

CONSPECTUS
It has been over half a century since the last class of antibiotics active against the most problematic Gram-negative bacteria was approved by the Food and Drug Administration (FDA). The major challenge with developing antibiotics to treat these infections is not drug—target engagement, but rather the inability of most small molecules to traverse the Gram-negative membranes, be retained, and accumulate within the cell. Despite an abundance of lead compounds, limited understanding of the physicochemical properties needed for compound accumulation (or avoidance of efflux) in Gram-negative bacteria has precluded a generalizable approach for developing Gram-negative antibiotics. Indeed, in many instances, despite years of intensive derivatization efforts and the synthesis of hundreds of compounds aimed at building-in Gram-negative activity, little or no progress has been made in expanding the spectrum of activity for many Gram-positive-only antibiotics. In this Account, we describe the discovery and successful applications of a promising strategy for enhancing accumulation of Gram-positive-only antibiotics as a means of imbuing compounds with broad-spectrum activity.
Utilizing a prospective approach examining accumulation in Escherichia coli for over 180 diverse compounds, we found that small molecules have an increased likelihood to accumulate in E. coli when they contain an ionizable Nitrogen, have low Three-dimensionality, and are Rigid. Implementing these guidelines, codified as the “eNTRy rules” and assisted by the web application www.entry-way.org, we have facilitated compound entry and systematically built Gram-negative activity into Gram-positive-only antibiotics. Though each antibiotic will have case-specific considerations, we describe a set of important criteria to consider when selecting candidate Gram-positive-only antibiotics for conversion to Gram-negative active versions via the eNTRy rules. As detailed herein, using this blueprint the spectrum of activity was expanded for three antibiotic classes that engage three different biological targets: DNA gyrase inhibitor 6DNM, FabI inhibitor Debio-1452, and FMN riboswitch inhibitor Ribocil C. In each scenario, the eNTRy rules guided the synthesis of key analogues predisposed to accumulate in Gram-negative bacteria leading to compounds that display antibiotic activity (minimum inhibitory concentrations ≤ 8 μg mL−1) against E. coli and other Gram-negative ESKAPE pathogens. While the eNTRy rules will continue to be refined and enhanced as more accumulation data is gathered, based on these collective results and on other examples not covered herein, it is clear that the eNTRy rules are actionable for the development of novel broad-spectrum antibiotics from Gram-positive-only compounds. By enabling prediction of compound accumulation, the eNTRy rules should facilitate the process of discovering and developing novel antibiotics active against Gram-negative bacteria.
Graphical Abstract
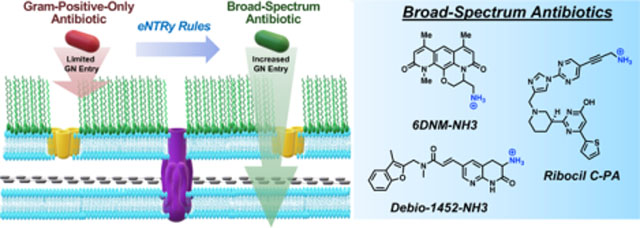
INTRODUCTION
The rising threat of multidrug-resistant Gram-negative bacterial infections is a major public health concern.4–7 In 2017 the World Health Organization (WHO) introduced a list of priority pathogens for which novel antimicrobial therapies are urgently needed and all those considered “critical priority” are Gram-negative bacteria, namely drug-resistant Enterobacteriaceae, Pseudomonas aeruginosa, and Acinetobacter baumannii.8, 9 Especially troubling is the fact that no new classes of antibiotics active against Gram-negative ESKAPE pathogens10, 11 have been approved by the Food and Drug Administration (FDA) in over 50 years;12 while new drugs from already approved classes are welcome, such compounds are often susceptible to pre-exisiting resistance mechanisms. In the absence of novel antibiotics and regulatory approvals, treating these problematic pathogens is becoming more challenging; for example the antibiotic colistin, known to cause nephrotoxicity in 60% of patients,13 has been recently reintroduced to the clinic for the treatment of multidrug resistant infections after years of not being needed.14
While retrospective studies focusing on antibacterial activity have been used in an attempt to define structural properties favorable for Gram-negative activity,15, 16 they have not led to generalizable approaches for producing new Gram-negative-active antibiotics, as has been discussed.17 The major challenge in discovering small molecules to develop into new drugs for Gram-negative pathogens has been traced to the limited understanding of compound accumulation in these bacteria.16 Despite the discovery of lead compounds with either outstanding on-target activity in biochemical assays or outstanding whole-cell activity against Gram-positive pathogens, the inability to build whole-cell Gram-negative activity into these leads has been noted time and again.4, 18 In a similar vein, even massive screens of millions of compounds, followed-up by detailed structure-activity relationship (SAR) campaigns, typically fail to provide compounds with whole-cell Gram-negative activity.4, 12, 16, 18–22 Detailed herein is the description of an approach whereby data from a prospective study on compound accumulation in Escherichia coli has been used to develop a generalized blueprint enabling interested parties to systematically integrate Gram-negative activity into antibiotics that are only active against Gram-positive bacteria.
PROBLEMATIC PERMEATION BARRIER IN GRAM-NEGATIVE BACTERIA
A major difficulty with developing antibiotics for Gram-negative pathogens is the complex cell membrane structure and embedded efflux pumps of these organisms.23–25 Unlike Gram-positive bacteria, Gram-negative bacteria have two cellular membranes, with the outermost membrane containing densely-packed lipopolysaccharides that prevents passive diffusion of most small molecules. Compounds able to cross through the outer membrane usually do so through porins,26 which are water-filled ß-barrel proteins lined with charged amino acids, or through the self-promoted uptake pathway (typically polycationic molecules).27 However once inside the cell, compounds are subject to ejection by highly promiscuous efflux pumps.18, 28 Therefore, in order to accumulate in Gram-negative bacteria and exert the desired antibacterial activity, a small molecule must cross the membranes and engage its biological target faster than it is pumped out. Thus far, the requirement for this rare combination of characteristics has prevented the accumulation of many antimicrobials in Gram-negative pathogens, designating them as Gram-positive-only antibiotics.
CONVERSION STRATEGIES IN ANTIMICROBIALS
One potential method for developing Gram-negative active compounds would be to extend the spectrum of activity of existing Gram-positive-only antimicrobials. This strategy is attractive as scores of such compounds have been identified through screening against Gram-positive bacteria (such as Staphylococcus aureus) and most antibacterial targets are highly conserved across both Gram-positive and Gram-negative pathogens.11 Indeed, it is common for antibiotics to be active against Gram-negative bacteria with a permeabilized outer membrane or that are efflux pump deficient, but not against the corresponding wild-type strains.29–31 We have compiled a list of ~70 such Gram-positive-only antibiotics and have detailed their physicochemical features,2 but there are numerous other potential conversion targets. By enhancing compound entry and accumulation while maintaining target engagement, Gram-positive-only antibiotics can be converted into broad-spectrum agents. There are only scattered examples in the literature of antibiotics having their spectrum of activity expanded through derivative synthesis;32–35 more commonly, the enormous undertaking of synthesizing hundreds of derivatives of important antibiotics without any notable success in incorporating Gram-negative activity proves frustrating.36–43
DISCOVERY OF THE eNTRy RULES
We performed a systematic analysis to gain insight into small-molecule accumulation in E. coli with the aim of constructing a knowledge base that could enable the discovery of physicochemical features that facilitate compound accumulation in Gram-negative bacteria.1 An assay utilizing liquid chromatography tandem mass-spectrometry (LC-MS/MS) was developed from progenitor protocols,44–46 validated using antibiotic controls, and used to quantify the accumulation of over 180 structurally diverse non-antibiotic compounds in the wild-type strain E. coli MG1655.1 Critical for success here was the use of compound accumulation (and not antibacterial activity) as the key metric, which allowed our analysis to extend beyond antibiotics. Equally important was the use of a structurally diverse collection of compounds, both drug-like compounds, and natural product-like compounds generated through the complexity-to-diversity (CtD) paradigm47–50 to enable facile SAR analysis.
As there are multiple factors that can influence small-molecule accumulation, such as the presence of porins and efflux pumps,26, 28 the accumulation studies were performed in whole cells. To begin the analysis, a diverse set of 100 compounds, including positively charged, negatively charged, and neutral compounds, were selected for evaluation. Since most antibacterial drugs are natural products or derivatives thereof, it was crucial that many members of this compound set possessed natural product-like properties yet were synthetically accessible to allow for systematic modification to probe the importance of individual structural features. Accumulation results suggested that charge is a primary factor that contributes to accumulation in E. coli, with positively charged compounds being the most likely to accumulate.1 Of these accumulating compounds, a majority contained primary amines. To further evaluate the role of the primary amine, SAR studies were performed on multiple classes of accumulating compounds. Replacing the amine with other functional groups such as an amide, ester, carboxylic acid, nitrile, azide, or alcohol resulted in dramatically reduced accumulation, demonstrating the positive influence of the amine on accumulation.1 However, from the total set of 68 primary amines tested, 32 primary amines did not accumulate, suggesting that while the presence of a primary amine was critical for accumulation in E. coli, it alone was not sufficient.
