

Abstract
Broad‐spectrum antibiotics target multiple gram‐positive and gram‐negative bacteria, and can collaterally damage the gut microbiota. Yet, our knowledge of the extent of damage, the antibiotic activity spectra, and the resistance mechanisms of gut microbes is sparse. This limits our ability to mitigate microbiome‐facilitated spread of antibiotic resistance. In addition to antibiotics, non‐antibiotic drugs affect the human microbiome, as shown by metagenomics as well as in vitro studies. Microbiome–drug interactions are bidirectional, as microbes can also modulate drugs. Chemical modifications of antibiotics mostly function as antimicrobial resistance mechanisms, while metabolism of non‐antibiotics can also change the drugs’ pharmacodynamic, pharmacokinetic, and toxic properties. Recent studies have started to unravel the extensive capacity of gut microbes to metabolize drugs, the mechanisms, and the relevance of such events for drug treatment. These findings raise the question whether and to which degree these reciprocal drug–microbiome interactions will differ across individuals, and how to take them into account in drug discovery and precision medicine. This review describes recent developments in the field and discusses future study areas that will benefit from systems biology approaches to better understand the mechanistic role of the human gut microbiota in drug actions.
Keywords: antibiotics, antimicrobials, human gut microbiome, metagenomics, microbial community
Subject Categories: Microbiology, Virology & Host Pathogen Interaction
Bidirectional interactions have been reported between drugs and the gut microbiome. This Review discusses recent developments in the field as well as future study areas to better understand the mechanistic role of the human gut microbiota in drug actions.

Introduction
Our understanding of how the human gut microbiota contributes to health and disease, and how it changes over time, life stages, different geographic regions, and in response to environmental factors has increased dramatically over the last decade (The Integrative HMP (iHMP) Research Network Consortium, 2019; Pasolli et al, 2019; Nayfach et al, 2019; Falony et al, 2016). The current consensus is that the gut microbiome has a highly individualized composition, especially at the bacterial strain level (Franzosa et al, 2015). Further, healthy individuals retain a largely stable microbiota composition for most of their adulthood (Sommer et al, 2017; Mehta et al, 2018). This composition is established in early stages of life (Bäckhed et al, 2015; Wampach et al, 2017) and is dependent more on the environment than on host genetics (Rothschild et al, 2018). Hence strong perturbations, such as dietary shifts and antibiotic consumption, can unbalance microbiome stability, with so far unpredictable recovery (Willing et al, 2011; Falony et al, 2016; Lynn et al, 2018). On the other hand, chemical modification of therapeutic compounds by intestinal bacteria can influence the therapeutic effect of drugs (Fig 1). We have only recently begun to explore these complex, bidirectional interactions between our resident microbes and medication. In this review, we provide an overview of the different systems‐level approaches that can be employed to gain insights into the drug–microbiome–host triad (Fig 2). A better and more systematic understanding of these interactions and their underlying molecular constituents can be instrumental for diagnostic, prognostic, and ultimately, therapeutic applications.
Figure 1. Overview on the drug–microbiome–host triad and their interactions.
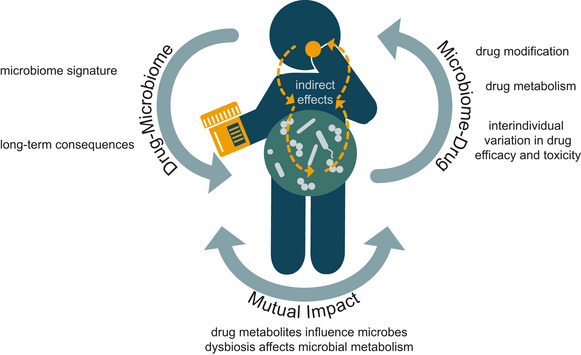
Left: The intake of drugs can have a direct influence on individual members of the gut microbiome (classic example: antibiotics) but can also change the composition and functionality of the microbiome through indirect, host‐mediated ways (example: proton‐pump inhibitors, which might alter the microbiome composition by increasing the gastric pH). Right: Intestinal bacteria can modify and metabolise drugs. In addition, the microbiome can indirectly modulate host xenobiotic metabolism in the liver. Furthermore, there is crosstalk between all these interactions. Ultimately, these complex interactions can possibly have negative health consequences and cause interpersonal differences in treatment outcomes.
Figure 2. Systems approaches to study drug–microbiome–host interactions.
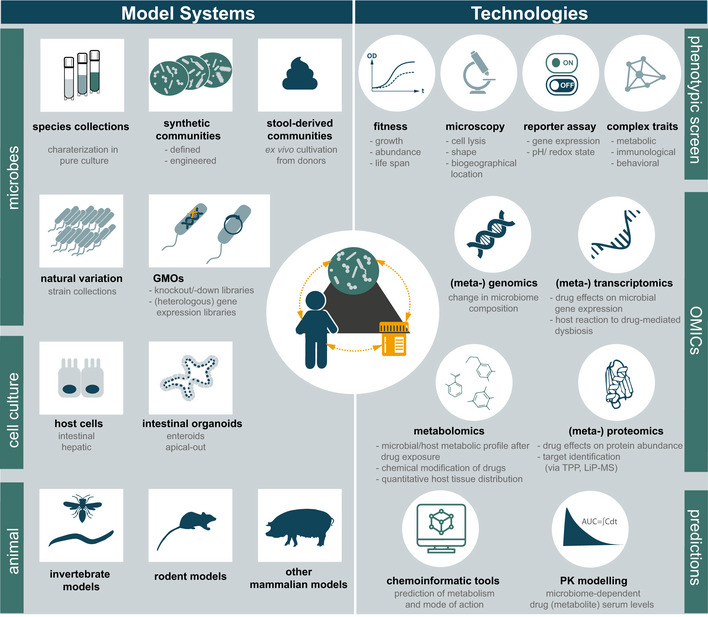
Left: A wide variety of model systems can be used to study drug–microbiome–host interactions. On the microbial side, (possibly genetically modified) isolates in pure culture or synthetic or stool‐derived microbial communities are applied. On the host side, simple cell culture systems, intestinal organoids but also different animal models can be employed. Right: Diverse technologies help to decipher drug–microbiome–host interactions. Approaches can be broadly divided into phenotypic characterization, OMICs approaches, and model‐based predictions. Depending on the research question, appropriate model systems and suitable technologies can be combined. TPP: thermal proteome profiling, LiP‐MS: limited proteolysis‐coupled mass spectrometry.
Therapeutic drugs alter the gut microbiome composition
Evidence from metagenomic‐based cohorts and clinical studies—the top‐down approach
Exploring the factors that explain inter‐individual differences in the intestinal microbiome composition across large population cohorts have repeatedly identified medication as a main contributor (Falony et al, 2016; Ticinesi et al, 2017; Jackson et al, 2018; Vich Vila et al, 2020). Although such studies have been insightful and have revealed the cumulative and dramatic impact medication has on the gut microbiome composition, they are still underpowered for separating the effects of individual drug classes. To begin stratifying these effects, one can broadly separate drugs to antimicrobials, developed to target microbes, and to drugs designed to interact with human/host targets, here referred to as human‐targeted drugs.
Antimicrobial drugs comprise antibiotics, antifungals, antiprotozoals, antivirals, and anti‐archaeals. These compounds target proteins that are typically absent in the host or are clearly distinguishable from their human homologues, yet they are often present in commensal microbes colonizing the human body. As a consequence, antimicrobials can “collaterally damage” the microbiome and thereby have mild to severe side effects to patients (Kuhn et al, 2016). This has been best studied for antibiotics, with clinical and animal studies illustrating changes in the gut microbiome composition and physiological host parameters, such as metabolic, cognitive, and immune functions (Cho et al, 2012; Cox et al, 2014; Hwang et al, 2015; Fröhlich et al, 2016; Hagan et al, 2019). Initial data indicate that the microbiota of healthy patients can partially rebound post‐antibiotic treatment (Rashid et al, 2015; Palleja et al, 2018). However, it remains unclear whether this is true for a broader and/or more diverse population, and what are the links to antibiotic classes, initial microbiome composition and treatment duration. Similarly, our knowledge on the target spectra, mode of action, and resistance mechanisms of the different classes of antibiotics and their specific effect on gut commensal bacterial species is scarce (preprint: Maier et al, 2020). To gain mechanistic insights into these matters, assays, tools, and test systems from decades of antibiotic research on pathogens can be capitalized and adapted to study gut commensal species in pure culture, within microbial communities and within the host, especially at a systematic level (Fig 2) (Maier & Typas, 2017). Such detailed mechanistic knowledge can help design better and more precise strategies to prevent or revert antibiotics‐caused "collateral damage," which at the moment are based on generic processes with limited success and/or adverse outcomes, such as fecal transplantation or administration of probiotics (Zmora et al, 2018; Suez et al, 2018; DeFilipp et al, 2019) (Box 2).
