

SUMMARY
To identify genes whose loss confers resistance to CHK1 inhibitors, we perform genome-wide CRISPR-Cas9 screens in non-small-cell lung cancer (NSCLC) cell lines treated with the CHK1 inhibitor prexasertib (CHK1i). Five of the top six hits of the screens, MYBL2 (B-MYB), LIN54, FOXM1, cyclin A2 (CCNA2), and CDC25B, are cell-cycle-regulated genes that contribute to entry into mitosis. Knockout of MMB-FOXM1 complex components LIN54 and FOXM1 reduce CHK1i-induced DNA replication stress markers and premature mitosis during Late S phase. Activation of a feedback loop between the MMB-FOXM1 complex and CDK1 is required for CHK1i-induced premature mitosis in Late S phase and subsequent replication catastrophe, indicating that dysregulation of the S to M transition is necessary for CHK1 inhibitor sensitivity. These findings provide mechanistic insights into small molecule inhibitors currently studied in clinical trials and provide rationale for combination therapies.
Graphical Abstract
In brief
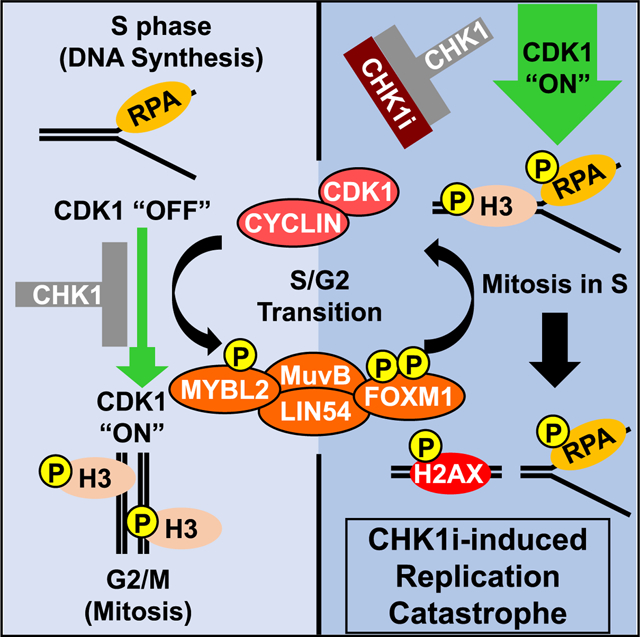
Branigan et al., by using genome-wide CRISPR screens, identify the MMB-FOXM1 complex as being required for CHK1 inhibitor (CHK1i) sensitivity. Their study shows that CHK1i-induced premature activation of the G2/M transcriptional program by this complex triggers a breakdown in the separation of DNA synthesis and mitosis, leading to replication catastrophe.
INTRODUCTION
Duplication of the eukaryotic cellular genome occurs in an orderly process during the S phase of cell division. Replication of the genome must be completed prior to mitosis to avoid events that result in genomic instability. DNA replication stress can occur when oncogenes, genotoxic agents, or inhibitors of DNA replication cause stalled replication forks, leading to activation of DNA damage response (DDR) pathways. Checkpoint Kinase 1 (CHK1) is activated by Ataxia Telangiectasia and Rad3-related protein (ATR) phosphorylation, at SQ sites S317 and S345 (Kim et al., 1999; Zhao and Piwnica-Worms, 2001), to resolve incomplete DNA replication structures consisting at least partly of single-stranded DNA (Lupardus et al., 2002; Saldivar et al., 2017; Zou and Elledge, 2003). Replication stress also leads to the phosphorylation of the replication fork component RPA32 on S4, S8, and S33 and the regulator of heterochromatin KAP1 (also known as TRIM28 or TIF1B) on S824 to remedy this altered DNA replication (Ashley et al., 2014; Liu et al., 2012; Olson et al., 2006; Yajima et al., 2009). Unresolved replication stress can lead to cell death by replication catastrophe, in which exhaustion of replication factors, such as RPA, triggers widespread DNA double-strand breaks (DSBs) marked by γ-H2AX (Furuta et al., 2003; Toledo et al., 2013). Understanding how the ATR-CHK1 pathway prevents replication catastrophe is critical, as inhibitors of ATR or CHK1 are being considered for clinical use.
The ATR-CHK1 pathway limits DNA replication stress during S phase by coordinating origin firing with CDK activity. ATR-activated CHK1 antagonizes CDK1 activity by activating the CDK1-inhibitory WEE1 kinase and suppressing the CDK1-activating CDC25 phosphatases (Furnari et al., 1997; O’Connell et al., 1997). Suppression of CDK1 activity during S phase by ATR and CHK1 prevents premature activation of the transcription factor FOXM1 and expression of mitotic genes (Saldivar et al., 2018). Cyclin A (CCNA2)/CDK1 activity however is required for late origin firing (Katsuno et al., 2009). Therefore, complete suppression of CDK activity would decrease origin firing and subsequent activation of ATR. During S phase, the ATR-CHK1 pathway maintains a careful balance of sufficient CDK1 activity to complete DNA replication but not enough to trigger mitosis.
Transcriptional regulation also plays a key role in managing cell cycle progression. During cell cycle entry, an E2F-dependent wave of gene expression during the G1/S transition facilitates DNA replication, and the MYBL2/MuvB (MMB)-FOXM1 complex drives a second wave of gene expression during G2/M that promotes mitosis. During S phase, the MuvB complex, comprised of LIN54, LIN9, LIN37, LIN52, and RBBP4, together with MYBL2 binds to cell cycle gene homology region (CHR) DNA motifs, which are LIN54 binding sites, at promoters of genes whose expression is required for mitosis (Knight et al., 2009; Litovchick et al., 2007; Müller et al., 2012; Pilkinton et al., 2007; Schmit et al., 2009). FOXM1 is subsequently recruited to these promoters in an MMB-dependent fashion. This recruitment coincides with an increase in the levels of several hundred G2/M cell cycle genes, including CCNA2, CCNB1, PLK1, and CDC25B (Chen et al., 2013; Down et al., 2012; Fischer et al., 2016; Sadasivam et al., 2012).
Due to oncogene-driven acceleration of cell division, cancer cells may become hyper-dependent on the ATR-CHK1 pathway to manage replication stress and maintain sufficient genomic integrity for continued survival. Inhibitors of ATR and CHK1 have been developed as anti-cancer agents to exploit this vulnerability. Monotherapy with the CHK1 inhibitor (CHK1i) prexasertib has shown safety and tolerability (Hong et al., 2016, 2018). Prexasertib and CHK1 depletion have little to no effect on non-transformed cells such as RPE-1, whereas CHK1i mono-therapy exhibits activity in multiple cancer cell lines (Blosser et al., 2020; Cole et al., 2011; King et al., 2015; Koppenhafer et al., 2018). However, intrinsic or acquired resistance to CHK1i may limit clinical efficacy. Given the myriad roles of CHK1, such resistance could potentially emerge by a wide range of possible mechanisms. CRISPR-Cas9 pooled whole-genome single guide RNA (sgRNA) knockout (KO) screens have emerged as particularly useful tools for unbiased discovery of gene losses that confer a phenotype of interest (Hanna and Doench, 2020). Prior CRISPR screens with ATR inhibitors (ATRis) have implicated both the cell cycle and DDR regulatory roles of the ATR-CHK1 pathway in modulating inhibitor sensitivity. Loss of CDC25 family members conferred resistance to ATR inhibition, and the loss of RNaseH2 increased sensitivity (Ruiz et al., 2016; Wang et al., 2019). Mechanistic efforts to understand how different roles of the ATR-CHK1 pathway mediate drug sensitivity can increase understanding of the underlying biology of these inhibitors, identify potential markers, and improve strategies to overcome drug resistance.
RESULTS
MMB-FOXM1 complex components are required for CHK1i sensitivity in NSCLC cells
To identify genes whose monogenic loss confers resistance to the CHK1i prexasertib, genome-wide CRISPR-Cas9 KO screens were performed in two non-small-cell lung cancer (NSCLC) cell lines, namely, A549 and NCI-H460. Both lines showed comparable CHK1i sensitivity, with half maximal inhibitory concentration (IC50) values in the 50- to 100-nM range (Figure S1A). When treated at these concentrations, cells showed increases in markers of replication stress including phosphorylation of RPA32 on S4/S8 and S33 and KAP1 on S824 (pKAP1) (Figure 1A). There was also a concomitant increase in the DNA DSB marker γ-H2AX. CHK1 phosphorylation reached maximum levels near the CHK1i IC50 dose, and the overall levels of CHK1 decreased at higher doses (Figure 1A). The decrease in total CHK1 is consistent with the induction of ubiquitin-proteasome-dependent degradation in response to genotoxic stress (Zhang et al., 2005). The decreased viability and induction of replication stress and γ-H2AX indicate that CHK1 inhibition in NSCLC cells leads to replication-stress-induced catastrophe.
Figure 1. MMB-FOXM1 complex components are required for CHK1i sensitivity in NSCLC cells.
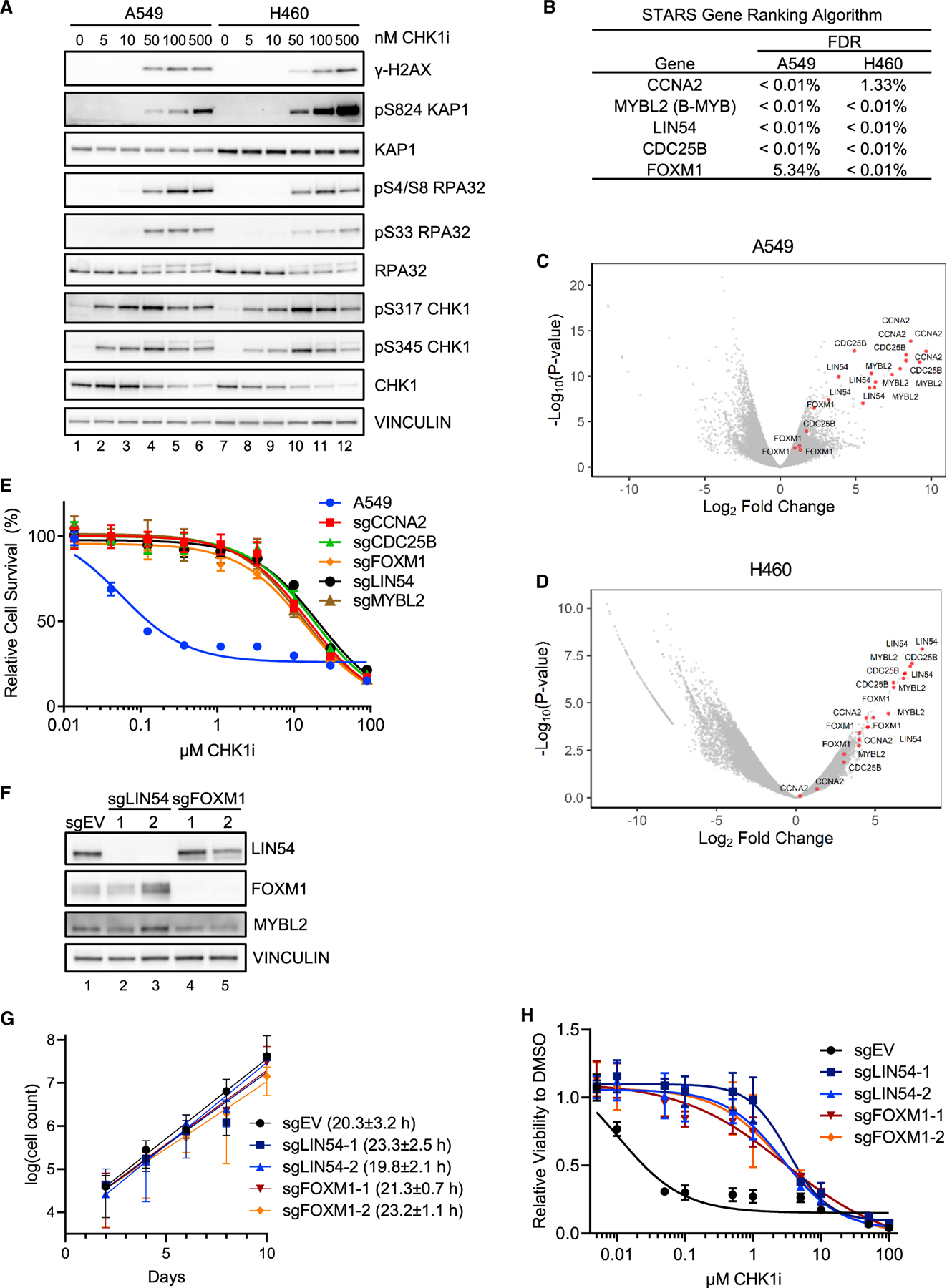
(A) Immunoblot of replication stress markers in A549 and NCI-H460 cells after treatment with increasing CHK1i concentrations for 24 h.
(B) Table of genes identified by the STARS algorithm as having multiple sgRNAs significantly enriched after CHK1i treatment.
(C and D) Volcano plots of sgRNA enrichment in CHK1i-positive selection screen in A549 (C) and NCI-H460 (D) cells.
(E) Viability curves of A549 cells expressing sgRNAs in the presence of increasing CHK1i concentrations for 72 h. n = 3.
(F) Immunoblot of MMB-FOXM1 components in A549 LIN54 KO and FOXM1 KO cells lines.
(G) A 10-day growth curve and doubling time of A549 EV, LIN54 KO, and FOXM1 KO cells. n = 3.
(H) Viability curves of A549 EV, LIN54 KO, and FOXM1 KO cells in the presence of increasing CHK1i concentrations for 72 h. n = 3.
Mean ± SD.
Screens to identify genes required for CHK1i sensitivity were performed in A549 and H460 cells using the Brunello human-genome-wide lentiviral guide RNA (gRNA) pooled library (Doench et al., 2016). CCNA2, CDC25B, FOXM1, LIN54, and MYBL2 emerged as the top hits in both cell lines (Figures 1B and S1B; Tables S1 and S2). All four of the individual sgRNAs targeting each of these genes showed substantial enrichment in both cell lines (Figures 1C and 1D). The presence of a CDC25 family member and CCNA2 is consistent with previous studies (King et al., 2015; Ruiz et al., 2016; Wang et al., 2019), but having multiple MMB-FOXM1 complex components as hits, including LIN52 in A549 cells (Figure S1B), stood out, as this finding implicated mitotic gene expression in CHK1i sensitivity.
