

Abstract
Enterococci are Gram-positive, opportunistic pathogens that reside throughout the gastrointestinal tracts of most terrestrial organisms. Enterococci are resistant to many antibiotics, which makes enterococcal infections difficult to treat. Enterococci are also particularly hardy bacteria that can tolerate a variety of environmental stressors. Understanding how enterococci sense and respond to the extracellular environment to enact adaptive biological responses may identify new targets that can be exploited for development of treatments for enterococcal infections. Bacterial eukaryotic-like serine/threonine kinases (eSTKs) and cognate phosphatases (STPs) are important signaling systems that mediate biological responses to extracellular stimuli. Some bacterial eSTKs are transmembrane proteins that contain a series of extracellular repeats of the penicillin-binding and Ser/Thr kinase-associated (PASTA) domain, leading to their designation as “PASTA kinases”. Enterococcal genomes encode a single PASTA kinase and its cognate phosphatase. Investigations of the enterococcal PASTA kinase revealed its importance in resistance to antibiotics and other cell wall stresses, in enterococcal colonization of the mammalian gut, clues about its mechanism of signal transduction, and its integration with other enterococcal signal transduction systems. In this review, we describe the current state of knowledge of PASTA kinase signaling in enterococci and describe important gaps that still need to be addressed to provide a better understanding of this important signaling system.
Introduction
Enterococci are Gram-positive, opportunistic pathogens and inhabitants of the gastrointestinal tract of most terrestrial organisms. Enterococci are considered to be commensals, but in immunocompromised or debilitated individuals, they can cause deadly infections (Hidron et al., 2008; Sievert et al., 2013; Weiner et al., 2016). Two enterococcal species, Enterococcus faecalis and Enterococcus faecium, are the main culprits of healthcare-associated enterococcal infections. Vancomycin-resistant enterococci have been classified by the Centers for Disease Control and Prevention as a serious threat (CDC, 2019; Weiner et al., 2016). Prior antibiotic therapy is a known risk factor for acquisition of a subsequent enterococcal infection (Carmeli et al., 2002; Shepard and Gilmore, 2002). Disruption of the intestinal microbial ecosystem by antibiotics enables enterococci to proliferate to abnormally high numbers in the gut, then disseminate to cause infections elsewhere in the body (Brandl et al., 2008; Carmeli et al., 2002; Donskey et al., 2000; Shepard and Gilmore, 2002; Ubeda et al., 2010). Antibiotic treatment of such infections is often ineffective due to intrinsic and acquired mechanisms of resistance (Hollenbeck and Rice, 2012; Miller et al., 2014; Miller et al., 2016).
Enterococci can also be found in the oral cavity and saliva (Sedgley et al., 2006; Wang et al., 2012; Zhu et al., 2010) and are frequently associated with oral diseases such as caries, periodontitis, endodontic infections (Komiyama et al., 2016; Kouidhi et al., 2011; Rams et al., 2013; Wang et al., 2012). E. faecalis has been known for decades to be one of the most frequently isolated species from root canal infections, especially persistent infections associated with apical periodontitis (Ferrari et al., 2005; Gomes et al., 2008; Hancock III et al., 2001; Molander et al., 1998; Peciuliene et al., 2000; Peciuliene et al., 2001; Rôças et al., 2004a; Rôças et al., 2004b; Schirrmeister et al., 2009; Sedgley et al., 2006; Sedgley et al., 2004; Siqueira Jr and Rôças, 2004; Stuart et al., 2006; Sundqvist, 1992; Sundqvist et al., 1998; Tennert et al., 2014). The hardy nature of enterococci makes it difficult to use dental pre-treatments effectively (e. g. chlorhexidine and sodium hypochlorite) contributing to failed dental procedures (Estrela et al., 2008; Hancock III et al., 2001; Peciuliene et al., 2001; Pinheiro et al., 2003; Rôças et al., 2004b; Sedgley et al., 2005; Zerella et al., 2005). Conventional methods for treatment of endodontic infections, including the use of intracanal medicaments, often do not effectively eliminate E. faecalis (Ferrari et al., 2005), likely due to the intrinsic resistance of the organism to the stresses imposed by these treatments. There is also evidence that sublethal concentrations of certain disinfectants used in dentistry (such as chlorhexidine) can lead to enhanced resistance of enterococci to certain antibiotics (Bhardwaj et al., 2017; Gadea et al., 2017a; Gadea et al., 2017b; Kampf, 2019). Lipoteichoic acid, a cell surface component of E. faecalis, is a potent stimulator of pro-inflammatory responses (Bhakdi et al., 1991; Sipert et al., 2010). E. faecalis is a prolific producer of extracellular superoxide (Huycke et al., 2002; Huycke et al., 1996; Huycke et al., 2001; Huycke and Moore, 2002), which has been proposed to contribute to periapical tissue injury and bone loss in apical periodontitis (Marton et al., 1993). In addition, E. faecalis binds to host molecules (Hubble et al., 2003; Kowalski et al., 2006; Nallapareddy and Murray, 2008) (e.g. collagen) and invades dentinal tubules (Akpata and Blechman, 1982; Haapasalo and Orstavik, 1987; Love, 2001; Orstavik and Haapasalo, 1990), facilitating its persistence. Overall, the collective picture that emerges is one in which the ability of E. faecalis to cause periradicular disease derives from its ability to survive the effects of root canal treatment and persist in the root canals and dentinal tubules of teeth (Stuart et al., 2006). Thus, understanding the mechanisms that enable enterococci to overcome antimicrobials and proliferate may reveal new therapeutic targets to prevent or treat enterococcal infections throughout the body.
Most bacteria possess numerous signaling systems that sense and respond to the surrounding environment to promote survival. The most ubiquitous are the now-familiar two-component signal transductions systems, consisting in the simplest form of a histidine kinase sensor and its cognate response regulator (often a transcription factor), but over the past ~15 years there has been a growing recognition of the importance of eukaryotic-like serine/threonine kinases (eSTKs) and their cognate phosphatases (STPs) as mediators of a variety of critical bacterial processes. Most of these kinases belong to the same superfamily of Ser/Thr kinases that are widespread in eukaryotic organisms (i.e. “Hanks-type” kinases). Although many soil-dwelling bacteria contain multiple Ser/Thr kinases with diverse domain architectures and functions, oral streptococci and enterococci generally contain only 1 such kinase encoded in a given genome, which is the focus of this review. The genomes of clinically relevant enterococci encode a single eSTK and its cognate STP, called IreK and IreP, respectively (originally PrkC and PrpC), in E. faecalis (Stk and StpA, respectively, in E. faecium) (Kristich et al., 2007; Sacco et al., 2014).
IreK belongs to a family of eSTKs found throughout Actinobacteria and Firmicutes that exhibit a conserved bipartite domain architecture (Figure 1) in which the cytoplasmic kinase domain is separated by a transmembrane segment from an extracellular module containing a variable number of penicillin-binding and Ser/Thr kinase-associated (PASTA) domains, leading to their designation as “PASTA kinases” (Hanks and Hunter, 1995; Hanks et al., 1988; Janczarek et al., 2018; Krupa and Srinivasan, 2005; Pereira et al., 2011; Stancik et al., 2018). The number of PASTA domains in a given PASTA kinase (anywhere from 1 to 5) varies in different bacterial species (Calvanese et al., 2017; Jones and Dyson, 2006; Yeats et al., 2002). For example, IreK of E. faecalis possesses 5 PASTA domains, while the PASTA kinase ortholog in E. faecium possesses 4 and that of Streptococcus pneumoniae possesses 3 PASTA domains. Sequence analyses by Calvanese et al. (2017) identified PASTA kinase homologs in the databases containing only 1 or 2 PASTA domains, but thus far most experimental research has focused on PASTA kinases that contain between 3 and 5 PASTA domains. It is unclear why most PASTA kinases possess multiple PASTA domains, or why different PASTA kinases possess different numbers of PASTA domains, but a bioinformatic analysis by Jones and Dyson (2006) reported that among mycobacterial PASTA kinases a given PASTA domain of a PASTA kinase has a closer phylogenetic relationship with the equivalent PASTA domain from homologous PASTA kinases than with the other PASTA domains within the same kinase, suggesting that there may be evolutionary pressure to maintain functional differences among distinct PASTA domains of PASTA kinases.
Figure 1. Domain architecture of PASTA kinases.
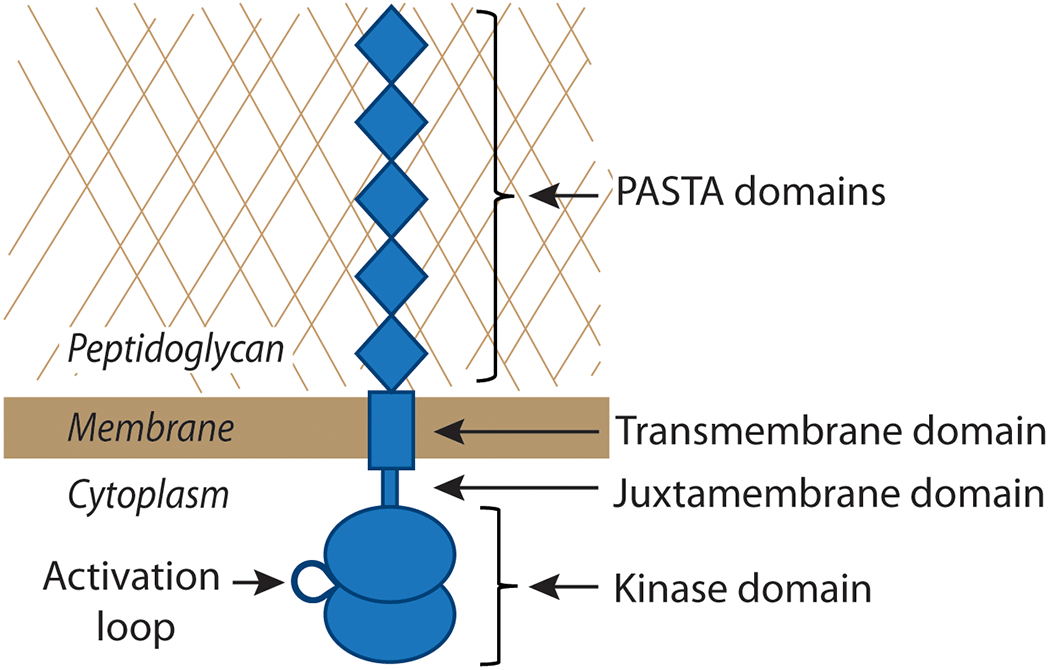
PASTA kinases contain a cytoplasmic, N-terminal two-lobed kinase domain, a juxtamembrane domain (sometimes referred to as a ‘linker’) of ~60 amino acids, a single transmembrane domain, and a variable number of extracellular PASTA domains. Shown is a representation of IreK from E. faecalis, which contains 5 PASTA domains. The activation loop is a centrally located structural feature of the well-conserved kinase domains that becomes phosphorylated upon activation of the kinase.