For host‐targeted drugs, increasing evidence suggests that they are associated with shifts in gut microbiome composition. Known examples span a broad range of therapeutic classes and include the antidiabetic metformin, proton‐pump inhibitors, antipsychotics, non‐steroidal anti‐inflammatory drugs, paracetamol, opioids, selective serotonin reuptake inhibitors, laxatives, and statins (Le Bastard et al, 2018; Jackson et al, 2018; Kummen et al, 2020; MetaCardis Consortium et al, 2020). These shifts are not necessarily unfavorable for the host. In certain cases, host‐targeted drugs can diversify the gut microbiome (MetaCardis Consortium et al, 2020)—a feature generally linked to a healthy microbiome. However, the functional implications of these taxonomic shifts, for example in terms of altered metabolic capacities and/or antibiotic resistance repertoires, need to be assessed separately for each compound (Vich Vila et al, 2020).
Current clinical studies of the effects of medication on the gut microbiome have mostly been cross‐sectional, while interventional or longitudinal approaches and comparisons to treatment‐naïve but diseased control groups are often missing. As a result, it is difficult to differentiate between disease‐mediated and drug‐related effects. This issue is exemplified by the antidiabetic drug metformin. The drug shows limited oral bioavailability, resulting in high intestinal drug concentration. It was one of the first non‐antibiotic drugs that was shown to influence gut microbiome composition (Napolitano et al, 2014) and revealed the need to stratify for treatment when interpreting microbiome signatures (Forslund et al, 2015). At the same time, this finding stimulated causal studies that directly linked compositional shifts to the improvement of metabolic dysfunction and hyperglycemia (Wu et al, 2017). One proposed mechanism involves metformin decreasing the relative abundance of Bacteroides fragilis and downregulating its associated bile salt hydrolase activity. This leads to an accumulation of glycoursodeoxycholic acid, which inhibits the intestinal farnesoid X receptor (FXT) signaling and thereby improves various metabolic outcomes in mice, including hyperglycemia (Sun et al, 2018). Other proposed mechanisms to explain the microbiome‐mediated hypoglycemic effect of metformin include the microbial production of short‐chain fatty acids, promotion of gut barrier integrity and increased secretion of gut hormones such as glucagon‐like peptide 1 and peptide YY (PYY) (reviewed in Pryor et al, 2020). Remarkably, several model systems such as Caenorhabditis elegans (Cabreiro et al, 2013), mice (Shin et al, 2014), and rats (Bauer et al, 2018) were instrumental in elucidating these metformin–microbiome–host interactions, highlighting the translation of these phenomena between evolutionarily distant organisms and demonstrating the utility of different model organisms to study these interactions. In contrast to metformin, we are far from dissecting the interaction of the vast majority of host‐targeted drugs with gut microbes. It remains unclear whether these drugs act directly on the microbes, what is their spectrum and underlying molecular interactions, and what is the impact on the microbiome as a whole, on the drug’s therapeutic action and on the host. To close this knowledge gap and optimize drug therapies, further well‐designed clinical studies are needed, which must be seamlessly coordinated with bottom‐up approaches (Fig 2).
Ex vivo studies—accelerating mechanistic understanding of drug–microbiome interactions by reducing the complexity and increasing the throughput—the bottom‐up approach
While clinical studies provide an excellent global picture of drug effects on the microbiome, ex vivo approaches allow for a systematic, controlled, and question‐specific dissection of these interactions at various scales ranging from molecules to inter‐organismal interactions. Recent advances in high‐throughput approaches for the cultivation of fastidious anaerobes (Box 1) allowed the first systematic studies of the effects of drugs on intestinal microbes. A large‐scale in vitro screen of 1,200 marketed drugs showed direct impact on the growth of at least one of forty tested human gut commensal species for 78% of the antibacterial drugs, 53% of other antimicrobials, and 24% of the human‐targeted drugs (Maier et al, 2018). Although drugs across all therapeutic classes had a direct impact on gut commensal species, the effect was most pronounced for antimetabolites, antipsychotics, and calcium‐channel blockers. Some of these compounds, such as antimetabolites, target conserved enzymes and pathways in prokaryotes and eukaryotes and thus, likely have the same mode of action in gut commensals as in host cells. However, for the vast majority of human‐targeted drugs with activity against gut bacteria, their bacterial targets remain obscure. Identifying microbial targets for these drugs will open new possibilities for repurposing them as antibacterials and/or for mitigating their collateral damage on gut bacteria. Intriguingly, human‐targeted drugs impacting microbes in vitro resembled antibiotics with respect to their reported side effects in clinics, providing initial evidence that they also impact gut commensals in vivo. Moreover, antibiotic‐resistant microbes were in general also more resistant to human‐targeted drugs, suggesting that resistance mechanisms against antibiotics and non‐antibiotics at least partially overlap. Initial profiling of these common resistance mechanisms revealed efflux pumps, transporters and detoxifications mechanisms. Other activities, such as cell envelope properties, stress responses and target modification are also likely involved. Precisely mapping this level of cross‐resistance and collateral sensitivity (i.e., resistance to one drug providing sensitivity to another) is vital to mitigate the risks human‐targeted drugs may entail for antibiotic resistance and to exploit collateral sensitivity opportunities to delay, prevent or revert antibiotic resistance (Pál et al, 2015; Baym et al, 2016). To this end, a number of established systems approaches can be specifically geared to deconvolute drug targets and reveal resistance mechanism, as demonstrated for chemical genetics (Cacace et al, 2017; Kintses et al, 2019), proteomics (thermal proteome profiling (Mateus et al, 2020), limited proteolysis‐coupled mass spectrometry (Schopper et al, 2017), and metabolomics (Zampieri et al, 2018) (Fig 2).
Box 1. Representative microbes and microbiomes.
A: Representative microbes
The significance of systemic mapping of drug–microbiome interactions increases with the number of representative microbes tested. Consequently, comprehensive species and strain collections are essential. The benefit of such collections further increases, the better the isolates are characterized (e.g., genome sequence), and the more detailed metadata information is provided (e.g., health status of the host).
Gut microbiome isolate collections
The compilation of such collections usually follows certain selection criteria—such as being representative for the gut microbiome of healthy individuals—and focuses on type strains, which are obtained from publicly available strain collections such as DSMZ, ATCC/BEI Resources, etc. (www.dsmz.de, http://www.atcc.org, www.beiresources.org) (e.g., Tramontano et al, 2018). Further collections are needed that are representative for other body sites, certain diseases, age‐groups, ethnicities, food preferences, etc.. While most concentrate on maximizing phylogenetic diversity of prevalent and abundant species, for a global picture it is also important to capture rare species and species diversity (i.e., strain‐level variation).
Strain‐level variation
Current studies only phenotype one or few strains per species, usually starting with type strains. For most tested species, it is unknown how representative they are. Although pangenomes can be estimated for many gut species (Zou et al, 2019), it is unclear how this translates into phenotypic variation. However, previous work suggests that drug metabolism and drug sensitivity are strain‐specific traits (Koppel et al, 2018; preprint: Maier et al, 2020) and that functional strain differences can impact human health. Such observations underline the importance of sampling many strains per bacterial species. Several efforts have been recently made toward this aim by collecting hundreds of human gut bacterial isolates. In the future, such collections need to continue expanding to cover strain and species diversity—for example, many unknown species are predicted from metagenome‐assembled genomes (Almeida et al, 2019; Pasolli et al, 2019; Nayfach et al, 2019).