To validate these hits, KO lines targeting each gene were generated using sgRNAs as well as an empty vector (EV) control. The KO lines were highly resistant to CHK1i compared to the EV cells (Figures 1E and S1C). To determine how the loss of mitosis-promoting genes would confer CHK1i resistance, clonal A549 cell lines with two independent sgRNAs targeting FOXM1 and LIN54 were generated (Figure 1F). There were no apparent differences in the growth rate of the LIN54 and FOXM1 KO versus EV cells over 10 days (Figure 1G). LIN54 and FOXM1 KO cells showed increased viability in the presence of CHK1i and two different ATRi (Figures 1H, S1D, and S1E).
Perturbation of LIN54 and FOXM1 reduces replication stress markers during Late S phase
To determine if LIN54 and FOXM1 were required for replication stress induced by CHK1i, LIN54 and FOXM1 KO cells were treated with CHK1i for up to 24 h (Figure 2A). Phosphorylated CHK1 (pS317 and pS345) appeared within 2 h of treatment in EV as well as in LIN54 and FOXM1 KO cells. In EV cells, phosphorylated RPA (pS4/S8) appeared within 2 h of treatment (Figure 2A, lanes 2 and 17), followed by pKAP1 at 4 and 8 h (Figure 2A, lanes 3, 4, 18, and 19). Eventually, γ-H2AX accumulated after 8 and 24 h of treatment (Figure 2A, lanes 4, 5, 19, and 20). In contrast, LIN54 and FOXM1 KO cells showed greatly reduced levels and delayed appearance of the replication stress markers in response to CHK1i. Detectable levels of pS4/S8-RPA were delayed until 8 and 24 h after CHK1i and were diminished in both LIN54 and FOXM1 KO cells compared to EV (Figure 2A, lanes 9, 10, 14, 15, 24, 25, 29, and 30). Levels of pKAP1 and γ-H2AX were also decreased in LIN54 and FOXM1 KO cells (Figure 2A, lanes 10, 15, 25, and 30). These results indicate that, although the LIN54 and FOXM1 KO cells respond in a similar manner as EV cells to CHK1i as indicated by phosphorylation of CHK1 at ATR target sites, they showed greatly reduced DNA replication stress and DNA damage responses.
Figure 2. Loss of MMB-FOXM1 complex components reduces replication stress markers during Late S phase.
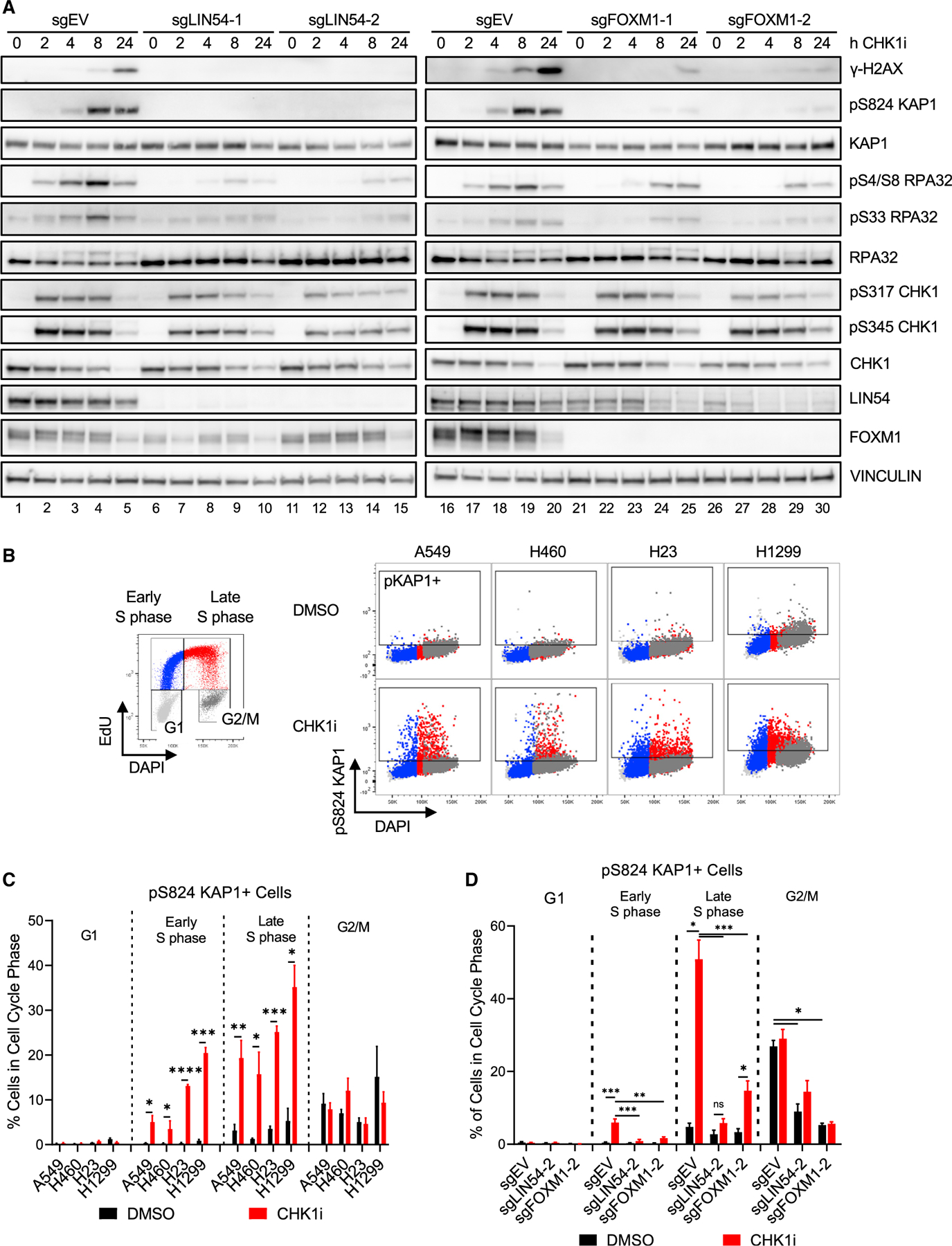
(A) Immunoblot of replication stress markers in EV, LIN54 KO, and FOXM1 KO A549 cells treated with 100 nM CHK1i over a 24-h time course.
(B–D) Flow cytometry to assess pKAP1 levels in different cell cycle phases.
(B) NSCLC cells were treated with DMSO or 100 nM CHK1i for 2 h and pulsed with 10 μM EdU for the last hour. pKAP1 versus DNA content with cell cycle populations denoted by the colors in the legend. Box denotes the gate for pKAP1-positive cells.
(C) Quantification of (B), with replicates. n = 3.
(D) EV, LIN54 KO, and FOXM1 KO cells were treated and labeled as in (B). pKAP1-positive cells in each cell cycle phase. n = 3.
Mean ± SEM; ANOVA with Tukey correction; *p < 0.05; **p < 0.01; ***p < 0.001.
To determine if CHK1i-induced replication stress was a cell-cycle-dependent event, pKAP1 levels were assessed in four NSCLC cell lines by flow cytometry. Of note, both A549 and NCI-H460 harbor KRAS mutations and are p53 wild-type, whereas NCI-H23 contains both KRAS and p53 mutations, and NCI-H1299 has wild-type KRAS and is p53 null. CHK1i treatment of the NSCLC cell lines did not lead to substantial changes in cell cycle distribution regardless of KRAS or p53 status except for a slight but significant decrease in the G2/M phase of H460 cells (Figures S2A and S2B). pKAP1 was induced by CHK1i during Late S phase and, to a lesser extent, Early S phase, but not during G1 or G2/M in NSCLC cells regardless of their KRAS or p53 status (Figures 2B and 2C), indicating that CHK1i-induced replication stress peaks during late DNA synthesis.
Next, pKAP1 levels and cell cycle distribution were assessed in LIN54 and FOXM1 KO cells or cells depleted for CCNA2, MYBL2, or LIN54 to determine if perturbation of these genes prevented the induction of pKAP1 in Late S phase. MYBL2 and cyclin A have been previously implicated in DNA synthesis (García and Frampton, 2006; Katsuno et al., 2009), raising the possibility that altered S phase progression could explain why perturbation of these loci conferred CHK1i resistance. CHK1 inhibition did not consistently alter the cell cycle distribution of the LIN54 or FOXM1 KO cells except for increased G2/M populations (Figure S2C). Similarly, depletion of LIN54, CCNA2, or MYBL2 led to diverging changes in S phase populations but increased G2/M populations, consistent with impaired G2/M progression (Figures S2D and S2E).
Loss of either LIN54 or FOXM1 significantly reduced the induction of pKAP1 in Late S phase as well as small but significant populations of pKAP1-positive cells in Early S phase (Figure 2D). pKAP1 levels in G2/M were similar in CHK1i compared to vehicle-treated cells, but the overall levels of pKAP1 in G2/M were lower in the LIN54 and FOXM1 KO cells than those in EV cells. Depletion of LIN54 reduced the appearance of pKAP1 in Late S phase similar to the LIN54 KO cells, whereas perturbation of CCNA2 resulted in increased pKAP1 levels that were insensitive to CHK1i (Figure S2F). In contrast, pKAP1 was significantly induced by CHK1i in siMYBL2 samples in both Early and Late S phase. These results suggest that induction of replication stress may not be sufficient for CHK1i sensitivity.
Induction of pKAP1 by CHK1i during Late S phase is ATR dependent
To understand the role of replication stress in CHK1i sensitivity, the requirements for pKAP1 induction were assessed. Ataxia Telangiectasia Mutated (ATM), ATR, and DNA-dependent Protein Kinase (DNA-PK) activities have been linked to KAP1 phosphorylation at S824 (White et al., 2006). Subsequent studies predominantly focused on ATM-dependent KAP1 phosphorylation in response to DNA DSBs (White et al., 2012; Ziv et al., 2006) or more recently DNA-PK-dependent KAP1 phosphorylation in response to ATR inhibition (Buisson et al., 2015; Dunlop et al., 2020). To assess the contribution of ATM and DNA-PK activity to KAP1 phosphorylation after treatment with CHK1i or agents that induce DNA damage, i.e., hydroxyurea (HU) or doxorubicin (DOXO), ATM and DNA-PK autophosphorylation was assessed in relation to pKAP1 levels by immunoblot and by flow cytometry in the case of ATM. pDNA-PK was induced to a similar extent by all three treatments, whereas pATM was preferentially induced by DOXO (Figures 3A and S3C). HU led to reduced DNA synthesis, whereas CHK1i or DOXO did not alter DNA synthesis appreciably (Figure S3A). pKAP1-positive cells were induced modestly after CHK1i or HU and to a greater extent after DOXO treatment (Figures 3A and S3B). Similarly, pATM levels in pKAP1-positive cells were elevated to a lesser extent in CHK1i- or HU-treated cells than DOXO-treated cells (Figures 3B and 3C), raising the possibility that another kinase led to CHK1i-induced activation of pKAP1 during Late S phase.
Figure 3. Induction of pKAP1 by CHK1i during Late S phase is ATR dependent.
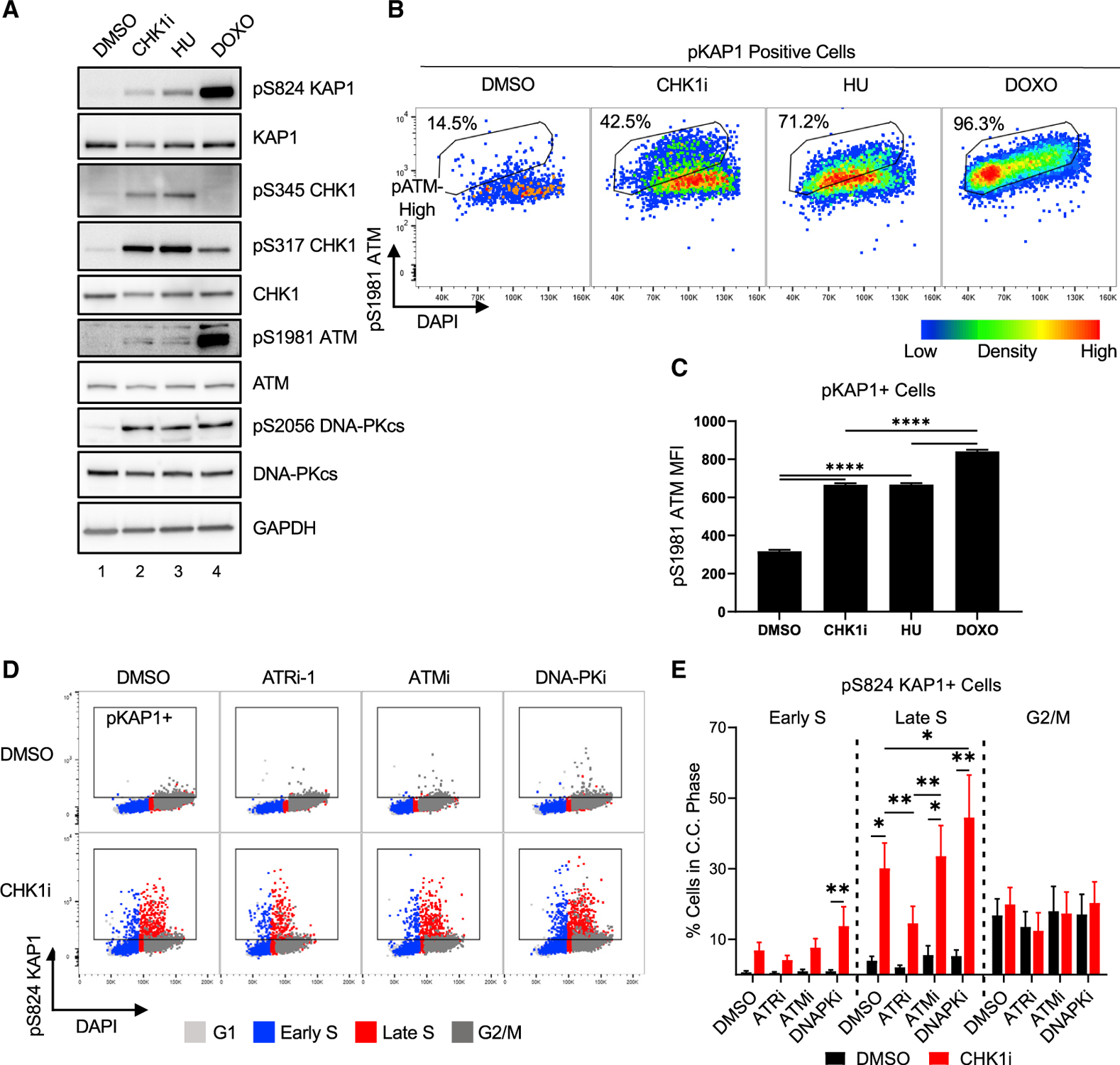
(A) Immunoblot of replication stress markers in A549 cells treated with 100 nM CHK1i, 2 mM HU, or 500 nM DOXO for 2 h.