A chemoinformatic approach was taken to further investigate the factors contributing to amine accumulation. For the set of 68 primary amines, a total of 297 molecular descriptors were calculated for each compound. Using these descriptors, a random forest classification model was trained to predict amine accumulation. From this computational analysis arose (in addition to positive charge) two other major factors that influence accumulation – flexibility and globularity. A compound that is less flexible and more rigid (as measured by the number of rotatable bonds) and that has low three-dimensionality (as measured by a globularity score) is more likely to accumulate in E. coli; key comparisons of structurally-related compounds are shown in Figure 1A. Combining these findings, guidelines for compound accumulation in E. coli were developed (Figure 1B), and the first Gram-positive-only compound they were applied to was the natural product deoxynybomycin (DNM, Figure 1C). These guiding principles, – later codified as the “eNTRy rules”35 – stipulate that small molecules are likely to accumulate in E. coli if they possess an ionizable Nitrogen, low Three-dimensionality (as measured by globularity), and are Rigid (low number of rotatable bonds).1 These three parameters have the advantage of being chemical traits easily recognized and understood by medicinal chemists/microbiologists/chemical biologists and as such can be readily integrated into new compounds, especially with the assistance of a free web tool described further below. In addition to these major traits, as shown in Figure 1A amphiphilic moment (vsurf_A) and amine steric hindrance also appear to influence accumulation.1 While primary amines are able to facilitate accumulation in a manner superior to secondary, tertiary, or quaternary amines (Figure 1A),1 recent unbiased accumulation studies show that guanidiniums, and in some cases N-alkylpyridiniums, are also able to facilitate compound accumulation in E. coli.51
Figure 1.
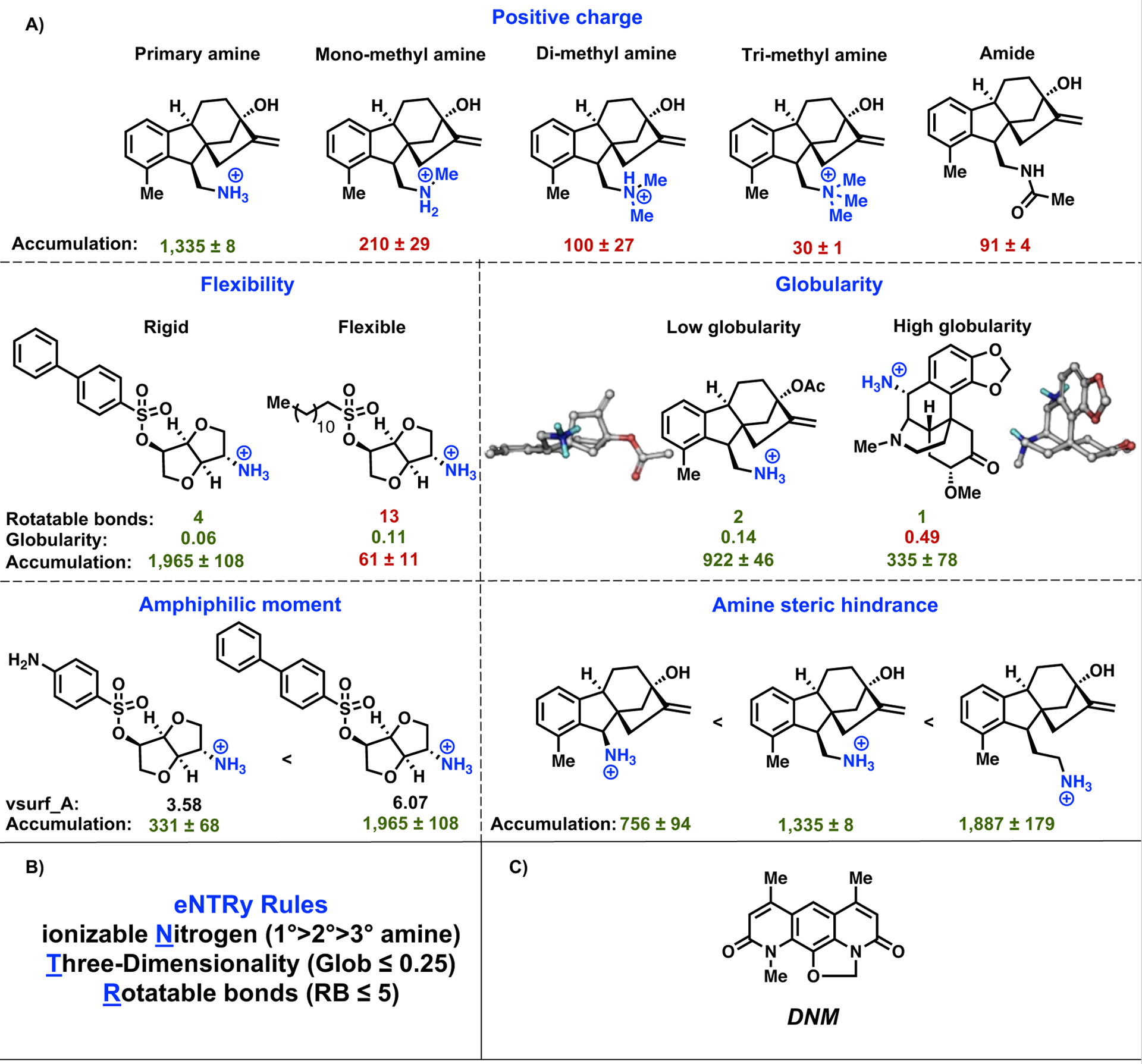
(A) Effect of positive charge, flexibility, globularity, amphiphilic moment, and amine steric hindrance on compound accumulation in E. coli MG1655. Accumulation is reported in nmol per 1012 colony-forming units (CFUs). The s.e.m. is reported for accumulation values, experiments were performed in biological triplicate. (B) Predictive guidelines for compound accumulation in E. coli, specified as the eNTRy rules. Molecules are more likely to accumulate in E. coli if they possess an ionizable nitrogen, have low globularity, and are relatively rigid. (C) Structure of natural product deoxynybomycin (DNM). Reproduced with permission from ref.1. Copyright 2017 Nature.
BLUEPRINT FOR BUILDING BROAD-SPECTRUM ACTIVITY
The true test for any predictive guidelines is their actionability, that is, their ability to be used to rapidly design and generate novel Gram-negative-active antibiotics. While each situation is necessarily case-specific, some generalizable aspects of our approach have been codified, with our blueprint for building Gram-negative antibiotic activity into Gram-positive-only compounds detailed herein. Of critical importance is selecting an appropriate starting point, and for this the following criteria should be considered:
Candidate antibiotics should have outstanding activity (minimum inhibitory concentration (MIC) ≤ 1 μg mL−1) in Gram-negative strains where permeability or efflux have been compromised (such as E. coli ΔrfaC or ΔtolC), indicating that the compound would kill wild-type strains of these bacteria if it could accumulate. There are scores of such compounds; a listing of ~70 is given in a recent publication,2 but there are many others as well.
Ideally, candidate antibiotics should have rotatable bonds and globularity that fit the eNTRy rules, and these parameters can be calculated using the free web app www.entry-way.org. Alternatively, rotatable bonds and/or globularity can be reduced through derivative synthesis.
If there is an X-ray crystal structure of the candidate antibiotic in complex with its biological target, this will be very useful in determining where to make modifications (for example, placement of a positive charge), and compound design can also be facilitated through computational docking studies.
In addition to the X-ray structure, the presence of significant SAR data can greatly facilitate the design of new compounds where accumulation is optimized (via the eNTRy rules) while maintaining target engagement.
Below is summarized work on three structurally different antibiotic classes, compounds that engage with three different biological targets, for which we have produced Gram-negative-active (MICs ≤ 8 μg mL−1) derivatives guided by the eNTRy rules. In each case, the progenitor compound is analyzed through the four points above. Not covered below are important applications of the eNTRy rules by others to engineer Gram-negative activity into inhibitors of DNA gyrase,52 microbial ribosomes,53 LpxC,54 topoisomerase IV,55 and biotin carboxylase,17 and to create a high accumulating version of a CpxA phosphatase inhibitor.56 While there is no doubt that the guidelines for compound accumulation in Gram-negative bacteria will continue to be refined, based on these collective results it is clear that in many cases the eNTRy rules, as they currently exist, provide an excellent path forward for imbuing antibiotics with activity against Gram-negative pathogens.
