structural basis for substrate promiscuity
The structural underpinnings of promiscuity that affords access to both small and bulky substrates[52–54, 93] became apparent from the structure of human SQOR. An early model of human SQOR[81] based on a bacterial enzyme structure presumed an active site comprising a cysteine disulfide on the re face of the FAD cofactor that was protected from solvent by capping loops that exist in some bacterial SQORs.[69–70] However, the crystal structure of human SQOR revealed that on the mitochondrial matrix side, a large electropositive cavity leads into the active site, and the orientation of the cysteine trisulfide causes 379Cys to protrude into the cavity (Figure 8A).[72–73] This exposure to bulk solvent favors 379Cys as the site of sulfide addition. The FAD cofactor is buried and shielded from solvent by a loop that spans residues 195Pro to 203Gly and includes 201Cys, which is positioned proximal to the isoalloxazine ring of FAD. Based on substrate docking models, the cavity can accommodate the GSH tripeptide with its thiol moiety positioned to accept the sulfane sulfur on 379Cys[72–73] (Figure 8B). Consequently, this cavity, which is large enough to accommodate GSH or CoA, also allows facile access to lower molecular weight acceptors including sulfite[53–54, 72] and methanethiol.[93] Presumably, substrate promiscuity is limited by the lower abundance of these acceptors relative to GSH and by the membrane environment of SQOR.[93]
Figure 8. Structural basis for catalytic promiscuity in SQOR.
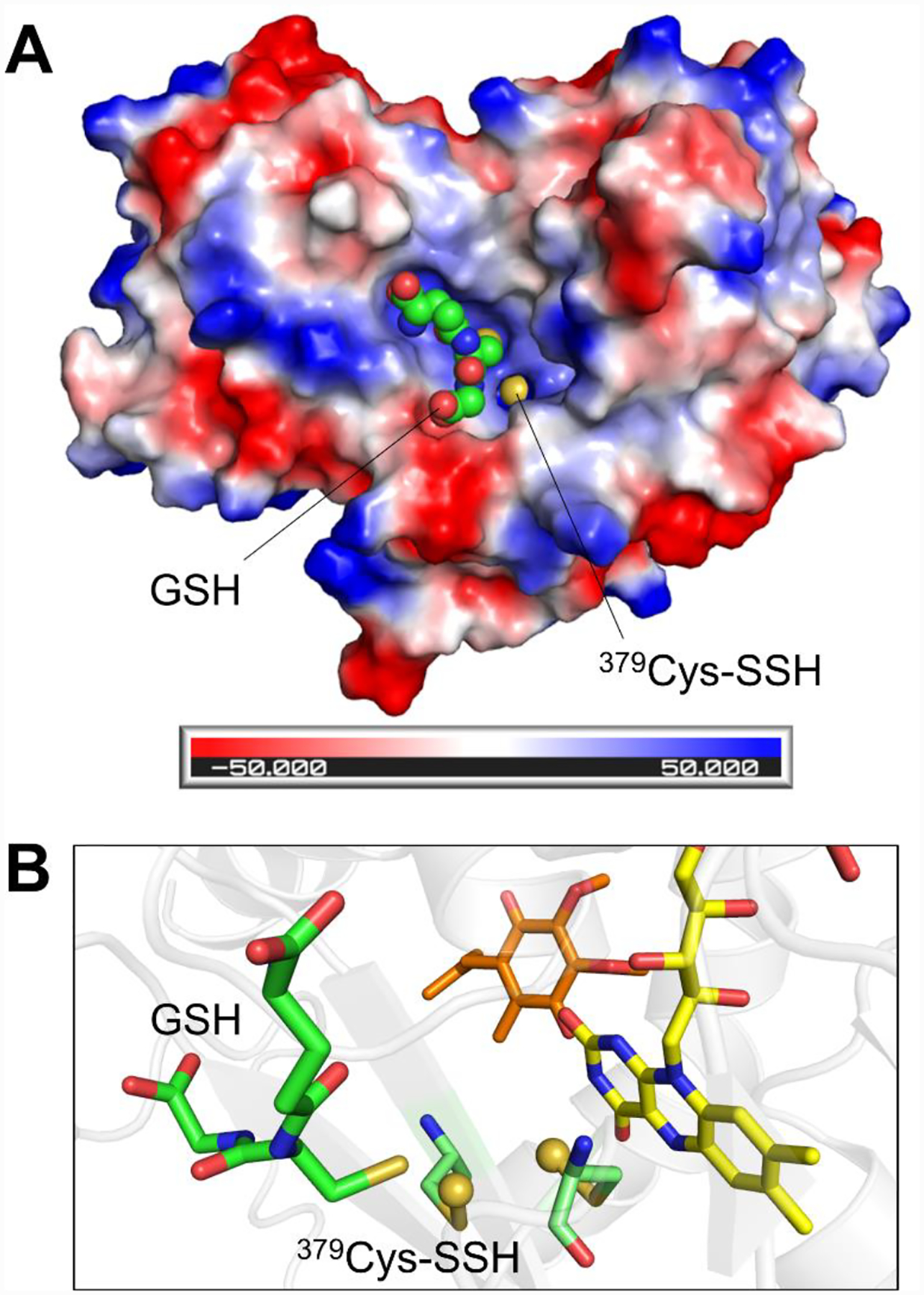
A, Electrostatic surface potential map of the SQOR monomer, revealing a large electropositive cavity containing the exposed 379Cys-SSH persulfide, shown as a yellow sphere. GSH is docked in the cavity and shown as green spheres. B, Orientation of the docked GSH, shown as green sticks, relative to the active site cysteine persulfides, shown as pale green sticks with the persulfide sulfurs shown as yellow spheres. The thiol moiety of GSH is oriented proximal to 379Cys-SSH, which would facilitate sulfur transfer (PDB: 6OIB).
