

Abstract
Ferroptosis is a type of programmed cell death induced by the accumulation of lipid peroxidation and lipid reactive oxygen species (ROS) in cells. It has been recently demonstrated that cancer cells are vulnerable to ferroptosis inducers (FIN). However, the therapeutic potential of ferroptosis inducers in prostate cancer in pre-clinical settings has not been explored. In this study, we demonstrate that mediators of ferroptosis SLC7A11, SLC3A2 and GPX4 are expressed in treatment-resistant prostate cancer. We further demonstrate that treatment-resistant prostate cancer cells are sensitive to two ferroptosis inducers, erastin and RSL3. Treatment with erastin and RSL3 led to a significant decrease in prostate cancer cell growth and migration in vitro and significantly delayed the tumor growth of treatment-resistant prostate cancer in vivo, with no measurable side effects. Combination of erastin or RSL3 with standard-of-care second-generation anti-androgens for advanced prostate cancer halted prostate cancer cell growth and migration in vitro and tumor growth in vivo. These results demonstrate the potential of erastin or RSL3 independently and in combination with standard-of-care second-generation anti-androgens as novel therapeutic strategies for advanced prostate cancer.
Introduction
Prostate cancer is the most commonly diagnosed noncutaneous malignancy in US men (1). Prostate cancer accounts for 30,000 deaths annually in the US, almost always from metastatic disease (1). The mainstay of treatment for advanced prostate cancer is androgen deprivation therapy (ADT) (2). Although ADT is initially effective in nearly all men, the disease commonly recurs referred to as castration resistant prostate cancer (CRPC) (3), which is largely responsible for prostate cancer associated deaths. Current therapeutic approaches for CRPC include second-generation anti-androgens such as enzalutamide, abiraterone, apalutamide and darolutamide, taxane-based chemotherapy, immunotherapy and targeted therapies (4–13). While adenocarcinoma positive for androgen receptor (AR) is the predominant histological variant of CRPC (adeno-CRPC), 10–15% of metastatic CRPCs present neuroendocrine phenotype called neuroendocrine prostate cancer (NEPC) and 20–25% present double-negative phenotype (DNPC) (14–19). NEPC and DNPC are characterized with loss of expression of AR resulting in resistance to therapies that target the AR pathway and aggressive clinical behavior (14–18). Presence of neuroendocrine markers is a characteristic of NEPC, while DNPC exhibits lack of neuroendocrine markers (14–18). Currently, there are no long-term effective or curative treatments available for adeno-CRPC, NEPC and DNPC and thus exploring novel therapeutic approaches for advanced prostate cancer is critical.
Ferroptosis is an iron-dependent programmed cell death mechanism that is induced by the accumulation of lipid peroxidation (20–24). Previous studies revealed that ferroptosis is characterized by accumulation of peroxidation of phospholipids enriched with polyunsaturated fatty acids and reactive oxygen species (ROS) (25). Glutathione peroxidase (GPX4) and solute carrier family 7 member 11 (SLC7A11) are two major regulators of ferroptosis (6, 25–29). GPX4 utilizes reduced glutathione (GSH) to convert lipid hydroperoxides to lipid alcohols, thereby alleviating lipid peroxidation and inhibiting ferroptosis while SLC7A11 is a transmembrane transporter that exchanges extracellular cystine for intracellular glutamate (6, 25–29). Loss or pharmacological inhibition of GPX4 or SLC7A11 leads to ferroptosis induction (26, 27, 30). Ferroptosis is involved in pathophysiological processes of various diseases including cancers, and can act as a natural barrier to tumor progression (31, 32). In cancer, ferroptosis inducers have shown a promising anti-cancer activity in models of multiple cancer types (24, 33, 34). Ferroptosis was recognized as a distinct mechanism of non-apoptotic programmed cell death through a small molecule screen, and the identification of erastin and RSL3 as compounds that induce selective lethality in cancer cells that express mutant HRAS (24, 35–38). RSL3 is known to inhibit GPX4, and loss or inhibition of GPX4 leads to induction of ferroptosis in cancer cells (24, 26). Erastin inhibits the cystine/glutamate transporter system Xc- composed of SLC7A11 and SLC3A2 amino acid transporters, and has been shown to induce ferroptosis across cancer types (24, 38, 39). Previous studies have shown that drug-resistant cancer cells are vulnerable to GPX4 inhibition and ferroptosis induction (33, 34). Moreover, induction of ferroptosis enhances the therapeutic efficacy of cisplatin in cancer cells (40, 41), suggesting that ferroptosis inducers may be even more potent in combination therapy settings. In the context of prostate cancer, it has been shown that DECR1 is an androgen-regulated survival protein that protects cells from ferroptosis and targeting DECR1 induces ferroptosis (42). In addition, treatment with enzalutamide induces lipid peroxidation and leads to sensitivity to GPX4 inhibition and ferroptosis in vitro (43). However, the therapeutic potential of ferroptosis inducers erastin and RSL3 in prostate cancer has not been tested in vivo.
Herein, we performed pre-clinical assessment of the therapeutic potential of two FINs, erastin and RSL3, in treatment-resistant prostate cancer. We demonstrate that ferroptosis mediators, SLC7A11, SLC3A2 and GPX4 are expressed in adeno-CRPC, DNPC and NEPC xenografts. Erastin and RSL3 increase ROS production and impair cell viability, growth, and migration of prostate cancer cells in vitro. Erastin and RSL3 also significantly delay prostate cancer tumor growth in vivo. Treatment with FINs in combination with second-generation anti-androgens, enzalutamide and abiraterone, halted prostate cancer cell growth and migration in vitro and tumor growth in vivo. Our study demonstrates that prostate cancer cells are vulnerable to ferroptosis induction, and FINs may represent a new class of therapeutic agents for advanced prostate cancer as single agents and in combination with standard-of-care therapies for CRPC.
Materials and Methods
Immunohistochemical staining
Indicated cell line derived xenograft tumor samples were fixed in 10% formalin overnight at 4° C, followed by immersion in 70% ethanol and subsequently embedded in paraffin. Samples were sectioned at 4 microns and affixed to slides. Sections from cell line-derived xenograft or PDX tumor samples were heated to 65°C for one hour, then moved to clarify to remove paraffin, followed by rehydration in sequential ethanol (100%, 95% and 70%) to rehydrate. After 10-minute water incubation, antigen retrieval was performed in 10 mM sodium citrate buffer pH 6.0 at 95°C for 20 minutes. Samples were allowed to cool to room temperature and rinsed, and then incubated with 3% hydrogen peroxide for 10 minutes to block endogenous peroxidase activity. Tumor samples were blocked with 2.5% goat serum for one hour followed by incubation with primary antibodies, anti-SLC7A11 (PA5-33050, 1:100), anti-GPX4 (sc-166120, 1:100) overnight at 4° C in a humidifying chamber. After three washes with 1X PBS, samples were incubated with anti-mouse HRP-conjugated secondary antibody for one hour at room temperature followed by developing with DAB reagent (DAKO), and counter-staining with hematoxylin. The PDX tissue samples were subjected to manual blinded scoring for intensity of staining as low, moderate and high as shown in Supplementary Figure S1A, B.
Cell lines and cell culture
Human prostate cancer cell lines DU145, PC3, 22Rv1, LNCaP and NCI-H660 were purchased from American Type Culture Collection (ATCC). ARCaP was purchased from Novicure Biotechnology. C4-2 cells were a kind gift from Dr. Owen Witte (University of California Los Angeles). DU145, PC3, 22RV1, ARCaP, C4-2, and LNCaP were cultured and maintained in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum, 100 U/mL penicillin, and 0.1% streptomycin, and 1% GlutaMAX. NCI-H660 cells were maintained in RPMI-1640 Medium supplemented with 5% fetal bovine serum, 0.005 mg/ml Insulin, 0.01 mg/ml transferrin, 30 nM sodium selenite, 10 nM hydrocortisone, 10 nM beta-estradiol, and 1% GlutaMAX. Cell lines were authenticated at the Stanford functional genomics facility for short tandem repeat (STR) profiling and routinely assayed for mycoplasma using MycoAlert Mycoplasma Detection kit (Lonza).