DNA GYRASE INHIBITOR 6DNM INTO 6DNM-NH3
As detailed above, the ideal candidate would already meet the flexibility and shape parameters necessary to accumulate in Gram-negative bacteria and would only require the installation of a primary amine on a position that does not disrupt cellular target engagement. As such, the natural product deoxynybomycin (DNM, Figure 1C) is an outstanding conversion target. DNM shows antibacterial activity by inhibiting wild-type and mutant DNA gyrase,57, 58 with derivatives displaying activity only against Gram-positive organisms.58 DNM has no rotatable bonds and a low globularity score of 0.04, suggesting that the addition of a primary amine could yield a derivative that accumulates and is active against Gram-negative bacteria.
While there is no X-ray structure of DNM with DNA gyrase, a reasonably extensive SAR does exist,58 meaning that DNM meets most of the Blueprint criteria, as outlined in Figure 2. The SAR data suggested that simple alkyl extensions off each of the three methyl groups were tolerated, as was an appendage of a methyl group off the methylene bridge in the 5-membered ring.58 Guided by this SAR, an analogue of DNM was first synthesized by expanding the five-membered ring in DNM to a six-membered ring, creating the compound 6DNM (Figure 2).1 6DNM shows antimicrobial activity against S. aureus strains (MIC = 0.06–1 μg mL−1), but has no activity against wild-type E. coli MG1655 (MIC > 32 μg mL−1) (Figure 2). Additionally, 6DNM does not accumulate in E. coli MG1655, and importantly, 6DNM shows good antibacterial activity against an E. coli strain with compromised efflux (MIC = 0.125 μg mL−1). Inspired by this observation and guided by the eNTRy rules, a derivative of 6DNM that contains a primary amine while also maintaining low rotatable bonds and globularity was synthesized, the racemic compound 6DNM-NH3 (Figure 2). Not only does 6DNM-NH3 retain activity against S. aureus, but it also accumulates in E. coli at high levels. Consistent with this enhanced accumulation, 6DNM-NH3 shows considerable activity against E. coli MG1655 (MIC = 0.5 μg mL−1). 6DNM and 6DNM-NH3 were further evaluated against an expanded panel of Gram-negative bacteria, laboratory strains, and clinical isolates of ESKAPE pathogens (E. coli, K. pneumoniae, A. baumannii, Enterobacter cloacae, and P. aeruginosa). Unlike 6DNM, 6DNM-NH3 shows notable activity against all Gram-negative bacteria, except against P. aeruginosa (Figure 2).
Figure 2.
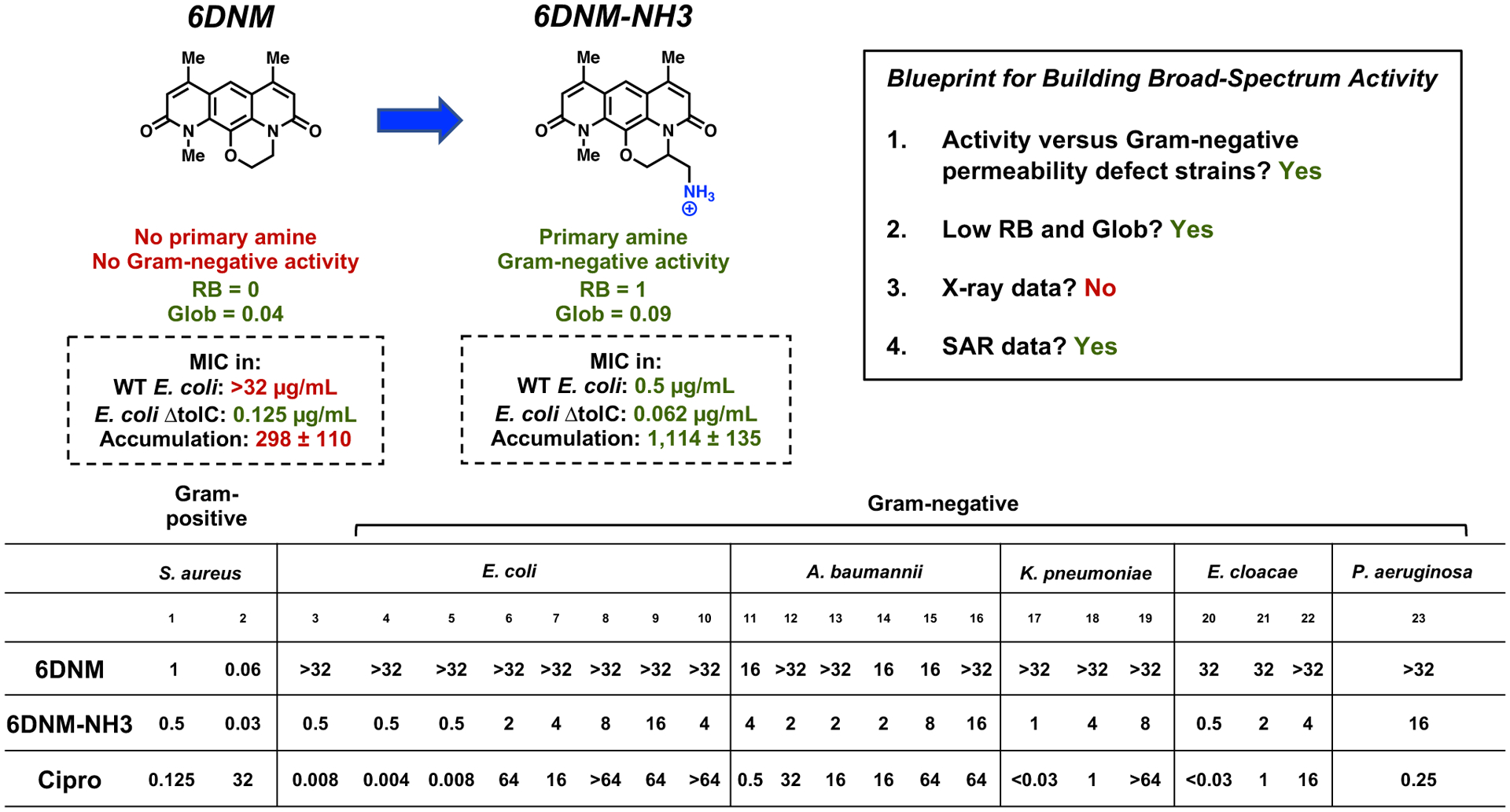
Conversion of 6DNM into 6DNM-NH3. WT E. coli is MG1655. Accumulation is reported in nmol per 1012 colony-forming units (CFUs). The s.e.m. is reported for accumulation values. Evaluation of 6DNM, 6DNM-NH3, and ciprofloxacin against a panel of Gram-positive and Gram-negative pathogens. MIC values were determined using the micro-dilution broth method as outlined by the Clinical and Laboratory Standards Institute (http://clsi.org) and are listed in μg mL−1. All experiments were performed in biological triplicate. Reproduced with permission from ref. 1. Copyright 2017 Nature.
FABI INHIBITOR DEBIO-1452 INTO DEBIO-1452-NH3
To assist in the implementation of the eNTRy rules a web application, www.entry-way.org, was developed and utilized to evaluate the conversion potential of almost 70 Gram-positive-only antibiotics;2 these are FDA-approved antibiotics, drugs in clinical development, and lead compounds in pre-clinical stages of development. Through this analysis, Debio-1452 emerged as an attractive conversion target. Debio-1452 has a low globularity (0.093) and four rotatable bonds, meeting two of the three eNTRy rules and missing only a primary amine (Figure 3A).
Figure 3.