Recent examples for such libraries include:
Broad Institute‐OpenBiome Microbiome Library (Poyet et al, 2019).
Culturable Genome Reference (CGR) Collection (Zou et al, 2019).
Human Gastrointestinal Bacteria Culture Collection (HBC) (Forster et al, 2019).
Global Microbiome Conservancy (http://microbiomeconservancy.org).
Collection of coexisting isolates from the same host
Instead of collecting and phenotyping strains from a large number of different individuals, strain collections can originate from a single person (Goodman et al, 2011; Coyne et al, 2014). As these co‐resident strains are collected from the same human host, they capture the co‐evolved and coexisting strain‐level diversity within one individual. Personalized collections are of particular value for investigations of inter‐individual differences in drug–microbiome interactions.
B: Microbiomes
The number of different community compositions to be examined scales almost infinitely. To tackle this challenge, two fundamentally different approaches can be pursued: synthetic communities can be assembled starting from axenic bacterial cultures (bottom‐up approach) or natural, self‐assembled communities, e.g., derived from human stool can be utilized (top‐down approach).
Synthetic communities
Reductionist consortia of defined organisms are assembled in modular ways, either donor‐specific or pooled. Individual community members are usually well‐characterized and ideally genetically tractable. Systematic manipulations of the strain and genetic composition of synthetic communities enable the identification of causal links between the composition and observed community phenotypes (Shetty et al, 2019).
Stoolbanks
Stool samples provide a non‐invasive starting point for studying the complex, self‐assembled human microbiome (Bolan et al, 2016) and can be incubated with drugs ex vivo (Maurice et al, 2013; van de Steeg et al, 2018). Recently, so‐called “stoolbanks” became more sophisticated in order to promote accessibility to fecal microbiota transplantation in clinical practice (Cammarota et al, 2019). But they can also be used for research purposes, especially if they are open‐access and non‐profit, such as OpenBiome. Subsequent microbiome preservation efforts aim for long‐term storage: for example, the “The Microbiota Vault” (www.microbiotavault.org) is a project to conserve the microbial diversity associated with our bodies and environments for future generations.
In both setups, key functional and compositional profiles of the gut microbiota need to be maintained, for example in continuous flow bioreactor systems or microfluidic gut models (Guzman‐Rodriguez et al, 2018). As these technically laborious systems are challenging to adapt to high‐throughput workflows, continuous dilution batch cultures in multi‐well formats have been successfully applied to screen drug effects on microbial communities (Venturelli et al, 2018; Li et al, 2019).
The numerous interactions observed between human‐targeted drugs and gut microbes in vitro beg the question of whether they are relevant in vivo. For example, it is unclear whether microbes alone similarly respond to drugs as when part of a community, and how the spatially structured intestinal environments and drug concentration gradients inside the host affect drug response. One way to leverage drug–microbiome interactions to the community level is to test assembled (“synthetic”) communities (Box 1). Microbes can behave the same in communities as in an axenic culture (the drug being as effective against them) or can have communal emergent properties: be more protected (cross‐protection) or sensitized (cross‐sensitization) to the drug. It is currently unclear how often such emerging communal properties occur and/or what drives them. Drug chemical modification can lead to both cross‐protection (Vega & Gore, 2014) and cross‐sensitization (Roemhild et al, 2020), but also other less direct effects could elicit similar results: the change in physiological stage of the bacterial cells (e.g., stress responses and transporters induced at the community level), changes of environment (i.e., pH changes (Ratzke & Gore, 2018)), or the opening of niches in a competitive environment. To investigate such responses systematically, robust high‐throughput ways are needed to grow communities (Box 1) and to follow species abundance, ideally at an absolute quantification level (e.g., by metaproteomics (Li et al, 2020), Fig 2). Understanding the frequency and molecular drivers of such interactions will be of paramount importance to exploit or mitigate microbiome‐mediated drug effects in clinics (Fig 3).
Figure 3. Applications of knowledge gain from studying drug–microbiome–host interactions.
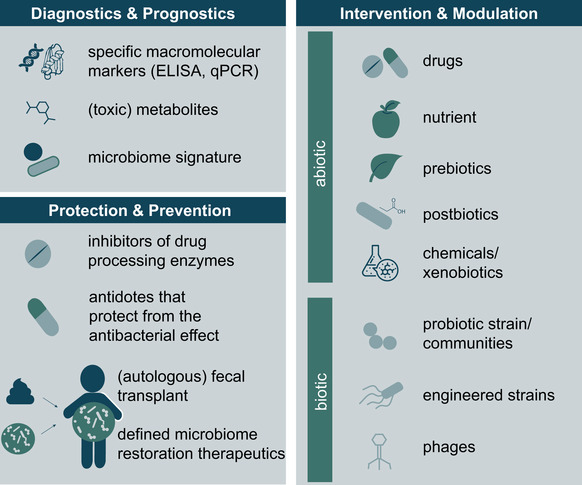
Diagnostics and Prognostics: Microbiome‐derived biomarkers (macromolecules, metabolites and compositions) can be used to diagnose diseases, but also for prognosis of the disease course or to predict treatment success. Protection and Prevention: Various measures can be applied to reduce undesired drug effects on the microbiome or to suppress chemical drug modifications by intestinal bacteria. With better understanding of the drug–microbiome–host triad, interventions of increased specificity can be employed (i.e., from fecal transplants to defined restoration therapeutics). Intervention and Modulation: There are both abiotic and biotic approaches to influence the microbiome, its functional output and consequently drug–microbiome–host interactions. For more detailed explanations, see Box 2.
Microbiome effects on drugs
Microbes alter the chemistry of drugs and drug metabolites
Given the structural similarity between small molecule drugs and endogenous metabolites, the fact that many drugs are derived from natural products, and the large enzymatic potential of the microbiome, microbial drug metabolism is to be expected. Indeed, already in the early 20th century the drug prontosil was found to require bacterial conversion to unfold its antibiotic effects (Fuller, 1937). Since then, accumulating evidence suggests that microbial modification of drugs and drug metabolites seems to be the rule rather than the exception. Such microbial drug metabolism can result in the same or different chemical products as the human metabolic enzymes, leading to drug activation (e.g., sulfasalazine, Sousa et al, 2014), inactivation (e.g., L‐dopa and digoxin (Lindenbaum et al, 1981; Haiser et al, 2013; Maini Rekdal et al, 2019)) or toxicity (e.g., sorivudine and brivudine, (Zimmermann et al, 2019a; Nakayama et al, 1997). In addition to drug molecules, drug metabolites are also subject to microbial metabolism. Phase II drug metabolites (produced by conjugation reactions) have been found to be deconjugated to their precursor molecules (i.e., phase I metabolites (Wallace et al, 2010) or original drug molecules (Taylor et al, 2019)) by microbes. More importantly, these types of microbial metabolism can impact pharmacokinetics, in particular the intestinal abundance of drug and drug metabolites, and thereby alter drug response and toxicity (Wallace et al, 2010; Taylor et al, 2019). Since differences in microbiome‐encoded genetic contents far exceed genetic differences between human individuals, it is very likely that the microbiota composition may be behind a large fraction of person‐to‐person variation in drug response, especially in terms of drug side effects. In the following paragraphs, we will discuss various approaches to investigate microbiome drug metabolism, its impact on drug response and potential avenues to harness microbiome drug metabolism to improve therapeutic drug interventions. The latter would undoubtedly present an opportunity for the pharmaceutical industry and precision medicine applications in clinics.
Systematic studies reveal extensive microbial drug metabolism
A compound’s metabolism in the human body is a decisive factor for its success during preclinical and clinical drug development. To assess drug metabolism early in drug discovery pipelines, numerous in vitro and in silico protocols have been developed and standardized. New technologies, such as microfluidics screens and machine learning predictions have been recently incorporated in such pipelines (Kirchmair et al, 2015; Eribol et al, 2016). The use of cellular or cell‐free enzyme preparation (e.g., cytosolic and microsome isolations) enables systematic ex vivo high‐throughput screens for the metabolism of hundreds of compounds in parallel (Williamson et al, 2017; Underhill & Khetani, 2018). The results of such systematic assays, together with insights from in vivo drug metabolism, are the basis for rule‐based and machine learning computational methods to predict xenobiotic metabolism (Djoumbou‐Feunang et al, 2019; de Bruyn Kops et al, 2019).