IreP belongs to a superfamily of PP2C-type phosphatases that require divalent cations (e.g. manganese) for activity. Like PASTA kinases, IreP homologs are encoded in the genomes of most Gram-positive bacteria, where the phosphatase gene is typically located immediately upstream of the PASTA kinase gene. In E. faecalis, the genes encoding IreP and IreK overlap by a few codons, suggesting they are translationally coupled to maintain the appropriate stoichiometry in the cell. Indeed, the phosphatase- and PASTA kinase-encoding genes have been found to be co-transcribed in numerous bacteria, including oral streptococci (Absalon et al., 2009; Agarwal et al., 2011; Beltramini et al., 2009; Gaidenko et al., 2002; Hussain et al., 2006; Jin and Pancholi, 2006; Madec et al., 2002; Novakova et al., 2005; Rajagopal et al., 2003) suggesting that they comprise key components of a finely tuned regulatory circuit. IreP homologs are known to dephosphorylate both their cognate PASTA kinase as well as other kinase substrates, regulating the activation of the PASTA kinase itself as well as the activity of the downstream signaling network as a whole (Beltramini et al., 2009; Debarbouille et al., 2009; Ohlsen and Donat, 2010; Pereira et al., 2011; Shi, 2009). Mutations in the phosphatases lead to defects in cell division, growth, survival and virulence (Burnside et al., 2010; Gaidenko et al., 2002; Rajagopal et al., 2003; Sharma et al., 2016), phenotypes which are probably mediated by dysregulated and unconstrained activity of the cognate PASTA kinase.
This review will discuss what is known about the PASTA kinase of enterococci, including its role in regulating enterococcal physiology and antimicrobial resistance, mechanisms of IreK-mediated signal transduction, and will discuss gaps in knowledge to be addressed in future research.
IreK in enterococcal physiology
IreK of E. faecalis (originally known as PrkC) was first described by Kristich et al. (2007) and shown to be important for cellular morphology, antibiotic resistance and short-term persistence in the intestine of mice. A deletion of ireK resulted in decreased resistance to a number of cell-wall targeting antimicrobial agents, especially cephalosporins (members of the beta-lactam family of cell wall-targeting antibiotics). The ΔireK mutant also exhibited decreased resistance to other agents that impact the cell envelope such as bile, sodium cholate, and sodium dodecyl sulfate (SDS). Later studies demonstrated that the ΔireK mutant also exhibited decreased resistance to lysozyme and nisin, agents that target the peptidoglycan (PG) and its biosynthesis, respectively (Banla et al., 2018; Hall et al., 2013; Labbe and Kristich, 2017). Nisin is a bacteriocin, and although a role for IreK in resistance to bacteriocins has not been investigated broadly, it seems likely given the importance of IreK in cell wall stress resistance that IreK would also be critical for survival of E. faecalis in the oral cavity that is rich in bacteriocin-producing streptococci. Overall, the diversity in molecular structure and mechanism of action of these antimicrobials suggests that IreK likely responds to perturbation of the membrane or the cell wall, rather than to a specific antimicrobial compound. The kinase activity of IreK per se is required for resistance to cell-wall stress and long-term mammalian gut colonization (Banla et al., 2018; Hall et al., 2013; Kristich et al., 2011), because a mutant version of the kinase carrying a lysine-to-arginine substitution of a critical lysine residue within the kinase domain that impairs kinase activity phenocopied the ireK deletion. Substitution of the equivalent lysine residue in PASTA kinases from other organisms also impairs their activity (Duran et al., 2005; Kang et al., 2005; Madec et al., 2002; Novakova et al., 2005). Together, these findings suggest IreK senses environmental stresses that impact the cell wall, resulting in upregulation of IreK kinase activity to promote an adaptive biological response.
PASTA kinases of other bacteria also appear to play a role in cell wall homeostasis and may respond to PG fragments or lipid II (precursor used for extracellular PG synthesis) to elicit a signaling cascade (Hardt et al., 2017; Lee et al., 2010; Maestro et al., 2011; Mir et al., 2011; Shah et al., 2008; Squeglia et al., 2011). The role for IreK in resistance to antimicrobials, in particular antimicrobials targeting the cell wall, is also widely conserved among homologous PASTA kinases of other bacteria; the pattern of specific antibiotics and extent of resistance that depends on the PASTA kinase varies among different species (Beltramini et al., 2009; Cameron et al., 2012; Cuenot et al., 2019; Pensinger et al., 2016). The E. faecalis ΔireK mutant exhibits altered morphology (i.e. chains of bacteria instead of the typical diplococci), suggesting a role for IreK in cell shape and possibly cell division. PASTA kinases of other bacteria, including oral streptococci, have also been implicated in cell division and morphology, because the kinase deletion mutants often exhibit aberrant division and cell shape phenotypes (Banu et al., 2010; Burnside et al., 2010; Chaba et al., 2002; Giefing et al., 2008; Hussain et al., 2006; Jin and Pancholi, 2006; Rajagopal et al., 2003) PASTA kinases have been shown to interact with cell division machinery and to localize to sites of cell division (Beilharz et al., 2012; Giefing et al., 2010; Rued et al., 2017), suggesting a mechanism by which PASTA kinases could influence cell division and morphology.
Additionally, PASTA kinases have been implicated in virulence, toxin production, biofilm production, and even cellular viability among low-GC Gram-positive bacteria, including oral streptococci (Beilharz et al., 2012; Beltramini et al., 2009; Burnside et al., 2010; Chawla et al., 2014; Fernandez et al., 2006; Hussain et al., 2006; Jin and Pancholi, 2006; Kang et al., 2005; Pensinger et al., 2014; Rajagopal et al., 2003). IreK is required for long-term enterococcal colonization of the mammalian gut (Banla et al., 2018), and other PASTA kinases have been shown to be important for persistence and virulence in the host as well (Banu et al., 2010; Bugrysheva et al., 2011; Debarbouille et al., 2009; Hussain et al., 2006; Pensinger et al., 2016; Rajagopal et al., 2003). Together, the central nature of PASTA kinases in a variety of critical cellular processes collectively suggest that PASTA kinases might be valuable therapeutic targets. The activity of PASTA kinases, including IreK, can be modulated pharmacologically (Fernandez et al., 2006; Hall et al., 2013; Lougheed et al., 2011; Pensinger et al., 2014; Schaenzer et al., 2017; Shah et al., 2008; Wang et al., 2017b; Xu et al., 2017), stimulating efforts to identify specific inhibitors with the goals of using them therapeutically. Generally, this has been done by screening libraries of small molecule kinase inhibitors against wild-type strains of bacteria to identify inhibitors that lead to reduced resistance to beta-lactam antibiotics. One of the main considerations is that the therapeutic must be specific for a PASTA kinase of a given species to avoid targeting other bacteria and, especially, host eSTKs. Some compounds fitting that criteria have already been identified and are currently under investigation (Pensinger et al., 2014; Schaenzer et al., 2018; Schaenzer et al., 2017).
IreK architecture
IreK and other PASTA kinases exhibit a conserved bipartite domain architecture that defines the family (Figure 1). These kinases consist of (i) an intracellular catalytic kinase domain that belongs to the Hanks-type superfamily; (ii) a ‘linker’ segment (often referred to as a ‘juxtamembrane’ domain) of ~60 residues that connects the kinase domain to the transmembrane helix; (iii) a transmembrane domain; and (iv) a series of extracellular PASTA domains (5 PASTAs in IreK). The precise molecular function of PASTA domains is not well understood; however, the prevailing model proposes that PASTA domains of PASTA kinases serve as ligand-binding domains that recognize the molecular signal(s) responsible for triggering kinase activity. As the name implies, PASTA domains are generally found in eSTKs (i.e. PASTA kinases) and penicillin-binding proteins (PBPs; enzymes that catalyze the final steps of PG assembly) of Gram-positive bacteria. There are only low levels of primary sequence similarity between distinct PASTA domains in a given PASTA kinase, but strong structural similarity, suggesting that there may be functional similarity as well (Yeats et al., 2002). Structural studies, including NMR, X-ray crystallography and modeling studies, on isolated PASTA modules (or fragments thereof) from several PASTA kinases have suggested that successive PASTA domains adopt an extended, rigid conformation that has been hypothesized to facilitate ligand binding and dimerization of the kinase (Barthe et al., 2010; Paracuellos et al., 2010; Pereira et al., 2011; Prigozhin et al., 2016; Ruggiero et al., 2012). However, structural evidence for ligand-mediated changes in PASTA module conformation has not yet been reported.
