

Abstract
Introduction:
Despite diverse treatment modalities and novel therapies, many cancers and patients are not effectively treated. Cancer immunotherapy has recently achieved breakthrough status yet is not effective in all cancer types or patients and can generate serious adverse effects. Oncolytic viruses (OVs) are a promising new therapeutic modality that harnesses virus biology and host interactions to treat cancer. OVs, genetically engineered or natural, preferentially replicate in and kill cancer cells, sparing normal cells/tissues, and mediating anti-tumor immunity.
Areas covered:
This review focuses on OVs as cancer therapeutic agents from a historical perspective, especially strategies to boost their immunotherapeutic activities. OVs offer a multifaceted platform, whose activities are modulated based on the parental virus and genetic alterations. In addition to direct viral effects, many OVs can be armed with therapeutic transgenes to also act as gene therapy vectors, and/or combined with other drugs or therapies.
Expert opinion:
OVs are an amazingly versatile and malleable class of cancer therapies. They tend to target cellular and host physiology as opposed to specific genetic alterations, which potentially enables broad responsiveness. The biological complexity of OVs have hindered their translation; however, the recent approval of talimogene laherparepvec (T-Vec) has invigorated the field.
Keywords: cancer, clinical trials, immunotherapy, immunovirotherapy, oncolytic virus
1. Introduction
Cancer arises from the accumulation of genetic and epigenetic alterations that transform normal cells toward abnormal cell growth. During the transformation process, each tumor cell can evolve in distinct ways, often leading to heterogeneity that complicates therapy. Conventional cancer therapies/standards-of-care include surgical resection to remove solid tumors, often leaving behind malignant cells; radiotherapy that disrupts and kills dividing cells, but does not specifically target tumor cells and exerts minimal direct effects on metastatic tumors and hematological malignancies; and chemotherapy, a systemic cytotoxic therapy with a limited therapeutic index. More recently, molecularly targeted drugs and monoclonal antibodies have entered the clinical armament (Figure 1). Molecular targeted drugs can exhibit exquisite specificity but are dependent on genetic alterations of proteins that drive cancer cell survival and growth and thus are very susceptible to resistance. Monoclonal antibodies target extracellular molecules / antigens that are unique to or overexpressed in tumors, acting directly, through cellular or complement dependent cytotoxicity, or as drug-conjugates, but limited by the presence of suitable antigens. Oncolytic viruses (OVs) are large, complex biologics that often have ill-defined mechanisms of action and are dependent on inherent virus biology and virus-host interactions. In this review, we will describe how viruses are endowed with oncolytic activity, their connection to immunotherapy, and ways to enhance immunovirotherapy, OV-mediated immunotherapy.
Figure 1.
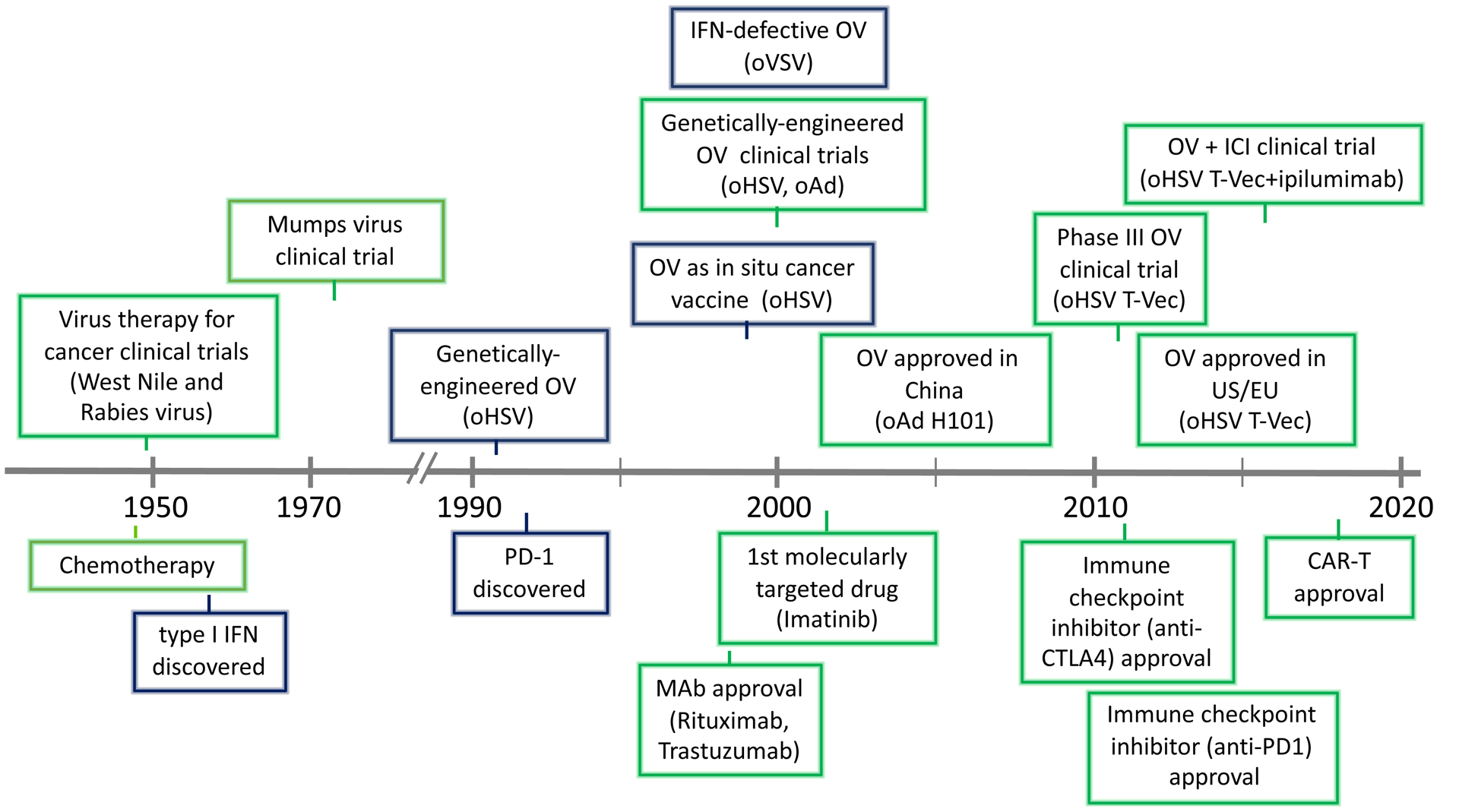
Historical timeline of oncolytic viruses and immunotherapy used for cancer treatment, with preclinical (blue box) and clinical (green box) highlights.
1.1. Immunotherapy
In 2013, Science magazine named cancer immunotherapy, including immune checkpoint inhibition / blockade and adoptive T cell transfer/chimeric antigen receptor T cells (CAR-T), as breakthrough of the year. Since then immune checkpoint inhibitors (ICIs) have been widely employed for a variety of solid tumors, while CAR-T cells have been approved for a number of hematological tumors [1]. Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) functions as an immune checkpoint to downregulate adaptive immune responses [2], so that blocking antibodies (i.e., ipilimumab) removed the brake on effector anti-tumor immune cells and led to dramatic cures in a subset of patients, leading to FDA approval (Figure 1) [2,3]. Following this, antibodies against the receptor, programmed cell death protein 1 (PD-1; e.g., cemiplimab, nivolumab and pembrolizumab) and its ligand, programmed death-ligand 1 (PD-L1; e.g., atezoliziumab) were developed as additional ICIs and approved by the FDA in 2014 and 2016, respectively (Figure 1) [3]. Combination of CTLA-4 and PD-1/PD-L1 ICIs improved outcomes in patients, but significantly increased serious adverse events. Unfortunately, a large proportion of patients and cancers respond poorly to ICIs [3]. ICIs against other immune checkpoints (e.g., LAG3, TIM3, Vista, TIGIT), as well as agonist antibodies against immune co-stimulatory molecules like OX40 and CD137 are also entering the clinic [2]. Additional cancer immunotherapies include; cytokines (e.g., IL-2, IL-12, IFNα) [4], vaccines, adoptively transferred immune cells (tumor infiltrating lymphocytes (TILs), NK cells) [1], small molecules attacking immune suppressive pathways (e.g., inhibitors of indoleamine-2,3-dioxygenase (IDO), chemokines, purinergic signaling), and oncolytic viruses (OVs) [5].
Despite thrilling clinical results from some of these novel therapies, non-responsiveness, resistance, and toxicity remain key roadblocks [1,3]. In general, ICI responsive tumors are considered immunologically “hot”, with a high level of TILs, increased PD-L1 expression, and high mutational burden [2,3,6]. In contrast, poorly responsive tumors are considered immunologically “cold”, due to a lack of tumor-associated antigen (TAA) expression or presentation, low density of TILs, infiltration by immune suppressive cells (Tregs, M2 macrophages, myeloid-derived suppressor cells, and neutrophils), expression of inhibitory cytokines (e.g., TGF-β and IL-10), and/or overexpressing alternate immune checkpoints (e.g., TIM-3, LAG-3, BTLA and VISTA) [2,6]. Therefore, tremendous effort has focused on converting ‘cold’ to ‘hot’ tumors to expand the number of immune responsive cancers and to improve the efficacy of cancer immunotherapy.
1.2. History of oncolytic viruses (OVs)
Oncolytic virotherapy is a therapeutic strategy that exploits OV’s selective replication in cancer cells and cytotoxicity. OVs are endowed with cancer selectivity, naturally or by genetic modification, enabling them to spread throughout tumors [7]. This in situ amplification of OV is unique for a pharmacological agent. The view that viruses could be used for cancer therapy arose in the late 1890s when it was noted the a “flu-like” disease coincided with a reduction in tumor cells in a leukemic patient [8]. An early definition of viruses that could be used to treat cancer in clinical trials was: “A virus should be oncolytic in some host and infective, but of low virulence, in man.” [9]. Based on this a number of clinical trials of different wild-type RNA viruses (Bunyamwera (bunyaviridae), Dengue (flaviviridae), Ilheus (flaviviridae), Newcastle disease (paramyxoviridae), Semliki Forest (togaviridae), and West Nile (flaviviridae)) were initiated at Memorial Sloan-Kettering in 1949 (Figure 1), with over 150 patients treated [10]. Of these, West Nile virus Egypt 101 seemed most active, with transient inhibitory effects [11]. Around the same time a trial with rabies vaccine for melanoma was conducted in 30 patients [12]. A later study in 1956 with adenovirus (Ad) in cervical carcinoma patients identified a set of core OV principles: (i) “selectively produced oncolytic effects in carcinoma tissue”; (ii) “lack of response after inoculation of…heat-inactivated virus”; and (iii) “recovery of virus from tumor…suggests virus multiplication” [13], that still hold. Unfortunately, despite extensive necrosis selective to the tumor, but not metastases, there was no appreciable effect on tumor progression [13]. In the 1970’s, an additional number of viruses were tested for oncolytic activity in patients with a variety of solid tumors [14]. In a clinical trial of mumps virus, about 40% of patients experienced some tumor regression, but typically succumbed to metastases [15]. Notably, some tumor responses were attributed to anti-tumor immunity [15]. These early studies laid the foundation for virotherapy (Figure 1), however, with their limited success and advances with radio- and chemo-therapy, interest in OV declined.
The advent of genetic-engineering and molecular virology enabled viruses to be tailored for tumor specificity and safety. This revival in the study of OVs was initiated in 1991 with a demonstration that herpes simplex virus (HSV) with the thymidine kinase (TK) gene deleted was attenuated for neurovirulence, yet efficacious in inhibiting the growth of human glioma xenografts [16]. This led to huge growth in the field. A broad diversity of virus types and genetic alterations were pursued and entered clinical trial, both DNA viruses (Ad, HSV, parvovirus, vaccinia virus (VV), Myxoma virus (MYXV)) and RNA viruses (coxsackievirus, Maraba virus, measles virus (MV), Newcastle disease virus (NDV), poliovirus, reovirus, retrovirus, Seneca Valley virus, Semliki forest virus, vesicular stomatitis virus (VSV)) [7,17] (Table 1). This culminated in 2015 with FDA and EMA approval of oncolytic (o)HSV talimogene laherparepvec (T-Vec, Imlygic™) for the treatment of advanced melanoma [18]. Almost all cancer types have been shown to be susceptible to OVs pre-clinically, with a broad range of cancers evaluated in the clinic (e.g., carcinomas, glioma, melanoma, sarcomas, neuroblastoma, myeloma) using a variety of OVs [7,17] (Table 1, 2).
Table 1.
Selection of OVs in clinical trials.
| Virus | OV name | Genetic modifications | Cancer | Drug combination | Clinical Trial Number |
|---|---|---|---|---|---|
| DNA Viruses | |||||
| Herpesvirus | NV1020 | HSV1-HSV2 recombinant / UL56Δ / internal repeat Δ | Colon cancer liver mets | NCT00149396 | |
| 1716 (Seprehvir) |
γ34.5Δ | Mesothelioma Non-CNS solid |
NCT01721018 NCT00931931 |
||
| G207 | γ34.5Δ / LacZ→ICP6− | Recurrent HGG Recurrent HGG/children Recurrent GBM Pediatric cerebellar brain tumors |
Radiation Radiation Radiation |
NCT00028158 NCT04482933 NCT00157703 NCT03911388 |
|
| G47Δ | γ34.5Δ / LacZ→ICP6− / ICP47Δ | Recurrent glioblastoma Prostate |
UMIN000002661 (Japan) | ||
| HF10 (TBI-1401) |
IRLΔ / UL56-LATΔ / gBsyn / UL53-55 duplicated | SCC, melanoma Pancreatic Melanoma Melanoma |
Chemo Nivolumab Ipi |
NCT01017185 NCT03252808 NCT03259425 NCT03153085 |
|
| rQNestin34.5v.2 | γ34.5Δ / nestin enhancer-γ34.5 / ICP6− | Recurrent HGG | CPA | NCT03152318 | |
| C134 | γ34.5Δ / HCMV-IRS1 | Recurrent HGG | NCT03657576 | ||
| Adenovirus | Onyx-015 (dl1520) |
Ad5 E1B-55kDΔ / E3BΔ | Head and neck | Chemo | NCT00006106 |
| H101 | Ad5 E1BΔ / E3BΔ | HCC | FOLFOX | NCT03780049 | |
| DNX-2401 (Delta-24-RGD) |
Ad5 E1A-24 bp Δ / RGD-fiber | Recurrent HGG Recurrent HGG Recurrent HGG |
TMZ Pembro |
NCT00805376 NCT01956734 NCT02798406 |
|
| ICOVIR-5 | Ad5 E2Fpro-E1A-24 bp Δ / RGD-fiber | Melanoma | NCT01864759 | ||
| OBP-301 (Telomelysin) |
Ad5 hTERTpro-E1A-IRES-E1B | Gastroeosphageal HCC |
Pembro |
NCT03921021 NCT02293850 |
|
| ColoAd1 (enadenotucirev, EnAd) | Ad3-Ad11p chimeric / E3Δ / E4orf4Δ | Metastatic colorectal, bladder Rectal |
Nivolumab Capecitabine + Radiation |
NCT02028442 NCT02636036 NCT03916510 |
|
| Poxvirus (vaccinia) | GL-ONC1 (GLV-1h68) |
Lister VV GFP→F14.5LΔ / LacZ→J2R (TK)Δ / GUS→A56RΔ | Head and neck Ovarian |
Radiation+Cisplatin Chemo |
NCT01584284 NCT02759588 |
| JX-929 (vvDD-CDSR) |
Western reserve VV CD-somatostatin receptor→TKΔ / LacZ→VGFΔ | Melanoma, breast, head and neck | NCT00574977 | ||
| Parvovirus | H-1PV (ParvOryx01) |
none | Pancreatic Recurrent HGG |
NCT02653313 NCT01301430 |
|
| RNA Viruses | |||||
| Paramyxovirus | NDV-HUJ | none | Recurrent HGG | NCT00348842 | |
| NDV PV701 |
none | Peritoneal | NCT00055705 | ||
| Measles MV-NIS |
none | Multiple myeloma Gynecological |
CPA Chemo |
NCT00450814 NCT02364713 |
|
| Picornavirus | Poliovirus PVS-RIPO |
replace IRES with HRV2-IRES | Recurrent HGG Recurrent HGG Melanoma |
Pembro |
NCT02986178 NCT04479241 NCT03712358 |
| Coxsackie CVA21 (CAVATAK) |
none | Bladder Melanoma NSCLC, bladder Melanoma |
Mitomycin C Ipi Pembro Pembro+MK-7684 |
NCT02316171 NCT03408587 NCT02043665 NCT04303169 |
|
| Seneca Valley virus (NTX-010) |
none | Small cell lung Neuroblastoma, Rhabdomyosarcoma |
CPA |
NCT01017601 NCT01048892 |
|
| Reovirus | Reolysin (Pelareorep) | none | Breast Pancreatic Pancreatic |
Pembro Chemo |
NCT01656538 NCT03723915 NCT02620423 |
Abbreviations: Chemo, chemotherapy; CPA, cyclophosphamide; Δ, deletion; HCC, hepatocellular carcinoma; HGG, high grade glioma; Ipi, ipilimumab; NSCLC, non-small cell lung cancer; Pembro, pembrolizumab; pro, promoter; SCC, squamous cell carcinoma; TMZ, temozolomide.