In contrast to human drug metabolism, comparable large‐scale data sets for microbiome drug metabolism are mostly lacking, limiting the information available to build predictive models of microbial drug modifications. To circumvent this limitation, several research groups have used information on primary and secondary metabolism to infer potential drug modification reactions based on biochemical reactions and substrate structures (Klünemann et al, 2014; Guthrie et al, 2019). Although this approach is consistent with the chemical similarity between drugs and endogenous compounds, it suffers from the fact that the genes, biochemistry, and lifestyle of most gut microbiome members are poorly characterized (Almeida et al, 2019). This makes it also challenging to define a (standardized) set of microbiome‐derived species/strains/enzymes to test their activity against drug molecules, as it exists for human drug‐metabolizing enzymes. As a workaround, two recent studies have cultured complete human fecal communities to test their drug‐metabolizing capacity ex vivo with a panel of up to 438 different compounds (van de Steeg et al, 2018; Javdan et al, 2020). This experimental setup has the advantage that microbial community members do not have to be selected a priori and encompasses microbial interactions that can impact drug metabolism, as shown for sequential L‐dopa metabolism by two different species (Maini Rekdal et al, 2019). A challenge of this approach is the uneven strain distribution in isolated microbial communities, which may mask and underestimate the metabolic potential of microbes found at low abundance ex vivo, but may very well be active and relevant in vivo. Comparable to the described systematic bottom‐up approach to test drug activity on representative panels of bacteria in isolation (Maier et al, 2018), similar efforts have been employed to deduce their metabolic activity against a large panel of drugs (Zimmermann et al, 2019b). Testing microbial communities or single bacterial strains, up to 65% of the assayed drugs were metabolized, suggesting that the microbial drug metabolism is a far more common phenomenon than the few anecdotal examples collected over the last few decades (reviewed in Wilson & Nicholson, 2017).
Gaining molecular insights into microbial drug metabolism
Ex vivo drug transformation assays with fecal communities isolated from different individuals have demonstrated vast interpersonal differences in the communities’ drug‐metabolizing capacity (Zimmermann et al, 2019b) (Fig 2), which are corroborated by differences in the drug‐metabolizing potential for different bacterial species and strains (Lindenbaum et al, 1981; Haiser et al, 2013; Zimmermann et al, 2019b). These findings suggest that the molecular mechanisms of microbial drug transformation need to be identified to predict the drug‐metabolizing capacity of an individual's microbiome. To identify microbial enzymes and pathways responsible for drug conversion, several systems approaches have been applied. Based on the assumption that metabolic pathways are often transcriptionally induced by their substrates, transcriptional comparison in the presence and absence of a given drug can be performed. This approach was successfully applied to identify the enzymes of Eggerthella lenta (DSM 2243) and Escherichia coli (K12) that metabolize digoxin (Haiser et al, 2013) and 5‐fluoruracil (preprint: Spanogiannopoulos et al, 2019), respectively. Gain‐of‐function and loss‐of‐function genetic screens have been combined with mass spectrometry‐based analytics to systematically identify genes involved in microbial drug metabolism (Zimmermann et al, 2019a, 2019b) (Fig 2). Drug‐specific chemical probes have also been employed to probe enzyme activity and to pull down enzymes conveying a drug conversion of interest, as elegantly applied for the identification of beta‐glucuronidases (Jariwala et al, 2020). Finally, computational approaches based on metabolic reaction networks, comparative genomics of bacterial isolates, or microbiome composition have been employed to identify possible genetic factors responsible for drug metabolism (Klünemann et al, 2014; Mallory et al, 2018; Guthrie et al, 2019). Once identified, microbial genes involved in drug metabolism can serve as potential biomarkers to quantitatively predict the drug metabolic capacity of a given microbial community (Zimmermann et al, 2019b) (Fig 3), opening new paths for understanding the impact of microbial drug metabolism on the host and eventually its role in the interpersonal variability in drug response.
The role of the host
Interactions between drugs and microbes identified in vitro need to be validated in the host context, to establish that microbes and drug meet at relevant concentrations and at the same location. Additional interactions that are usually not adequately reflected by in vitro systems but are relevant in the host context include dietary interactions, host drug metabolism, immune responses, and the presence of endogenous host molecules. Trying to understand the molecular mechanisms that govern the mutual interactions between microbiome and host and trying to explain the compositional adaptations of the microbial community and altered physiology of the host is at the very heart of microbiome research. Which environmental and host factors shape the composition and the functional output of the microbiome? How do altered microbiome composition and functions affect the host? Altogether, the consequences of microbiome–drug–host interactions need to be understood at a molecular level in order to allow harnessing them and applying them to improve therapy (Fig 3). Below, we discuss suitable approaches for studying microbiome–drug–host interactions (Fig 2).
In vitro approaches
Microbial communities can interact with and affect the host with peptides/proteins (Gil‐Cruz et al, 2019), RNA (Liu et al, 2016), and metabolites (Uchimura et al, 2018; Koh & Bäckhed, 2020). In the context of microbiome–drug–host interactions, in particular in the case of small molecule drugs, metabolite‐based interactions seem natural. Decades of pharmacological research have led to the development of in vitro approaches to systematically screen for molecules with a potential effect on the host. Some of which have also been successfully applied to study metabolic microbiome–host interactions. Membrane‐bound G‐protein‐coupled receptors (GPCR) are a prime target for pharmacological interventions, currently representing more than one‐third of the targets for prescribed drugs (Rask‐Andersen et al, 2011). These molecular sensors are omni‐present in mammalian hosts, bind ligands from their environment, and transduce the signal through molecular cascades to change cell physiology. Several studies have recently been published employing high‐throughput GPCR activation assays to screen for microbiome‐produced GPCR ligands (Cohen et al, 2017; Colosimo et al, 2019; Chen et al, 2019). Each of these studies started with metabolites extracted from microbial cultures, which were then tested on engineered GPCR‐reporter cell lines to pinpoint receptor activation. Strikingly, these studies identified microbiome‐derived ligands for yet uncharacterized, so‐called orphan GPCR, which are of particular interest to potentially expand the drug target space. Following the same principle, reporter cell lines for the activation of nuclear receptors, another major target class of drug targets, have been employed to identify microbiome‐derived ligands of human receptors (Estrela et al, 2019). These studies illustrate the applicability and power of systematic screens based on human cell lines, initially developed in drug discovery pipelines, to map the chemical interactome between the microbiome and the host. Following these examples, similar screening approaches could be applied to the analysis of different receptor classes, metabolic activity, and transporter specificity. Clear strengths of these assays include their reductionist character, mechanistic insights, and high‐throughput capacity, whereas the lack of tissue context and physiological relevance represent obvious limitations.
Intestinal enteroids and organoids overcome this limitation through differentiation of stem cells into specific intestinal cell types, such as enterocytes and goblet cells, forming crypt macrostructures, and encompass intestinal properties, such as barrier functions (Sato et al, 2009; Yin et al, 2014; Pearce et al, 2018). Such organ culture systems have been used to study the interactions of human enteric tissue with pathogenic and commensal bacteria (Lukovac et al, 2014; Pleguezuelos‐Manzano et al, 2020). Furthermore, these systems have been successfully employed to study the effect of bacterial surface or secreted molecules, such as lipopolysaccharides and muramyl‐dipeptides (Nigro et al, 2014; Naito et al, 2017), and of microbiome‐derived small molecules, such as short‐chain fatty acids and indolacrylic acid (Park et al, 2016; Wlodarska et al, 2017; Pleguezuelos‐Manzano et al, 2020). Paired with microfluidic technology, assays co‐culturing microbes with host tissues have been developed to create artificial gut systems on a chip, enabling systematic measurements under controlled conditions, while maximally mimicking the microbiome–host interface (Jalili‐Firoozinezhad et al, 2019). Such in vitro approaches simulating microbiome–host interactions can in the future propel our mechanistic understanding of drug–microbiome–host interactions.