PASTA domains have been proposed to sense cell envelope stress , possibly through interactions with PG fragments or cell wall precursor lipid II, or by directly binding to antibiotics (Hardt et al., 2017; Kaur et al., 2019; Lee et al., 2010; Maestro et al., 2011; Mir et al., 2011; Righino et al., 2018; Shah et al., 2008; Squeglia et al., 2011; Wang et al., 2017a). Studies using either in vitro techniques to assess interactions or docking models proposed interactions between PASTA domains and PG fragments (Lee et al., 2010; Maestro et al., 2011; Mir et al., 2011; Paracuellos et al., 2010; Righino et al., 2018; Squeglia et al., 2011). A specific residue at the third position of the peptide side chain of PG fragments (m-DAP) was required to initiate a signal for exiting dormancy by the PASTA kinases of Bacillus subtilis and Mycobacterium tuberculosis (Lee et al., 2010; Mir et al., 2011; Shah et al., 2008), supporting the model that PG fragments serve as specific ligands for recognition by PASTA domains. However, in many cases the reported Kd values for PG fragment-PASTA interactions are in the high micromolar, and in some cases millimolar, range (Kaur et al., 2019; Lee et al., 2010; Maestro et al., 2011; Mir et al., 2011; Squeglia et al., 2011), although some interactions in the lower micromolar range (~12-70 μM) have been identified as well (Mir et al., 2011; Paracuellos et al., 2010; Wang et al., 2017a). It remains to be determined whether interactions with relatively low binding affinities are biologically relevant for the function of the kinase. Studies of Staphylococcus aureus and M. tuberculosis PASTA kinases revealed that the cell wall precursor lipid II acts as a ligand, and that its effects are dependent on the composition of the stem peptide (Hardt et al., 2017; Kaur et al., 2019). In these cases, interaction with lipid II resulted in activation of the kinase.
To assess the importance of PASTA domains for function of E. faecalis IreK, Labbe et al. (2017) constructed a panel of IreK mutants lacking individual PASTA domains. The panel included a series of mutants in which PASTA domains were successively truncated, as well as mutants in which each of the 5 individual PASTA domains was uniquely removed. Analysis of IreK function in the collection of mutants revealed several findings: (i) no individual PASTA domain is essential for IreK function; (ii) a mutant with as few as one PASTA domain retained the ability to respond to cell wall stress and mediate cephalosporin resistance; (iii) a mutant lacking all 5 PASTA domains was unable to respond to cephalosporin stress or mediate wild-type cephalosporin resistance; and (iv) the mutant lacking all 5 PASTA domains retained normal activity during exponential growth conditions in the absence of stress (Labbe and Kristich, 2017). Collectively these results indicate that the extracellular PASTA domains of IreK are, at least partly, functionally redundant with each other, and are required for IreK to be activated by cell wall stress imposed by antibiotics that impair PG synthesis. However, the PASTA domains do not appear to be necessary for IreK to respond to all stimuli. Labbe et al. (2017) showed that growth inhibition, independent of the presence of any antibiotic, leads to deactivation of IreK signaling. Hence, in addition to antibiotic-mediated cell wall stress, IreK is activated by another signal related to ongoing cell growth or division. Because the IreK mutant lacking all 5 PASTA domains retained this ability, it appears IreK is able to receive sensory input via both PASTA-dependent and PASTA-independent means. It remains unclear how, mechanistically, IreK is activated by growth or division, or what domain of IreK is important for this function. Whether or not the transmembrane segment possesses specific functions, such as responding to a growth-related signal, remains unknown for IreK and other PASTA kinases. However, other PASTA kinases are known to localize to sites of cell division and physically interact with the cell division machinery (Beilharz et al., 2012; Fleurie et al., 2014; Giefing et al., 2010; Hardt et al., 2017; Jarick et al., 2018; Mir et al., 2011; Morlot et al., 2013; Pompeo et al., 2018; Pompeo et al., 2015), suggesting that such interactions could be a source of sensory input to activate IreK.
The juxtamembrane linker is a conserved feature of the architecture of PASTA kinases, but its function is poorly understood. Phosphorylation at sites within the juxtamembrane linker of PASTA kinases has been described, but the contribution of phosphorylation at these sites to signal transduction has, in most cases, not been elucidated (Boitel et al., 2003; Duran et al., 2005; Madec et al., 2003; Young et al., 2003). When phosphorylated on its juxtamembrane linker, the PASTA kinase of M. tuberculosis was shown to interact with FhaA, a ForkHead Associated (FHA)-domain containing protein that is involved in PG synthesis (Roumestand et al., 2011; Viswanathan et al., 2017). However, it is unclear how, or if, that interaction modulates the activity of FhaA or the PASTA kinase itself. Labbe et al. (2019) identified 2 sites of phosphorylation on the juxtamembrane linker of IreK (Labbe et al., 2019). Mutations (either phospho-ablative: threonine to alanine, or phospho-mimetic: threonine to glutamate) at the two phosphorylation sites in the IreK juxtamembrane linker did not have an impact on enterococcal cephalosporin resistance, indicating that phosphorylation of those residues does not play a role in this response. Moreover, sequence analysis indicates the E. faecalis genome does not encode any FHA domain-containing proteins. Thus, additional work is required to elucidate the function of the IreK juxtamembrane linker domain.
The cytoplasmic kinase domains of PASTA kinases are the most highly conserved domains and belong to the superfamily of Hanks-type kinases that are widespread in eukaryotic organisms. These kinases adopt a two-lobed structure with a flexible ‘activation loop’ positioned at the interface of the two lobes, near the kinase active site located in the cleft between the lobes. Phosphorylation of the activation loop is a common feature of PASTA kinases and has been thought to be required for kinase activation in vivo (Boitel et al., 2003; Bryant-Hudson et al., 2011; Madec et al., 2002; Nolen et al., 2004; Shakir et al., 2010; Young et al., 2003), although most studies of PASTA kinase phosphorylation focus on the kinase in vitro, with an exception where a surrogate strain is used as an in vivo host (Zheng et al., 2018). Labbe et al. (2019) identified 5 sites of phosphorylation on the kinase domain of IreK: three in the activation loop (T163, T166, T168) and 2 in the kinase catalytic core (T148 and T218). Mutations at T148 in the catalytic core of IreK did not have an impact on cephalosporin resistance, and the function of phosphorylation at this site remains unknown. Phospho-ablative mutations at sites on the activation loop (T163, T166, T168) resulted in loss of both IreK phosphorylation and activity (i.e. phosphorylation of substrates), and decreased cephalosporin resistance, indicating that phosphorylation of the activation loop residues is important for kinase function in vivo (Kristich et al., 2011; Labbe et al., 2019). No specific site on the activation loop was obviously more important than any other, and the effect of phosphorylation at multiple sites appeared to be additive (Labbe et al., 2019).
Modifications of IreK at T218 in the helical lobe exhibited reciprocal effects on phosphorylation of IreK, on IreK activity, as well as on cephalosporin resistance. In contrast to substitutions on the activation loop, phospho-ablative mutation of T218 to alanine resulted in increased phosphorylation of the kinase and increased cephalosporin resistance, while phospho-mimetic mutation of T218 to glutamate resulted in decreased phosphorylation of the kinase and a subsequent decrease in cephalosporin resistance. Analysis of combinatorial substitutions at both T218 and the activation loop indicated that phosphorylation at T218 serves to modulate the extent of phosphorylation on the activation loop, thereby regulating overall activity of the kinase. This data supports the idea that phosphorylation of T218 represents a negative feedback mechanism to regulate IreK activity. More studies are necessary to further explore the mechanism of T218 regulation of IreK activity in vivo.
Mechanism of IreK signal transduction
The prevailing model in the literature for signaling by PASTA kinases invokes ligand binding by the extracellular PASTA domain(s) to promote kinase dimerization. Upon dimerization, the kinases are proposed to autophosphorylate efficiently on their activation loops, resulting in kinase activation and subsequent phosphorylation of downstream substrates (Figure 2). The model is based in part on studies of PASTA kinase fragments in vitro where it has been demonstrated that dimerization of the kinase stabilizes the unit and enhances autophosphorylation (Gay et al., 2006; Greenstein et al., 2007; Lombana et al., 2010; Mieczkowski et al., 2008; Young et al., 2003) and mutations within those dimerization domains resulted in decreased substrate phosphorylation both in vitro and in vivo (Lombana et al., 2010). However, it is important to note that many aspects of this model have not been tested explicitly, especially using full-length PASTA kinases in live cells under physiological conditions. Our work on IreK has begun to define the mechanism of PASTA kinase signaling under physiologically relevant conditions.
Figure 2. Model for PASTA kinase activation and signal transduction.
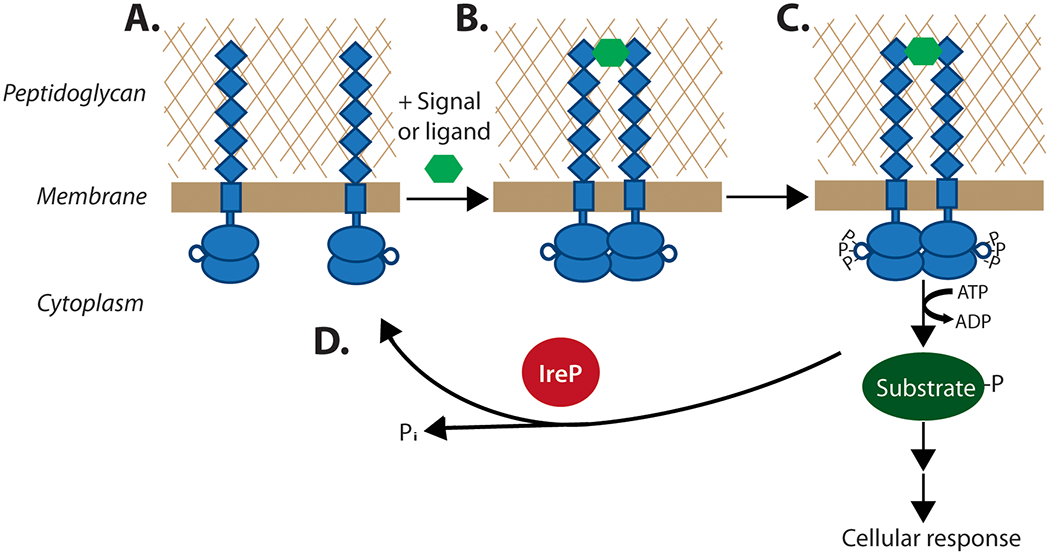
(A) In the absence of a signal, PASTA kinases are proposed to occupy an inactive state (i.e. unphosphorylated, monomeric). Upon binding to an extracellular ligand (hexagon), the kinase dimerizes (B) which results in autophosphorylation on the activation loops of intracellular kinase domains, activating the kinase for robust phosphorylation of downstream substrates (C) to elicit an adaptive biological response. The cognate phosphatase (IreP in E. faecalis) dephosphorylates IreK and phosphorylated substrates, shutting off the signaling cascade (D).
