

Abstract
The synthesis of bona fide organometallic CeIV complexes is a formidable challenge given the typically oxidizing properties of the CeIV cation and reducing tendencies of carbanions. Herein, we report a pair of compounds comprising a CeIV − Caryl bond [Li(THF)4][CeIV(κ2-ortho-oxa)(MBP)2] (3-THF) and [Li(DME)3][CeIV(κ2-ortho-oxa)(MBP)2] (3-DME), ortho-oxa = dihydro-dimethyl-2-[4-(trifluoromethyl)phenyl]-oxazolide, MBP2– = 2,2′-methylenebis(6-tert-butyl-4-methylphenolate), which exhibit CeIV − Caryl bond lengths of 2.571(7) – 2.5806(19) Å and strongly-deshielded, CeIV − Cipso 13C{1H} NMR resonances at 255.6 ppm. Computational analyses reveal the Ce contribution to the CeIV − Caryl bond of 3-THF is ~12%, indicating appreciable metal-ligand covalency. Computations also reproduce the characteristic 13C{1H} resonance, and show a strong influence from spin-orbit coupling (SOC) effects on the chemical shift. The results demonstrate that SOC-driven deshielding is present for CeIV − Cipso 13C{1H} resonances and not just for diamagnetic actinide compounds.
Subject terms: Chemical bonding, Chemical bonding
Ce(IV) organometallic compounds are rare due to Ce(IV) being a powerful oxidant. Herein, the authors explore the covalency of a pair of organocerium complexes bearing a Ce(IV)-C(aryl) bond and examine their structure by NMR spectroscopy, X-ray diffraction analysis, and computational calculations.
Introduction
The bonding between lanthanides and ligands has been described as purely ionic1,2. However, modern spectroscopic and computational techniques have challenged this simple assumption3–6. The covalency in M − X interactions can result in anomalous and diagnostic nuclear magnetic resonance shifts, X = 13C, 15N, 19F, 77Se, 125Te, resulting from participation of f-element orbital angular momentum7–13. Another important aspect of detailed f-element electronic structure is multiconfigurational character, as described in the model example of cerocene, Ce(COT)2, COT = cyclooctatetraene ligand14. The case for multiconfigurational character in cerocene has been made through X-ray absorption, SQUID magnetometry, and multi-reference computational studies and tied strongly to the characteristics of the cerium-carbon bonding15. Despite the interest surrounding f-element covalency and multiconfigurational effects, there are few examples of organometallic CeIV complexes16. The current literature is limited to metallocene CeIV complexes or α-heteroatom stabilized CeIV − C σ-bonds. Reported examples of CeIV − metallocene bonding include CeIV complexes of cyclopentadienide, cyclooctatetraene dianion, and bispentalene dianion ligands17–21. Complexes containing a CeIV − C σ-bond, however, are limited to either an N-heterocyclic carbene (NHC) complexes, e.g., Ce[L4] (Fig. 1a), or a bis(iminophosphorano)methandiide complex, e.g., [Ce(BIPMTMS)(ODipp)2] (Fig. 1a)22,23. The latter examples are expected to have electronic structures that deviate significantly from typical organometallic alkyl, aryl, or alkynyl ligands due to the heteroatom α-substitution22–25. The scarcity of CeIV − C containing-complexes likely arises from the unstable combination of strongly reducing carbanions and the oxidizing CeIV cation16. As a result, the formation of reactive, carbon-centered radicals and CeIII species is observed. Our team has a long-standing interest in the isolation of redox stable CeIV species to elucidate the relationship between ligand field and the CeIII/CeIV couple26–28.
Fig. 1. Examples of CeIV − C σ bonds.

a Previous examples of complexes with formally CeIV − C σ bonds, which are stabilized by either nitrogen22 or phosphorus heteroatoms23. b This work detailing the synthesis and characterization of a CeIV − Caryl bond, including computational analysis. Carbon atoms bound to cerium are indicated with a C.
Herein, we expand our studies for the isolation of a pair of CeIV − Caryl compounds. These compounds display unusually high 13C NMR shifts compared to other diamagnetic MIV − Caryl compounds. Relativistic density functional calculations verify that the high NMR shifts are due to large SOC effects supported by the increased covalency of the CeIV − Caryl bond.
Results
Synthesis and structures of CeIV − Caryl
Considering strategies to stabilize a CeIV − Caryl bond, we hypothesized that tethering the aryl group to the Ce center would kinetically inhibit homolysis of the Ce − C bond. In addition, we sought a sterically-protected Ce center to prevent reactivity at the ipso-carbon. Lastly, we chose a supporting ligand that would stabilize the CeIV oxidation state to prevent charge transfer and subsequent Ce − C bond homolysis. With these considerations in mind, we aimed to prepare a CeIV − Caryl bond from the CeIV bis(methylene bisphenolate) complex Ce(THF)2(MBP)2, that was previously synthesized by members of the Schelter laboratory (1, Fig. 2)29. Aryloxide ligands have been previously shown to both stabilize the CeIV oxidation state and high valent organometallic species of other metal species28–34. Addition of a yellow solution of ortho-lithiated oxazoline 2 (Li-ortho-oxa) to a purple benzene solution of Ce(THF)2(MBP)2 (1) at room temperature resulted in an immediate color change of the solution to dark red. The 1H NMR spectrum of the reaction mixture revealed loss of the pseudo C2v symmetry of 1 and formation of a C1 symmetric product. Likewise, there was also a shift in both the 7Li and 19F NMR resonances of 2, and the two methylene protons and methyl groups of the oxazoline were no longer degenerate in the 1H NMR spectrum. All 1H, 7Li, and 19F NMR resonances were well within the range of diamagnetic signals, leading to the assignment of the product of the reaction as [Li(THF)4][Ce(κ2-ortho-oxa)(MBP)2] (3-THF). Dark red X-ray quality crystals of 3-THF were grown over 3 days from a cooled (−25 °C) mixture of 3-THF in toluene and THF layered with pentane. The crystals were collected in 66% yield (Fig. 2). Alternatively, crystallizing from a cooled (−25 °C) solution of crude 3-THF in DME layered with pentane resulted in dark-red X-ray quality crystals of 3-DME over 3 days (Fig. 3). Crystals of 3-DME were collected in a slightly higher 75% yield. The differences in NMR data between 3-THF and 3-DME are negligible compared to experimental error. While compound 3-DME crystallizes with a single molecule in the asymmetric unit, compound 3-THF crystallizes with two independent molecules in the asymmetric unit, with only minor differences between the structures. The Ce − O(phenoxide) bond distances of 3-THF and 3-DME (2.1636(13)–2.202(4) Å) compare well with the Ce − O(phenoxide) bond distances observed in the reported structure of 1 (2.113(2)–2.152(2) Å)29. The Ce − C bond distances of 3-THF and 3-DME are 2.571(7) – 2.5806(19) Å and are shorter than reported CeIII − Caryl bond lengths: 2.621(4) – 2.64 ± 0.02 Å35,36. The difference in ionic radii between 6-coordinate CeIII and CeIV is 0.14 Å; however, the difference between 3-THF and 3-DME and previous CeIII − Caryl complexes is only 0.04 – 0.07 Å37. We ascribe this difference to the steric demand by the MTB ligands vs. the pentamethylcyclopentadienyl ligands used in the prior work. Previously reported complexes containing CeIV − C σ-bonds are 2.652(7) – 2.705(2) Å and 2.385(2) – 2.441(5) Å for the Ce − C NHC and Ce − C(bis(iminophosphorano)methandiide) ligands, respectively22,23,25. With this data in hand, we assign this complex as a CeIV − Caryl complex.