Altogether, in vitro screening approaches adapted from pharmaceutical research and novel technology‐driven platforms can both facilitate the systematic study of drug‐microbiome–host interactions. As shown for GPCRs, such approaches can provide functional insights into the molecular interactions between microbes and the host, caused by drug administration. A systematic dissection of the underlying interactions will pave the way for future in vivo studies in animal models and clinical settings.
Animal models
Ultimately, we aim at understanding the effects of drug intake on the whole organism. To this end, invertebrate models can represent powerful models bridging cell culture systems to complex model organisms and cohort studies. Both the fruit fly Drosophila melanogaster and nematode worm C. elegans, two model organisms with well‐established genetic and genomic resources, are often overlooked in microbiome research (Norvaisas & Cabreiro, 2018). This originates from the fact that their associated microbes poorly reflect the taxonomic and functional diversity of the human microbiome and that in the case of C. elegans, the impact of microbes on the host is rather of nutritional than of symbiotic nature (Trinder et al, 2017; Douglas, 2019; Zimmermann et al, 2020). Both organisms have long‐standing traditions of high‐throughput screening and permit a variety of different biomedical readouts ranging from fluorescence reporters of gene function to lifespan, fertility, and behavioral investigations. Furthermore, they both present valuable advantages for microbiome studies, such as effortless large‐scale production of germ‐free or gnotobiotic animals and facile genetic manipulation, allowing for scalable, cost‐ and time‐efficient studies of the host–drug–microbiome interface (Diot et al, 2018; Douglas, 2018). These two invertebrate models have proven their worth for drug discovery research (Pandey & Nichols, 2011; O’Reilly et al, 2014; Fernández‐Hernández et al, 2016) and have lately been successfully employed to study interactions at the drug–microbiome–host triad.
The low diversity gut microbiome of Drosophila melanogaster has recently been advantageous in revealing general principles of antibiotic tolerance that are mediated by metabolic interspecies interactions (Aranda‐Díaz et al, 2020). In a series of elegant studies, the C. elegans model allowed to identify bacterial nucleotide metabolism genes that affect chemotherapeutic efficacy on the host (Scott et al, 2017; García‐González et al, 2017) or to understand how diet can affect metformin’s positive effect on lifespan by gut microbes (Pryor et al, 2019). In summary, invertebrate models can be instrumental in pre‐selecting the most relevant of the many possible drug–microbe combinations for a given question.
In contrast to invertebrate models, rodent models have been the standard for pharmaceutical and microbiome research for decades (Nguyen et al, 2015). They are suited for pharmacokinetic studies, allow using established disease models and are more relevant to human host physiology and microbiota bio‐geography. In the microbiome field, rodent models are valued for the controlled experimental manipulation of host (knockouts), microbiome (gnotobiology), and environment (e.g., diet) and their genetic, anatomical, and physiological relatedness to humans. These are ideal starting points to address questions on drug–microbiome–host interactions. Historically, microbiome‐mediated drug metabolism was first discovered in rats: while the anti‐inflammatory drug, salicylazosulfapyridine was metabolized in conventional animals, the parent compound remained unchanged in aseptic (antibiotic treated) rats (Peppercorn & Goldman, 1972). This was the starting point for analogous studies with other drugs under the assumption of comparable metabolic functionalities between rodent‐ and human‐associated microbes. Likewise, many decades later, the combination of genetically engineered gut commensals and gnotobiotic mice provided a system to quantitatively separate host and microbiome contribution to shared drug metabolism and assess the role of a single microbial enzyme in this interaction (Zimmermann et al, 2019a). Other researchers employed combinatorial therapies, i.e., antibiotics combined with the drug under investigation to unravel the influence of the microbiome on the drug’s pharmacokinetic parameters (Malfatti et al, 2020). Furthermore, rodent models are helpful to investigate possible therapeutic strategies to mitigate microbiome‐induced drug toxicity, such as inhibitors of the bacterial beta‐glucuronidase enzymes (Wallace et al, 2010; Bhatt et al, 2020).
There are numerous rodent studies on drug‐mediated compositional microbiome changes and their consequences on host physiology. A number have examined the short‐ and long‐term effects of antibiotics (e.g., Cox et al, 2014; Cho et al, 2012; Nobel et al, 2015; Ruiz et al, 2017). Increasingly, such studies also investigate the effects of non‐antibiotic drugs and diet on drug susceptibility and recovery (Ng et al, 2019; Cabral et al, 2019; Garland et al, 2020). While humanized mice (colonized with human microbiota) have become a cornerstone model to demonstrate causality between altered microbiome composition and host phenotype in various diseases, this strategy has so far found little use to assess whether a drug’s therapeutic effect is mediated through the microbiome. One exception is again the antidiabetic drug metformin, where fecal transplantation of metformin‐treated patients into germ‐free mice was shown to be sufficient to improve glucose tolerance of recipient mice (Wu et al, 2017). This approach provides a powerful tool to investigate signaling along the drug–microbiome–host axis with many conceivable ways for improvement (e.g., enrichment and purification steps, defined microbial consortia, ex vivo incubation of drugs and microbes) (Walter et al, 2020). Rodent models have further contributed to our understanding of how the gut microbiome impacts anticancer immunotherapy by PD‐1 (Tanoue et al, 2019), CTLA‐4 blockage (Vétizou et al, 2015; Sivan et al, 2015; Mager et al, 2020) or in cyclophosphamide therapy (Viaud et al, 2013), all resulting in findings of high transferability to humans (reviewed in (Zitvogel et al, 2018).
Comparative systems‐level analyses of gnotobiotic and conventionally raised mice make it possible to map the effects of microbial colonization at the organismal scale (Mills et al, 2020). Such approaches have revealed that numerous host xenobiotic processing genes, i.e., P450 cytochromes (CYPs), phase II enzymes and transporters are influenced by the microbiome, both at the RNA and protein level and at various body sites (Selwyn et al, 2016; Kuno et al, 2016, 2019; Fu et al, 2017). Hence, the microbiome can also have an indirect impact on drug pharmacokinetics by modulating xenobiotic metabolism of the host (Dempsey & Cui, 2019).
Well‐designed approaches that allow parallelizing the performed analyses and thus reducing the amount of experimental animals will tremendously accelerate our understanding of drug–microbiome–host interactions in both directions, namely those of drugs on microbes as well as those of microbes on drugs.
Translation to human
A better mechanistic understanding of the drug–microbiome–host interactions opens the translational possibility to harness the microbiome and its interpersonal variability in composition to improve drug treatments in both general and personalized manners. Such microbiome‐based treatments could encompass a wide range of different applications (Fig 3). Analogous to human genetic markers guiding drug dosing and potential drug‐drug interaction risks, microbiome biomarkers could be used to predict drug response and guide treatment regimens, as showcased for digoxin (Haiser et al, 2013). The identification of microbiome‐encoded enzymes that negatively impact drug response is the basis for the development of specific inhibitors targeting these microbial processes. Such inhibitors have been developed to inhibit microbial metabolism of L‐dopa and deglucuronidation of drug metabolites (Wallace et al, 2010; Maini Rekdal et al, 2019). Although conceptually interesting, adding additional bioactive compounds to a given drug formulation comes with new challenges, such as regulatory hurdles, increased polypharmacy, and target delivery to the microbiome. Furthermore, targeting microbial enzymes bears the inherent risk of altering microbiome composition and potentially function. However, this risk also presents an opportunity. In contrast to the human genomes, the gut microbiome can be rapidly modified, uniquely allowing both sides of the patient‐drug interaction to be optimized for maximum therapeutic benefit (Taylor et al, 2019). Interventions such as dietary changes, antibiotic administration, or fecal microbiota transplantation (FMT), induce general shifts in microbiome composition; whereas prebiotics, probiotics and phage therapies have the potential of introducing targeted changes to the microbiome (Box 2). Aside from these interventions which aim at altering the microbiome composition more permanently, approaches to temporarily change the functional output of the microbiome have also been envisioned. Such transient changes could be achieved through the administration of probiotics that do not stably colonize the gut, but that change gut physiology during their intestinal passage. Another promising avenue is the use of postbiotics, which are the functional output of beneficial microbes, such as metabolites, that are administered abiotically.
Box 2. Microbiome modulations.