4. Sulfide oxidation and its influence on metabolic pathways
4.1. Sulfide has bimodal effects on oxidative phosphorylation
A role for H2S oxidation in energy production has been known for decades with SQOR contributing to membrane-bound electron transport in autotrophic and chemolithotrophic bacteria,[116–118] archaea,[119] and eukarya.[120] Sulfide-driven quinone reduction attributable to SQOR activity was first reported in membranes of the green sulfur bacterium Chlorobium thiosulfatophilum in which H2S oxidation is coupled to the electron transport chain and reduction of NADP+.[121] Similarly, coupling of sulfide oxidation to quinone reduction and energy production in Rhodobacter sulfidophilus[122] and the cyanobacterium Oscillatoria limnetica,[123] have been reported. The link between H2S oxidation and oxidative phosphorylation was initially investigated in eukaryotes that are adapted to high environmental sulfide exposure, such as the bivalve Solemya reidi,[124–125] the killifish Fundulus parvipinnis,[126] and the lugworm Arenicola marina.[127–128] Studies on A. marina SQOR revealed that sulfide oxidation leads to CoQ reduction and entry into the electron transfer chain at the level of Complex III.[129]
H2S stimulates mitochondrial respiration at lower concentrations but inhibits it at higher concentrations, as seen in studies with isolated human mitochondria and in cell lines by monitoring oxygen consumption kinetics.[8, 82, 130] Cellular capacity for H2S oxidation is dependent in part on SQOR levels, and its overexpression in Chinese hamster ovary cells decreases sensitivity to respiratory poisoning by H2S.[130] Conversely, SQOR knockdown in HT-29 cells increases susceptibility to respiratory poisoning by H2S.[8] SQOR and the downstream sulfide oxidation enzymes, ETHE1 and rhodanese, are expressed at higher levels in colorectal cancer tissue as well as in multiple human colon cancer cell lines, which are more resistant to H2S versus non-malignant colon epithelial cells.[8] Increased expression of the sulfide oxidation pathway enzymes in cancer cells may thus counteract the anti-proliferative effects of H2S exposure.[8] Inhibition of Complex IV by H2S with consequent build-up of the CoQH2 pool, could reverse electron flow through Complex II, leading to conversion of fumarate to succinate.[82] Succinate accumulation is observed in ischemia, and restoration of Complex II activity in the forward direction during reperfusion leads to rapid oxidation of succinate, with concomitant reduction of the CoQ pool, which drives reverse electron transfer and ROS production at Complex I, contributing to tissue injury.[131] It has not been clearly established whether complex IV inhibition induces H2S oxidation via reversal of electron flow through Complex II and/or whether CoQH2 can drive reversal of electron flow through SQOR itself, leading to ROS production. Other mitochondrial flavoenzymes that connect to the electron transport chain, including dihydroorotate dehydrogenase[132] and glycerol-3-phosphate dehydrogenase,[133] can oxidize CoQH2, forming ROS.
4.2. Sulfide induces a reprogramming of mitochondrial bioenergetics
Sulfide oxidation by SQOR can exert a direct influence on bioenergetics via the mitochondrial CoQ pool, which represents a major redox nexus. Coupling of SQOR activity to the CoQ pool creates intersections between sulfide metabolism and: (i) Complex I, which oxidizes NADH, (ii) Complex II, which oxidizes succinate and FADH2, (iii) dihydroorotate dehydrogenase, which is involved in de novo pyrimidine biosynthesis,[134] (iv) glycerol-3-phosphate dehydrogenase, which links to both carbohydrate and lipid metabolism,[135] and (v) the electron-transferring flavoprotein dehydrogenase, which is involved in fatty acid and branched chain amino acid metabolism.[136] Acute exposure to high H2S levels in the colon, with a consequent decrease in the CoQ/CoQH2 ratio could limit other activities that rely on an oxidized CoQ pool. Thus, in addition to respiratory inhibition, H2S oxidation can also inhibit other oxidative metabolic pathways.
Thus, hyper-reduction of the CoQ pool, like mitochondrial dysfunction,[137] can lead to a decrease in the mitochondrial NAD+/NADH ratio, limiting electron acceptor and uridine availability. Indeed, treatment of malignant colon cell lines with H2S restricts cell proliferation, which can be restored by exogenous pyruvate and uridine, demonstrating that acute exposure to high H2S leads to electron acceptor insufficiency.[8] These cells also become deficient in aspartate synthesis, which also requires NAD+ for synthesis from carbon sources such as glucose or glutamine as demonstrated in cells with mitochondrial dysfunction.[138–139] The reductive stress in H2S-treated cells is partially alleviated by metabolic rewiring of the citric acid cycle via reductive carboxylation of α-ketoglutarate to citrate. SQOR activity can therefore trigger metabolite signaling via alterations in mitochondrial redox homeostasis.[8]
4.3. Inherited Deficiency of SQOR
Inborn errors in SQOR metabolism have been identified recently in three children from two unrelated families and were associated with severe metabolic abnormalities resembling Leigh disease.[140] Disease onset varied between ~4 to 8 years of age, and the acute symptoms were triggered by fasting or infection. The symptoms included lactic acidosis, multi-organ failure, neurological disorders, and Leigh-like brain lesions. Two patients who were siblings were homozygous for the Glu213Lys mutation, affecting a residue that is remote from the active site, but predicted to disrupt hydrogen bonding with neighboring arginine residues. The third patient was homozygous for a single base pair deletion (c446delT) in the SQOR gene, predicted to lead to mRNA degradation or production of non-functional enzyme due to the resulting frameshift. The mutations led to greatly diminished SQOR levels in tissues expressing the Glu213Lys mutation while SQOR was not detected in fibroblasts carrying the deletion mutation. Complex IV activity but not its assembly was adversely affected by SQOR deficiency. Notably, the gastrointestinal defects and accumulation of acylcarnitines reported for ETHE1 deficiency[141] were not seen in SQOR deficiency,[140] although elevated H2S was reported for both conditions.