(B and C) Flow cytometry to assess ATM activity and pKAP1 levels in cells treated as in (A) and pulsed with 10 μM EdU for the last hour.
(B) Density dot plot of pS1981 ATM levels versus DAPI stain of pKAP1-positive cells. Box denotes pS1981 ATM high based on the DOXO-treated sample.
(C) pS1981 ATM mean fluorescence intensity (MFI) in pKAP1-positive cells identified in Figure S3B. n = 4.
(D and E) Flow cytometry of pKAP1 levels after treatment as in Figure 2B ± ATRi, ATMi, or DNA-PKi. (D) pKAP1 versus DAPI levels with cell cycle phases overlaid in the colors denoted in the legend. Box denotes the pKAP1-positive population.
(E) Quantification of (D), with replicates. n = 5. ANOVA-mixed effects analysis with Tukey correction.
Mean ± SEM; ANOVA with Tukey correction; *p < 0.05; **p < 0.01; ***p < 0.001.
Given that increased pCHK1 after CHK1i treatment is a marker of ATR activity, it was possible that ATR could be phosphorylating KAP1 instead of ATM. To determine the relative contribution of ATM, ATR, and DNA-PK to KAP1 phosphorylation, cells were treated with an ATR, ATM, or DNA-PK inhibitor alone or in combination with CHK1i. Similar to CHK1i, ATRi treatment led to a modest decrease in the G2/M population, whereas ATMi or DNA-PKi treatment did not significantly perturb cell cycle distribution (Figure S3D). However, ATRi did not increase levels of pKAP1 during S phase, indicating that this increase was specific to CHK1i (Figures 3D and 3E). ATRi but not ATMi or DNA-PKi, when combined with CHK1i, led to a significant reduction in pKAP1-positive cells during Late S phase. The requirement of ATR activity for CHK1i-induced KAP1 phosphorylation during Late S phase suggests that DNA replication was deregulated.
CHK1i increases DNA replication regardless of loss of LIN54 or FOXM1
CHK1 inhibition can lead to increased origin firing in a CDK-dependent manner (Petermann et al., 2010). To assess the levels of DNA synthesis after CHK1 inhibition, 5-ethynyl-2′-deoxyuridine (EdU) incorporation was quantified in EV, LIN54 KO, and FOXM1 KO cells (Figure 4A). The levels of DNA synthesis in both Early and Late S phase increased significantly in response to CHK1i compared to those in response to DMSO in EV, LIN54 KO, and FOXM1 KO cells. Although the increased levels of DNA synthesis after CHK1i treatment were similar in EV, LIN54 KO, and FOXM1 KO cells during Early S phase, DNA synthesis during Late S phase was significantly reduced in the CHK1i-treated LIN54 KO and FOXM1 KO cells compared to those in EV cells.
Figure 4. CHK1i increases DNA replication regardless of the loss of LIN54 or FOXM1.
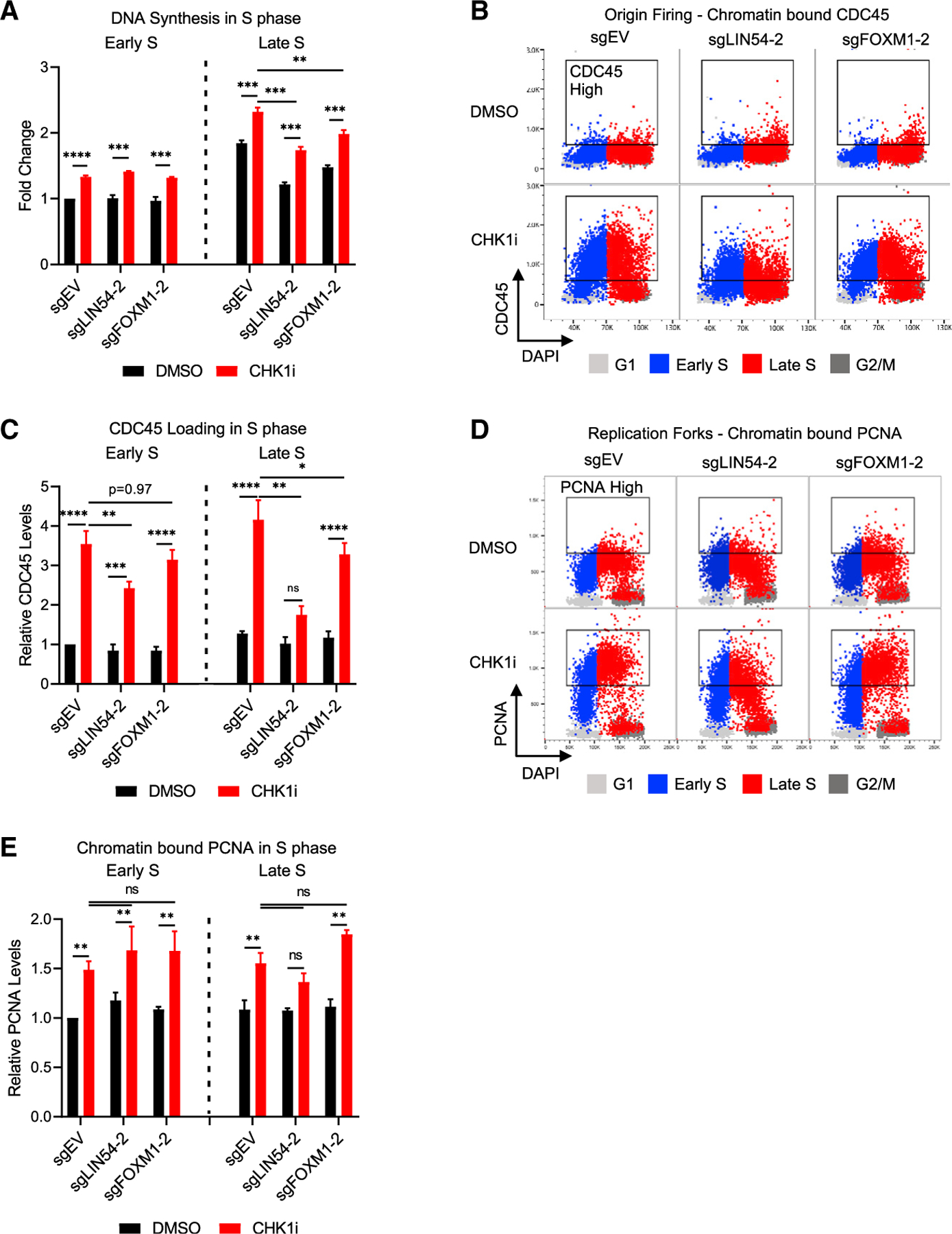
(A) Relative EdU incorporation in Early S and Late S phase in LIN54 or FOXM1 KO cells after CHK1 inhibition. MFI of S phase populations per gates in Figure S2A were normalized to EV DMSO Early S populations. n = 3.
(B and C) Chromatin flow cytometry for chromatin-bound CDC45.
(B) CDC45 versus DAPI levels with cell cycle phases overlaid in the colors denoted. Box denotes >98th percentile of CDC45 in the sgEV-DMSO sample.
(C) MFI of CDC45 in Early and Late S phase. n = 3.
(D and E) Chromatin flow cytometry for chromatin-bound PCNA.
(D) PCNA versus DAPI levels with cell cycle phases overlaid in the colors denoted. Box denotes >98th percentile of PCNA in the sgEV-DMSO sample.
(E) MFI of PCNA in Early and Late S phase. n = 3. Mean ± SEM; ANOVA with Tukey correction; *p < 0.05; **p < 0.01; ***p < 0.001.
To understand the impact of CHK1 inhibition on DNA replication, levels of chromatin-bound CDC45 and Proliferating Cell Nuclear Antigen (PCNA) were assessed by chromatin flow cytometry. CDC45 loading on DNA can provide a measure of origin firing (Köhler et al., 2016), and PCNA bound to chromatin can be used to assess active DNA replication (Janke et al., 2018; Sirbu et al., 2013). Chromatin-bound CDC45 and PCNA increased significantly in response to CHK1i in EV cells as well as LIN54 and FOXM1 KO cells during Early S phase, and in EV and FOXM1 KO cells during Late S phase (Figures 4B–4E), consistent with increased origin firing throughout S phase. Because CHK1i-induced increases in DNA replication and origin firing were not blunted by the loss of LIN54 or FOXM1, particularly in Early S phase, another function of the MMB-FOXM1 complex was required for CHK1i sensitivity.
Activation of G2/M genes after CHK1i is MMB-FOXM1 dependent
Phosphorylation of the MMB-FOXM1 components MYBL2 and FOXM1 by cyclin-CDK complexes promotes their transcriptional activity in an ATR-CHK1-dependent manner (Laoukili et al., 2008; Saldivar et al., 2018; Ziebold et al., 1997). The induction of FOXM1 phosphorylation on T600 (pFOXM1) after CHK1 inhibition was assessed in synchronized (Figure S4A) and asynchronously replicating A549 cells by flow cytometry (Figure 5A). pFOXM1 appeared within 2 h post-release from a thymidine block after 1 h of CHK1i treatment compared to 9 h post-release in untreated cells (Figure S4A). pFOXM1 was induced by CHK1i in Late S phase in all four NSCLC cell lines tested (Figures 5B and 5C), consistent with perturbation of the ATR-CHK1 pathway leading to early MMB-FOXM1 activity.
Figure 5. Activation of G2/M genes after CHK1i is MMB-FOXM1 dependent.
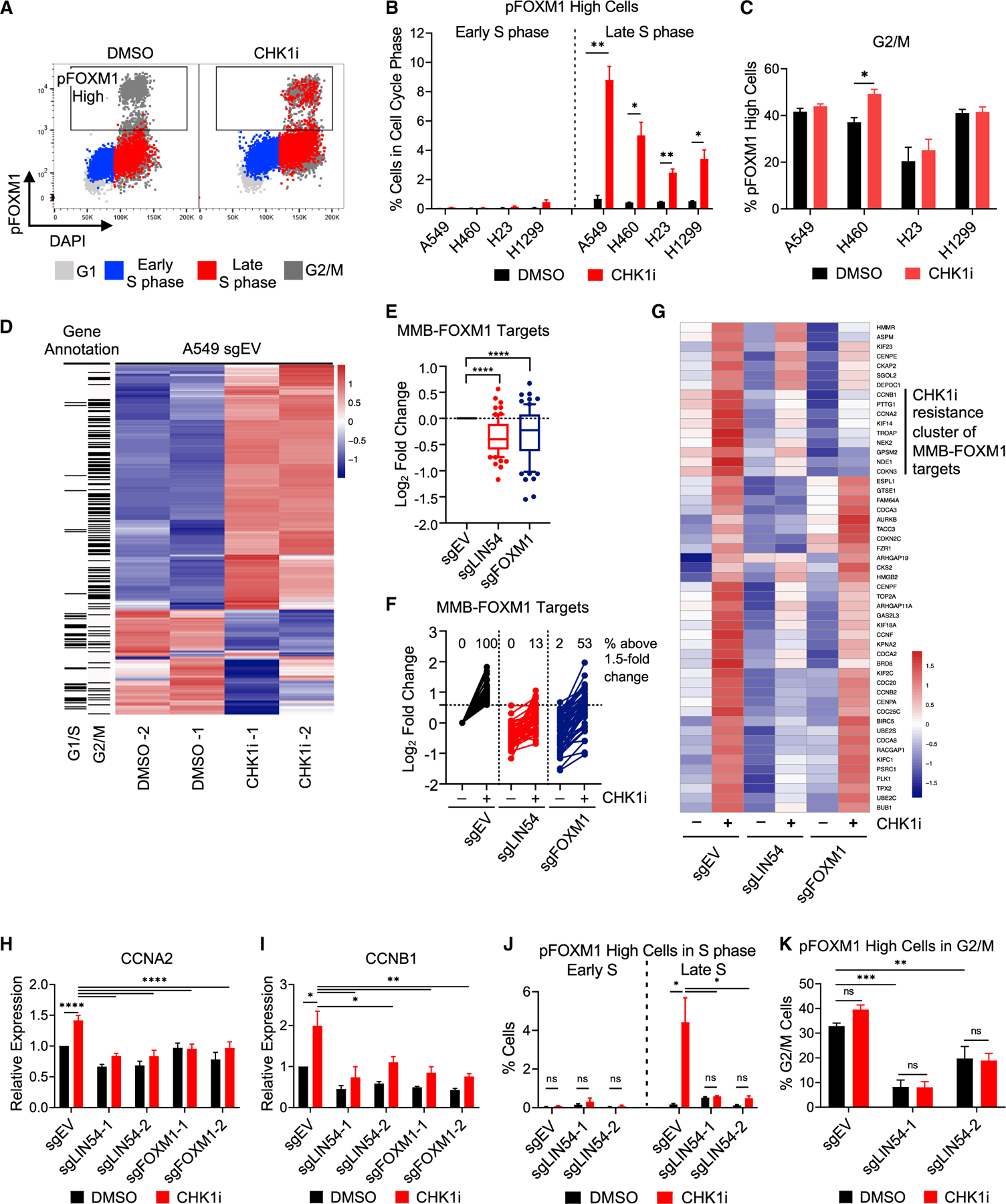
(A–C) pFOXM1 levels in NSCLC cell lines after CHK1i. Cells were treated as in Figure 2B.
(A) pFOXM1 versus DAPI levels with cell cycle phase overlaid in the colors denoted. Box denotes high pT600 FOXM1 levels found in G2/M cells of the DMSO sample.
(B and C) Percentage of pFOXM1-High cells in Early and Late S phase (B) and G2/M (C). n = 4.
(D–G) Gene expression during S phase. EV, LIN54 KO, and FOXM1 KO cells were synchronized by single thymidine block. Samples were treated with DMSO or 100 nM CHK1i at 1 h post-release and samples were collected at 3 h post-release. Total RNA was extracted and sequenced.