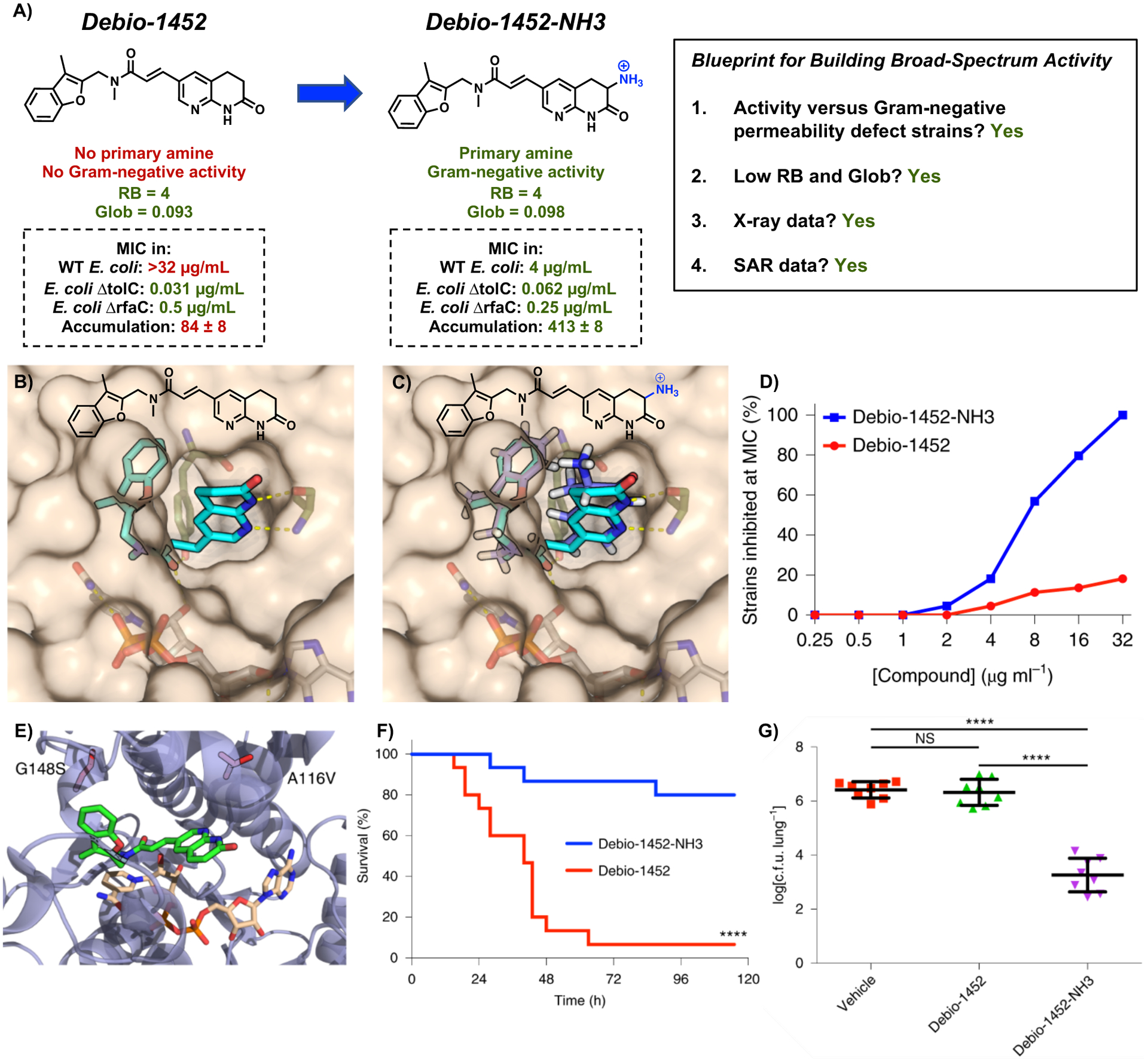
(A) Conversion of Debio-1452 into Debio-1452-NH3. WT E. coli is strain MG1655. Accumulation is reported in nmol per 1012 colony-forming units (CFUs). The s.e.m. is reported for accumulation values. (B) Solvent exposure of the naphthyridinone ring of Debio-1452 when bound to S. aureus FabI suggests sites for modification. (C) Computational docking of Debio-1452-NH3 overlaid with the crystal structure of Debio-1452. (D) Evaluation of Debio-1452 and Debio-1452-NH3 against a panel of 44 Enterobacteriaceae clinical isolates. All experiments were performed in biological triplicate. (E) Locations of E. coli FabI amino acid substitutions conferring resistance to Debio-1452-NH3. (F) Kaplan-Meier survival curve of mouse efficacy model of sepsis induced with A. baumannii W41979. Statistical significance was determined by two-tailed log-rank (Mantel-Cox) test. (G) Bacterial burden model of acute pneumonia in mice infected with K. pneumoniae BAA-1705. Data are shown as means ± s.d. and statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons. NS, not significant (P > 0.05); ****P < 0.0001. Reproduced with permission from ref. 2. Copyright 2020 Nature Microbiology.
Originating from a high-throughput biochemical screen at GlaxoSmithKline,59, 60 Debio-1452 inhibits the enoyl-acyl carrier protein reductase enzyme FabI, critical to the elongation cycle of the bacterial fatty acid biosynthesis pathway (FAS-II). This bacterial FAS-II pathway is not only essential for Gram-positive and Gram-negative bacteria, but its components are distinct from those found in the analogous mammalian FAS-I pathway. Debio-1452 displays remarkable antibiotic activity against S. aureus (MIC = 0.008 μg mL−1) but does not show any activity against Gram-negative ESKAPE pathogens up to its solubility limit (MIC > 32 μg mL−1). However, when tested against E. coli with compromised efflux or permeability defects (ΔtolC or ΔrfaC), Debio-1452 is very potent (MIC = 0.031 μg mL−1 and MIC = 0.5 μg mL−1, respectively)2, 61 suggesting that the lack of whole-cell activity results from low cellular accumulation and not from lack of target engagement. Thus, possessing favorable globularity and flexibility parameters, demonstrating potent activity in permeability-defect strains of E. coli, having an X-ray structure bound to its target, and an extensive SAR,36–38 Debio-1452 met all the Blueprint criteria in Figure 3A and thus appeared as an outstanding conversion target. The strategic addition of a primary amine was sought to convert Debio-1452 into an analogue with increased E. coli accumulation and Gram-negative antibiotic activity.
In examining the co-crystal structure of Debio-1452 bound to S. aureus FabI (which has sequence similarity with FabI of Gram-negative bacteria), the 3-position of the naphthyridinone ring (adjacent to the lactam carbonyl) was revealed to be the most solvent-exposed region of the molecule (Figure 3B),62 and as such, this position was identified as a promising point for amine introduction (Figure 3C). Amine-containing compounds were synthesized and evaluated for their whole-cell accumulation in E. coli MG1655 using the standard LC-MS/MS assay.2 While Debio-1452 did not accumulate when compared to negative controls, derivatives containing ionizable nitrogens accumulated.2 Debio-1452-NH3 (Figure 3A, as a racemic mixture) emerged as the most active of the modified compounds synthesized. Not only does Debio-1452-NH3 retain potent antibacterial activity in S. aureus similar to that of parent Debio-1452, but it also shows antibacterial activity in E. coli strains with intact outer membranes (MIC = 4 μg mL−1), which was not seen with Debio-1452 (MIC > 32 μg mL−1). Debio-1452-NH3 is also active against strains of the Gram-negative bacteria E. cloacae, K. pneumoniae, and A. baumannii, with MIC values from 4–8 μg mL−1. Evaluation against a panel of 61 multidrug-resistant clinical isolates revealed that Debio-1452-NH3 displays markedly more potent antimicrobial activity than Debio-1452 (data for 44 Enterobacteriaceae clinical isolates shown in Figure 3D). Despite its ability to accumulate in P. aeruginosa,2 Debio-1452-NH3 was not active against P. aeruginosa as these bacteria possess an additional FabI isoform, FabV, which enables rescue of the FAS-II pathway from inhibition of FabI.63
To assess the antibacterial mode of action of Debio-1452-NH3, resistant mutants were generated in E. coli MG1655, and sequencing of these mutants revealed changes in fabI leading to amino acid substitutions near the active site of FabI, at positions A116 and G148 (Figure 3E). As Debio-1452-NH3 showed little toxicity to mammalian cells at 72 h and minimal activity shift in the presence of human serum, its efficacy in mouse infection models was investigated. Debio-1452 and Debio-1452-NH3 were evaluated head-to-head in 10 mouse models of infection with A. baumannii, K. pneumoniae, and E. coli with overall survival and bacterial burden for each type of infection determined. Debio-1452-NH3-treated mice showed significant improved survival and reduced bacterial burden, rescuing mice from lethal infections with clinical isolates of these Gram-negative bacteria (Figure 3F, G). While Debio-1452 originated in the early 2000s and over 100 derivatives have been synthesized,36–38 none displayed robust activity against Gram-negative ESKAPE pathogens until the development of Debio-1452-NH3.
FMN RIBOSWITCH INHIBITOR RIBOCIL C INTO RIBOCIL C-PA
The final example covered herein is the conversion of Ribocil C into a Gram-negative active derivative.3 Discovered through a cell-based screen conducted at Merck & Co, Ribocil C was identified as a compound with potent activity against S. aureus (MIC = 0.5 μg mL−1) and permeabilized E. coli (MIC = 0.25 – 1 μg mL−1) (Figure 4A).64 Ribocil C targets the flavin mononucleotide (FMN) riboswitch, inducing bacterial death through riboflavin starvation. There are no human homologues of the FMN riboswitch or known riboswitches in mammalian systems, making the FMN riboswitch a compelling target for antibiotics.65 While Gram-positive organisms can scavenge riboflavin from their environment, this does not appear to be the case in E. coli.64, 66 This result suggests that the FMN riboswitch may be an even better antibiotic target in Gram-negative bacteria relative to Gram-positives; however, this possibility had not been explored as Ribocil C and its derivatives do not possess activity against wild-type Gram-negative bacteria.64, 66
Figure 4.

