

Abstract
Neuroinflammation is implicated in the pathogenesis of alcohol use disorders. We investigated the role of Gut-Brain interactions in alcohol-induced neuroinflammation by probiotic-mediated manipulation of intestinal dysbiosis in mice. Chronic ethanol feeding induced dysbiosis, as evidenced by an increase in Firmicutes/Bacteroidetes ratio and depletion of Lactobacillus species in the colon. Ethanol increased the levels of IL-1β, IL-6, and TNFα in plasma and the mRNA for IL-1β, IL-6, TNFα, and MCP1 genes in the cerebral cortex and hippocampus. Ethanol feeding increased inulin flux from the circulation into different brain regions, accompanied by the increase in TLR4 mRNA levels in the cerebral cortex and hippocampus. The immunofluorescence confocal microscopy showed that ethanol elevates the expression of microglial activation marker TMEM119 in the cerebral cortex. Feeding L. plantarum suppressed the ethanol-induced dysbiosis to some extent, as evidenced by attenuation of ethanol effects on Firmicutes/Bacteroidetes ratio and abundance of Lactobacillus spp. L. plantarum blocked ethanol-induced elevation of plasma cytokines, inulin permeability to the brain, mRNA for TLR4, IL-1β, IL-6, TNFα, and MCP1 in brain regions, and the expression of TMEM119 in the cerebral cortex. The L. plantarum effect was absent in mice that express a dominant-negative EGFR, suggesting that the EGFR receptor plays an essential role in the protective effect of L. plantarum against ethanol-induced neuroinflammation. L. plantarum, when administered after chronic ethanol-induced injury, rescued the ethanol-induced systemic inflammation and neuroinflammation. This study demonstrates that L. plantarum in the gut prevents and mitigates ethanol-induced neuroinflammation by an EGFR-dependent mechanism.
Keywords: Dysbiosis, probiotic, ethanol, microglia, alcohol use disorder
Introduction
Alcohol use disorders (AUD) and alcohol-related organ damage are significant global concerns [1, 2]. The hallmark of chronic alcohol use-mediated brain disorders includes neuroinflammation and neurodegeneration [3–6]. Although the direct impact of alcohol and its metabolites plays a significant role in neuroinflammation, the mounting evidence indicates that interactions at the Gut-Microbiome-Brain axis play a crucial role in alcohol use-related behavioral disorders [7–9]. The neurochemical mechanisms of Gut-Microbiome-Brain Axis are unclear. The bidirectional interactions between the gut and brain are likely involved in the pathogenesis of AUD [10].
A common denominator in the interactions between the gut and other organs, including Gut-Brain interactions, in alcohol-related pathologies is the gut microbiome. Recent studies have implicated gut microbiota in the pathogenesis of many neuropsychiatric disorders [9, 11]. Evidence suggests that alcohol-induced changes in gut microbiota may mediate neuroinflammation involving microglia and astrocytes in the amygdala and may contribute to withdrawal behaviors and symptoms [12]. Therefore, dysbiosis of the gut microbiome may contribute to alcohol abuse, alcohol-related brain dysfunction, and behavioral disorders. A piece of direct evidence to this hypothesis is provided by a recent study reporting that alcohol-induced neuroinflammation is ameliorated by the reduction of intestinal bacterial load by antibiotic treatment [13]. Therefore, further validation of gut-microbiome-brain interactions during alcoholism is crucial to identify targets for the development of the therapeutic intervention in AUD.
A significant body of evidence indicates that alcohol consumption leads to disruption of intestinal epithelial tight junctions, mucosal barrier dysfunction, and increased diffusion of bacterial toxins from the gut lumen into the intestinal mucosa and to the systemic circulation causing endotoxemia [14]. Alcohol-induced endotoxemia leads to systemic inflammation and targets multiple organs, including the liver, pancreas, and brain. At this time, the mechanism involved in gut microbiome-mediated neuroinflammation is unclear. Our recent study demonstrated that supplementation of the diet with the probiotic, L. plantarum blocked ethanol-induced gut barrier dysfunction, endotoxemia, and liver damage [15]. Therefore, probiotic-mediated prevention or rescue of alcohol-induced intestinal dysbiosis is a valid approach to dissect the gut-brain interactions during alcoholism. Epidermal growth factor (EGF) is a well-established gastrointestinal mucosal protective factor. EGF prevents acetaldehyde-induced barrier disruption in the mouse and human colonic mucosa [16–18]. The previous study indicated that EGF receptor (EGFR) was required for L-glutamine and L. plantarum-mediated prevention of ethanol-induced gut barrier dysfunction and endotoxemia [15, 19].
In the current study, we investigated the preventive and mitigating effects of L. plantarum on alcohol-induced neuroinflammation and examined the role of EGFR in this probiotic effect of L. plantarum.
Methods
Materials
Maltodextrin was purchased from Bioserv (Flemington, NJ, USA). Regular Lieber-DeCarli alcohol diet was purchased from Dyets Inc (Bethlehem, PA, USA; Cat #710260). Anti-GFAP antibody was purchased from Invitrogen (Carlsbad, CA), and the anti-TMEM119 antibody was purchased from Abcam (Cambridge, MA). AlexaFlour‐488‐conjugated anti‐mouse IgG and Cy3‐conjugated anti‐rabbit IgG were purchased from Molecular Probes (Eugene, OR).
Animals and diets
All animal experiments were performed according to the protocols approved by the UTHSC Institutional Animal Care and Use Committee. Animals were housed in an institutional animal care facility with 12:12 hours of light-dark cycles. All mice had free access to regular laboratory chow and water until the start of experiments. During the experiments, they were fed Lieber-DeCarli liquid diet. The animals and the experimental protocols were the same as those described previously [15].