(D) Heatmap showing the 350 most differentially expressed genes in EV cells between the DMSO- and CHK1-treated samples.
(E) Expression of MMB-FOXM1 target genes in relation to EV cells. Log2 fold change calculated using DESeq2.
(F and G) Expression of MMB-FOXM1 target genes upregulated in EV cells after CHK1i by ≥1.5-fold change (51 genes), depicted as log2 fold change determined in relation to EV DMSO-treated cells, with % of genes upregulated above 1.5-fold change in each sample reported (F), and as a heatmap (G).
(H and I) qRT-PCR of MMB-FOXM1 target genes CCNA2 (H) and CCNB1 (I) during S phase in EV, LIN54 KO, and FOXM1 KO cells after CHK1 inhibition. Samples were synchronized, treated, and collected as in (D)–(G). n = 3.
(J and K) pFOXM1 levels in LIN54 KO cells. EV or LIN54 KO cells were treated as in Figure 2B. Samples were stained for pFOXM1, EdU, and DAPI. Percentage of pFOXM1-High cells in Early and Late S phase (J) and G2/M. (K) n = 4.
Mean ± SEM; ANOVA with Tukey correction; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
To determine if disruption of the MMB-FOXM1 complex alters gene expression in response to CHK1 inhibition, RNA sequencing (RNA-seq) profiling was performed in EV, LIN54 KO, and FOXM1 KO cells synchronized in S phase and treated with CHK1i for 2 h. Gene set enrichment analysis (GSEA) revealed that DNA repair genes were not significantly perturbed by CHK1 inhibition (Figure S4B). In contrast, G1/S genes, comprised of genes involved in DNA replication, were significantly decreased (Figure S4C), whereas G2/M genes, containing mitotic regulator genes, were significantly increased following CHK1 inhibition (Figure S4D). G1/S and G2/M genes were well represented in the top 350 most differentially expressed genes in the EV cells after CHK1 inhibition (Figure 5D). G1/S genes comprised the downregulated genes (Figure S4E), whereas G2/M genes were among the upregulated genes (Figure S4F), consistent with the GSEA. Compared to EV cells, a loss of LIN54 restored the expression of downregulated genes to a greater extent than the loss of FOXM1 (Figure S4G). The induction of upregulated genes, however, was impaired to a similar extent in both LIN54 and FOXM1 KO cells (Figure S4H). The induction of MMB-FOXM1 target genes by CHK1i was significantly reduced in both the LIN54 and FOXM1 KO cells (Figures 5E–5G). Of note, CCNA2, a top screen hit, and CCNB1, a significant hit in H460 cells (Figure S1B), were both present in a group of highly expressed MMB-FOXM1 target genes not induced by CHK1i in either LIN54 or FOXM1 KO cells. Levels of these genes, as well as the top hit CDC25B, were significantly increased in S phase by CHK1 inhibition in EV but not in LIN54 and FOXM1 KO cells (Figures 5H, 5I, and S4I–S4K). Cyclin B1 protein levels were reduced and not induced by CHK1i in S-phase LIN54 KO cells (Figure S4L). Thus, perturbing the MMB-FOXM1 complex impairs the induction of CDK1 activators during S phase after CHK1 inhibition.
The decrease in CDK1 activators with MMB-FOXM1 complex perturbation suggests a positive feedback loop between the MMB-FOXM1 complex and cyclin/CDK1 complexes. Consistent with this, CCNA2 small interfering RNA (siRNA) knockdown substantially decreased the percentages of high pFOXM1 cells in Late S and G2/M (Figures S4M and S4N). The fraction of G2/M cells with high levels of pFOXM1 was not significantly affected by CHK1i but was significantly decreased in LIN54 KO and si-MYBL2 cells (Figures 5K and S4N) Perturbation of LIN54 or MYBL2 also prevented the induction of pFOXM1 in Late S phase by CHK1i (Figures 5J, S4L, and S4M), indicating that an intact MMB complex is required. Taken together, these results indicate that an MMB-FOXM1-CDK1 feedback loop drives the CHK1i-dependent induction of the G2/M transcription program during S phase.
MMB-FOXM1 complex components are required for CHK1i-induced premature mitosis in Late S phase
The premature activation of the mitotic program in response to CHK1i raised the possibility that essential S phase and mitotic processes were occurring simultaneously. Increased or premature histone H3 S10 phosphorylation (pH3), a mitotic marker, has been previously observed following perturbation of the ATR-CHK1 pathway (Ruiz et al., 2016; Zuazua-Villar et al., 2014). A limitation of these studies is that the cell cycle phase was assessed by DNA content, leaving it unclear whether pH3 induced by these inhibitors coincided with active DNA synthesis. Simultaneous pH3 and BrdU staining, however, was observed in CHK1 haploinsufficient mice (Lam et al., 2004). If DNA synthesis and mitosis were occurring simultaneously, mitotic events could sufficiently disrupt DNA synthesis and trigger replication catastrophe.
To determine if ATR or CHK1 inhibition led to a breakdown in the separation of DNA synthesis from mitosis, levels of pH3 were assessed by flow cytometry during each cell cycle phase in four NSCLC cell lines (Figure 6A). Levels of pH3 were increased particularly in kate S phase in all four lines (Figures 6B and 6C). An increase in pH3-positive cells was observed during Late S phase following inhibition of ATR or CHK1 but not ATM (Figure 6D). Similarly, ATR and CHK1 but not ATM inhibition led to some increase in pH3 in G2/M cells (Figure 6E). Dual inhibition of ATR and CHK1 led to a significant increase, relative to either alone, in pH3-positive cells during Late S phase but not in G2/M (Figures 6D and 6E). These results indicate that inhibition of the ATR-CHK1 pathway leads to DNA replication and mitotic events occurring simultaneously particularly during Late S phase.
Figure 6. MMB-FOXM1 complex components are required for CHK1i-induced premature activation of mitosis in Late S phase.
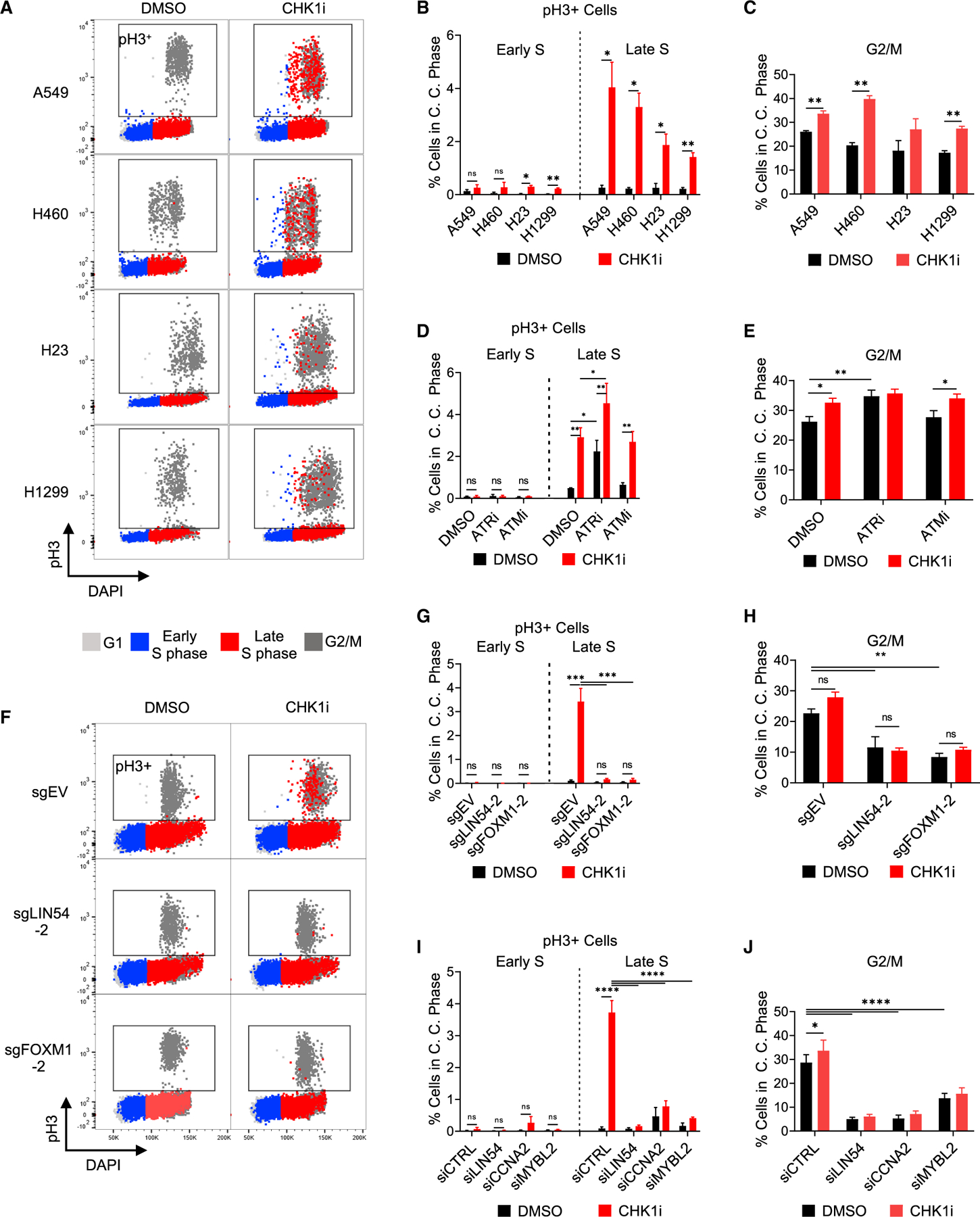
(A–C) Flow cytometry analysis of pH3 levels by cell cycle phase in NSCLC cells. Cells were treated as in Figure 2B.
(A) Dot plot of pH3 levels versus DAPI levels with cell cycle phases overlaid in the colors denoted. Box denotes the pH3+ gate.
(B and C) Percent pH3 positive cells in Early and Late S phase (B) and G2/M (C). n = 3.
(D and E) Flow cytometry analysis of pH3 levels in A549 cells treated as in Figure 2B ± 100 nM ATRi or 1 μM ATMi.
(F–H) Flow cytometry analysis of pH3 levels by phase of the cell cycle in LIN54 KO or FOXM1 KO A549 cells after CHK1i. Samples treated as in Figure 2B.
(F) pH3 levels versus DAPI levels with cell cycle phases overlaid in the colors denoted. Box denotes the pH3+ gate.
(G and H) Percent pH3 positive cells in Early and Late S phase (G) and G2/M (H). n = 3.
(I and J) Flow cytometry analysis of pH3 levels by phase of the cell cycle in A549 cells following LIN54, CCNA2, or MYBL2 siRNA knockdown. Percent pH3 positive cells in Early and Late S phase (I) and G2/M (J). n = 4.
Mean ± SEM; ANOVA with Tukey correction; *p < 0.05; **p < 0.01; ***p < 0.001.
Next, we assessed the role of the MMB-FOXM1 complex in regulating CHK1i-induced pH3 during S phase (Figure 6F). CHK1 inhibition led to increased levels of pH3 during Late S phase in EV cells, whereas the loss of LIN54 or FOXM1 abolished this effect (Figures 6F and 6G). LIN54 and FOXM1 KO cells also showed a significantly smaller fraction of G2/M cells positive for pH3 independent of CHK1i treatment (Figure 6H). This was similarly observed following siRNA knockdown of LIN54, CCNA2, or MYBL2 (Figures 6I and 6J). This result indicates that a functional MMB-FOXM1 complex is required for CHK1i-induced breakdown in the separation between DNA replication and mitosis.
Activation of an MMB-FOXM1 complex CDK1-positive feedback loop is required for CHK1i-induced replication catastrophe
CDK1 activity has been previously implicated in ATR-CHK1i sensitivity (Aarts et al., 2012; Ruiz et al., 2016). CDK1 activity was restricted in CHK1i-resistant FAM122A KO cells by WEE1 stabilization (Li et al., 2020), suggesting that CDK1 activities during the S/G2 transition influence CHK1i sensitivity. Which activities of CDK1 are required for replication catastrophe, however, remained unclear. To identify the levels of CDK1 activity necessary to trigger an MMB-FOXM1-CDK1 feedback loop and premature mitosis in S phase, A549 cells were treated with CHK1i and increasing concentrations of the CDK1 inhibitor RO3306 (CDK1i). As levels of CDK1i were increased, the fraction of G2/M cells increased independently of CHK1 inhibition (Figure S5D) and the fraction of G1 and Early S phase cells decreased correspondingly (Figures S5A and S5B), whereas the levels of Late S phase cells did not change (Figure S5C). Increased DNA synthesis in response to CHK1i was not diminished by less than 5 μM CDK1i (Figures 7A and 7B).
Figure 7. Activation of an MMB-FOXM1 complex CDK1-positive feedback loop is required for CHK1i-induced replication catastrophe.