Labbe et al. (2017) analyzed phosphorylation and activation (i.e. substrate phosphorylation) of full-length IreK in live cells of E. faecalis. This study demonstrated that IreK is activated by extracellular stressors that perturb the integrity of the cell wall (i.e. antimicrobials) in vivo. Wild-type E. faecalis cells treated with cephalosporins, vancomycin, or lysozyme exhibited increased phosphorylation of IreK itself as well as an IreK substrate (IreB), consistent with the proposed model. Mutagenesis studies of IreK revealed that sites of phosphorylation on the activation loop of IreK, as well as presence of the extracellular PASTA domains, are required both for elevated IreK phosphorylation as well as enhanced phosphorylation of substrate to occur (Labbe et al., 2019; Labbe and Kristich, 2017). Moreover, a genetic defect that compromised cell wall integrity also led to activation of IreK. In contrast, treatment with an antimicrobial that does not affect cell wall synthesis did not result in activation of IreK. Hence, these results indicate that the mechanism of IreK signaling aligns with the proposed model, an important validation using full-length kinase in vivo. Ligand-dependent dimerization of IreK (or other PASTA kinases) has not yet been demonstrated in vivo, and remains one of the important untested features of the model shown in Figure 2.
Although PASTA kinases are key components required to respond to cell wall stress, the kinases do not function alone. Rather, the output of the signaling system is coordinately regulated in partnership with the cognate phosphatase. In enterococci, the cognate phosphatase directly regulates the activity of the kinase by dephosphorylating it, as demonstrated by in vitro assays with purified proteins as well as in vivo (Kristich et al., 2011; Sacco et al., 2014). Absence of the IreP phosphatase in E. faecalis cells leads to highly phosphorylated and overly active IreK (Labbe and Kristich, 2017), indicating there is not another phosphatase capable of dephosphorylating IreK. Absence of IreP leads to elevated resistance to antimicrobials, and to a marked loss of fitness relative to the wild-type in the absence of antimicrobials (Desbonnet et al., 2016; Kristich et al., 2011). Hence, unregulated IreK activity is detrimental to the cells, providing a rationale for the evolution of a tightly controlled signaling circuit. The phosphatase is also capable of dephosphorylating substrates that have been phosphorylated by IreK (Hall et al., 2013; Kellogg and Kristich, 2018; Kristich et al., 2011; Sacco et al., 2014) to terminate the biological response once it is no longer needed. Studies of PASTA kinases and their phosphatases in other bacteria support the hypothesis that an important role of the phosphatase is to regulate the activity of the kinase to ensure efficient functioning of this signaling system (Beltramini et al., 2009; Debarbouille et al., 2009; Ohlsen and Donat, 2010; Rajagopal et al., 2003). Whether (or how) the activity of the phosphatase itself is subject to regulation by environmental stimuli remains unknown. Our work (Kristich et al., 2011) suggested that the activity of IreP is modulated by IreK; in lysates from cells with a kinase in the “locked” active state, the IreP phosphatase activity observed was consistently lower, although a specific molecular mechanism to explain this effect has not been defined. In M. tuberculosis, there is evidence that the phosphatase can be directly phosphorylated by its cognate PASTA kinase (Sajid et al., 2011), but in that case the phosphorylation event enhanced the phosphatase activity instead of dampening it.
Substrates for phosphorylation by IreK
Numerous substrates have been described for PASTA kinases in the literature, which are involved in processes such as protein synthesis, cell division, cell wall synthesis, cell morphology, control of gene expression, and metabolism (Absalon et al., 2009; Hardt et al., 2017; Hirschfeld et al., 2019; Leiba et al., 2012; Lima et al., 2011; Lomas-Lopez et al., 2007; Novakova et al., 2010; Novakova et al., 2005; Pensinger et al., 2016; Pereira et al., 2015; Rajagopal et al., 2005). Often these substrates have been identified using in vitro approaches, and there is variability with respect to the extent that they have been validated as authentic physiological substrates in vivo. For enterococci, two PASTA kinase substrates have been described: IreB and CroS. IreB is a negative regulator of cephalosporin resistance, as its deletion results in increased cephalosporin resistance (Hall et al., 2013). IreK phosphorylates IreB both in vitro and in vivo, and absence of phosphorylation due to phospho-ablative substitutions in IreB or to the absence of IreK resulted in reduced cephalosporin resistance, indicating that IreK-mediated phosphorylation of IreB relieves the inhibitory effect of IreB to promote cephalosporin resistance. IreB forms a dimer in solution, and oligomerization appears to be required for its function (Hall et al., 2017). The specific biochemical mechanism by which IreB influences cephalosporin resistance in enterococci has not been defined, although a recent study indicated that an IreB homolog in Listeria monocytogenes regulates proteolytic degradation of a key enzyme in the PG synthesis pathway (Wamp et al., 2020).
IreK also phosphorylates CroS, the membrane-bound sensor kinase from a classical two-component signaling system that mediates responses to cell wall stress in enterococci (Comenge et al., 2003; Hancock and Perego, 2004; Kellogg and Kristich, 2016; Kellogg et al., 2017; Muller et al., 2018). CroS phosphorylates its cognate DNA-binding response regulator, CroR, to regulate gene expression in response to a variety of antibiotics that trigger cell wall stress (Comenge et al., 2003; Kellogg and Kristich, 2016, 2018; Kellogg et al., 2017). Our work revealed that IreK enhances CroS/R-mediated gene expression in response to cell wall stress (and influences cephalosporin resistance) by phosphorylating a threonine in the CroS kinase domain to boost CroS-mediated phosphorylation of CroR (Kellogg and Kristich, 2018). PASTA kinases have been shown to influence two-component signaling systems in other bacteria (Canova et al., 2014; Fridman et al., 2013; Hardt et al., 2017; Jers et al., 2011; Libby et al., 2015; Lin et al., 2009; Ulijasz et al., 2009), although in these cases typically it is the response regulator that is directly phosphorylated by the kinase rather than the sensor histidine kinase.
Gaps in knowledge
PASTA kinases are understudied signaling systems that regulate critical aspects of bacterial biology across a wide spectrum of diverse bacterial species, including antibiotic-resistant pathogens. Hence, there exists a compelling need to elucidate the underlying molecular mechanisms by which PASTA kinases monitor the condition or homeostasis of the bacterial cell wall to drive adaptive biological responses and preserve cell wall integrity and cell division. Several areas in need of further study are noted here.
Although we have validated several features of the prevailing model for PASTA kinase signaling, much remains to be learned about the molecular actions used by PASTA kinases to detect their stimuli and transmit that information across the membrane to trigger signaling inside the cell. For example, it is unclear what the physiological ligand(s) is that is recognized by IreK (PG fragments, lipid II, etc.?), or whether IreK recognizes multiple different ligands via distinct PASTA domains. The dynamics of the IreK PASTA module in vivo (i.e. the conformation(s) the PASTA module adopts in cells), has not been investigated. Moreover, it is unclear if, or how, IreK dimerization impacts signaling. We need to determine if IreK dimerizes at all, if it does so in a ligand-dependent manner, and if dimerization is indeed necessary for signaling. Beyond dimerization, investigation into whether IreK localizes to specific subcellular sites, and if that localization is dynamic or important for signaling is needed. Further study on the role of phosphorylation of IreK at sites other than the kinase activation loop are needed to decipher the role of these modifications, and more broadly the function of the juxtamembrane linker domain remains largely a mystery.
Given the critical requirement for careful tuning of PASTA kinase vs. phosphatase activity to maintain appropriate signaling output, regulation of phosphatase activity is another area that requires further investigation. Early studies suggest that the phosphatases may be regulated, but there is currently no clear evidence to suggest what controls phosphatase activity. The phosphatases are challenging to study due to the fact that they can dephosphorylate the PASTA kinases themselves (reducing kinase activity) but also dephosphorylate downstream substrates. Deconvoluting those effects from each other must be done to get a better understanding of how phosphatase regulation occurs and how that contributes to the overall regulation of the PASTA kinase signaling pathway.
Lastly, although we have identified 2 physiological substrates for phosphorylation by IreK in E. faecalis, our preliminary data suggest that there are likely to be additional substrates. Identifying those substrates and elucidating the functional effects of IreK-mediated phosphorylation on their activities will be necessary to fully understand the integrated role of IreK in enterococcal biology.
Acknowledgments
Funding Information
This work was supported in part by grants AI134660, AI128219, GM135256 and AI132927 from the National Institutes of Health (NIH). The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
The authors confirm that there are no conflicts of interest to disclose.