4.4. The role of SQOR in proteostasis
Studies on Caenorhabditis elegans have implicated SQOR as a mediator of the unfolded protein response that was induced by exposure to H2S,[142] which also led to enhanced thermotolerance and increased lifespan.[143–144] Significant effects on protein translation due to H2S treatment are observed in C. elegans upon deletion of sqrd-1 encoding a putative SQOR. These effects include reduced incorporation of 35S-labeled methionine into proteins and an increase in free ribosomal subunits and translation factors.[142] Translational perturbations in the sqrd-1 mutant C. elegans were traced to an increase in eIF2α phosphorylation upon H2S exposure, leading to the induction of ER and mitochondrial stress responses.[142] Based on these results, SQOR was proposed to play a role in proteostasis.
In mammalian cells, accumulation of phosphorylated eIF2α and decreased protein synthesis were observed upon exposure to exogenous H2S, or stimulation of CSE-dependent H2S synthesis.[145] Phosphorylation of eIF2α was reversible and traced to the protein phosphatase PP1c, which was persulfidated upon H2S treatment. H2S-induced accumulation of eIF2α phosphorylation led to pre-conditioning, which partially protected cells from subsequent ER stress-induced cell death. While the role of SQOR was not investigated, protective persulfidation might play a role in the H2S-based modulation of the integrated stress response, shielding proteins from irreversible oxidative damage.
4.5. Interplay of sulfide oxidation and butyrate oxidation
H2S concentrations in the colon are higher than in other tissues by several orders of magnitude,[42–43] due to microbial H2S production.[44–45] Correspondingly, colonocytes are adapted to withstand acute exposure to high H2S.[106] The primary fuel source for these cells is butyrate, a short-chain fatty acid produced by gut microbiota via fermentation of insoluble fiber remnants.[146–148] The first step in butyrate oxidation is catalyzed by butyryl-CoA dehydrogenase (ACADS), a flavoenzyme residing in the mitochondrial matrix. ACADS oxidizes butyryl-CoA to crotonyl-CoA, with the resulting electrons relayed to the electron transferring flavoprotein, ETF. ETF subsequently reduces ETF dehydrogenase in the inner mitochondrial membrane, where electrons enter the CoQ pool. As discussed above, because sulfide oxidation by SQOR also drives electrons into the CoQ pool, acute H2S exposures can be challenging for energy production in colonocytes.
Sulfide inhibits butyrate oxidation in colonocytes, which mimics the conditions seen in ulcerative colitis and implicates sulfide as a factor in its pathology.[49–50] This inhibition is marked by increased butyryl-CoA levels and decreased crotonyl-CoA levels,[50] suggesting the involvement of ACADS. A mysteriously stable “green” ACADS was isolated decades ago from bovine, ovine, and porcine liver[149–152] and bacteria.[153] While the green color was later assigned to a CT complex between FAD and a tightly-bound coenzyme A persulfide (CoA-SSH),[105] the intracellular source of CoA-SSH remained unknown. We recently demonstrated that the substrate promiscuity of SQOR extends to CoA, supporting the synthesis of CoA-SSH, which in turn inhibits ACADS forming a CT complex.[73] We proposed that SQOR-catalyzed CoA-SSH production and subsequent inhibition of ACADS, thereby prioritizes the CoQ pool for sulfide over butyrate oxidation (Figure 9).
Figure 9. Interplay of sulfide and butyrate oxidation.
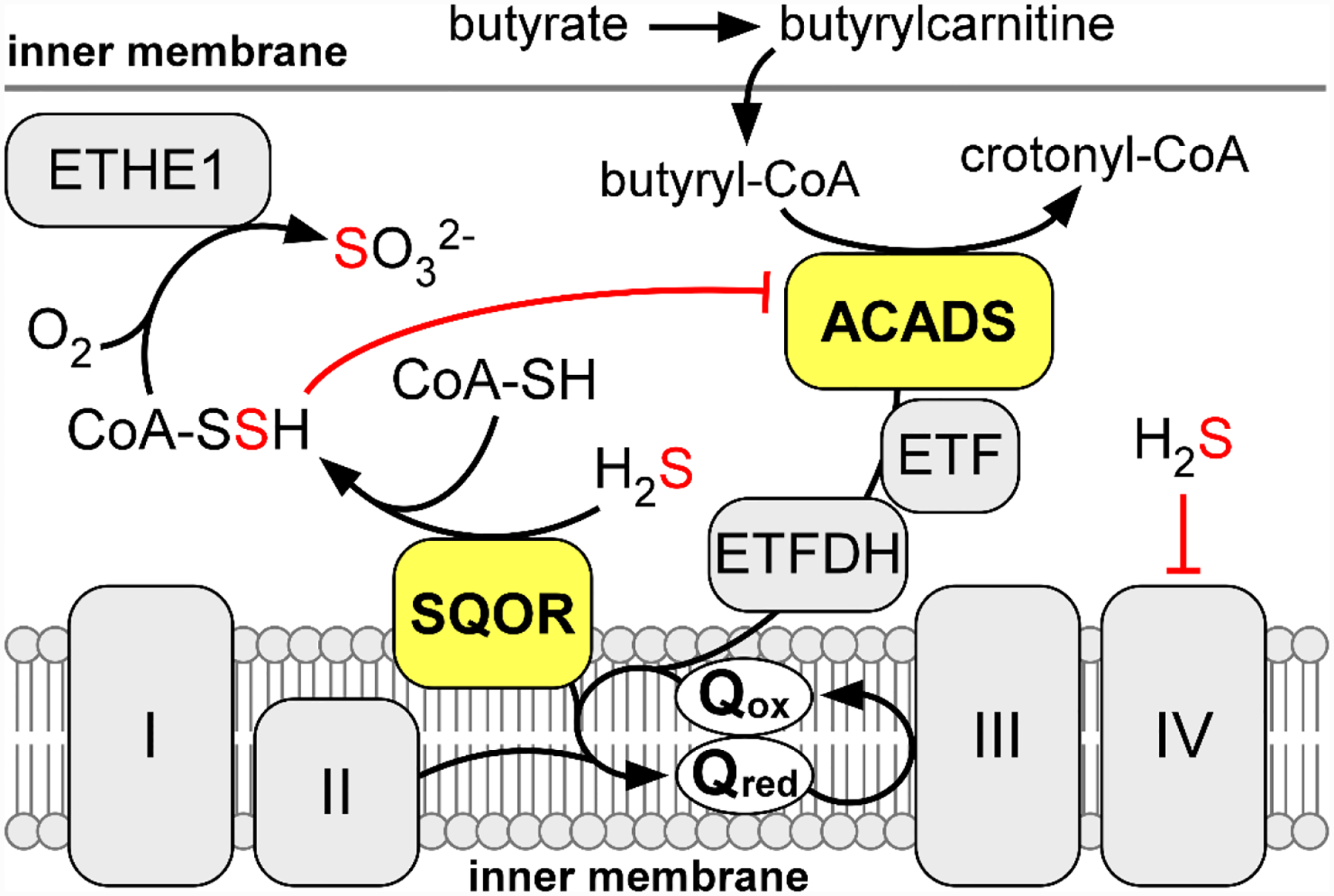
The activities of SQOR and ACADS both drive electrons into the mitochondrial Q pool, which restricts the capacity of sulfide oxidation during acute H2S exposures. As a countermeasure, SQOR can catalyze the formation of CoA-SSH, a tight-binding inhibitor of ACADs. Inhibition of ACADS by CoA-SSH relieves competition for the Q pool to prioritize sulfide oxidation.
Summary and Outlook
