

Abstract
p53 is one of the most well-studied tumor suppressors. It is mutated or deleted in half of all cancers. In the other half carrying wild type p53, the p53 signaling pathway is disrupted by abnormalities of other components in the pathway. Due to its paramount role in tumor suppression, p53 has attracted great interest in drug development as any clinically successful therapeutic agent to target the p53 pathway will save millions of lives. However, designing therapeutics targeting the pathway has been extremely challenging, despite more than forty years of research. This review will summarize past and current efforts of developing p53-based gene therapy and targeted therapies for cancer treatment. In addition, the current efforts of exploiting the immunogenicity of p53 protein for cancer immunotherapy will be reviewed. Challenges and future directions for targeting the p53 pathway will be discussed.
Keywords: p53, tumor suppressor, gain of function, cancer, cancer therapy, immunotherapy
1. Introduction
In 1979, several groups that worked on the SV40 virus independently identified a cellular binding partner of the SV40 large T-antigen. The binding partner later was named p53 and has emerged as one of the centers of cancer research1–5. In the beginning, p53 was thought to be an oncogene because most of the p53 cDNA clones were obtained from transformed cells, in which the p53 gene (TP53) was mutated6–8. Ten years later, researchers started to realize that p53 is instead a tumor suppressor, and it is mutated and deleted in more than half of human tumors9,10. In 1990, germline p53 mutations were found to be associated with the Li-Fraumeni syndrome (LFS) 11, a rare inherited familial disorder that greatly increases the risk of early onset of a wide range of cancer types12. Since then, the research of the p53 field has exploded and p53 becomes one of the most well-studied molecules in history. As of writing this review, there are some 100,000 articles cited in PubMed about p53. The publishing pace on p53 has been steadily increasing since its discovery and reached around 13 articles per day in 2019.
After more than four decades of research, it is well accepted that p53 mainly acts as a stress-activated transcription factor (Figure 1). Under unstressed conditions, p53 is ubiquitylated by E3 ligases, such as MDM213, COP114, PIRH215, and TRIM2416, and subsequently degraded by the proteasome machinery. The expression of MDM2 and PIRH2 is induced by p53 and therefore they form a negative feedback loop to control the steady-state levels of p53 in cells. Due to ubiquitylation, the half-life of p53 in cells is several minutes and the levels of p53 are kept low. Another important negative regulator of p53 activity is MDMX (also called MDM4), a homologue of MDM2. Unlike MDM2, MDM4 has no detectable E3 ligase activity and instead inhibits p53 activity together with MDM2 17 When cells encounter various stresses, such as genotoxic stresses, virus infection, hypoxia, and oncogene overexpression, p53 is activated and binds to chromatin to regulate the transcription of its downstream targets. The activation process is delicately regulated by numerous cofactors and post-translational modifications, such as phosphorylation, acetylation, sumoylation, and methylation18,19. The downstream targets mediate diverse cellular functions of p53 in apoptosis, ferroptosis, cell cycle arrest, metabolism, autophagy, stem cell differentiation, DNA repair, and senescence19. All these cellular pathways are thought to contribute to the tumor suppressive function of p53. However, recent studies using genetically engineered mouse models (GEMMs) and in vivo screens raised the possibility that some of these pathways are more important than others in tumor suppression. Using a mouse model called p5325,26, Attardi and colleagues showed that acute DNA damage-induced apoptosis and cell cycle arrest are dispensable for the tumor suppressive function of p53 20. Using several GEMMs of acetylation-defective mutants of p53, Gu and colleagues demonstrated that apoptosis, cell cycle arrest, and senescence are not the mediators of p53 in tumor suppression 21. They further provided evidence showing that metabolism regulation may be one of the mediators. Herold, Strasser, and colleagues performed in vivo screens of p53 targets in mice. They found that DNA repair genes, such as Mlh1, Msh2, Rnf144b, Cav1, and Ddit4, are critical for the tumor suppressive functions of p53, at least in the context of leukemia and lymphoma 22 Overall, these studies suggest that the relative contributions of each p53-regulated cellular pathway to tumor suppression need to be reassessed.
Figure 1. A schematic showing the p53 signaling pathway under unstressed and stressed conditions.

Under the unstressed condition, p53 is ubiquitylated by MDM2 and other E3 ligases and subsequently degraded by the proteasome machinery. Under stressed conditions, p53 is activated and post-translationally modified. Some of these modifications prevent p53 from E3-mediated degradation and increase the steady-state levels of p53. Activated p53 binds to chromatin and regulates transcription of its targets, which mediates various cellular functions of p53. These cellular functions are thought to contribute to the tumor suppressive function of p53. Red lollipops: post-translational modifications (PTMs); Blue beads on a string: nucleosomes.
Regardless of the cellular mechanisms, p53 is an indisputably important tumor suppressor. About half of all tumors have p53 mutations or deletions. In the other half, although p53 protein is intact, either upstream regulators or downstream mediators are dysregulated, which leads to the disruption of the whole p53 pathway. With the dysregulation of the p53 signaling pathway present in almost all tumors, the development of therapeutics targeting the p53 pathway has attracted great interest. Any therapeutic that is successful in the clinical setting will benefit millions of cancer patients. This review will not comprehensively review the molecular mechanisms of action of p53. Instead, it will focus on the history, strategies, and current status of developing therapeutic targets of the p53 network. The challenges of developing p53-based therapeutics and future directions will be discussed.
2. Gene Therapy
Since most cancer cells have a defective p53 signaling pathway, a straightforward concept is to put wild type (WT) p53 back into cancer cells. The WT p53 will then lead to tumor regression (Figure 2). This concept led to the development of a recombinant adenovirus expressing WT p53 (rAd5-p53) (Gendicine™), which received approval by the Chinese Food and Drug Administration (CFDA) in 2003 for treating head and neck carcinoma23. There were also reports showing clinical benefits of Gendicine™ in other types of cancer24. A similar virus (Advexin) developed by Introgen, however, failed to receive approval from the FDA in the United States in 2008. Gendicine™ is often cited as the first gene therapy treatment. It has not received approval for clinical use in other countries. Recently, another similar adenovirus expressing p53 (Ad-p53), developed by MultiVir, in combination with immune checkpoint inhibitors is in a clinical trial for recurrent or metastatic head and neck cancer (www.clinicaltrials.gov, NCT03544723).
Figure 2. Strategies of targeting cancer cells carrying WT p53, mutant p53, or p53 deletion.

Strategies for each type of cancer cells are highlighted in red. Adenovirus-based gene therapy, chemo and radiotherapy, MDM32 and MDMX inhibitors can be used to target cancer cells carrying wild type p53. Gene therapy, compounds restoring WT conformation, HSP inhibitors, immunotherapy, and inhibition of synthetic lethal genes can be used for targeting cancer cells carrying mutant p53. For targeting cancer cells with p53 deletions, gene therapy and inhibition of synthetic lethal genes can be used.