Reagents
Erastin, (1S,3R)-RSL3, and ferrostatin-1 were purchased from APExBIO for in vitro experiments. The compounds were dissolved in dimethyl sulfoxide (DMSO) at 10 mM concentration and stored at −20° C. For in vivo experiments, erastin was obtained from BOC Sciences and (1S,3R)-RSL3 was purchased from Cayman Chemical (Ann Arbor, Michigan, USA). Enzalutamide and abiraterone acetate were purchased from Targetmol (Boston, Massachusetts, USA). Enzalutamide was dissolved in DMSO, and abiraterone was dissolved in either absolute ethanol or DMSO for in vitro as previously described (44–48) and in vivo studies, respectively.
CellTiter-Blue cell viability assay
5000 cells/well were seeded into 96-well plates. After incubation for 24 hours at 37° C, cells were treated with erastin (1.25, 2.5, 5, 10, and 20 μM), or (1S,3R)-RSL3 (0.125, 0.25, 0.5, 1, 2, and 4 μM) and matched volumes of DMSO were used as a vehicle control. 72 hours post-treatment, the viability of cells was assayed with the CellTiter-Blue® Cell Viability Assay (Promega) kit according to the manufacturer’s instructions and measured with a Tecan plate reader. Cell viability was calculated as percentage (%) compared to the control (0 or vehicle treatment) for each cell line as follows: The mean absorbance of the control (0 or vehicle treatment) was set to 100%. Percentage viability in each control technical replica was calculated: each control technical replicate/mean absorbance control × 100. For the treatment arms the % cell viability is equal to the absorbance of each treatment technical replicate/mean absorbance control × 100.
7AAD and Trypan blue assays
5×104 DU145, PC3 or C4-2 cells were cultured in 24-well plates overnight at 37° C. The next day, cells were treated with erastin (1.25, 2.5, 5 μM) or (1S,3R)-RSL3 (0.125, 0.25, 0.5 μM) for 72 hours. Cells were washed with PBS and harvested with Trypsin/EDTA (0.25%) followed by resuspension in PBS with 0.5 μg/ml 7AAD (BioLegend, 420403) for 10 minutes in the dark before analysis using flow cytometry. To analyze live-dead cell ratios, treated cells were stained with 0.4% trypan blue solution. The percentage of live-dead cells was quantified using Countess II Automated Cell Counter (Thermo Fisher Scientific).
Colony formation assays
DU145, PC3, ARCaP, 22Rv1, C4-2 (500 cells per well) and LNCaP (5000 cells per well) cell lines were grown in 6-well plates overnight to allow cells to attach. Cells were treated with erastin (5 μM), or (1S,3R)-RSL3 (0.5 μM) for single treatment experiments. For combination treatment experiments, C4-2 cells were treated with erastin (2 μM), (1S,3R)-RSL3 (50 nM), enzalutamide (2 μM), and abiraterone acetate (2 μM) in vitro as previously described (44–48). Cells were cultured for nine days, and media containing 10% FBS and compounds were exchanged every three days. Colonies were fixed with methanol and stained with 0.01% crystal violet solution for one hour at room temperature and washed with water. An equal volume of DMSO was used as a control.
Migration assay
48 hours prior seeding into 24-well transwell inserts, pore size 8.0 um, 6.5 mm diameter (Transwell™ Permeable Polyester Membrane Inserts, Corning Inc, Corning, New York), cells were treated with erastin (1.25 μM), (1S,3R)-RSL3 (0.125 μM) or an equal volume of DMSO as a control. 5×104 cells were then seeded in serum-free media in 24-well transwell inserts. The inserts were incubated in 10% FBS supplemented media in 24-well plates for 20 hours. The bottom chamber was filled with 10% FBS supplemented media containing erastin (1.25 μM), (1S,3R)-RSL3 (0.125 μM) or an equal volume of DMSO as a vehicle control. Cells that passed through the membrane were fixed and stained with 0.01% crystal violet solution. For migration assays with higher concentrations of compounds, cells were seeded in serum-free medium including erastin (5 μM), or (1S,3R)-RSL3 (0.5 μM) in 24-well transwell inserts. The inserts were then incubated in 10% FBS supplemented medium including erastin (5 μM), or (1S,3R)-RSL3 (0.5 μM) in 24-well plates for 20 hours.
3D Matrigel drop invasion assay
5×104 DU145, PC3 or C4-2 cells were suspended in 10 μl matrigel and pipetted as a droplet into a 24-well plate for 20 minutes to form Matrigel drop prior to adding media and compounds as described previously (49, 50). DU145 and PC3 tumoroids were treated with erastin (1.25 μM and 5 μM) or (1S,3R)-RSL3 (0.125 μM and 0.5 μM) every three days for six days. C4-2 tumoroids were treated with erastin (5 μM) or (1S,3R)-RSL3 (0.5 μM). The radial distance the cell had migrated away from the edge of tumoroids was measured as radial migration on day six. For combination therapy experiments, erastin or (1S,3R)-RSL3 were combined with enzalutamide or abiraterone acetate at the following doses: erastin (5 μM), (1S,3R)-RSL3 (0.5 μM), enzalutamide (5 μM), and abiraterone acetate (5 μM). Media containing 10% FBS and compounds were changed once at Day 3. DMSO and ethanol were used as vehicle controls.
Cellular reactive oxygen species (ROS) measurement
Briefly, 1×105 cells were grown in 12-well plates overnight at 37°C. The next day, cells were treated with erastin (5 μM) or (1S,3R)-RSL3 (1 μM) for 6 hours followed by the addition of 1 μM H2DCF for 20 minutes. Cells were washed with PBS and harvested with Trypsin/EDTA (0.25%) followed by washing twice with PBS. Cells were subjected to flow cytometry to measure the levels of cellular ROS.
Western blotting
50 mg of xenograft tissues were homogenized and lysed using RIPA lysis buffer containing protease inhibitor cocktails (Thermo Scientific). Protein was quantified using the BCA assay, and an equal amount of protein for each lysate (50 ug) was resolved by 4–12% gradient SDS-PAGE followed by transfer onto nitrocellulose membranes. Membranes were blocked with 5% milk for 1 hour in tris buffered saline and probed with primary antibodies for overnight at 4° C in tris buffered saline. Washing was followed by incubation with secondary antibody for 1 hour at room temperature in tris buffered saline containing 0.1% Tween-20. Anti-GPX4 (sc-166120, 1:1000 for WB) and anti-GAPDH antibodies (sc-32233, 1:3000) were obtained from Santa Cruz Biotechnology. Anti-SLC7A11 antibody was purchased from Invitrogen, Thermo Fisher Scientific (PA1-16775, 1:500 for WB). Anti-SLC3A2 was purchased from Santa Cruz (sc-376815). Secondary antibodies with HRP were obtained from Fisher Scientific (anti-mouse PI31432, and anti-rabbit PI31462, 1:2000). Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific, PI32106) was used to develop chemiluminescent signals that were measured on an IVIS Lumina imager.
Xenograft models
All animal studies and procedures have been approved and performed in accordance with Stanford Administrative Panel on Laboratory Animal Care (APLAC), IACUC, as well as the USAMRMC Animal Care and Use Review Office (ACURO). DU145, PC3, ARCaP, or C4-2 (5×105) and NCI-H660 (1×106) cells were suspended in 50 μl of 80% matrigel. Tumor cells were implanted subcutaneously into the flank of 6–8 weeks old NSG (NOD-SCID-IL2R γ) (Jackson Laboratory) male mice. Mice with established tumors with ~50–80 mm3 average volumes (measured by calipers and calculated as (length × width × height) / 2) were randomized into treatment groups including vehicle (DMSO), erastin (20 mg/kg in 20 μl DMSO plus 130 μl corn oil, ip, daily) as previously described (51), RSL3 (100 mg/kg in 20 μl DMSO plus 80 μl corn oil, ip, biweekly) (26). For combination therapy experiments, mice were randomized into different treatment groups including vehicle, erastin (20 mg/kg), RSL3 (100 mg/kg), enzalutamide (10 mg/kg in 5% DMSO, 30% PEG 300, 65% H20, oral gavage, daily), the combination of erastin with enzalutamide, or the combination of RSL3 with enzalutamide.
Results
SLC7A11 and GPX4 are expressed in advanced prostate cancer.