The high efficiency of sulfide oxidation catalyzed by SQOR serves as a protective shield against respiratory poisoning and triggers transient reprogramming of mitochondrial metabolism via inhibition of Complex IV. Inherited deficiency of SQOR presents as a newly described cause of Leigh disease with decreased Complex IV activity. On the other hand, H2S oxidation leads to the formation of highly reactive persulfides, which could be important for protein persulfidation, a modification that is increased in cells following H2S exposure. It is presently not known whether the primary role of SQOR is to activate H2S by forming persulfides while simultaneously preventing its toxic accumulation or signaling via perturbations in electron transport chain, or both.
Structural and biochemical characterization of SQOR continues to surprise, revealing a unique catalytic cysteine trisulfide, and a persulfide-based CT complex with FAD. The promiscuity of SQOR can be traced to the large electropositive entrance to the active site that accommodates a range of substrates and has the potential to generate a variety of low molecular weight persulfides. The production of CoA-SSH, long known as a tight binding inhibitor of ACADS, has recently been traced to the relaxed substrate specificity of SQOR and could prioritize sulfide over butyrate oxidation during acute colonic exposure to H2S. It is not known whether other persulfides are produced via SQOR in a tissue-specific manner and in response to intra- or extra-cellular triggers. A deeper understanding of the structure and mechanism of SQOR will facilitate its therapeutic targeting in H2S-related pathologies.
Acknowledgements
This work was supported by the National Institutes of Health (GM130183 to R.B.).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Yant WP, Am. J. Public Health 1930, 20, 598–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Evans CL, Exp QJ. Physiol. Cogn. Med. Sci 1967, 52, 231–248 [DOI] [PubMed] [Google Scholar]
- [3].Bouillaud F, Blachier F, Antioxid. Redox Signal 2011, 15, 379–391 [DOI] [PubMed] [Google Scholar]
- [4].Beauchamp RO Jr., Bus JS, Popp JA, Boreiko CJ, Andjelkovich DA, Crit. Rev. Toxicol 1984, 13, 25–97 [DOI] [PubMed] [Google Scholar]
- [5].Abe K, Kimura H, J. Neurosci 1996, 16, 1066–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kimura H, Antioxid. Redox Signal 2010, 12, 1111–1123 [DOI] [PubMed] [Google Scholar]
- [7].Vitvitsky V, Kabil O, Banerjee R, Antioxid. Redox Signal 2012, 17, 22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Libiad M, Vitvitsky V, Bostelaar T, Bak DW, Lee HJ, Sakamoto N, Fearon E, Lyssiotis CA, Weerapana E, Banerjee R, J. Biol. Chem 2019, 294, 12077–12090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Peter EA, Shen X, Shah SH, Pardue S, Glawe JD, Zhang WW, Reddy P, Akkus NI, Varma J, Kevil CG, Am J. Heart Assoc. 2013, 2, e000387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Szabo C, Coletta C, Chao C, Modis K, Szczesny B, Papapetropoulos A, Hellmich MR, Proc. Natl. Acad. Sci. U. S. A 2013, 110, 12474–12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ostrakhovitch EA, Akakura S, Sanokawa-Akakura R, Goodwin S, Tabibzadeh S, Exp. Cell Res 2015, 330, 135–150 [DOI] [PubMed] [Google Scholar]
- [12].Coletta C, Szabo C, Curr. Vasc. Pharmacol 2013, 11, 208–221 [PubMed] [Google Scholar]
- [13].Chen D, Pan H, Li C, Lan X, Liu B, Yang G, Huazhong Univ J. Sci. Technol. (Health Sci.) 2011, 31, 632–636 [DOI] [PubMed] [Google Scholar]
- [14].Ang SF, Moochhala SM, MacAry PA, Bhatia M, PLoS One 2011, 6, e24535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun L, Sun S, Li Y, Pan W, Xie Y, Wang S, Zhang Z, Chin. Med. J 2014, 127, 893–899 [PubMed] [Google Scholar]
- [16].Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC, Dickhout JG, Lhotak S, Meng QH, Wang R, Circulation 2013, 127, 2523–2534 [DOI] [PubMed] [Google Scholar]
- [17].Fang L, Li H, Tang C, Geng B, Qi Y, Liu X, Can. J. Physiol. Pharmacol 2009, 87, 531–538 [DOI] [PubMed] [Google Scholar]
- [18].Tan G, Pan S, Li J, Dong X, Kang K, Zhao M, Jiang X, Kanwar JR, Qiao H, Jiang H, Sun X, PLoS One 2011, 6, e25943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Blackstone E, Morrison M, Roth MB, Science 2005, 308, 518. [DOI] [PubMed] [Google Scholar]
- [20].Szabo C, Nat. Rev. Drug. Discov 2007, 6, 917–935 [DOI] [PubMed] [Google Scholar]
- [21].Wallace JL, Blackler RW, Chan MV, Da Silva GJ, Elsheikh W, Flannigan KL, Gamaniek I, Manko A, Wang L, Motta JP, Buret AG, Antioxid. Redox Signal 2015, 22, 398–410 [DOI] [PubMed] [Google Scholar]