(A) Conversion of Ribocil C into Ribocil C-PA. WT E. coli is strain BW25113. Accumulation is reported in nmol per 1012 colony-forming units (CFUs). The s.e.m. is reported for accumulation values. (B) X-ray crystal structure of Ribocil B in complex with the FMN riboswitch aptamer of F. nucleatum suggests sites for amine installation. (C) Assessment of Ribocil C and Ribocil C-PA against a panel of multidrug-resistant clinical isolates of K. pneumoniae. MICs were performed in M9-MOPS media per CLSI guidelines. All experiments were performed in biological triplicate. (D) Sequencing of Ribocil C-PA resistant strains of E. coli reveals mutations map to the FMN riboswitch aptamer or expression platform. (E) Kaplan-Meier survival curve of mouse efficacy model of E. coli AR0493 sepsis. Statistical significance was determined by two-tailed log-rank (Mantel-Cox) test. (F) Bacterial burden model of acute pneumonia in mice infected with E. coli AR0493. Data are shown as means ± s.d. and statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons. NS, not significant (P > 0.05); ***P < 0.001; ****P < 0.0001. Reproduced with permission from ref. 3. Copyright 2020 Journal of American Chemical Society and ref. 64. Copyright 2015 Nature.
With notable antimicrobial activity in permeability-defect strains of E. coli (MIC = 0.25 μg mL−1 in ΔtolC and MIC = 1 μg mL−1 in ΔrfaC), the lack of whole-cell Gram-negative activity for Ribocil C was attributed to low cellular accumulation. As such, optimization of Ribocil C using the eNTRy rules was performed with the goal of producing a variant that is active against Gram-negative bacteria. With Ribocil C already meeting two of the three proposed eNTRy rules – low three-dimensionality (globularity = 0.099) and five rotatable bonds – strategic installation of a primary amine was the key objective (Figure 4A). Examination of the X-ray crystal structure of a progenitor compound, Ribocil B, in a complex with the Fusobacterium nucleatum FMN riboswitch aptamer64 (which has sequence similarity with the Enterobacteriaceae version) revealed the northern pyrimidine ring to be the most solvent-exposed site, and thus this position was prioritized for introducing amines (Figure 4B). Although an extensive SAR did not exist for Ribocil C, the compound did meet the other Blueprint criteria in Figure 4A, establishing itself as a promising target for conversion to a Gram-negative-active version.
Key derivatives were synthesized with amines at this target position and the resulting compounds were evaluated for their ability to accumulate in E. coli and exert antimicrobial activity. From this evaluation, three derivatives emerged as next-generation leads, led by Ribocil C-PA (Figure 4A); these compounds have notably higher accumulation than Ribocil C in E. coli MG1655, and display antibacterial activity against membrane-compromised Gram-negative strains. In addition, these compounds display whole-cell activity in Enterobacteriaceae strains including E. coli, E. cloacae, and K. pneumoniae. Unsurprisingly, these compounds do not show any activity against A. baumannii or P. aeruginosa10 as there is low homology between the FMN riboswitch target of these pathogens when compared to that of Enterobacteriaceae. With the best balance of antibacterial activity and low toxicity to mammalian cells amongst the derivatives, Ribocil C-PA was prioritized and evaluated further. Ribocil C-PA was assessed against multidrug-resistant clinical isolates of E. coli (n=42) and K. pneumoniae (n=54) (Figure 4C), revealing that the clinical isolates were markedly more susceptible to Ribocil C-PA than Ribocil C.
Two separate experiments were conducted to investigate the antibacterial mode of action of Ribocil C-PA. The first examined E. coli production of flavins in the presence of Ribocil C-PA. An antibiotic targeting the FMN riboswitch would inhibit the downstream transcription and translation of key enzymes involved in riboflavin biosynthesis, resulting in the depletion of the FMN and flavin adenosine dinucleotide (FAD) intracellular flavin levels.64 An LC-MS/MS assay was used to monitor these flavin levels in E. coli treated with Ribocil C-PA, and these experiments showed a reduction in flavins, consistent with Ribocil C-PA engaging and inhibiting the FMN riboswitch. Secondly, spontaneous mutants resistant to Ribocil C-PA were generated in E. coli BW25113, K. pneumoniae ATCC 27736, and E. coli ΔtolC (JW5503). Sequencing of these strains revealed mutations that mapped back to the FMN riboswitch aptamer or expression platform (Figure 4D). Efficacy in mouse models of infection with E. coli was assessed, evaluating overall survival with septicemia and bacterial burden with an acute pneumonia infection (Figure 4E, F). Ribocil C-PA-treated mice showed significant increase in survival as 80% (12/15 mice) burdened by septic infection were rescued (Figure 4E). Ribocil C-PA-treated mice also showed a 2-log reduction in bacterial burden (Figure 4F). In contrast, Ribocil C-treated mice did not exhibit significant reductions in bacterial burden nor was Ribocil C able to rescue mice in the survival studies.
Ribocil C-PA is an interesting case where the new compound has six rotatable bonds, just outside the boundaries of the eNTRy rules, possibly due to it adopting a preferable conformation that reduces rotational freedom.3 The availability of Ribocil C-PA now allows a full exploration of the potential of targeting the FMN riboswitch as a means to kill Gram-negative pathogens. As this target does not have high conservation across Gram-negative bacteria, there is the possibility of developing additional compounds that might possess narrow-spectrum activity against specific Gram-negative pathogens.
CONCLUSIONS AND OUTLOOKS
The successful conversions of 6DNM, Debio-1452, and Ribocil C (along with other analogous conversions from other laboratories)17, 52–56 into Gram-negative-active compounds highlight the considerable promise of the eNTRy rules to engineer improved compound accumulation and Gram-negative antimicrobial activity into Gram-positive-only antibiotics. Moving forward, of the three novel Gram-negative active antibiotics discussed herein, Debio-1452-NH3 (or a follow-on derivative) has the highest probability for advancement given its potent antibacterial activity, low frequency of resistance, and the divergent nature of the FabI target relative to any human homologues. While the eNTRy rules were discovered from E. coli accumulation data, the antibacterial activity seen in other Gram-negative ESKAPE pathogens such as K. pneumoniae, E. cloacae, and A. baumannii suggests some overlap between the physicochemical properties needed for compound accumulation in these problematic bacteria that are also difficult to permeate. However, P. aeruginosa has a higher permeability barrier16 and a larger evolutionary divergence compared to the aforementioned Gram-negative species,67 suggesting that target conservation may become a more relevant issue for P. aeruginosa. While biochemical screens have been performed against P. aeruginosa targets, with fewer permeabilized P. aeruginosa strains (relative to E. coli) it can be challenging to validate these targets;68 availability of additional permeabilized P. aeruginosa strains will be helpful for the development of separate and/or more specific accumulation rules for P. aeruginosa.
While the ability to develop broad-spectrum agents from Gram-positive-only antibiotics is critical, also important is the discovery of narrow-spectrum antibiotics, which, if coupled with an appropriate diagnostic or other information about the precise bacteria causing an infection, could provide infection control with minimal disruption to the microbiome.69 One avenue for narrow-spectrum development are compounds that target components of the Gram-negative outer membrane or periplasm,70 as these structures are specific to Gram-negative bacteria. These types of cellular targets remain underexplored and underexploited,71–73 and (with the exception of colistin) have yet to be developed into clinically used antibiotics for Gram-negative pathogens.
In addition to eNTRy rule success stories, examples where applying the eNTRy rules has failed to produce Gram-negative active compounds can also be instructive. In our recently reported efforts with 6-azaindazole,51 we were able to produce high accumulating derivatives of the parent antibiotic, but these compounds lost antibacterial activity, including against permeability-defect strains, suggesting that target engagement was disrupted.51 Indeed, retaining target engagement for the “converted” (high-accumulating) compound is likely to be the greatest challenge in such work, and while options beyond primary amines open up more possibilities,51 some ligands likely fit so tightly in their target’s binding site that any modification will be deleterious to target engagement.