To examine the clinical relevance of ferroptosis in prostate cancer, we first assessed SLC7A11 and GPX4 protein levels in LuCaP patient-derived xenograft (PDX) models derived from metastatic prostate cancer (52) (Figure 1A, B and Supplementary Figure S1A, B). High levels of GPX4 were detected in adeno-CRPC (PDX n=36, sample n=108) as well as NEPC PDX samples (PDX n=3, sample n=9) (Figure 1B), while high levels of SLC7A11 were predominantly observed in adeno-CRPC (PDX n=35, sample n=105) (Figure 1A). The protein levels of SLC7A11 and GPX4 were further tested in prostate cancer cell lines in vitro (Supplementary Figure S1C, D). We also assessed SLC7A11 and SLC3A2 subunits of the xCT amino acid transporter along with GPX4 protein levels in prostate cancer cell line-derived xenografts including LNCaP (androgen sensitive), ARCaP AR low (adeno-CRPC), C4-2, 22Rv1 (adeno-CRPC), PC3 (NEPC-like characterized with lack of AR and expression of some of the NE markers), DU145 (DNPC), H660 (NEPC), and Trop2 derived-neuroendocrine prostate cancer (TD-NEPC) (49) by immunohistochemistry (IHC) (Figure 1C–E). SLC7A11, SLC3A2 and GPX4 were expressed across all tested xenografts (Figure 1C–E) but slightly different from what we have observed by western blot (Supplementary Figure S1C, D). Taken together, our results reveal that SLC7A11, SLC3A2 and GPX4 are expressed across androgen sensitive, adeno-CRPC, NEPC and DNPC xenografts.
Figure 1. SLC7A11, SLC3A2 and GPX4 are expressed in advanced prostate cancer.
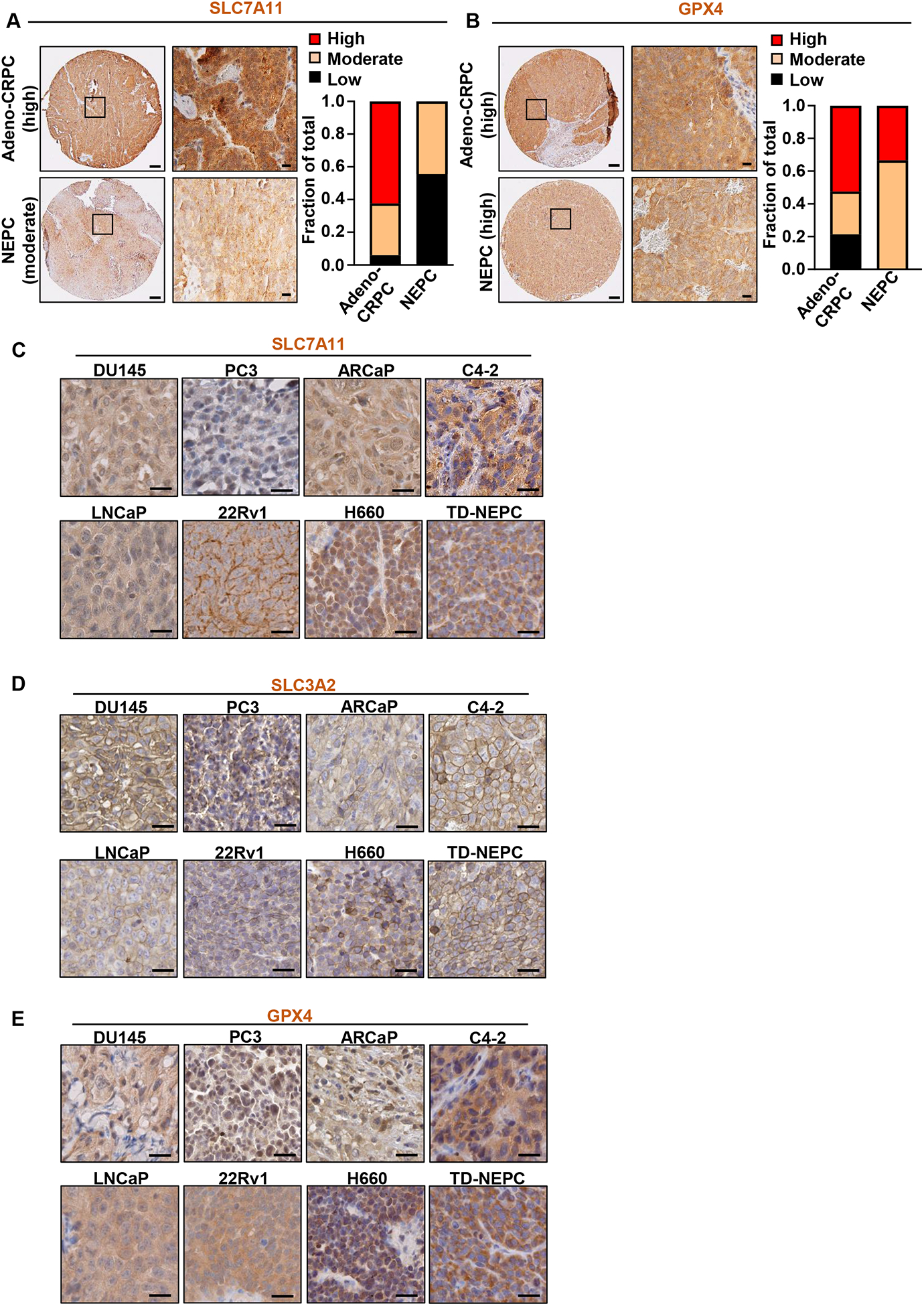
(A, B) IHC staining of SLC7A11 (A) and GPX4 (B) in previously described PDX TMAs TD-NEPC (52). LuCaP PDX TMAs contained 3 sample cores per PDX from adeno-CRPC (PDX n=36, sample n=108 or n=35, sample n=105) and NEPC PDXs (PDX n=3, sample n=9). Cores with insufficient tissue were excluded from the analyses. Intensities of IHC staining for SLC7A11 and GPX4 were blind scored as low, moderate, and high as shown in Supplementary Figure S1A and S1B and plotted. Scale bars represent 200 μm and 20 μm respectively. (C, D, E) IHC staining of SLC7A11 (C) SLC3A2 (D) and GPX4 (E) on different cell line-derived prostate cancer xenografts including DU145, PC3, ARCaP, C4-2, LNCaP, 22Rv1, H660, and previously described TD-NEPC (Trop2-driven NEPC) (49) Scale bars represent 20 μm.
Ferroptosis induction inhibits prostate cancer cell growth, invasion and migration in vitro.
To test whether prostate cancer is sensitive to ferroptosis induction, we treated prostate cancer cell representing different variants of prostate cancer including AR positive androgen sensitive (LNCaP), AR positive adeno-CRPC (C4-2), AR and AR-V7 isoform positive adeno-CRPC (22Rv1), NEPC (H660), NEPC-like (PC3), AR low adeno-CRPC (ARCaP) and DNPC (DU145) in vitro with different doses of erastin and RSL3 (Figure 2A, B). All prostate cancer cell lines were vulnerable to ferroptosis induction mediated by erastin and RSL3 (Figure 2A, B). Treatment with Ferrostatin-1 (Fer-1), an inhibitor of ferroptosis (21), rescued the cells from erastin-induced ferroptosis in vitro (Supplementary Figure S2A). Treatment with erastin and RSL3 led to an increase in intracellular ROS levels, a hallmark of ferroptosis induction, in all prostate cancer cell lines except H660 at 6 hrs post treatment initiation (Figure 2C, D). As H660 are sensitive to erastin and RSL3 on viability assay at 72 hrs post treatment (Figure 2A, B), it is plausible that in H660 ROS occurs later and immediately preceding the effects on viability or their sensitivity is potentially due to high levels of iron in these cells. In addition, H660 are slow growing non-adherent cells, which could be possibly associated with the time at which they release ROS. Both, erastin and RSL3 diminished colony formation of all tested prostate cancer cell lines (Figure 2E, F). Together, these results suggest that ferroptosis induction is a promising approach to impair the growth of prostate cancer cells independent of their phenotype and AR status in vitro.
Figure 2. Erastin and RSL3 induce ROS and inhibit prostate cancer cell growth in vitro.