Adult C57BL/6 (wildtype; 8–10 weeks old; Harlan, Houston, TX), rtTA, and rtTA-Egfr*Tg mice were used in these studies. The rtTA-Egfr*Tg mice were generated by crossing B6;SJL-Tg(tetO-Egfr*)2–9Jek/J (Egfr*Tg) with B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae/ (rtTA) mice (Jackson Laboratories, Sacramento, CA). The Egfr*Tg mice express a dominant-negative, cytoplasmic tyrosine kinase domain-deleted mutant form of EGFR (EGFR*) regulated by the tetracycline operator (tetO). The rtTA mice have a widespread expression of an optimized form of reverse tetracycline-controlled trans-activator protein. When Egfr*Tg mice are bred to rtTA mice, the offspring that are positive for both rtTA and Egfr* genes (rtTA-Egfr*Tg) express EGFR* in the presence of doxycycline. These mice were fed doxycycline in a liquid diet to induce Egfr* gene, which was confirmed by PCR. EGFR* expression disrupts subsequent activation of EGFR signal transduction pathways that are involved in regulating cell proliferation, differentiation, and survival.
Three independent studies were performed, as illustrated in the schematic in Figure 1. Adult female wildtype mice were used in Studies #1 and #3, as female mice are known to be more sensitive to alcohol. However, in study #2, we included both male and female rtTA and rtTA-Egfr*Tg mice. Male mice showed a pattern of changes in response to alcohol and L. plantarum, similar to female mice.
Figure 1: Schematic illustration of experimental design.
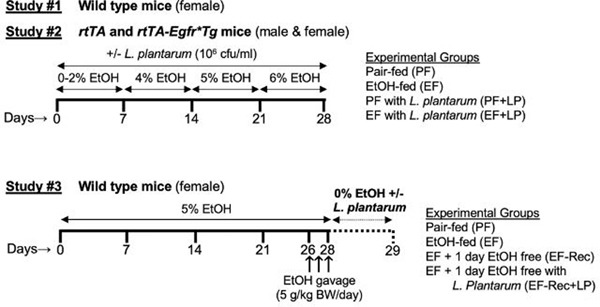
The types of mice used and the treatment conditions in different groups of mice in different experiments are outlined.
Ethanol Feeding
Two types of ethanol feeding models were used: 1) chronic ethanol feeding and 2) chronic + binge ethanol feeding as outlined in the schematic (Fig. 1). Chronic ethanol feeding was performed in Study #1 and #2, whereas the Chronic + Binge feeding method was used in Study #3.
Chronic ethanol feeding:
This ethanol feeding method was performed as described before [15, 19]. Briefly, mice were fed Lieber–DeCarli liquid diet containing varying concentrations of ethanol with or without L. plantarum (strain 256, 106 cfu/ml) or isocaloric maltodextrin for 4 weeks. Animals were gradually adapted to the ethanol-containing diet (0% for 2 days, 1% for 2 days, and 2% ethanol for 3 days followed by 4% ethanol for 1 week, 5% for 1 week, and 6% for 1 week). Control animals were pair-fed diets with isocaloric substitution of ethanol with maltodextrin.
Chronic + binge ethanol feeding:
This method of ethanol feeding model was adopted from a previous study [20]. Mice were fed Lieber–DeCarli liquid diet as above but with 5% ethanol for 4 weeks. During the last 3 days, animals received additional ethanol gavage (5 g/kg BW/day). Following ethanol feeding, animals were divided into 3 groups. One group was euthanized the same day. The other two groups were switched to an ethanol-free diet with (Group 3) or without (Group 2) L. plantarum (106 cfu/ml), and after 24 hours, animals in groups 2 and 3 were euthanized. The pair-fed control group was maintained as described above in the chronic ethanol feeding method. In all experiments, animals were maintained in pairs to facilitate body temperature maintenance and prevent social isolation. Animals were pair-fed to maintain a similar diet intake in different groups. During the last week of the experiments, the average diet intake per mouse per day was 0.7 ml. Therefore, the L. plantarum intake by a mouse was about 0.7 × 106 cfu/day.
One hour before euthanasia, mice were injected with FITC-inulin as described below. Blood samples were collected by cardiac puncture and centrifuged at 3,000 x g for 10 minutes at 4°C to prepare plasma. Colonic flushing was used for microbiome analysis. Brain samples (cerebral cortex) were fixed in buffered formalin or cryofixed in OCT; the remainder of the tissues was frozen for RNA extraction.
Microbiome analysis
Colonic flushings were collected for microbiome analysis. DNA was extracted with TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. 16S ribosomal RNA was analyzed by qPCR for different bacterial phyla or species using SYBR Green/ROX master mix (Qiagen) in an Applied Biosystems QuantStudio 6 FlexReal-Time PCR instrument. (Norwalk, CT, USA). Primer sequences for 16S ribosomal RNA genes for Lactobacillus, Lactobacillus species (plantarum, rhamnosus, acidophilus, casei, and gasseri), Bacteroidetes, and Firmicutes were chosen according to previous publications [21].
Blood-brain barrier permeability measurement.
Blood-brain barrier dysfunction was evaluated by measuring the vascular-to-brain flux of FITC-inulin (6 kDa) [22]. On the last day of the experiment, mice were intravenously injected with FITC-inulin (50 mg/ml solution; 2 μl/g body weight) via the tail vein. One hour after FITC-inulin administration, blood samples were collected by cardiac puncture under isoflurane anesthesia for plasma preparation. Brain regions were dissected and homogenized in 0.3 ml of PBS (pH 7.6). Homogenates were centrifuged at 10,000 × g, 40°C for 20 minutes to collect supernatants. Fluorescence in plasma and extracts of brain regions were measured using a fluorescence plate reader. Fluorescence values in the supernatant were normalized to fluorescence values in corresponding plasma samples and calculated as the percent of the amount injected.