References
- Absalon C, Obuchowski M, Madec E, Delattre D, I.Holland B, and Seror JS (2009). CpgA, EF-Tu and the stressosome protein YezB are substrates of the Ser/Thr kinase/phosphatase couple, PrkC/PrpC, in Bacillus subtilis. Microbiology 155, 932–943. [DOI] [PubMed] [Google Scholar]
- Agarwal S, Agarwal S, Pancholi P, and Pancholi V (2011). Role of serine/threonine phosphatase (SP-STP) in Streptococcus pyogenes physiology and virulence. J Biol Chem 286, 41368–41380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akpata ES, and Blechman H (1982). Bacterial invasion of pulpal dentin wall in vitro. J Dent Res 61, 435–438. [DOI] [PubMed] [Google Scholar]
- Banla IL, Kommineni S, Hayward M, Rodrigues M, Palmer KL, Salzman NH, and Kristich CJ (2018). Modulators of Enterococcus faecalis Cell Envelope Integrity and Antimicrobial Resistance Influence Stable Colonization of the Mammalian Gastrointestinal Tract. Infect Immun 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu LD, Conrads G, Rehrauer H, Hussain H, Allan E, and van der Ploeg JR (2010). The Streptococcus mutans serine/threonine kinase, PknB, regulates competence development, bacteriocin production, and cell wall metabolism. Infect Immun 78, 2209–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthe P, Mukamolova GV, Roumestand C, and Cohen-Gonsaud M (2010). The structure of PknB extracellular PASTA domain from mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure 18, 606–615. [DOI] [PubMed] [Google Scholar]
- Beilharz K, Novakova L, Fadda D, Branny P, Massidda O, and Veening JW (2012). Control of cell division in Streptococcus pneumoniae by the conserved Ser/Thr protein kinase StkP. Proc Natl Acad Sci 109, E905–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramini AM, Mukhopadhyay CD, and Pancholi V (2009). Modulation of cell wall structure and antimicrobial susceptibility by a Staphylococcus aureus eukaryote-like serine/threonine kinase and phosphatase. Infect Immun 77, 1406–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhakdi S, Klonisch T, Nuber P, and Fischer W (1991). Stimulation of monokine production by lipoteichoic acids. Infect Immun 59, 4614–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhardwaj P, Hans A, Ruikar L, Guan Z, and Palmer KL (2017). Reduced Chlorhexidine and Daptomycin Susceptibility in Vancomycin-Resistant Enterococcus faecium after Serial Chlorhexidine Exposure. Antimicrob Agents Chemother 62, e01235–01217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitel B, Ortiz-Lombardia M, Duran R, Pompeo F, Cole ST, Cervenansky C, and Alzari PM (2003). PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol Microbiol 49, 1493–1508. [DOI] [PubMed] [Google Scholar]
- Brandl K, Plitas G, Mihu CN, Ubeda C, Jia T, Fleisher M, Schnabl B, DeMatteo RP, and Pamer EG (2008). Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature 455, 804–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant-Hudson KM, Shakir SM, and Ballard JD (2011). Autoregulatory characteristics of a Bacillus anthracis serine/threonine kinase. J Bacteriol 193, 1833–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugrysheva J, Froehlich BJ, Freiberg JA, and Scott JR (2011). Serine/threonine protein kinase Stk is required for virulence, stress response, and penicillin tolerance in Streptococcus pyogenes. Infect Immun 79, 4201–4209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnside K, Lembo A, de Los Reyes M, Iliuk A, Binhtran NT, Connelly JE, Lin WJ, Schmidt BZ, Richardson AR, Fang FC, et al. (2010). Regulation of hemolysin expression and virulence of Staphylococcus aureus by a serine/threonine kinase and phosphatase. PLoS One 5, e11071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvanese L, Falcigno L, Squeglia F, D’Auria G, and Berisio R (2017). PASTA in Penicillin Binding Proteins and Serine/Threonine Kinases: A Recipe of Structural, Dynamic and Binding Properties. Curr Med Chem 24, 4038–4056. [DOI] [PubMed] [Google Scholar]
- Cameron DR, Ward DV, Kostoulias X, Howden BP, Moellering RC Jr., Eliopoulos GM, and Peleg AY (2012). Serine/threonine phosphatase Stp1 contributes to reduced susceptibility to vancomycin and virulence in Staphylococcus aureus. J Infect Dis 205, 1677–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canova MJ, Baronian G, Brelle S, Cohen-Gonsaud M, Bischoff M, and Molle V (2014). A novel mode of regulation of the Staphylococcus aureus Vancomycin-resistance-associated response regulator VraR mediated by Stk1 protein phosphorylation. Biochem Biophys Res Commun 447, 165–171. [DOI] [PubMed] [Google Scholar]
- Carmeli Y, Eliopoulos MG, and Samore HM (2002). Antecedent Treatment with Different Antibiotic Agents as a Risk Factor for Vancomycin Resistant Enterococcus. Emerg Infecti Dis 8, 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC (2019). Antibiotic Resistance Threats in the United States, 2019. US Department of Health and Human Services, CDC. [Google Scholar]
- Chaba R, Raje M, and Chakraborti PK (2002). Evidence that a eukaryotic-type serine/threonine protein kinase from Mycobacterium tuberculosis regulates morphological changes associated with cell division. Eur J Biochem 269, 1078–1085. [DOI] [PubMed] [Google Scholar]
- Chawla Y, Upadhyay S, Khan S, Nagarajan SN, Forti F, and Nandicoori VK (2014). Protein kinase B (PknB) of Mycobacterium tuberculosis is essential for growth of the pathogen in vitro as well as for survival within the host. J Biol Chem 289, 13858–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comenge Y, Quintiliani R Jr., Li L, Dubost L, Brouard JP, Hugonnet JE, and Arthur M (2003). The CroRS two-component regulatory system is required for intrinsic beta-lactam resistance in Enterococcus faecalis. J Bacteriol 185, 7184–7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenot E, Garcia-Garcia T, Douche T, Gorgette O, Courtin P, Denis-Quanquin S, Tremblay YDN, Hoys S, Candela T, and Martin-Verstraet I (2019). The Ser/Thr Kinase PrkC Participates in Cell Wall Homeostasis and Antimicrobial Resistance in Clostridium difficile. Infect Immun 87, e00005–00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debarbouille M, Dramsi S, Dussurget O, Nahori MA, Vaganay E, Jouvion G, Cozzone A, Msadek T, and Duclos B (2009). Characterization of a serine/threonine kinase involved in virulence of Staphylococcus aureus. J Bacteriol 191, 4070–4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desbonnet C, Tait-Kamradt A, Garcia-Solache M, Dunman P, Coleman J, Arthur M, and Rice LB (2016). Involvement of the Eukaryote-Like Kinase-Phosphatase System and a Protein That Interacts with Penicillin-Binding Protein 5 in Emergence of Cephalosporin Resistance in Cephalosporin-Sensitive Class A Penicillin-Binding Protein Mutants in Enterococcus faecium. mBio 7, e02188–02115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donskey CJ, Chowdhry TK, Hecker MT, Hoyen CK, Hanrahan JA, Hujer AM, Hutton-Thomas RA, Whalen CC, Bonomo RA, and Rice LB (2000). Effect of antibiotic therapy on the density of vancomycin-resistant enterococci in the stool of colonized patients. N Engl J Med 343, 1925–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran R, Villarino A, Bellinzoni M, Wehenkel A, Fernandez P, Boitel B, Cole ST, Alzari PM, and Cervenansky C (2005). Conserved autophosphorylation pattern in activation loops and juxtamembrane regions of Mycobacterium tuberculosis Ser/Thr protein kinases. Biochem Biophys Res Commun 333, 858–867. [DOI] [PubMed] [Google Scholar]
- Estrela C, Silva JA, Gonçalves de Alencar AH, Leles CR, and Decurcio DA (2008). Efficacy of sodium hypochlorite and chlorhexidine against Enterococcus faecalis -a systematic review. J Appl Oral Sci 16, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez P, Saint-Joanis B, Barilone N, Jackson M, Gicquel B, Cole ST, and Alzari PM (2006). The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J Bacteriol 188, 7778–7784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari PHP, Cai S, and Bombana AC (2005). Effect of endodontic procedures on enterococci, enteric bacteria and yeasts in primary endodontic infections. Int Endod J 38, 372–380. [DOI] [PubMed] [Google Scholar]
- Fleurie A, Manuse S, Zhao C, Campo N, Cluzel C, Lavergne JP, Freton C, Combet C, Guiral S, Soufi B, et al. (2014). Interplay of the serine/threonine-kinase StkP and the paralogs DivIVA and GpsB in pneumococcal cell elongation and division. PLoS Genet 10, e1004275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman M, Williams GD, Muzamal U, Hunter H, Siu KW, and Golemi-Kotra D (2013). Two unique phosphorylation-driven signaling pathways crosstalk in Staphylococcus aureus to modulate the cell-wall charge: Stk1/Stp1 meets GraSR. Biochem 52, 7975–7986. [DOI] [PubMed] [Google Scholar]
- Gadea R, Fernandez Fuentes MA, Perez Pulido R, Galvez A, and Ortega E (2017a). Effects of exposure to quaternary-ammonium-based biocides on antimicrobial susceptibility and tolerance to physical stresses in bacteria from organic foods. Food Microbiol 63, 58–71. [DOI] [PubMed] [Google Scholar]
- Gadea R, Glibota N, Pérez Pulido R, Gálvez A, and Ortega E (2017b). Adaptation to Biocides Cetrimide and Chlorhexidine in Bacteria from Organic Foods: Association with Tolerance to Other Antimicrobials and Physical Stresses. J Agric Food Chem 65, 1758–1770. [DOI] [PubMed] [Google Scholar]
- Gaidenko TA, Kim TJ, and Price CW (2002). The PrpC serine-threonine phosphatase and PrkC kinase have opposing physiological roles in stationary-phase Bacillus subtilis cells. J Bacteriol 184, 6109–6114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gay LM, Ng HL, and Alber T (2006). A conserved dimer and global conformational changes in the structure of apo-PknE Ser/Thr protein kinase from Mycobacterium tuberculosis. J Mol Biol 360, 409–420. [DOI] [PubMed] [Google Scholar]