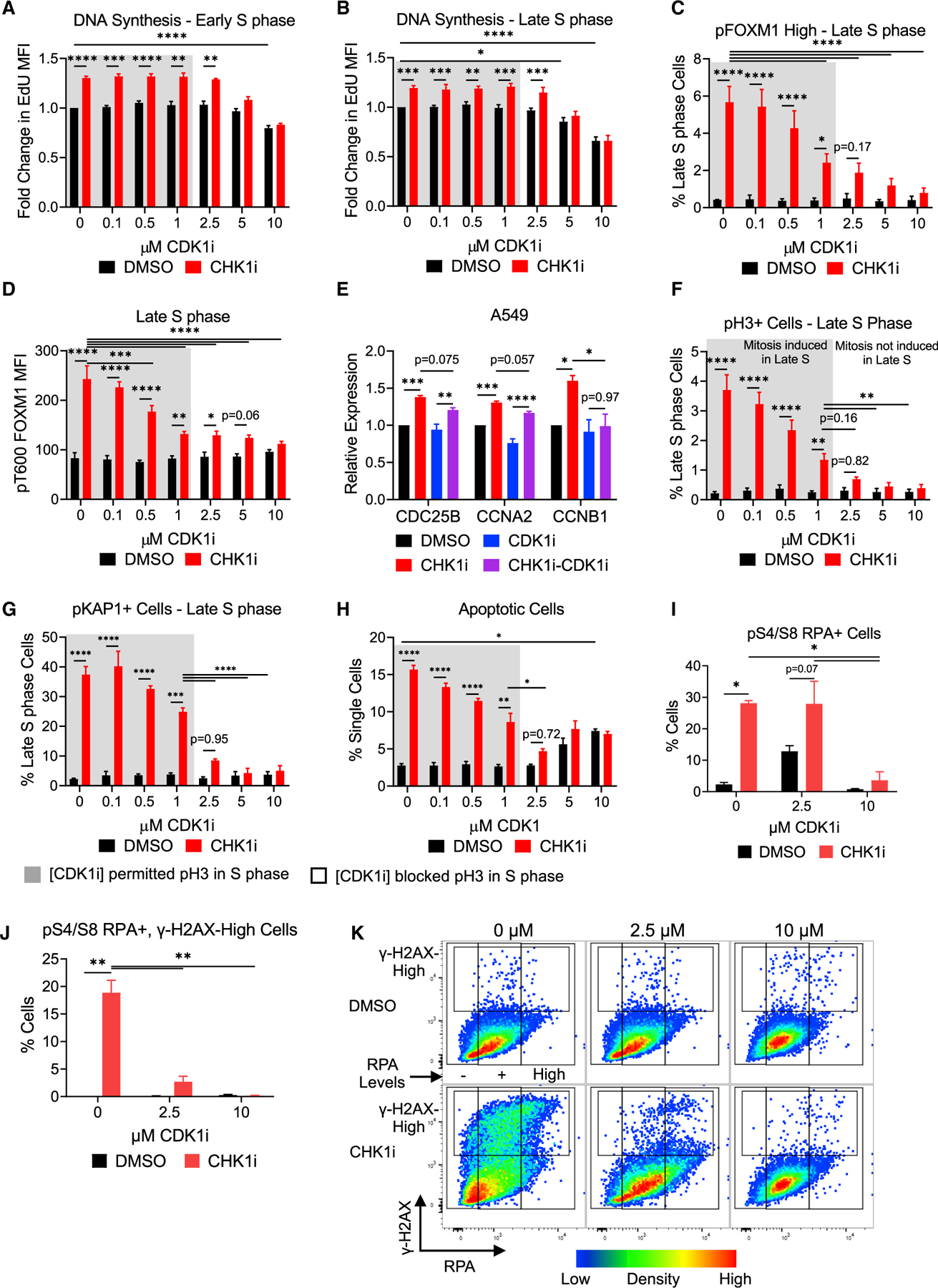
(A and B) Relative EdU levels in Early (A) and late (B) S phase. EdU levels were normalized to the 0 μM CDK1i-DMSO sample. n = 5.
(C and D) pFOXM1 levels in Late S phase after dual CHK1 (100 nM) and CDK1 inhibition with % pFOXM1-High cells in Late S phase (C) and pFOXM1 MFI (D). n = 4.
(E) qRT-PCR for MMB-FOXM1 target genes CDC25B, CCNA2, and CCNB1 in S phase A549 cells. A549 cells treated as in Figure 5D ± 2.5 μM CDK1i. n = 3.
(F) pH3 levels after dual CHK1 and CDK1 inhibition in Late S phase. n = 5.
(G) pKAP1 levels after dual CHK1 and CDK1 inhibition in Late S phase. n = 5; ANOVA mixed effects analysis with Tukey correction.
(H) Percent Annexin V+ PI− apoptotic cells treated with CHK1i ± CDK1i for 24 h. n = 3.
(I–K) Chromatin-associated phospho-S4/S8 RPA or RPA compared to γ-H2AX in cells treated with CHK1i ± CDKi for 24 h.
(I) Percentage of pS4/S8 RPA+ cells.
(J) Percentage of pS4/S8 RPA+ and γ-H2AX-High cells.
(K) Density dot plot of γ-H2AX High versus chromatin-associated RPA. n = 3.
Gray boxes denote concentrations of CDK1i that showed a significant increase in pH3 levels with CHK1i in (F). Mean ± SEM; ANOVA with Tukey correction. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.
In contrast to DNA replication, significant induction of pFOXM1-High cells by CHK1i in Late S phase was prevented by 2.5 μM CDK1i (Figure 7C). Although lower concentrations of CDK1i also significantly reduced the fraction of pFOXM1-High cells in Late S phase after CHK1i in a dose-dependent manner, pFOXM1-High cells were still significantly induced at these concentrations by CHK1i. A small but significant increase in pFOXM1 after CHK1i treatment was observed at 2.5 μM CDK1i (Figure 7D), suggesting that, although FOXM1 can still be phosphorylated, the level of CDK1 activity blocked by 2.5 μM CDK1i was necessary for significant accumulation of pFOXM1. Increasing doses of CDK1i did not affect the fraction of pFOXM1-High cells in G2/M but led to a decrease in pT600 FOXM1 levels in a dose-dependent manner, with a significant drop as the concentration of CDK1i increased from 1 μM to 2.5 μM (Figures S5E and S5F). A total of 2.5 μM CDK1i reduced the accumulation of pFOXM1 in S-phase-synchronized A549 and H460 cells (Figure S5G). CDK1 inhibition did not alter pMYBL2 levels but caused a shift in the gel migration of the pMYBL2 band, suggesting that other MYBL2 phosphorylation sites may be CDK1 dependent. The degradation of cyclin E in S phase is dependent on CDK2 activity (Clurman et al., 1996). Doses up to 10 μM of RO-3306 did not affect cyclin E degradation after CHK1 inhibition in either A549 or H460 cells, suggesting that CDK2 activity was not impaired by these levels of CDK1i.
To determine if 2.5 μM CDK1i also blocked the induction of G2/M genes by CHK1i, the expression of MMB-FOXM1 target genes was assessed by qRT-PCR in S phase A549 and H460 cells. A total of 2.5 μM CDK1i blocked the induction of CCNB1 but not CDC25B or CCNA2 in A549 cells (Figure 7E) and significantly reduced the induction of CCNB1 and CCNA2 but not CDC25B in the more sensitive H460 cells (Figure S5H). The differential effect of CDK1i on these MMB-FOXM1 target genes could reflect the differing number of CHR motifs in the gene promoters (Fischer et al., 2016) or the recently identified regulation of epigenetic factors by CDK1 (Michowski et al., 2020). The consistent result is that CDK1 activity remaining after 2.5 μM CDK1i was sufficient to block CHK1i-induced MMB-FOXM1 activity and increased expression of CCNB1, the mitotic activator of CDK1, during S phase.
Similarly, 2.5 μM and higher doses of CDK1i reduced the appearance of the mitotic marker pH3 in response to CHK1i during Late S phase and, in general, during G2/M (Figures 7F and S5I). Both the fraction of pKAP1-positive G2/M cells as well as CHK1i-induced pKAP1-positive S phase cells was dependent on CDK1 activity (Figures 7G, S5J, and S5K). Increasing levels of CDK1i reduced the appearance of pKAP1 in Early S, Late S, and G2. A total of 2.5 μM CDK1i was sufficient to reduce pKAP1 levels in G2/M and block pKAP1 induction in Late S phase (Figures 7G, S5J, and S5K). Furthermore, the same concentration of CDK1i prevented pH3 induction, suggesting that the breakdown in the separation of DNA synthesis and mitosis is a required step for DNA replication stress after CHK1 inhibition.
To establish if activation of the MMB-FOXM1-CDK1 feedback loop is required for CHK1i-induced replication catastrophe, the amount of CDK1 activity required for apoptosis 24 h after CHK1 inhibition was assessed by Annexin V/propidium iodide (PI) staining. CHK1i induced an apoptotic population that was dependent on CDK1 activity (Figure 7H). At 2.5 μM, CDK1i blocked CHK1i-induced apoptosis, whereas 10 μM CDK1i significantly induced apoptosis by itself. The induction of replication stress markers was also examined at 24-h post-CHK1i treatment (Figure S6A). pKAP1, as well as pRPA and pCHK1, were induced in the presence of 2.5 μM but not 10 μM CDK1i. A total of 2.5 μM or higher of CDK1i reduced CHK1i-induced γ-H2AX, although 10 μM CDK1i induced low levels of γ-H2AX on its own.
Replication catastrophe occurs when replication factors such as RPA become exhausted, leading to unrepairable DNA damage (Toledo et al., 2017). Prolonged CHK1i treatment induced a pS4/S8 RPA-positive population that was also γ-H2AX positive (Figures 7I, 7J, and S6B), indicating the accumulation of DNA damage in replication stressed cells. A pS4/S8 RPA-positive population was observed with 2.5 μM CDK1i, consistent with a mitotic arrest where RPA is hyperphosphorylated (Liu et al., 2012; Oakley et al., 2003). Treatment with CHK1i and 2.5 μM or higher CDK1i reduced levels of pS4/S8 RPA-positive/γ-H2AX-High cells, but only 10 μM CDK1i completely blocked the induction of pS4/S8 RPA in response to CHK1i. This finding indicated that blocking sufficient CDK1 activity to induce mitosis during DNA replication prevented the accumulation of DNA damage but not replication stress. Chromatin-bound RPA and γ-H2AX were also examined to determine if this DNA damage was due to RPA exhaustion. CHK1i induced an RPA-High population that was γ-H2AX-High (Figures 7K, S6C, and S6D), consistent with replication catastrophe. An RPA-positive/γ-H2AX-High population was also induced, suggesting a low RPA exhaustion threshold or exhaustion of an alternative replication factor. Similar to pS4/S8 RPA, 2.5 μM CDK1i blocked the induction of the RPA-High, γ-H2AX-High population, indicating that premature activation of the mitotic program is required for the accumulation of DNA damage in RPA exhausted cells.
The requirement for mitotic levels of CDK1 activity in CHK1i-induced replication catastrophe raised the possibility that replication catastrophe could be induced in LIN54 or FOXM1 KO cells by aberrantly activating CDK activity by targeting an alternate negative regulator. Levels of WEE1, which phosphorylates an inhibitory site on CDK1 (McGowan and Russell, 1993), were induced by CHK1 inhibition during S phase in EV, LIN54 KO, and FOXM1 KO cells (Figure S7A). An inhibitor of WEE1, adavosertib (WEE1i), has shown synergy in combination with CHK1 is in vivo (Carrassa et al., 2012; Chilà et al., 2015; Russell et al., 2013). Treatment of EV and LIN54 KO cells with WEE1i resulted in the appearance of a slower migrating FOXM1 species consistent with phosphorylation resulting from increased CDK activity (Figure S7B). The ATR-CHK1 pathway was activated after WEE1i treatment in EV, LIN54 KO, and FOXM1 KO cells, as denoted by induction of pCHK1 in the WEE1i-treated samples. Thus, WEE1 and the ATR-CHK1 pathway work to oppose CDK activation when the other is inhibited. γ-H2AX was induced in IN54 or FOXM1 KO cells after WEE1i but not CHK1i treatment (Figure S7B), suggesting that cells that become resistant to CHK1i by perturbation of the MMB-FOXM1 pathway may still be sensitive to WEE1i. Consistent with this observation, EV cells and three out of four LIN54 or FOXM1 KO cell lines had similar sensitivities to WEE1i (Figure S7C). Viability in LIN54 KO and FOXM1 KO cells was not further reduced by the addition of CHK1i to WEE1i (Figure S7D), in keeping with the dependency on MMB-FOXM1 complex-induced premature mitosis for CHK1i sensitivity.
DISCUSSION
The results in this study describea positive feedback loop between the MMB-FOXM1 complex, the G2/M mitotic transcriptional program, and CDK1 activity that is restrained during S phase by CHK1 (Figure S7E). The ATR-CHK1 pathway has previously been observed to regulate FOXM1 by CDK1 activity (Saldivar et al., 2018), whereas cyclin A or B complexes, typically expressed as part of the mitotic transcription program, also promote MMB-FOXM1 activity (Fu et al., 2008; Lane et al., 1997; Laoukili et al., 2008; Ziebold et al., 1997). In addition, a loss of cyclin F, a negative regulator of CDK phosphorylation of MYBL2, sensitizes cells to inhibitors of the ATR-CHK1 pathway (Burdova et al., 2019; Klein et al., 2015; Mavrommati et al., 2018), underscoring that interplay between cyclin/CDK and MMB-FOXM1 activities in ATR/CHK1i sensitivity. This study builds on these prior observations by demonstrating the interdependent nature of increased MMB-FOXM1 complex activity, G2/M gene expression, and CDK1-dependent phosphorylation required for CHK1i sensitivity.
The CHK1i-induced phosphorylation of KAP1 at S824 is an ATR-and CDK1-dependent event that reflects dysregulation of the S/G2/M transition. A CHK1i-independent population of pS824 KAP1-positive cells was observed basally in G2/M. The presence of this G2/M population and its dependency on CDK1 activity indicate that KAP1 S824 phosphorylation is a cell-cycle-dependent event. KAP1 phosphorylation promotes chromatin relaxation to facilitate DNA damage repair and coincides with the loss of HP1 foci on chromatin (White et al., 2012; Ziv et al., 2006). HP1 chromatin dynamics are altered during mitosis to facilitate chromosome segregation (Abe et al., 2016; Chu et al., 2014), so the observed pKAP1 in G2/M could result from the regulation of heterochromatin during G2/M. Surprisingly, the induction of pKAP1 in Late S by CHK1i was ATR dependent but not ATM or DNA-PK dependent. Increased pKAP1 after CHK1 inhibition could reflect altered heterochromatin dynamics, given that pKAP1 promotes chromatin relaxation, as the cell attempts to complete DNA replication prior to mitosis. Further study of KAP1 phosphorylation will be needed to understand the function of KAP1 phosphorylation in the S/G2 transition and in response to CHK1 inhibition.
Premature mitosis during DNA replication is necessary for replication catastrophe and stress after CHK1 inhibition. Loss of MMB-FOXM1 components reduced the induction of the mitotic marker pH3 during Late S phase after CHK1 inhibition. CDK1 inhibition restored the separation of DNA replication and mitosis with 2.5 μM of CDK1i, blocking the induction of a pH3-positive population in Late S phase. The induction of pKAP1, RPA-High/γ-H2AX-High cells, and apoptosis by CHK1i treatment was also blocked by 2.5 μM of CDK1i, demonstrating that premature mitosis was necessary for DNA replication stress and subsequent replication catastrophe. A total of 2.5 μM CDK1i was previously shown to partially rescue cell death after depletion of ATR by an inducible degron (Eykelenboom et al., 2013), suggesting that this threshold of CDK1 activity also may be relevant for ATR inhibition as well. The MMB-FOXM1 target gene CCNB1 appears to be a key gene for CHK1i resistance, as it was identified as a significant hit in H460 cells and depletion of CCNB1 reduced CHK1i sensitivity in recently published work in ovarian cancer cells (Nair et al., 2020). Cyclin B1 levels however were not predictive of CHK1i sensitivity in a panel of head and neck squamous cell lines (van Harten et al., 2019), indicating that other components of the mitotic program may be involved in CHK1i sensitivity. Further investigation will be needed to identify the key components of the mitotic program that promote CHK1i-induced replication catastrophe.