- [22].Maines MD, Annu. Rev. Pharmacol. Toxicol 1997, 37, 517–554 [DOI] [PubMed] [Google Scholar]
- [23].Ignarro LJ, Biosci. Rep 1999, 19, 51–71 [DOI] [PubMed] [Google Scholar]
- [24].Mustafa AK, Gadalla MM, Snyder SH, Sci. Signal 2009, 2, re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH, Sci. Signal 2009, 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen KY, Morris JC, Environ. Sci. Technol 1972, 6, 529–537 [Google Scholar]
- [27].Mishanina TV, Libiad M, Banerjee R, Nat. Chem. Biol 2015, 11, 457–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Libiad M, Motl N, Akey DL, Sakamoto N, Fearon ER, Smith JL, Banerjee R, J. Biol. Chem 2018, 293, 2675–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gao XH, Krokowski D, Guan BJ, Bederman I, Majumder M, Parisien M, Diatchenko L, Kabil O, Willard B, Banerjee R, Wang B, Bebek G, Evans CR, Fox PL, Gerson SL, Hoppel CL, Liu M, Arvan P, Hatzoglou M, eLife 2015, 4, e10067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cuevasanta E, Moller MN, Alvarez B, Arch. Biochem. Biophys 2017, 617, 9–25 [DOI] [PubMed] [Google Scholar]
- [31].Filipovic MR, Zivanovic J, Alvarez B, Banerjee R, Chem. Rev 2018, 118, 1253–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Beatty PW, Reed DJ, Arch. Biochem. Biophys 1980, 204, 80–87 [DOI] [PubMed] [Google Scholar]
- [33].Mosharov E, Cranford MR, Banerjee R, Biochemistry 2000, 39, 13005–13011 [DOI] [PubMed] [Google Scholar]
- [34].Singh S, Banerjee R, Biochim. Biophys. Acta 2011, 1814, 1518–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chiku T, Padovani D, Zhu W, Singh S, Vitvitsky V, Banerjee R, J. Biol. Chem 2009, 284, 11601–11612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Singh S, Padovani D, Leslie RA, Chiku T, Banerjee R, J. Biol. Chem 2009, 284, 22457–22466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Kabil O, Vitvitsky V, Xie P, Banerjee R, Antioxid. Redox Signal 2011, 15, 363–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Yadav PK, Yamada K, Chiku T, Koutmos M, Banerjee R, J. Biol. Chem 2013, 288, 20002–20013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Yadav PK, Vitvitsky V, Carballal S, Seravalli J, Banerjee R, J. Biol. Chem 2020, 295, 6299–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Furne J, Saeed A, Levitt MD, Am. J. Physiol. Regul. Integr. Comp. Physiol 2008, 295, R1479–1485 [DOI] [PubMed] [Google Scholar]
- [41].Levitt MD, Abdel-Rehim MS, Furne J, Antioxid. Redox Signal 2011, 15, 373–378 [DOI] [PubMed] [Google Scholar]
- [42].Macfarlane GT, Gibson GR, Cummings JH, J. Appl. Bacteriol 1992, 72, 57–64 [DOI] [PubMed] [Google Scholar]
- [43].Deplancke B, Finster K, Graham WV, Collier CT, Thurmond JE, Gaskins HR, Exp. Biol. Med. (Maywood) 2003, 228, 424–433 [DOI] [PubMed] [Google Scholar]
- [44].Turnbaugh PJ, Gordon JI, J. Physiol 2009, 587, 4153–4158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Linden DR, Antioxid. Redox Signal 2014, 20, 818–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mathai JC, Missner A, Kugler P, Saparov SM, Zeidel ML, Lee JK, Pohl P, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 16633–16638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Atuma C, Strugala V, Allen A, Holm L, Am. J. Physiol. Gastrointest. Liver Physiol 2001, 280, G922–929 [DOI] [PubMed] [Google Scholar]
- [48].Johansson ME, Thomsson KA, Hansson GC, J. Proteome Res 2009, 8, 3549–3557 [DOI] [PubMed] [Google Scholar]
- [49].Roediger WE, Duncan A, Kapaniris O, Millard S, Clin. Sci 1993, 85, 623–627 [DOI] [PubMed] [Google Scholar]
- [50].Babidge W, Millard S, Roediger W, Mol. Cell. Biochem 1998, 181, 117–124 [DOI] [PubMed] [Google Scholar]
- [51].Hildebrandt TM, Grieshaber MK, FEBS J 2008, 275, 3352–3361 [DOI] [PubMed] [Google Scholar]
- [52].Jackson MR, Melideo SL, Jorns MS, Biochemistry 2012, 51, 6804–6815 [DOI] [PubMed] [Google Scholar]
- [53].Libiad M, Yadav PK, Vitvitsky V, Martinov M, Banerjee R, J. Biol. Chem 2014, 289, 30901–30910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Landry AP, Ballou DP, Banerjee R, J. Biol. Chem 2017, 292, 11641–11649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Tiranti V, Viscomi C, Hildebrandt T, Di Meo I, Mineri R, Tiveron C, Levitt MD, Prelle A, Fagiolari G, Rimoldi M, Zeviani M, Nat. Med 2009, 15, 200–205 [DOI] [PubMed] [Google Scholar]