Fig. 2. Syntheses of title CeIV − Caryl compounds 3-THF and 3-DME.

The complex 1 was treated with the isolable aryl lithium 2 to generate the title complexes 3-THF or 3-DME.
Fig. 3. Crystal structure of 3-DME.
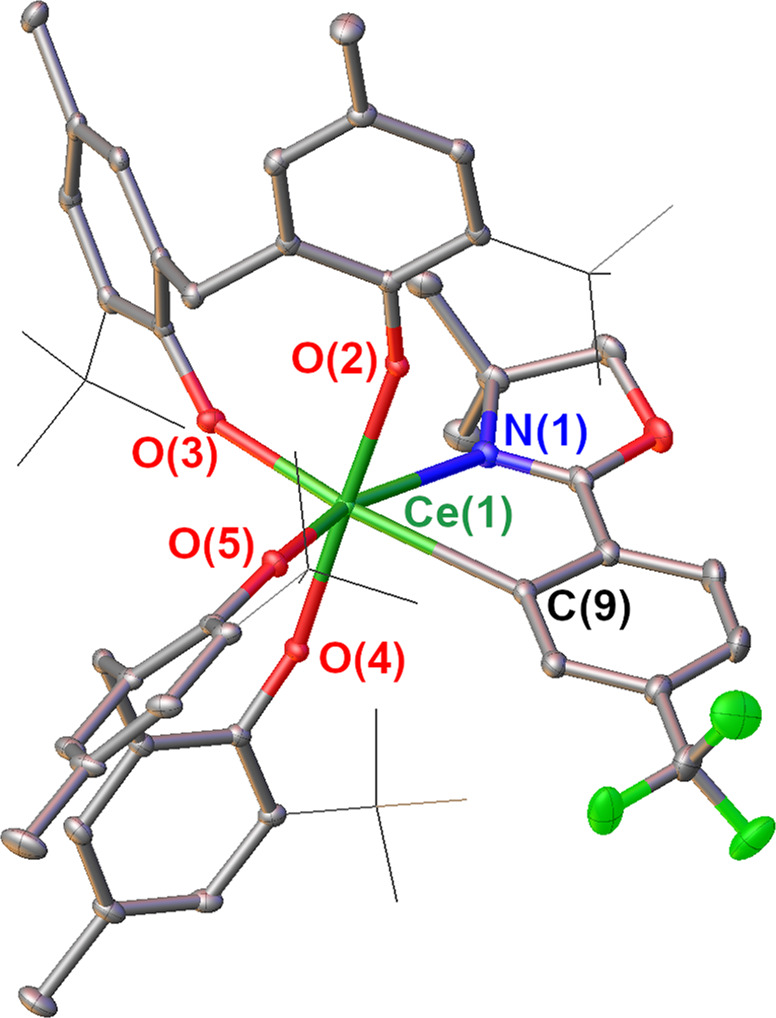
Thermal ellipsoid plot of the X-ray crystal structure of 3-DME at the 30% probability level. For clarity, C–H hydrogens and the Li(DME)3+ cation were removed; In addition tert-butyl groups are displayed in wireframe. Selected bond lengths in Å: Ce(1)–C(9): 2.5806(19); Ce(1)–N(1): 2.6176(16); Ce(1)–O(2): 2.1750(12); Ce(1)–O(3): 2.2062(13); Ce(1)–O(4): 2.1640(12); Ce(1)–O(5): 2.1636(13).
Electrochemical analysis
To better understand how the ortho-oxa− group impacts the stability of the CeIV cation, electrochemistry was performed on 3-THF. The Epa of 3-THF, −1.67 V vs. Fc/Fc+, shifts by −0.72 V relative to the E1/2 of 1 (−0.94 V vs. Fc/Fc+), indicating that the ortho-oxa− moiety significantly stabilizes the CeIV couple in THF. The reduction of 3-THF is not reversible under the electrochemical conditions, although the event precedes a reversible oxidation at E1/2 = −0.94 V vs. Fc/Fc+ and an irreversible oxidation at Epa = −0.43 V (Supplementary Figs. 18–19). We postulate that the reduction of the CeIV center is followed by dissociation of the ortho-oxa− fragment, producing 1 and 2. Indeed, the return anodic scan comprises waves at E1/2 = −0.94 V vs. Fc/Fc+ and Epa = −0.43 V respectively, consistent with the previous assignment for compound 1 and inferred for compound 2 (Fig. 4)29.
Fig. 4. Cyclic voltammagram of 3-THF.
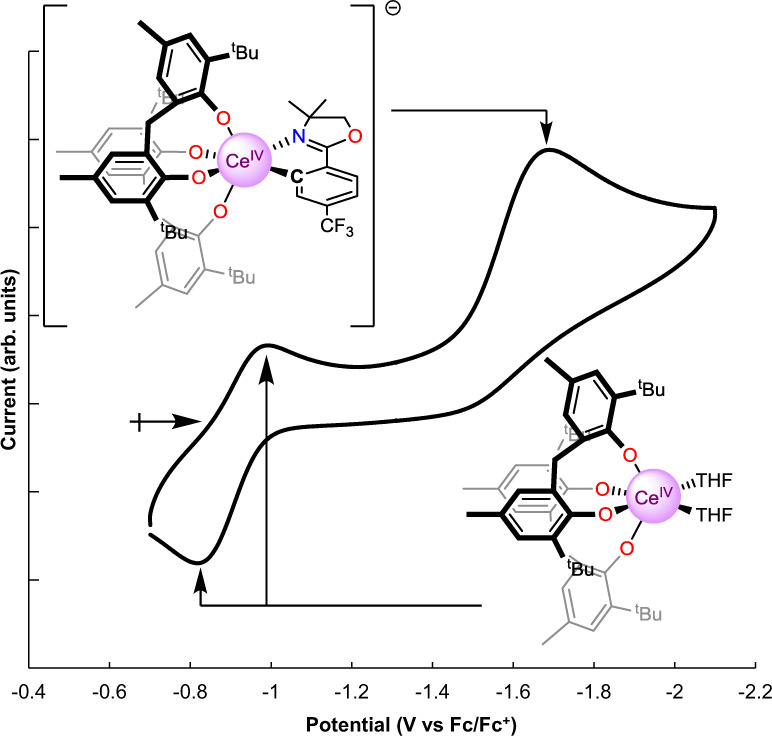
Solvent: THF; electrolyte 0.1 M [nPr4N][BArF4]; [analyte] = 0.001 M; OCP = −0.815 V vs. Fc/Fc+, noted by the right-facing arrow; ν = 100 mV s−1. The trace shows the reduction of 3 at Epc = −1.67 V vs. Fc/Fc+ with a return wave at E1/2 = −0.94 V vs. Fc/Fc+, which is the CeIII/CeIV couple of 129. The waves corresponding to 1 do not appear in the first scan (Supplementary Figs. 18–19).