Another type of viral gene therapy related to p53 is oncolytic adenoviruses. It was known that several DNA viruses encode oncoproteins to inactivate p53. Several examples of these viral oncoproteins are polyomavirus SV40 large T-antigen, adenovirus E1B, and human papillomavirus. A mutant adenovirus called ONYX-015, which has the E1B gene deleted, was developed aiming to specifically kill tumor cells carrying defective p53 (mutations or deletions). The rationale behind ONYX-015’s putative specificity for p53 defective cancer cells is that this mutant virus will rapidly amplify in these cells and eventually lyse the cells due to the defective cell cycle arrest and apoptosis caused by p53 deficiency. On the other hand, in cancer cells or normal cells carrying WT p53, the virus cannot efficiently amplify because wild type p53 elicits stress responses and limits the spread of the virus. It is noteworthy that the specificity of ONYX-015 toward killing cancer cells with defective p53 has remained controversial. Some reports supported this concept while others showed that ONYX-015 kills cancer cells regardless of p53 status25–29. Some reports even showed the opposite: p53 is required for the killing of ONYX-01529,30. The clinical development of ONYX-015 was suspended due to financial reasons. A similar oncolytic adenovirus called H101, however, was approved by CFDA in 2005 to treat head and neck cancer31. Similar to rAd5-p53, H1010 has not received any clinical approval for cancer treatment outside of China.
In theory, the reintroduction of WT p53 may also inhibit certain tumors carrying the WT p53 gene. As described above, although these tumors have intact p53, the activity of the p53 pathway may be dysregulated, for example, through the overexpression of its negative inhibitors, MDM2 or MDMX. A common issue for virus-based gene therapies for p53 is delivery efficiency. Since not every cell in a tumor is transduced by the virus, tumor relapse is very common after treatment31.
3. Activate p53 by MDM2 and MDMX Inhibitors
3.1. MDM2 inhibitors
MDM2 and MDMX are major negative regulators for p53. Mdm2 or MdmX deletion in mice causes early embryonic lethality, which is completely rescued by p53 deletion, suggesting that the main functions of these two proteins are to inhibit p5332–34. In cancer cells carrying WT TP53, the MDM2 and MDMX genes are frequently overexpressed either through gene amplification or transcriptional upregulation35,36. Therefore, MDM2 and MDMX are good drug targets for cancer treatment (Figure 2), as inhibition of MDM2 or MDMX will increase the levels and/or transcriptional activity of p53 in WT p53-carrying cancer cells.
The first MDM2 inhibitor, Nutlin 3a, was designed by Roche 37 Nutlin 3a binds to MDM2 at the p53 interacting domain and therefore blocks the interaction between p53 and MDM2, which leads to the accumulation and elevated transcription activity of p53. Nutlin 3a has been shown to induce cell cycle arrest and apoptosis in cancer cells in vitro and xenograft tumors in vivo37. Studies by other groups reported that Nutlin 3a had p53-independent effects in cells, probably due to the off-target effect or the fact that MDM2 also has other targets besides p53. For example, p73, RB1, and E2F1 all are degraded by MDM238–40.
Although Nutlin 3a had shown a good efficacy of killing cancer cells in vitro, its suboptimal pharmacological properties prevented its further clinical development. The second generation of Nutlin compound, RG7112 (RO5045337), was developed by Roche and tested in clinical trials41. Compared to Nutlin 3a, RG7112 showed lower IC50 in killing cancer cells and higher selectivity for MDM241. Two registered Phase I trials for cancer treatments (www.clinicaltrials.gov, NCT00623870, NCT00559533) were completed, in which the maximum tolerated dose of RG7112 was determined in hematologic neoplasms and advanced solid tumors. However, no phase II or III trials have been conducted for RG7112.
Thus far, RG7388 (RO5503781, Idasanutlin) is probably the most potent and selective Nutlin 3a derivative42. It inhibited the growth of SJSA1 (WT p53) human osteosarcoma xenograft tumors at a dose of about four times lower than RG711242. Currently, there are 15 registered clinical trials of RG7112 either as monotherapy or in combination with other anticancer agents. The most advanced clinical trial of RG7112 is a phase III trial (www.clinicaltrials.gov, NCT02545283) in relapsed or refractory acute myeloid leukemia (AML), which has finished recruiting patients. The result of this trial is expected in 202243. In the trial, the primary endpoint is to evaluate the effect of RG7112 in combination with the chemotherapy drug, Cytarabine, versus Cytarabine alone on the overall survival of patients with WT p53.
Other companies or institutes have also developed their MDM2 inhibitors, such as AMG232 from Kartos Therapeutics, SAR405838 (MI-77301) from Sanofi, and MK-8242 (SCH-900242) from Merck44. These MDM2 inhibitors are also being evaluated in clinical trials for various types of cancer either alone or in combination.
3.2. MDMX inhibitors
Like MDM2, MDMX has also attracted much attention for inhibitor development, although fewer inhibitors have been identified to date. Most earlier efforts were devoted to designing stapled peptides to inhibit MDMX activity. Stapled peptides generally have an alpha helix structure and a hydrocarbon bond (staple) between two non-adjacent amino acid residues. These hydrocarbon-stapled peptides have shown biological activity toward inhibiting protein-protein interactions45. Compared to small-molecule compounds, stapled peptides can target protein-protein interactions (PPIs) with greater specificity because of their ability of binding to large PPI surfaces. In addition, stapled peptides do not generate toxic metabolite intermediate during degradation as most small-molecule compounds do. Therefore, stapled peptides offer a new way of therapeutic intervention.
A highly specific stapled peptide (Stabilized Alpha Helix of p53, SAH-p53-8) to inhibit the MDMX:p53 interaction has been designed46. SAH-p53-8 was found to activate p53 in MDMX-dependent cancer cells and induce apoptosis in vivo. It also showed activity to inhibit tumor growth in a xenograft model of JEG-3. However, later studies showed that SAH-p53-8 binds to serum avidly, a property limiting its entry to tumor cells and further clinical development47,48. Based on a different peptide sequence, ALRN-6924, a dual stapled peptide inhibitor for MDM2 and MDMX, was developed by Aileron Therapeutics. ALRN-6924 simultaneously inhibits MDM2 and MDMX and shows a promising anti-tumor effect in several xenograft tumor models overexpressing MDM2 or MDMX49. This dual inhibitor is being evaluated in five Phase I and Phase II clinical trials for AML and several solid tumors.
3.3. Side effects of MDM2 and MDMX inhibitors
One interesting observation of MDM2 and MDMX inhibitors is that reactivation of p53 by these inhibitors is generally well tolerated by most normal tissues, although these tissues also express WT p53. In both xenograft mouse models and clinical trials, most normal tissues seem to have a higher threshold for p53-induced killing than tumor tissues. This concept is underscored by studies using GEMMs, in which WT p53 is restored in p53 defective tumor cells by genetic approaches50–52. In these whole animal studies, restoration of WT p53 is well tolerated in normal tissues.
It is not completely understood why WT p53 tumors are more sensitive to p53 reactivation than most normal tissues. One possibility is that although the TP53 gene is intact in WT p53 tumors, the activity of the p53 pathway is dysregulated. The survival signaling pathways in these tumor cells are significantly reprogramed in such a way that makes these cells addicted to the downregulation of p53 activity. Therefore, p53 reactivation disrupts reprogrammed survival pathways and causes cell death. The other possibility is that p53 has non-cell-autonomous functions. In the tumor settings, p53 activation affects the functions of tumor infiltrated immune cells and/or stromal cells, which have effects on tumor growth. A third possibility is that p53 reactivation preferentially kills rapidly proliferating cells, such as cancer cells and certain blood cells. Indeed, the most common side effect of the MDM2 inhibitor RG7112 in clinical trials is hematological toxicity53,54. Understanding the mechanism(s) underlying the preference of p53 activation to cancer cells may help to develop better inhibitors with increased specificity and decreased side effects.