- Giefing C, Jelencsics KE, Gelbmann D, Senn BM, and Nagy E (2010). The pneumococcal eukaryotic-type serine/threonine protein kinase StkP co-localizes with the cell division apparatus and interacts with FtsZ in vitro. Microbiol 156, 1697–1707. [DOI] [PubMed] [Google Scholar]
- Giefing C, Meinke AL, Hanner M, Henics T, Bui MD, Gelbmann D, Lundberg U, Senn BM, Schunn M, Habel A, et al. (2008). Discovery of a novel class of highly conserved vaccine antigens using genomic scale antigenic fingerprinting of pneumococcus with human antibodies. J Exp Med 205, 117–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes BPFA, Pinheiro ET, Jacinto RC, Zaia AA, Ferraz CCR, and Souza-Filho FJ (2008). Microbial analysis of canals of root-filled teeth with periapical lesions using polymerase chain reaction. J Endod 34, 537–540. [DOI] [PubMed] [Google Scholar]
- Greenstein AE, Echols N, Lombana TN, King DS, and Alber T (2007). Allosteric activation by dimerization of the PknD receptor Ser/Thr protein kinase from Mycobacterium tuberculosis. J Biol Chem 282, 11427–11435. [DOI] [PubMed] [Google Scholar]
- Haapasalo M, and Orstavik D (1987). In vitro infection and disinfection of dentinal tubules. J Dent Res 66, 1375–1379. [DOI] [PubMed] [Google Scholar]
- Hall CL, Lytle BL, Jensen D, Hoff JS, Peterson FC, Volkman BF, and Kristich CJ (2017). Structure and Dimerization of IreB, a Negative Regulator of Cephalosporin Resistance in Enterococcus faecalis. J Mol Biol 429, 2324–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CL, Tschannen M, Worthey EA, and Kristich CJ (2013). IreB, a Ser/Thr kinase substrate, influences antimicrobial resistance in Enterococcus faecalis. Antimicrob Agents Chemother 57, 6179–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancock III HH, Sigurdsson A, Trope M, and Moiseiwitsch J (2001). Bacteria isolated after unsuccessful endodontic treatment in a North American population. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 91, 579–586. [DOI] [PubMed] [Google Scholar]
- Hancock LE, and Perego M (2004). Systematic inactivation and phenotypic characterization of two-component signal transduction systems of Enterococcus faecalis V583. J Bacteriol 186, 7951–7958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, and Hunter T (1995). Protein kinases 6. The eukaryotic protein kinase superfamily: kinase (catalytic) domain structure and classification. FASEB 9, 576–596. [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, and Hunter T (1988). The Protein Kinase Family: Conserved Features and Deduced Phylogeny of the Catalytic Domains. Science 241, 42–52. [DOI] [PubMed] [Google Scholar]
- Hardt P, Engels I, Rausch M, Gajdiss M, Ulm H, Sass P, Ohlsen K, Sahl HG, Bierbaum G, Schneider T, et al. (2017). The cell wall precursor lipid II acts as a molecular signal for the Ser/Thr kinase PknB of Staphylococcus aureus. Int J Med Microbiol 307, 1–10. [DOI] [PubMed] [Google Scholar]
- Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, Pollock DA, and Fridkin SK (2008). NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006-2007. Infect Control and Hosp Epidemiol 29, 996–1011. [DOI] [PubMed] [Google Scholar]
- Hirschfeld C, Gomez-Mejia A, Bartel J, Hentschker C, Rohde M, Maass S, Hammerschmidt S, and Becher D (2019). Proteomic Investigation Uncovers Potential Targets and Target Sites of Pneumococcal Serine-Threonine Kinase StkP and Phosphatase PhpP. Front Microbiol 10, 3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck BL, and Rice LB (2012). Intrinsic and acquired resistance mechanisms in enterococcus. Virulence 3, 421–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubble TS, Hatton JF, Nallapareddy SR, Murray BE, and Gillespie MJ (2003). Influence of Enterococcus faecalis proteases and the collagen-binding protein, Ace, on adhesion to dentin. Oral Microbiol Immunol 18, 121–126. [DOI] [PubMed] [Google Scholar]
- Hussain H, Branny P, and Allan E (2006). A eukaryotic-type serine/threonine protein kinase is required for biofilm formation, genetic competence, and acid resistance in Streptococcus mutans. J Bacteriol 188, 1628–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huycke MM, Abrams V, and Moore DR (2002). Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis 23, 529–536. [DOI] [PubMed] [Google Scholar]
- Huycke MM, Joyce W, and Wack MF (1996). Augmented production of extracellular superoxide by blood isolates of Enterococcus faecalis. J Infect Dis 173, 743–746. [DOI] [PubMed] [Google Scholar]
- Huycke MM, Moore D, Joyce W, Wise P, Shepard L, Kotake Y, and Gilmore MS (2001). Extracellular superoxide production by Enterococcus faecalis requires demethylmenaquinone and is attenuated by functional terminal quinol oxidases. Mol Microbiol 42, 729–740. [DOI] [PubMed] [Google Scholar]
- Huycke MM, and Moore DR (2002). In vivo production of hydroxyl radical by Enterococcus faecalis colonizing the intestinal tract using aromatic hydroxylation. 33, 818–826. [DOI] [PubMed] [Google Scholar]
- Janczarek M, Vinardell JM, Lipa P, and Karas M (2018). Hanks-Type Serine/Threonine Protein Kinases and Phosphatases in Bacteria: Roles in Signaling and Adaptation to Various Environments. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarick M, Bertsche U, Stahl M, Schultz D, Methling K, Lalk M, Stigloher C, Steger M, Schlosser A, and Ohlsen K (2018). The serine/threonine kinase Stk and the phosphatase Stp regulate cell wall synthesis in Staphylococcus aureus. Sci Rep 8, 13693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jers C, Kobir A, Sondergaard EO, Jensen PR, and Mijakovic I (2011). Bacillus subtilis two-component system sensory kinase DegS is regulated by serine phosphorylation in its input domain. PLoS One 6, e14653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, and Pancholi V (2006). Identification and biochemical characterization of a eukaryotic-type serine/threonine kinase and its cognate phosphatase in Streptococcus pyogenes: their biological functions and substrate identification. J Mol Biol 357, 1351–1372. [DOI] [PubMed] [Google Scholar]
- Jones G, and Dyson P (2006). Evolution of transmembrane protein kinases implicated in coordinating remodeling of gram-positive peptidoglycan: inside versus outside. J Bacteriol 188, 7470–7476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampf G (2019). Antibiotic ResistanceCan Be Enhanced in Gram-Positive Species by Some Biocidal Agents Used for Disinfection. Antibiotics (Basel) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang CM, Abbott DW, Park ST, Dascher CC, Cantley LC, and Husson RN (2005). The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev 19, 1692–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur P, Rausch M, Malakar B, Watson U, Damle NP, Chawla Y, Srinivasan S, Sharma K, Schneider T, Jhingan GD, et al. (2019). LipidII interaction with specific residues of Mycobacterium tuberculosis PknB extracytoplasmic domain governs its optimal activation. Nat Commun 10, 1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg SL, and Kristich CJ (2016). Functional Dissection of the CroRS Two-Component System Required for Resistance to Cell Wall Stressors in Enterococcus faecalis. J Bacteriol 198, 1326–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg SL, and Kristich CJ (2018). Convergence of PASTA Kinase and Two-Component Signaling in Response to Cell Wall Stress in Enterococcus faecalis. J Bacteriol 200, e00086–00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg SL, Little JL, Hoff JS, and Kristich CJ (2017). Requirement of the CroRS Two-Component System for Resistance to Cell Wall-Targeting Antimicrobials in Enterococcus faecium. Antimicrob Agents Chemother 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komiyama EY, Lepesqueur LS, Yassuda CG, Samaranayake LP, Parahitiyawa NB, Balducci I, and Koga-Ito CY (2016). Enterococcus Species in the Oral Cavity: Prevalence, Virulence Factors and Antimicrobial Susceptibility. PLoS One 11, e0163001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouidhi B, Zmantar T, Mahdouani K, Hentati H, and Bakhrouf A (2011). Antibiotic resistance and adhesion properties of oral Enterococci associated to dental caries. BMC Microbiol 11, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski WJ, Kasper EL, Hatton JF, Murray BE, Nallapareddy SR, and Gillespie MJ (2006). Enterococcus faecalis adhesin, Ace, mediates attachment to particulate dentin. J Endod 32, 634–637. [DOI] [PubMed] [Google Scholar]
- Kristich CJ, Little JL, Hall CL, and Hoff JS (2011). Reciprocal regulation of cephalosporin resistance in Enterococcus faecalis. mBio 2, e00199–00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristich CJ, Wells CL, and Dunny GM (2007). A eukaryotic-type Ser/Thr kinase in Enterococcus faecalis mediates antimicrobial resistance and intestinal persistence. PNAS 104, 3508–3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupa A, and Srinivasan N (2005). Diversity in domain architectures of Ser/Thr kinases and their homologues in prokaryotes. BMC Genomics 6, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe BD, Hall CL, Kellogg SL, Chen Y, Koehn O, Pickrum AM, Mirza SP, and Kristich CJ (2019). Reciprocal Regulation of PASTA Kinase Signaling by Differential Modification. J Bacteriol 201, e00016–00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe BD, and Kristich CJ (2017). Growth- and Stress-Induced PASTA Kinase Phosphorylation in Enterococcus faecalis. J Bacteriol 199, e00363–00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M, Hesek D, Shah IM, Oliver AG, Dworkin J, and Mobashery S (2010). Synthetic peptidoglycan motifs for germination of bacterial spores. Chembiochem 11, 2525–2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiba J, Hartmann T, Cluzel ME, Cohen-Gonsaud M, Delolme F, Bischoff M, and Molle V (2012). A novel mode of regulation of the Staphylococcus aureus catabolite control protein A (CcpA) mediated by Stk1 protein phosphorylation. J Biol Chem 287, 43607–43619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby EA, Goss LA, and Dworkin J (2015). The Eukaryotic-Like Ser/Thr Kinase PrkC Regulates the Essential WalRK Two-Component System in Bacillus subtilis. PLoS Genet 11, e1005275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima A, Duran R, Schujman GE, Marchissio MJ, Portela MM, Obal G, Pritsch O, de Mendoza D, and Cervenansky C (2011). Serine/threonine protein kinase PrkA of the human pathogen Listeria monocytogenes: biochemical characterization and identification of interacting partners through proteomic approaches. J Proteomics 74, 1720–1734. [DOI] [PubMed] [Google Scholar]