13C NMR analysis
While the 1H, 7Li, and 19F NMR of 3-THF and 3-DME showed minimal changes compared to the starting materials, the 13C{1H} NMR of the ipso-carbon of both 3-THF and 3-DME showed a significant shift (difference between 3-THF and 3-DME is less than >0.05 ppm which is within error). Indeed, the ipso-13C{1H} resonance was located at 255.6 ppm, well outside of the typical range for aryl resonances (100–170 ppm) and shifted by ~50 ppm downfield relative to the Li − Caryl resonance for the starting material, 2 (Fig. 5). This 13C{1H} shift is further downfield than observed for other characterized, diamagnetic MIV − Caryl interactions; the highest being Th(2-C6H4CH2NMe2)4, which exhibits a ThIV − C 13C{1H} signal at 230.8 ppm38,39. The shift of the 13C{1H} signal for the carbon atom bound to metal cations has been implicated as a reporter for the degree of covalency in f-element cation-carbon bonds7–9. In this light, 3-THF and 3-DME have an anomalously high covalency for a MIV-aryl interaction. As with the bond distance metrics, there are few salient examples of CeIV − C σ-bonds for comparison of the 13C{1H} NMR shifts. The compounds isolated by P. Arnold and co-workers display a 13C{1H} shift at ~213 ppm for the CeIV–NHC22. The compounds reported by Liddle contain CeIV − C (bis(iminophosphorano)methandiide) 13C{1H} shifts in the range of 324.6–343.5 ppm, depending on the secondary ligands bound to the CeIV cation; [Ce(BIPMTMS)(ODipp)2] exhibits a 13C{1H} shift of 324.6 ppm23,25. Notably, these compounds contain substantially different substituents attached to the Ce–C carbon, diminishing the significance of their comparison.
Fig. 5. Identification of the unusual 13C resonance.
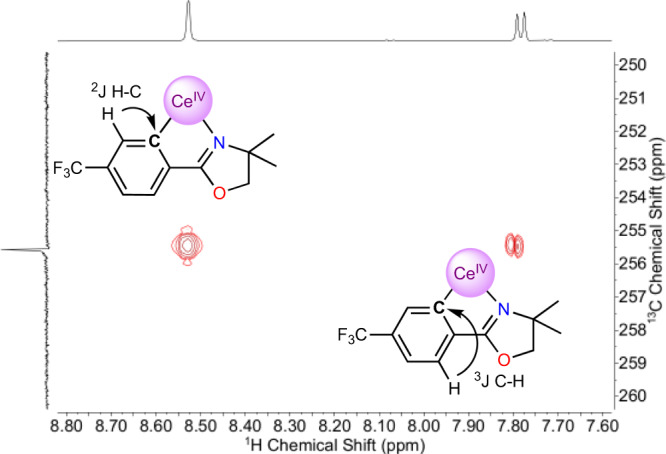
A portion of the HMBC spectrum showing the long-range C-H coupling to the 13C resonance at 255.6 ppm. An interpretation of the spectrum is inlaid, indicating that the signal at 255.6 ppm corresponds to the ipso-carbon.
Computational bonding analysis
To further understand the nature of the CeIV − Caryl interaction, we turned to computations to assess the electronic structure of the anionic, cerium-containing portion of 3-THF (referred to as 3). The geometry of 3 was optimized starting from the structure of 3-THF determined by X-ray crystallography, using density functional theory (DFT) with the B3LYP functional, all-electron Slater-type basis sets for all atoms, and other standard settings as detailed in the SI. The agreement between experiment and theory was excellent, with only minor differences in the CeIV − ligand bond lengths (≤0.02 Å). The MOs with the most Ce 4f character remain largely metal-centered and span the seven lowest unoccupied molecular orbitals (LUMO to LUMO + 6, Supplementary Figs. 13–19) of the complex, a common feature for CeIV compounds as well as for cerium species with a debated CeIV/CeIII oxidation state3,15. HOMO to HOMO − 3 (Supplementary Figs. 26–29), for 3 are mostly delocalized phenoxide-centered orbitals, while HOMO − 4 (Fig. 6) corresponds to the highest occupied orbital showing significant ligand-metal (CeIV − Caryl) hybridization.
Fig. 6. MO’s of 3 depicting the Ce–Caryl bond.
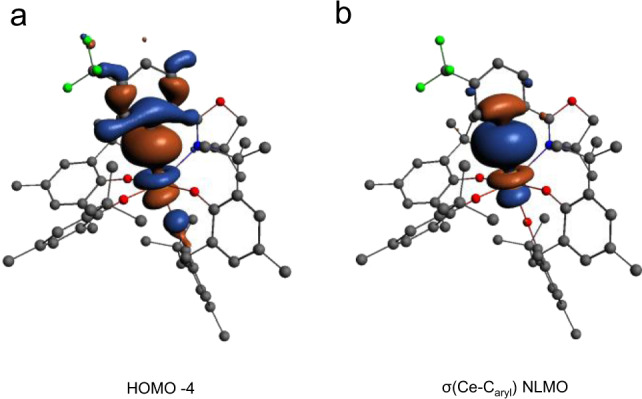
a The DFT/B3LYP frontier Kohn-Sham molecular orbital of 3 (HOMO − 4). b The two-center two-electron bonding NLMO. Atomic orbital contributions of the NLMOs and other bond properties of all the Ce containing bonds are listed in Table 1.
The metal-ligand bonding in 3 is characterized in Fig. 6 and Table 1 in terms of natural localized molecular orbitals (NLMOs) and bond orders. There are two two-center two-electron σ bonds describing the donation bonding between the aryl carbon and oxazolinide nitrogen and Ce, and σ + 2π bonds describing the bonding between each of the O atoms and Ce (Supplementary Figs. 37–40). Among these, the CeIV − Caryl σ bond shown in Fig. 6 has the largest covalency, in terms of shared density, with 12% Ce contribution of which 32/62% involve 5d/4f. Previously reported CeIV − C bonds contain 8–13% Ce contribution25. Ce tends to be less covalent than UVI (UVI − C bonds 22–29% U) but similar to ThIV (ThIV − Caryl bonds 10–15%)9,25,38,40. In the remaining σ and π bonds with the N and O atoms, Ce contributes about 3–4% weight, suggesting that these bonds are mainly ionic. The bond ionicities are also reflected in the bond orders compiled in Table 1, all being significantly smaller than one (which would indicate a full single bond). In the sum of all interactions, however, Ce receives sizable electron donation from the surrounding ligands. For instance, the calculated Ce charge is +2.26 (Mulliken charge), +2.46 (Bader charge), and +2.44 (natural charge from a natural bond orbital (NBO) analysis) instead of the formal +4. The Ce natural electron configuration retrieved from the NBO analysis is 4f0.76d0.60, which deviates considerably from the formal 4f05d0. The large Ce 4f electron count of 3 (0.76), associated mainly with the sizable Ce − Caryl bonding, is comparable to the calculated and experimentally-determined Ce 4f electron counts in CeO2 and Ce(C8H8)241–43. We anticipate that this similarity has important implications regarding the electronic structure of 3, in the sense that it may potentially exhibit a multi-configurational ground-state wavefunction with CeIII/IV character, similar to cerocene. However, further spectroscopic studies are needed, and are under way, to confirm this assignment for 3.