Table 2. Selection of armed OVs.
For cancer, preclinical studies indicate cell line used, prefixed with h if human cell line.
| Therapeutic Transgene | Virus Name |
Modifications | Immunologic Effects (preclinical) | Cancer | Combinations | Clinical trial Reference |
|---|---|---|---|---|---|---|
| Cytokine | ||||||
| GM-CSF | HSV Talimogene laherparepvec (T-Vec) |
GM-CSF→γ34.5Δ / ICP47Δ | Produce granulocytes and monocytes | Melanoma Melanoma Breast Breast Breast Sarcoma Sarcoma Rectal |
Ipi Pembrolizumab Nivolumab Nivolumab +Ipi Paclitaxel Trabectedin Radiation Chemo +Radiation |
NCT01740297 NCT02263508 NCT04185311 NCT04185311 NCT02779855 NCT03886311 NCT03300544 NCT02923778 |
| HSV RP1 |
GM-CSF-GALV-GP-R→γ34.5Δ / ICP47Δ | Cutaneous SCC Cutaneous SCC Melanoma |
Cemiplimab Nivolumab |
NCT04349436 NCT04050436 NCT03767348 |
||
| VV JX-594 (Pexa-Vec) |
Wyeth VV LacZ-hGM-CSF→TKΔ | Differentiate monocytes into DCs | Solid tumors Solid tumors HCC Colorectal Breast, sarcoma |
Ipi Sorafenib Irinotecan CPA |
NCT01636284 NCT02977156 NCT02562755 NCT01394939 NCT02630368 |
|
| Ad CG0070 (Ad5/3-D24-GMCSF) |
Ad5 E2F-1pro-E1A / GM-CSF→E3-19kΔ | Recruit NK cells, induce tumor-specific CTLs | Bladder Bladder |
Pembrolizumab |
NCT02365818 NCT04387461 |
|
| Ad ONCOS-102 |
Ad5/3 E1AΔ24 / GM-CSF→E3-19kΔ | Peritoneal Melanoma Mesothelioma |
Durvalumab Pembrolizumab CPA + carboplatin |
NCT02963831 NCT03003676 NCT02879669 |
||
| NDV MEDI5395 |
F mutation / GM-CSF→P-M | Myeloid cell infection, proinflammatory cytokines | Solid tumors | Durvalumab | NCT03889275 | |
| IL-12 | HSV G47Δ-IL12 |
G47Δ / LacZ -mIL-12→ICP6− | Increased M1 macrophages, Teff/Treg | Glioblastoma | anti-PD-1, anti-CTLA-4 | [142] |
| HSV M032 |
hIL-12→γ34.5Δ | Activate T & NK cells | Glioma | NCT02062827 | ||
| HSV NV1042 |
HSV1-HSV2 recombinant / UL56Δ / IRΔ | Prostate, breast | [96] | |||
| Ad Ad-TD-nsIL-12 |
Ad5 E1A CR2Δ / E1B-19kΔ / non-secreting IL-12→E3-19kΔ | NK and CTL activities | Pancreatic (Syrian hamster) | [98] | ||
| VV vvvDD-IL-12FG |
Tethered IL-12, YFP→TKΔ / VGFΔ | Increased TILs, decreased Treg, MDSCs | Colon MC38 | anti-PD-1 | [99] | |
| VSV rVSV-IL12 |
p35-IRES-p40 | murine SSC VII | [94] | |||
| IL-2 | VV vvDD-IL2-RG |
GPI-IL-2, YFP→TKΔ / VGFΔ | Increased CD8+/Treg ratio | MC38, | anti-ICI | [100] |
| NDV rLaSota/IL12 |
Increased TILs and CTL | B16 melanoma, H22 HCC | [101] | |||
| IL-15 | VV vvDD-IL5Rα |
IL-15Rα-IL15, YFP→TKΔ / VGFΔ | Increased CTL and memory | B16, ID8 ovarian | anti-PD-1 | [178] |
| MYXV vMyx-IL5Rα-tdTr- |
IL-15Rα-IL15, tdTomatoRed | Increased T and NK cell | B16 | [179] | ||
| Influenza A (IAV) aT116-IL-15 |
IL-15-2A FMDV-NS1Δ | Increased NK, memory CD8+ T cells, CTL | B16 | [30] | ||
| VSV opt.hIL-15 |
MΔM51 / Ig leader-hIL-15 | Increased NK and T cell responses | CT26 lung mets | [30] | ||
| IFNα / β | VSV VSV-hINFβ |
Liver | NCT01628640 | |||
| VSV VSV-hINFβ-NIS (Voyager-V1) |
Endometrial Myeloma, lymphoma NSCLC, head & neck Solid tumors |
Ruxolitinib Pembrolizumab Cemiplimab |
NCT03120624 NCT03017820 NCT03647163 NCT04291105 |
|||
| Ad KD3-IFNα |
Ad5 E1A-2 small Δ / E3Δ / ADPhi / hIFNα | h Hep3B | [104] | |||
| RGD-Cox2-ΔE3-ADP-ham-IFN | Ad5 Cox2pro-E1A / E3Δ / ADPhi / hamster IFN /RGD fiber | Syrian hamster pancreatic | [180] | |||
| VV JX-795 |
B18RΔ / mIFNβ→TKΔ | CMT-93 | [105] | |||
| TRAIL | Ad H5CmTERT-AdTRAIL |
HRE-Myc site-TERTpro-E1A-RbΔ / E1B-19kΔ / stTRAIL→E3 | Increased apoptosis | h U87 | [181] | |
| HSV G47Δ-TRAIL |
γ34.5Δ / LacZ, sTRAIL→ICP6− / ICP47Δ | Increased apoptosis | h GSCs | [182] | ||
| NDV NDV/Anh-TRAIL |
sTRAIL | Increased apoptosis, T cells | H22 hepatoma | [183] | ||
| Chemokine | ||||||
| CCL5 (RANTES) | VV vvCCL5 |
mCCL5, DsRed→TKΔ / VGFΔ | Increased TIL and DC | MC-38 | [108] | |
| Ad Ad-RANTES-E1A |
CCL5-IRES-E1A / E1BΔ | Increased CTL, infiltrating/activate DC | JC mammary, E.G-7 lymphoma | [107] | ||
| CCL19 | VV vvCCL19 |
mCCL19, DsRed→TKΔ / VGFΔ | Increased CD4+ TILs and DCs | MC-38 | [109] | |
| CXCL9 | VSV VSV-CXCL9 |
M51R / hCXCL9 mCXCL9 |
No significant effects | h FaDu 5TGM1 |
[112] | |
| Immune Checkpoint Inhibitors (ICI) | ||||||
| CTLA-4 | Ad Ad5/3-Δ24aCTLA4 |
Ad5/3 E1AΔ24 / anti-hCTLA-4 mAb→E3Δ | Increased hPBMC IL2/IFNγ | [30] | ||
| MV MV-aCTLA-4 |
anti-mCTLA-4 scFv / EGFP | B16-CD20 | [117] | |||
| PD-1 | HSV oHSV2-aPD1 |
anti-hPD-1 mAb→ γ34.5Δ / ICP47Δ | Increased effector splenocytes | B16R | [118] | |
| HSV OVH-aMPD-1 |
anti-mPD-1 scFv→γ34.5Δ / ICP0Δ /hTERTpro-ICP27 | Increased effector T cells, MDSCs | Hepa1-6 | anti-TIGIT | [116] | |
| VV WR-mAb1 WR-scFv |
WR VV RR (I4L)Δ / anti-hamster PD-1→TKΔ | MCA 205 | [115] | |||
| MYXV vPD1 |
sPD1-extracellular, GFP | Enhanced tumor-specific CD8+ activation | B16 | [30] | ||
| PD-L1 | VSV VSVM51R-PD-L1 |
M51R / anti-hPD-L1 scFv | Increased activated CD8+ splenocytes | LLC-hPD-L1 | [114] | |
| Co-stimulatory agonists | ||||||
| CD40L | Ad AdCD40L (CGTG-401) |
Ad5/3 hTERTpro-E1A / CD40L→E3Δ | Melanoma | CPA | NCT01455259 | |
| CD40 | Ad NG-350A |
EnAd anti-CD40 agonist | Epithelial | NCT03852511 | ||
| OX40L | Ad DNX-2440 (Delta-24-RGDOX) |
Ad5 E1AΔ24 / OX40L→E3Δ / RGD-fiber | Glioblastoma | NCT03714334 | ||
| 4-1BBL | VV rV-4-1BBL |
LacZ, 4-1BBL→TK− | Increased CD8+ TILs | B16 | Lymphodepletion | [30] |
| ICOSL | NDV NDV-ICOSL |
NDV LaSota mICOSL | Increased TILs | B16 | anti-CTLA-4 | [125] |
| GITRL | Ad Delta-24-GREAT |
Ad5 E1AΔ24 / mGITRL→E3Δ / RGD-fiber | Increased CD8+ TILs | GL261 | [122] | |
|
T-cell engager (BiTE) |
||||||
| VV EphA2-TEA-VV |
anti-EphA2 scFV-anti-CD3 scFv, DsRed→TKΔ / VGFΔ | Bystander killing, T cell activation | h A549 + PBMCs | [129] | ||
| Ad oAd-BiTE (ICO15K-cBiTE) |
Ad5 E2F pro-E1AΔ24 / anti-hEGFR scFv-anti-CD3 scFv / RGD-fiber | Enhances CAR-T | h Panc-1, HCT116 | CAR-T cells | [134] | |
| Ad EnAd-SA-FAP |
EnAd anti-hFAP scFv-anti-CD3 scFv-RFP→E3Δ | Increased T cell activation, M1-macrophages, killed FAP+ | h malignant ascites | [132] | ||
| MV MV-mCD3xCEA MV-mCD3xCD20 |
anti-mCD3 scFv-anti-CEA (or CD20) scFv | Increased TIL, CD8+/Treg ratio | MC38-CEA, B16-CD20-CD46 | [130] | ||
| Tumor-associated antigens (TAAs) | ||||||
| Prostatic acid phosphatase (PAP) | HSV bPΔ6-hPAP |
HSV hPAP, LacZ→ICP6− | hPAP not mPAP more efficacious. | TRAMP-C2 | [138] | |
| MAGE-A3 | Maraba virus MG1-MA3 |
Maraba Gmut / Mmut / hMAGE-A3 | Ad-MAGEA3 prime - MG1 boost | MAGE-A3+ solid tumors Melanoma, SCC |
Ad-MAGEA3 Pembrolizumab |
NCT02285816 NCT03773744 |
| HPV E6, E7 | Maraba virus MG1-E6E7 |
Maraba Gmut / Mmut / HPV17, 18 E6–E7 | Ad-E6E7 prime - MG1 boost | HPV+ cancer | Ad-E6E7, Atezolizumab | NCT03618953 |
| Combinations | ||||||
| Ad LOAd703 |
Ad5/35 E2Fpro-E1AΔ24 / E3AΔ / trimerized membrane-bound hCD40L-T2A-h4-1BBL | Solid tumors Melanoma Pancreatic |
Atezolizumab Gem, Pac, Atezolizumab |
NCT03225989 NCT04123470 NCT02705196 |
||
| Ad TILT-123 |
Ad5/3 E2Fpro-E1AΔ24 / TNFα-IRES-IL2→E3Δ | NCT04217473 | ||||
| VV TBio-6517 |
IL-12, anti-CTLA-4, Flt3L | Solid tumors | Pembrolizumab | NCT04301011 | ||
| HSV ONCR-177 |
anti-PD-1-IL12, anti-CTLA-4-Flt3L-CCL4 / miR-T-UL8 / gBmut / UL37mut / miR-T-ICP27 / JointΔ / ICP47Δ / miR-T-ICP4 / miR-T-γ | Solid tumors | Pembrolizumab | NCT04348916 | ||
| Ad NG641 |
EnAd / CXCL9, CXCL10, IFNα, FAP-BiTE | Epithelial | NCT04053283 |
Abbreviations: Ad, adenovirus; Chemo, chemotherapy; CPA, cyclophosphamide; Δ, deletion; GSCs, glioblastoma stem-like cells; h, human; HCC, hepatocellular carcinoma; HGG, high grade glioma; HSV, herpes simplex virus; Ipi, ipilimumab; MV, measles virus; MYXV, Myxoma virus; NDV, Newcastle disease virus; NSCLC, non-small cell lung cancer; pro, promoter; SCC, squamous cell carcinoma; TMZ, temozolomide; VSV, vesicular stomatitis virus; VV, vaccinia virus
1.3. OV features.
OVs target tumors through four distinct mechanisms (Figure 2). (i) The primary and defining feature is selective virus replication in cancer cells and OV amplification in situ and spread through the tumor [7]. With the exception of retroviruses, OV replication is associated with cell death (oncolysis). Direct selective cytotoxicity and spread enables tumor destruction in a fashion more targeted and safer than chemotherapy. For safety purposes, it is crucial that replication and cytotoxicity are restricted to cancer cells, especially with pathogenic viruses. The modes of OV induced cancer cell death are important for the types and degree of anti-tumor activity [19] (see section 3). (ii) Immunovirotherapy (also OV immunotherapy or viroimmunotherapy), where OV infection induces an inflammatory tumor microenvironment (TME) and anti-tumor immune responses [5,20]. The ability of OV to induce tumor-specific immune responses in preclinical models was first demonstrated in 1999 with oHSV (Figure 1), where the virus behaved as an ‘in situ cancer vaccine’; inhibiting the growth of non-inoculated tumors (abscopal effect), inducing tumor-specific CTLs, and requiring T cells for efficacy [21]. This antigen-agnostic vaccine effect is advantageous compared to targeting specific TAAs or neoantigens because it doesn’t require a priori knowledge about the tumor or generation of patient-specific reagents [22]. Furthermore, OVs can potentially turn an immunologically ‘cold’ TME into an ‘hot’ one through the induction of chemokines and cytokines, as seen in cancer patients after oHSV T-Vec therapy [23] and preclinically with NDV [24]. Optimizing immunovirotherapy is a balance between beneficial anti-tumor immunity and detrimental anti-virus immune responses [25]. (iii) OVs can also target tumor-associated stroma cells, such as endothelial cells causing vascular collapse in the tumor, reported to occur with oNDV, oVV, ooVSV, and HSV [26–28] and cancer-associated fibroblasts [29] to modulate the TME. In addition, OV-induced influx of neutrophils can lead to clot formation and vascular collapse [26]. (iv) OVs can be armed with therapeutic transgenes or sequences for a cancer gene therapy strategy [7,30]. This provides a platform to deliver and express these sequences locally in the tumor and target non-infected cells, thus enhancing therapy. In this review, we focus on OVs as immunotherapeutic agents, and strategies to boost anti-cancer immunity, and discuss challenges and future directions of this promising new cancer therapy.