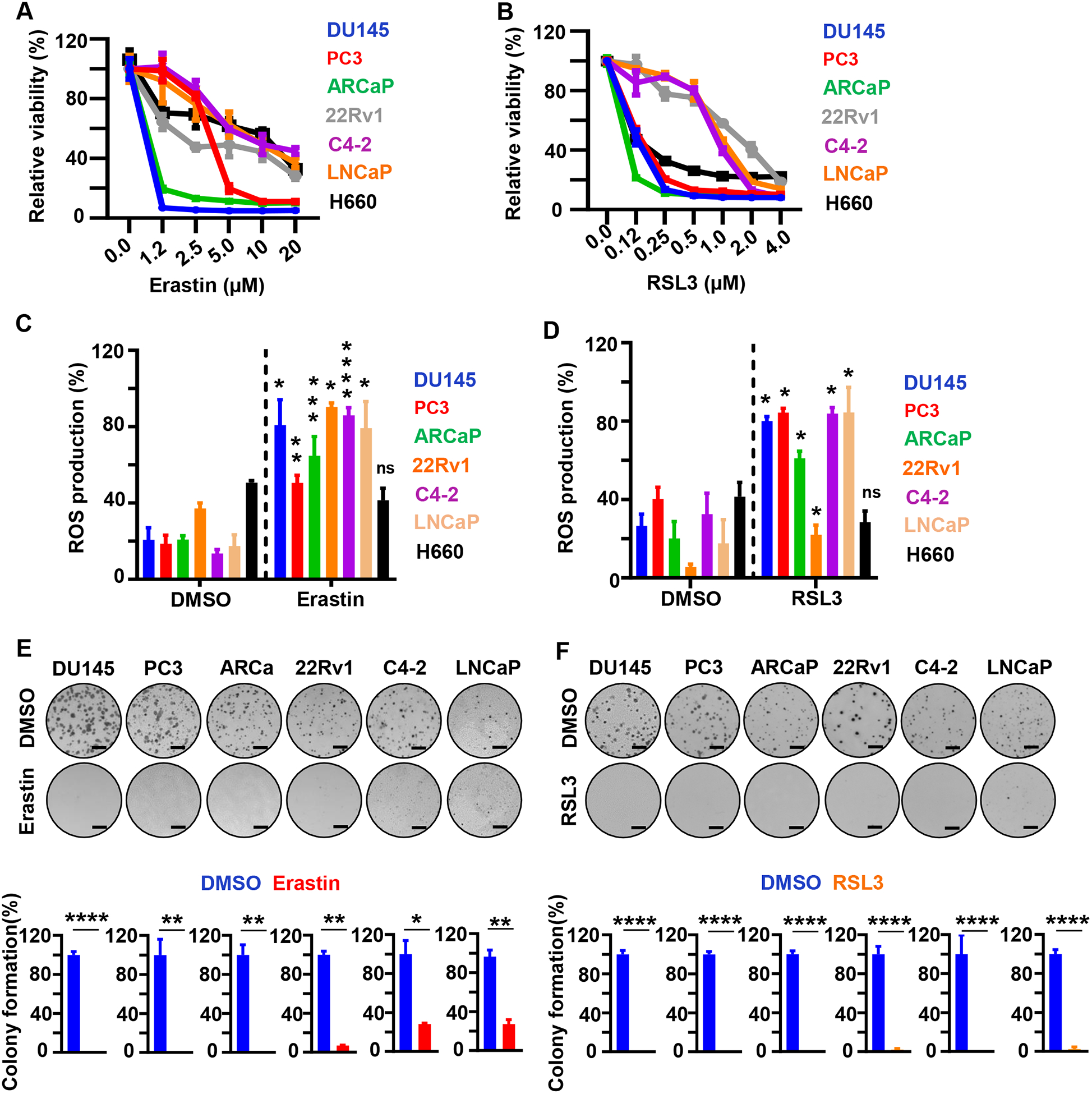
(A, B). Viability (%) of prostate cancer cell lines (DU145, PC3, ARCaP, 22RV1, C4-2, LNCaP and H660) following erastin (0, 1.25, 2.5, 5, 10, and 20 μM) (A) or RSL3 (0, 0.125, 0.25, 0.5, 1.0, 2.0, and 4.0 μM) (B) treatment for 72 hours. Experiments were repeated twice independently to verify the reproducibility of the data. Representative experiments are shown. (C, D) ROS measurement by flow cytometry. Cells were treated with erastin (5 μM) (C) or RSL3 (1 μM) (D) for 6 hours followed by incubation with H2DCF for 20 min at 37°C. Relative fluorescence was normalized to untreated control cells and represented as relative percentage of ROS production. Experiments were performed in triplicate and shown. (E, F) Colony formation assay. Prostate cancer cells were grown for nine days in presence of erastin (5 μM) (E) or RSL3 (500 nM) (F). Representative experiments and images are shown. Media containing the indicated compounds was changed every three days. Three independent experiments were performed with triplicate wells. Representative experiments and images are shown. Scale bars represent 4 mm. * P<0.05, **P<0.01, *** P<0.001, **** P<0.0001, ns-no significance, Student’s t-test. Error bars represent mean ± SEM.
Cell motility and migration are critical for cancer progression, invasiveness, and metastasis. We investigated whether ferroptosis induction affected prostate cancer migration and invasion utilizing a 3D Matrigel drop invasion assay (49, 50) and a transwell chamber migration assay. To ensure the rate of migration was not affected by cell death, we treated DU145, PC3 and C4-2 cells with erastin and RSL3 at concentrations that did not affect cell viability (Supplementary Figure S2B, C). For 3D Matrigel drop assay, the radial distance the cells had migrated cells from edge of the matrigel drop were measured on Day 6 after plating the cells (Figure 3A–D). Treatment of prostate cancer cells with two different doses of erastin or RSL3 significantly inhibited prostate cancer cell migration and invasion (Figure 3A–D and Supplementary Figure S3A, B). Likewise, treatment with erastin and RSL3 decreased the migration of PC3 and DU145 cells in a transwell migration assay (Figure 3E, F and Supplementary Figure S3C, D). Taken together, the results suggest that ferroptosis induction induces intracellular ROS production and reduces prostate cancer cell growth and migration.
Figure 3. Erastin and RSL3 inhibit prostate cancer cell invasion and migration in vitro.
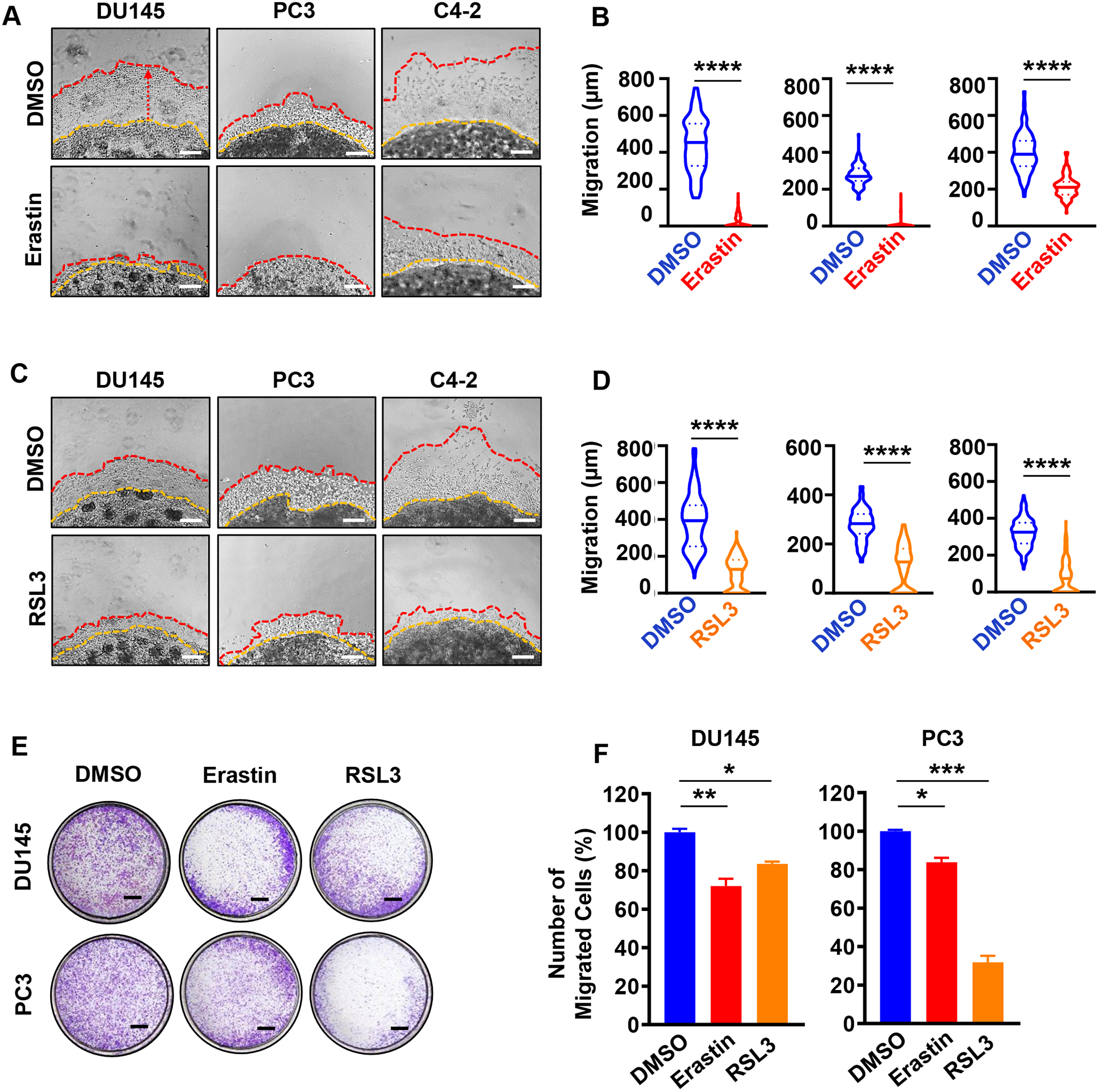
(A-D) 3D Matrigel drop invasion assay for DU145, PC3 and C4-2 cells upon erastin (A, B) or RSL3 (C, D) treatment. DU145 and PC3 matrigel drops were treated with erastin (1.25 μM) or RSL3 (125 nM). C4-2 matrigel drops were treated with higher doses of erastin (5 μM) or RSL3 (500 nM) because of their sensitivity to the compounds at higher doses on viability assay (Figure 2A and B). Media and treatments were exchanged every three days for six days incubation. The distances of migrated cells away from edge of the matrigel drop were measured as migration (μm) on Day 6. Experiments were performed in duplicate with triplicate wells. Representative experiments and images are shown. Scale bars represent 200 μm. (E-F) Migration assay for DU145 and PC3 cells. Cells were pretreated with erastin (1.25 μM) or RSL3 (125 nM) for 48 hours. Then, 5×104 viable cells were plated into transwell chambers for 20 hours upon erastin (1.25 μM) or RSL3 (125 nM) treatment, then fixed and stained with methanol and 0.01% crystal violet solution. Scale bar=1 mm. Experiments were performed in duplicate with two wells for each condition. Representative experiments and images are shown. For all experiments, DMSO was used as a vehicle control. For treated cells, the relative percentage of migrated cells was calculated relative to the DMSO treated control. Statistical analysis was performed with Student’s t-test (* P<0.05, **P<0.01, *** P<0.001, **** P<0.0001, ns-no significance) and error bars represent mean ± SEM.
Erastin and RSL3 decrease prostate tumor growth in vivo.