Table 1.
NLMO compositions and bond orders for 3.
| Bond/NLMO | %Ce (%s; %d; %f) | WBO[a] | MBO[b] | DIQTAIM[c] |
|---|---|---|---|---|
| σ(Ce–C) | 12 (5; 32; 62) | 0.41 | 0.46 | 0.50 |
| σ(Ce–N) | 4 (14; 49; 37) | 0.15 | 0.11 | 0.25 |
| σ(Ce–O)[d] | 3 (14; 50; 37) | 0.41 | 0.46 | 0.62 |
| 2xπ(Ce–O)[d] | 4 (1; 38; 61) |
aWiberg bond order in the natural atomic orbital (NAO) basis.
bMayer–Mulliken bond order.
cDelocalization index based on QTAIM Bader analysis.
dThe data are given as averages over the four Ce-O bonds.
Computational chemical shift analysis
Computed 13C NMR chemical shifts for the ipso-carbon, with various approaches, are compiled in Supplementary Table 2. The NMR shift was sensitive to the applied DFT approximations, a common observation in NMR shift calculations for compounds containing lanthanides and actinides. The best agreement with the experiment (256 ppm) for the ipso-13C chemical shift in 3 was obtained with a PBE hybrid with 40% exact exchange, PBEh-40, which gave 259 ppm. The same functional previously provided excellent ligand chemical shifts in actinide complexes40,44. PBEh-40 produced a similar cerium electronic structure (Mulliken/natural charge of +2.43/+2.66, 4f0.585d0.59 NBO natural electron configuration, Ce − Caryl WBO of 0.37) as B3LYP. Reasonable agreement with the experimental chemical shift was obtained also with the KT2 functional (265 ppm), which is known to perform well in NMR calculations45. The comparison between the ipso-13C chemical shift calculated without and with SOC, with PBEh-40/KT2, reveals a 40/51 ppm downfield shift caused by SOC, which is largely triggered by the Ce 4f and 5d involvement in the CeIV − Caryl σ bond.
In order to rationalize the anomalous 13C shift, we carried out an analysis of the DFT/PBEh-40 13C isotropic shielding (σiso) in terms of NLMOs46,47. The NMR shielding data are gathered in Table 2 and the relevant NLMOs are shown in Supplementary Fig. 41. Note that these NLMOs are equivalent to those obtained with DFT/B3LYP. The analysis shows that the σ(Ce − Caryl) covalent bond is the principal cause of the SOC-induced deshielding of the ipso-carbon. According to Table 2, the core orbital of the Ce-bound Caryl brings a diamagnetic contribution of roughly 200 ppm to σiso. This value is counterbalanced by two large, negative shielding contributions generated by the σ bonds involving the ipso-carbon, namely the σ(Ce − Caryl) bond and the two σ(Caryl − C) bonds involving the two neighboring carbons of the aryl ring, denoted as C1 and C2. From Table 2 it is evident that the σ(Ce − Caryl) contribution to σiso is as important (without SOC) or even more important (with SOC) than the combined σiso contributions generated by the σ(Caryl − C) NLMOs.
Table 2.
NLMO contributions to the 13C isotropic nuclear shielding (σiso) in 3a.
| σisoSR, SR-ZORA | σisoSO, SO-ZORA | ΔSO d | |||||||
|---|---|---|---|---|---|---|---|---|---|
| NLMOb | Lc | NLc | L + NL | L | NL | L + NL | L | NL | L + NL |
| σ(Ce-Caryl) | −103 | 2 | −102 | −156 | 2 | −154 | −52 | 0 | −52 |
| Σσ(Caryl-C1,2) | −106 | −5 | −110 | −94 | −5 | −99 | 12 | −1 | 11 |
| 1score (Caryl) | 201 | 0 | 201 | 202 | 0 | 202 | 1 | 0 | 1 |
| Σother | −26 | 11 | −15 | −25 | 11 | −14 | 1 | 0 | 1 |
| Σ(all of above) | −34 | 8 | −26 | −73 | 8 | −65 | −38 | −1 | −39e |
aDFT/PBEh-40 calculations. SR = scalar relativistic. SO = relativistic calculation including SOC. All shielding contributions are in ppm. The geometry orientation is such that the 13C corresponds to the origin of the cartesian axes and C–Ce bond aligns with the z-axis.
bThe NLMOs are shown in Supplementary Fig. 41.
cL and NL indicate contributions from the Lewis and non-Lewis parts of the NLMO.
dDefined as σisoSO − σisoSR.
eSOC effects of +1/−39 ppm on the reference/probe shielding cause the +40 ppm SOC change in the chemical shift quoted in the text.
Indeed, with SOC in particular, the σ(Ce − Caryl) NLMO yields a dominant negative contribution to σiso of −154 ppm, of which −169 ppm is paramagnetic and 15 ppm is diamagnetic (not shown separately in Table 2), while the combined σiso contribution from the σ(Caryl − C1,2) NLMOs is −99 ppm. The overall SOC effects on the total isotropic shielding, σiso, add up to −39 ppm. The largest contribution to this value is generated by the σ(Ce − Caryl) NLMO, −52 ppm, and there are secondary contributions from the σ(Caryl − C1,2) NLMOs, 11 ppm (Table 2, last column), indicating that these orbitals are somewhat delocalized onto the metal. Hence, the covalent bonding between the Ce center and Caryl is the main contributor of the 13C SOC deshielding and this aspect is strongly related to the sizable Ce 4f and 5d character of the σ(Ce − Caryl) NLMO: 53% f and 41% d with PBEh-40, or 62% f and 32% d with B3LYP, with the 4f likely generating most of the SOC deshielding.
Discussion
We have synthesized, characterized, and crystallized complexes featuring a CeIV − Caryl bond. The synthesis of the title complexes 3-THF and 3-DME was realized from the reaction of the lithium-aryl salt 2 with 1. Electrochemical analysis revealed that the aryl interaction resulted in a notable stabilization of the CeIV oxidation state, shifting the Epc of CeIV reduction by 720 mV relative to the THF adduct. A combination of 13C{1H} NMR and DFT was used to investigate the covalency of the CeIV − Caryl bonding. 13C{1H} NMR analysis revealed that the ipso-carbon was shifted to 255.6 ppm, an indicator of metal-ligand covalency in the CeIV − Caryl bond. This result was supported by NLMO analysis, which showed a 12% metal contribution to the CeIV − Caryl bond. We expect that these results will further inform fundamental bonding in high valent f-elements and be effective in guiding the preparation of other f-element organometallic complexes.