Figure 2. Schematic illustration of the mechanisms of OVs for cancer therapy.
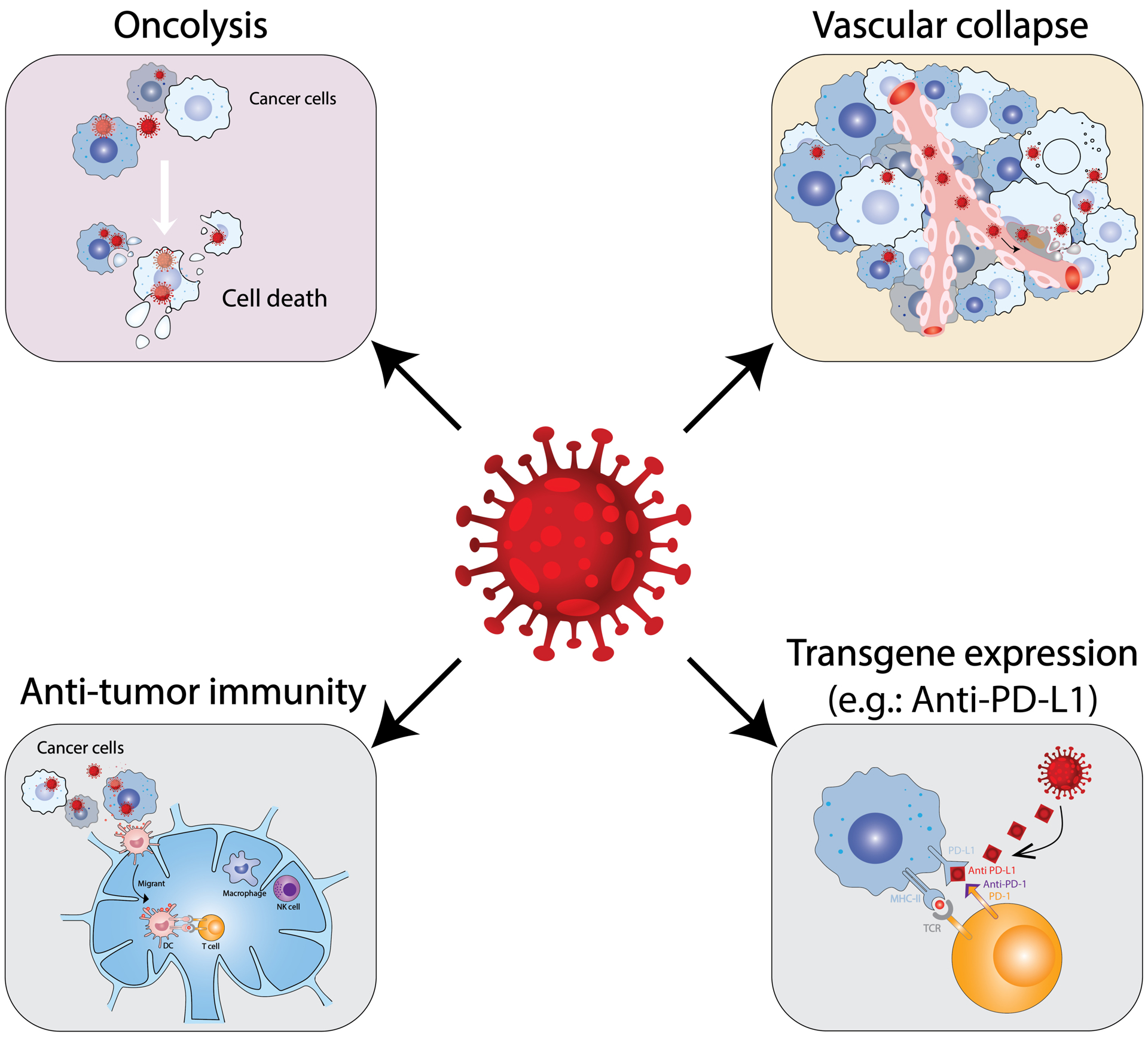
OVs can directly destroy cancer cells (oncolysis) and tumor-associated endothelial cells, which leads to tumor mass reduction and vascular collapse. The killed tumor cells release DAMPs and tumor-associated antigens that are presented to T cells and induce anti-tumor immunity. Lastly, OVs can be armed to express different transgenes that target non-infected cells and improve immunovirotherapy.
2. Designing oncolytic viruses: endowing cancer selectivity and safety
The initial application of virotherapy to cancer patients in the mid-1900s used wild-type viruses such as Ad, mumps, West Nile, etc, or infected cell lysates [11,13,15]. However, their selectivity for cancer was not understood, they weren’t specific to cancer cells only, and could cause adverse effects. In order to resurrect virotherapy, it was necessary to identify naturally-occurring OVs, utilize safer vaccine strains (VV, MV), and/or genetically modify viruses to make them cancer selective and safe [7,20]. Some viruses, especially from non-human hosts, are non-pathogenic in humans with inherent tumor tropism (naturally-occurring OVs): MYXV, due to active Akt and PKR, and SAMD9 inhibition [31]; coxsackievirus, due to receptor expression; NDV, due to Ras activation and defects in antiviral / type I IFN signaling pathways; rodent parvovirus, due to dysregulated signaling pathways [32]; murine retrovirus, due to proliferation; reovirus, due to activated Ras signaling and PKR inhibition; and VSV, due to defective type I IFN signaling [20,33]. It is important to note from a safety standpoint that direct administration of high doses of virus for cancer therapy is very different from natural exposure or vaccination of individuals.
Many viruses, especially human pathogens, are insufficiently safe to administer to patients or are not naturally cancer selective, so they must be attenuated by passage in culture, as with live vaccines (MV, poliovirus, VV), or genetically modified. Viruses can encode proteins that are complemented in particular cell types, for example cancer cells express genes not expressed in non-dividing post-mitotic cells, like those involved in nucleotide metabolism. This was exploited in the first genetically-engineered OV, HSV-1 dlsptk, with TK deleted [16], and in oVV [34] and a chimeric poxvirus [35]. However, TK is not specific to cancer cells and oHSV-TK− is resistant to anti-viral therapy, a safety mechanism. HSV has several additional genes involved in nucleotide metabolism, such as ribonucleotide reductase and uracil DNA glycosylase, which confers specificity for cancer cells and also attenuates viral pathogenicity [36].
2.1. Apoptosis and type 1 interferon (IFN)
Other pathways dysregulated in cancer cells that can enable cancer specificity of OV mutants include apoptosis, innate immune responses and type I IFN, and oncogenic pathways. Viruses encode anti-apoptotic genes to avoid cell death-mediated inhibition in normal cells, while apoptosis is deficient in most cancer cells, so OVs don’t need anti-apoptotic genes [37]. Examples of viral anti-apoptotic genes deleted in OVs include: HSV-1 Us3, which inhibits virus-induced apoptosis, endowing cancer selectivity to oHSV [38]; Ad E1B-19K, functional homologue of Bcl-2, with improved cancer selectivity and virus replication [37]; MYXV M011L, a Bcl2 homologue [39]; VV F1L, binds Bak and Bim and blocks inflammasome NLRP1, increasing safety and efficacy [40]; and VV serpin SPI-1 (B22R) and SPI-2 (B13R), improving safety and efficacy [41]. NDV has natural cancer selectivity due to tumor resistance to apoptosis [42].
The type I IFN signaling pathway and innate antiviral responses are often defective/suppressed in cancer [43], providing an important discriminator with normal cells that can be exploited by naturally-occurring OVs and to generate genetically modified OVs. The first OV shown to be cancer selective due to IFN defects was VSV, which replicated in human cancer cell lines even with IFN doses protective in normal cells [33]. While VSV reduced tumors, it was pathogenic in mice. To attenuate pathogenicity, yet retain cancer selectivity, VSV was mutated in the M protein (VSVΔM51), which blocks nuclear export of IFNβ mRNA and increased type I IFN [44] or engineered to express IFNβ (VSV-IFNbeta) (section 4.2.4) [45]. NDV is selective for glioblastoma cells lacking type I IFN, so that expression of influenza NS1, IFN signaling inhibitor, overcame NDV-resistance of type I IFN-positive cells [46].
2.2. Protein translation and tumor suppressors
Dysregulation of protein translation control is a common feature of cancer cells, with regulation of eIF2α being a major hub for responding to cellular perturbations, including virus infection. Double-strand RNA-dependent protein kinase (PKR) is often downregulated by oncogenes (RAS, MYC, PI3K, EGFR) in cancer cells. HSV encodes 2 genes that interfere with PKR signaling; γ34.5, a co-factor for PP1α to dephosphorylate inactive p-eIF2α, and Us11, a PKR inhibitor [36]. γ34.5 also binds beclin 1 blocking autophagy, inhibits TBK1 to counter IFN signaling, and is the major viral neuropathogenicity factor [36]. Therefore, it has been deleted in most oHSVs entering clinical trial, including 1716, G207, C134, rQNestin34.5v.2 (Table 1), and armed T-Vec, M032, RP1 (Table 2) [20]. Unfortunately, γ34.5Δ oHSVs are attenuated for replication even in tumor cells and poorly permissive in some cancer stem cells [47]. The following genetic alterations overcome this deficiency: expressing the late Us11 gene early by deleting ICP47, as in T-Vec and G47Δ [36]; driving glioma-specific γ34.5 expression with the nestin enhancer [48]; or through expression of the HCMV PKR inhibitor gene IRS1 [49]. All these oHSV constructs have entered clinical trial (Table 1). Because ICP47 blocks MHC I presentation, its deletion makes oHSV-infected cancer cells visible to effector T cells [36].
Many viruses require unregulated proliferation for growth, as in cancer cells, and thus express factors that block tumor suppressors or promote proliferation. While wild type Ad was examined in early clinical trials [13], it was important to construct oAd that were conditionally replicative (CRAd). The first genetically-engineered oAd was designed to target p53-deficient tumors cells through deletion of E1B, which inactivates p53 (dl1520, ONXY-015, H101) [20,50]. ONYX-015 was the first genetically engineered OV moved into clinical trials in the late 1990s, while a similar construct, H101, was the first OV approved, in China (Figure 1) [20]. A second tumor suppressor blocked by Ad is Rb, so deleting the Rb binding region of E1A generates a cancer-selective oAd (dl922–947 and delta24) [50]. As opposed to inhibiting tumor suppressors, VV encodes vaccinia growth factor (VGF), which binds EGFR and activates Ras signaling. Deletion of VGF limits oVV replication to cancer cells and improves safety [51].
2.3. Transductional targeting
Another approach to increase cancer selectivity, enabling more efficient systemic delivery and minimizing off-target toxicity, is to target virus binding to cell surface molecules/receptors expressed by cancer cells (transductional targeting) [52]. This involves altering virus tropism by mutating/detargeting and/or retargeting viral attachment proteins using glycoproteins from other viruses with different tropism, inserting peptide ligands/antibodies into viral attachment proteins, or use of soluble bispecific adapters [20,53]. For example, the Ad5 fiber binds CAR, which is not highly expressed on many cancer cells. Incorporating the Arg-Gly-Asp (RGD) motif into the Ad fiber knob (Delta-24-RGD (DNX-2401)) allows the virus to bind to integrins that are highly expressed on cancer cells [50]. MV H glycoprotein can be detargeted by mutations in the CD46 and nectin-4 binding domains and retargeted by insertion of single chain antibodies to carcinoembryonic antigen (CEA) in adenocarcinomas and CD38 on myeloma [54].
HSV-1 is more complex because entry is a multistep process involving 4 essential (gB, gD, gH/gL) and 1 nonessential glycoprotein (gC) and a ubiquitous set of cell surface receptors/binding proteins (HVEM, nectin-1, integrins ανβ3/ανβ6/ανβ8, 3-OS-heparan sulfate, PILRα, heparan sulfate) [53]. Complete detargeting involves mutations in gC and gD [55], while mutations in gB can improve virus entry [53]. To retarget oHSV, gC, gB, gD, and gH can accommodate insertions of ligands (e.g., IL-13, EPO) or single chain antibodies (e.g., against EpCAM, EGFR, and HER2) [55]. This strategy requires cell surface molecules expressed on all and only the targeted cancer cells, with resistance due to receptor downregulation, similar to other therapies targeting cell surface molecules, such as monoclonal antibodies and CAR-T. HSV glycoproteins contain the major antibody neutralization domains [55], so appropriate mutation/deletion of these sites could enable systemic administration of oHSV, and similarly for other OVs.
2.4. Transcriptional targeting
The third approach to endow cancer selectivity is to regulate viral essential early gene expression with tissue- or tumor-specific promoters to restrict virus replication to those cells expressing the appropriate factors to initiate transcription from those sequences [52]. This strategy was first demonstrated in oHSV [56] and oAd [57]. The first oAd example was the PSA promoter driving E1A only in prostate cells expressing PSA [57]. The hTERT promoter, selectively upregulated in a variety of tumors, was used to drive E1A-IRES-E1B in OBP-301 (Telomelysin), which has entered clinical trials for a range of tumors (Table 1) [58]. For oHSV, the albumin promoter/enhancer was used to drive expression of essential immediate-early gene ICP4, with virus yield over 1000-fold greater in hepatoma cells than non-albumin expressing cancer cells [56]. Other promoters driving ICP4 include, hTERT [59], calponin for sarcoma, and Wnt response elements for colorectal cancer [36]. Combinational strategies include; transcriptional regulation of essential immediate-early gene ICP27 by a prostate-specific promoter coupled with translational regulation using FGF-2 5’UTR [60], and survivin promoter-driven ICP4 with ERBB2 receptor retargeting [61]. An alternate strategy is to negatively regulate expression of ICP4 or other essential genes using miRNAs expressed in normal tissues, as was done with miR124 target sequences inserted into the 3’UTR of ICP4 for glioblastoma-specific replication [62]. MiRNA targets have also been used to regulate mRNAs in Ad, VV, VSV, MV, and IAV, and to directly target positive-strand RNA virus genomes, which was first described with coxsackievirus A21 [63].
3-. Mechanisms of inducing anti-tumor immunity by oncolytic viruses
Induction of anti-tumor immunity is a major contributor to OV efficacy, so a major goal of immunovirotherapy is to activate and redirect functional innate and adaptive immunity towards the tumor. After viral infection, host cells engage multiple mechanisms to stop virus replication. Initially, viral PAMPs are recognized by pattern recognition receptors (PRRs) [19]. In general, RNA viruses are recognized by retinoid acid-inducible receptors MDA5 and RIG-1, and TLR7 for single-stranded RNA or TLR3 for double-stranded RNA, while DNA viruses are recognized by double-stranded DNA sensors including cGAS/STING, IFI16, etc. [64]. Engagement of PRRs leads to activation of IFN signaling pathways, induction of inflammatory cytokines (e.g. IFN-α/-β, TNF-α, IL-6, NF-κB) and chemokines (CXCL9, CXCL10), and innate immune cell recruitment to viral infection sites [64]. Because OVs selectively kill tumor cells, key cellular participants of innate immunity (e.g., NK cells, DCs, macrophages, neutrophils) are recruited to tumor sites, priming adaptive immunity, and recruiting lymphoid cells [65]. NK cells recognize cancer cells due to upregulation of NK ligands and downregulation of MHCI, and kill them, however NK cells can also have detrimental effects on OV through clearance of infected cells [66].