Cytokine mRNA levels
Total RNA (1.5 μg) from cerebral cortex samples was used to generate cDNAs using the ThermoScript RT-PCR system for first-strand synthesis (Invitrogen). Quantitative PCR (qPCR) reactions were performed using cDNA mix (cDNA corresponding to 35 ng RNA) with 300 nmoles of primers in a final volume of 25 μl of 2× concentrated RT2 Real-Time SYBR Green/ROX master mix (Qiagen) in an Applied Biosystems QuantStudio 6 Flex Real-Time PCR instrument (Norwalk, CT, USA). The cycle parameters were: 50°C for 2min, one denaturation step at 95 °C for 10 min and 40 cycles of denaturation at 95°C for 10 seconds followed by annealing and elongation at 60°C. The relative gene expression of each transcript was normalized to GAPDH using the ΔΔCt method. Primer sequences for TLR-4, IL-1β, IL-6, TNF-α, and MCP-1 were chosen according to the previous publication [15].
Plasma cytokine assay
Plasma cytokine levels were measured using commercially available immunoassay ELISA kits for mice (R&D System, Minneapolis, MN, USA). IL-1β (Cat# DY401), TNF-α (Cat# DY410), and interleukin-6 (Cat# DY406) levels were estimated according to the manufacturer’s instructions. The results are expressed as picograms of cytokine per milliliter of plasma.
Immunofluorescence microscopy
Immunofluorescence staining was performed as described previously for colon [15]. Cryosections (10 μm) of the cerebral cortex were fixed in acetone: methanol (1:1), permeabilized with 0.2% Triton X-100 in PBS for 10 minutes and blocked in 4% nonfat milk in Triton-Tris buffer. It was incubated with primary antibodies (FITC-conjugated mouse monoclonal anti-GFAP and rabbit polyclonal anti-TMEM119 antibodies) for one hour. It was then incubated for another hour with the secondary antibodies (Cy3-conjugated anti-rabbit IgG antibodies). Hoechst 33342 dye was added during the last 10 minutes of incubation. Sections were washed three times after permeabilization, incubation with primary antibodies, and after incubation with secondary antibodies. Fluorescence was examined using a Zeiss 710 confocal microscope (Carl Zeiss GmbH, Jena, Germany) and processed as previously described [15, 19]. All images for tissue samples from different groups were collected and processed under identical conditions of laser, gain, and magnification.
Statistical Analyses
All data are expressed as Mean ± SEM. The differences among multiple groups were first analyzed by ANOVA (Prism 6.0). When a statistical significance was detected, Tukey’s t-test was used to determine the statistical significance between multiple testing groups and the corresponding control. Statistical significance was established at 95%.
Results
L. plantarum attenuates ethanol-induced intestinal dysbiosis
Alcohol-induced alteration of the bacterial population in the intestine is well-established [23]. Our recent study showed that feeding L. plantarum attenuates and mitigates chronic ethanol-induced intestinal mucosal barrier dysfunction [15]. In the current study, we evaluated the effect of L. planatarum on ethanol-induced intestinal dysbiosis by RT-qPCR. The relative abundance of both Firmicutes (Fig. 2A) and Bacteriodetes (Fig. 2B) was significantly reduced by ethanol feeding, whereas Firmicutes/Bacteriodetes (F/B) ratio was increased by several folds (Fig. 2C). Feeding L. plantarum further reduced Firmicutes in the presence or absence of ethanol, but it blocked ethanol-induced downregulation of Bacteroidetes abundance and upregulation of the F/B ratio.
Figure 2: L. plantarum prevents ethanol-induced alteration of the intestinal microbiome population.

Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) with or without L. plantarum (106 CFU/ml) (LP). Bacterial 16S RNA extracted from colonic fecal samples was subjected to qPCR for Firmicutes (A) and Bacteroidetes (B). Firmicutes/Bacteroidetes (F/B) ratios were calculated (C). Values are means ± SEM (n = 6). Asterisks indicate the “EF” values that are significantly (p-value < 0.05) different from corresponding “PF” values, and the hashtags indicate the “EF+LP” values that are different from corresponding “EF” values.
Chronic ethanol feeding depletes intestinal Lactobacillus and feeding L. plantarum ameliorates this effect of ethanol
Probiotics are beneficial bacteria in the intestinal lumen that protect the intestinal mucosa [24]. In this study, we examined the abundance of different Lactobacillus genus in pair-fed and ethanol-fed mouse colon. The relative abundance of Lactobacillus spp was dramatically reduced by chronic ethanol feeding (Fig. 3A). Ethanol depleted L. plantarum (Fig. 3B), L. rhamnosus (Fig. 3C), L. acidophilus (Fig. 3D) and L. casei (Fig. 3E), but it did not alter the abundance of L. gasseri (Fig. 3F). Feeding L. plantarum blocked ethanol-induced depletion of L. plantarum, L. acidophilus, and L. casei, but it further reduced L. rhamnosus and had no effect on L. gasseri.
Figure 3: L. plantarum prevents ethanol-induced down-regulation of Lactobacillus species.
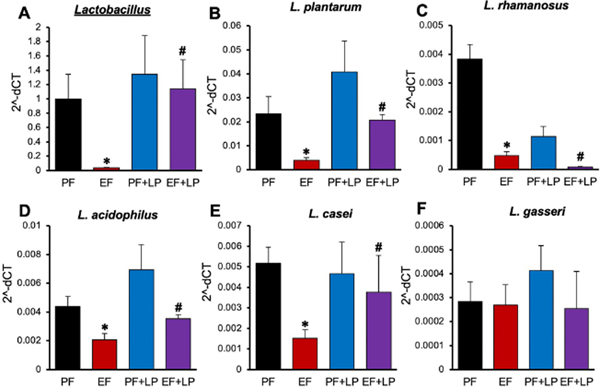
Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) with or without L. plantarum (106 CFU/ml) (LP). Bacterial 16S RNA extracted from colonic fecal samples was subjected to qPCR for Lactobacillus (A), L. plantarum (B), L. rhamnosus (C), L. acidophilus (D), L. casei (E) and L. gasseri (F). Values are means ± SEM (n = 6). Asterisks indicate the “EF” values that are significantly (p-value < 0.05) different from corresponding “PF” values, and the hashtags indicate the “EF+LP” values that are different from corresponding “EF” values.