Understanding the relationship of compound structure with pharmacokinetic parameters and solubility has enabled medicinal chemists to routinely tune these features through derivative synthesis. As more nuance is added to the eNTRy rules – analysis of the exact nature of the positive charge, variants for different Gram-negative organisms, etc. – the number of success stories using medicinal chemistry to tailor compounds for whole-cell accumulation and activity against Gram-negative bacteria will continue to grow. Wide applicability of the blueprint described herein could have ramifications for screening and discovery of lead compounds. For example, biochemical (in vitro) screens as a means to discover lead compounds have been eschewed74 in the antibiotic arena due to the well-known difficulty of engineering whole-cell activity into leads;4, 16, 22, 75 however, the example of Debio-1452-NH3 (whose parent compound was discovered in a biochemical screen59, 60) shows that this approach can be fruitful. In addition, the futility of whole-cell screening of standard compound collections against Gram-negative bacteria has been recognized, given that experts in antibacterial drug discovery have screened >8 million compounds in this fashion with little success.4, 16, 18, 19, 22, 35 While screening for whole-cell activity against Gram-negative bacteria utilizing compound collections comprised of members that comply with the eNTRy rules can be envisioned and has been suggested,1 in practice the synthesis of large numbers of such compounds may be challenging; it might be much more feasible to screen compounds versus permeability-defect Gram-negative strains and then build-in activity against wild-type Gram-negative bacteria via the eNTRy rules. The demonstration that the eNTRy rules can be successful in multiple contexts opens up new possibilities and suggests re-thinking some standard practices and dogma in antibacterial drug discovery and development.
ACKNOWLEDGMENTS
We thank the University of Illinois and the NIH (AI136773) for funding this work. K.A.M. is a member of the NIH Chemistry-Biology Interface Training Grant (T32-GM136629).
BIOGRAPHIES
Paul J. Hergenrother was born in Akron, OH in 1972, received a B.S. in chemistry from the University of Notre Dame in 1994, and a Ph.D. in chemistry from the University of Texas, Austin in 1999. After a stint as an American Cancer Society postdoctoral fellow at Harvard University, he joined the faculty at the University of Illinois, Urbana-Champaign in 2001 where he is currently the Kenneth L. Rinehart Endowed Chair in Natural Products Chemistry. His laboratory seeks to use small molecules to identify novel biological targets for the treatment of cancer and drug-resistant bacteria.
Kristen A. Muñoz was born in West Covina, California in 1995, obtained a B.A. in chemistry and Spanish from Claremont McKenna College in 2017 and is currently in her fourth year of the Ph.D. program in chemistry at the University of Illinois at Urbana-Champaign in the laboratory of Prof. Paul J. Hergenrother. Her research interests include the development of novel antibiotics for the treatment of antibiotic-resistant Gram-negative bacteria.
Footnotes
The authors declare the following competing financial interest(s): The University of Illinois has filed patents on some of the compounds described in this manuscript.
REFERENCES
- 1.Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ, Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]; Whole-cell accumulation data was gathered for a structurally diverse compound set, enabling unbiased discernment of physiochemical features that facilitate compound accumulation in E. coli. These guidelines were applied to convert deoxynybomycin, a Gram-positive-only natural product, into an antibiotic with broad-spectrum activity.
- 2.Parker EN; Drown BS; Geddes EJ; Lee HY; Ismail N; Lau GW; Hergenrother PJ, Implementation of permeation rules leads to a FabI inhibitor with activity against Gram-negative pathogens. Nat. Microbiol 2020, 5, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]; A web application that predicts small molecule accumulation in E. coli – eNTRyway – was developed and utilized to re-design Debio-1452, a Gram-positive-only antibiotic, into Debio-1452-NH3, a variant that accumulates in E. coli and possesses antimicrobial activity against a panel of high-priority Gram-negative pathogens including activity in ten mouse infection models.
- 3.Motika SE; Ulrich RJ; Geddes EJ; Lee HY; Lau GW; Hergenrother PJ, Gram-Negative Antibiotic Active through Inhibition of an Essential Riboswitch. J. Am. Chem. Soc 2020, 142, 10856–10862. [DOI] [PMC free article] [PubMed] [Google Scholar]; The eNTRy rules were implemented to convert Ribocil C, a Gram-positive-only antibiotic, into Ribocil C-PA, which accumulates to high levels in E. coli. Ribocil C-PA is active against Gram-negative clinical isolates and displays efficacy in mouse models of Gram-negative infection.
- 4.Payne DJ; Gwynn MN; Holmes DJ; Pompliano DL, Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug. Discov 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- 5.Cassini A; Högberg LD; Plachouras D; Quattrocchi A; Hoxha A; Simonsen GS; Colomb-Cotinat M; Kretzschmar ME; Devleesschauwer B; Cecchini M; Ouakrim DA; Oliveira TC; Struelens MJ; Suetens C; Monnet DL, Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect. Dis 2019, 19, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Periasamy H; Gnanamani A, Polymyxins resistance among Gram-negative pathogens in India. Lancet Infect. Dis 2020, 20, 1362–1363. [DOI] [PubMed] [Google Scholar]
- 7.Huh K; Chung DR; Ha YE; Ko J-H; Kim S-H; Kim M-J; Huh HJ; Lee NY; Cho SY; Kang C-I; Peck KR; Song J-H, Impact of Difficult-to-Treat Resistance in Gram-negative Bacteremia on Mortality: Retrospective Analysis of Nationwide Surveillance Data. Clin. Infect. Dis 2020, 71, e487–e496. [DOI] [PubMed] [Google Scholar]
- 8.Willyard C, The drug-resistant bacteria that pose the greatest health threats. Nature 2017, 543, 15–15. [DOI] [PubMed] [Google Scholar]
- 9.CDC. Antibiotic Resistance Threats in the United States, 2019. https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed December 17, 2020).
- 10.Rice LB, Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis 2008, 197, 1079–1081. [DOI] [PubMed] [Google Scholar]
- 11.Lewis K, Platforms for antibiotic discovery. Nat. Rev. Drug. Discov 2013, 12, 371–387. [DOI] [PubMed] [Google Scholar]
- 12.Hoffman PS, Antibacterial discovery: 21st century challenges. Antibiotics 2020, 9, 213–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zavascki AP; Nation RL, Nephrotoxicity of polymyxins: Is there any difference between colistimethate and polymyxin B? Antimicrob. Agents Chemother 2017, 61, e02319–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Biswas S; Brunel JM; Dubus JC; Reynaud-Gaubert M; Rolain JM, Colistin: An update on the antibiotic of the 21st century. Expert Rev. Anti. Infect. Ther 2012, 10, 917–934. [DOI] [PubMed] [Google Scholar]
- 15.O’Shea R; Moser HE, Physicochemical properties of antibacterial compounds: Implications for drug discovery. J. Med. Chem 2008, 51, 2871–2878. [DOI] [PubMed] [Google Scholar]
- 16.Brown DG; May-Dracka TL; Gagnon MM; Tommasi R, Trends and exceptions of physical properties on antibacterial activity for gram-positive and gram-negative pathogens. J. Med. Chem 2014, 57, 10144–10161. [DOI] [PubMed] [Google Scholar]
- 17.Andrews LD; Kane TR; Dozzo P; Haglund CM; Hilderbrandt DJ; Linsell MS; Machajewski T; McEnroe G; Serio AW; Wlasichuk KB; Neau DB; Pakhomova S; Waldrop GL; Sharp M; Pogliano J; Cirz RT; Cohen F, Optimization and Mechanistic Characterization of Pyridopyrimidine Inhibitors of Bacterial Biotin Carboxylase. J. Med. Chem 2019, 62, 7489–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Silver LL, Challenges of antibacterial discovery. Clin. Microbiol. Rev 2011, 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan PF; Holmes DJ; Payne DJ, Finding the gems using genomic discovery: Antibacterial drug discovery strategies - The successes and the challenges. Drug Discov. Today Ther. Strateg 2004, 1, 519–527. [Google Scholar]