- Lin WJ, Walthers D, Connelly JE, Burnside K, Jewell KA, Kenney LJ, and Rajagopal L (2009). Threonine phosphorylation prevents promoter DNA binding of the Group B Streptococcus response regulator CovR. Mol Microbiol 71, 1477–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomas-Lopez R, Paracuellos P, Riberty M, Cozzone AJ, and Duclos B (2007). Several enzymes of the central metabolism are phosphorylated in Staphylococcus aureus. FEMS Microbiol Lett 272, 35–42. [DOI] [PubMed] [Google Scholar]
- Lombana TN, Echols N, Good MC, Thomsen ND, Ng HL, Greenstein AE, Falick AM, King DS, and Alber T (2010). Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknB. Structure 18, 1667–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lougheed KE, Osborne SA, Saxty B, Whalley D, Chapman T, Bouloc N, Chugh J, Nott TJ, Patel D, Spivey VL, et al. (2011). Effective inhibitors of the essential kinase PknB and their potential as anti-mycobacterial agents. Tuberculosis (Edinb) 91, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love RM (2001). Enterococcus faecalis--a mechanism for its role in endodontic failure. Int Endod J 34, 399–405. [DOI] [PubMed] [Google Scholar]
- Madec E, Laszkiewicz A, Iwanicki A, Obuchowski M, and Séror S (2002). Characterization of a membrane-linked Ser/Thr protein kinase in Bacillus subtilis, implicated in developmental processes. Mol Microbiol 46, 571–586. [DOI] [PubMed] [Google Scholar]
- Madec E, Stensballe A, Kjellstrom S, Cladiere L, Obuchowski M, Jensen ON, and Seror SJ (2003). Mass spectrometry and site-directed mutagenesis identify several autophosphorylated residues required for the activity of PrkC, a Ser/Thr kinase from Bacillus subtilis. J Mol Biol 330, 459–472. [DOI] [PubMed] [Google Scholar]
- Maestro B, Novakova L, Hesek D, Lee M, Leyva E, Mobashery S, Sanz JM, and Branny P (2011). Recognition of peptidoglycan and beta-lactam antibiotics by the extracellular domain of the Ser/Thr protein kinase StkP from Streptococcus pneumoniae. FEBS Lett 585, 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton IJ, Balla G, Hegedus C, Redi P, Szilagyi Z, Karmazsin L, and Kiss C (1993). The role of reactive oxygen intermediates in the pathogenesis of chronic apical periodontitis. Oral Microbiol Immunol 8, 254–257. [DOI] [PubMed] [Google Scholar]
- Mieczkowski C, Iavarone AT, and Alber T (2008). Auto-activation mechanism of the Mycobacterium tuberculosis PknB receptor Ser/Thr kinase. EMBO J 27, 3186–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WR, Munita JM, and Arias CA (2014). Mechanisms of antibiotic resistance in enterococci. Expert Rev Anti Infect Ther 12, 1221–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller WR, Murray BE, Rice LB, and Arias CA (2016). Vancomycin-Resistant Enterococci: Therapeutic Challenges in the 21st Century. Infect Dis Clin North Am 30, 415–439. [DOI] [PubMed] [Google Scholar]
- Mir M, Asong J, Li X, Cardot J, Boons GJ, and Husson RN (2011). The extracytoplasmic domain of the Mycobacterium tuberculosis Ser/Thr kinase PknB binds specific muropeptides and is required for PknB localization. PLoS Pathog 7, e1002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molander A, Reit C, Dahlén G, and Kvist T (1998). Microbiological status of root-filled teeth with apical periodontitis. Int Endod J 31, 1–7. [PubMed] [Google Scholar]
- Morlot C, Bayle L, Jacq M, Fleurie A, Tourcier G, Galisson F, Vernet T, Grangeasse C, and Di Guilmi AM (2013). Interaction of Penicillin-Binding Protein 2x and Ser/Thr protein kinase StkP, two key players in Streptococcus pneumoniae R6 morphogenesis. Mol Microbiol 90, 88–102. [DOI] [PubMed] [Google Scholar]
- Muller C, Massier S, Le Breton Y, and Rince A (2018). The role of the CroR response regulator in resistance of Enterococcus faecalis to D-cycloserine is defined using an inducible receiver domain. Mol Microbiol 107, 416–427. [DOI] [PubMed] [Google Scholar]
- Nallapareddy SR, and Murray BE (2008). Role played by serum, a biological cue, in the adherence of Enterococcus faecalis to extracellular matrix proteins, collagen, fibrinogen, and fibronectin. J Infect Dis 197, 1728–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolen B, Taylor S, and Ghosh G (2004). Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell 15, 661–675. [DOI] [PubMed] [Google Scholar]
- Novakova L, Bezouskova S, Pompach P, Spidlova P, Saskova L, Weiser J, and Branny P (2010). Identification of multiple substrates of the StkP Ser/Thr protein kinase in Streptococcus pneumoniae. J Bacteriol 192, 3629–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakova L, Saskova L, Pallova P, Janecek J, Novotna J, Ulrych A, Echenique J, Trombe MC, and Branny P (2005). Characterization of a eukaryotic type serine/threonine protein kinase and protein phosphatase of Streptococcus pneumoniae and identification of kinase substrates. FEBS J 272, 1243–1254. [DOI] [PubMed] [Google Scholar]
- Ohlsen K, and Donat S (2010). The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int J Med Microbiol 300, 137–141. [DOI] [PubMed] [Google Scholar]
- Orstavik D, and Haapasalo M (1990). Disinfection by endodontic irrigants and dressings of experimentally infected dentinal tubules. Endod Dent Traumatol 6, 142–149. [DOI] [PubMed] [Google Scholar]
- Paracuellos P, Ballandras A, Robert X, Kahn R, Herve M, Mengin-Lecreulx D, Cozzone AJ, Duclos B, and Gouet P (2010). The extended conformation of the 2.9-A crystal structure of the three-PASTA domain of a Ser/Thr kinase from the human pathogen Staphylococcus aureus. J Mol Biol 404, 847–858. [DOI] [PubMed] [Google Scholar]
- Peciuliene V, Balciuniene I, Eriksen HM, and Haapasalo M (2000). Isolation of Enterococcus faecalis in previously root-filled canals in a Lithuanian population. J Endod 26. [DOI] [PubMed] [Google Scholar]
- Peciuliene V, Reynaud AH, Balciuniene I, and Haapasalo M (2001). Isolation of yeasts and enteric bacteria in root-filled teeth with chronic apical periodontitis. Int Endod J 34, 429–434. [DOI] [PubMed] [Google Scholar]
- Pensinger DA, Aliota MT, Schaenzer AJ, Boldon KM, Ansari IU, Vincent WJ, Knight B, Reniere ML, Striker R, and Sauer JD (2014). Selective pharmacologic inhibition of a PASTA kinase increases Listeria monocytogenes susceptibility to beta-lactam antibiotics. Antimicrob Agents Chemother 58, 4486–4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensinger DA, Boldon KM, Chen GY, Vincent WJ, Sherman K, Xiong M, Schaenzer AJ, Forster ER, Coers J, Striker R, et al. (2016). The Listeria monocytogenes PASTA Kinase PrkA and Its Substrate YvcK Are Required for Cell Wall Homeostasis, Metabolism, and Virulence. PLoS Pathog 12, e1006001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira SF, Gonzalez RL Jr., and Dworkin J (2015). Protein synthesis during cellular quiescence is inhibited by phosphorylation of a translational elongation factor. Proc Natl Acad Sci U S A 112, E3274–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira SF, Goss L, and Dworkin J (2011). Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol Mol Biol Rev 75, 192–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro ET, Gomes BPFA, Ferraz CCR, Teixeira FB, Zaia AA, and Souza Filho FJ (2003). Evaluation of root canal microorganisms isolated from teeth with endodontic failure and their antimicrobial susceptibility. Oral Microbiol Immunol 18, 100–103. [DOI] [PubMed] [Google Scholar]
- Pompeo F, Byrne D, Mengin-Lecreulx D, and Galinier A (2018). Dual regulation of activity and intracellular localization of the PASTA kinase PrkC during Bacillus subtilis growth. Sci Rep 8, 1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pompeo F, Foulquier E, Serrano B, Grangeasse C, and Galinier A (2015). Phosphorylation of the cell division protein GpsB regulates PrkC kinase activity through a negative feedback loop in Bacillus subtilis. Mol Microbiol 97, 139–150. [DOI] [PubMed] [Google Scholar]
- Prigozhin DM, Papavinasasundaram KG, Baer CE, Murphy KC, Moskaleva A, Chen TY, Alber T, and Sassetti CM (2016). Structural and Genetic Analyses of the Mycobacterium tuberculosis Protein Kinase B Sensor Domain Identify a Potential Ligand-binding Site. J Biol Chem 291, 22961–22969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal L, Clancy A, and Rubens CE (2003). A eukaryotic type serine/threonine kinase and phosphatase in Streptococcus agalactiae reversibly phosphorylate an inorganic pyrophosphatase and affect growth, cell segregation, and virulence. J Biol Chem 278, 14429–14441. [DOI] [PubMed] [Google Scholar]
- Rajagopal L, Vo A, Silvestroni A, and Rubens CE (2005). Regulation of purine biosynthesis by a eukaryotic-type kinase in Streptococcus agalactiae. Mol Microbiol 56, 1329–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rams TE, Feik D, Mortensen JE, Degener JE, and van Winkelhoff AJ (2013). Antibiotic susceptibility of periodontal Enterococcus faecalis. J Periodontol 84, 1026–1033. [DOI] [PubMed] [Google Scholar]
- Righino B, Galisson F, Pirolli D, Vitale S, Rety S, Gouet P, and De Rosa MC (2018). Structural model of the full-length Ser/Thr protein kinase StkP from S. pneumoniae and its recognition of peptidoglycan fragments. J Biomol Struct Dyn 36, 3666–3679. [DOI] [PubMed] [Google Scholar]
- Rôças IN, Jung I-Y, Lee C-Y, and Siqueira JF Jr (2004a). Polymerase chain reaction identification of microorganisms in previously root-filled teeth in a South Korean population. J Endod 30, 504–508. [PubMed] [Google Scholar]