OVs typically kill tumor cells by triggering immunogenic cell death (ICD), which is key to inducing immunity and involves different programmed cell death pathways (immunogenic apoptosis, necroptosis, pyroptosis, and autophagic cell death) [19]. This results in cell surface expression or release of DAMPs, such as HMGB1, ATP, heat shock proteins and calreticulin, and TAAs for cross-presentation, all contributing to induction of adaptive antitumor immunity [19,64]. OVs overcome some of the defects for T cell priming in the tumor by upregulating MHC I expression, BATF3+ DC maturation and antigen presentation, and T cell activation signals [64]. However, virus infection itself induces antiviral adaptive immune responses, with viral antigens often dominant over TAAs, which can redirect the immune response and limit virus spread through neutralizing antibodies and/or T cell immune responses [64,65]. The fine balance between anti-virus and anti-tumor immunity is key in determining the outcome of OV treatment, however, it is poorly understood [66].
Pre-existing or OV-induced anti-viral immunity can have variable impacts on OV efficacy. For intratumoral OV administration, this can be negligible or even beneficial. After intratumoral or intraperitoneal oHSV administration, there was no difference in inhibition of syngeneic tumor growth between mice that were seropositive or seronegative [67,68]. Ad pre-immunization did not affect intratumoral treatment with oAd, but reduced virus transit to normal liver and lungs [69]. NDV anti-tumor activity was shown to be significantly enhanced, both locally and at distal lesions, in mice that were pre-immunized, an effect that was CD8+, but not CD4+, -dependent [70]. Hepatic arterial delivery of oHSV was not altered by pre-immunization, while it was for low-dose intravenous [71]. In an intravenous MV-NIS clinical trial of multiple myeloma there was no detectable difference between baseline measles titers (seropositive and negative) and responders [72]. Pre-existing reovirus neutralizing antibodies did not limit systemic virus delivery to the tumor in a liver metastases clinical trial due to virus carriage and shielding by blood myeloid cells [73]. In many clinical trials with human OVs, patient eligibility has been limited to seropositive patients [74–76], or included an initial low dose to induce anti-virus immunity [77], for safety purposes.
4. Oncolytic viruses armed with immunomodulatory or reporter transgenes
OVs are unlikely to infect and kill all cancer or detrimental ‘normal’ cells in a tumor, so bystander effects are important. OVs typically induce anti-tumoral immune responses, but these are often insufficient to control or eradicate tumors. As many OVs can accommodate exogenous sequences, inserting therapeutic immunomodulatory transgenes into the viral genome for localized expression in the tumor is an effective way to boost anti-tumoral immunity (Table 2) [78]. For potent cytokines, localized expression can also be key to reducing toxicity arising from systemic administration. In addition to therapeutic transgenes, OVs can be engineered to express reporter genes that enable imaging of virus infection [79]. Non-invasive imaging is a potent tool to understand the dynamics of virus spread, especially in the clinic.
4.1. Armed OVs expressing reporter genes
A number of reporter genes with different imaging modalities have been encoded in OVs [79]. E.coli LacZ has been inserted into oHSVs (G207, G47Δ) and oVVs (JX-594, GLV-1h68) (Table 1, 2). In addition to histochemical detection, it provided a unique sequence to distinguish administered oHSV from patients’ endogenous HSV in clinical trials [80]. Fluorescent proteins can be optically imaged non-invasively, with the most commonly used being GFP [79]. An oHSV encoding GFP (NV1066) has been used diagnostically for intraoperative detection of lymph node metastases [79] and micro-metastatic disease in peritoneal washes from pancreatic cancer patients [81]. Similarly, oVV GLV-1h68 and oAd OBP-301 (TelomeScan), derived from OBP-301 (Table 1), have detected metastases in vivo preclinically [82,83]. OVs expressing luciferase (oVV, oHSV, oAd, oVSV) have been used to non-invasively follow virus replication and biodistribution in animal models [79]. The sodium iodine symporter (NIS) is a useful radiotracer for non-invasive imaging of OV (oAd, oHSV, oVV, oMV, oVSV) spread in mice and patients after administration of 123I or 99mTcO4, which are approved for human use, and SPECT/CT imaging [84]. A number of clinical trials of NIS-expressing OVs, MV-NIS and VSV-IFNβ-NIS (Table 1, 2), revealed a threshold for detection and high variability [84]. In addition, NIS facilitates 131I accumulation and cytotoxicity, and thus can be used for radiovirotherapy [85].
4.2. Armed OVs expressing cytokines
4.2.1. Granulocyte-macrophage colony-stimulating factor (GM-CSF)
Granulocyte-macrophage colony-stimulating factor (GM-CSF) plays a critical role in stimulating myeloid lineage progenitor cells to differentiate, and recruiting and activating DCs, macrophages, and MDSCs [86]. Early gene therapy studies showed that GM-CSF expressing cancer cells, compared to other immunomodulatory molecules, exhibited the greatest efficacy as tumor vaccines [87]. Based on the vaccine studies, GM-CSF was one of the earliest cytokines to be expressed from OVs, first with oHSV, including OncoVEXmGM-CSF [88]. However, GM-CSF expression from oHSV had only modest or no effect in syngeneic mouse models [88,89].
OncoVEX expressing human GM-CSF went on to clinical trial and was approved, as T-Vec, for advanced melanoma in 2015 [18]. Based on the phase I results, with tumor selective virus replication, local GM-CSF expression, but low-grade “flu-like” symptoms in HSV-1 seronegative patients, a multi-dosing schedule was followed with an initial low-dose (106 pfu/ml) to seroconvert, followed by higher dose (108 pfu/ml) repeated biweekly [77]. The phase III OPTiM trial compared intratumoral T-Vec with subcutaneous GM-CSF in unresectable stage IIIB-IV melanoma and resulted in a CR of 16.9% versus 0.7% and median OS of 24.5 versus 18.9 months [90]. Importantly, decreases in the size of non-injected tumor lesions were detected, strongly supporting immunotherapeutic abscopal effects [91]. A phase III trial in head and neck melanoma found an even higher response rate [18]. The oHSV clinical trial results did not reveal any outcome differences between seropositive and seronegative patients [90]. The clinical trials with T-Vec did not evaluate OncoVEX without GM-CSF, so it remains unclear how much GM-CSF expression contributed to its efficacy. Other OVs have been constructed expressing GM-CSF (oAd, oVV, oMV, oNDV), with a number entering clinical trial; oAd CG0070 (E2F-1 driven E1A) for bladder cancer [92], oAd ONCOS-102 (Ad5/3-E1A Δ24) for solid tumors [93], and oVV Pexa-Vec (JX-549; TK-deleted) for hepatocellular carcinoma [30] (Table 2). Unfortunately, a phase III clinical of Pexa-Vec with sorafenib was halted when it failed an interim futility analysis.
4.2.2. IL-12
IL-12 is a potent anti-tumor heterodimeric cytokine that promotes the growth/activity of NK, T, and B cells, differentiation of Th1 cells, production of IFNγ and CXCL10, and inhibits tumor angiogenesis [86,94]. Unfortunately IL12 is very toxic systemically [86]. OVs provide an advantageous platform for localized and limited expression to overcome toxicity, yet maintain efficacy. A number of oHSVs expressing murine IL12 (M002, NV1042, G47Δ-IL12, and R-115) have exhibited superior efficacy, with no toxicity in syngeneic mouse tumor models [94]. For example, NV1042 (derived from NV1020; Table 1), delayed tumor progression in transgenic spontaneously arising breast cancer after intratumoral administration, and prostate cancer after intravenous administration [95,96]. Efficacy of oHSV expressing IL12 was associated with increased TILs, tumor associated macrophages (TAMs), IFNγ, and decreased Tregs [94]. IL12 also has anti-angiogenic properties, so that oHSV treated glioblastoma had reduced neovasculature and VEGF expression [94,97]. In light of these promising preclinical studies, an oHSV expressing human IL12, M032, was found to be safe in nonhuman primates and entered clinical trial (Table 2).
A large number of OVs expressing IL12 have been constructed (oAd, oVV, oVSV,o MV, oMaraba, oNDV) [94]. In some of these cases, high levels of IL12 were toxic in vivo. To overcome toxicity, non-secreted IL12 was encoded in oAd (Ad-TD-nsIL-12; Table 2). Ad-TD-nsIL-12 treatment of Syrian hamster pancreatic cancer models enhanced survival, yet lacked the severe toxicity of Ad-TD with secreted IL12 [98]. An alternate strategy is to tether IL12 to the cell membrane, as with oVV (vvDD-IL-12FG) [99]. In general, IL-12 has proven to be one of the most effective immunomodulatory genes encoded in OVs.
4.2.3. IL-2
IL-2 is a growth factor for effector T and NK cells, as well as Tregs, and recombinant IL-2 is approved for cancer therapy but use is limited by toxicity [86]. It has been used for arming OVs to enhance effector T cells and reduce systemic toxicity. For example, oVV expressing membrane-bound IL-2 (vvDD-IL2-RG) was as effective as secreted IL-2 in inhibiting syngeneic tumor growth, but did not cause systemic toxicity [100]. Tumor growth inhibition was dependent on CD8+, but not CD4+ T cells, and IFNγ [100]. NDV expressing IL-2 was more efficacious than NDV by stimulating T cell proliferation and CTL [101]. To condition the TME for adoptive T cell therapy, TNFα-IRES-IL2 was inserted into oAd Ad5/3-E2F-E1AΔ24 (TILT-123; Table 2), which could substitute for lymphodepleting preconditioning in syngeneic tumor models [102].
4.2.4. Type I IFNs
Type I IFNs are critical for anti-viral innate responses (section 2.1), and also play important roles in anti-tumor immunity; upregulating MHC I expression, promoting DC maturation, survival and activation of CTL and NK cells, and polarization towards Th1 responses [86]. Therefore, OV armed with type I IFN should improve safety due to normal cellular IFN responses and efficacy, as was the case with oncolytic VSV, MV, Ad, and VV. Conversely, IFNβ expression from NDV inhibited virus replication and didn’t enhance efficacy [103]. Expression of mouse IFNβ from VSV greatly improved efficacy in a mouse syngeneic, but not xenograft, tumor model compared to expression of human IFNβ (not active in mouse) and did not induce neurologic toxicity [45]. VSV-mIFNβ treatment induced VSV-specific, but not tumor-specific, T cell responses [45]. VSV-hIFNβ-NIS was the first oVSV to enter clinical trial (Table 2). Similarly, oAd expressing IFNα (KD3-IFN) was more efficacious in both a human xenograft and Syrian hamster tumor models, with reduced hepatotoxicity [104], and JX-795, an IFNβ expressing oVV with a deletion of B18R, a secreted decoy for type I IFNs, was more efficacious than parental oVV, with improved tumor restricted biodistribution [105].
Other cytokines have also been encoded in OVs, including IL-15 (oVSV, oNDV, oInfluenza, oVV, oMYXV, oAd, oHSV), IL-18 (oHSV, oAd), and TRAIL (oAd, oHSV, oNDV) (Table 2) [30].
4.3. Armed OVs expressing chemokines
Chemokines are a family of secreted chemoattractant proteins that mediate immune cell trafficking to influence immune responses, both beneficially and detrimentally, promoting tumorigenesis and/or immunosuppression [106]. OVs have been armed with various chemokines (Table 2). CCL5 (RANTES) is a proinflammatory chemokine recruiting T cells, DCs, macrophages, and NK cells to the TME [106]. An oAd expressing RANTES increased tumors-specific TILs and NK cells and inhibited primary and distal tumors [107]. CCL5 expressing oVV (vvCCL5) had decreased pathogenicity, increased immune cell infiltration (effector CD4+ T cells and DCs), virus in the tumor, and inhibition of tumor growth [108]. vvDD-CCL19 treatment of mouse tumors also resulted in increased DC and effector T cell infiltration and inhibition of tumor growth [109]. In contrast, intravenous injection of oVV expressing CXCL11 (vvDD-CXCL11) was no better than parental vvDD in a subcutaneous model, despite increasing TILs [110], while it greatly extended survival in an intraperitoneal tumor model [111]. OVSV expressing CXCL9 did not increase TILs nor improve inhibition of tumor growth in mouse syngeneic tumors [112].
4.4. Arming OVs with immune checkpoint inhibitors (ICIs)
The TME is inherently immunosuppressive, including up-regulation of negative regulators, such as immune checkpoints, resulting in exhaustion of effector T cells [2]. ICI therapy, blocking negative regulation of T cell function, is a major breakthrough for cancer immunotherapy [3]. Thus, arming OVs with ICIs for local intratumoral expression may have better efficacy and/or less toxicity than the combination of OVs with ICIs (section 5.3), since systemic ICIs can cause adverse effects [3]. A number of OVs (oHSV, oVV, oMYXV, oMV, oVSV) have been constructed expressing anti-PD-1 or -PD-L1 single chain antibodies (scFvs) [113] (Table 2), although for some their efficacy was not greater than the parental OV not expressing an ICI [114–116]. MVs expressing either anti-PD-L1 or -CTLA-4 were the first antibody expressing negative-strand RNA viruses [117]. Unfortunately, MV-aCTLA-4 was less effective than systemic anti-CTLA-4 antibody with oMV, while MV-aPD-L1 had similar outcomes to systemic anti-PD-L1 with oMV [117]. The lack of improved efficacy over ICI antibodies + OV was also seen with oHSV and oVSV expressing anti-PD-1 or -PD-L1 [114,118]. Further addition of systemic anti-TIGIT ICI to oHSV expressing anti-PD-1 significantly improved inhibition of injected and non-injected tumors [116]. Overall, these results raise concerns about the value of ICI armed OVs.
4.5. Arming OVs with co-stimulatory checkpoints
In addition to ICI, co-stimulatory checkpoints can also be targeted for immunotherapy, using ligands or agonistic antibodies [30]. CD40 ligand (CD40L), encoded in CGTG-401 (Table 2), was used to treat a small number of patients with advanced solid tumors, who then displayed immune-mediated effects [119]. OX40L and GITRL were also encoded in oAd Delta-24-RGD (DNX-2240 and Delta-24-GREAT, respectively) (Table 2). Compared to Delta-24-RGD, DNX-2240 treatment of orthotopic tumors increased activated TILs and significantly extended survival, but not in immunodeficient mice, which was further extended by anti-PD-L1 treatment [120]. In a metastatic melanoma model, Delta-24-RGDOX injection of a subcutaneous tumor also inhibited an intracerebral tumor, an abscopal effect [121]. Treatment induced systemic increases in effector CD4+ and CD8+ T cells and reduced the frequency of exhausted TILs and regulatory T cells [121]. Delta-24-GREAT elicited similar anti-tumor efficacy in the same glioma model and both oAds protected ‘cured’ mice from tumor rechallenge [121,122]. To enhance efficacy, two co-stimulatory ligands (trimerized-CD40L and 4–1BBL) were encoded by oAd LOAd-703 and evaluated in clinical trials (Table 2). Because LOAd doesn’t infect or replicate in mouse cells, preclinical studies were performed mostly in human cells in vitro, with no striking differences detected between LOAd-703 and LOAd or LOAd-CD40L [123,124].
NDV injection induced upregulation of co-stimulatory receptors, with ICOS prominent in both injected and non-injected tumors [125]. Therefore, oNDV expressing ICOSL was constructed, which improved inhibition of non-injected, but not injected, tumor growth compared to oNDV [125]. Anti-CTLA-4 synergized with both oNDV and NDV-ICOSL, however combination with NDV-ICOSL was better than with oNDV [125], illustrating how ICIs and co-stimulatory ligands can be layered onto OVs to improve anti-tumor activity. In contrast, oNDV expressing an agonistic anti-CD28 scFv in combination with anti-CTLA-4 was no better than oNDV [126]. oHSV can accommodate multiple therapeutic transgenes, as exemplified by the Replimune vectors expressing GM-CSF and GALVR fusion protein (oHSV RP1; Table 2), and a panel of co-stimulatory ligands (CD40L, 4–1BBL, or OX40L). In a bilateral tumor model, all co-stimulatory ligands behaved similarly and were only modestly better than parental RP1 at inhibiting non-injected tumor growth [127]. This raises the question whether meaningful differences in efficacy are detectable with OVs expressing multiple immunomodulatory factors in mouse immunocompetent models.