Methods
General considerations
See Supplementary Methods for further details.
Ce(THF)2(MBP)2 (1)
We previously reported a synthesis of 1 that could not be separated from the lithium halide byproducts29. This revised method provides clean 1. In an N2 filled drybox, to a clear, colorless solution of H2MBP (0.270 g, 0.793 mmol, 2 equiv) in 4 mL of THF in a 20 mL scintillation vial with a Teflon coated stir bar, was added a yellow solution of Ce(OtBu)4(THF)2 (0.200 g, 0.396 mmol, 1 equiv) in a 6 mL solution of 2:1 THF:benzene at room temperature with stirring. The reaction immediately turned an intense purple color and was stirred for 1 h. The volatile materials were removed under reduced pressure, the residue was triturated with 2 mL of benzene to liberate the tert-butanol byproduct, and the volatile materials were again removed under reduced pressure. The resulting purple solid was transferred onto a medium porosity fritted filter and washed with 5 × 2 mL of pentane. The purple solid was dried under reduced pressure for 3 h. Yield: 0.311 g, 0.324 mmol, 82%.
NMR data for this complex was not previously reported and is provided here:
1H NMR (400 MHz, THF-d8) δ: 7.15 (s, 4H), 6.79 (s, 4H), 5.01 (d, J = 13.4 Hz, 2H) 3.51 (d, J = 14.0 Hz, 2H), 2.31 (s, 12 H), 1.44 (s, 36 H).
13C{1H} NMR (100 MHz, THF-d8) δ: 168.12, 137.25, 134.45, 129.13, 128.17, 124.12, 35.40, 34.99, 31.15, 20.93.
The quantity of THF present for 1 was verified by 1H-NMR in C6D6.
[Li(THF)][ortho-oxa] (2)
Synthesis adapted from similar compounds48. In a N2 filled drybox, a solution containing H-ortho-oxa (1.217 g, 5.0 mmol, 1 equiv) and 10 mL of hexanes in a 20 mL scintillation vial with a Teflon coated stir bar was placed in a −30 °C freezer for 30 mins. The vial was removed from the freezer and, while stirring, a solution of n-butyl lithium (2.5 M, 5 mmol, 2 mL) was added dropwise over 5 min. The solution turned from colorless to yellow to brown and a yellow solid precipitated. The reaction mixture was stirred for 50 min at room temperature, after which the solid was collected by filtration over a coarse-porosity fritted-filter and subsequently washed with 3 × 2 mL of hexanes and 1 × 2 mL of pentane. The tan solid was then dried under reduced pressure for 2 h. The solid was then dissolved in minimal THF at rt and then placed in a −30 °C freezer overnight. Yellow crystalline blocks formed and were collected over a coarse-porosity fritted-filter and washed with 3 × 2 mL of pentane. The yellow blocks were dried for 2 h under reduced pressure. Yield: 0.831 g, 2.59 mmol, 52%.
1H NMR (400 MHz, THF-d8) δ: 8.28 (s, 1H), 7.57 (d, J = 8.0 Hz, 1H), 7.07 (dd, J = 8.0, 2.7 Hz, 1H), 4.17 (s, 2H), 1.34 (s, 6H).
13C{1H} NMR (101 MHz, THF-d8) δ: 203.98, 172.88, 143.18 (q, J = 2.2 Hz), 137.45 (q, J = 3.3 Hz), 127.47 (q, J = 274.1 Hz), 127.31 (q, J = 28.2 Hz), 124.46, 119.28 (q, J = 4.0 Hz), 80.39, 66.58, 29.03.
19F NMR (376 MHz, THF-d8) δ: –64.29
7Li NMR (156 MHz, THF-d8) δ: 2.08
Anal. Cal. for C12H11F3LiNO•(C4H8O)0.5: C, 58.96; H, 5.30; N, 4.91. Found C, 59.41; H, 5.41; N, 4.75.
The quantity of THF present for 2 was verified by 1H-NMR in C6D6.
[Li(THF)4][Ce(ortho-oxa)(MBP)2 (3-THF)
In an N2 filled drybox, two 20 mL scintillation vials were placed in a −30 °C freezer. One contained a dark purple solution of 1 (0.200 g, 0.208 mmol, 1 equiv) in 4 mL of benzene with a Teflon coated stir bar and the other contained a yellow solution of 2 (0.067 g, 0.208 mmol, 1 equiv) in 4 mL of benzene. After cooling for 30 min, the now frozen solution of 2 was removed from the freezer and allowed to thaw. Immediately upon thawing, the frozen solution of 1 was removed from the freezer and the solution of 2 was added dropwise at rt over 2 min. Upon mixing, the solution immediately changed from a dark purple to a dark red color and was allowed to stir for 5 min at rt. At this point the volatile materials were removed under vacuum. The resulting solid was redissolved in a mixture of 3 mL of toluene and 8 drops of THF in an 8 mL scintillation vial. This solution was layered with 5 mL of pentane and placed in a −30 °C freezer for 3 days. During this time, red crystals formed, and were collected by filtration over a medium porosity fritted filter and washed with cold pentane 5 × 2 mL. Yield: 0.198 g, 0.137 mmol, 66%.
1H NMR (500 MHz, THF-d8) δ: 8.53 (s, 1H), 7.78 (d, J = 8.0 Hz, 1H), 7.12 (d, J = 7.5 Hz, 1H), 7.01 (s, 2H), 6.92 (s, 2H), 6.75 (s, 1H), 6.66 (s, 1H), 6.62 (s, 1H), 6.58 (s, 1H), 5.10 (d, J = 13.4 Hz, 1H), 4.72 (d, J = 13.5 Hz, 1H), 4.32 (s, 1H), 4.01 (s, 1H), 3.23 (d, J = 13.4 Hz, 1H), 3.10 (d, J = 13.6 Hz, 1H), 2.27 − 2.05 (m, 12H), 1.57 (s, 3H), 1.45 (s, 9H), 1.39 (s, 9H), 1.24 (s, 3H), 1.16 (s, 9H), 1.08 (s, 9H).
13C{1H} NMR (126 MHz, THF-d8) δ: 255.58, 174.49, 168.21, 167.40, 166.81, 137.97, 137.72, 137.33, 137.11, 136.98, 136.79, 135.03, 134.90, 134.58, 132.58 (q, J = 3.1 Hz), 131.11 (q, J = 29.3 Hz), 128.85, 128.75, 128.61, 127.02, 126.64 (q, J = 177 Hz), 126.08, 124.25, 124.11, 123.95, 123.60, 120.18 (q, J = 4.1 Hz), 82.35, 68.88, 35.89, 35.71, 35.60, 35.03, 32.46, 31.54, 31.09, 30.97, 30.16, 21.31, 21.17.