4. Target p53 Mutants
Most tumor cells contain TP53 genetic alterations including mutations, deletions, and translocations. The type and penetrance of these alterations vary among tumor types. For example, serous ovarian cancer has a high frequency (almost 100%) of TP53 mutations while teratomas have a very low frequency (about 2%). Osteosarcoma has a high frequency of deletions instead of mutations. Because TP53 mutations are much more prevalent than deletions and translocations55, this review will mainly focus on cancer cells with TP53 mutations. Most TP53 mutations are clustered in the DNA binding domain. Within the DNA binding domain, several amino acid residues have a much higher frequency of mutations than others, and these residues are generally called hotspots TP53 mutations. R175, G245, R248, R249, R273, and R282 are the major hotspots. The top eight hotspots represent about one-third of all TP53 mutations56. TP53 mutations not only cause loss of function in p53 mutants but also endow these mutants with new functions, dubbed gain of functions (GOFs), which will be described below. To target cancer cells with TP53 mutations, in addition to the viral gene therapy described above, there are at least three other major approaches, which will be reviewed as follows (Figure 2).
4.1. Restore WT p53 activity
CP-31398 is the first reported small-molecule compound that stabilizes and increases the transcriptional activities of mutant p5357. It was discovered by screening compounds that can restore the WT conformation to a mutated p53 DNA binding domain. The assay was based on the recognition by a monoclonal antibody (mAb1620), which preferentially binds to the WT conformation. CP-31398 increased mRNA levels of a p53 target, p21 (CDKN1A), in SAOS2 cells (p53 null) exogenously expressing p53 mutants (C173A and R249S). However, the mechanism of action of CP-31398 was questioned by another study58. After extensive testing, Fersht and colleagues found no evidence to support that CP-31398 bound to the DNA binding domain of p53. Instead, they found that the compound bound to DNA. Furthermore, p53 mutant-independent activity was observed for CP-31398. The team did find that the compound increased the recognition of R175 mutant by mAbl620. Studies from others reported that CP-31398 also affected the activity of WT p53 59,60. One study raised the possibility that CP-31398 may regulate the activity of p53 by alkylating the thiol groups in the DNA binding domain of p5361. These studies suggest that further studies are needed to delineate the mechanism of action of CP-31398.
Following CP-31398, several compounds were reported to reactivate mutant p53. STIMA-1, a compound with a similar structure to CP-31398, was found to induce apoptosis of p53 mutant (R175H and R273H)-carrying cancer cells than WT or p53 null cells61. Like CP-31398, STIMA-1 was shown to alkylate the thiol group in the DNA binding domain of p53. In vitro, STIMA-1 stimulated the expression of p21 and PUMA in a p53 mutant-dependent manner. Another compound PRIMA-1 and its derivative PRIMA-1met (APR246) have been developed by Wiman and colleagues62,63,64 PRIMA-1met has been shown to covalently modify residues C124 and C277 and reactivate mutant p53. Both PRIMA-1 and PRIMA-1met (APR246) have been and are being evaluated in the clinical trials (www.clinicaltrials.gov, NCT03268382, NCT03931291, NCT04214860) 65. RITA is another compound that was shown to reactivate several p53 mutants, such as R175H, R248W, and R273H66.
Using a rational designing approach based on p53 structure, small molecule compounds PK083 and PK7080 were found to target a unique surface cleft created by the Y220C mutation67–70. Both PK083 and PK7080 bound to the Y220C mutant, restored the WT conformation, and induced Y220C-dependent cell cycle arrest and apoptosis67,70.
Correct folding of p53 requires zinc, and lack of zinc in the DNA binding domain leads to misfolding of the protein. By supplying zinc to certain p53 mutants, such as R175H, these mutants can restore WT p53 conformation and function71,72. Through screening the NCI-60 cell line panel, Carpizo and his colleagues identified a compound called NSC319726/ZMC1 that specifically reactivated R175H mutant73. Mechanistic studies revealed that NSC319726 did not bind to p53. Instead, it increased the intracellular zinc concentration and therefore enhanced the folding of R175H mutants.
It has been shown that not all p53 mutants have the same function74. In the same vein, the mechanisms underlying the DNA binding loss of p53 mutants are not the same. In a simplified way, TP53 mutations are categorized into two groups, DNA contact and structural75. Contact mutations, such as R273H, block the binding of p53 to DNA while the overall structure of the mutant remains largely the same as WT. Structural mutations, such as R175H, disrupt the local or global structure of p53. Therefore, it is highly unlikely for one compound to restore WT conformation to every p53 mutant. For designing clinical trials using these compounds, it is critical to select a subset of patients whose p53 mutants can respond to each compound.
4.2. Inhibit the gain of functions of p53 mutants
The gain of function (GOF) of mutant p53 was first reported by Levine and colleagues76. In that study, several hotspot p53 mutants were introduced into p53 null cancer cells (SAOS2) or mouse fibroblasts [10(3)]. 10(3) cells expressing these p53 mutants were more tumorigenic in nude mice than parental cells. SAOS2 cells expressing some of the p53 mutants formed more colonies in the anchorage-independent assay than parental cells but failed to generate tumors in nude mice. Using the enhancer/promoter of multi-drug resistant gene (MDR) as a reporter, p53 mutants were shown to induce the expression of MDR while the WT p53 did not, demonstrating that hotspot p53 mutants have a GOF compared to WT p53. The GOF concept was further underscored by genetically engineered mouse models (GEMMs). R172H (R175H in human) and R270H (R273H in human) knockin mice showed similar overall survival compared to p53 knockout mice77,78. However, tumors from the knockin mice were more metastatic than those from the knockout mice, indicating that pro-metastasis is another GOF.
The discovery of the GOFs of p53 mutants provides a basis for developing therapeutics to inhibit the oncogenic effects resulting from the GOFs of p53 mutants. Since the discovery, many mechanisms of mutant p53 GOFs have been proposed. The pro-metastatic effects of R172H and R270H were attributed to p63 inhibition77,78. Further, it was later shown that hotspot p53 mutants enhanced metastasis through the SMAD/TGF-beta pathway79, EGFR/integrin recycling80, or snoRNA upregulation through ETS2 81. Besides cell migration and metastasis, several other tumor cell behaviors have been reported to be affected by p53 mutants. For example, p53 mutants have been shown to affect mammary acinar structure by disrupting the mevalonate pathway82 and to promote breast cancer cell survival via binding to ETS2 and inducing the expression of several epigenetic enzymes or modifier, such as MLL1, MLL2, and MOZ83.
A majority of the proposed molecular mechanisms of p53 mutant GOFs involve the binding of p53 mutants to transcription factors, such as p63, ETS, and NF-Y. The binding of p53 mutants to the new interacting transcription factors allows p53 mutants to regulate new targets. Notably, there are thousands of publications about GOFs of p53 in the literature. To gain detailed mechanisms of GOFs of mutant p53, readers are encouraged to read several recent reviews84–90.