4.6. Armed OVs expressing bispecific T-cell engagers (BiTEs)
Directing T cells to kill uninfected target cancer or TME cells, a bystander effect, should enhance immunovirotherapy. Bispecific T-cell engagers (BiTEs) consist of a T-cell engager (e.g., anti-CD3 scFv) and a scFv specific for a cell surface antigen to redirect T cells to kill those target cells, irrespective of TCR specificity, antigen presentation, or co-stimulation [128]. Recombinant BiTEs have demonstrated some success in hematological tumors but not solid tumors [128]. Arming OVs with secretory BiTEs may overcome some BiTE drawbacks, such as short half-life in serum and off-target autoimmunity, and improve therapeutic efficacy through increased intratumoral concentrations.
The first OV-BiTE was an oVV targeting EphA2, which was more effective than oVV-GFP in a human xenograft model when human PBMCs were transferred [129]. Oncolytic MVs encoding a BiTE targeting CD20 was more effective in syngeneic B16-CD20 tumors than CEA-BiTE, while human PBMCs significantly enhanced the efficacy of MV-CEA-BiTE in patient-derived xenografts [130]. To target immunosuppressive cancer associated fibroblasts, an anti-FAP-BiTE expressed in oAd (ICO15K-FBiTE and EnAd-SA-FAP) increased TILs and decreased levels of FAP [131], and depleted FAP+ fibroblasts and activated T cells in human malignant ascites [132]. Folate receptor (FR) β is a cell surface marker of pro-tumorigenic M2-like tumor-associated macrophages that was targeted by an oAd-BiTE that activated T cells and depleted macrophages in human ascites [133]. Local expression of BiTEs can overcome CAR-T problems with solid tumor heterogeneity, low frequency of CAR-transduced T cells, and immune-suppression, for example by targeting a different tumor antigen than the CAR-T. An oAd expressing an EGFR-targeting BiTE (ICO15K-cBiTE) was used in combination with FR-CAR-T cells to target FRα+/EGFR+ tumor cells. The oAd-BiTE and CAR-T combination was synergistic in inhibiting tumor growth and had better cancer cell killing than a combination of FR-CAR-T and Cetux-CAR-T (targeting EGFR) cells [134].
4.7. Armed OVs expressing tumor-associated antigens (TAAs) and combinations
An essential component of an anti-tumor response is the priming and recognition of a TAA, the basis for vaccination. One means to induce robust adaptive immunity is with an OV expressing a TAA,as an oncolytic vaccine or in a heterologous prime (replication-deficient Ad-TAA)-boost (OV-TAA) strategy. This has been demonstrated with oVSV-TAA [135] and oMaraba MG1-TAA [136]. MG1 expressing HPV E6–E7 and MAGE-A3 are in clinical trials (Table 2). A TAA discovery strategy used OVs expressing a tumor-derived cDNA library to screen for functional TAAs. Such an oVSV human cDNA library screen identified 3 TAAs that in combination eliminated mouse tumors [137]. Vaccination with a xenogenic self-antigen can break tolerence, as seen with an oHSV expressing human PAP, which inhibited mouse prostate tumor growth, where mouse PAP was no better than parental oHSV [138].
To further enhance anti-tumor activity, OVs have been constructed that express combinations of multiple immunomodulatory transgenes. Some of these are already in clinical trial, including: oHSV ONCR-177 expressing IL-12, anti-CTLA4, anti-PD-1, CCL4, and Flt3L [139]; oAd TILT-123 expressing IL-2 and TNFα [140]; oAd NG-641 expressing FAP-targeted BiTE, CXCL9, CXCL10, and IFNα [141]; oAd LOAd703 expressing CD40L and 4–1BBL [124]; and oVV TBio-6517 expressing IL-12, anti-CTLA-4, and Flt3L (Table 2).
5. OVs in combination with other immunomodulatory agents
Because of the heterogeneous nature of cancer, the TME, and treatment resistance, most successful cancer therapies are multimodal, comprising multiple therapeutic strategies and/or agents. This is likely also true for OVs, as most OVs act locally in tumors and additional immunological boosts to overcome tumor immunosuppression will be necessary [142]. Synergy between an OV and immunomodulatory therapeutic is clinically important, not only because of increased efficacy but because it may permit lower and less toxic therapeutic doses. Evaluating any interactions, whether synergistic and antagonistic, is also important as the therapeutic may be a standard-of-care. It is easier to implement a combination clinical trial with an approved or in clinical trial agent than to translate a new agent in combination with OV. A range of therapeutics have immunomodulatory effects, both direct and indirect; chemotherapy and radiotherapy through induction of ICD, vascular normalizers to promote immune cell infiltration, small molecule inhibitors of suppressor immune cells, and ICI to overcome immunosuppression [113,143,144]. The following sections represent only a subset of pharmacological agents evaluated in combination with OVs. Additional agents include; topoisomerase inhibitors (mitoxantrone, irinotecan), cisplatin, nucleotide analogues (gemcitabine, capecitabine), proteasome inhibitors (bortezomib), microtubule disrupting agents (paclitaxel, vincristine), antibiotics (doxorubicin, mitomycin-C), mTOR inhibitors (rapamycin), and kinase inhibitors (sunitinib, trametinib, sorafenib, ruxolitinib) [144,145]. Some of these combinations have entered clinical trial (Table 1, 2).
5.1. OV combined with Chemotherapy
In contrast to OVs, chemotherapeutics have a narrow therapeutic index with severe dose-limiting toxicities. Chemotherapeutic drugs impact anti-tumor immune responses beneficially through induction of ICD or detrimentally through depletion of select immune cell subtypes [145]. They are often standards-of-care, so their combination with OVs in clinical trials may be required or easier from a regulatory perspective. Among OV clinical trials, 21% were combined with chemotherapy (clinicaltrials.gov).
cyclophosphamide (CPA) is an alkylating agent with complex dose-dependent immune modulating effects, for example, transient depletion of Tregs, proinflammatory cytokine production, and suppression of innate immune responses, including reduction in neutralizing antibodies that limit OV spread. This prompted preclinical combination studies with many OVs (oHSV, oAd, oVV, oMYXV, oMV, oVSV, oReovirus) that often demonstrated enhanced anti-tumor activity [144]. Mice treated with oVSV or oMV and concurrent clinically-relevant CPA suppressed primary and anamnestic anti-viral antibody responses [146]. In a mouse glioblastoma model, CPA treatment synergized with oMYXV, which was associated with a lack of infiltrating leukocytes and macrophages and sustained virus infection [147].
In patients, effects of CPA with OV have been more limited or lacking. CPA preconditioning and reovirus didn’t decrease reovirus neutralizing antibodies or Tregs, contrary to what was seen in murine models [148]. Similarly, metronomic CPA with Seneca Valley virus NTX-010 or MV-NIS (Table 1) didn’t decrease neutralizing antibodies or inhibit virus clearance [72,149]. Treatment of patients with advanced solid tumors with metronomic CPA and oAd-GMCSF was associated with a decrease in Tregs, no change in anti-tumor or anti-virus T cell responses, and better disease control [150]. A number of clinical trials ongoing or unreported combining CPA with oVV JX-594, oAd ONCOS-102, oHSV rQNestin34.5v.2, and MV-NIS (Table 1, 2) may provide insight into the potential value of CPA with OV.
Temozolomide (TMZ) is another alkylating agent that is a standard-of-care for glioblastoma patients. TMZ synergizes with oHSV in human glioblastoma stem-like cells due to inhibition of DNA damage responses [151], and also affects immune responses. In a mouse glioblastoma model, where oHSV G47Δ-IL12 treatment extended survival, the combination with concurrent TMZ, which reduced the number of TILs and macrophages, abrogated the beneficial effects of oHSV [152]. oMYXV (M011L deleted) synergized with TMZ in treating murine glioma tumors in immunocompetent, but not immunodeficient mice, although the ‘cured’ mice were not protected from tumor rechallenge, indicating no immune memory [39]. The sequence of TMZ and oAd administration in a mouse glioma model played a role in T cell infiltration and treatment efficacy; TMZ after oAd extended survival with an increase in glioma-specific T cells, while TMZ before oAd was not significantly better than oAd alone but decreased tumor infiltrating DCs and CD8+ T cells [153]. This illustrates the complex dynamics between alkylating agents, OVs, and immunity, and the prominent impact of treatment timing and dosing. It remains unclear how representative mouse models are of the clinical situation, but underscores the need for dose and timing consideration in clinical trials.
5.2. OV combined with histone deacetylase (HDAC) inhibitors
Histone deacetylases (HDACs), often upregulated in cancer, deacetylate histones, leading to epigenetic silencing, and non-histones, such as transcription, DNA repair, and stress response factors [154]. HDAC inhibitors induce cancer cell growth arrest, differentiation, and cell death, but also modulate immune responses, increasing antigen presentation and NK ligand expression, and decreasing Tregs [154]. Four inhibitors have been approved in the US for cancer treatment; vorinostat (SAHA, Zolinza), romidepsin (FK228, Istodax), belinostat (PXD-101, Beleodaq), and panobinostat (LBH589) [144]. A number of HDAC inhibitors (VPA, TSA, vorinostat, LBH589, entinostat (MS-275)) have been used in combination with OVs (oHSV, oVV, oAd, oVSV, oMV, oReovirus, oParvovirus) in human cancer models where they inhibit IFN responses, and increase virus receptor levels, virus replication, cancer cell killing, and anti-tumor activity in immunodeficient mice [144,154]. There are only a few studies examining the combination of HDAC inhibitor and OV in immunocompetent models. In human PBMC-melanoma cell co-cultures, VPA with oHSV T-Vec enhanced NK cell killing of melanoma cells and priming of melanoma-specific CTLs by virus-infected melanoma cells [155]. MS-275 increased oVSV intratumoral replication and inhibited tumor growth in a mouse breast cancer model refractory to oVSV [156]. In a heterologous prime-OV boost strategy, MS-275 synergized with VSV-TAA; inducing transient lymphopenia, reduced anti-VSV neutralizing antibody, Tregs, and autoimmunity, and enhanced CTL [157]. In a combination of adoptive T cell therapy with oVSV-TAA, addition of MS-275 cured mice with syngeneic tumors expressing the TAA due to polarization of immunosuppressive myeloid cells to a pro-inflammatory M1 phenotype [158].
5.3. OV combined with immune checkpoint inhibitors (ICIs)
Stand-alone ICI therapy has revolutionized cancer treatment in a subset of patients in some cancers [2,3]. Both OV and ICI immunotherapies advanced in the clinic at similar times, with the pivotal phase III clinical trial of oHSV T-Vec overlapping ICI FDA approval (Figure 1). The combination of OV with ICI was obvious, as both treatments involve different mechanisms of action and responses to ICI are usually dependent on a ‘hot’ TME [2,6]. Thus, a large number of OVs (oHSV, oVV, oAd, oCoxsackie, oReo, oMaraba, oPolio, and oVSV) combined with ICIs advanced in the clinic in parallel with pre-clinically studies [65,113] (Table 1, 2). In a first-in-human OV+ICI clinical trial in advanced melanoma, oHSV T-Vec followed by pembrolizumab (anti-PD-1) resulted in the conversion of a ‘cold’ TME to a ‘hot’ one, so that responding patients did not have baseline CD8+ T cell infiltration or an IFNγ signature, contrary to what had been seen in ICI alone trials, and then increased CD8+ T cell infiltration and PD-L1 expression after treatment [23]. A phase III clinical trial of this combination for advanced melanoma has completed enrollment (Table 2). The first OV + ICI clinical trial combined oHSV T-Vec with ipilimumab (anti-CTLA-4 antibody) in advanced melanoma patients, where both agents were approved. In the phase II clinical trial, the combination significantly improved the objective response rate compared with ipilimumab monotherapy (39% versus 18%, respectively) [159].
In a window-of-opportunity clinical trial for glioblastoma, resected tumors after systemic delivery of reovirus exhibited increased CD8+ T cell infiltration, PD-L1 protein, and upregulated IFN-regulated gene (IRG) expression, potentially priming patients for ICI treatment [160]. It should be noted that upregulation of type I IFN and PD-L1 can have detrimental effects on OV therapy, for example, inhibition of abscopal effects in non-inoculated tumors after NDV treatment in mice [161]. Because of OV priming properties, timing of ICI treatment may affect efficacy. Administration of anti-CTLA-4 after oVV significantly improved efficacy compared to coincident administration [162], while combination of oVV with anti-PD-L1 was most effective when administered simultaneously [163]. In general, delayed or simultaneous ICI administration was best with all OVs tested in mouse models. There have been clinical trial designs with both schedules, although the scientific rationale and clinical advantages for each design are lacking [65].
Anti-CTLA-4 and anti-PD-1/PD-L1 promote anti-tumor immunity through independent pathways, and thus their combination can enhance efficacy, as seen in clinical trials, although with elevated toxicity [3]. In a preclinical study in glioblastoma, an ICI non-responsive cancer, with a mouse cancer stem cell model, curative therapy required IL-12 armed oHSV (G47Δ-IL12) and both anti-CTLA-4 and anti-PD-1 antibodies, which was dependent on CD4+ and CD8+ T cells and macrophages [142]. The combination of oVV JX-594 with both anti-PD-1 and anti-CTLA-4 significantly inhibited tumor growth and survival compared to JX-594 with individual ICIs in a spontaneously-arising breast cancer model [164]. This illustrates the likely necessity for multimodal therapy and dual checkpoints, and the interconnected responses involved in treating ICI non-responsive tumors. Interactions between OV and ICI are complex, often unique to each OV and tumor, so the therapeutic outcomes of combination therapy can vary and it is still unclear what features underlie an active combination.
Rather than blocking negative signaling, one can increase positive signaling with co-stimulatory checkpoint (ICOS, OX40, 4–1BB, GITR) agonists [2]. When oVSV-IFNβ was combined with OX40 agonist antibody, it increased T cell responses but towards the virus, and had no effect on survival, whereas the combination with anti-PD-1 extended survival [165]. Oncolytic vvDD combined with agonistic anti-4–1BB antibody improved efficacy compared to single treatments, that was somewhat dependent on T or NK cells, or neutrophils [166].
5.4. OV combined with radiotherapy
Radiotherapy is a standard-of-care for many locally advanced cancers. While direct cytotoxicity caused by ionizing radiation is considered the predominant mechanism, immune responses play an important role [167]. Radiation impacts immunotherapy in a variety of ways, both positive and negative, for example: inducing immunogenic cell death; the abscopal effect; altered gene expression, including immune checkpoints, TGFβ, and chemokines; tumor debulking and TME reprogramming; lymphocyte depletion; and inducing DNA damage and cGAS/STING [167]. OVs can also induce DNA damage and inhibit DNA repair pathways, and have been shown to synergize with radiation in human tumor models [168]. It has been proposed that OVs can behave as radiosensitizers, leading to enhanced efficacy at potentially lower and less toxic doses of radiation, as well as anti-tumor immunity.