19F NMR (470 MHz, THF-d8) δ: −62.44
7Li NMR (194 MHz, THF-d8) δ: −0.57
X-ray quality crystals were obtained from a vapor diffusion of pentane into concentrated solutions of 3 in a solution consisting of 1:2 THF:tolulene in a −30 °C freezer.
Anal. Cal. for C74H99CeF3LiNO9•(C7H8): C, 65.61; H, 7.66; F, 3.95; N, 1.03. Found C, 65.21; H, 6.65; N, 1.30. Best result of three attempts.
UV-Vis: λ = 460 nm (ε = 7533 Lmol−1 cm−1), λ = 292 nm (ε = 24,426 Lmol−1 cm−1).
[Li(DME)3][Ce(ortho-oxa)(MBP)2 (3-DME)
In an N2 filled drybox, two 20 mL scintillation vials were placed in a − 30 °C freezer. One contained a dark purple solution of 1 (0.100 g, 0.104 mmol, 1 equiv) in 2 mL of benzene with a Teflon coated stir bar and the other contained a yellow solution of 2 (0.034 g, 0.104 mmol, 1 equiv) in 2 mL of benzene. After cooling for 30 min, the now frozen solution of 2 was removed from the freezer and allowed to thaw. Immediately upon thawing, the frozen solution of 1 was removed from the freezer and the solution of 2 was added dropwise at rt over 2 min. Upon mixing, the solution immediately changed from a dark purple to a dark red color and was allowed to stir for 5 min at rt. At this point the volatile materials were removed under reduced pressure. The resulting solid was dissolved in of 3 mL of DME in an 8 mL scintillation vial. This solution was layered with 5 mL of pentane and placed in a −30 °C freezer for 3 days. During this time, red crystals formed, and were collected by filtration over a medium porosity fritted filter and washed with cold pentane 5 × 2 mL. Yield: 0.111 g, 0.079 mmol, 75%.
1H NMR (500 MHz, THF-d8) δ: 8.53 (s, 1H), 7.80 (d, J = 7.9 Hz, 1H), 7.13 (dd, J = 8.1 Hz, 2.6 Hz, 1H), 7.01 (s, 2H), 6.91 (s, 2H), 6.76 (s, 1H), 6.68 (s, 1H), 6.64 (s, 1H), 6.59 (s, 1H), 5.12 (d, J = 13.3 Hz, 1H), 4.72 (d, J = 13.4 Hz, 1H), 4.33 (d, J = 7.6 Hz, 1H), 4.01 (d, J = 6.6 Hz 1H), 3.24 (d, J = 13.6 Hz, 1H), 3.11 (d, J = 13.6 Hz, 1H), 2.24 (s, 3H), 2.19 (s, 9H), 1.58 (s, 3H), 1.46 (s, 9H), 1.40 (s, 9H), 1.25 (s, 3H), 1.17 (s, 9H), 1.09 (s, 9H).
13C{1H} NMR (126 MHz, THF-d8) δ: 255.61, 174.55, 168.23, 167.46, 166.87, 138.03, 137.77, 137.40, 137.18, 137.05, 136.84, 135.08, 134.94, 134.63, 132.65 (q, J = 3.5 Hz), 131.18 (q, J = 29.5 Hz), 129.03, 128.82, 128.66, 127.22 (q, J = 177 Hz), 127.06, 126.14, 124.29, 124.18, 123.99, 123.66, 120.24 (q, J = 4.0 Hz), 82.41, 68.93, 35.94, 35.76, 35.68, 35.09, 32.52, 31.59, 31.14, 31.03, 30.22, 21.36, 21.21.
19F NMR (470 MHz, THF-d8) δ: −62.44
7Li NMR (194 MHz, THF-d8) δ: −0.57
X-ray quality crystals were obtained from a layering of pentane on top of a saturated solution of 3-DME in DME (1:1, DME:Pentane).
Anal. Cal. for C70H101CeF3LiNO11: C, 62.90; H, 7.62; N, 1.05. Found C, 62.45; H, 7.32; N, 1.55.
Supplementary information
Acknowledgements
E.J.S., P.J.W., and G.B.P. gratefully acknowledge the U.S. National Science Foundation (CHE-1955724 to E.J.S., CHE-1902509 to P.J.W., and Graduate Research Fellowship Program NSF-GRFP to G.B.P.). We also thank the University of Pennsylvania for support of this work. D.C.S. and J.A. acknowledge support from the U.S. Department of Energy, Office of Basic Energy Sciences, Heavy Elements program, under grant DE-SC0001136. We thank the Center for Computational Research (CCR) at the University of Buffalo for providing computational resources.
Author contributions
G.B.P. and E.J.S conceived this project. G.B.P. performed the synthesis, electrochemical, NMR, and UV-Vis experiments. M.R.G. and P.J.C. collected and solved the X-ray structures. G.B.P., P.J.W., and E.J.S. analyzed the experimental data. D.-C.S. and J.A. conducted the theoretical computations and analyzed the results. G.B.P., D.-C.S., J.A., P.J.W. and E.J.S. participated in drafting the paper. All authors discussed the results and contributed the preparation of the final paper.
Data availability
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition nos. CCDC 1998883 (3-THF) and 2043597 (3-DME). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available within the Article and its Supplementary Information and from the corresponding authors upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks Justin Walensky and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Jochen Autschbach, Email: jochena@buffalo.edu.
Patrick J. Walsh, Email: pwalsh@sas.upenn.edu
Eric J. Schelter, Email: schelter@sas.upenn.edu
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-021-21766-4.
References
- 1.Raymond KN, Eigenbrot CW. Structural criteria for the mode of bonding of organoactinides and -lanthanides and related compounds. Acc. Chem. Res. 1980;13:276–283. doi: 10.1021/ar50152a005. [DOI] [Google Scholar]
- 2.Kaltsoyannis, N. & Scott, P. The f elements. (Oxford University Press, 1999).