Taken together, the results of this study add to the understanding of the factors that play a role in the S/G2/M checkpoint, which delays entry into mitosis until after DNA synthesis is complete, and was first described in fission yeast (Enoch and Nurse, 1990). Subsequently, vertebrate cells including DT40 B-lymphoma cells lacking Chk1 were shown to enter mitosis with incompletely replicated DNA when DNA synthesis is blocked (Zachos et al., 2005). Like this study, these prior studies highlighted the crucial role of regulation of CDK1 activity. Here, we demonstrated the impact of differences in CDK1 activity regulated by an MMB-FOXM1-dependent feedback loop in triggering replication catastrophe after CHK1 inhibition. LIN54 and FOXM1 were separately shown to modulate CHK1i sensitivity in ovarian and pancreatic cancer, respectively (Blosser et al., 2020; Chung et al., 2019). This result suggests that the insights about the S/G2/M transition derived from this study could inform the use of CHK1is in additional cancer types.
Synergism of CHK1 or ATR inhibition with WEE1 inhibition has been observed in several preclinical studies (Bukhari et al., 2019; Busch et al., 2017; Carrassa et al., 2012; Chaudhuri et al., 2014; Chilà et al., 2015; Chung et al., 2019; Davies et al., 2011; Ghelli Luserna Di Rorà et al., 2019; Hauge et al., 2017; Magnussen et al., 2015; Mak et al., 2015; Russell et al., 2013), albeit with incomplete understanding as to why. Aberrant activation of CDK1 and CDK2, by inhibition of WEE1, may bypass the attenuation of CDK1 activation due to the loss of the MMB-FOXM1 complex. The addition of CHK1i to WEE1i may not be beneficial in cells that become resistant to CHK1i due to this mechanism, but other approaches, such as the addition of a DNA damaging agent such as gemcitabine, may be more effective (Chung et al., 2019; Kausar et al., 2015).
Several CHK1 and ATRis as well as WEE1 inhibitors have been the subject of ongoing and completed clinical trials. In at least one trial (NCT02873975), patients were selected based on replication stress markers, which stems from the idea that tumors prone to replication stress due to genetic abnormalities would be overwhelmed by additional DNA replication stress induced by CHK1 inhibition. Rather, it appears that premature activation of the mitotic program, which requires an intact MMB-FOXM1-CDK1 axis, triggers cell death by replication catastrophe. Our work suggests novel biomarkers that may inform our understanding of patients with responding tumors versus those with intrinsic resistance.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James A. DeCaprio (james_decaprio@dfci.harvard.edu).
Materials availability
Cell lines generated in this study are available upon request.
Data and code availability
RNA-seq data have been deposited in the NCI Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO: GSE154545, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154545.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human NSCLC cell lines A549 and NCI-H460 cells stably transfected with pXPR_311 for constitutive expression of the Cas9 endonuclease (A549-Cas9 and NCI-H460-Cas9) were obtained from the Genetic Perturbation Platform at the Broad Institute, Cambridge, MA (Broad GPP). Human NSCLC cell lines A549, NCI-H460, NCI-H23, and NCI-H1299 were obtained from ATCC, Manassas, VA.
METHOD DETAILS
Cell Culture
Human NSCLC cells were propagated in RPMI 1640 medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) and 1% Pen/Strep (GIBCO) or 100 μg/ml Normocin (InvivoGen, San Diego, CA) at 37°C in 5% CO2. Sequences of sgRNA used for further studies listed in Table S3. KO populations of sgLIN54 and sgFOXM1 cell lines were generated by serial dilution and confirmed by immunoblot.
For single thymidine blocks, 500,000 (60 mm plate) or 1,200,000 (100 mm plate) cells were plated into 2 mM thymidine-containing RPMI media for 16 h. To release cells, cells were washed twice with prewarmed PBS and grown in prewarmed RPMI media to which inhibitors were added at the times denoted. For siRNA experiments, siRNA were reverse transfected at a final concentration of 20 nM into ~106 A549 cells using Lipofectamine RNAiMAX in 100 mm dish. The next day cells were plate into 2 100 mm dishes for drug treatment the following day.
CRISPR positive selection screens for drug resistance
A549-Cas9 and NCI-H460-Cas9 cells were infected with the Brunello human genome-wide lentiviral gRNA pooled library (Broad Institute, Cambridge, MA). The library consists of 76,441 sgRNAs targeting 19,114 different genes (Doench et al., 2016). A minimum of 135 million cells of each cell line was cultured in 12-well tissue culture plates containing 3 million cells per well to ensure adequate representation of each sgRNA, e.g., an average of 500 cells per sgRNA, when infected at a multiplicity of infection of 0.3 in media supplemented with 8 μg/ml polybrene. Plates were centrifuged at 2000 rpm at 37°C for 2 h, and then the media was replaced with fresh media without polybrene for incubation overnight. The cells were then transferred to 15 cm tissue culture dishes and infected cells were selected with 2 μg/mL puromycin for 4 days. After selection, 40 million cells were cultured for each of three replicates in 15 cm dishes containing 5 million cells per dish. An additional 40 million cells per replicate were harvested and frozen for subsequent genomic DNA extraction. Cultured cells were then treated with either 100 nM (A549) or 50 nM (NCI-H460) prexasertib for 19 days. These concentrations were selected to achieve roughly no net increase or decrease in the total number of cells during the 19 days. Cells were re-plated every 3–4 days and maintained in prexasertib-containing media, each time plating a minimum of 40 million cells per replicate. At the end of the screen, the cells were harvested and frozen from genomic DNA extraction.
Genomic DNA isolation, sgRNA sequencing and identification of top hits
Genomic DNA was isolated using QIAamp DNA blood midi and maxi kits (QIAGEN, Hilden, Germany) per manufacturer’s instructions. PCR was performed to attach sequencing adaptors and barcodes for multiplexed sgRNA sequencing, as previously described (Doench et al., 2016). Samples were sequenced on an Illumina HiSeq2000. Reads per million + 1 were log2 transformed, and then the log2-fold change of each sgRNA after 19 days of prexasertib treatment was determined by comparing its abundance to that in Brunello human genome-wide lentiviral sgRNA pooled library plasmid DNA. The average (mean) values of three replicates were used for subsequent analyses. The STARS gene-ranking algorithm was used to determine the top 10% of genes for which 2 or more sgRNAs became enriched in cells treated with prexasertib (Doench et al., 2016). Top gene hits were identified as those whose false discovery rates were < 10% in both NSCLC cell lines. Additionally, the normalized data were also analyzed with the hypergeometric distribution method (https://github.com/mhegde/volcano_plots). The volcano plots of these results were created using R (https://www.r-project.org).
Growth Curves
A549 cells were seeded at 30,000 cells in 60 mm dishes and harvested and fixed with cold ethanol (70%) every two days for ten days. The media was refreshed on days 4, 8, and 10, and cells were re-plated into 150 mm dishes on day 6 to prevent contact arrest. Cell count was determined using Countbright Beads (Life Technologies) and flow cytometry (BD FACS Canto II).
Clonogenic Assays
500–1000 cells were seeded per well into 6-well plates. After 24 h, cells were continuously treated with the indicated compounds for about 2 weeks until discrete colonies could be readily counted in control wells. Colonies were fixed with fixation solution (5:1 methanol: acetic acid) at room temperature for 20 min and then stained with a solution of 0.5% crystal violet in methanol for 2 h.
Viability Assays
1000 cells in 100 μL media were placed in each well of a 96 well plate the day before drug treatment. 100 μL of media with 2X drug concentration was added to achieve the final concentrations shown. 72 h post-treatment, cells were incubated at room temperature for 30 mins before quantification of viability using CellTiter-Glo (Promega) on an M200 Infinite plate reader (Tecan).
Immunoblotting
Whole-cell extracts were generated by incubating cell pellets in EBC buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.5 mM EDTA, 10% Glycerol, 0.5% NP-40, Protease Cocktail Set I and Phosphatase Cocktail Set I) and cleared by centrifugation. Relative protein levels in lysates were normalized by Bradford assay and samples were boiled in 6x SDS buffer. Samples were then loaded on 4%–20% TGX gels (Bio-Rad) for electrophoresis which were then transferred to a nitrocellulose membrane (Bio-Rad) by wet transfer. Membranes were blocked in 5% nonfat milk (Boston Biochem) for 1 h at room temperature and incubated in primary antibody overnight at 4C. Primary antibodies were diluted as follows: CDK1 and cyclin E were diluted 1:200 in 3% nonfat milk in TBST; pS1981 ATM was diluted 1:500 in 3% nonfat milk in TBST; γ-H2AX, pS317 CHK1, CHK1, ATM, DNA-PKcs and MYBL2 were diluted 1:1000 in 3% nonfat milk in TBST; pS824 KAP1, KAP1, pS4/S8 RPA, pS33 RPA, RPA, cyclin B1, and GAPDH were diluted 1:2000 in 3% nonfat milk TBST; pT487 MYBL2 was diluted 1:5000 in 3% nonfat milk; LIN54 was diluted 1:1000 in 5% nonfat milk in TBST; FOXM1 was diluted 1:2000 in 5% nonfat milk TBST; vinculin was diluted 1:30000 in 5% nonfat milk in TBST; pS345 CHK1, pT161 CDK1, pY15 CDK1/2 and pS10 histone H3 were diluted 1:1000 in 5% BSA in TBST; pS2056 DNA-PKcs was diluted 1:2000 in 5% BSA in TBST. Membranes were incubated in secondary antibodies, diluted 1:5000 in 5% nonfat milk, for 1 h at room temperature. Bands were visualized using Super Signal West Pico (Thermo Scientific) or Immobilon Western Chemiluminescent (Millipore) HRP substrate on the G-box imaging system (Syngene).
Flow Cytometry
Trypsinized cells were washed with PBS and fixed in 10% formalin in PBS for 15 min at room temperature. Cells were then washed three times with 1% BSA in PBS and permeabilized in 70% ethanol at −20°C for 30 min to overnight. After three washes with 1% BSA in PBS, incorporated EdU was labeled with a CLICK reaction (2 mM CuSO4, 100 μM THPTA, 100 mM sodium ascorbate, and 2 μM Alexa 647 Azide or Calfluor 647 Azide in PBS) by rotating for 30 min at room temperature. In the case of pATM, instead of the CLICK reaction, samples were incubated in 200 ug/ml DNaseI in DPBS for 1 hour at 37°C. Samples were then washed thrice with 1% BSA in PBS and incubated with the primary antibody in 1% BSA in PBS overnight at 4°C. Primary antibodies were diluted as follows: 1:250, pT600 FOXM1; 1:500, pS824 KAP1; 1: 500, pS1981 ATM; and 1:50, pS10 histone H3-Alexa Fluor 488 conjugate. After 3 washes with 1% BSA in PBS, samples were incubated with secondary antibodies, which were diluted 1:250 in 1% BSA in PBS, for 1 h at room temperature in darkness. pATM 488 was incubated with mouse-Alexa Fluor 488 conjugate, pT600 FOXM1 was incubated with a rabbit-Alexa Fluor 488 conjugate and pS824 KAP1 was incubated with a rabbit-PE conjugate. After three more 1% BSA in PBS washes, DNA staining was then conducted by incubating samples in darkness and at room temperature for 1 h in a 1 μg/ml DAPI, 100 ng/ml RNase A staining solution. pATM samples were incubated with anti-BrdU-APC (1:50 in 1% BSA in PBS) for 1 hour at room temperature followed by three washes between secondary and DAPI staining. Samples were analyzed on Canto II or Fortessa analyzers (BD Biosciences) and 50,000 total events were captured for each sample. A negative control where an EdU pulse and primary antibodies were omitted but otherwise normally processed was used to establish negative populations and perform background subtraction on MFIs. A rabbit-Alexa 488 isotype control was used for the negative control for experiments with pH3-Alexa 488 staining. The Early and Late S phase gates were determined by dividing the EdU positive population of the EV or DMSO control in half based on DAPI signal.
The chromatin flow cytometry protocol was based upon approaches used in previous studies (Hauge et al., 2017; Matson et al., 2017). Briefly, trypsinized cells were incubated on ice in 750 μL of Buffer A (300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 10 mM PIPES pH 7.0, 0.5% Triton x-100, Protease Cocktail Set I, and Phosphatase Cocktail Set I) for 5 min. 250 μL of Buffer B (300 mM sucrose, 100 mM NaCl, 3 mM MgCl2, 10 mM PIPES pH 7.0, 0.5% Triton x-100, 10% Formalin, Phosphatase Cocktail Set I, and Phosphatase Cocktail Set I) was immediately added, and samples were incubated for an additional 15 min on ice. Cells were then pelleted (1000 × g, 5 min, 4°C) and washed with 1% BSA in PBS 3 times before proceeding onto EdU staining as with other samples. Primary antibodies for chromatin flow cytometry were diluted as follows: CDC45, 1:50; PCNA, 1:500; RPA32, 1:500; pS4/S8 RPA32, 1:1000; and H2AX, 1:1000. For CDC45 samples, AFDye 546 Azide and anti-rabbit Alexa 647 secondary were used. An anti-mouse Alexa 488 secondary antibody and Calfluor 647 Azide was used with PCNA samples. For RPA32/pRPA32 and γ-H2AX samples, anti-rabbit Alexa 647 secondary and anti-mouse Alexa 488 secondary were used for RPA32/pRPA32 and γ-H2AX respectively. Otherwise, the CLICK reaction, primary and secondary as well as DAPI staining were conducted as described above.
For Annexin V/PI staining, cells were dissociated with StemPro Accutase (GIBCO) and stained using the FITC Annexin V Apoptosis Detect Kit II.