A general strategy of targeting the p53 mutant GOFs is to “piggyback” on existing therapeutic agents targeting the pathways or genes affected by mutant p53, for example, EGFR inhibitors , statins for the mevalonate pathway82, and inhibitors for MLLs83. All these inhibitors either have already been approved or are being evaluated in clinical trials. It is worth mentioning that GOFs are highly context-dependent and diverse. The unique genetic makeup of each tumor cell may determine or affect the GOFs of p53 mutants. Due to these reasons, the translation of these exciting bench observations into the clinic is challenging. In addition, there is emerging evidence showing that interactions between mutant p53 and the tumor microenvironment may shape the GOFs of mutant p5391. Nonetheless, a comprehensive picture of the GOFs of p53 may contribute to personalized medicine in cancer treatment.
4.3. Target p53 mutant stability
One strategy of targeting the p53 mutant is to reduce its stability. This strategy is based on the concept of oncogene addiction. Oncogene addiction describes the phenomenon that some cancer cells, despite their complex genetic and epigenetic alterations, rely on a single or a small set of oncogenes for survival or growth92. As described above, certain p53 mutants are pro-oncogenic6–8. If cancer cells are addicted to mutant p53, reduction of mutant p53 levels will cause cancer cell death.
The stability of mutant p53 is enhanced by heat shock proteins (HSPs), such as HSP90, HSP70, and its cofactor HDAC693,94. Binding of the HSP complex prevents degradation of mutant p53 by ubiquitin E3 ligases, MDM2, and CHIP95,96. HSP inhibitors, such as Geldanamycin, have been shown to destabilize mutant p5397. Treating mice carrying germline p53 R172H or R248Q mutation with HSP90 inhibitors, Ganetespib or Alvespimycin (17DMAG) plus SAHA (histone deacetylase inhibitor), significantly extended the life span of these mice, suggesting that HSP inhibition is a feasible strategy to target mutant p53. HSP inhibition has long been investigated in clinical trials, although so far none of the HSP inhibitors has received FDA approval98,99. It is worth noting that HSPs have many clients besides mutant p53. Indeed, the stability of WT p53 is also regulated by HSPs. Therefore, HSP inhibition will have pleiotropic effects on cancer cells100. Therefore, p53 mutation status alone may not be a good predictor for the clinical benefits of HSPs inhibitors.
As described above, MDM2 inhibitors are being evaluated in clinical trials to inhibit WT p53 tumors. The fact that mutant p53 is also degraded by MDM2 creates a conundrum for these inhibitors because these inhibitors may promote tumorigenesis and/or metastasis in tumors carrying mutant p53. This idea was supported by a study from Lozano and colleagues101. Using a GEMM of p53 mutant R172H (R175H in humans), they found that Mdm2 deletion in the context of R172H mutant promoted tumorigenesis and metastasis. It will be interesting to see in clinical trials whether long-term treatment with MDM2 inhibitors selects p53 mutant clones that are more aggressive than WT clones.
5. Synthetic Lethality of p53 Loss
As described above, targeting the GOFs of p53 mutants is extremely challenging due to the diverse and context-dependent mechanisms of GOFs. Another strategy is to exploit the synthetic lethality mechanism(s) in cancer cells carrying TP53 mutations. The basis of synthetic lethality is that cancer cells develop two compensatory survival pathways. Removal of either pathway does not kill the cell while simultaneous deletion of the two pathways (synthetic) causes cell death (lethality)102. A clinically proven example of synthetic lethality in cancer treatment is the use of poly(ADP-ribose) polymerases (PARP) inhibitors in BRCA1- or BRCA2-defective breast and ovarian cancers. PAPR is involved in repairing single-strand breaks. When PARP is inhibited, single-strand breaks will turn into double-strand breaks. In cells with proficient BRCA1 and BRCA2, these double-strand breaks will be repaired by BRCA1 and BRCA2, and cells survive. However, in cells with defective BRCA1 and BRCA2, unrepaired double-strand breaks kill the cells. Therefore, synthetic lethality is extremely useful for targeting the loss of a tumor suppressor. Synthetic lethality screens have been performed in cancer cells with TP53 mutations or deletions. In a study using a computational approach in NCI-60, TCGA, CCLE datasets, synthetic lethality candidates to p53 loss were identified, such as PLK1, PLK4, CDK1, CDK16, MTOR, AURKA, etc103. Using an RNAi kinome viability screen in head and neck squamous cell carcinoma cells, several kinases, such as WEE1, AURKA, FYN, were found to be synthetic lethal to p53104. The WEE1 inhibitor, MK-1775, was selected for further preclinical studies and showed activity as a single agent or in combination with cisplatin in p53 mutated head and neck tumors. Phase I and II clinical trials using another WEE1 inhibitor, AZD1775, either as a monotherapy or in combination with gemcitabine, cisplatin, or carboplatin, have shown clinical benefits for patients with advanced solid tumors105,106. Although these Phase I and II trials were completed in 2015, no registered Phase III clinical trial has been initiated.
As cancer genomes are extremely genetically and epigenetically heterogeneous, the genes synthetically lethal to p53 are likely cell type- and tissue type-dependent. Due to the throughput limitation, most synthetic lethal screens were done in one or two cell lines. Therefore, it remains unclear whether the hits from the screens are cell type-specific. For this reason, future screens using more diverse cell lines are needed to identify more general synthetic lethal genes to p53.
6. Cancer Immunotherapy for p53
6.1. Mutant p53-based adoptive cell transfer
Adoptive cell transfer (ACT) is one category of cancer immunotherapy and has remarkable clinical benefits for advanced cancers, such as metastatic melanomas 107,108 . For patients with metastatic melanomas, the response rate is 50%, and some patients have achieved very long term remissions 108. There are several types of ACTs. One type is called tumor infiltrating lymphocytes (TILs), in which T cells are isolated from cancer patients, propagated in large quantities ex vivo, and then infused back into the same patients to fight cancer (Figure 3). Another type of ACT is the T cell receptor (TCR) T cell transfer. In TCR-ACT, TCRs are cloned from T cells that recognize a small peptide from tumor cells, called tumor antigen. Cloned TCRs are packaged into a retrovirus, lentivirus, or Sleeping Beauty (a baculovirus system), which are subsequently used to transduce T cells. These engineered T cells (TCR-T) now can specifically target the tumor cells and mediate tumor regression after being reinfused into the patient. The third type of ACT is the chimeric antigen receptor T (CAR-T) cells. CAR-T cells are different from TCR-T cells in that the TCR in CAR-T is engineered to have both antigen binding and T cell stimulation modules (chimeric). Three CAR-T cell therapies have received FDA approvals for treating B-cell precursor acute lymphoblastic leukemia, relapsed or refractory large B-cell lymphoma, and relapsed or refractory mantle cell lymphoma (MCL).
Figure 3. A framework of exploiting the immunogenicity of p53 mutants for the adoptive cell transfer.