- 3.Löble MW, et al. Covalency in Lanthanides. An X-ray Absorption Spectroscopy and Density Functional Theory Study of LnCl6x– (x = 3, 2) J. Am. Chem. Soc. 2015;137:2506–2523. doi: 10.1021/ja510067v. [DOI] [PubMed] [Google Scholar]
- 4.Cheisson T, et al. Multiple Bonding in Lanthanides and Actinides: Direct Comparison of Covalency in Thorium(IV)- and Cerium(IV)-Imido Complexes. J. Am. Chem. Soc. 2019;141:9185–9190. doi: 10.1021/jacs.9b04061. [DOI] [PubMed] [Google Scholar]
- 5.Kerridge A. Quantification of f-element covalency through analysis of the electron density: insights from simulation. Chem. Commun. 2017;53:6685–6695. doi: 10.1039/C7CC00962C. [DOI] [PubMed] [Google Scholar]
- 6.Kaindl G, Schmiester G, Sampathkumaran EV, Wachter P. Pressure-induced changes in LIII x-ray-absorption near-edge structure of CeO2 and CeF4: Relevance to 4f-electronic structure. Phys. Rev. B.: Condens. Matter. 1988;38:10174–10177. doi: 10.1103/PhysRevB.38.10174. [DOI] [PubMed] [Google Scholar]
- 7.Williams UJ, et al. Synthesis, Bonding, and Reactivity of a Cerium(IV) Fluoride Complex. Inorg. Chem. 2014;53:27–29. doi: 10.1021/ic402769u. [DOI] [PubMed] [Google Scholar]
- 8.Lewis AJ, Carroll PJ, Schelter EJ. Stable Uranium(VI) Methyl and Acetylide Complexes and the Elucidation of an Inverse Trans Influence Ligand Series. J. Am. Chem. Soc. 2013;135:13185–13192. doi: 10.1021/ja406610r. [DOI] [PubMed] [Google Scholar]
- 9.Mullane KC, et al. 13C NMR Shifts as an Indicator of U–C Bond Covalency in Uranium(VI) Acetylide Complexes: An Experimental and Computational Study. Inorg. Chem. 2019;58:4152–4163. doi: 10.1021/acs.inorgchem.8b03175. [DOI] [PubMed] [Google Scholar]
- 10.Smiles DE, Wu G, Hrobárik P, Hayton TW. Use of 77Se and 125Te NMR Spectroscopy to Probe Covalency of the Actinide-Chalcogen Bonding in [Th(En){N(SiMe3)2}3]− (E = Se, Te; n = 1, 2) and Their Oxo-Uranium(VI) Congeners. J. Am. Chem. Soc. 2016;138:814–825. doi: 10.1021/jacs.5b07767. [DOI] [PubMed] [Google Scholar]
- 11.Staun SL, Sergentu D-C, Wu G, Autschbach J, Hayton TW. Use of 15N NMR spectroscopy to probe covalency in a thorium nitride. Chem. Sci. 2019;10:6431–6436. doi: 10.1039/C9SC01960J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smiles DE, Wu G, Hrobárik P, Hayton TW. Synthesis, Thermochemistry, Bonding, and 13C NMR Chemical Shift Analysis of a Phosphorano-Stabilized Carbene of Thorium. Organometallics. 2017;36:4519–4524. doi: 10.1021/acs.organomet.7b00202. [DOI] [Google Scholar]
- 13.Rungthanaphatsophon P, Huang P, Walensky JR. Phosphorano-Stabilized Carbene Complexes with Short Thorium(IV)– and Uranium(IV)–Carbon Bonds. Organometallics. 2018;37:1884–1891. doi: 10.1021/acs.organomet.8b00137. [DOI] [Google Scholar]
- 14.Walter MD, Booth CH, Lukens WW, Andersen RA. Cerocene Revisited: the Electronic Structure of and Interconversion Between Ce2(C8H8)3 and Ce(C8H8)2. Organometallics. 2009;28:698–707. doi: 10.1021/om7012327. [DOI] [Google Scholar]
- 15.Smiles DE, et al. The duality of electron localization and covalency in lanthanide and actinide metallocenes. Chem. Sci. 2020;11:2796–2809. doi: 10.1039/C9SC06114B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anwander R, Dolg M, Edelmann FT. The difficult search for organocerium(iv) compounds. Chem. Soc. Rev. 2017;46:6697–6709. doi: 10.1039/C7CS00147A. [DOI] [PubMed] [Google Scholar]
- 17.Greco A, Cesca S, Bertolini W. New π-cyclooctatetraenyl and π-cyclopentadienyl complexes of cerium. J. Organomet. Chem. 1976;113:321–330. doi: 10.1016/S0022-328X(00)96143-6. [DOI] [Google Scholar]
- 18.Kilimann U, Herbst-Irmer R, Stalke D, Edelmann FT. An Efficient Access to Organocerium(IV) Complexes: Synthesis and Structure of Bis[1,3,6-tris(trimethylsilyl)cyclooctatetraene]cerium(IV) Angew. Chem. Int. Ed. 1994;33:1618–1621. doi: 10.1002/anie.199416181. [DOI] [Google Scholar]
- 19.Balazs G, et al. Cerium(III) and Cerium(IV) Bis(η8-pentalene) Sandwich Complexes:synthetic, Structural, Spectroscopic, and Theoretical Studies. Organometallics. 2007;26:3111–3119. doi: 10.1021/om7002738. [DOI] [Google Scholar]
- 20.Schneider D, Harmgarth N, Edelmann FT, Anwander R. Ceric Cyclopentadienides Bearing Alkoxy, Aryloxy, Chlorido, or Iodido Co-Ligands. Chem. -Eur. J. 2017;23:12243–12252. doi: 10.1002/chem.201700743. [DOI] [PubMed] [Google Scholar]
- 21.Sutton AD, Clark DL, Scott BL, Gordon JC. Synthesis and Characterization of Cerium(IV) Metallocenes. Inorganics. 2015;3:589–596. doi: 10.3390/inorganics3040589. [DOI] [Google Scholar]
- 22.Casely, I. J., Liddle, S. T., Blake, A. J., Wilson, C. & Arnold, P. L. Tetravalent cerium carbene complexes. Chem. Commun., 5037–5039, (2007). [DOI] [PubMed]
- 23.Gregson M, et al. A Cerium(IV)–Carbon Multiple Bond. Angew. Chem. Int. Ed. 2013;52:13016–13019. doi: 10.1002/anie.201306984. [DOI] [PubMed] [Google Scholar]
- 24.Arnold PL, Casely IJ, Zlatogorsky S, Wilson C. Organometallic Cerium Complexes from Tetravalent Coordination Complexes. Helv. Chim. Acta. 2009;92:2291–2303. doi: 10.1002/hlca.200900152. [DOI] [Google Scholar]
- 25.Gregson M, et al. The inverse-trans-influence in tetravalent lanthanide and actinide bis(carbene) complexes. Nat. Commun. 2017;8:14137. doi: 10.1038/ncomms14137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson JR, Carroll PJ, Walsh PJ, Schelter EJ. The Impact of Ligand Reorganization on Cerium(III) Oxidation Chemistry. Angew. Chem. Int. Ed. 2012;51:10159–10163. doi: 10.1002/anie.201203481. [DOI] [PubMed] [Google Scholar]
- 27.Levin JR, Dorfner WL, Dai AX, Carroll PJ, Schelter EJ. Density Functional Theory as a Predictive Tool for Cerium Redox Properties in Nonaqueous Solvents. Inorg. Chem. 2016;55:12651–12659. doi: 10.1021/acs.inorgchem.6b01779. [DOI] [PubMed] [Google Scholar]