RNA Expression
Total RNA was extracted from cell pellets using the RNeasy Plus kit (QIAGEN). RNA was checked for quality using TapeStation and Qubit before library preparation using Kappa stranded mRNA Hyper Prep and sequencing using Illumina NS500. Reads were mapped to Hg19 using STAR alignment (Dobin et al., 2013) and counts were generated using HTseq (Anders et al., 2015). Differential gene expression analysis was perform using DESeq2 (Love et al., 2014) and heatmaps were generated using R package pheatmap. Mean counts were generated using DESeq2 mean-ratio normalization method. Gene set enrichment analysis was conducted using GSEA software (https://www.broad.mit.edu/gsea/downloads.jsp) (Mootha et al., 2003; Subramanian et al., 2005). RNA-seq datasets have been deposited in the GEO database (accession number GSE154545). Gene annotations in Figure 5D for G1/S and G2/M genes are from Schade et al. (2019). RT-qPCR to validate RNA expression data was done using cDNA generated from total RNA isolated by the RNeasy Plus kit and the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). qPCR was performed on the cDNA using the primers listed in Table S3 and Brilliant III Ultra-Fast qPCR Master Mix (Agilent) using a MxAria (Agilent)
QUANTIFICATION AND STATISTICAL ANALYSIS
Flow cytometry data was analyzed using FlowJo. GraphPad Prism was used to generate graphs and conduct statistical analyses for flow cytometry, viability, growth curves and RT-qPCR. Statistical details of experiments can be found in the figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| pS1981 ATM | Biolegend | Cat# 651201; RRID: AB_10900815 |
| ATM | Cell Signaling Technologies | Cat# 2873T; RRID: AB_2062659 |
| MYBL2 | Millipore | Cat# MABE866 |
| pT487 MYBL2 | Abcam | Cat# Ab76009; RRID: AB_1309969 |
| CDC45 | Cell Signaling Technologies | Cat# 11881S; RRID: AB_2715569 |
| CDK1 | Santa Cruz Biotechnology | Cat# sc-54; RRID: AB_627224 |
| pT161 CDK1 | Cell Signaling Technologies | Cat# 9114S; RRID: AB_2074652 |
| pY15 CDK1/2 | Cell Signaling Technologies | Cat# 9111S; RRID: AB_331460 |
| CHK1 | Santa Cruz Biotechnology | Cat# sc-8408; RRID: AB_627257 |
| pS317 CHK1 | Cell Signaling Technologies | Cat# 2344S; RRID: AB_331488 |
| pS345 CHK1 | Cell Signaling Technologies | Cat# 2341S; RRID: AB_330023 |
| Cyclin E (CCNE) | Santa Cruz Biotechnology | Cat# sc-247; RRID: AB_627357 |
| Cyclin B1 | Santa Cruz Biotechnology | Cat # sc-752; RRID: AB_2072134 |
| pS2056 DNA-PKcs | Cell Signaling Technologies | Cat# 68716S |
| DNA-PKcs | Cell Signaling Technologies | Cat# 38168S; RRID: AB_2799128 |
| FOXM1 | Santa Cruz Biotechnology | Cat# sc-520;RRID: AB_631523 |
| pT600 FOXM1 | Cell Signaling Technologies | Cat# 14655S; RRID: AB_2798557 |
| GAPDH | Santa Cruz Biotechnology | Cat# sc-47724; RRID: AB_627678 |
| γ-H2AX | Millipore | Cat# 05–636-I; RRID: AB_2755003 |
| pS10 histone H3 | Cell Signaling Technologies | Cat# 3377T; RRID: AB_1549592 |
| pS10 histone H3 - Alexa 488 conjugate | Cell Signaling Technologies | Cat# 3465S; RRID: AB_10695860 |
| KAP1 | Bethyl Laboratories | Cat# A300–274A-M; RRID: AB_185559 |
| pS824 KAP1 | Bethyl Laboratories | Cat# A300–767A-M; RRID: AB_669740 |
| LIN54 | Bethyl Laboratories | Cat# A300–799A-M; RRID: AB_11218173 |
| PCNA | Cell Signaling Technologies | Cat# 2586S; RRID: AB_2160343 |
| RPA32 | Bethyl Laboratories | Cat# A300–244A-M; RRID: AB_185548 |
| pS4/S8 RPA32 | Bethyl Laboratories | Cat# A300–245A-M; RRID: AB_210547 |
| pS33 RPA32 | Bethyl Laboratories | Cat# A300–246A-M; RRID: AB_210547 |
| VINCULIN | Sigma-Aldrich | Cat# V9264; RRID: AB_10603627 |
| anti-BrdU-APC conjugate | Biolegend | Cat# 339808; RRID: AB_10895898 |
| anti-mouse IgG - HRP | Bethyl Laboratories | Cat# A90–116P; RRID: AB_67183 |
| anti-rabbit IgG - HRP | Bethyl Laboratories | Cat# A120–101P; RRID: AB_67264 |
| anti-mouse IgG - Alexa 488 conjugate | Cell Signaling Technologies | Cat# 4408S; RRID: AB_10694704 |
| Rabbit IgG isotype control | Cell Signaling Technologies | Cat# 4340S; RRID: AB_561545 |
| anti-Rabbit IgG - Alexa 488 conjugate | Life Technologies | Cat# A11008; RRID: AB_143165 |
| anti-Rabbit IgG - PE conjugate | Cell Signaling Technologies | Cat# 8885S; RRID: AB_2797677 |
| anti-Rabbit IgG - Alexa 647 conjugate | Life Technologies | Cat# A21245; RRID: AB_141775 |
| Biological samples | ||
| Brunello human genome-wide lentiviral sgRNA pooled library | Broad Institute (Doench et al., 2016) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Prexasertib (LY2606368, CHK1i) | Eli Lilly & Co. | S7178 |
| AZ20 (ATRi-2) | Santa Cruz Biotechnology | sc-503186 |
| VE-822 (ATRi-1) | MedChem Express | HY-13902 |
| KU-55933 (ATMi) | Selleck | S1092 |
| Nedisertib (DNA-PKi) | MedChem Express | HY-10570 |
| RO-3306 (CDK1i) | Santa Cruz Biotechnology | sc-358700 |
| Adavosertib (AZD1775, WEE1i) | Selleck | S1525 |
| Doxorubicin (DOXO) | Cell Signaling Technologies | 5927S |
| Countbright Beads | Life Technologies | C36950 |
| AFDye 546 Azide | Click Chemistry Tools | 1283 |
| Alexa 647 Azide | Life Technologies | A10277 |
| CalFluor 647 Azide | Click Chemistry Tools | 1372 |
| 5-ethynyl-2′-deoxyuridine (EdU) | Life Technologies | A10044 |
| 5-bromo-2′-deoxyuridine (BrdU) | Sigma-Aldrich | B5002 |
| 4′,6-diamidino-2-phenylindole (DAPI) | Invitrogen | D3571 |
| Lipofectamine RNAiMAX | Invitrogen | 13778030 |
| DNaseI | Sigma Aldrich | D4513–1VL |
| Protease Cocktail Set I | Millipore | 539131 |
| Phosphatase Cocktail Set I | Millipore | 524624 |
| Critical commercial assays | ||
| FITC Annexin V Apoptosis Detection Kit II | BD Biosciences | 556570 |
| Deposited data | ||
| RNA-seq Data | This Paper | GEO: GSE154545 |
| Experimental models: cell lines | ||
| A549 | ATCC | CCL-185 |
| A549-Cas9 (pXPR_311) | Broad Institute | N/A |
| NCI-H460 | ATCC | HTB-177 |
| NCI-H460-Cas9 (pXPR_311) | Broad Institute | N/A |
| NCI-H23 | ATCC | CRL-5800 |
| NCI-H1299 | ATCC | CRL-5803 |
| Oligonucleotides: siRNA and other | ||
| siCTRL | Dharmacon | D-001810–10 |
| siLIN54 | Dharmacon | L-019325–01 |
| siCCNA2 | Dharmacon | L-003205–00 |
| siMYBL2 | Dharmacon | L-010444–01 |
| sgRNA and qPCR primers | This Paper | Table S3 |
Highlights.
The MMB-FOXM1 complex components are required for CHK1i sensitivity
CHK1i prematurely activates the G2/M transcriptional program by MMB-FOXM1
Mitotic pH3 induced by CHK1i during S phase requires the MMB-FOXM1 complex
Premature mitosis is required for replication catastrophe after CHK1 inhibition
ACKNOWLEDGMENTS
We thank Huy Nguyen for assistance with bioinformatics analysis and visualization for Figures 1B and 1C. We thank Mona Ahmed for technical assistance regarding the pATM staining and DNA-PKcs reagents in Figure 3. This research was supported by the US Public Health Service grants F31CA189328 to T.B.B.; R35CA232128, R01CA63113, R01CA173023, and P01CA203655 to J.A.D.; and K08CA172354 to D.K. This work was also supported by the Joint Center for Radiation Therapy (to D.K.) and in part by a Sponsored Research Agreement from Eli Lilly to G.I.S. and A.D.D.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108808.
DECLARATION OF INTERESTS
D.K. has served as a consultant to Vertex. G.I.S. is a consultant/advisory board member for Lilly, Sierra Oncology, Merck-EMD Serono, Pfizer, Astex, Almac, Roche, Bicycle Therapeutics, Fusion Pharmaceuticals, G1 Therapeutics, Bayer, Ipsen, Cybrexa Therapeutics, Angiex, Daiichi Sankyo, and Seattle Genetics and reports receiving commercial research grants from Lilly, Sierra Oncology, Merck-EMD Serono, and Merck & Co. A.D.D. is a consultant/advisory board member for Lilly Oncology, Merck-EMD Serono, Intellia Therapeutics, Sierra Oncology, Cyteir Therapeutics, Third Rock Ventures, AstraZeneca, Ideaya Inc., and Cedilla Therapeutics Inc.; a stockholder in Ideaya Inc., Cedilla Therapeutics Inc., and Cyteir; and received research support from Lilly Oncology and Merck-EMD Serono. J.A.D. is a consultant to Merck & Co. and EMD Serono and received research support from Constellation Pharmaceuticals.
REFERENCES
- Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, and Turner NC (2012). Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2, 524–539. [DOI] [PubMed] [Google Scholar]
- Abe Y, Sako K, Takagaki K, Hirayama Y, Uchida KS, Herman JA, De-Luca JG, and Hirota T (2016). HP1-Assisted Aurora B Kinase Activity Prevents Chromosome Segregation Errors. Dev. Cell 36, 487–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashley AK, Shrivastav M, Nie J, Amerin C, Troksa K, Glanzer JG, Liu S, Opiyo SO, Dimitrova DD, Le P, et al. (2014). DNA-PK phosphorylation of RPA32 Ser4/Ser8 regulates replication stress checkpoint activation, fork restart, homologous recombination and mitotic catastrophe. DNA Repair (Amst.) 21, 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blosser WD, Dempsey JA, McNulty AM, Rao X, Ebert PJ, Lowery CD, Iversen PW, Webster YW, Donoho GP, Gong X, et al. (2020). A pan-cancer transcriptome analysis identifies replication fork and innate immunity genes as modifiers of response to the CHK1 inhibitor prexasertib. Oncotarget 11, 216–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Boisvert JL, Benes CH, and Zou L (2015). Distinct but Concerted Roles of ATR, DNA-PK, and Chk1 in Countering Replication Stress during S Phase. Mol. Cell 59, 1011–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhari AB, Lewis CW, Pearce JJ, Luong D, Chan GK, and Gamper AM (2019). Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Invest 129, 1329–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdova K, Yang H, Faedda R, Hume S, Chauhan J, Ebner D, Kessler BM, Vendrell I, Drewry DH, Wells CI, et al. (2019). E2F1 proteolysis via SCF-cyclin F underlies synthetic lethality between cyclin F loss and Chk1 inhibition. EMBO J. 38, e101443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch CJ, Kröger MS, Jensen J, Kriegs M, Gatzemeier F, Petersen C, Münscher A, Rothkamm K, and Rieckmann T (2017). G2-checkpoint targeting and radiosensitization of HPV/p16-positive HNSCC cells through the inhibition of Chk1 and Wee1. Radiother. Oncol 122, 260–266. [DOI] [PubMed] [Google Scholar]
- Carrassa L, Chilà R, Lupi M, Ricci F, Celenza C, Mazzoletti M, Broggini M, and Damia G (2012). Combined inhibition of Chk1 and Wee1: in vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 11, 2507–2517. [DOI] [PubMed] [Google Scholar]
- Chaudhuri L, Vincelette ND, Koh BD, Naylor RM, Flatten KS, Peterson KL, McNally A, Gojo I, Karp JE, Mesa RA, et al. (2014). CHK1 and WEE1 inhibition combine synergistically to enhance therapeutic efficacy in acute myeloid leukemia ex vivo. Haematologica 99, 688–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Müller GA, Quaas M, Fischer M, Han N, Stutchbury B, Shar-rocks AD, and Engeland K (2013). The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell. Biol 33, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilà R, Basana A, Lupi M, Guffanti F, Gaudio E, Rinaldi A, Cascione L, Restelli V, Tarantelli C, Bertoni F, et al. (2015). Combined inhibition of Chk1 and Wee1 as a new therapeutic strategy for mantle cell lymphoma. Oncotarget 6, 3394–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu L, Huo Y, Liu X, Yao P, Thomas K, Jiang H, Zhu T, Zhang G, Chaudhry M, Adams G, et al. (2014). The spatiotemporal dynamics of chromatin protein HP1α is essential for accurate chromosome segregation during cell division. J. Biol. Chem 289, 26249–26262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Vail P, Witkiewicz AK, and Knudsen ES (2019). Coordinately Targeting Cell-Cycle Checkpoint Functions in Integrated Models of Pancreatic Cancer. Clin. Cancer Res 25, 2290–2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clurman BE, Sheaff RJ, Thress K, Groudine M, and Roberts JM (1996). Turnover of cyclin E by the ubiquitin-proteasome pathway is regulated by cdk2 binding and cyclin phosphorylation. Genes Dev. 10, 1979–1990. [DOI] [PubMed] [Google Scholar]
- Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, Diskin SJ, Attiyeh EF, Sennett R, Norris G, et al. (2011). RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. USA 108, 3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies KD, Cable PL, Garrus JE, Sullivan FX, von Carlowitz I, Huerou YL, Wallace E, Woessner RD, and Gross S (2011). Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol. Ther 12, 788–796. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down CF, Millour J, Lam EW, and Watson RJ (2012). Binding of FoxM1 to G2/M gene promoters is dependent upon B-Myb. Biochim. Biophys. Acta 1819, 855–862. [DOI] [PubMed] [Google Scholar]
- Dunlop CR, Wallez Y, Johnson TI, Bernaldo de Quirós Fernández S, Durant ST, Cadogan EB, Lau A, Richards FM, and Jodrell DI (2020). Complete loss of ATM function augments replication catastrophe induced by ATR inhibition and gemcitabine in pancreatic cancer models. Br. J. Cancer 123, 1424–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch T, and Nurse P (1990). Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell 60, 665–673. [DOI] [PubMed] [Google Scholar]
- Eykelenboom JK, Harte EC, Canavan L, Pastor-Peidro A, Calvo-Asensio I, Llorens-Agost M, and Lowndes NF (2013). ATR activates the S-M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep. 5, 1095–1107. [DOI] [PubMed] [Google Scholar]