Only a small number of studies have examined the combination of OVs with radiotherapy in immunocompetent rodent models. Reovirus is not significantly inactivated by radiation (<25 Gy), while the combination greatly extended survival in syngeneic B16 tumors [169]. In a rat model of advanced extremity sarcoma, oVV (GLV-1h68) in combination with radiation significantly extended survival compared to single treatments alone, due to increased intrinsic apoptosis [170]. In an Ad-permissive immunocompetent Syrian hamster model, oAd combined with radiation was more effective than radiation alone, and further improved when oAd expressed hamster IFNα [171]. The combination of an armed oAd (dB7) expressing IL-12 and GM-CSF with radiation delayed tumor growth in a mouse syngeneic hepatocarcinoma model, with an increase in apoptotic cells and TILs [172]. Radiotherapy synergized with VSV-IFNβ in the syngeneic RM9 prostate cancer model with increased CD8+ and CD4+ TILs and resistance to tumor rechallenge [173]. The combination of NDV with a single dose of radiation (dose-dependent) and anti-PD-1 reduced tumor growth compared to the combination without NDV, including non-treated contralateral tumors [174]. Expressing anti-CTLA-4 scFv from NDV or systemic administration of antibody were similarly efficacious in combination with radiation [174].
A number of clinical trials evaluating the combination of OV with radiation have been initiated. A phase I trial for locally advanced rectal cancer combined intravenous EnAd with chemoradiation (NCT03916510). oHSV G207 has been combined with radiation (5 Gy within 24 hr of G207) in recurrent GBM (NCT00157703) and in children with brain tumors (NCT03911388) (Table 1). In advanced solid tumor patients receiving palliative radiotherapy, reovirus was injected intratumorally, without toxicity [175].
6. Conclusion
OVs are promising agents for cancer therapy because of their selective replication/killing (oncolysis) and induction of inflammation. Many strategies have been applied to modify OVs in order to improve their selectivity and therapeutic efficacy. To specifically target cancer cells instead of normal cells, three general approaches have been used for viruses that are not naturally tumor-tropic: (i) deleting genes that are essential for viral replication in normal cells but not in tumor cells, and/or pathogenicity; (ii) arming OVs to target cell surface molecules expressed on cancer cells (transductional targeting); and (iii) regulating essential OV gene expression with tumor-specific promoters/enhancers (transcriptional targeting). All these approaches need to also engender safety. Importantly, a broad range of viruses can be endowed with oncolytic activity, each with unique properties. As it became clear that the therapeutic outcomes of OV treatment were often immune mediated, there was a switch from oncolytic to immunologic studies. Approaches to enhance immunovirotherapy are now a major focus of research in the field. OVs are an outstanding platform for cancer therapy because of their: in situ amplification, multiple forms of ICD to induce inflammation, unique mechanisms of action affording beneficial interactions with other therapies, ability to accommodate gene/sequence insertions (arming) for tumor delivery/expression, and diverse host-virus interactions that can be genetically altered. These properties have been taken into account when developing new OVs and strategies for immunovirotherapy, including arming OVs with therapeutic transgenes (cytokines, chemokines, co-stimulatory molecules, ICIs, BiTEs, TAA) that can target the myriad players comprising a complex heterogeneous tumor, and combining OVs, armed or not, with a broad array of therapeutic agents (e.g., chemotherapy, HDAC inhibitors, ICIs), all of which have demonstrated efficacy in preclinical models. Many of these OVs and combinations have entered clinical trial and the approval of T-Vec has validated this approach and energized the field.
7. Expert opinion
OVs provide a unique therapeutic modality for cancer. They harness the inherent biology of viruses to target cancer for destruction through selective virus replication, cytotoxicity, spread, and host anti-viral responses. This selectivity is based on cancer cell physiology and the hallmarks of cancer, many of which disable anti-viral responses. Viruses that are not naturally tumor tropic must be genetically modified / engineered to be cancer selective. Confining the virus and associated pathogenicity to tumors is a key requirement, as safety is paramount for clinical translation. The boundless diversity of viruses that can be oncolytic affords extensive opportunities for manipulation and therapeutic development, depending upon our understanding of virus and tumor biology. In addition to the activities of viral gene products, OVs provide a platform for gene delivery to the tumor and production of factors to alter the TME and enhance efficacy. It is important to constantly bear in mind the ‘dark side’ of virus genetic modification, where the OV becomes more pathogenic. This is especially relevant with strategies that dampen anti-viral immune responses, expand virus tropism, or highly express toxic molecules. For example, oncolytic Semliki Forest virus was armed with VV B18R gene, a type I IFN decoy receptor that neutralizes IFN responses, which increased efficacy but also neurotoxicity [176].
T-Vec, the only FDA approved OV, is administered by direct intratumoral injection. As the virus doesn’t seem to spread from infected tumor lesions, delivery is limited to those lesions that can be accessed from the periphery, as opposed to visceral lesions, as in the clinical trials [91]. For metastatic disease, it might be better to deliver OV systemically so it can potentially infect all tumor lesions, even those not visible radiographically. How the means of OV delivery affects anti-tumor activity and immunity is important to unravel. If the predominant therapeutic modality is immune-mediated and not oncolytic, infecting only a subset of tumor lesions might be sufficient. Unfortunately, there is extensive tumor heterogeneity, immunologic and genetic, within each patient, both spatial and temporal as tumors evolve in response to physiological conditions and treatment, which creates significant roadblocks to therapy. In this case, systemic delivery might expose the full range of TAAs. However, antigen cross-presentation and antigen/epitope spread to generate effector immune cells recognizing non-infected tumor cells should facilitate immunity of even heterogeneous tumors. Boosting immunovirotherapy to sufficiently activate immune effector cells and/or repress immunosuppressive cells or factors and eliminate the tumors will not be simple. We require a better understanding of how to overcome the roadblocks to anti-tumor immunity. Tumor heterogeneity will also affect OV targeting, especially those based on specific genetic alterations, cell surface markers, or transcriptionally regulated sequences. Again, this will require generating immune responses against non-targeted tumor cells.
Major concerns in translating preclinical results to the clinic are the paucity of representative immunocompetent animal models and the species-specificity of different viruses. Most preclinical tumor models are in mice, whose immune system and response to pathogens differs from humans and varies between inbred strains. There are only a limited number of syngeneic mouse cancer cell lines compared to the multitude of human cancer cell lines, often only a couple per cancer. Spontaneously-arising transgenic mouse models have advantages in arising in situ, however, they usually only contain a few genetic modifications compared to the heterogeneity of human tumors, are costly, and sporadic development means difficulty in generating groups of similar tumor size, time course, and specific location for treatment comparisons and to enable intratumoral OV delivery [96]. As the number of therapeutic transgenes in individual OVs and OV combinations increases, it is getting difficult to distinguish biologically-relevant differences in efficacy because of the limited measurement range, i.e., tumor size or survival. From the virus standpoint, many OVs are human specific due to receptor specificity (i.e, CD46 for MV), replication (Ad is human specific while HSV is attenuated in mouse cells), or protein activity (i.e., HSV ICP47, inhibitor of class I presentation, is not active in mice [36]), which impacts the interpretation of results in immunocompetent mouse models.
Anti-OV neutralizing antibodies, due to human natural exposure or vaccination (e.g., HSV, Ad, VV, MV, poliovirus, reovirus), or multiple OV administrations can impede systemic delivery. There are a number of approaches to deal with this problem: (i) cell carriers to transport OV through the circulation [73]; (ii) shield the virion surface or encapsulate in nanoparticles, for example polymer-coated oMV [177]; and (iii) genetically modify dominant exposed epitopes recognized by neutralizing antibodies [50,55]. For any carrier or shielding process, the OV needs to be able to exit or be released in the tumor so it can productively infect cancer cells. The success of these strategies depends greatly on virus biology and cellular interactions. Some OVs naturally associate with immune cells, as exemplified by reovirus in patients [73].
We currently lack biomarkers for patient- or cancer-specific OV sensitivity and for efficacy of immunovirotherapy in patients. This is important not only to target clinical treatment to the appropriate patients and identify responding/non-responding patients early in the course of treatment, but also to improve the preclinical models and identify targets for therapeutic development. Even with well-studied ICI therapy, biomarker development remains a challenge and the mechanisms of action and resistance are not fully understood [2]. We are reaching a point where there are more OVs and combinations with robust preclinical activity to be tested in the clinic than reasonable numbers of appropriate patients. The goal of immunotherapy is a durable complete response, a ‘cure’, not just an improvement in progression-free or overall survival, so we need ways to prioritize different OVs and strategies to identify the most potentially potent ones for translation. The field is in need of additional approved OVs for clinical use to demonstrate that T-Vec is not a fluke and that the promise and preclinical successes of OVs are clinically relevant.
Article highlights.
Oncolytic viruses (OVs) selectively replicate in and kill cancer cells, but not normal tissue, and induce anti-tumor immunity.
Several approaches have been used to generate OVs, such as deleting viral genes not necessary in cancer cells, targeting unique cancer-cell surface receptors, or regulating essential viral gene expression with tumor-specific promoters.
Oncolytic herpes simplex virus T-Vec the first OV approved in the US and Europe.
OVs armed with immunomodulatory transgenes such as cytokines, chemokines, immune checkpoint inhibitors (ICI), co-stimulatory checkpoint agonists, bispecific T-cell engagers (BiTE), and tumor-associated antigens (TAA) provide a gene therapy platform to target the tumor microenvironment and enhance immunovirotherapy.
Combining OVs with immunomodulatory pharmacological agents such as ICIs, chemotherapy, radiotherapy, and histone deacetylase (HDAC) inhibitors can improve efficacy.
Multiple OVs are in clinical trials for a broad range of cancers, including those expressing therapeutic transgenes and in combination with ICIs and chemotherapeutics.
Funding:
This work was supported in part by a grant from NIH (R01 CA160762) and the Thomas A. Pappas Chair in Neuroscience.
Abbreviations
- Ad
adenovirus
- BiTE
bispecific T-cell engager
- CAR-T
chimeric antigen receptor T cells
- CPA
cyclophosphamide
- CRAd
conditionally-replicative adenovirus
- CTLA-4
cytotoxic T lymphocyte-associated antigen 4
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- HDAC
histone deacetylase
- HSV
herpes simplex virus
- ICD
immunogenic cell death
- ICI
immune checkpoint inhibitor
- IFN
interferon
- IL
interleukin
- MV
measles virus
- MYXV
myxoma virus
- NDV
Newcastle disease virus
- NK
natural killer
- o
oncolytic
- OV
oncolytic virus
- PAMP
pathogen-associated molecular pattern
- PD-1
programmed cell death protein 1
- PD-L1
programmed cell death-ligand 1
- PPR
pattern recognition receptor
- TAA
tumor-associated antigen
- TK
thymidine kinase
- TIL
tumor infiltrating lymphocytes
- TME
tumor microenvironment
- TMZ
temozolomide
- T-Vec
talimogene laherparepvec
- VSV
vesicular stomatitis virus
- VV
vaccinia virus
Footnotes
Declaration of Interest
S. Rabkin is a co-inventor on patents relating to oHSV, owned and managed by Georgetown University and Massachusetts General Hospital, that were licensed to Amgen and ActiVec Inc, for which royalties have been received and has received honoraria from Replimune Inc. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer Disclosures:
One Peer Reviewer declares affiliation with the oncolytic virotherapy company, OncoMyx. Peer reviewers on this manuscript have no other relevant financial relationships or otherwise to disclose.
References
- 1.Miller JF, Sadelain M. The journey from discoveries in fundamental immunology to cancer immunotherapy. Cancer Cell. 2015; 27:439–49. [DOI] [PubMed] [Google Scholar]; * Excellent review of the history of immunology as it related to immunotherapy.
- 2.Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018; 8:1069–1086. [DOI] [PubMed] [Google Scholar]
- 3.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018; 359:1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waldmann TA. Cytokines in Cancer Immunotherapy. Cold Spring Harb Perspect Biol. 2018; 10:a028472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiocca EA, Rabkin SD. Oncolytic viruses and their application to cancer immunotherapy. Cancer Immunol Res. 2014; 2:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Galon J, Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat Rev Drug Discov. 2019; 18:197–218. [DOI] [PubMed] [Google Scholar]
- 7.Harrington K, Freeman DJ, Kelly B, et al. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019; 18:689–706. [DOI] [PubMed] [Google Scholar]
- 8.Dock G The influence of complicating diseases upon leukaemia. The Am J Med Sci (1827–1924). 1904;127:563. [Google Scholar]
- 9.Southam CM, Moore AE. West Nile, Ilheus, and Bunyamwera virus infections in man. Am J Trop Med Hyg. 1951; 31:724–41. [DOI] [PubMed] [Google Scholar]
- 10.Southam CM. Present status of oncolytic virus studies. Trans N Y Acad Sci. 1960; 22:657–73. [DOI] [PubMed] [Google Scholar]
- 11.Southam CM, Moore AE. Clinical studies of viruses as antineoplastic agents with particular reference to Egypt 101 virus. Cancer. 1952; 5:1025–34. [DOI] [PubMed] [Google Scholar]
- 12.Higgins GK, Pack GT. Virus therapy in the treatment of tumors. Bull Hosp Joint Dis. 1951; 12:379–82. [PubMed] [Google Scholar]
- 13.Smith RR, Huebner RJ, Rowe WP, et al. Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer. 1956; 9:1211–1218. [DOI] [PubMed] [Google Scholar]
- 14.Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007; 4:101–17. [DOI] [PubMed] [Google Scholar]
- 15.Asada T Treatment of human cancer with mumps virus. Cancer. 1974; 34:1907–28. [DOI] [PubMed] [Google Scholar]
- 16.Martuza RL, Malick A, Markert JM, et al. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991; 252:854–6. [DOI] [PubMed] [Google Scholar]; ** First report of a genetically engineered OV (HSV) and its efficacy in an orthotopic tumor model
- 17.Zheng M, Huang J, Tong A, et al. Oncolytic Viruses for Cancer Therapy: Barriers and Recent Advances. Mol Ther Oncolytics. 2019; 15:234–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bommareddy PK, Peters C, Saha D, et al. Oncolytic Herpes Simplex Viruses as a Paradigm for the Treatment of Cancer. Ann Revi Cancer Biology. 2018; 2:155–173. [Google Scholar]
- 19.Guo ZS, Liu Z, Bartlett DL. Oncolytic Immunotherapy: Dying the Right Way is a Key to Eliciting Potent Antitumor Immunity. Front Oncol. 2014; 4:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015; 14:642–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toda M, Rabkin SD, Kojima H, et al. Herpes simplex virus as an in situ cancer vaccine for the induction of specific anti-tumor immunity. Hum Gene Ther. 1999; 10:385–93. [DOI] [PubMed] [Google Scholar]
- 22.Russell SJ, Barber GN. Oncolytic Viruses as Antigen-Agnostic Cancer Vaccines. Cancer Cell. 2018; 33:599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ribas A, Dummer R, Puzanov I, et al. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017; 170:1109–1119.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** First clinical demonstratiion of OV (oHSV) conversion of ‘cold’ to ‘hot’ TME, turning anti-PD-1 nonresponders into responders.
- 24.Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014; 6:226ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Intratumoral OV (NDV) induces inflammatory responses that sensitize tumors to treatment with anti-CTLA-4 ICI.
- 25.Gujar S, Pol JG, Kim Y, et al. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018; 39:209–221. [DOI] [PubMed] [Google Scholar]
- 26.Breitbach CJ, De Silva NS, Falls TJ, et al. Targeting tumor vasculature with an oncolytic virus. Mol Ther. 2011; 19:886–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Breitbach CJ, Arulanandam R, De Silva N, et al. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 2013; 73:1265–75. [DOI] [PubMed] [Google Scholar]
- 28.Matuszewska K, Santry LA, van Vloten JP, et al. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin Cancer Res. 2019; 25:1624–1638. [DOI] [PubMed] [Google Scholar]
- 29.Ilkow CS, Marguerie M, Batenchuk C, et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat Med. 2015; 21:530–6. [DOI] [PubMed] [Google Scholar]
- 30.de Graaf JF, de Vor L, Fouchier RAM, et al. Armed oncolytic viruses: A kick-start for anti-tumor immunity. Cytokine Growth Factor Rev. 2018; 41:28–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rahman MM, McFadden G. Oncolytic Virotherapy with Myxoma Virus. J Clin Med. 2020; 9:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bretscher C, Marchini A. H-1 Parvovirus as a Cancer-Killing Agent: Past, Present, and Future. Viruses. 2019; 11:562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stojdl DF, Lichty B, Knowles S, et al. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000; 6:821–5. [DOI] [PubMed] [Google Scholar]; * First demonstration that tumor defects in IFN signaling enable VSV cancer cell targeting and inhibition of tumor growth.