Currently, there are no effective therapies for NEPC and DNPC. Hence, we further tested the effect of erastin and RSL3 on DNPC, adeno-CRPC with low AR and NEPC. To test the therapeutic efficacy of erastin and RSL3 in vivo, we established subcutaneous xenograft tumor models of DU145 (DNPC), ARCaP (adeno-CRPC with low AR), PC3 (NEPC-like), and H660 (NEPC) human prostate cancer cell lines in the flanks of immunocompromised (NSG) male mice. When tumor volumes reached an average size of ~50–80 mm3, animals were treated with erastin (20 mg/kg) or vehicle, administered intraperitoneally once daily (Figure 4A). There were no measurable side effects observed in erastin-treated animals as assessed by animal body weight and lack of distressed behavior (Supplementary Figure 4A). Treatment of mice with erastin led to a significant decrease in tumor growth with an increase in tumor necrosis in DU145, ARCaP, PC3, and H660 (P< 0.05) xenografts (Figure 4B, C and Supplementary Figure S4B). Similarly, treatment with RSL3 significantly decreased tumor growth and tumor weight at end point of DU145 (P<0.0001) and PC3 (P<0.01) xenografts with no measurable side effects assessed by animal body weight and any signs of distress (Figure 4D–F and Supplementary Figure S4C, D). These data indicate that erastin and RSL3 decrease prostate cancer tumor growth in vivo.
Figure 4. Treatment with erastin and RSL3 delays prostate cancer growth in vivo.

(A) Schematic diagram of experimental design using erastin treatment. 5×105 DU145, ARCaP or PC3 (5×105) and NCI-H660 (1×106) cells were mixed in 50 μl of 80% matrigel and implanted subcutaneously (sc) into the flanks of male NSG mice. When tumors averaged 50 to 80 mm3, mice were randomized into vehicle or erastin treatment (20 mg/kg, IP, daily) groups. (B) DU145, ARCaP, PC3, and H660 tumors volumes were measured by caliper (1/2 × (Length × Width × Height)) every three days and presented as fold change over the tumor volume at Day 1. (C) Tumor weights (g) were measured following tumor excision at the experimental end point and graphed as violin plots. (D) Schematic diagram of experimental design upon RSL3 treatment. (E) DU145 and PC3 (5×105) cells were mixed in 80% Matrigel and implanted subcutaneously into flank of NSG male mice. Once the average of tumor volume reached to 50 to 80 mm3, mice were randomized into vehicle or RSL3 treatment (100 mg/kg, IP, biweekly) groups. Tumor volumes (1/2 (Length × Width × Height)) were measured every third day and shown as fold change over Day 1 tumor volume. (F) Tumor weights (g) were measured at the experimental end point. Statistical analysis was performed with Student’s t-test at each time point (* P<0.05, **P<0.01, *** P<0.001, and **** P<0.0001). Error bars signify mean ± SEM.
Combination of ferroptosis inducers with second-generation anti-androgens impedes tumor cell growth in vitro and in vivo.
We further tested the therapeutic potential of erastin and RSL3 in combination therapies with second-generation anti-androgens enzalutamide or abiraterone. C4-2 adeno-CRPC cells that express AR were used for the studies as they are responsive to enzalutamide and abiraterone. Cells that are AR low or AR negative were not used for testing combinations, due to their resistance to agents targeting AR signaling axis. Erastin in combination with either enzalutamide or abiraterone dramatically reduced colony formation when compared to cells treated with either agent alone (Figure 5A, B). Likewise, RSL3 in combination with enzalutamide or abiraterone decreased colony formation of prostate cancer cells when compared to treatment with RSL3, enzalutamide or abiraterone alone (Figure 5C, D). Treatment of C4-2 cells with erastin in combination with either enzalutamide or abiraterone significantly reduced C4-2 cell migration and invasion in vitro (Figure 5E, F). Similarly, RSL3 in combination with enzalutamide or abiraterone inhibited C4-2 cell migration and invasion when compared to RSL3, enzalutamide or abiraterone alone (Figure 5G, H).
Figure 5. Combination of erastin or RSL3 with second generation anti-androgens, enzalutamide and abiraterone, inhibits prostate cancer cell growth and invasion in vitro.