- Fischer M, Grossmann P, Padi M, and DeCaprio JA (2016). Integration of TP53, DREAM, MMB-FOXM1 and RB-E2F target gene analyses identifies cell cycle gene regulatory networks. Nucleic Acids Res. 44, 6070–6086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, Tindall DJ, and Chen J (2008). Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat. Cell Biol 10, 1076–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari B, Rhind N, and Russell P (1997). Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science 277, 1495–1497. [DOI] [PubMed] [Google Scholar]
- Furuta T, Takemura H, Liao ZY, Aune GJ, Redon C, Sedelnikova OA, Pilch DR, Rogakou EP, Celeste A, Chen HT, et al. (2003). Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem 278, 20303–20312. [DOI] [PubMed] [Google Scholar]
- García P, and Frampton J (2006). The transcription factor B-Myb is essential for S-phase progression and genomic stability in diploid and polyploid mega-karyocytes. J. Cell Sci 119, 1483–1493. [DOI] [PubMed] [Google Scholar]
- Ghelli Luserna Di Rorà A, Bocconcelli M, Ferrari A, Terragna C, Bruno S, Imbrogno E, Beeharry N, Robustelli V, Ghetti M, Napolitano R, et al. (2019). Synergism Through WEE1 and CHK1 Inhibition in Acute Lymphoblastic Leukemia. Cancers (Basel) 11, 1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna RE, and Doench JG (2020). Design and analysis of CRISPR-Cas experiments. Nat. Biotechnol 38, 813–823. [DOI] [PubMed] [Google Scholar]
- Hauge S, Naucke C, Hasvold G, Joel M, Rødland GE, Juzenas P, Stokke T, and Syljuåsen RG (2017). Combined inhibition of Wee1 and Chk1 gives synergistic DNA damage in S-phase due to distinct regulation of CDK activity and CDC45 loading. Oncotarget 8, 10966–10979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong D, Infante J, Janku F, Jones S, Nguyen LM, Burris H, Naing A, Bauer TM, Piha-Paul S, Johnson FM, et al. (2016). Phase I Study of LY2606368, a Checkpoint Kinase 1 Inhibitor, in Patients With Advanced Cancer. J. Clin. Oncol 34, 1764–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong DS, Moore K, Patel M, Grant SC, Burris HA 3rd, William WN Jr., Jones S, Meric-Bernstam F, Infante J, Golden L, et al. (2018). Evaluation of Prexasertib, a Checkpoint Kinase 1 Inhibitor, in a Phase Ib Study of Patients with Squamous Cell Carcinoma. Clin. Cancer Res 24, 3263–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke R, King GA, Kupiec M, and Rine J (2018). Pivotal roles of PCNA loading and unloading in heterochromatin function. Proc. Natl. Acad. Sci. USA 115, E2030–E2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno Y, Suzuki A, Sugimura K, Okumura K, Zineldeen DH, Shimada M, Niida H, Mizuno T, Hanaoka F, and Nakanishi M (2009). Cyclin A-Cdk1 regulates the origin firing program in mammalian cells. Proc. Natl. Acad. Sci. USA 106, 3184–3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kausar T, Schreiber JS, Karnak D, Parsels LA, Parsels JD, Davis MA, Zhao L, Maybaum J, Lawrence TS, and Morgan MA (2015). Sensitization of Pancreatic Cancers to Gemcitabine Chemoradiation by WEE1 Kinase Inhibition Depends on Homologous Recombination Repair. Neoplasia 17, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ST, Lim DS, Canman CE, and Kastan MB (1999). Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem 274, 37538–37543. [DOI] [PubMed] [Google Scholar]
- King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D, and Marshall MS (2015). LY2606368 Causes Replication Catastrophe and Antitumor Effects through CHK1-Dependent Mechanisms. Mol. Cancer Ther 14, 2004–2013. [DOI] [PubMed] [Google Scholar]
- Klein DK, Hoffmann S, Ahlskog JK, O’Hanlon K, Quaas M, Larsen BD, Rolland B, Rösner HI, Walter D, Kousholt AN, et al. (2015). Cyclin F suppresses B-Myb activity to promote cell cycle checkpoint control. Nat. Commun 6, 5800. [DOI] [PubMed] [Google Scholar]
- Knight AS, Notaridou M, and Watson RJ (2009). A Lin-9 complex is recruited by B-Myb to activate transcription of G2/M genes in undifferentiated embryonal carcinoma cells. Oncogene 28, 1737–1747. [DOI] [PubMed] [Google Scholar]
- Köhler C, Koalick D, Fabricius A, Parplys AC, Borgmann K, Pospiech H, and Grosse F (2016). Cdc45 is limiting for replication initiation in humans. Cell Cycle 15, 974–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppenhafer SL, Goss KL, Terry WW, and Gordon DJ (2018). mTORC1/2 and Protein Translation Regulate Levels of CHK1 and the Sensitivity to CHK1 Inhibitors in Ewing Sarcoma Cells. Mol. Cancer Ther 17, 2676–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam MH, Liu Q, Elledge SJ, and Rosen JM (2004). Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 6, 45–59. [DOI] [PubMed] [Google Scholar]
- Lane S, Farlie P, and Watson R (1997). B-Myb function can be markedly enhanced by cyclin A-dependent kinase and protein truncation. Oncogene 14, 2445–2453. [DOI] [PubMed] [Google Scholar]
- Laoukili J, Alvarez M, Meijer LA, Stahl M, Mohammed S, Kleij L, Heck AJ, and Medema RH (2008). Activation of FoxM1 during G2 requires cyclin A/Cdk-dependent relief of autorepression by the FoxM1 N-terminal domain. Mol. Cell. Biol 28, 3076–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Kozono D, Deraska P, Branigan T, Dunn C, Zheng XF, Parmar K, Nguyen H, DeCaprio J, Shapiro GI, et al. (2020). CHK1 Inhibitor Blocks Phosphorylation of FAM122A and Promotes Replication Stress. Mol. Cell 80, 410–422.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, and DeCaprio JA (2007). Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol. Cell 26, 539–551. [DOI] [PubMed] [Google Scholar]
- Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, Troksa K, Shrivastav M, Nickoloff JA, and Oakley GG (2012). Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res. 40, 10780–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupardus PJ, Byun T, Yee MC, Hekmat-Nejad M, and Cimprich KA (2002). A requirement for replication in activation of the ATR-dependent DNA damage checkpoint. Genes Dev. 16, 2327–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnussen GI, Emilsen E, Giller Fleten K, Engesæter B, Nähse-Kumpf V, Fjær R, Slipicevic A, and Flørenes VA (2015). Combined inhibition of the cell cycle related proteins Wee1 and Chk1/2 induces synergistic anti-cancer effect in melanoma. BMC Cancer 15, 462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak JP, Man WY, Chow JP, Ma HT, and Poon RY (2015). Pharmacological inactivation of CHK1 and WEE1 induces mitotic catastrophe in nasopharyngeal carcinoma cells. Oncotarget 6, 21074–21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matson JP, Dumitru R, Coryell P, Baxley RM, Chen W, Twaroski K, Webber BR, Tolar J, Bielinsky AK, Purvis JE, and Cook JG (2017). Rapid DNA replication origin licensing protects stem cell pluripotency. eLife 6, e30473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrommati I, Faedda R, Galasso G, Li J, Burdova K, Fischer R, Kessler BM, Carrero ZI, Guardavaccaro D, Pagano M, and D’Angiolella V (2018). β-TrCP- and Casein Kinase II-Mediated Degradation of Cyclin F Controls Timely Mitotic Progression. Cell Rep. 24, 3404–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan CH, and Russell P (1993). Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 12, 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michowski W, Chick JM, Chu C, Kolodziejczyk A, Wang Y, Suski JM, Abraham B, Anders L, Day D, Dunkl LM, et al. (2020). Cdk1 Controls Global Epigenetic Landscape in Embryonic Stem Cells. Mol. Cell 78, 459–476.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Müller GA, Quaas M, Schümann M, Krause E, Padi M, Fischer M, Litovchick L, DeCaprio JA, and Engeland K (2012). The CHR promoter element controls cell cycle-dependent gene transcription and binds the DREAM and MMB complexes. Nucleic Acids Res. 40, 1561–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair J, Huang TT, Murai J, Haynes B, Steeg PS, Pommier Y, and Lee JM (2020). Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 39, 5520–5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MJ, Raleigh JM, Verkade HM, and Nurse P (1997). Chk1 is a wee1 kinase in the G2 DNA damage checkpoint inhibiting cdc2 by Y15 phosphorylation. EMBO J. 16, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley GG, Patrick SM, Yao J, Carty MP, Turchi JJ, and Dixon K (2003). RPA phosphorylation in mitosis alters DNA binding and protein-protein interactions. Biochemistry 42, 3255–3264. [DOI] [PubMed] [Google Scholar]
- Olson E, Nievera CJ, Klimovich V, Fanning E, and Wu X (2006). RPA2 is a direct downstream target for ATR to regulate the S-phase checkpoint. J. Biol. Chem 281, 39517–39533. [DOI] [PubMed] [Google Scholar]
- Petermann E, Woodcock M, and Helleday T (2010). Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl. Acad. Sci. USA 107, 16090–16095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilkinton M, Sandoval R, Song J, Ness SA, and Colamonici OR (2007). Mip/LIN-9 regulates the expression of B-Myb and the induction of cyclin A, cyclin B, and CDK1. J. Biol. Chem 282, 168–175. [DOI] [PubMed] [Google Scholar]
- Ruiz S, Mayor-Ruiz C, Lafarga V, Murga M, Vega-Sendino M, Ortega S, and Fernandez-Capetillo O (2016). A Genome-wide CRISPR Screen Identifies CDC25A as a Determinant of Sensitivity to ATR Inhibitors. Mol. Cell 62, 307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MR, Levin K, Rader J, Belcastro L, Li Y, Martinez D, Pawel B, Shumway SD, Maris JM, and Cole KA (2013). Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 73, 776–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadasivam S, Duan S, and DeCaprio JA (2012). The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev. 26, 474–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros-Soberanis F, Samejima K, Xie L, Paulson JR, Earnshaw WC, et al. (2018). An intrinsic S/G2 checkpoint enforced by ATR. Science 361, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schade AE, Fischer M, and DeCaprio JA (2019). RB, p130 and p107 differentially repress G1/S and G2/M genes after p53 activation. Nucleic Acids Res. 47, 11197–11208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmit F, Cremer S, and Gaubatz S (2009). LIN54 is an essential core subunit of the DREAM/LINC complex that binds to the cdc2 promoter in a sequence-specific manner. FEBS J. 276, 5703–5716. [DOI] [PubMed] [Google Scholar]
- Sirbu BM, McDonald WH, Dungrawala H, Badu-Nkansah A, Kavanaugh GM, Chen Y, Tabb DL, and Cortez D (2013). Identification of proteins at active, stalled, and collapsed replication forks using isolation of proteins on nascent DNA (iPOND) coupled with mass spectrometry. J. Biol. Chem 288, 31458–31467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Toledo L, Neelsen KJ, and Lukas J (2017). Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell 66, 735–749. [DOI] [PubMed] [Google Scholar]
- van Harten AM, Buijze M, van der Mast R, Rooimans MA, Martens-de Kemp SR, Bachas C, Brink A, Stigter-van Walsum M, Wolthuis RMF, and Brakenhoff RH (2019). Targeting the cell cycle in head and neck cancer by Chk1 inhibition: a novel concept of bimodal cell death. Oncogenesis 8, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, Chen Z, Sri-vastava M, McLaughlin ME, Navone NM, et al. (2019). Genome-wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 38, 2451–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White DE, Negorev D, Peng H, Ivanov AV, Maul GG, and Rauscher FJ III. (2006). KAP1, a novel substrate for PIKK family members, colocalizes with numerous damage response factors at DNA lesions. Cancer Res. 66, 11594–11599. [DOI] [PubMed] [Google Scholar]
- White D, Rafalska-Metcalf IU, Ivanov AV, Corsinotti A, Peng H, Lee SC, Trono D, Janicki SM, and Rauscher FJ III. (2012). The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol. Cancer Res 10, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yajima H, Lee KJ, Zhang S, Kobayashi J, and Chen BP (2009). DNA double-strand break formation upon UV-induced replication stress activates ATM and DNA-PKcs kinases. J. Mol. Biol 385, 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos G, Rainey MD, and Gillespie DA (2005). Chk1-dependent S-M checkpoint delay in vertebrate cells is linked to maintenance of viable replication structures. Mol. Cell. Biol 25, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Otterness DM, Chiang GG, Xie W, Liu YC, Mercurio F, and Abraham RT (2005). Genotoxic stress targets human Chk1 for degradation by the ubiquitin-proteasome pathway. Mol. Cell 19, 607–618. [DOI] [PubMed] [Google Scholar]
- Zhao H, and Piwnica-Worms H (2001). ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol 21, 4129–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebold U, Bartsch O, Marais R, Ferrari S, and Klempnauer KH (1997). Phosphorylation and activation of B-Myb by cyclin A-Cdk2. Curr. Biol 7, 253–260. [DOI] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, and Shiloh Y (2006). Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol 8, 870–876. [DOI] [PubMed] [Google Scholar]
- Zou L, and Elledge SJ (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]
- Zuazua-Villar P, Rodriguez R, Gagou ME, Eyers PA, and Meuth M (2014). DNA replication stress in CHK1-depleted tumour cells triggers premature (S-phase) mitosis through inappropriate activation of Aurora kinase B. Cell Death Dis. 5, e1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited in the NCI Gene Expression Omnibus database (https://www.ncbi.nlm.nih.gov/geo/) and are accessible through GEO: GSE154545, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154545.