- 34.Zeh HJ, Bartlett DL. Development of a replication-selective, oncolytic poxvirus for the treatment of human cancers. Cancer Gene Ther. 2002; 9:1001–12. [DOI] [PubMed] [Google Scholar]
- 35.Chaurasiya S, Chen NG, Lu J, et al. A chimeric poxvirus with J2R (thymidine kinase) deletion shows safety and anti-tumor activity in lung cancer models. Cancer Gene Ther. 2020; 27:125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peters C, Rabkin SD. Designing Herpes Viruses as Oncolytics. Mol Ther Oncolytics. 2015; 2:15010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu TC, Kirn D. Viruses with deletions in antiapoptotic genes as potential oncolytic agents. Oncogene. 2005; 24:6069–79. [DOI] [PubMed] [Google Scholar]
- 38.Liu TC, Wakimoto H, Martuza RL, et al. Herpes simplex virus Us3(−) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt targeting molecular therapeutics. Clin Cancer Res. 2007; 13:5897–902. [DOI] [PubMed] [Google Scholar]
- 39.Pisklakova A, McKenzie B, Zemp F, et al. M011L-deficient oncolytic myxoma virus induces apoptosis in brain tumor-initiating cells and enhances survival in a novel immunocompetent mouse model of glioblastoma. Neuro Oncol. 2016; 18:1088–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelin A, Foloppe J, Petryk J, et al. Deletion of Apoptosis Inhibitor F1L in Vaccinia Virus Increases Safety and Oncolysis for Cancer Therapy. Mol Ther Oncolytics. 2019; 14:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo ZS, Naik A, O’Malley ME, et al. The enhanced tumor selectivity of an oncolytic vaccinia lacking the host range and antiapoptosis genes SPI-1 and SPI-2. Cancer Res. 2005; 65:9991–8. [DOI] [PubMed] [Google Scholar]
- 42.Mansour M, Palese P, Zamarin D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resistant cells. J Virol. 2011; 85:6015–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matveeva OV, Chumakov PM. Defects in interferon pathways as potential biomarkers of sensitivity to oncolytic viruses. Rev Med Virol. 2018; 28:e2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stojdl DF, Lichty BD, tenOever BR, et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell. 2003; 4:263–75. [DOI] [PubMed] [Google Scholar]
- 45.Willmon CL, Saloura V, Fridlender ZG, et al. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 2009; 69:7713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zamarin D, Martínez-Sobrido L, Kelly K, et al. Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol Ther. 2009; 17:697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peters C, Paget M, Tshilenge KT, et al. Restriction of Replication of Oncolytic Herpes Simplex Virus with a Deletion of gamma34.5 in Glioblastoma Stem-Like Cells. J Virol. 2018; 92:e00246–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiocca EA, Nakashima H, Kasai K, et al. Preclinical Toxicology of rQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol Ther Methods Clin Dev. 2020; 17:871–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cassady KA. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol. 2005; 79:8707–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baker AT, Aguirre-Hernández C, Halldén G, et al. Designer Oncolytic Adenovirus: Coming of Age. Cancers (Basel). 2018; 10:201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McCart JA, Ward JM, Lee J, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001; 61:8751–7. [PubMed] [Google Scholar]
- 52.Cattaneo R, Miest T, Shashkova EV, et al. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008; 6:529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Goins WF, Hall B, Cohen JB, et al. Retargeting of herpes simplex virus (HSV) vectors. Curr Opin Virol. 2016; 21:93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aref S, Bailey K, Fielding A. Measles to the Rescue: A Review of Oncolytic Measles Virus. Viruses. 2016; 8:294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu XQ, Xin HY, Lyu YN, et al. Oncolytic herpes simplex virus tumor targeting and neutralization escape by engineering viral envelope glycoproteins. Drug Deliv. 2018; 25:1950–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miyatake S, Iyer A, Martuza RL, et al. Transcriptional targeting of herpes simplex virus for cell-specific replication. J Virol. 1997; 71:5124–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rodriguez R, Schuur ER, Lim HY, et al. Prostate attenuated replication competent adenovirus (ARCA) CN706: a selective cytotoxic for prostate-specific antigen-positive prostate cancer cells. Cancer Res. 1997; 57:2559–63. [PubMed] [Google Scholar]
- 58.Nemunaitis J, Tong AW, Nemunaitis M, et al. A phase I study of telomerase-specific replication competent oncolytic adenovirus (telomelysin) for various solid tumors. Mol Ther. 2010; 18:429–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang W, Ge K, Zhao Q, et al. A novel oHSV-1 targeting telomerase reverse transcriptase-positive cancer cells via tumor-specific promoters regulating the expression of ICP4. Oncotarget. 2015; 6:20345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee CY, Bu LX, DeBenedetti A, et al. Transcriptional and translational dual-regulated oncolytic herpes simplex virus type 1 for targeting prostate tumors. Mol Ther. 2010; 18:929–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sasso E, Froechlich G, Cotugno G, et al. Replicative conditioning of Herpes simplex type 1 virus by Survivin promoter, combined to ERBB2 retargeting, improves tumour cell-restricted oncolysis. Sci Rep. 2020; 10:4307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mazzacurati L, Marzulli M, Reinhart B, et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol Ther. 2015; 23:99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ruiz AJ, Russell SJ. MicroRNAs and oncolytic viruses. Curr Opin Virol. 2015; 13:40–8. [DOI] [PubMed] [Google Scholar]
- 64.Bommareddy PK, Shettigar M, Kaufman HL. Integrating oncolytic viruses in combination cancer immunotherapy. Nat Rev Immunol. 2018; 18:498–513. [DOI] [PubMed] [Google Scholar]; * Excellent review of immunovirotherapy.
- 65.Russell L, Peng KW, Russell SJ, et al. Oncolytic Viruses: Priming Time for Cancer Immunotherapy. BioDrugs. 2019; 33:485–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alvarez-Breckenridge CA, Choi BD, Suryadevara CM, et al. Potentiating oncolytic viral therapy through an understanding of the initial immune responses to oncolytic viral infection. Curr Opin Virol. 2015; 13:25–32. [DOI] [PubMed] [Google Scholar]
- 67.Chahlavi A, Rabkin S, Todo T, et al. Effect of prior exposure to herpes simplex virus 1 on viral vector-mediated tumor therapy in immunocompetent mice. Gene Ther. 1999; 6:1751–8. [DOI] [PubMed] [Google Scholar]
- 68.Lambright ES, Kang EH, Force S, et al. Effect of preexisting anti-herpes immunity on the efficacy of herpes simplex viral therapy in a murine intraperitoneal tumor model. Mol Ther. 2000; 2:387–93. [DOI] [PubMed] [Google Scholar]
- 69.Dhar D, Spencer JF, Toth K, et al. Effect of preexisting immunity on oncolytic adenovirus vector INGN 007 antitumor efficacy in immunocompetent and immunosuppressed Syrian hamsters. J Virol. 2009; 83:2130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ricca JM, Oseledchyk A, Walther T, et al. Pre-existing Immunity to Oncolytic Virus Potentiates Its Immunotherapeutic Efficacy. Mol Ther. 2018; 26:1008–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delman KA, Bennett JJ, Zager JS, et al. Effects of preexisting immunity on the response to herpes simplex-based oncolytic viral therapy. Hum Gene Ther. 2000; 11:2465–72. [DOI] [PubMed] [Google Scholar]
- 72.Dispenzieri A, Tong C, LaPlant B, et al. Phase I trial of systemic administration of Edmonston strain of measles virus genetically engineered to express the sodium iodide symporter in patients with recurrent or refractory multiple myeloma. Leukemia. 2017; 31:2791–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Adair RA, Roulstone V, Scott KJ, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012; 4:138ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Geevarghese SK, Geller DA, de Haan HA, et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther. 2010; 21:1119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zeh HJ, Downs-Canner S, McCart JA, et al. First-in-man study of western reserve strain oncolytic vaccinia virus: safety, systemic spread, and antitumor activity. Mol Ther. 2015; 23:202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Desjardins A, Gromeier M, Herndon JE 2nd, et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med. 2018; 379:150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hu JC, Coffin RS, Davis CJ, et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res. 2006; 12:6737–47. [DOI] [PubMed] [Google Scholar]
- 78.Chaurasiya S, Fong Y, Warner SG. Optimizing Oncolytic Viral Design to Enhance Antitumor Efficacy: Progress and Challenges. Cancers (Basel). 2020; 12:1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Haddad D, Fong Y. Molecular imaging of oncolytic viral therapy. Mol Ther Oncolytics. 2015; 1:14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000; 7:867–74. [DOI] [PubMed] [Google Scholar]
- 81.Kelly KJ, Wong J, Gönen M, et al. Human Trial of a Genetically Modified Herpes Simplex Virus for Rapid Detection of Positive Peritoneal Cytology in the Staging of Pancreatic Cancer. EBioMedicine. 2016; 7:94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kelly KJ, Brader P, Woo Y, et al. Real-time intraoperative detection of melanoma lymph node metastases using recombinant vaccinia virus GLV-1h68 in an immunocompetent animal model. Int J Cancer. 2009; 124:911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yano S, Takehara K, Miwa S, et al. Improved Resection and Outcome of Colon-Cancer Liver Metastasis with Fluorescence-Guided Surgery Using In Situ GFP Labeling with a Telomerase-Dependent Adenovirus in an Orthotopic Mouse Model. PLoS One. 2016; 11:e0148760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Miller A, Russell SJ. The use of the NIS reporter gene for optimizing oncolytic virotherapy. Expert Opin Biol Ther. 2016; 16:15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dingli D, Peng KW, Harvey ME, et al. Image-guided radiovirotherapy for multiple myeloma using a recombinant measles virus expressing the thyroidal sodium iodide symporter. Blood. 2004; 103:1641–6. [DOI] [PubMed] [Google Scholar]
- 86.Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the Treatment of Cancer. J Interferon Cytokine Res. 2019; 39:6–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dranoff G, Jaffee E, Lazenby A, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993; 90:3539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003; 10:292–303. [DOI] [PubMed] [Google Scholar]
- 89.Varghese S, Rabkin SD, Liu R, et al. Enhanced therapeutic efficacy of IL-12, but not GM-CSF, expressing oncolytic herpes simplex virus for transgenic mouse derived prostate cancers. Cancer Gene Ther. 2006; 13:253–65. [DOI] [PubMed] [Google Scholar]
- 90.Andtbacka RHI, Collichio F, Harrington KJ, et al. Final analyses of OPTiM: a randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III-IV melanoma. J Immunother Cancer. 2019; 7:145. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** Final results from first successful phase III OV clinical trial, demonstrating immune effects and leading to first approved OV (HSV) in the US and Europe.
- 91.Andtbacka RH, Ross M, Puzanov I, et al. Patterns of Clinical Response with Talimogene Laherparepvec (T-VEC) in Patients with Melanoma Treated in the OPTiM Phase III Clinical Trial. Ann Surg Oncol. 2016; 23:4169–4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Packiam VT, Lamm DL, Barocas DA, et al. An open label, single-arm, phase II multicenter study of the safety and efficacy of CG0070 oncolytic vector regimen in patients with BCG-unresponsive non-muscle-invasive bladder cancer: Interim results. Urol Oncol. 2018; 36:440–447. [DOI] [PubMed] [Google Scholar]
- 93.Ranki T, Pesonen S, Hemminki A, et al. Phase I study with ONCOS-102 for the treatment of solid tumors - an evaluation of clinical response and exploratory analyses of immune markers. J Immunother Cancer. 2016; 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nguyen HM, Guz-Montgomery K, Saha D. Oncolytic Virus Encoding a Master Pro-Inflammatory Cytokine Interleukin 12 in Cancer Immunotherapy. Cells. 2020; 9:400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu R, Varghese S, Rabkin SD. Oncolytic herpes simplex virus vector therapy of breast cancer in C3(1)/SV40 T-antigen transgenic mice. Cancer Res. 2005; 65:1532–40. [DOI] [PubMed] [Google Scholar]
- 96.Varghese S, Rabkin SD, Nielsen GP, et al. Systemic therapy of spontaneous prostate cancer in transgenic mice with oncolytic herpes simplex viruses. Cancer Res. 2007; 67:9371–9. [DOI] [PubMed] [Google Scholar]
- 97.Cheema TA, Wakimoto H, Fecci PE, et al. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013; 110:12006–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang P, Li X, Wang J, et al. Re-designing Interleukin-12 to enhance its safety and potential as an anti-tumor immunotherapeutic agent. Nat Commun. 2017; 8:1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ge Y, Wang H, Ren J, et al. Oncolytic vaccinia virus delivering tethered IL-12 enhances antitumor effects with improved safety. J Immunother Cancer. 2020; 8:e000710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu Z, Ge Y, Wang H, et al. Modifying the cancer-immune set point using vaccinia virus expressing re-designed interleukin-2. Nat Commun. 2018; 9:4682. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Example of armed OV where expression of membrane-tethered cytokine overcame toxicity of secreted cytokine, yet maintained efficacy.