- 20.Fernandes P, Antibacterial discovery and development—the failure of success? Nat. Biotechnol 2006, 24, 1497–1503. [DOI] [PubMed] [Google Scholar]
- 21.Gwynn MN; Portnoy A; Rittenhouse SF; Payne DJ, Challenges of antibacterial discovery revisited. Ann. N. Y. Acad. Sci 2010, 1213, 5–19. [DOI] [PubMed] [Google Scholar]
- 22.Tommasi R; Brown DG; Walkup GK; Manchester JI; Miller AA, ESKAPEing the labyrinth of antibacterial discovery. Nat. Rev. Drug. Discov 2015, 14, 529–542. [DOI] [PubMed] [Google Scholar]
- 23.Nikaido H; Vaara M, Molecular basis of bacterial outer membrane permeability. Microbiol. Rev 1985, 49, 1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikaido H, Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev 2003, 67, 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao S; Adamiak JW; Bonifay V; Mehla J; Zgurskaya HI; Tan DS, Defining new chemical space for drug penetration into Gram-negative bacteria. Nat. Chem. Biol 2020, 16, 1293–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cowan SW; Schirmer T; Rummel G; Steiert M; Ghosh R; Pauptit RA; Jansonius JN; Rosenbusch JP, Crystal structures explain functional properties of two E. coli porins. Nature 1992, 358, 727–733. [DOI] [PubMed] [Google Scholar]
- 27.Hancock REW, Peptide antibiotics. Lancet 1997, 349, 418–422. [DOI] [PubMed] [Google Scholar]
- 28.Du D; Wang-Kan X; Neuberger A; van Veen HW; Pos KM; Piddock LJV; Luisi BF, Multidrug efflux pumps: structure, function and regulation. Nat. Rev. Microbiol 2018, 16, 523–539. [DOI] [PubMed] [Google Scholar]
- 29.Sulavik MC; Houseweart C; Cramer C; Jiwani N; Murgolo N; Greene J; Didomenico B; Shaw KJ; Miller GH; Hare R; Shimer G, Antibiotic susceptibility profiles of Escherichia coli strains lacking multidrug efflux pump genes. Antimicrob. Agents Chemother 2001, 45, 1126–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pérez A; Poza M; Fernández A; Del Carmen Fernández M; Mallo S; Merino M; Rumbo-Feal S; Cabral MP; Bou G, Involvement of the AcrAB-TolC efflux pump in the resistance, fitness, and virulence of Enterobacter cloacae. Antimicrob. Agents Chemother 2012, 56, 2084–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamae C; Liu A; Kim K; Sitz D; Hong J; Becket E; Bui A; Solaimani P; Tran KP; Yang H; Miller JH, Determination of antibiotic hypersensitivity among 4,000 single-gene-knockout mutants of Escherichia coli. J. Bacteriol 2008, 190, 5981–5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Acred P; Brown DM; Turner DH; Wilson MJ, PHARMACOLOGY AND CHEMOTHERAPY OF AMPICILLIN—A NEW BROAD‐SPECTRUM PENICILLIN. Br. J. Pharmacol. Chemother 1962, 18, 356–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thirring K; Heilmayer W; Reidl R; Kollman H; Ivezic-Schoenfeld Z; Wicha W; Paukner S; Strickman D 12-EPI-PLEUROMUTILINS. U. S. Patent WO2015110481A1, 2015. [Google Scholar]
- 34.Jelić D; Antolović R, From erythromycin to azithromycin and new potential ribosome-binding antimicrobials. Antibiotics 2016, 5, 29–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richter MF; Hergenrother PJ, The challenge of converting gram-positive-only compounds into broad-spectrum antibiotics. Ann. N. Y. Acad. Sci 2019, 1435, 18–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Takhi M; Sreenivas K; Reddy CK; Munikumar M; Praveena K; Sudheer P; Rao BNVM; Ramakanth G; Sivaranjani J; Mulik S; Reddy YR; Narasimha Rao K; Pallavi R; Lakshminarasimhan A; Panigrahi SK; Antony T; Abdullah I; Lee YK; Ramachandra M; Yusof R; Rahman NA; Subramanya H, Discovery of azetidine based ene-amides as potent bacterial enoyl ACP reductase (FabI) inhibitors. Eur. J. Med. Chem 2014, 84, 382–394. [DOI] [PubMed] [Google Scholar]
- 37.Christie SM; Ren J; Johnson ME Enoyl reductase inhibitors with antibacterial activity. U.S. Patent 20180072666A1, 2018. [Google Scholar]
- 38.Gerusz V; Escaich S; Oxoby M; Denis A Novel Heterocyclic Acrylamides And Their Use As Pharmaceuticals. U.S. Patent 8846711B2, 2011. [Google Scholar]
- 39.De Jonge BLM; Walkup GK; Lahiri SD; Huynh H; Neckermann G; Utley L; Nash TJ; Brock J; San Martin M; Kutschke A; Johnstone M; Laganas V; Hajec L; Gu RF; Ni H; Chen B; Hutchings K; Holt E; McKinney D; Gao N; Livchak S; Thresher J, Discovery of inhibitors of 4’-phosphopantetheine adenylyltransferase (PPAT) to validate PPAT as a target for antibacterial therapy. Antimicrob. Agents Chemother 2013, 57, 6005–6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qiu X; Janson CA; Smith WW; Green SM; McDevitt P; Johanson K; Carter P; Hibbs M; Lewis C; Chalker A; Fosberry A; Lalonde J; Berge J; Brown P; Houge-Frydrych CSV; Jarvest RL, Crystal structure of Staphylococcus aureus tyrosyl-tRNA synthetase in complex with a class of potent and specific inhibitors. Protein Sci. 2001, 10, 2008–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takrouri K; Cooper HD; Spaulding A; Zucchi P; Koleva B; Cleary DC; Tear W; Beuning PJ; Hirsch EB; Aggen JB, Progress against Escherichia coli with the oxazolidinone class of antibacterials: Test case for a general approach to improving whole-cell gram-negative activity. ACS Infect. Dis 2016, 2, 405–426. [DOI] [PubMed] [Google Scholar]
- 42.Uria-Nickelsen M; Neckermann G; Sriram S; Andrews B; Manchester JI; Carcanague D; Stokes S; Hull KG, Novel topoisomerase inhibitors: Microbiological characterisation and in vivo efficacy of pyrimidines. Int. J. Antimicrob. Agents 2013, 41, 363–371. [DOI] [PubMed] [Google Scholar]
- 43.Keating TA; Newman JV; Olivier NB; Otterson LG; Andrews B; Boriack-Sjodin PA; Breen JN; Doig P; Dumas J; Gangl E; Green OM; Guler SY; Hentemann MF; Joseph-Mccarthy D; Kawatkar S; Kutschke A; Loch JT; McKenzie AR; Pradeepan S; Prasad S; Martínez-Botella G, In vivo validation of thymidylate kinase (TMK) with a rationally designed, selective antibacterial compound. ACS Chem. Biol 2012, 7, 1866–1872. [DOI] [PubMed] [Google Scholar]
- 44.Bazile S; Moreau N; Bouzard D; Essiz M, Relationships among antibacterial activity, inhibition of DNA gyrase, and intracellular accumulation of 11 fluoroquinolones. Antimicrob. Agents Chemother 1992, 36, 2622–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Davis TD; Gerry CJ; Tan DS, General platform for systematic quantitative evaluation of small-molecule permeability in bacteria. ACS Chem. Biol 2014, 9, 2535–2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai H; Rose K; Liang LH; Dunham S; Stover C, Development of a liquid chromatography/mass spectrometry-based drug accumulation assay in Pseudomonas aeruginosa. Anal. Biochem 2009, 385, 321–325. [DOI] [PubMed] [Google Scholar]
- 47.Huigens RW; Morrison KC; Hicklin RW; Timothy TA; Richter MF; Hergenrother PJ, A ring-distortion strategy to construct stereochemically complex and structurally diverse compounds from natural products. Nat. Chem 2013, 5, 195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rafferty RJ; Hicklin RW; Maloof KA; Hergenrother PJ, Synthesis of complex and diverse compounds through ring distortion of abietic acid. Angew. Chem. Int. Ed 2014, 53, 220–224. [DOI] [PubMed] [Google Scholar]
- 49.Garcia A; Drown BS; Hergenrother PJ, Access to a Structurally Complex Compound Collection via Ring Distortion of the Alkaloid Sinomenine. Org. Lett 2016, 18, 4852–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hicklin RW; Lõpez Silva TL; Hergenrother PJ, Synthesis of bridged oxafenestranes from pleuromutilin. Angew. Chem. Int. Ed 2014, 53, 9880–9883. [DOI] [PubMed] [Google Scholar]