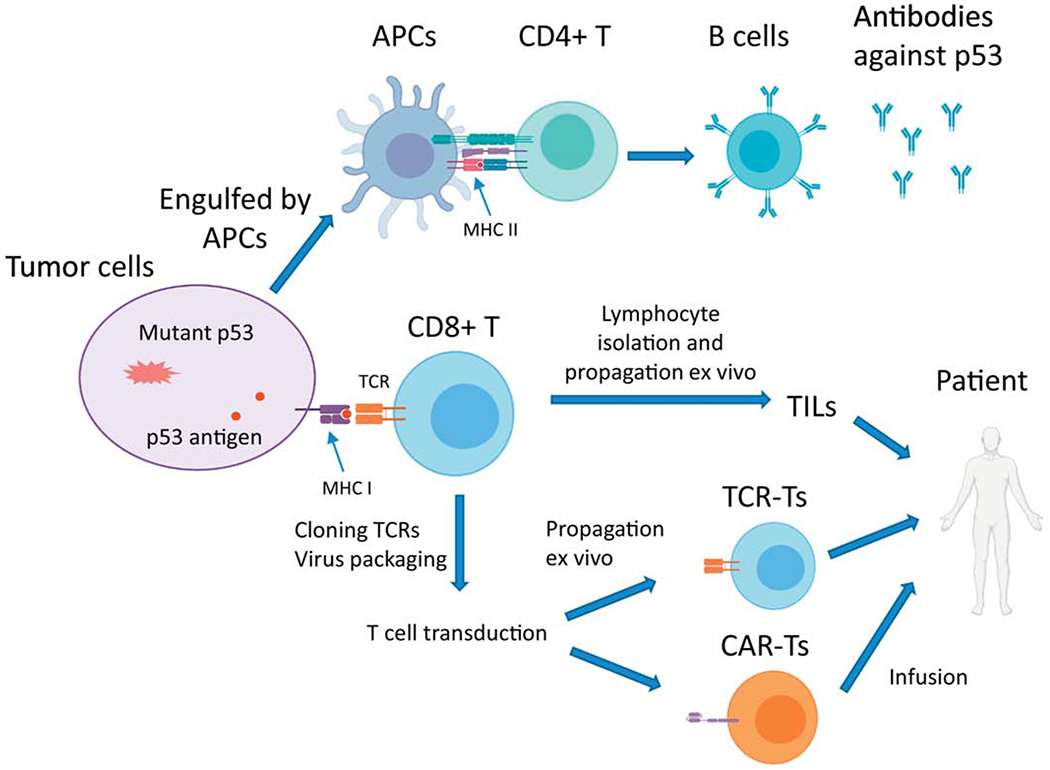
Tumor cells carrying mutant p53 or apoptotic bodies can be engulfed by antigen presenting cells (APCs). The MHC II of APCs presents mutant p53-derived neoantigens to CD4+ T cells, which help B cells produce antibodies against the neoantigens. Most tumor cells express MHC I, which self-presents mutant p53-derived neoantigens. CD8+ T cells are activated through the interaction between MHC I and T cell receptors (TCRs). Activated CD8+ T cells (cytotoxic T cells) can attack tumor cells. The tumor infiltrated lymphocytes (TILs), containing mostly CD8+ T cells and natural killer (NK) cells, can be isolated from tumors, propagated to a large number ex vivo, and given back to the patients to attack the tumors. TCRs specific to mutant p53-neoantigens can be cloned and packaged into viral particles to generate either TCR-T cells or CAR-T cells, which are infused back into the same patients.
A critical step for generating effective TCR-Ts or CAR-Ts is to clone/engineer a TCR that is highly specific to neoantigens from a tumor. Neoantigens are antigens from mutated proteins in tumor cells. Due to the high expression levels in tumors, p53 mutants are good candidates to generate neoantigens. In 1979, Levine and colleagues found that sera from mice with transformed tumors had antibodies against endogenous p53 2. This is the first report showing that the p53 protein is immunogenic and can activate CD4+ T (T helper) cell response. Later studies showed that both human and murine WT p53 and certain p53 mutants have immunogenicity and can activate CD8+ T cells (cytotoxic T cells) 109–111, although these studies were performed under non-endogenous conditions. Therefore, it had remained unclear whether endogenous mutant p53 in human tumors can activate T cell responses and whether peptides containing p53 mutations are immunogenic.
A recent study investigating immunogenicity of mutant p53 in 140 patients with various tumor types showed that endogenous p53 mutants could elicit CD4+ (helper) and CD8+ (cytotoxic) T cell responses 112. Strikingly, 39% (11 out of 28) of screened patients had TILs that recognized autologous mutant p53 neoantigens. In addition, isolated TILs or TCR-engineered T cells recognized cancer cell lines that endogenously express p53 mutants. Although this study did not show whether these TILs and TCR-engineered T cells have therapeutic benefits in tumor regression, it demonstrated that endogenous p53 mutants in human tumor cells are immunogenic and marked the first step for mutant p53 based ACT approaches. It is worth noting that not every peptide containing a TP53 mutation is immunogenic, probably due to the sequence requirement for neoantigens. Peptides containing hotspot mutations R175H, Y220C, G245S, R248Q, R248W, and R282 were shown to activate T cell responses, albeit with a wide range of frequencies 112.
Going forward, it is conceivable that a collection of TCRs can be cloned and used to recognize neoantigens from certain immunogenic TP53 mutations. This collection of TCRs can then be used either in TCR-ACT or engineered for autologous or allogeneic CAR-T cell therapy. There are several Phase I clinical trials for evaluating the safety and efficacy (response rate) of anti-p53 TCR-engineered lymphocytes in metastatic tumors (for example, www.clinicaltrials.gov, NCT00393029, NCT00496860). It is currently unclear whether these engineered lymphocytes have a clinical benefit of inhibiting tumor growth 113.
6.2. The roles of p53 in immune checkpoint regulation
Another category of cancer immunotherapy is the immune checkpoint blockade. Tumors develop various mechanisms to escape immune surveillance by T cells or natural killer (NK) cells in the host. One mechanism is through the upregulation of the molecules involved in the immune checkpoint, such as the programmed cell death ligand 1 (PD-L1) on tumor cells 114–116. PD-L1 suppresses the activation of T cells and induces their apoptosis. Inhibition of the interaction between PD-L1 and its receptor PD-1 has proved to have huge clinical benefits, as the FDA has approved several antibodies against PD-L1 or PD-1 to treat various cancers 117.
The roles of p53 in immune checkpoint regulation have been observed. A recent study showed that activated p53, either by genotoxic or non-genotoxic stress, induces the expression of PD-L1 and PD-1 in several cancer cell lines in vitro. However, the functional consequence of this molecular observation remains to be determined. On the contrary, two independent studies have shown that p53 downregulates PD-L1 expression through inducing microRNA 34a (miR34a) 118,119. miR34a binds to the 3’ untranslated region (UTR) of PD-L1 to decrease its expression, suggesting that p53 has a role in immune checkpoint regulation. A phase I clinical trial using liposomes loaded with miR34a showed that this agent increased cytotoxic CD8+ T cells while reduced the CD8+; PD-L1+ subpopulation, suggesting that the p53-miR34a-PD-L1 axis may be clinically exploited to inhibit PD-L1 118. It is worth mentioning that the miR34 family members have many targets including CDK4, RUNX2, and a myriad of other pro-oncogenic or survival factors 120–122. Therefore, activation of the p53-miR34 axis will inhibit PD-L1 and several oncogenic pathways simultaneously.
Another study explored the predictive value of TP53 and KRAS mutation status in the response of lung adenocarcinoma to PD-L1 blockade. TP53 or KRAS mutation status was found to be correlated with higher PD-L1 expression and better response to PD-L1 blockade 123. Reanalysis of public clinical trial data revealed that the co-mutation of TP53 and KRAS has a remarkable predictive value to PD-L1 blockade response of lung cancer. The mechanism underlying this correlation remained unknown. Nonetheless, these studies established a potential connection between TP53 mutation status and tumor responses to immune checkpoint blockade. Future mechanistic studies will help to firmly determine whether TP53 mutation status can serve as a clinical biomarker and guide immune checkpoint blockade therapy.