- 101.Bai F, Niu Z, Tian H, et al. Genetically engineered Newcastle disease virus expressing interleukin 2 is a potential drug candidate for cancer immunotherapy. Immunol Lett. 2014; 159:36–46. [DOI] [PubMed] [Google Scholar]
- 102.Santos JM, Cervera-Carrascon V, Havunen R, et al. Adenovirus Coding for Interleukin-2 and Tumor Necrosis Factor Alpha Replaces Lymphodepleting Chemotherapy in Adoptive T Cell Therapy. Mol Ther. 2018; 26:2243–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buijs P, van Nieuwkoop S, Vaes V, et al. Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma. Viruses. 2015; 7:2980–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Shashkova EV, Spencer JF, Wold WS, et al. Targeting interferon-alpha increases antitumor efficacy and reduces hepatotoxicity of E1A-mutated spread-enhanced oncolytic adenovirus. Mol Ther. 2007; 15:598–607. [DOI] [PubMed] [Google Scholar]
- 105.Kirn DH, Wang Y, Le Boeuf F, et al. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007; 4:e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Strazza M, Mor A. The Complexity of Targeting Chemokines to Promote a Tumor Immune Response. Inflammation. 2020; 43:1201–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lapteva N, Aldrich M, Weksberg D, et al. Targeting the intratumoral dendritic cells by the oncolytic adenoviral vaccine expressing RANTES elicits potent antitumor immunity. J Immunother. 2009; 32:145–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li J, O’Malley M, Urban J, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. 2011; 19:650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Li J, O’Malley M, Sampath P, et al. Expression of CCL19 from oncolytic vaccinia enhances immunotherapeutic potential while maintaining oncolytic activity. Neoplasia. 2012; 14:1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moon EK, Wang LS, Bekdache K, et al. Intra-tumoral delivery of CXCL11 via a vaccinia virus, but not by modified T cells, enhances the efficacy of adoptive T cell therapy and vaccines. Oncoimmunology. 2018; 7:e1395997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Liu Z, Ravindranathan R, Li J, et al. CXCL11-Armed oncolytic poxvirus elicits potent antitumor immunity and shows enhanced therapeutic efficacy. Oncoimmunology. 2016; 5:e1091554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Eckert EC, Nace RA, Tonne JM, et al. Generation of a Tumor-Specific Chemokine Gradient Using Oncolytic Vesicular Stomatitis Virus Encoding CXCL9. Mol Ther Oncolytics. 2020; 16:63–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chiu M, Armstrong EJL, Jennings V, et al. Combination therapy with oncolytic viruses and immune checkpoint inhibitors. Expert Opin Biol Ther. 2020; 20:635–652. [DOI] [PubMed] [Google Scholar]
- 114.Wu C, Wu M, Liang M, et al. A novel oncolytic virus engineered with PD-L1 scFv effectively inhibits tumor growth in a mouse model. Cell Mol Immunol. 2019; 16:780–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kleinpeter P, Fend L, Thioudellet C, et al. Vectorization in an oncolytic vaccinia virus of an antibody, a Fab and a scFv against programmed cell death −1 (PD-1) allows their intratumoral delivery and an improved tumor-growth inhibition. Oncoimmunology. 2016; 5:e1220467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Lin C, Ren W, Luo Y, et al. Intratumoral Delivery of a PD-1-Blocking scFv Encoded in Oncolytic HSV-1 Promotes Antitumor Immunity and Synergizes with TIGIT Blockade. Cancer Immunol Res. 2020; 8:632–647. [DOI] [PubMed] [Google Scholar]
- 117.Engeland CE, Grossardt C, Veinalde R, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. 2014; 22:1949–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhu Y, Hu X, Feng L, et al. Enhanced Therapeutic Efficacy of a Novel Oncolytic Herpes Simplex Virus Type 2 Encoding an Antibody Against Programmed Cell Death 1. Mol Ther Oncolytics. 2019; 15:201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schiza A, Wenthe J, Mangsbo S, et al. Adenovirus-mediated CD40L gene transfer increases Teffector/Tregulatory cell ratio and upregulates death receptors in metastatic melanoma patients. J Transl Med. 2017; 15:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jiang H, Rivera-Molina Y, Gomez-Manzano C, et al. Oncolytic Adenovirus and Tumor-Targeting Immune Modulatory Therapy Improve Autologous Cancer Vaccination. Cancer Res. 2017; 77:3894–3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jiang H, Shin DH, Nguyen TT, et al. Localized Treatment with Oncolytic Adenovirus Delta-24-RGDOX Induces Systemic Immunity against Disseminated Subcutaneous and Intracranial Melanomas. Clin Cancer Res. 2019; 25:6801–6814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Rivera-Molina Y, Jiang H, Fueyo J, et al. GITRL-armed Delta-24-RGD oncolytic adenovirus prolongs survival and induces anti-glioma immune memory. Neurooncol Adv. 2019; 1:vdz009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Wenthe J, Naseri S, Hellström AC, et al. Immunostimulatory oncolytic virotherapy for multiple myeloma targeting 4–1BB and/or CD40. Cancer Gene Ther. 2020. May 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Eriksson E, Milenova I, Wenthe J, et al. Shaping the Tumor Stroma and Sparking Immune Activation by CD40 and 4–1BB Signaling Induced by an Armed Oncolytic Virus. Clin Cancer Res. 2017; 23:5846–5857. [DOI] [PubMed] [Google Scholar]
- 125.Zamarin D, Holmgaard RB, Ricca J, et al. Intratumoral modulation of the inducible co-stimulator ICOS by recombinant oncolytic virus promotes systemic anti-tumour immunity. Nat Commun. 2017; 8:14340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Vijayakumar G, McCroskery S, Palese P. Engineering Newcastle Disease Virus as an Oncolytic Vector for Intratumoral Delivery of Immune Checkpoint Inhibitors and Immunocytokines. J Virol. 2020; 94:e016677–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Thomas S, Kuncheria L, Roulstone V, et al. Development of a new fusion-enhanced oncolytic immunotherapy platform based on herpes simplex virus type 1. J Immunother Cancer. 2019; 7:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guo ZS, Lotze MT, Zhu Z, et al. Bi- and Tri-Specific T Cell Engager-Armed Oncolytic Viruses: Next-Generation Cancer Immunotherapy. Biomedicines. 2020; 8:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yu F, Wang X, Guo ZS, et al. T-cell engager-armed oncolytic vaccinia virus significantly enhances antitumor therapy. Mol Ther. 2014; 22:102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Speck T, Heidbuechel JPW, Veinalde R, et al. Targeted BiTE Expression by an Oncolytic Vector Augments Therapeutic Efficacy Against Solid Tumors. Clin Cancer Res. 2018; 24:2128–2137. [DOI] [PubMed] [Google Scholar]
- 131.de Sostoa J, Fajardo CA, Moreno R, et al. Targeting the tumor stroma with an oncolytic adenovirus secreting a fibroblast activation protein-targeted bispecific T-cell engager. J Immunother Cancer. 2019; 7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Freedman JD, Duffy MR, Lei-Rossmann J, et al. An Oncolytic Virus Expressing a T-cell Engager Simultaneously Targets Cancer and Immunosuppressive Stromal Cells. Cancer Res. 2018; 78:6852–6865. [DOI] [PubMed] [Google Scholar]
- 133.Scott EM, Jacobus EJ, Lyons B, et al. Bi- and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J Immunother Cancer. 2019; 7:320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wing A, Fajardo CA, Posey AD Jr., et al. Improving CART-Cell Therapy of Solid Tumors with Oncolytic Virus-Driven Production of a Bispecific T-cell Engager. Cancer Immunol Res. 2018; 6:605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bridle BW, Stephenson KB, Boudreau JE, et al. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010; 18:1430–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Pol JG, Zhang L, Bridle BW, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014; 22:420–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Pulido J, Kottke T, Thompson J, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012; 30:337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Castelo-Branco P, Passer BJ, Buhrman JS, et al. Oncolytic herpes simplex virus armed with xenogeneic homologue of prostatic acid phosphatase enhances antitumor efficacy in prostate cancer. Gene Ther. 2010; 17:805–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kennedy EM, Farkaly T, Behera P, et al. Abstract 1455: Design of ONCR-177 base vector, a next generation oncolytic herpes simplex virus type-1, optimized for robust oncolysis, transgene expression and tumor-selective replication. Cancer Res. 2019; 79(13 Supplement):1455–1455. [Google Scholar]
- 140.Santos JM, Heiniö C, Cervera-Carrascon V, et al. Oncolytic adenovirus shapes the ovarian tumor microenvironment for potent tumor-infiltrating lymphocyte tumor reactivity. J Immunother Cancer. 2020; 8:e000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Champion BR, Besneux M, Patsalidou M, et al. Abstract 5013: NG-641: An oncolytic T-SIGn virus targeting cancer-associated fibroblasts in the stromal microenvironment of human carcinomas. Cancer Res. 2019; 79(13 Supplement):5013–5013. [Google Scholar]
- 142.Saha D, Martuza RL, Rabkin SD. Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer Cell. 2017; 32:253–267. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Demonstration that ICI non-responsive cancer can be cured preclinically, but requires two ICIs and oHSV expressing IL12 to overcome immunosuppressive TME.
- 143.Harrington K, Freeman DJ, Kelly B, et al. Optimizing oncolytic virotherapy in cancer treatment. Nat Rev Drug Discov. 2019; 18:689–706. [DOI] [PubMed] [Google Scholar]
- 144.Malfitano AM, Di Somma S, Iannuzzi CA, et al. Virotherapy: From single agents to combinatorial treatments. Biochem Pharmacol. 2020; 177:113986. [DOI] [PubMed] [Google Scholar]
- 145.Phan M, Watson MF, Alain T, et al. Oncolytic Viruses on Drugs: Achieving Higher Therapeutic Efficacy. ACS Infect Dis. 2018; 4:1448–1467. [DOI] [PubMed] [Google Scholar]
- 146.Peng KW, Myers R, Greenslade A, et al. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 2013; 20:255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zemp FJ, McKenzie BA, Lun X, et al. Cellular factors promoting resistance to effective treatment of glioma with oncolytic myxoma virus. Cancer Res. 2014; 74:7260–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Roulstone V, Khan K, Pandha HS, et al. Phase I Trial of Cyclophosphamide as an Immune Modulator for Optimizing Oncolytic Reovirus Delivery to Solid Tumors. Clin Cancer Res. 2015; 21:1305–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Burke MJ, Ahern C, Weigel BJ, et al. Phase I trial of Seneca Valley Virus (NTX-010) in children with relapsed/refractory solid tumors: a report of the Children’s Oncology Group. Pediatr Blood Cancer. 2015; 62:743–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Cerullo V, Diaconu I, Kangasniemi L, et al. Immunological effects of low-dose cyclophosphamide in cancer patients treated with oncolytic adenovirus. Mol Ther. 2011; 19:1737–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kanai R, Rabkin SD, Yip S, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012; 104:42–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Saha D, Rabkin SD, Martuza RL. Temozolomide antagonizes oncolytic immunovirotherapy in glioblastoma. J Immunother Cancer. 2020; 8:e000345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Kleijn A, van den Bossche W, Haefner ES, et al. The Sequence of Delta24-RGD and TMZ Administration in Malignant Glioma Affects the Role of CD8(+)T Cell Anti-tumor Activity. Mol Ther Oncolytics. 2017; 5:11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Conte M, De Palma R, Altucci L. HDAC inhibitors as epigenetic regulators for cancer immunotherapy. Int J Biochem Cell Biol. 2018; 98:65–74. [DOI] [PubMed] [Google Scholar]
- 155.Jennings VA, Scott GB, Rose AMS, et al. Potentiating Oncolytic Virus-Induced Immune-Mediated Tumor Cell Killing Using Histone Deacetylase Inhibition. Mol Ther. 2019; 27:1139–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nguyên TL, Abdelbary H, Arguello M, et al. Chemical targeting of the innate antiviral response by histone deacetylase inhibitors renders refractory cancers sensitive to viral oncolysis. Proc Natl Acad Sci U S A. 2008; 105:14981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bridle BW, Chen L, Lemay CG, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013; 21:887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nguyen A, Ho L, Workenhe ST, et al. HDACi Delivery Reprograms Tumor-Infiltrating Myeloid Cells to Eliminate Antigen-Loss Variants. Cell Rep. 2018; 24:642–654. [DOI] [PubMed] [Google Scholar]
- 159.Chesney J, Puzanov I, Collichio F, et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J Clin Oncol. 2018; 36:1658–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Samson A, Scott KJ, Taggart D, et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci Transl Med. 2018; 10:eaam7577. [DOI] [PMC free article] [PubMed] [Google Scholar]; * Clinical trial of intravenous reovirus followed by tumor resection to examine immune effects of OV in brain tumors.
- 161.Zamarin D, Ricca JM, Sadekova S, et al. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J Clin Invest. 2018; 128:1413–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Rojas JJ, Sampath P, Hou W, et al. Defining Effective Combinations of Immune Checkpoint Blockade and Oncolytic Virotherapy. Clin Cancer Res. 2015; 21:5543–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Liu Z, Ravindranathan R, Kalinski P, et al. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat Commun. 2017;8:14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chon HJ, Lee WS, Yang H, et al. Tumor Microenvironment Remodeling by Intratumoral Oncolytic Vaccinia Virus Enhances the Efficacy of Immune-Checkpoint Blockade. Clin Cancer Res. 2019; 25:1612–1623. [DOI] [PubMed] [Google Scholar]
- 165.Durham NM, Mulgrew K, McGlinchey K, et al. Oncolytic VSV Primes Differential Responses to Immuno-oncology Therapy. Mol Ther. 2017; 25:1917–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.John LB, Howland LJ, Flynn JK, et al. Oncolytic virus and anti-4–1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res. 2012; 72:1651–60. [DOI] [PubMed] [Google Scholar]
- 167.Weichselbaum RR, Liang H, Deng L, et al. Radiotherapy and immunotherapy: a beneficial liaison? Nat Rev Clin Oncol. 2017; 14:365–379. [DOI] [PubMed] [Google Scholar]
- 168.Touchefeu Y, Vassaux G, Harrington KJ. Oncolytic viruses in radiation oncology. Radiother Oncol. 2011; 99:262–70. [DOI] [PubMed] [Google Scholar]
- 169.Twigger K, Vidal L, White CL, et al. Enhanced in vitro and in vivo cytotoxicity of combined reovirus and radiotherapy. Clin Cancer Res. 2008; 14:912–23. [DOI] [PubMed] [Google Scholar]
- 170.Wilkinson MJ, Smith HG, McEntee G, et al. Oncolytic vaccinia virus combined with radiotherapy induces apoptotic cell death in sarcoma cells by down-regulating the inhibitors of apoptosis. Oncotarget. 2016; 7:81208–81222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Salzwedel AO, Han J, LaRocca CJ, et al. Combination of interferon-expressing oncolytic adenovirus with chemotherapy and radiation is highly synergistic in hamster model of pancreatic cancer. Oncotarget. 2018; 9:18041–18052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim W, Seong J, Oh HJ, et al. A novel combination treatment of armed oncolytic adenovirus expressing IL-12 and GM-CSF with radiotherapy in murine hepatocarcinoma. J Radiat Res. 2011; 52:646–54. [DOI] [PubMed] [Google Scholar]
- 173.Udayakumar TS, Betancourt DM, Ahmad A, et al. Radiation Attenuates Prostate Tumor Antiviral Responses to Vesicular Stomatitis Virus Containing IFNβ, Resulting in Pronounced Antitumor Systemic Immune Responses. Mol Cancer Res. 2020; 18:1232–1243. [DOI] [PubMed] [Google Scholar]
- 174.Vijayakumar G, Palese P, Goff PH. Oncolytic Newcastle disease virus expressing a checkpoint inhibitor as a radioenhancing agent for murine melanoma. EBioMedicine. 2019; 49:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Harrington KJ, Vile RG, Melcher A, et al. Clinical trials with oncolytic reovirus: moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev. 2010; 21:91–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Sarén T, Ramachandran M, Martikainen M, et al. Insertion of the Type-I IFN Decoy Receptor B18R in a miRNA-Tagged Semliki Forest Virus Improves Oncolytic Capacity but Results in Neurotoxicity. Mol Ther Oncolytics. 2017; 7:67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Nosaki K, Hamada K, Takashima Y, et al. A novel, polymer-coated oncolytic measles virus overcomes immune suppression and induces robust antitumor activity. Mol Ther Oncolytics. 2016;3:16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Kowalsky SJ, Liu Z, Feist M, et al. Superagonist IL-15-Armed Oncolytic Virus Elicits Potent Antitumor Immunity and Therapy That Are Enhanced with PD-1 Blockade. Mol Ther. 2018; 26:2476–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Tosic V, Thomas DL, Kranz DM, et al. Myxoma Virus Expressing a Fusion Protein of Interleukin-15 (IL15) and IL15 Receptor Alpha Has Enhanced Antitumor Activity. PLOS ONE. 2014; 9:e109801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.LaRocca CJ, Han J, Gavrikova T, et al. Oncolytic adenovirus expressing interferon alpha in a syngeneic Syrian hamster model for the treatment of pancreatic cancer. Surgery. 2015; 157:888–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Oh E, Hong J, Kwon OJ, et al. A hypoxia- and telomerase-responsive oncolytic adenovirus expressing secretable trimeric TRAIL triggers tumour-specific apoptosis and promotes viral dispersion in TRAIL-resistant glioblastoma. Sci Rep. 2018; 8:1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Jahan N, Lee JM, Shah K, et al. Therapeutic targeting of chemoresistant and recurrent glioblastoma stem cells with a proapoptotic variant of oncolytic herpes simplex virus. Int J Cancer. 2017; 141:1671–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wu Y, He J, Geng J, et al. Recombinant Newcastle disease virus expressing human TRAIL as a potential candidate for hepatoma therapy. Eur J Pharmacol. 2017; 802:85–92. [DOI] [PubMed] [Google Scholar]