L. plantarum attenuates ethanol-induced systemic inflammation
Chronic alcohol consumption leads to systemic inflammation and multiple organ damage [14]. We evaluated the effects of ethanol and L. plantarum on plasma levels of proinflammatory cytokines as a readout for systemic inflammation. Plasma levels of IL-1β (Fig. 4A), IL-6 (Fig. 4B), and TNFα (Fig. 4C) were significantly elevated by ethanol feeding by several folds. Feeding L. plantarum significantly reduced ethanol-induced upregulation of IL-1β, IL-6, and TNFα. L. plantarum by itself did not affect basal levels of these cytokines in plasma.
Figure 4: L. plantarum blocks ethanol-induced systemic inflammation.
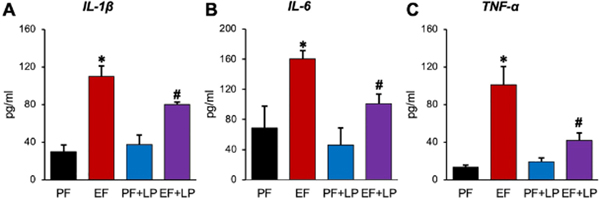
Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) with or without L. plantarum (106 CFU/ml) (LP). Plasma levels of IL-1β (A), IL-6 (B), and TNFα (C) were measured by ELISA. Values are means ± SEM (n = 6). Asterisks indicate the “EF” values that are significantly (P < 0.05) different from corresponding “PF” values, and the hashtags indicate the “EF+LP” values that are different from corresponding “EF” values.
L. plantarum attenuates Ethanol-induced increases in inulin permeability into brain regions
Ethanol feeding has been shown to disrupt the blood-brain barrier (BBB) integrity [25]. Vascular-to-brain flux of FITC-inulin was measured as an index of permeability through BBB. Ethanol feeding significantly increased FITC-inulin flux into the cortex, hippocampus, cerebellum, and corpus striatum (Fig. 5A). Feeding L. plantarum significantly reduced the effects of ethanol on BBB inulin permeability.
Figure 5: L. plantarum prevents ethanol-induced blood-brain barrier permeability and TLR4 expression.
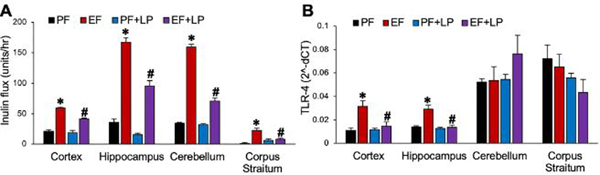
Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) with or without L. plantarum (106 CFU/ml) (LP). A: Vascular-to-brain flux of FITC-inulin was measured in different regions of the brain. B: The levels of TLR4 mRNA were measured by qPCR in different regions of the brain. Values are means ± SEM (n = 6). Asterisks indicate the “EF” values that are significantly (p-value < 0.05) different from corresponding “PF” values, and the hashtags indicate the “EF+LP” values that are different from corresponding “EF” values.
Ethanol feeding elevates TLR4 mRNA in brain cortex and hippocampus, which is blocked by L. plantarum
Chronic alcohol consumption leads to endotoxemia due to bacterial lipopolysaccharides (LPS) absorption from the gut [14]. An increase in inulin permeability to the brain suggest that ethanol may increase LPS flux into brain regions. The limitation in the sensitivity of LPS assay prevents the measurement of LPS in the brain regions. Elevated LPS has been consistently shown to be associated with increased expression of TLR-4, the LPS receptor, and other downstream signaling elements such as MYD88. Therefore, we measured TLR-4 mRNA as a readout for elevated endotoxin flux into the brain. TLR-4 mRNA expression in the brain cortex and hippocampus was significantly higher in ethanol-fed mice than in pair-fed control mice (Fig. 5B). Feeding L. plantarum completely blocked the ethanol-induced increase in TLR-4 mRNA. TLR-4 mRNA in the cerebellum and corpus striatum were unaffected.
L. plantarum attenuates ethanol-induced inflammatory responses in brain cortex and hippocampus
Higher inulin permeability and TLR-4 mRNA expression in the ethanol-fed mouse brain raised the question of whether ethanol induces inflammatory responses in the brain and whether L. plantarum blocks this effect. We measured the mRNA for selected proinflammatory cytokines and chemokine in the brain regions. The levels of IL-1β (Fig. 6A), IL-6, (Fig. 6B), TNFα (Fig. 6C), and MCP-1 (Fig. 6D) mRNA were significantly higher in the cortex and hippocampus in ethanol-fed mice compared to that in pair-fed control mice. Feeding L. plantarum blocked ethanol-induced elevation of IL-1β, IL-6, TNFα, and MCP-1 mRNA in both cortex and hippocampus (Fig. 6A, 6B, 6C, and 6D). IL-1β and MCP-1 mRNA levels were significantly higher in the cerebellum of ethanol-fed mice than in pair-fed control mice. L. plantarum feeding blocked ethanol-induced elevation of MCP-1 mRNA but had no significant effect on IL-1β mRNA. Ethanol or L. plantarum had no effect on cytokine mRNA expression in the corpus striatum region of the brain.
Figure 6: L. plantarum attenuates ethanol-induced neuroinflammation.

Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) with or without L. plantarum (106 CFU/ml) (LP). The levels of mRNA for IL-1β (A), IL-6 (B), TNFα (C), and MCP1 (D) genes were measured by qPCR in different regions of the brain. Values are Mean ± SEM (n = 6). Asterisks indicate the “EF” values that are significantly (p-value < 0.05) different from corresponding “PF” values, and the hashtags indicate the “EF+LP” values that are different from corresponding “EF” values.
L. plantarum attenuates ethanol-induced microglial and astrocyte markers in the brain
Evidence suggests that acute or chronic alcohol consumption modulates the function and morphology of microglia and astrocyte in the brain [26]. We examined the effect of ethanol and L. plantarum on microglia and astrocytes by staining the sections of the brain cortex for TMEM119 and GFAP, respectively. Both TMEM119 and glial fibrillary acidic protein (GFAP) stains were increased in the ethanol-fed mouse brain compared to that in pair-fed control mice (Fig. 7A). Feeding L. plantarum blocked the ethanol-induced increase in TMEM119 and GFAP stains. These effects of ethanol and L. plantarum on TMEM119 fluorescence were confirmed by densitometric analysis (Fig. 7B).
Figure 7: L. plantarum blocks Ethanol-induced elevation of microglia and astrocyte activation markers in the cerebral cortex.
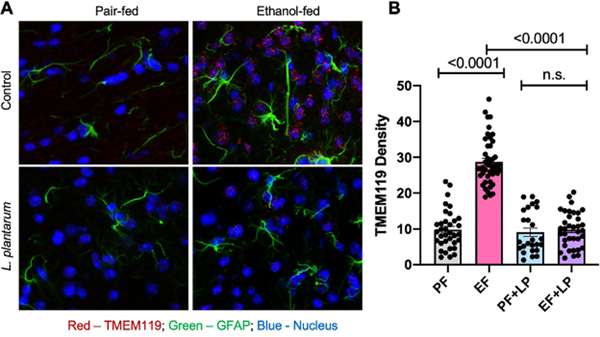
Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) and with or without L. plantarum (106 CFU/ml) (LP). A: Cryosections of the cerebral cortex were stained for TMEM119 (red), and GFAP (green) proteins and confocal fluorescence images were captured under similar conditions. B: The TMEM119 fluorescence is evaluated by using ImageJ software presented as arbitrary values. Values are Mean ± SEM. The dots represent the values for individual cells from 2–4 mouse brains (3 for PF, 4 for EF, 2 for PF+LP, and 3 for EF+LP). The numbers above the bars are p-values for differences between the groups indicated by the horizontal bars underneath; “n.s.” indicates no significant difference (p-value >0.05) between indicated groups.
EGFR plays a role in L. plantarum–mediated attenuation of the ethanol-induced inflammatory response in the hippocampus
We evaluated the role of EGFR in L. plantarum effect on ethanol-induced neuroinflammation by using the transgenic mice. Ethanol feeding increased the levels of mRNA for IL-1β (Fig. 8A), IL-6 (Fig. 8B), TNFα (Fig. 8C), and MCP-1 (Fig. 8D) genes in the hippocampus of rtTA mice; L. plantarum blocked these effects of ethanol on IL-1β, IL-6, TNFα, and MCP-1 mRNA in hippocampus in the absence or presence of doxycycline. In rtTA-Egfr*Tg mice, however, L. plantarum failed to block ethanol-induced elevation of mRNA for these cytokine and chemokine genes in the presence of doxycycline. In this study, we included both male and female mice; however, the individual values indicated no sex-dependent differences in the effects of alcohol or L. plantarum.
Figure 8: EGFR plays a role in L. plantarum-mediated prevention of ethanol-induced neuroinflammation.

Adult rtTA and rtTA-EGFR*-Tg mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF), with or without L. plantarum (106 CFU/ml) (LP) and with or without doxycycline (Dox). RNA extracted from the cerebral cortex was subjected to qPCR for IL-1β (A), IL-6 (B), TNF-α (C), and MCP1 (D) genes. Values are means ± sem (n = 4 for PF and EF and 6 for LP+EF and Dox+LP+EF groups). Asterisks indicate the “EF” values that are significantly (p-value < 0.05) different from corresponding “PF” values. The hashtags indicate the “EF+LP” values that are different from corresponding “EF” values; the symbol Δ indicates the “EF+LP+Dox” value that is different from corresponding values for “EF+LP without Dox” values.
L. plantarum rescues ethanol-induced systemic inflammation and neuroinflammation
Studies above demonstrate that L. plantarum feeding prevents ethanol-induced neuroinflammation by an EGFR-dependent mechanism. We conducted a study to determine whether L. plantarum can rescue the brain from ethanol-induced neuroinflammation. The plasma levels of proinflammatory cytokines, IL-1β (Fig. 9A), IL-6 (Fig. 9B), and TNFα (Fig. 9C), were elevated by ethanol feeding. These plasma cytokine levels were further increased at 24 hours after withdrawal from ethanol by maintaining the ethanol-free diet. However, the supplementation of the ethanol-free diet with L. plantarum significantly reversed the ethanol-induced elevation of plasma cytokines.
Figure 9: L. plantarum rescues ethanol-induced systemic inflammation.