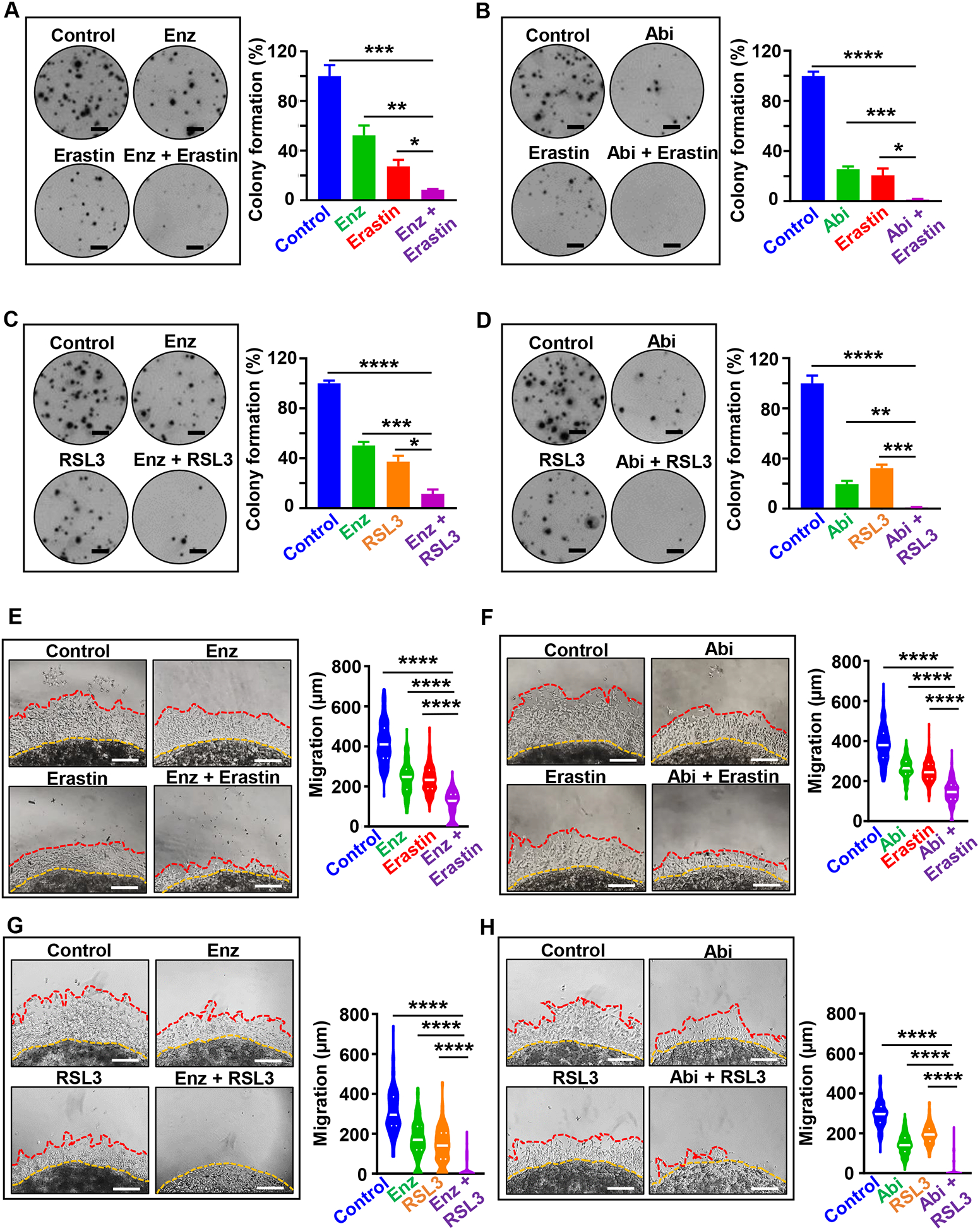
(A-D) Colony formation assay. (A, B) C4-2 cells were grown for nine days in presence of erastin (2 μM), enzalutamide (2 μM), abiraterone acetate (2 μM), erastin plus enzalutamide, and erastin plus abiraterone acetate. Media containing the compounds was exchanged every three days. Colonies were then fixed in methanol and stained with crystal violet. After washing and drying plates, plates were scanned on Celigo Imaging Cytometer (Nexcelom Bioscience) and the percentage of the well covered by colonies was quantified in ImageJ. (C, D) Colony formation assay for C4-2 cells upon treatment with RSL3 (50 nM), enzalutamide (2 μM), abiraterone acetate (2 μM), RSL3 plus enzalutamide, and RSL3 plus abiraterone acetate for nine days. Three independent experiments were performed with triplicate wells. Representative experiments and images are shown. Scale bars represent 4 mm. (E-H) 3D Matrigel drop invasion assay. (E, F) C4-2 cells were plated in matrigel drop invasion assay and treated with erastin (5 μM), enzalutamide (5 μM), abiraterone acetate (5 μM), erastin plus enzalutamide, and erastin plus abiraterone acetate (G, H) C4-2 prostate cancer cells were plated in matrigel drop invasion assay and treated with RSL3 (500 nM), enzalutamide (5 μM), abiraterone acetate (5 μM), RSL3 plus enzalutamide, and RSL3 plus abiraterone acetate. Media and treatment were exchanged every three days for six days. Radial migration distance (μm) was measured on Day 6. Experiments were performed in duplicate with triplicate wells. Statistical analysis was performed with Student’s t-test (* P< 0.05, **P<0.01, *** P < 0.001, **** P<0.0001, ns-no significance) and error bars represent mean ± SEM.
To evaluate the therapeutic efficacy of erastin and RSL3 in combination with anti-androgens in vivo, we treated established C4-2 xenografts with erastin (20 mg/kg, IP, daily) and enzalutamide (10 mg/kg, oral gavage, daily) when tumor volumes reached ~50–80 mm3 on average (Figure 6A). Consistent with the effects of erastin and RSL3 on adeno-CRPC with low AR, DNPC and NEPC xenografts (Figure 4), erastin and RSL3 significantly delayed the tumor growth of C4-2 xenografts (Figure 6). Furthermore, combined treatment with erastin and enzalutamide significantly inhibited tumor growth assessed by tumor volumes and tumor weights at end point when compared to treatment with vehicle, erastin or enzalutamide alone (Figure 6B, C). We did not observe any significant differences in body weight of animals treated with erastin and enzalutamide when compared to vehicle control and single therapy arms (Figure 6D). Furthermore, we tested the therapeutic potential of RSL3 in combination with enzalutamide in vivo using C4-2 xenograft model (Figure 6E–H). Consistent with erastin, RSL3 in combination with enzalutamide halted tumor growth and was more potent than RSL3 and enzalutamide alone in vivo (Figure 6E–G). We did not detect any measurable side effects assessed by animal body and signs of distress in any of the treatments when compared to vehicle control (Figure 6H). Therefore, the combination of erastin or RSL with second generation anti-androgens represents a potent therapeutic strategy when compared to single agent treatments for adeno-CRPC.
Figure 6. Combination of erastin or RSL3 with second generation anti-androgens, enzalutamide and abiraterone, inhibits prostate cancer xenograft growth in vivo.
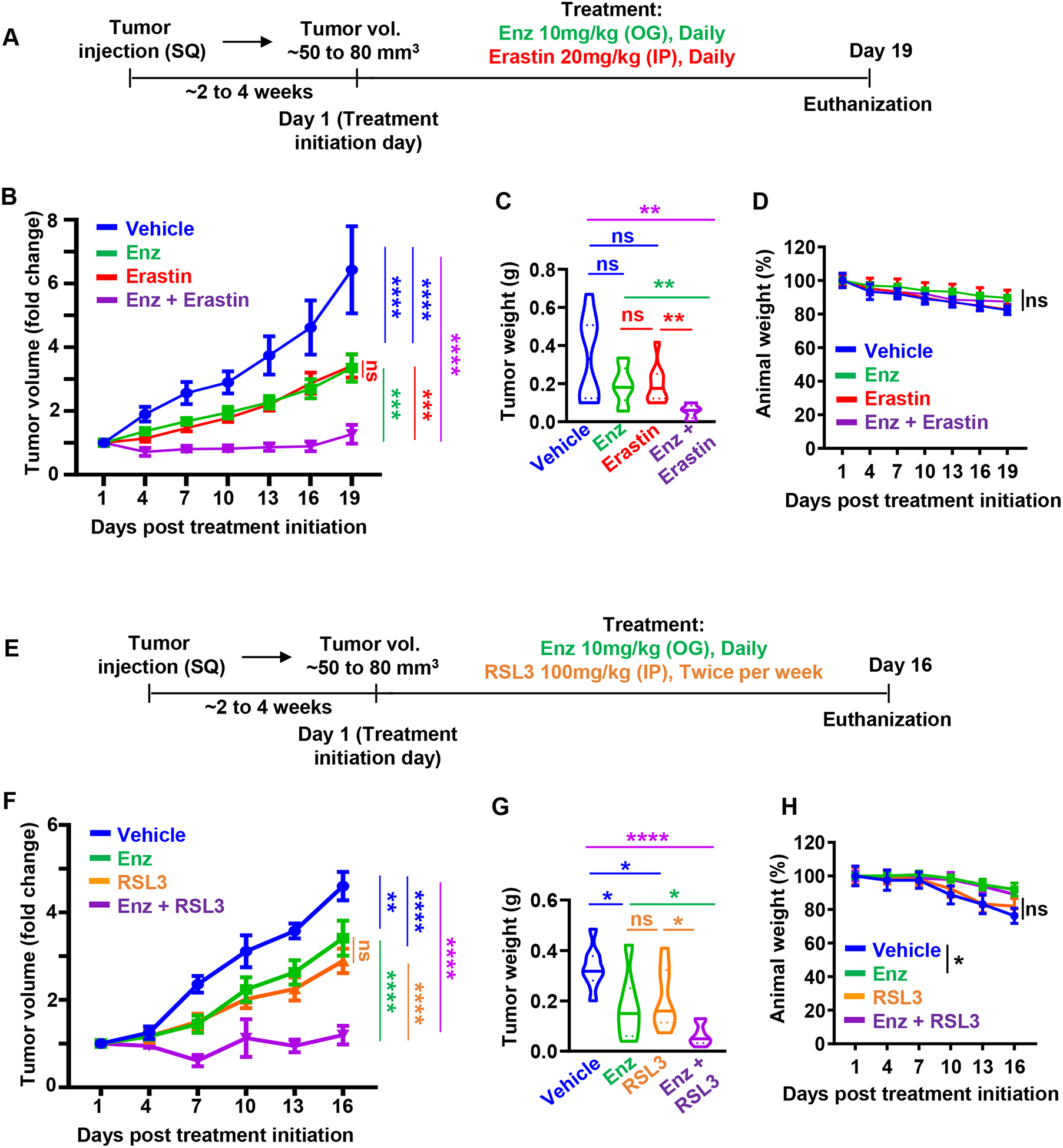
(A) Schematic diagram of prostate cancer xenograft models upon treatment with erastin (20 mg/kg, IP, daily) and enzalutamide (10 mg/kg, oral gavage, daily). (B) 5×105 C4-2 cells mixed with 80% Matrigel, were implanted bilaterally subcutaneously into the flanks of male NSG mice. Once the average of tumors volume reached to 50 to 80 mm3, mice were randomized into vehicle (n=6), erastin (n=6), enzalutamide (n=6), and erastin plus enzalutamide (n=6) treatment groups. Tumor volumes (1/2 (Length × Width × Height) were measured every third day and represented as fold change over tumor volume at Day 1. (C) Tumor weights (g) were measured after tissue resection at experimental end point. (D) Animal weights were measured every three days upon erastin and enzalutamide combination treatment and plotted. (E) Schematic diagram of prostate cancer xenograft models upon RSL3 (100 mg/kg, IP, twice per week) and enzalutamide (10 mg/kg, oral gavage, daily) treatments. (F) 5×105 C4-2 cells mixed with 80% matrigel, were implanted bilaterally subcutaneously into the flanks of NSG male mice. Mice with tumors with average volume of 50 to 80 mm3, were randomized into vehicle (n=7), RSL3 (n=5), enzalutamide (n=5), and RSL3 plus enzalutamide (n=5) treatment groups. Tumor volumes (1/2 × Length × Width × Height) were measured every 3 days and are shown as fold change when compared to Day 1. (G) Tumor weights (g) at experimental end point are shown. (H) Animal weights were measured every 3 days over the treatment course and plotted. Statistical analysis was performed with Student’s t-test (* P<0.05, **P<0.01, *** P<0.001, and **** P<0.0001) and error bars represent mean ± SEM.