- [56].Kabil O, Banerjee R, J. Biol. Chem 2012, 287, 44561–44567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Libiad M, Sriraman A, Banerjee R, J. Biol. Chem 2015, 290, 23579–23588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Johnson-Winters K, Nordstrom AR, Emesh S, Astashkin AV, Rajapakshe A, Berry RE, Tollin G, Enemark JH, Biochemistry 2010, 49, 1290–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Togawa T, Ogawa M, Nawata M, Ogasawara Y, Kawanabe K, Tanabe S, Chem. Pharm. Bull 1992, 40, 3000–3004 [DOI] [PubMed] [Google Scholar]
- [60].Cole DE, Evrovski J, J. Chromatogr. A 1997, 789, 221–232 [DOI] [PubMed] [Google Scholar]
- [61].Cole DE, Evrovski J, Pirone R, Chromatogr J. B Biomed. Appl 1995, 672, 149–154 [DOI] [PubMed] [Google Scholar]
- [62].Cole DE, Evrovski J, Clin. Chem 1996, 42, 654–655 [PubMed] [Google Scholar]
- [63].Kage S, Takekawa K, Kurosaki K, Imamura T, Kudo K, Int. J. Legal Med 1997, 110, 220–222 [DOI] [PubMed] [Google Scholar]
- [64].Durand M, Weinstein P, Forensic Toxicol. 2007, 25, 92–95 [Google Scholar]
- [65].Argyrou A, Blanchard JS, Prog. Nucleic Acid Res. Mol. Biol 2004, 78, 89–142 [DOI] [PubMed] [Google Scholar]
- [66].Miller SM, Handbook of Flavoproteins: Volume 2 Complex Flavoproteins, Dehydrogenases and Physical Methods (de Gruyter) 1st ed, Vol. 2, 2013, 165–202. [Google Scholar]
- [67].Claiborne A, Mallett TC, Yeh JI, Luba J, Parsonage D, Adv. Protein Chem 2001, 58, 215–276 [DOI] [PubMed] [Google Scholar]
- [68].Wallen JR, Mallett TC, Boles W, Parsonage D, Furdui CM, Karplus PA, Claiborne A, Biochemistry 2009, 48, 9650–9667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Marcia M, Ermler U, Peng G, Michel H, Proc. Natl. Acad. Sci. U. S. A 2009, 106, 9625–9630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Brito JA, Sousa FL, Stelter M, Bandeiras TM, Vonrhein C, Teixeira M, Pereira MM, Archer M, Biochemistry 2009, 48, 5613–5622 [DOI] [PubMed] [Google Scholar]
- [71].Cherney MM, Zhang Y, Solomonson M, Weiner JH, James MN, J. Mol. Biol 2010, 398, 292–305 [DOI] [PubMed] [Google Scholar]
- [72].Jackson MR, Loll PJ, Jorns MS, Structure 2019, 27, 794–805 e794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Landry AP, Moon S, Kim H, Yadav PK, Guha A, Cho US, Banerjee R, Cell Chem. Biol 2019, 26, 1515–1525 e1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Theissen U, Hoffmeister M, Grieshaber M, Martin W, Mol. Biol. Evol 2003, 20, 1564–1574 [DOI] [PubMed] [Google Scholar]
- [75].Pham VH, Yong JJ, Park SJ, Yoon DN, Chung WH, Rhee SK, Microbiology 2008, 154, 3112–3121 [DOI] [PubMed] [Google Scholar]
- [76].Marcia M, Ermler U, Peng G, Michel H, Proteins 2010, 78, 1073–1083 [DOI] [PubMed] [Google Scholar]
- [77].Sousa FM, Pereira JG, Marreiros BC, Pereira MM, Biochim. Biophys. Acta, Bioenerg 2018, 1859, 742–753 [DOI] [PubMed] [Google Scholar]
- [78].Chen ZW, Koh M, Van Driessche G, Van Beeumen JJ, Bartsch RG, Meyer TE, Cusanovich MA, Mathews FS, Science 1994, 266, 430–432 [DOI] [PubMed] [Google Scholar]
- [79].Reinartz M, Tschape J, Bruser T, Truper HG, Dahl C, Arch. Microbiol 1998, 170, 59–68 [DOI] [PubMed] [Google Scholar]
- [80].Frigaard NU, Dahl C, Adv. Microb. Physiol 2009, 54, 103–200 [DOI] [PubMed] [Google Scholar]
- [81].Mishanina TV, Yadav PK, Ballou DP, Banerjee R, J. Biol. Chem 2015, 290, 25072–25080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Goubern M, Andriamihaja M, Nubel T, Blachier F, Bouillaud F, FASEB J. 2007, 21, 1699–1706 [DOI] [PubMed] [Google Scholar]
- [83].Cherney MM, Zhang Y, James MN, Weiner JH, J. Struct. Biol 2012, 178, 319–328 [DOI] [PubMed] [Google Scholar]
- [84].Jespersen AM, Christensen T, Klausen NK, Nielsen F, Sorensen HH, Eur. J. Biochem 1994, 219, 365–373 [DOI] [PubMed] [Google Scholar]
- [85].Thomsen MK, Hansen BS, Nilsson P, Nowak J, Johansen PB, Thomsen PD, Christiansen J, Pharmacol. Toxicol 1994, 74, 351–358 [DOI] [PubMed] [Google Scholar]
- [86].Andersson C, Edlund PO, Gellerfors P, Hansson Y, Holmberg E, Hult C, Johansson S, Kordel J, Lundin R, Mendel-Hartvig IB, Noren B, Wehler T, Widmalm G, Ohman J, Int. J. Pept. Protein Res 1996, 47, 311–321 [DOI] [PubMed] [Google Scholar]