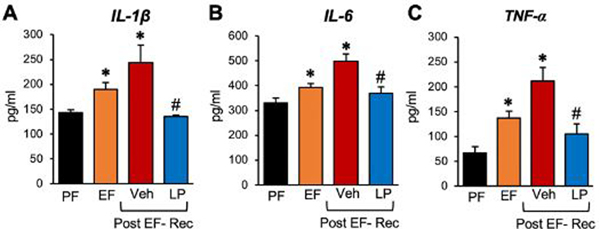
Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) for 4 weeks, following which ethanol was withdrawn for 24 hours by feeding the liquid diet without ethanol, but with (LP) or without (Veh) L. plantarum. The levels of IL-1β (A), IL-6 (B), and TNF-α (C) in the plasma were measured before and at 24 hours after ethanol withdrawal (Post EF-Rec) were measured. Values are means ± sem (n = 6). Asterisks indicate the “EF” (pre and post ethanol withdrawal) values that are significantly (p-value < 0.05) different from corresponding “PF” values. The hashtags indicate the “Post EF-Rec + LP” values different from corresponding “Post EF-Rec + Veh” values.
Ethanol feeding significantly elevated the mRNA for proinflammatory cytokines, IL-1β (Fig. 10A), IL-6 (Fig. 10B), and TNFα (Fig. 10C) in the hippocampus. The mRNA for these cytokines were further elevated at 24 hours after ethanol withdrawal. Dietary L. plantarum supplementation completely reversed the ethanol-induced expression of cytokine mRNA in the hippocampus.
Figure 10: L. plantarum reverses ethanol-induced neuroinflammation.
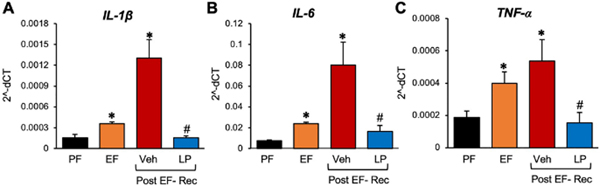
Adult female mice were fed a liquid diet with ethanol (EF) or isocaloric maltodextrin (PF) for 4 weeks, following which ethanol was withdrawn for 24 hours by feeding the liquid diet without ethanol, but with (LP) or without (Veh) L. plantarum. The mRNA levels for IL-1β (A), IL-6 (B), and TNF-α (C) in the plasma were measured before and at 24 hours after ethanol withdrawal (Post EF-Rec) were measured in the cerebral cortex. Values are means ± sem (n = 6). Asterisks indicate the “EF” (pre and post ethanol withdrawal) values that are significantly (p-value < 0.05) different from corresponding “PF” values. The hashtags indicate the “Post EF-Rec + LP” values different from corresponding “Post EF-Rec + Veh” values.
Discussion
Neuroinflammation and neurodegeneration are significant factors associated with the pathogenesis of AUD. Emerging evidence indicates that the bidirectional disturbances in the Gut-Brain axis are involved in alcohol-induced neuroinflammation and AUD. The gut microbiome is a crucial player in the regulation of Gut-Brain interactions. Therefore, understanding the alcohol-specific alterations in Gut-Microbiome-Brain interactions is essential to understand the pathogenesis of AUD and identify the therapeutic targets for the treatment of AUD. In the current study, we intervened alcohol-induced intestinal bacterial dysbiosis by feeding L. plantarum, a common probiotic that inhabits the human gut. The results of this study provide evidence of the role of L. plantarum in the prevention and mitigation of alcohol-induced neuroinflammation, the process involving functional EGFR.
The increase in F/B ratio by ethanol feeding indicated that chronic ethanol feeding induces dysbiosis in mouse colon under the experimental conditions of this study. The prevention of this ethanol-induced rise in the F/B ratio by L. plantarum indicated that L. plantarum suppresses ethanol-induced dysbiosis. In the current study, ethanol feeding reduced the overall abundance of Firmicutes. Surprisingly, L. plantarum further decreased the levels of Firmicutes, although itself belongs to Firmicutes phylum. The explanation for this effect of L. plantarum is unclear at this time. But, it is essential to note that the total 16S RNA was significantly reduced by ethanol in the absence of L. plantarum, but not in its presence. Firmicutes may be replaced by other phyla of bacteria, such as Actinobacteria and Proteobacteria. Although it is an oversimplified index, an altered F/B ratio is considered an indication of dysbiosis. Our study indicates that L. plantarum prevents ethanol-induced dysbiosis to some extent. For more detailed information, metagenomic analysis of 16S RNA is required.
The above conclusion is supported by the striking observation that ethanol feeding caused a dramatic depletion of bacteria under the Lactobacillus genus. The abundance of Lactobacillus spp., such as L. plantarum, L. acidophilus, L. rhamnosus and L. casei were all reduced by ethanol. Feeding L. plantarum, not only restored L. plantarum, but also restored the population of L. acidophilus and L. casei, suggesting that L. plantarum prevents ethanol-induced loss of other Lactobacillus spp. Therefore, L. plantarum may reduce the abundance of pathogenic Firmicutes while preventing the depletion of other probiotics under Firmicutes phylum. The results also suggest that L. plantarum is an excellent candidate probiotic for the intervention of alcoholic tissue injury.
Several-fold elevation of the proinflammatory cytokines, such as IL-1β, IL-6, and TNFα in plasma, demonstrate that chronic alcohol feeding, in the current model, caused systemic inflammation. Feeding L. plantarum significantly reduced this systemic inflammatory response to alcohol. Our recent study demonstrated that supplementation of the diet with L. plantarum blocks ethanol-induced disruption of tight junctions and adherens junctions in the colonic epithelium, increases in intestinal mucosal permeability to inulin, the elevation of plasma LPS, and liver damage [15]. Evidence indicates that plasma LPS plays a crucial role in the pathogenesis of alcoholic liver damage [14] and suggested to have a similar role of LPS in alcohol-induced brain injury [26]. Previous studies have indicated that alcohol disrupts BBB and hence likely to allow LPS to diffuse into brain tissue [25]. The present study results show that ethanol elevates the flux of intravenously administered FITC-inulin into the cerebral cortex, hippocampus, cerebellum, and corpus striatum. This observation confirms the previous suggestion that alcohol disrupts BBB and allows entry of LPS into brain tissue. Feeding L. plantarum significantly reduced ethanol-induced flux of inulin into brain regions. The plasma concentrations of FITC-inulin was similar in all groups, confirming that the increased levels of inulin in the brain of ethanol-fed mice and its reduction in L. plantarum-fed mice are indeed due to corresponding changes in the BBB permeability. The potential increase in LPS flux into the brain by ethanol is further supported by a significant elevation of TLR4 mRNA in the cerebral cortex and hippocampus of ethanol-fed mice. LPS is known to regulate the expression of TLR4 positively. We measured the mRNA for TLR4 as a readout for LPS flux into the brain regions. The prevention of ethanol-induced TLR4 expression in the cortex and hippocampus by L. plantarum is consistent with its effects on plasma cytokine levels and inulin flux into brain regions.