7. Tumor Microenvironment
In developing therapeutics targeting the p53 pathway, one critical aspect of tumor biology often underappreciated is the tumor microenvironment (TME). Although most p53-based therapeutic agents have remarkable efficacies of killing cancer cells in vitro and/or xenografted tumors in an immunocompromised setting, they are facing various unknown challenges when being tested in clinical trials, particularly for solid tumors. It has long been appreciated that solid tumors have an immunosuppressive microenvironment, which is infiltrated with immuno-suppressive cells and marked by T cell dysfunction 124. It is likely that the efficacies of these p53 targeting agents are significantly influenced by the TME in solid tumors 91. This possibility also provides a reasonable explanation as to why the most advanced clinical trial (a Phase III) of the MDM2 inhibitor, RG7388 (RO5503781, Idasanutlin), is for a hematological cancer type, AML. Although the preference of RG7388 to AML is sometimes being attributed to the fact that most AML patients have WT p53, it is also possible that the TME of solid tumors has limited the efficacy of RG7388. The influence of TME on p53 targeting agents starts to gain attention 91. For example, a Phase II clinical trial in head and neck cancer testing the effect of adenovirus expressing p53 in combination with immune checkpoint inhibitors, anti-PD-L1 antibodies, is ongoing (www.clinicaltrials.gov, NCT03544723). The result of this trial will help to determine whether immune checkpoint inhibitors and p53 gene therapy have additive or synergistic anti-tumor effects.
Although the vast majority of studies have been focused on TP53 mutations in tumor cells, reports are showing that stromal cells also contain a high frequency of TP53 mutations in human and mouse carcinoma 125–127, suggesting that the functions of p53 in stroma cells may contribute to its tumor suppressive function. It is worth noting that all cells, including stromal cells, in LFS patients carry TP53 mutations. How does the interaction between TP53 mutationbearing stromal cells and tumor cells affect tumorigenesis in LFS patients? Will the treatment strategies be different for LFS patients and those with somatic mutations? These questions have not been sufficiently studied both biologically and clinically.
8. Discussion and Closing Remarks
Developing therapeutics that target the p53 pathway has proven to be an extremely difficult quest. Although adenovirus-based gene therapy that reintroduces WT p53 into cancer cells has been approved for clinical use in China, this approach failed to gain wide traction, probably due to the intrinsic limitation of adenovirus-based gene delivery in tumors. Targeted therapy, as the fourth major approach in cancer treatment, has yet to receive FDA approval for the p53 pathway. There are several reasons for these futile endeavors. First, most cancer cells have a TP53 with loss of function mutations, which have low druggability. For example, compounds that restore WT p53 conformation to mutant p53 have yet made their ways to Phase III clinical trials. Second, the diverse mechanisms and context-dependence of GOFs of mutant p53 complicate the design of clinical trials for targeted therapies. Although this hurdle can be mitigated by identifying biomarkers to select patients who will most likely respond to each p53-based therapeutic agent, it requires a deeper understanding of the mechanisms underlying the GOFs of mutant p53 in each cancer type. Last and probably the most fundamental one is that we still do not understand exactly how p53 prevents tumorigenesis. Is cell cycle arrest, apoptosis, senescence, or metabolism essential for the tumor suppressive role of p53? Does p53 have a universal strategy to suppress tumorigenesis in various cell and tissue types? The answers to these key questions will not only help us gain insights into the basic biology of p53 but also guide the design of effective targeted therapy for the p53 pathway.
After more than four decades of research, it appears that we have only discovered a tip of the iceberg within the p53 ocean. Going forward, ACT-based immunotherapy approaches, such as TILs, TCR-T, or CAR-T cells, are very promising for designing effective therapeutics for the p53 pathway. These approaches largely rely on the expression of neoantigens from p53 mutants in tumors cells. Therefore, in theory, their efficacies are less affected by the diverse biology behind p53 mutants compared to other context-dependent approaches. Moreover, because most TP53 mutations cluster at several hotspot residues at the DNA binding domain, the diversity of neoantigens from these hotspot mutations is significantly decreased. The marriage of p53 biology to immunotherapy has the potential of revolutionizing the future design of p53-based cancer therapy.
Acknowledgment
This work was supported by the National Cancer Institute, USA, intramural grant, 1ZIABC011504-05 to Jing Huang.
Abbreviations List
- ACT
adoptive cell transfer
- CAR
chimeric antigen receptor
- GEMM
genetically engineered mouse model
- GOF
gain of functio
- PARP
poly(ADP-ribose) polymerase
- PD-L1
programmed cell death ligand 1
- PPI
protein-protein interaction
- PTM
post-translational modification
- TCR
T cell receptor
- TIL
tumor infiltrating lymphocyte
- WT
wild type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Submission Declaration
This paper has not been published and is not under consideration for publication elsewhere.
Conflict of Interest
The author has no conflict of interest.
References
- 1.Lane DP & Crawford LV T antigen is bound to a host protein in SV40-transformed cells. Nature 278, 261–263 (1979). [DOI] [PubMed] [Google Scholar]
- 2.Linzer DI & Levine AJ Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17, 43–52 (1979). [DOI] [PubMed] [Google Scholar]
- 3.Kress M, May E, Cassingena R & May P Simian virus 40-transformed cells express new species of proteins precipitable by anti-simian virus 40 tumor serum. J Virol 31, 472–483 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Melero JA, Stitt DT, Mangel WF & Carroll RB Identification of new polypeptide species (48-55K) immunoprecipitable by antiserum to purified large T antigen and present in SV40-infected and -transformed cells. Virology 93, 466–480 (1979). [DOI] [PubMed] [Google Scholar]
- 5.Smith AE, Smith R & Paucha E Characterization of different tumor antigens present in cells transformed by simian virus 40. Cell 18, 335–346 (1979). [DOI] [PubMed] [Google Scholar]
- 6.Eliyahu D, Raz A, Gruss P, Givol D & Oren M Participation of p53 cellular tumour antigen in transformation of normal embryonic cells. Nature 312, 646–649 (1984). [DOI] [PubMed] [Google Scholar]
- 7.Jenkins JR, Rudge K & Currie GA Cellular immortalization by a cDNA clone encoding the transformation-associated phosphoprotein p53. Nature 312, 651–654 (1984). [DOI] [PubMed] [Google Scholar]
- 8.Parada LF, Land H, Weinberg RA, Wolf D & Rotter V Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 312, 649–651 (1984). [DOI] [PubMed] [Google Scholar]
- 9.Finlay CA et al. Activating mutations for transformation by p53 produce a gene product that forms an hsc70-p53 complex with an altered half-life. Mol Cell Biol 8, 531–539 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halevy O, Rodel J, Peled A & Oren M Frequent p53 mutations in chemically induced murine fibrosarcoma. Oncogene 6, 1593–1600 (1991). [PubMed] [Google Scholar]
- 11.Malkin D et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250, 1233–1238 (1990). [DOI] [PubMed] [Google Scholar]
- 12.Li FP & Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 71, 747–752 (1969). [DOI] [PubMed] [Google Scholar]
- 13.Honda R, Tanaka H & Yasuda H Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 420, 25–27 (1997). [DOI] [PubMed] [Google Scholar]
- 14.Dornan D et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 429, 86–92 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Leng RP et al. Pirh2, a p53-induced ubiquitin-protein ligase, promotes p53 degradation. Cell 112, 779–791 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Allton K et al. Trim24 targets endogenous p53 for degradation. Proc Natl Acad Sci U S A 106, 11612–11616 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shvarts A et al. MDMX: a novel p53-binding protein with some functional properties of MDM2. EMBO J 15, 5349–5357 (1996). [PMC free article] [PubMed] [Google Scholar]