- [87].Canova-Davis E, Baldonado IP, Chloupek RC, Ling VT, Gehant R, Olson K, Gillece-Castro BL, Anal. Chem 1996, 68, 4044–4051 [DOI] [PubMed] [Google Scholar]
- [88].Santos AA, Venceslau SS, Grein F, Leavitt WD, Dahl C, Johnston DT, Pereira IA, Science 2015, 350, 1541–1545 [DOI] [PubMed] [Google Scholar]
- [89].Landry AP, Moon S, Bonanata J, Cho US, Coitino EL, Banerjee R, J. Am. Chem. Soc 2020, 142, 14295–14306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ida T, Sawa T, Ihara H, Tsuchiya Y, Watanabe Y, Kumagai Y, Suematsu M, Motohashi H, Fujii S, Matsunaga T, Yamamoto M, Ono K, Devarie-Baez NO, Xian M, Fukuto JM, Akaike T, Proc. Natl. Acad. Sci. U. S. A 2014, 111, 7606–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yadav PK, Martinov M, Vitvitsky V, Seravalli J, Wedmann R, Filipovic MR, Banerjee R, J. Am. Chem. Soc 2016, 138, 289–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Cuevasanta E, Lange M, Bonanata J, Coitino EL, Ferrer-Sueta G, Filipovic MR, Alvarez B, J. Biol. Chem 2015, 290, 26866–26880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Landry AP, Ballou DP, Banerjee R, ACS Chem. Biol 2018, 13, 1651–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Shih VE, Abroms IF, Johnson JL, Carney M, Mandell R, Robb RM, Cloherty JP, Rajagopalan KV, Engl N. J. Med 1977, 297, 1022–1028 [DOI] [PubMed] [Google Scholar]
- [95].Vincent AS, Lim BG, Tan J, Whiteman M, Cheung NS, Halliwell B, Wong KP, Kidney Int. 2004, 65, 393–402 [DOI] [PubMed] [Google Scholar]
- [96].Grings M, Moura AP, Parmeggiani B, Marcowich GF, Amaral AU, de Souza Wyse AT, Wajner M, Leipnitz G, Gene 2013, 531, 191–198 [DOI] [PubMed] [Google Scholar]
- [97].Iatropoulos A, Panis V, Mela E, Stefaniotis T, Madianos PN, Papaioannou W, J. Clin. Periodontol 2016, 43, 359–365 [DOI] [PubMed] [Google Scholar]
- [98].Pol A, Renkema GH, Tangerman A, Winkel EG, Engelke UF, de Brouwer APM, Lloyd KC, Araiza RS, van den Heuvel L, Omran H, Olbrich H, Oude Elberink M, Gilissen C, Rodenburg RJ, Sass JO, Schwab KO, Schafer H, Venselaar H, Sequeira JS, Op den Camp HJM, Wevers RA, Nat. Genet 2018, 50, 120–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Massey V, Palmer G, J. Biol. Chem 1962, 237, 2347–2358 [PubMed] [Google Scholar]
- [100].Massey V, Williams CH Jr., J. Biol. Chem 1965, 240, 4470–4480 [PubMed] [Google Scholar]
- [101].Abramovitz AS, Massey V, J. Biol. Chem 1976, 251, 5327–5336 [PubMed] [Google Scholar]
- [102].Matthews RG, Williams CH Jr., J. Biol. Chem 1976, 251, 3956–3964 [PubMed] [Google Scholar]
- [103].Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH Jr., Proc. Natl. Acad. Sci. U. S. A 1997, 94, 3621–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Meyer TE, Bartsch RG, Cusanovich MA, Biochemistry 1991, 30, 8840–8845 [DOI] [PubMed] [Google Scholar]
- [105].Williamson G, Engel PC, Mizzer JP, Thorpe C, Massey V, J. Biol. Chem 1982, 257, 4314–4320 [PubMed] [Google Scholar]
- [106].Levitt MD, Furne J, Springfield J, Suarez F, DeMaster E, J. Clin. Invest 1999, 104, 1107–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Szczepkowski TW, Skarzynski B, Weber M, Nature 1961, 189, 1007–1008 [DOI] [PubMed] [Google Scholar]
- [108].Koj A, Frendo J, Janik Z, Biochem. J 1967, 103, 791–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Ji AJ, Savon SR, Jacobsen DW, Clin. Chem 1995, 41, 897–903 [PubMed] [Google Scholar]
- [110].Augustyn KD, Jackson MR, Jorns MS, Biochemistry 2017, 56, 986–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Gunnison AF, Food Cosmet. Toxicol 1981, 19, 667–682 [DOI] [PubMed] [Google Scholar]
- [112].Stammati A, Zanetti C, Pizzoferrato L, Quattrucci E, Tranquilli GB, Food Addit. Contam 1992, 9, 551–560 [DOI] [PubMed] [Google Scholar]
- [113].Kosower NS, Kosower EM, Int. Rev. Cytol 1978, 54, 109–160 [DOI] [PubMed] [Google Scholar]
- [114].Meister A, J. Biol. Chem 1988, 263, 17205–17208 [PubMed] [Google Scholar]
- [115].Benchoam D, Semelak JA, Cuevasanta E, Mastrogiovanni M, Grassano JS, Ferrer-Sueta G, Zeida A, Trujillo M, Möller MN, Estrin DA, J. Biol. Chem 2020, jbc-RA120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Brune DC, Anoxygenic Photosynthetic Bacteria (Springer, Dordrecht: ) 1995, 847–870. [Google Scholar]