Discussion
Our study provides the first demonstration of the therapeutic potential of erastin and RSL3 in across prostate cancer variants including adeno-CRPC, NEPC and DNPC in a pre-clinical setting. Our results warrant further evaluation of the efficacy of ferroptosis inducers in clinical settings for treatment of prostate cancer. In fact, multiple agents approved by the food and drug administration (FDA) for treatment of malignancies and other conditions that have been shown to induce ferroptosis. For instance, the multi-kinase inhibitor, sorafenib, used for treatment of hepatocellular carcinoma, renal cell carcinoma and refractory differentiated thyroid carcinoma, has been shown to induce ferroptosis (39, 53). Sulfasalazine, a compound approved by the FDA for the treatment of rheumatoid arthritis and ulcerative colitis has also been demonstrated to induce ferroptosis in cancer cells (54). Other FDA approved agents such as altretamine used for treatment of ovarian cancer inhibits GPX4 and induces ferroptosis in cancer cells (55). Testing these clinically used agents as CRPC therapies should be further tested in preclinical models and if effective, they could be rapidly translated into clinical trials.
We have also demonstrated that erastin and RSL3 are more effective in inhibiting tumor growth when combined with enzalutamide or abiraterone in a preclinical model of adeno-CRPC. The combination of erastin or RSL3 with enzalutamide or abiraterone inhibited cell growth and migration. Furthermore, combination of erastin or RSL3 with enzalutamide halted tumor growth when compared to erastin, RSL3 or enzalutamide alone in vivo. Therefore, agents effective at inducing ferroptosis alone should be tested in combination with second-generation anti-androgens in pre-clinical and clinical trials.
Enzalutamide acts, in part, through preventing the nuclear translocation of AR (56) and can inhibit stabilization of AR mediated by heat shock proteins (HSPs) (57). Interestingly, several studies have shown that HSPs might inhibit ferroptosis induction. For example, overexpression of HSPB1 inhibits ferroptosis induction by erastin (58). HSPA5 also negatively regulates ferroptosis induction in human pancreatic ductal adenocarcinoma (PDAC) cells, and increased expression of HSPA5 represses ferroptosis induction by inhibition of GPX4 protein degradation (59). Therefore, enzalutamide might synergize with erastin and RSL3 by decreasing the expression of HSPs, negative regulator of ferroptosis. Another plausible mechanism of the increased efficacy of the combination of FINs with anti-androgens is that they act through separate programmed cell death pathways, namely apoptosis ferroptosis. Indeed, enzalutamide has been shown to induce apoptosis via increased expression of BAX, and decreased Bcl-2 expression (57). Further studies need to be conducted to delineate the precise mechanisms underlying the efficacy of FINs and anti-androgens combination.
In summary, our study demonstrates that erastin and RSL3 decrease the viability, growth, migration, and invasion of multiple prostate cancer cells in vitro. Furthermore, erastin and RSL3 significantly delay tumor growth of adeno-CRPC, NEPC and DNPC xenografts in vivo with no measurable side effects. The combination of erastin or RSL3 with second-generation anti-androgens inhibited prostate cancer cell growth and migration in vitro and halted tumor growth of adeno-CRPC xenografts when compared to erastin, RSL3 or second-generation anti-androgens alone in vivo. Overall, our finding suggests that ferroptosis induction may represent a promising therapeutic strategy across prostate cancer variants either as a monotherapy or in combination with standard-of-care second generation anti-androgens used for treatment of CRPC.
Supplementary Material
Statement of significance.
Findings reveal that induction of ferroptosis is a new therapeutic strategy for advanced prostate cancer as a monotherapy and in combination with second-generation anti-androgens.
Acknowledgements
T.S. is supported by the Canary Foundation, the National Institutes of Health/National Cancer Institute (NCI) R03CA230819, R37CA240822, and R01CA244281. The establishment and characterization of the LuCaP PDX models have been supported by the PNW Prostate Cancer SPORE P50CA097186 and P01CA163227. J.D.B. is supported by NIH CA196387. A.G. is supported by is supported by the U.S. Army Medical Research Acquisition Activity, through the CDMRP Award No. W81XWH-19-1-0333. The work was also supported by NIH S10 OD023518-01A1 for the Celigo S Imaging Cytometer (200- BFFL-S). Opinions, interpretation, conclusions, and recommendations are those of the authors and not necessarily endorsed by the funding agencies.
Footnotes
Conflicts of interest:
The authors declare no competing interests.