- 18.Kruse JP & Gu W Modes of p53 regulation. Cell 137, 609–622 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Tavana O & Gu W p53 modifications: exquisite decorations of the powerful guardian. J Mol Cell Biol 11, 564–577 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brady CA et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145, 571–583 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li T et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Janic A et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat Med 24, 947–953 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Pearson S, Jia H & Kandachi K China approves first gene therapy. Nat Biotechnol 22, 3–4 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang WW et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum Gene Ther 29, 160–179 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Heise C et al. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med 3, 639–645 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Bischoff JR et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science 274, 373–376 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Rothmann T, Hengstermann A, Whitaker NJ, Scheffner M & zur Hausen H Replication of ONYX-015, a potential anticancer adenovirus, is independent of p53 status in tumor cells. J Virol 72, 9470–9478 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodrum FD & Ornelles DA p53 status does not determine outcome of E1B 55-kilodalton mutant adenovirus lytic infection. J Virol 72, 9479–9490 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall AR, Dix BR, O’Carroll SJ & Braithwaite AW p53-dependent cell death/apoptosis is required for a productive adenovirus infection. Nat Med 4, 1068–1072 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Dix BR, O’Carroll SJ, Myers CJ, Edwards SJ & Braithwaite AW Efficient induction of cell death by adenoviruses requires binding of E1B55k and p53. Cancer Res 60, 2666–2672 (2000). [PubMed] [Google Scholar]
- 31.Garber K China approves world’s first oncolytic virus therapy for cancer treatment. J Natl Cancer Inst 98, 298–300 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Jones SN, Roe AE, Donehower LA & Bradley A Rescue of embryonic lethality in Mdm2-deficient mice by absence of p53. Nature 378, 206–208 (1995). [DOI] [PubMed] [Google Scholar]
- 33.Montes de Oca Luna R, Wagner DS & Lozano G Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Parant J et al. Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat Genet 29, 92–95 (2001). [DOI] [PubMed] [Google Scholar]
- 35.Wade M, Li YC & Wahl GM MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat Rev Cancer 13, 83–96 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Momand J, Jung D, Wilczynski S & Niland J The MDM2 gene amplification database. Nucleic Acids Res 26, 3453–3459 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vassilev LT et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Xiao ZX et al. Interaction between the retinoblastoma protein and the oncoprotein MDM2. Nature 375, 694–698 (1995). [DOI] [PubMed] [Google Scholar]
- 39.Lau LM, Nugent JK, Zhao X & Irwin MS HDM2 antagonist Nutlin-3 disrupts p73-HDM2 binding and enhances p73 function. Oncogene 27, 997–1003 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Zhang Z et al. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene 24, 7238–7247 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Vu B et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med Chem Lett 4, 466–469 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ding Q et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 56, 5979–5983 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Montesinos P et al. MIRROS: An ongoing randomized phase 3 trial of idasanutlin + ARA-C in patients with relapsed or refractory acute myeloid leukemia. Journal of Clinical Oncology 37, TPS7063–TPS7063 (2019). [Google Scholar]
- 44.Tisato V, Voltan R, Gonelli A, Secchiero P & Zauli G MDM2/X inhibitors under clinical evaluation: perspectives for the management of hematological malignancies and pediatric cancer. Journal of hematology & oncology 10, 133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ali AM, Atmaj J, Van Oosterwijk N, Groves MR & Domling A Stapled Peptides Inhibitors: A New Window for Target Drug Discovery. Comput Struct Biotechnol J 17, 263–281 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bernal F et al. A stapled p53 helix overcomes HDMX-mediated suppression of p53. Cancer Cell 18, 411–422 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wachter F et al. Mechanistic validation of a clinical lead stapled peptide that reactivates p53 by dual HDM2 and HDMX targeting. Oncogene 36, 2184–2190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chang YS et al. Stapled alpha-helical peptide drug development: a potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc Natl Acad Sci U S A 110, E3445–3454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carvajal LA et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci Transl Med 10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martins CP, Brown-Swigart L & Evan GI Modeling the therapeutic efficacy of p53 restoration in tumors. Cell 127, 1323–1334 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Ventura A et al. Restoration of p53 function leads to tumour regression in vivo. Nature 445, 661–665 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Xue W et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445, 656–660 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ray-Coquard I et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 13, 1133–1140 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Andreeff M et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin Cancer Res 22, 868–876 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Soussi T & Wiman KG Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell 12, 303–312 (2007). [DOI] [PubMed] [Google Scholar]
- 56.Baugh EH, Ke H, Levine AJ, Bonneau RA & Chan CS Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ 25, 154–160 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Foster BA, Coffey HA, Morin MJ & Rastinejad F Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510 (1999). [DOI] [PubMed] [Google Scholar]
- 58.Rippin TM et al. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21, 2119–2129 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Takimoto R et al. The mutant p53-conformation modifying drug, CP-31398, can induce apoptosis of human cancer cells and can stabilize wild-type p53 protein. Cancer Biol Ther 1, 47–55 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Wang W, Takimoto R, Rastinejad F & El-Deiry WS Stabilization of p53 by CP-31398 inhibits ubiquitination without altering phosphorylation at serine 15 or 20 or MDM2 binding. Mol Cell Biol 23, 2171–2181 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zache N et al. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol Oncol 2, 70–80 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bykov VJ et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 8, 282–288 (2002). [DOI] [PubMed] [Google Scholar]
- 63.Lambert JM et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388 (2009). [DOI] [PubMed] [Google Scholar]
- 64.Lambert JM, Moshfegh A, Hainaut P, Wiman KG & Bykov VJ Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 29, 1329–1338 (2010). [DOI] [PubMed] [Google Scholar]
- 65.Bykov VJ & Wiman KG Mutant p53 reactivation by small molecules makes its way to the clinic. FEBS Lett 588, 2622–2627 (2014). [DOI] [PubMed] [Google Scholar]
- 66.Burmakin M, Shi Y, Hedstrom E, Kogner P & Selivanova G Dual targeting of wild-type and mutant p53 by small molecule RITA results in the inhibition of N-Myc and key survival oncogenes and kills neuroblastoma cells in vivo and in vitro. Clin Cancer Res 19, 5092–5103 (2013). [DOI] [PubMed] [Google Scholar]
- 67.Liu X et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res 41, 6034–6044 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bromley D, Bauer MR, Fersht AR & Daggett V An in silico algorithm for identifying stabilizing pockets in proteins: test case, the Y220C mutant of the p53 tumor suppressor protein. Protein Eng Des Sel 29, 377–390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Joerger AC, Ang HC & Fersht AR Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci U S A 103, 15056–15061 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Boeckler FM et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A 105, 10360–10365 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Margalit O et al. Zinc supplementation augments in vivo antitumor effect of chemotherapy by restoring p53 function. International journal of cancer. Journal international du cancer 131, E562–568 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Pintus SS et al. The substitutions G245C and G245D in the Zn(2+)-binding pocket of the p53 protein result in differences of conformational flexibility of the DNA-binding domain. J Biomol Struct Dyn 31, 78–86 (2013). [DOI] [PubMed] [Google Scholar]