The microbiome has become a primary therapeutic target, with many ongoing clinical trials for multiple medical indications. These studies typically aim at modulating the microbiome toward a health‐promoting state for its human host (e.g., for colorectal cancer (Fong et al, 2020), for atherosclerosis (Chen et al, 2020)). The means to do so vary immensely and include a range of interventions that can be separated in biotic and abiotic agents leading to either global or targeted changes of the microbiome composition. Furthermore, some of these microbiome‐targeted therapies aim at permanently altering the microbiome, whereas others aim at a transient effect. All of these interventions have in common that they alter the functional output of the microbial community and hence the microbiome–host interactions. Although, microbiome modulations have not yet been extensively explored to alter microbiome–drug–host interactions to improve drug response and alleviate adverse effects, we provide an overview of the potential means to do so (see also Fig 3).
Abiotic interventions consist of dietary changes that shift microbiome composition and prebiotics, which are specific compounds, such as certain sugars, that are preferred by microbiome subpopulations leading to their increase in abundance. Additional abiotic agents include peptides, drugs, and other xenobiotics, of which antibiotics are intuitive microbiome modifiers. More recently, postbiotics have gained increasing attention (Wegh et al, 2019). The term summarizes a variety of different bioactive fermentation products such as short‐chain fatty acids or secondary bile acids. In contrast to the other agents, postbiotics do not act via compositional microbiome changes but directly mimic an altered functional microbiome output.
Biotic interventions are based on biological agents, such as entire gut communities or specific microbes to modify the function of a person’s microbiota. Fecal microbiota transplantations (FMT) transfer the entire microbial gut community from one person to another. Due to the challenges to standardize and regulate fecal material (Giles et al, 2019), many efforts aim at engineering synthetic communities of defined quality and properties that can be transplanted. Probiotics describe specific bacterial strains intended for therapeutic purposes (Suez et al, 2019). They are GRAS‐certified (generally regarded as safe) by the Food and Drug Administration and include microbes from different phyla such as Lactobacillus reuteri and Bifidobacterium casei. Further, probiotic bacteria can be genetically modified to express specific therapeutic properties (e.g., therapeutic proteins (Gurbatri et al, 2020)), whereas next‐generation probiotics are based on microbial isolates from the gut microbiota that are re‐inserted, possibly after in vitro modification (O’Toole et al, 2017). The discovery of increasing numbers of microbiome‐associated bacteriophages opens the opportunity to apply phage‐therapy to eliminate unwanted bacterial species and strains from a microbial community in a very specific manner (Sausset et al, 2020). In preclinical models of colorectal cancer, phages of the tumor‐associated Fusobacterium nucleatum have even been used for targeted drug delivery (Zheng et al, 2019).
Microbiome‐targeting interventions could also be employed to counteract the compositional shifts introduced by medicinal drug treatments. Autologous fecal microbiota transplantation (auto‐FMT) has improved treatment outcome of patients undergoing allogeneic hematopoietic stem cell transplantation to reconstitute their microbiome after the antibiotic regimens during hematopoietic stem cell replacement (Taur et al, 2018). Similar approaches could be applied to treat dysbiosis induced by human‐targeted drug treatments. Two recent studies co‐administered small molecule drugs with antibiotics to alleviate the effects of antibiotics on gut commensals (preprint: Maier et al, 2020; Garland et al, 2020), providing new ways to avoid or revert the collateral damage of drug treatment on healthy microbiomes.
Apart from direct drug–microbiome interactions, indirect effects can also influence microbiome and host functions and interactions. For example, proton‐pump inhibitors (PPI)s are among the drugs with the most pronounced effect on the gut microbiome composition (Imhann et al, 2016). It is thought that drug‐induced changes in intestinal physiology, such as increased gastric pH, contribute to this effect. Analogously, drugs that alter intestinal motility have also been shown to impact microbiome composition (Vich Vila et al, 2020). The microbiome can also indirectly influence human drug metabolism and impact pharmacokinetics through regulation or inhibition of human enzymes. For example, the microbiome‐produced metabolite p‐cresol has been shown to competitively inhibit O‐sulfonation enzymes in the liver, which interferes with acetaminophen clearance (Clayton et al, 2009). Given the complexity of these indirect effects and our current lack of their understanding, systematic approaches are needed to comprehensively map these interactions at the molecular level.
When translating laboratory findings, in particular from high‐throughput in vitro screens, to human studies, it is essential to take pharmacokinetic principles into account. For example, oral drugs, which are still the major form of formulations used, are typically absorbed in the small intestine where bacterial densities are rather low. In contrast, the large intestine harbors the most dense and diverse microbial communities of the human body, but the absorption capacity of the colon is limited due to reduced surface area and number of transport proteins. This raises the question of which bacteria–drug interactions are actually relevant to drug pharmacokinetics.
To overcome these limitations, physiology‐based pharmacokinetic (PBPK) models that take into account the microbiome–drug interactions for pharmacokinetics have been recently developed (Zimmermann et al, 2019a; Zimmermann‐Kogadeeva et al, 2020). Limited bioavailability, altered intestinal transit and slow release drug formulations directly influence drug concentrations in the large intestine, which can affect reciprocal effects between the drug and the microbiome. In order to directly impact drug pharmacokinetics, microbial metabolism and/or bioaccumulation has to occur in the intestinal tube, where absorption of drug or its metabolites also takes place. An example of such direct competition between microbial drug metabolism and intestinal absorption was shown for L‐dopa in the small intestine, which reduces the absorbed drug in a microbiome‐dependent manner (Maini Rekdal et al, 2019). Another example is the microbial conversion of sorivudine and brivudine to the liver‐toxic metabolite bromovinyluracil in the large intestine, which leads to increased serum levels and liver toxicity following large intestinal absorption (Zimmermann et al, 2019a). The microbiome‐specific PBPK model further predicts that extensive biliary excretion of drug and drug metabolites can lead to their accumulation in the intestine and hence result in enhanced drug–microbiome interactions. This provides a route and explanation for non‐orally administered drugs to interact with the gut microbiome, as shown for the intravenously administered anticancer drug irinotecan (CPT‐11), for which microbiome‐caused adverse effects are dose and treatment limiting (Wallace et al, 2010). CPT‐11 is a prodrug that gets converted to its active compound SN‐38 by human enzymes, before phase II liver reactions (i.e., glucuronidation) inactivate the compound before biliary secretion into the intestine. The human gut microbiome encodes for hundreds of different beta‐glucuronidases that remove glucuronate from liver‐derived (drug) metabolites (Pollet et al, 2017). In the case of SN38, this leads to local accumulation of the cytotoxic compound resulting in intestinal complications, such as severe diarrhea. In other cases, microbiome‐produced deglucuronidation products can be either directly or following additional microbial conversion re‐absorbed from the intestine impacting systemic drug or drug metabolite exposure, as shown for mycophenolate and clonazepam, respectively (Elmer & Remmel, 1984; Ishizaki et al, 2012; Zimmermann et al, 2019a). Although, systematic approaches are needed to comprehensively map possible drug–microbiome–host interactions at the molecular level, these examples emphasize the importance of keeping an organismal view on the results and evaluating their relevance for translatability to clinics.
Conclusions and future perspectives
Systematic approaches have started to provide new insights into drug–microbiome–host interactions. A molecular understanding of the drug–microbiome–host triad will open up strategies to fine‐tune existing clinical drug treatments, improving drug efficacy and reducing adverse effects. Such strategies could encompass both microbiome‐inspired compound modifications and modulations of the microbiome composition. It is conceivable that approaches geared toward a systematic assessment of drug–microbiome interactions will become part of preclinical drug development, complementing current in vitro assays and in silico modeling to predict human drug metabolism and toxicity. In addition to the need of taking into account drug–microbiome–host interactions for new regulatory requirements, such knowledge can be key at preclinical drug development stages. It can improve early compound triaging and target studies on specific patient groups, foreseeing complications that may arise in the clinical phase due to interpersonal microbiome differences.