- 51.Perlmutter SJ; Geddes EJ; Drown BS; Motika SE; Lee MRL; Hergenrother PJ, Compound Uptake into E. coli Can Be Facilitated by N-Alkyl Guanidiniums and Pyridiniums. ACS Infect. Dis 2020, 7, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Masci D; Hind C; Islam MK; Toscani A; Clifford M; Coluccia A; Conforti I; Touitou M; Memdouh S; Wei X; La Regina G; Silvestri R; Sutton JM; Castagnolo D, Switching on the activity of 1,5-diaryl-pyrrole derivatives against drug-resistant ESKAPE bacteria: Structure-activity relationships and mode of action studies. Eur. J. Med. Chem 2019, 178, 500–514. [DOI] [PubMed] [Google Scholar]
- 53.Lukežič T; Fayad AA; Bader C; Harmrolfs K; Bartuli J; Groß S; Lešnik U; Hennessen F; Herrmann J; Pikl Š; Petković H; Müller R, Engineering Atypical Tetracycline Formation in Amycolatopsis sulphurea for the Production of Modified Chelocardin Antibiotics. ACS Chem. Biol 2019, 14, 468–477. [DOI] [PubMed] [Google Scholar]
- 54.Cohen F; Aggen JB; Andrews LD; Assar Z; Boggs J; Choi T; Dozzo P; Easterday AN; Haglund CM; Hildebrandt DJ; Holt MC; Joly K; Jubb A; Kamal Z; Kane TR; Konradi AW; Krause KM; Linsell MS; Machajewski TD; Miroshnikova O; Moser HE; Nieto V; Phan T; Plato C; Serio AW; Seroogy J; Shakhmin A; Stein AJ; Sun AD; Sviridov S; Wang Z; Wlasichuk K; Yang W; Zhou X; Zhu H; Cirz RT, Optimization of LpxC Inhibitors for Antibacterial Activity and Cardiovascular Safety. ChemMedChem 2019, 14, 1560–1572. [DOI] [PubMed] [Google Scholar]
- 55.Hu Y; Shi H; Zhou M; Ren Q; Zhu W; Zhang W; Zhang Z; Zhou C; Liu Y; Ding X; Shen HC; Yan SF; Dey F; Wu W; Zhai G; Zhou Z; Xu Z; Ji Y; Lv H; Jiang T; Wang W; Xu Y; Vercruysse M; Yao X; Mao Y; Yu X; Bradley K; Tan X, Discovery of Pyrido[2,3- b]indole Derivatives with Gram-Negative Activity Targeting Both DNA Gyrase and Topoisomerase IV. J. Med. Chem 2020, 63, 9623–9649. [DOI] [PubMed] [Google Scholar]
- 56.Li Y; Gardner JJ; Fortney KR; Leus IV; Bonifay V; Zgurskaya HI; Pletnev AA; Zhang S; Zhang ZY; Gribble GW; Spinola SM; Duerfeldt AS, First-generation structure-activity relationship studies of 2,3,4,9-tetrahydro-1H-carbazol-1-amines as CpxA phosphatase inhibitors. Bioorg. Med. Chem. Lett 2019, 29, 1836–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hiramatsu K; Igarashi M; Morimoto Y; Baba T; Umekita M; Akamatsu Y, Curing bacteria of antibiotic resistance: reverse antibiotics, a novel class of antibiotics in nature. Int. J. Antimicrob. Agents 2012, 39, 478–85. [DOI] [PubMed] [Google Scholar]
- 58.Parkinson EI; Bair JS; Nakamura BA; Lee HY; Kuttab HI; Southgate EH; Lezmi S; Lau GW; Hergenrother PJ, Deoxynybomycins inhibit mutant DNA gyrase and rescue mice infected with fluoroquinolone-resistant bacteria. Nat. Comm 2015, 6, 6947–6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seefeld MA; Miller WH; Newlander KA; Burgess WJ; Payne DJ; Rittenhouse SF; Moore TD; DeWolf WE; Keller PM; Qiu X; Janson CA; Vaidya K; Fosberry AP; Smyth MG; Jaworski DD; Slater-Radosti C; Huffman WF, Inhibitors of bacterial enoyl acyl carrier protein reductase (FabI): 2,9-disubstituted 1,2,3,4-tetrahydropyrido[3,4-b]indoles as potential antibacterial agents. Bioorg. Med. Chem. Lett 2001, 11, 2241–2244. [DOI] [PubMed] [Google Scholar]
- 60.Payne DJ; Miller WH; Berry V; Brosky J; Burgess WJ; Chen E; DeWolf WE; Fosberry AP; Greenwood R; Head MS; Heerding DA; Janson CA; Jaworski DD; Keller PM; Manley PJ; Moore TD; Newlander KA; Pearson S; Polizzi BJ; Qiu X; Rittenhouse SF; Slater-Radosti C; Salvers KL; Seefeld MA; Smyth MG; Takata DT; Uzinskas IN; Vaidya K; Wallis NG; Winram SB; Yuan CCK; Huffman WF, Discovery of a novel and potent class of fabI-directed antibacterial agents. Antimicrob. Agents Chemother 2002, 46, 3118–3124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Karlowsky JA; Kaplan N; Hafkin B; Hoban DJ; Zhanel GG, AFN-1252, a FabI inhibitor, demonstrates a staphylococcus-specific spectrum of activity. Antimicrob. Agents Chemother 2009, 53, 3544–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaplan N; Albert M; Awrey D; Bardouniotis E; Berman J; Clarke T; Dorsey M; Hafkin B; Ramnauth J; Romanov V; Schmid MB; Thalakada R; Yethon J; Pauls HW, Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal fabI inhibitor. Antimicrob. Agents Chemother 2012, 56, 5865–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhu L; Lin J; Ma J; Cronan JE; Wang H, Triclosan resistance of Pseudomonas aeruginosa PAO1 is due to FabV, a triclosan-resistant enoyl-acyl carrier protein reductase. Antimicrob. Agents Chemother 2010, 54, 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howe JA; Wang H; Fischmann TO; Balibar CJ; Xiao L; Galgoci AM; Malinverni JC; Mayhood T; Villafania A; Nahvi A; Murgolo N; Barbieri CM; Mann PA; Carr D; Xia E; Zuck P; Riley D; Painter RE; Walker SS; Sherborne B; De Jesus R; Pan W; Plotkin MA; Wu J; Rindgen D; Cummings J; Garlisi CG; Zhang R; Sheth PR; Gill CJ; Tang H; Roemer T, Selective small-molecule inhibition of an RNA structural element. Nature 2015, 526, 672–677. [DOI] [PubMed] [Google Scholar]
- 65.Breaker RR, Riboswitches and translation control. Cold Spring Harb. Perspect. Biol 2018, 10, a032797–a032797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang H; Mann PA; Xiao L; Gill C; Galgoci AM; Howe JA; Villafania A; Barbieri CM; Malinverni JC; Sher X; Mayhood T; McCurry MD; Murgolo N; Flattery A; Mack M; Roemer T, Dual-Targeting Small-Molecule Inhibitors of the Staphylococcus aureus FMN Riboswitch Disrupt Riboflavin Homeostasis in an Infectious Setting. Cell Chem. Biol 2017, 24, 576–588. [DOI] [PubMed] [Google Scholar]
- 67.Brooks LE; Ul-Hasan S; Chan BK; Sistrom MJ, Quantifying the Evolutionary Conservation of Genes Encoding Multidrug Efflux Pumps in the ESKAPE Pathogens To Identify Antimicrobial Drug Targets. mSystems 2018, 3, e00024–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fernández-Piñar R; Lo Sciuto A; Rossi A; Ranucci S; Bragonzi A; Imperi F, In vitro and in vivo screening for novel essential cell-envelope proteins in Pseudomonas aeruginosa. Sci. Rep 2015, 5, 17593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ribeiro CFA; Silveira GGDOS; Cândido EDS; Cardoso MH; Espínola Carvalho CM; Franco OL, Effects of Antibiotic Treatment on Gut Microbiota and How to Overcome Its Negative Impacts on Human Health. ACS Infect. Dis 2020, 6, 2544–2559. [DOI] [PubMed] [Google Scholar]
- 70.Pandeya A; Ojo I; Alegun O; Wei Y, Periplasmic Targets for the Development of Effective Antimicrobials against Gram-Negative Bacteria. ACS Infect. Dis 2020, 2, 2337–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang G; Baidin V; Pahil KS; Moison E; Tomasek D; Ramadoss NS; Chatterjee AK; McNamara CW; Young TS; Schultz PG; Meredith TC; Kahne D, Cell-based screen for discovering lipopolysaccharide biogenesis inhibitors. Proc. Natl. Acad. Sci. U.S.A 2018, 115, 6834–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smith PA; Koehler MFT; Girgis HS; Yan D; Chen Y; Chen Y; Crawford JJ; Durk MR; Higuchi RI; Kang J; Murray J; Paraselli P; Park S; Phung W; Quinn JG; Roberts TC; Rougé L; Schwarz JB; Skippington E; Wai J; Xu M; Yu Z; Zhang H; Tan M-W; Heise CE, Optimized arylomycins are a new class of Gram-negative antibiotics. Nature 2018, 561, 189–194. [DOI] [PubMed] [Google Scholar]
- 73.Imai Y; Meyer KJ; Iinishi A; Favre-Godal Q; Green R; Manuse S; Caboni M; Mori M; Niles S; Ghiglieri M; Honrao C; Ma X; Guo JJ; Makriyannis A; Linares-Otoya L; Böhringer N; Wuisan ZG; Kaur H; Wu R; Mateus A; Typas A; Savitski MM; Espinoza JL; O’Rourke A; Nelson KE; Hiller S; Noinaj N; Schäberle TF; D’Onofrio A; Lewis K, A new antibiotic selectively kills Gram-negative pathogens. Nature 2019, 576, 459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jackson N; Czaplewski L; Piddock LJV, Discovery and development of new antibacterial drugs: learning from experience? J. Antimicrob. Chemoth 2018, 73, 1452–1459. [DOI] [PubMed] [Google Scholar]
- 75.Tommasi R; Iyer R; Miller AA, Antibacterial Drug Discovery: Some Assembly Required. ACS Infect Dis 2018, 4 (5), 686–695. [DOI] [PubMed] [Google Scholar]