- [117].Friedrich CG, Adv. Microb. Physiol 1998, 39, 235–289 [DOI] [PubMed] [Google Scholar]
- [118].Kelly DP, Shergill JK, Lu WP, Wood AP, Antonie Van Leeuwenhoek 1997, 71, 95–107 [DOI] [PubMed] [Google Scholar]
- [119].Stetter KO, FEMS Microbiol. Rev 1996, 18, 149–158 [Google Scholar]
- [120].Grieshaber MK, Volkel S, Annu. Rev. Physiol 1998, 60, 33–53 [DOI] [PubMed] [Google Scholar]
- [121].Knaff DB, Buchanan BB, Biochim. Biophys. Acta 1975, 376, 549–560 [DOI] [PubMed] [Google Scholar]
- [122].Brune DC, Trüper HG, Arch. Microbiol 1986, 145, 295–301 [Google Scholar]
- [123].Arieli B, Padan E, Shahak Y, J. Biol. Chem 1991, 266, 104–111 [PubMed] [Google Scholar]
- [124].O’Brien J, Vetter RD, J. Exp. Biol 1990, 149, 133–148 [DOI] [PubMed] [Google Scholar]
- [125].Powell MA, Somero GN, Science 1986, 233, 563–566 [DOI] [PubMed] [Google Scholar]
- [126].Bagarinao T, Vetter RD, J. Comp. Physiol., B 1990, 160, 519–527 [Google Scholar]
- [127].Völkel S, Grieshaber MK, Mar. Biol 1994, 118, 137–147 [Google Scholar]
- [128].Völkel S, Grieshaber MK, Physiologist 1994, 37, A88 [Google Scholar]
- [129].Volkel S, Grieshaber MK, Eur. J. Biochem 1996, 235, 231–237 [DOI] [PubMed] [Google Scholar]
- [130].Lagoutte E, Mimoun S, Andriamihaja M, Chaumontet C, Blachier F, Bouillaud F, Biochim. Biophys. Acta 2010, 1797, 1500–1511 [DOI] [PubMed] [Google Scholar]
- [131].Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord ENJ, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa ASH, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP, Nature 2014, 515, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Forman HJ, Kennedy J, Arch. Biochem. Biophys 1976, 173, 219–224 [DOI] [PubMed] [Google Scholar]
- [133].Tretter L, Takacs K, Hegedus V, Adam-Vizi V, J. Neurochem 2007, 100, 650–663 [DOI] [PubMed] [Google Scholar]
- [134].Evans DR, Guy HI, J. Biol. Chem 2004, 279, 33035–33038 [DOI] [PubMed] [Google Scholar]
- [135].Harding JW Jr., Pyeritz EA, Copeland ES, White HB 3rd, Biochem. J 1975, 146, 223–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Watmough NJ, Frerman FE, Biochim. Biophys. Acta 2010, 1797, 1910–1916 [DOI] [PubMed] [Google Scholar]
- [137].King MP, Attardi G, Science 1989, 246, 500–503 [DOI] [PubMed] [Google Scholar]
- [138].Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM, Cell 2015, 162, 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG, Cell 2015, 162, 552–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Friederich MW, Elias AF, Kuster A, Laugwitz L, Larson AA, Landry AP, Ellwood‐Digel L, Mirsky DM, Dimmock D, Haven J, J. Inherited Metab. Dis 2020, 10.1002/jimd.12232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Tiranti V, D’Adamo P, Briem E, Ferrari G, Mineri R, Lamantea E, Mandel H, Balestri P, Garcia-Silva MT, Vollmer B, Rinaldo P, Hahn SH, Leonard J, Rahman S, Dionisi-Vici C, Garavaglia B, Gasparini P, Zeviani M, Am. J. Hum. Genet 2004, 74, 239–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Horsman JW, Miller DL, J. Biol. Chem 2016, 291, 5320–5325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Miller DL, Roth MB, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 20618–20622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Miller DL, Budde MW, Roth MB, PLoS One 2011, 6, e25476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Yadav V, Gao XH, Willard B, Hatzoglou M, Banerjee R, Kabil O, J. Biol. Chem 2017, 292, 13143–13153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Roediger WE, Gut 1980, 21, 793–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ, Aliment. Pharmacol. Ther 2008, 27, 104–119 [DOI] [PubMed] [Google Scholar]
- [148].den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud DJ, Bakker BM, J. Lipid Res 2013, 54, 2325–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Green DE, Mii S, Mahler HR, Bock RM, J. Biol. Chem 1954, 206, 1–12 [PubMed] [Google Scholar]
- [150].Mahler HR, J. Biol. Chem 1954, 206, 13–26 [PubMed] [Google Scholar]
- [151].Steyn-Parve EP, Beinert H, J. Biol. Chem 1958, 233, 853–861 [PubMed] [Google Scholar]
- [152].Shaw L, Engel PC, Biochem. J 1984, 218, 511–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Engel PC, Massey V, Biochem. J 1971, 125, 879–887 [DOI] [PMC free article] [PubMed] [Google Scholar]