An improved mechanistic understanding of reciprocal drug–microbiome interactions will also shed light on the potential role of the microbiome in drug–drug interactions, often observed in polypharmacy. More broadly, non‐drug compounds, such as other xenobiotics, nutrients, excipients, and even endogenous metabolites, likely influence drug–microbiome–host interactions as well. Therefore, we first need to systematically map these interactions and then to understand their mechanistic base to empower future (personalized) treatments. Big part of the future of drug discovery lies in harnessing the patients’ microbiome composition and function for an improved therapeutic outcome.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
The authors acknowledge funding from the German Research Foundation (Emmy‐Noether Program, CMFI Cluster of Excellence (EXE 2124)) to L.M., from ERC CoG uCARE to A.T., from UK Medical Research Council (project no. MC_UU_00025/11) to K.R.P., and from the Daimler‐und‐Benz‐Stiftung to M. Z.
Mol Syst Biol. (2021) 17: e10116
Contributor Information
Michael Zimmermann, Email: michael.zimmermann@embl.de.
Lisa Maier, Email: l.maier@uni-tuebingen.de.
References
- Almeida A, Mitchell AL, Boland M, Forster SC, Gloor GB, Tarkowska A, Lawley TD, Finn RD (2019) A new genomic blueprint of the human gut microbiota. Nature 568: 499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda‐Díaz A, Obadia B, Dodge R, Thomsen T, Hallberg ZF, Güvener ZT, Ludington WB, Huang KC (2020) Bacterial interspecies interactions modulate pH‐mediated antibiotic tolerance. Elife 9: e51493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva‐Datchary P, Li Y, Xia Y, Xie H, Zhong H et al (2015) Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe 17: 690–703 [DOI] [PubMed] [Google Scholar]
- Bauer PV, Duca FA, Waise TMZ, Rasmussen BA, Abraham MA, Dranse HJ, Puri A, O’Brien CA, Lam TKT (2018) Metformin alters upper small intestinal microbiota that impact a glucose‐SGLT1‐sensing glucoregulatory pathway. Cell Metab 27: 101–117.e5 [DOI] [PubMed] [Google Scholar]
- Baym M, Stone LK, Kishony R (2016) Multidrug evolutionary strategies to reverse antibiotic resistance. Science 351: aad3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt AP, Pellock SJ, Biernat KA, Walton WG, Wallace BD, Creekmore BC, Letertre MM, Swann JR, Wilson ID, Roques JR et al (2020) Targeted inhibition of gut bacterial β‐glucuronidase activity enhances anticancer drug efficacy. Proc Natl Acad Sci USA 117: 7374–7381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolan S, Seshadri B, Talley NJ, Naidu R (2016) Bio‐banking gut microbiome samples. EMBO Rep 17: 929–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruyn Kops C, Stork C, Šícho M, Kochev N, Svozil D, Jeliazkova N, Kirchmair J (2019) GLORY: generator of the structures of likely cytochrome P450 metabolites based on predicted sites of metabolism. Front Chem 7: 402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral DJ, Penumutchu S, Reinhart EM, Zhang C, Korry BJ, Wurster JI, Nilson R, Guang A, Sano WH, Rowan‐Nash AD et al (2019) Microbial metabolism modulates antibiotic susceptibility within the murine gut microbiome. Cell Metab 30: 800–823.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung K‐Y, Vergara‐Irigaray N, Cochemé HM, Noori T, Weinkove D, Schuster E, Greene NDE, Gems D (2013) Metformin retards aging in C. elegans by altering microbial folate and methionine metabolism. Cell 153: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cacace E, Kritikos G, Typas A (2017) Chemical genetics in drug discovery. Curr Opin Syst Biol 4: 35–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammarota G, Ianiro G, Kelly CR, Mullish BH, Allegretti JR, Kassam Z, Putignani L, Fischer M, Keller JJ, Costello SP et al (2019) International consensus conference on stool banking for faecal microbiota transplantation in clinical practice. Gut 68: 2111–2121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Nwe P‐K, Yang Y, Rosen CE, Bielecka AA, Kuchroo M, Cline GW, Kruse AC, Ring AM, Crawford JM et al (2019) A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell 177: 1217–1231.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PB, Black AS, Sobel AL, Zhao Y, Mukherjee P, Molparia B, Moore NE, Aleman Muench GR, Wu J, Chen W et al (2020) Directed remodeling of the mouse gut microbiome inhibits the development of atherosclerosis. Nat Biotechnol 38: 1288–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, Yamanishi S, Cox L, Methé BA, Zavadil J, Li K, Gao Z, Mahana D, Raju K, Teitler I et al (2012) Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488: 621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton TA, Baker D, Lindon JC, Everett JR, Nicholson JK (2009) Pharmacometabonomic identification of a significant host‐microbiome metabolic interaction affecting human drug metabolism. Proc Natl Acad Sci USA 106: 14728–14733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen LJ, Esterhazy D, Kim S‐H, Lemetre C, Aguilar RR, Gordon EA, Pickard AJ, Cross JR, Emiliano AB, Han SM et al (2017) Commensal bacteria make GPCR ligands that mimic human signalling molecules. Nature 549: 48–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colosimo DA, Kohn JA, Luo PM, Piscotta FJ, Han SM, Pickard AJ, Rao A, Cross JR, Cohen LJ, Brady SF (2019) Mapping interactions of microbial metabolites with human G‐protein‐coupled receptors. Cell Host Microbe 26: 273–282.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Cho I, Kim SG, Li H, Gao Z, Mahana D et al (2014) Altering the intestinal microbiota during a critical developmental window has lasting metabolic consequences. Cell 158: 705–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne MJ, Zitomersky NL, McGuire AM, Earl AM, Comstock LE (2014) Evidence of extensive DNA transfer between bacteroidales species within the human gut. MBio 5: e01305–e1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFilipp Z, Bloom PP, Torres Soto M, Mansour MK, Sater MRA, Huntley MH, Turbett S, Chung RT, Chen Y‐B, Hohmann EL (2019) Drug‐resistant E. coli bacteremia transmitted by fecal microbiota transplant. N Engl J Med 381: 2043–2050 [DOI] [PubMed] [Google Scholar]
- Dempsey JL, Cui JY (2019) Microbiome is a functional modifier of P450 drug metabolism. Curr Pharmacol Rep 5: 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diot C, Garcia‐Gonzalez AP, Walhout AJM (2018) C. elegans and its bacterial diet: an interspecies model to explore the effects of microbiota on drug response. Drug Discov Today Dis Models 28: 21–26 [Google Scholar]
- Djoumbou‐Feunang Y, Fiamoncini J, Gil‐de‐la‐Fuente A, Greiner R, Manach C, Wishart DS (2019) BioTransformer: a comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J Cheminformatics 11: 2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AE (2018) Drosophila and its gut microbes: a model for drug‐microbiome interactions. Drug Discov Today Dis Models 28: 43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas AE (2019) Simple animal models for microbiome research. Nat Rev Microbiol 17: 764–775 [DOI] [PubMed] [Google Scholar]
- Elmer GW, Remmel RP (1984) Role of the intestinal microflora in clonazepam metabolism in the rat. Xenobiotica 14: 829–840 [DOI] [PubMed] [Google Scholar]
- Eribol P, Uguz AK, Ulgen KO (2016) Screening applications in drug discovery based on microfluidic technology. Biomicrofluidics 10: 11502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrela AB, Nakashige TG, Lemetre C, Woodworth ID, Weisman JL, Cohen LJ, Brady SF (2019) Functional multigenomic screening of human‐associated bacteria for NF‐κB‐inducing bioactive effectors. MBio 10: e02587‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falony G, Joossens M, Vieira‐Silva S, Wang J, Darzi Y, Faust K, Kurilshikov A, Bonder MJ, Valles‐Colomer M, Vandeputte D et al (2016) Population‐level analysis of gut microbiome variation. Science 352: 560–564 [DOI] [PubMed] [Google Scholar]