References
- 1.Siegel RL, Miller KD, Jemal A, Cancer statistics, 2020. CA: A Cancer Journal for Clinicians 2020; 70: 7–30. [DOI] [PubMed] [Google Scholar]
- 2.Denis LJ, Griffiths K, Endocrine treatment in prostate cancer. Seminars in Surgical Oncology 2000; 18: 52–74. [DOI] [PubMed] [Google Scholar]
- 3.Sharifi N, Dahut WL, Steinberg SM, Figg WD, Tarassoff C, Arlen P et al. , A retrospective study of the time to clinical endpoints for advanced prostate cancer. BJU International 2005; 96: 985–989. [DOI] [PubMed] [Google Scholar]
- 4.Fizazi K, Shore N, Tammela TL, Ulys A, Vjaters E, Polyakov S et al. , Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. New England Journal of Medicine 2019; 380: 1235–1246. [DOI] [PubMed] [Google Scholar]
- 5.Scher HI, Fizazi K, Saad F, Taplin ME, Sternberg CN, Miller K et al. , Increased survival with enzalutamide in prostate cancer after chemotherapy. N Engl J Med 2012; 367: 1187–1197. [DOI] [PubMed] [Google Scholar]
- 6.Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS et al. , Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med 2014; 371: 424–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L et al. , Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med 2011; 364: 1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rathkopf DE, Antonarakis ES, Shore ND, Tutrone RF, Alumkal JJ, Ryan CJ et al. , Safety and Antitumor Activity of Apalutamide (ARN-509) in Metastatic Castration-Resistant Prostate Cancer with and without Prior Abiraterone Acetate and Prednisone. Clin Cancer Res 2017; 23: 3544–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF, Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol 2008; 26: 242–245. [DOI] [PubMed] [Google Scholar]
- 10.de Bono JS, Oudard S, Ozguroglu M, Hansen S, Machiels JP, Kocak I et al. , Prednisone plus cabazitaxel or mitoxantrone for metastatic castration-resistant prostate cancer progressing after docetaxel treatment: a randomised open-label trial. Lancet 2010; 376: 1147–1154. [DOI] [PubMed] [Google Scholar]
- 11.Mateo J, Carreira S, Sandhu S, Miranda S, Mossop H, Perez-Lopez R et al. , DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N Engl J Med 2015; 373: 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF et al. , Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 2010; 363: 411–422. [DOI] [PubMed] [Google Scholar]
- 13.Antonarakis ES, Piulats JM, Gross-Goupil M, Goh J, Ojamaa K, Hoimes CJ et al. , Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J Clin Oncol 2020; 38: 395–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vlachostergios PJ, Puca L, Beltran H, Emerging Variants of Castration-Resistant Prostate Cancer. Curr Oncol Rep 2017; 19: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nadal R, Schweizer M, Kryvenko ON, Epstein JI, Eisenberger MA, Small cell carcinoma of the prostate. Nat Rev Urol 2014; 11: 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY et al. , Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov 2011; 1: 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV et al. , Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J Clin Oncol 2018; 36: 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bluemn EG, Coleman IM, Lucas JM, Coleman RT, Hernandez-Lopez S, Tharakan R et al. , Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer Cell 2017; 32: 474–489 e476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Labrecque MP, Coleman IM, Brown LG, True LD, Kollath L, Lakely B et al. , Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J Clin Invest 2019; 129: 4492–4505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ et al. , Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017; 171: 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao JY, Dixon SJ, Mechanisms of ferroptosis. Cell Mol Life Sci 2016; 73: 2195–2209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magtanong L, Ko PJ, Dixon SJ, Emerging roles for lipids in non-apoptotic cell death. Cell Death Differ 2016; 23: 1099–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixon SJ, Stockwell BR, The role of iron and reactive oxygen species in cell death. Nat Chem Biol 2014; 10: 9–17. [DOI] [PubMed] [Google Scholar]
- 24.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE et al. , Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012; 149: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR, Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 2016; 113: E4966–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS et al. , Regulation of ferroptotic cancer cell death by GPX4. Cell 2014; 156: 317–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seibt TM, Proneth B, Conrad M, Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med 2019; 133: 144–152. [DOI] [PubMed] [Google Scholar]
- 28.Conrad M, Sato H, The oxidative stress-inducible cystine/glutamate antiporter, system × (c) (−) : cystine supplier and beyond. Amino Acids 2012; 42: 231–246. [DOI] [PubMed] [Google Scholar]
- 29.Feng H, Stockwell BR, Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol 2018; 16: e2006203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chi KN, Bjartell A, Dearnaley D, Saad F, Schroder FH, Sternberg C et al. , Castration-resistant prostate cancer: from new pathophysiology to new treatment targets. Eur Urol 2009; 56: 594–605. [DOI] [PubMed] [Google Scholar]
- 31.Bebber CM, Muller F, Prieto Clemente L, Weber J, von Karstedt S, Ferroptosis in Cancer Cell Biology. Cancers (Basel) 2020; 12: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu T, Ding W, Ji X, Ao X, Liu Y, Yu W et al. , Molecular mechanisms of ferroptosis and its role in cancer therapy. J Cell Mol Med 2019; 23: 4900–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A et al. , Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017; 551: 247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B et al. , Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 2017; 547: 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dolma S, Lessnick SL, Hahn WC, Stockwell BR, Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003; 3: 285–296. [DOI] [PubMed] [Google Scholar]
- 36.Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, MacPherson L et al. , Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 2012; 22: 1822–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang WS, Stockwell BR, Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008; 15: 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ et al. , RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 2007; 447: 864–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M et al. , Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014; 3: e02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roh JL, Kim EH, Jang H, Shin D, Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 2017; 11: 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C et al. , Ferroptosis: A Novel Anti-tumor Action for Cisplatin. Cancer Res Treat 2018; 50: 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nassar ZD, Mah CY, Dehairs J, Burvenich IJ, Irani S, Centenera MM et al. , Human DECR1 is an androgen-repressed survival factor that regulates PUFA oxidation to protect prostate tumor cells from ferroptosis. Elife 2020; 9: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tousignant KD, Rockstroh A, Poad BLJ, Talebi A, Young RSE, Taherian Fard A et al. , Therapy-induced lipid uptake and remodeling underpin ferroptosis hypersensitivity in prostate cancer. Cancer Metab 2020; 8: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao J, Ning S, Lou W, Yang JC, Armstrong CM, Lombard AP et al. , Cross-Resistance Among Next-Generation Antiandrogen Drugs Through the AKR1C3/AR-V7 Axis in Advanced Prostate Cancer. Mol Cancer Ther 2020; 19: 1708–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai S, Cao S, Jin L, Kobelski M, Schouest B, Wang X et al. , A positive role of c-Myc in regulating androgen receptor and its splice variants in prostate cancer. Oncogene 2019; 38: 4977–4989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu C, Yang JC, Armstrong CM, Lou W, Liu L, Qiu X et al. , AKR1C3 Promotes AR-V7 Protein Stabilization and Confers Resistance to AR-Targeted Therapies in Advanced Prostate Cancer. Mol Cancer Ther 2019; 18: 1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu C, Lou W, Yang JC, Liu L, Armstrong CM, Lombard AP et al. , Proteostasis by STUB1/HSP70 complex controls sensitivity to androgen receptor targeted therapy in advanced prostate cancer. Nat Commun 2018; 9: 4700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu C, Lou W, Zhu Y, Nadiminty N, Schwartz CT, Evans CP et al. , Niclosamide inhibits androgen receptor variants expression and overcomes enzalutamide resistance in castration-resistant prostate cancer. Clin Cancer Res 2014; 20: 3198–3210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hsu EC, Rice MA, Bermudez A, Marques FJG, Aslan M, Liu S et al. , Trop2 is a driver of metastatic prostate cancer with neuroendocrine phenotype via PARP1. Proc Natl Acad Sci U S A 2020; 117: 2032–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rice MA, Hsu EC, Aslan M, Ghoochani A, Su A, Stoyanova T, Loss of Notch1 Activity Inhibits Prostate Cancer Growth and Metastasis and Sensitizes Prostate Cancer Cells to Antiandrogen Therapies. Mol Cancer Ther 2019; 18: 1230–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oh BM, Lee SJ, Park GL, Hwang YS, Lim J, Park ES et al. , Erastin Inhibits Septic Shock and Inflammatory Gene Expression via Suppression of the NF-kappaB Pathway. J Clin Med 2019; 8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nguyen HM, Vessella RL, Morrissey C, Brown LG, Coleman IM, Higano CS et al. , LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease an--d Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017; 77: 654–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, Chauffert B et al. , Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer 2013; 133: 1732–1742. [DOI] [PubMed] [Google Scholar]
- 54.Gout PW, Buckley AR, Simms CR, Bruchovsky N, Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia 2001; 15: 1633–1640. [DOI] [PubMed] [Google Scholar]
- 55.Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, Nicoletti P et al. , Elucidating Compound Mechanism of Action by Network Perturbation Analysis. Cell 2015; 162: 441–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V et al. , Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 2009; 324: 787–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Abazid A, Martin B, Choinowski A, McNeill RV, Brandenburg LO, Ziegler P et al. , The androgen receptor antagonist enzalutamide induces apoptosis, dysregulates the heat shock protein system, and diminishes the androgen receptor and estrogen receptor β1 expression in prostate cancer cells. J Cell Biochem 2019; 120: 16711–16722. [DOI] [PubMed] [Google Scholar]
- 58.Sun X, Ou Z, Xie M, Kang R, Fan Y, Niu X et al. , HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015; 34: 5617–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu S, Zhang Q, Sun X, Zeh HJ 3rd, Lotze MT, Kang R et al. , HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res 2017; 77: 2064–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.