Neuroinflammation was assessed by measuring the levels of mRNA for proinflammatory cytokines such as IL-1β, IL-6, and TNFα and the chemokine, MCP1 in different brain regions. Several-fold elevation of mRNA for these cytokines and chemokine in the cerebral cortex and hippocampus indicates that ethanol induces inflammatory responses in these regions of the brain. The ethanol-induced neuroinflammation was almost entirely blocked by L. plantarum, suggesting that L. plantarum in the gut can regulate inflammation in the cerebral cortex and hippocampus. The mechanism of this remote influence of L. plantarum in the gut on the brain’s inflammatory responses is unclear at this time. The LPS absorption due to disruption of intestinal tight junction and the entry of LPS through leaky BBB may be involved in this interaction between the brain and gut microbiota. It is also likely that other bacterial factors may mediate the gut-brain interaction in the present model.
Microglia and astrocytes are the primary cells responsible for the immune response in the brain [26]. They recognize and respond to potential threats in the microenvironment in different brain regions [27]. Pathogen-associated and danger-associated molecular patterns target astrocytes and microglia, and activation of these cells releases proinflammatory cytokines creating a condition of neuroinflammation [27]. The activation marker of astrocytes is a glial GFAP that is upregulated by alcohol exposure in rats and the alcoholic human brain [4, 26]. In the current study, we examined the GFAP expression by immunofluorescence confocal microscopy. An increased GFAP stain in the cerebral cortex of ethanol-fed mice compared to that in pair-fed mice indicates that ethanol activates astrocytes in the current model. The microglia, the mononuclear macrophages in the brain, plays an essential role in immune surveillance and clearance of invading pathogens [28]. The transmembrane protein TMEM119 is exclusively expressed by a subset of microglia and is an excellent marker for microglia [29]. Ethanol feeding induced a dramatic elevation of TMEM119 stain in the cerebral cortex, suggesting that ethanol activates microglia. Interestingly, feeding L. plantarum almost completely attenuated ethanol-induced elevation of stain for both GFAP and TMEM119, suggesting that the intestinal L. plantarum blocks ethanol-induced activation of astrocytes and microglia. These data also indicate that the activation of microglia and astrocytes may mediate the increased expression of proinflammatory cytokines in the ethanol-fed mouse brain.
Our previous studies demonstrated that epidermal growth factor (EGF) and EGFR prevent ethanol-induced disruption of intestinal epithelial tight junctions and barrier dysfunction [17, 19]. Our recent study showed that L. plantarum-mediated prevention of ethanol-induced disruption of tight epithelial junctions, gut barrier dysfunction, endotoxemia, and liver damage was mediated by EGFR [15]. To determine the role of EGFR in L. plantarum effects on alcohol-induced neuroinflammation, we used rtTA-Egfr*Tg mice. Prevention of L. plantarum-mediated protection from ethanol by doxycycline in rtTA-Egfr*Tg mice, but not in rtTA mice indicated that the L. plantarum-mediated prevention of ethanol-induced neuroinflammation requires functional EGFR. The precise mechanism involved in this effect of L. plantarum on alcoholic neuroinflammation is unclear. It is likely that the EGFR-mediated protection of intestinal epithelial junctions and mucosal barrier function by L. plantarum is responsible for the prevention of neuroinflammation. Further investigation is required to delineate the precise mechanistic pathway involved in this Gut-Brain interaction.
Our previous study demonstrated that L. plantarum could reverse ethanol-induced disruption of intestinal epithelial tight junctions, mucosal barrier dysfunction, endotoxemia, and liver damage [15]. In the present study, we examined the effect of L. plantarum administered after the induction of ethanol-induced neuroinflammation. Twenty-four-hour withdrawal after four weeks of ethanol feeding resulted in a further increase in systemic inflammation and neuroinflammation. However, dietary supplementation during the ethanol withdrawal almost entirely reversed ethanol-induced systemic inflammation and neuroinflammation.
In summary, this study demonstrates that L. plantarum can prevent and mitigate the ethanol-induced neuroinflammation by an EGFR-dependent mechanism. We speculate that the prevention of ethanol-induced disruption of intestinal epithelial tight junctions, mucosal barrier dysfunction, and dysbiosis is involved in this neuroprotective effect of L. plantarum on alcoholic neuroinflammation. Overall, this study indicates the potential therapeutic benefit of L. plantarum in the prevention and treatment of AUD.
Acknowledgments
All authors contributed to study conception, data analysis, and interpretation.
Support: This study was supported by grants from the National Institutes of Health (AA12307) and Veterans Administration Merit Award (I01BX003014-01).
Footnotes
Disclosure statement
The authors declare no conflict of interest.