- Rôças IN, Siqueira JF Jr, and Santos KRN (2004b). Association of Enterococcus faecalis with different forms of periradicular diseases. J Endod 30, 315–320. [DOI] [PubMed] [Google Scholar]
- Roumestand C, Leiba J, Galophe N, Margeat E, Padilla A, Bessin Y, Barthe P, Molle V, and Cohen-Gonsaud M (2011). Structural insight into the Mycobacterium tuberculosis Rv0020c protein and its interaction with the PknB kinase. Structure 19, 1525–1534. [DOI] [PubMed] [Google Scholar]
- Rued BE, Zheng JJ, Mura A, Tsui HT, Boersma MJ, Mazny JL, Corona F, Perez AJ, Fadda D, Doubravova L, et al. (2017). Suppression and synthetic-lethal genetic relationships of DeltagpsB mutations indicate that GpsB mediates protein phosphorylation and penicillin-binding protein interactions in Streptococcus pneumoniae D39. Mol Microbiol 103, 931–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero A, De Simone P, Smaldone G, Squeglia F, and Berisio R (2012). Bacterial cell division regulation by Ser/Thr kinases: a structural perspective. Curr Protein and Pept Sci 13, 756–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacco E, Cortes M, Josseaume N, Rice LB, Mainardi JL, and Arthur M (2014). Serine/threonine protein phosphatase-mediated control of the peptidoglycan cross-linking L,D-transpeptidase pathway in Enterococcus faecium. mBio 5, e01446–01414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sajid A, Arora G, Gupta M, Upadhyay S, Nandicoori VK, and Singh Y (2011). Phosphorylation of Mycobacterium tuberculosis Ser/Thr phosphatase by PknA and PknB. PLoS One 6, e17871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaenzer AJ, Wlodarchak N, Drewry DH, Zuercher WJ, Rose WE, Ferrer CA, Sauer J-D, and Striker R (2018). GW779439X and Its Pyrazolopyridazine Derivatives Inhibit the Serine/Threonine Kinase Stk1 and Act As Antibiotic Adjuvants against β-Lactam-Resistant Staphylococcus aureus. ACS Infect Dis 4, 1508–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaenzer AJ, Wlodarchak N, Drewry DH, Zuercher WJ, Rose WE, Striker R, and Sauer JD (2017). A screen for kinase inhibitors identifies antimicrobial imidazopyridine aminofurazans as specific inhibitors of the Listeria monocytogenes PASTA kinase PrkA. J Biol Chem 292, 17037–17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirrmeister JF, Liebenow A-L, Pelz K, Wittmer A, Serr A, Hellwig E, and Al-Ahmad A (2009). New bacterial compositions in root-filled teeth with periradicular lesions. J Endod 35, 169–174. [DOI] [PubMed] [Google Scholar]
- Sedgley C, Buck G, and Appelbe O (2006). Prevalence of Enterococcus faecalis at multiple oral sites in endodontic patients using culture and PCR. J Endod 32, 104–109. [DOI] [PubMed] [Google Scholar]
- Sedgley CM, Lennan SL, and Clewell DB (2004). Prevalence, phenotype and genotype of oral enterococci. Oral Microbiol Immunol 19, 95–101. [DOI] [PubMed] [Google Scholar]
- Sedgley CM, Molander A, Flannagan SE, Nagel AC, Appelbe OK, Clewell DB, and Dahlén G (2005). Virulence, phenotype and genotype characteristics of endodontic Enterococcus spp. Oral Microbiol Immunol 20, 10–19. [DOI] [PubMed] [Google Scholar]
- Shah IM, Laaberki MH, Popham DL, and Dworkin J (2008). A eukaryotic-like Ser/Thr kinase signals bacteria to exit dormancy in response to peptidoglycan fragments. Cell 135, 486–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakir SM, Bryant KM, Larabee JL, Hamm EE, Lovchik J, Lyons CR, and Ballard JD (2010). Regulatory interactions of a virulence-associated serine/threonine phosphatase-kinase pair in Bacillus anthracis. J Bacteriol 192, 400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma AK, Arora D, Singh LK, Gangwal A, Sajid A, Molle V, Singh Y, and Nandicoori VK (2016). Serine/Threonine Protein Phosphatase PstP of Mycobacterium tuberculosis Is Necessary for Accurate Cell Division and Survival of Pathogen. J Biol Chem 291, 24215–24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard BD, and Gilmore MS (2002). Antibiotic-resistant enterococci: the mechanisms and dynamics of drug introduction and resistance. Microbes Infect 4, 215–224. [DOI] [PubMed] [Google Scholar]
- Shi Y (2009). Serine/threonine phosphatases: mechanism through structure. Cell 139, 468–484. [DOI] [PubMed] [Google Scholar]
- Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S, and Facilities, N.H.S.N.N.T.a.P.N. (2013). Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009–2010. Infect Control Hosp Epidemiol 34, 1–14. [DOI] [PubMed] [Google Scholar]
- Sipert CR, Moraes IG, Bernardinelli N, Garcia RB, Bramante CM, Gasparoto TH, Figueira EA, Dionísio TJ, Campanelli AP, Oliveira SHP, et al. (2010). Heat-killed Enterococcus faecalis alters nitric oxide and CXCL12 production but not CXCL8 and CCL3 production by cultured human dental pulp fibroblasts. J Endod 36, 91–94. [DOI] [PubMed] [Google Scholar]
- Siqueira JF Jr, and Rôças IN (2004). Polymerase chain reaction-based analysis of microorganisms associated with failed endodontic treatment. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 97, 85–94. [DOI] [PubMed] [Google Scholar]
- Squeglia F, Marchetti R, Ruggiero A, Lanzetta R, Marasco D, Dworkin J, Petoukhov M, Molinaro A, Berisio R, and Silipo A (2011). Chemical basis of peptidoglycan discrimination by PrkC, a key kinase involved in bacterial resuscitation from dormancy. J Am Chem Soc 133, 20676–20679. [DOI] [PubMed] [Google Scholar]
- Stancik IA, Sestak MS, Ji B, Axelson-Fisk M, Franjevic D, Jers C, Domazet-Loso T, and Mijakovic I (2018). Serine/Threonine Protein Kinases from Bacteria, Archaea and Eukarya Share a Common Evolutionary Origin Deeply Rooted in the Tree of Life. J Mol Biol 430, 27–32. [DOI] [PubMed] [Google Scholar]
- Stuart CH, Schwartz SA, Beeson TJ, and Owatz CB (2006). Enterococcus faecalis: its role in root canal treatment failure and current concepts in retreatment. J Endod 32, 93–98. [DOI] [PubMed] [Google Scholar]
- Sundqvist G (1992). Ecology of the root canal flora. J Endod 18, 427–430. [DOI] [PubMed] [Google Scholar]
- Sundqvist G, Figdor D, Persson S, and Sjögren U (1998). Microbiologic analysis of teeth with failed endodontic treatment and the outcome of conservative re-treatment Oral Surg Oral Med Oral Pathol Oral Radiol Endod 85, 86–93. [DOI] [PubMed] [Google Scholar]
- Tennert C, Fuhrmann M, Wittmer A, Karygianni L, Altenburger MJ, Pelz K, Hellwig E, and Al-Ahmad A (2014). New bacterial composition in primary and persistent/secondary endodontic infections with respect to clinical and radiographic findings. J Endod 40, 670–677. [DOI] [PubMed] [Google Scholar]
- Ubeda C, Taur Y, Jenq RR, Equinda MJ, Son T, Samstein M, Viale A, Socci ND, van den Brink MR, Kamboj M, et al. (2010). Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J Clin Invest 120, 4332–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulijasz AT, Falk SP, and Weisblum B (2009). Phosphorylation of the RitR DNA-binding domain by a Ser-Thr phosphokinase: implications for global gene regulation in the streptococci. Mol Microbiol 71, 382–390. [DOI] [PubMed] [Google Scholar]
- Viswanathan G, Yadav S, Joshi SV, and Raghunand TR (2017). Insights into the function of FhaA, a cell division-associated protein in mycobacteria. FEMS Microbiol Lett 364. [DOI] [PubMed] [Google Scholar]
- Wamp S, Rutter ZJ, Rismondo J, Jennings CE, Möller L, Lewis RJ, and Halbedel S (2020). PrkA controls peptidoglycan biosynthesis through the essential phosphorylation of ReoM. eLife 9, e56048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Marchettic R, Prisic S, Ishii K, Arai Y, Ohta I, Inuki S, Uchiyama S, Silipo A, Molinaro A, et al. (2017a). A Comprehensive Study of the Interaction between Peptidoglycan Fragments and the Extracellular Domain of Mycobacterium tuberculosis Ser/Thr Kinase PknB. Chembiochem 18, 2094–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QQ, Zhang CF, Chu CH, and Zhu XF (2012). Prevalence of Enterococcus faecalis in saliva and filled root canals of teeth associated with apical periodontitis. Int J Oral Sci 4, 19–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Bemis G, Hanzelka B, Zuccola H, Wynn M, Moody CS, Green J, Locher C, Liu A, Gao H, et al. (2017b). Mtb PKNA/PKNB Dual Inhibition Provides Selectivity Advantages for Inhibitor Design To Minimize Host Kinase Interactions. ACS Med Chem Lett 8, 1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner LM, Webb AK, Limbago B, Dudeck MA, Patel J, Kallen AJ, Edwards JR, and Sievert DM (2016). Antimicrobial-Resistant Pathogens Associated With Healthcare-Associated Infections: Summary of Data Reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2011–2014. Infect Control Hosp Epidemiol 37, 1288–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wang JX, Zhou JM, Xu CL, Huang B, Xing Y, Wang B, Luo R, Wang YC, You XF, et al. (2017). A novel protein kinase inhibitor IMB-YH-8 with anti-tuberculosis activity. Sci Rep 7, 5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeats C, Finn RD, and Bateman A (2002). The PASTA domain:a β-lactam-binding domain. Trends Biochem Sci 27, 438–440. [DOI] [PubMed] [Google Scholar]
- Young TA, Delagoutte B, Endrizzi JA, Falick AM, and Alber T (2003). Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat Struct Biol 10, 168–174. [DOI] [PubMed] [Google Scholar]
- Zerella JA, Fouad AF, and Spångberg LSW (2005). Effectiveness of a calcium hydroxide and chlorhexidine digluconate mixture as disinfectant during retreatment of failed endodontic cases. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 100, 756–761. [DOI] [PubMed] [Google Scholar]
- Zheng W, Cai X, Li S, and Li Z (2018). Autophosphorylation Mechanism of the Ser/Thr Kinase Stk1 From Staphylococcus aureus. Front Microbiol 9, 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Wang Q, Zhang C, Cheung GSP, and Shen Y (2010). Prevalence, phenotype, and genotype of Enterococcus faecalis isolated from saliva and root canals in patients with persistent apical periodontitis. J Endod 36. [DOI] [PubMed] [Google Scholar]