- 28.Piro NA, Robinson JR, Walsh PJ, Schelter EJ. The electrochemical behavior of cerium(III/IV) complexes: Thermodynamics, kinetics and applications in synthesis. Coord. Chem. Rev. 2014;260:21–36. doi: 10.1016/j.ccr.2013.08.034. [DOI] [Google Scholar]
- 29.Mahoney BD, Piro NA, Carroll PJ, Schelter EJ. Synthesis, Electrochemistry, and Reactivity of Cerium(III/IV) Methylene-Bis-Phenolate Complexes. Inorg. Chem. 2013;52:5970–5977. doi: 10.1021/ic400202r. [DOI] [PubMed] [Google Scholar]
- 30.Robinson JR, et al. Tuning Reactivity and Electronic Properties through Ligand Reorganization within a Cerium Heterobimetallic Framework. J. Am. Chem. Soc. 2013;135:19016–19024. doi: 10.1021/ja410688w. [DOI] [PubMed] [Google Scholar]
- 31.Halter DP, et al. Electrocatalytic H2O Reduction with f-Elements: mechanistic Insight and Overpotential Tuning in a Series of Lanthanide Complexes. J. Am. Chem. Soc. 2018;140:2587–2594. doi: 10.1021/jacs.7b11532. [DOI] [PubMed] [Google Scholar]
- 32.Watanabe T, Matsuo T, Kawaguchi H. A Tantalum(V) Carbene Complex:formation of a Carbene−Bis(phenoxide) Ligand by Sequential Proton and Hydride Abstraction. Inorg. Chem. 2006;45:6580–6582. doi: 10.1021/ic0608492. [DOI] [PubMed] [Google Scholar]
- 33.Akagi F, Ishida Y, Matsuo T, Kawaguchi H. Synthesis and reactivity of niobium complexes having a tripodal triaryloxide ligand in bidentate, tridentate, and tetradentate coordination modes. Dalton T. 2011;40:2375–2382. doi: 10.1039/c0dt01022g. [DOI] [PubMed] [Google Scholar]
- 34.Manz, T. A., Fenwick, A. E., Phomphrai, K., Rothwell, I. P. & Thomson, K. T. The nature of aryloxide and arylsulfide ligand bonding in dimethyltitanium complexes containing cyclopentadienyl ligation. Dalton Trans., 668–674, (2005). [DOI] [PubMed]
- 35.Maron L, Werkema EL, Perrin L, Eisenstein O, Andersen RA. Hydrogen for Fluorine Exchange in C6F6 and C6F5H by Monomeric [1,3,4-(Me3C)3C5H2]2CeH: Experimental and Computational Studies. J. Am. Chem. Soc. 2005;127:279–292. doi: 10.1021/ja0451012. [DOI] [PubMed] [Google Scholar]
- 36.Werkema EL, Andersen RA. Fluorine for Hydrogen Exchange in the Hydrofluorobenzene Derivatives C6HxF(6-2), where x = 2, 3, 4 and 5 by Monomeric [1,2,4-(Me3C)3C5H2]2CeH: the Solid State Isomerization of [1,2,4-(Me3C)3C5H2]2Ce(2,3,4,5-C6HF4) to [1,2,4-(Me3C)3C5H2]2Ce(2,3,4,6-C6HF4). J. Am. Chem. Soc. 2008;130:7153–7165. doi: 10.1021/ja800639f. [DOI] [PubMed] [Google Scholar]
- 37.Shannon R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A. 1976;32:751–767. doi: 10.1107/S0567739476001551. [DOI] [Google Scholar]
- 38.Pedrick EA, Hrobárik P, Seaman LA, Wu G, Hayton TW. Synthesis, structure and bonding of hexaphenyl thorium(iv): observation of a non-octahedral structure. Chem. Commun. 2016;52:689–692. doi: 10.1039/C5CC08265J. [DOI] [PubMed] [Google Scholar]
- 39.Seaman LA, et al. Comparison of the Reactivity of 2-Li-C6H4CH2NMe2 with MCl4 (M=Th, U): Isolation of a Thorium Aryl Complex or a Uranium Benzyne Complex. Angew. Chem. Int. Ed. 2013;52:10589–10592. doi: 10.1002/anie.201303992. [DOI] [PubMed] [Google Scholar]
- 40.Seaman LA, et al. A Rare Uranyl(VI)–Alkyl Ate Complex [Li(DME)1.5]2[UO2(CH2SiMe3)4] and Its Comparison with a Homoleptic Uranium(VI)–Hexaalkyl. Angew. Chem. Int. Ed. 2013;52:3259–3263. doi: 10.1002/anie.201209611. [DOI] [PubMed] [Google Scholar]
- 41.Mooßen O, Dolg M. Two interpretations of the cerocene electronic ground state. Chem. Phys. Lett. 2014;594:47–50. doi: 10.1016/j.cplett.2014.01.022. [DOI] [Google Scholar]
- 42.Hay PJ, Martin RL, Uddin J, Scuseria GE. Theoretical study of CeO2 and Ce2O3 using a screened hybrid density functional. J. Chem. Phys. 2006;125:034712. doi: 10.1063/1.2206184. [DOI] [PubMed] [Google Scholar]
- 43.Halbach RL, Nocton G, Booth CH, Maron L, Andersen RA. Cerium Tetrakis(tropolonate) and Cerium Tetrakis(acetylacetonate) Are Not Diamagnetic but Temperature-Independent Paramagnets. Inorg. Chem. 2018;57:7290–7298. doi: 10.1021/acs.inorgchem.8b00928. [DOI] [PubMed] [Google Scholar]
- 44.Hrobárik P, Hrobáriková V, Greif AH, Kaupp M. Giant Spin-Orbit Effects on NMR Shifts in Diamagnetic Actinide Complexes: Guiding the Search of Uranium(VI) Hydride Complexes in the Correct Spectral Range. Angew. Chem. Int. Ed. 2012;51:10884–10888. doi: 10.1002/anie.201204634. [DOI] [PubMed] [Google Scholar]
- 45.Keal TW, Tozer DJ. The exchange-correlation potential in Kohn–Sham nuclear magnetic resonance shielding calculations. J. Chem. Phys. 2003;119:3015–3024. doi: 10.1063/1.1590634. [DOI] [Google Scholar]
- 46.Autschbach J. Analyzing NMR shielding tensors calculated with two-component relativistic methods using spin-free localized molecular orbitals. J. Chem. Phys. 2008;128:164112. doi: 10.1063/1.2905235. [DOI] [PubMed] [Google Scholar]
- 47.Autschbach J, Zheng S. Analyzing Pt chemical shifts calculated from relativistic density functional theory using localized orbitals: The role of Pt 5d lone pairs. Magn. Reson. Chem. 2008;46:S45–S55. doi: 10.1002/mrc.2289. [DOI] [PubMed] [Google Scholar]
- 48.Jantzi KL, Guzei IA, Reich HJ. Solution and Solid-State Structures of Lithiated Phenyloxazolines. Organometallics. 2006;25:5390–5395. doi: 10.1021/om060551j. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Crystallographic data for the structures reported in this article have been deposited at the Cambridge Crystallographic Data Center (CCDC) under deposition nos. CCDC 1998883 (3-THF) and 2043597 (3-DME). These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. All other data supporting the findings of this study are available within the Article and its Supplementary Information and from the corresponding authors upon reasonable request.