References
- 1.Rajendram R, Lewison G, and Preedy VR, Worldwide alcohol-related research and the disease burden. Alcohol Alcohol, 2006. 41(1): p. 99–106. [DOI] [PubMed] [Google Scholar]
- 2.Rehm J, et al. , Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet, 2009. 373(9682): p. 2223–33. [DOI] [PubMed] [Google Scholar]
- 3.Choi DK, et al. , Differential effects of ethanol on glial signal transduction initiated by lipopolysaccharide and interferon-gamma. J Neurosci Res, 2005. 82(2): p. 225–31. [DOI] [PubMed] [Google Scholar]
- 4.Qin L and Crews FT, Chronic ethanol increases systemic TLR3 agonist-induced neuroinflammation and neurodegeneration. J Neuroinflammation, 2012. 9: p. 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis RL and Syapin PJ, Ethanol increases nuclear factor-kappa B activity in human astroglial cells. Neurosci Lett, 2004. 371(2–3): p. 128–32. [DOI] [PubMed] [Google Scholar]
- 6.Lee H, et al. , Ethanol selectively modulates inflammatory activation signaling of brain microglia. J Neuroimmunol, 2004. 156(1–2): p. 88–95. [DOI] [PubMed] [Google Scholar]
- 7.Leclercq S, et al. , Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun, 2012. 26(6): p. 911–8. [DOI] [PubMed] [Google Scholar]
- 8.Dubinkina VB, et al. , Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome, 2017. 5(1): p. 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodriguez-Gonzalez A and Orio L, Microbiota and alcohol use disorder: are psychobiotics a novel therapeutic strategy? Curr Pharm Des, 2020. [DOI] [PubMed] [Google Scholar]
- 10.Daulatzai MA, Chronic functional bowel syndrome enhances gut-brain axis dysfunction, neuroinflammation, cognitive impairment, and vulnerability to dementia. Neurochem Res, 2014. 39(4): p. 624–44. [DOI] [PubMed] [Google Scholar]
- 11.Hillemacher T, et al. , Alcohol, microbiome, and their effect on psychiatric disorders. Prog Neuropsychopharmacol Biol Psychiatry, 2018. 85: p. 105–115. [DOI] [PubMed] [Google Scholar]
- 12.Gorky J and Schwaber J, The role of the gut-brain axis in alcohol use disorders. Prog Neuropsychopharmacol Biol Psychiatry, 2016. 65: p. 234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lowe PP, et al. , Reduced gut microbiome protects from alcohol-induced neuroinflammation and alters intestinal and brain inflammasome expression. J Neuroinflammation, 2018. 15(1): p. 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao R, Endotoxemia and gut barrier dysfunction in alcoholic liver disease. Hepatology, 2009. 50(2): p. 638–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shukla PK, et al. , Lactobacillus plantarum prevents and mitigates alcohol-induced disruption of colonic epithelial tight junctions, endotoxemia, and liver damage by an EGF receptor-dependent mechanism. FASEB J, 2018: p. fj201800351R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sheth P, et al. , Epidermal growth factor prevents acetaldehyde-induced paracellular permeability in Caco-2 cell monolayer. Alcohol Clin Exp Res, 2004. 28(5): p. 797–804. [DOI] [PubMed] [Google Scholar]
- 17.Basuroy S, et al. , Acetaldehyde disrupts tight junctions and adherens junctions in human colonic mucosa: protection by EGF and L-glutamine. Am J Physiol Gastrointest Liver Physiol, 2005. 289(2): p. G367–75. [DOI] [PubMed] [Google Scholar]
- 18.Suzuki T, Seth A, and Rao R, Role of phospholipase Cgamma-induced activation of protein kinase Cepsilon (PKCepsilon) and PKCbetaI in epidermal growth factor-mediated protection of tight junctions from acetaldehyde in Caco-2 cell monolayers. J Biol Chem, 2008. 283(6): p. 3574–83. [DOI] [PubMed] [Google Scholar]
- 19.Meena AS, et al. , EGF receptor plays a role in the mechanism of glutamine-mediated prevention of alcohol-induced gut barrier dysfunction and liver injury. J Nutr Biochem, 2019. 64: p. 128–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrasek J, et al. , IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J Clin Invest, 2012. 122(10): p. 3476–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shukla PK, et al. , Human Defensin-5 Blocks Ethanol and Colitis-Induced Dysbiosis, Tight Junction Disruption and Inflammation in Mouse Intestine. Sci Rep, 2018. 8(1): p. 16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takeda S, et al. , Increased blood-brain barrier vulnerability to systemic inflammation in an Alzheimer disease mouse model. Neurobiol Aging, 2013. 34(8): p. 2064–70. [DOI] [PubMed] [Google Scholar]
- 23.Starkel P, et al. , Intestinal dysbiosis and permeability: the yin and yang in alcohol dependence and alcoholic liver disease. Clin Sci (Lond), 2018. 132(2): p. 199–212. [DOI] [PubMed] [Google Scholar]
- 24.Sanders ME, et al. , Probiotics and prebiotics in intestinal health and disease: from biology to the clinic. Nat Rev Gastroenterol Hepatol, 2019. 16(10): p. 605–616. [DOI] [PubMed] [Google Scholar]
- 25.Haorah J, et al. , Alcohol-induced blood-brain barrier dysfunction is mediated via inositol 1,4,5-triphosphate receptor (IP3R)-gated intracellular calcium release. J Neurochem, 2007. 100(2): p. 324–36. [DOI] [PubMed] [Google Scholar]
- 26.Szabo G and Lippai D, Converging actions of alcohol on liver and brain immune signaling. Int Rev Neurobiol, 2014. 118: p. 359–80. [DOI] [PubMed] [Google Scholar]
- 27.Guillemin GJ and Brew BJ, Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol, 2004. 75(3): p. 388–97. [DOI] [PubMed] [Google Scholar]
- 28.Erickson EK, et al. , Neuroimmune signaling in alcohol use disorder. Pharmacol Biochem Behav, 2019. 177: p. 34–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bennett ML, et al. , New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A, 2016. 113(12): p. E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]