- 73.Yu X, Vazquez A, Levine AJ & Carpizo DR Allele-specific p53 mutant reactivation. Cancer Cell 21, 614–625 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mello SS & Attardi LD Not all p53 gain-of-function mutants are created equal. Cell Death Differ 20, 855–857 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bullock AN & Fersht AR Rescuing the function of mutant p53. Nat Rev Cancer 1, 68–76 (2001). [DOI] [PubMed] [Google Scholar]
- 76.Dittmer D et al. Gain of function mutations in p53. Nat Genet 4, 42–46 (1993). [DOI] [PubMed] [Google Scholar]
- 77.Olive KP et al. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860 (2004). [DOI] [PubMed] [Google Scholar]
- 78.Lang GA et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872 (2004). [DOI] [PubMed] [Google Scholar]
- 79.Adorno M et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 137, 87–98 (2009). [DOI] [PubMed] [Google Scholar]
- 80.Muller PA et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 139, 1327–1341 (2009). [DOI] [PubMed] [Google Scholar]
- 81.Pourebrahim R et al. Integrative genome analysis of somatic p53 mutant osteosarcomas identifies Ets2-dependent regulation of small nucleolar RNAs by mutant p53 protein. Genes Dev 31, 1847–1857 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Freed-Pastor WA et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhu J et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhou X, Hao Q & Lu H Mutant p53 in cancer therapy-the barrier or the path. J Mol Cell Biol 11, 293–305 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Freed-Pastor WA & Prives C Mutant p53: one name, many proteins. Genes Dev 26, 1268–1286 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muller PA & Vousden KH Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25, 304–317 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bykov VJN, Eriksson SE, Bianchi J & Wiman KG Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 18, 89–102 (2018). [DOI] [PubMed] [Google Scholar]
- 88.Kim MP & Lozano G Mutant p53 partners in crime. Cell Death Differ 25, 161–168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sabapathy K & Lane DP Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat Rev Clin Oncol 15, 13–30 (2018). [DOI] [PubMed] [Google Scholar]
- 90.Di Agostino S et al. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10, 191–202 (2006). [DOI] [PubMed] [Google Scholar]
- 91.Amelio I & Melino G Context is everything: extrinsic signalling and gain-of-function p53 mutants. Cell Death Discov 6, 16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Weinstein IB & Joe AK Mechanisms of disease: Oncogene addiction--a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol 3, 448–457 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Hainaut P & Milner J Interaction of heat-shock protein 70 with p53 translated in vitro: evidence for interaction with dimeric p53 and for a role in the regulation of p53 conformation. EMBO J 11, 3513–3520 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hinds PW, Finlay CA, Frey AB & Levine AJ Immunological evidence for the association of p53 with a heat shock protein, hsc70, in p53-plus-ras-transformed cell lines. Mol Cell Biol 7, 2863–2869 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peng Y, Chen L, Li C, Lu W & Chen J Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J Biol Chem 276, 40583–40590 (2001). [DOI] [PubMed] [Google Scholar]
- 96.Li D et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res 9, 577–588 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Blagosklonny MV, Toretsky J & Neckers L Geldanamycin selectively destabilizes and conformationally alters mutated p53. Oncogene 11, 933–939 (1995). [PubMed] [Google Scholar]
- 98.Whitesell L & Lindquist SL HSP90 and the chaperoning of cancer. Nat Rev Cancer 5, 761–772 (2005). [DOI] [PubMed] [Google Scholar]
- 99.Shrestha L, Bolaender A, Patel HJ & Taldone T Heat Shock Protein (HSP) Drug Discovery and Development: Targeting Heat Shock Proteins in Disease. Curr Top Med Chem 16, 2753–2764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mahalingam D et al. Targeting HSP90 for cancer therapy. Br J Cancer 100, 1523–1529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Terzian T et al. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev 22, 1337–1344 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer 5, 689–698 (2005). [DOI] [PubMed] [Google Scholar]
- 103.Wang X & Simon R Identification of potential synthetic lethal genes to p53 using a computational biology approach. BMC Med Genomics 6, 30 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Moser R et al. Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res 20, 4274–4288 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leijen S et al. Phase I Study Evaluating WEE1 Inhibitor AZD1775 As Monotherapy and in Combination With Gemcitabine, Cisplatin, or Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol 34, 4371–4380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leijen S et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J Clin Oncol 34, 4354–4361 (2016). [DOI] [PubMed] [Google Scholar]
- 107.Rosenberg SA et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res 17, 4550–4557 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rosenberg SA, Restifo NP, Yang JC, Morgan RA & Dudley ME Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer 8, 299–308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Yanuck M et al. A mutant p53 tumor suppressor protein is a target for peptide-induced CD8+ cytotoxic T-cells. Cancer Res 53, 3257–3261 (1993). [PubMed] [Google Scholar]
- 110.Theoret MR et al. Relationship of p53 overexpression on cancers and recognition by anti-p53 T cell receptor-transduced T cells. Hum Gene Ther 19, 1219–1232 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu Z, Liu X, McCarty TM, Diamond DJ & Ellenhorn JD The use of transgenic mice to generate high affinity p53 specific cytolytic T cells. J Surg Res 69, 337–343 (1997). [DOI] [PubMed] [Google Scholar]
- 112.Malekzadeh P et al. Neoantigen screening identifies broad TP53 mutant immunogenicity in patients with epithelial cancers. J Clin Invest 129, 1109–1114 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fishman MN et al. Phase I trial of ALT-801, an interleukin-2/T-cell receptor fusion protein targeting p53 (aa264–272)/HLA-A*0201 complex, in patients with advanced malignancies. Clin Cancer Res 17, 7765–7775 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pardoll DM The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sharma P & Allison JP Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell 161, 205–214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zou W, Wolchok JD & Chen L PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci Transl Med 8, 328rv324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vaddepally RK, Kharel P, Pandey R, Garje R & Chandra AB Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers (Basel) 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Cortez MA et al. PDL1 Regulation by p53 via miR-34. J Natl Cancer Inst 108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang X et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal 27, 443–452 (2015). [DOI] [PubMed] [Google Scholar]
- 120.He L et al. A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Farooqi AA, Tabassum S & Ahmad A MicroRNA-34a: A Versatile Regulator of Myriads of Targets in Different Cancers. Int J Mol Sci 18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shin MH et al. A RUNX2-Mediated Epigenetic Regulation of the Survival of p53 Defective Cancer Cells. PLoS Genet 12, e1005884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dong ZY et al. Potential Predictive Value of TP53 and KRAS Mutation Status for Response to PD-1 Blockade Immunotherapy in Lung Adenocarcinoma. Clin Cancer Res 23, 3012–3024 (2017). [DOI] [PubMed] [Google Scholar]
- 124.Belli C et al. Targeting the microenvironment in solid tumors. Cancer Treat Rev 65, 22–32 (2018). [DOI] [PubMed] [Google Scholar]
- 125.Kurose K et al. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat Genet 32, 355–357 (2002). [DOI] [PubMed] [Google Scholar]
- 126.Matsumoto N, Yoshida T, Yamashita K, Numata Y & Okayasu I Possible alternative carcinogenesis pathway featuring microsatellite instability in colorectal cancer stroma. British journal of cancer 89, 707–712 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hill R, Song Y, Cardiff RD & Van Dyke T Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 123, 1001–1011 (2005). [DOI] [PubMed] [Google Scholar]