- Fernández‐Hernández I, Scheenaard E, Pollarolo G, Gonzalez C (2016) The translational relevance of Drosophila in drug discovery. EMBO Rep 17: 471–472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong W, Li Q, Yu J (2020) Gut microbiota modulation: a novel strategy for prevention and treatment of colorectal cancer. Oncogene 39: 4925–4943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira‐Silva S, Gudmundsdottir V, Krogh Pedersen H et al (2015) Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528: 262–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster SC, Kumar N, Anonye BO, Almeida A, Viciani E, Stares MD, Dunn M, Mkandawire TT, Zhu A, Shao Y et al (2019) A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat Biotechnol 37: 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, Huang K, Meadow JF, Gevers D, Lemon KP, Bohannan BJM, Huttenhower C (2015) Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci USA 112: E2930–E2938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich EE, Farzi A, Mayerhofer R, Reichmann F, Jačan A, Wagner B, Zinser E, Bordag N, Magnes C, Fröhlich E et al (2016) Cognitive impairment by antibiotic‐induced gut dysbiosis: analysis of gut microbiota‐brain communication. Brain Behav Immun 56: 140–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu ZD, Selwyn FP, Cui JY, Klaassen CD (2017) RNA‐Seq profiling of intestinal expression of xenobiotic processing genes in germ‐free mice. Drug Metab Dispos 45: 1225–1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller AT (1937) IS p‐aminobenzenesulphonamide the active agent in prontosil therapy? Lancet 229: 194–198 [Google Scholar]
- García‐González AP, Ritter AD, Shrestha S, Andersen EC, Yilmaz LS, Walhout AJM (2017) Bacterial metabolism affects the C. elegans response to cancer chemotherapeutics. Cell 169: 431–441.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland M, Hryckowian AJ, Tholen M, Oresic Bender K, Van Treuren WW, Loscher S, Sonnenburg JL, Bogyo M (2020) The clinical drug ebselen attenuates inflammation and promotes microbiome recovery in mice after antibiotic treatment for CDI. Cell Rep Med 1: 100005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil‐Cruz C, Perez‐Shibayama C, De Martin A, Ronchi F, van der Borght K, Niederer R, Onder L, Lütge M, Novkovic M, Nindl V et al (2019) Microbiota‐derived peptide mimics drive lethal inflammatory cardiomyopathy. Science 366: 881–886 [DOI] [PubMed] [Google Scholar]
- Giles EM, D’Adamo GL, Forster SC (2019) The future of faecal transplants. Nat Rev Microbiol 17: 719 [DOI] [PubMed] [Google Scholar]
- Goodman AL, Kallstrom G, Faith JJ, Reyes A, Moore A, Dantas G, Gordon JI (2011) Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci USA 108: 6252–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurbatri CR, Lia I, Vincent R, Coker C, Castro S, Treuting PM, Hinchliffe TE, Arpaia N, Danino T (2020) Engineered probiotics for local tumor delivery of checkpoint blockade nanobodies. Sci Transl Med 12: eaax0876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie L, Wolfson S, Kelly L (2019) The human gut chemical landscape predicts microbe‐mediated biotransformation of foods and drugs. Elife 8: e42866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman‐Rodriguez M, McDonald JAK, Hyde R, Allen‐Vercoe E, Claud EC, Sheth PM, Petrof EO (2018) Using bioreactors to study the effects of drugs on the human microbiota. Methods 149: 31–41 [DOI] [PubMed] [Google Scholar]
- Hagan T, Cortese M, Rouphael N, Boudreau C, Linde C, Maddur MS, Das J, Wang H, Guthmiller J, Zheng N‐Y et al (2019) Antibiotics‐driven gut microbiome perturbation alters immunity to vaccines in humans. Cell 178: 1313–1328.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, Turnbaugh PJ (2013) Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta . Science 341: 295–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I, Park YJ, Kim Y, Kim YN, Ka S, Lee HY, Seong JK, Seok Y, Kim JB (2015) Alteration of gut microbiota by vancomycin and bacitracin improves insulin resistance via glucagon‐like peptide 1 in diet‐induced obesity. FASEB J 29: 2397–2411 [DOI] [PubMed] [Google Scholar]
- Imhann F, Bonder MJ, Vich Vila A, Fu J, Mujagic Z, Vork L, Tigchelaar EF, Jankipersadsing SA, Cenit MC, Harmsen HJM et al (2016) Proton pump inhibitors affect the gut microbiome. Gut 65: 740–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki J, Tsuda T, Suga Y, Ito S, Arai K, Sai Y, Miyamoto K (2012) Change in pharmacokinetics of mycophenolic acid as a function of age in rats and effect of coadministered amoxicillin/clavulanate. Biol Pharm Bull 35: 1009–1013 [DOI] [PubMed] [Google Scholar]
- Jackson MA, Verdi S, Maxan M‐E, Shin CM, Zierer J, Bowyer RCE, Martin T, Williams FMK, Menni C, Bell JT et al (2018) Gut microbiota associations with common diseases and prescription medications in a population‐based cohort. Nat Commun 9: 2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili‐Firoozinezhad S, Gazzaniga FS, Calamari EL, Camacho DM, Fadel CW, Bein A, Swenor B, Nestor B, Cronce MJ, Tovaglieri A et al (2019) A complex human gut microbiome cultured in an anaerobic intestine‐on‐a‐chip. Nat Biomed Eng 3: 520–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jariwala PB, Pellock SJ, Goldfarb D, Cloer EW, Artola M, Simpson JB, Bhatt AP, Walton WG, Roberts LR, Major MB et al (2020) Discovering the microbial enzymes driving drug toxicity with activity‐based protein profiling. ACS Chem Biol 15: 217–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javdan B, Lopez JG, Chankhamjon P, Lee Y‐CJ, Hull R, Wu Q, Wang X, Chatterjee S, Donia MS (2020) Personalized mapping of drug metabolism by the human gut microbiome. Cell 181: 1661–1679.e22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kintses B, Jangir PK, Fekete G, Számel M, Méhi O, Spohn R, Daruka L, Martins A, Hosseinnia A, Gagarinova A et al (2019) Chemical‐genetic profiling reveals limited cross‐resistance between antimicrobial peptides with different modes of action. Nat Commun 10: 5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchmair J, Göller AH, Lang D, Kunze J, Testa B, Wilson ID, Glen RC, Schneider G (2015) Predicting drug metabolism: experiment and/or computation? Nat Rev Drug Discov 14: 387–404 [DOI] [PubMed] [Google Scholar]
- Klünemann M, Schmid M, Patil KR (2014) Computational tools for modeling xenometabolism of the human gut microbiota. Trends Biotechnol 32: 157–165 [DOI] [PubMed] [Google Scholar]
- Koh A, Bäckhed F (2020) From association to causality: the role of the gut microbiota and its functional products on host metabolism. Mol Cell 78: 584–596 [DOI] [PubMed] [Google Scholar]
- Koppel N, Bisanz JE, Pandelia M‐E, Turnbaugh PJ, Balskus EP (2018) Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. Elife 7: e33953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M, Letunic I, Jensen LJ, Bork P (2016) The SIDER database of drugs and side effects. Nucleic Acids Res 44: D1075–D1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummen M, Solberg OG, Storm‐Larsen C, Holm K, Ragnarsson A, Trøseid M, Vestad B, Skårdal R, Yndestad A, Ueland T et al (2020) Rosuvastatin alters the genetic composition of the human gut microbiome. Sci Rep 10: 5397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuno T, Hirayama‐Kurogi M, Ito S, Ohtsuki S (2016) Effect of intestinal flora on protein expression of drug‐metabolizing enzymes and transporters in the liver and kidney of germ‐free and antibiotics‐treated mice. Mol Pharm 13: 2691–2701 [DOI] [PubMed] [Google Scholar]
- Kuno T, Hirayama‐Kurogi M, Ito S, Ohtsuki S (2019) Proteomic analysis of small intestinal epithelial cells in antibiotic‐treated mice: changes in drug transporters and metabolizing enzymes. Drug Metab Pharmacokinet 34: 159–162 [DOI] [PubMed] [Google Scholar]
- Le Bastard Q, Al‐Ghalith GA, Grégoire M, Chapelet G, Javaudin F, Dailly E, Batard E, Knights D, Montassier E (2018) Systematic review: human gut dysbiosis induced by non‐antibiotic prescription medications. Aliment Pharmacol Ther 47: 332–345 [DOI] [PubMed] [Google Scholar]
- Li L, Abou‐Samra E, Ning Z, Zhang X, Mayne J, Wang J, Cheng K, Walker K, Stintzi A, Figeys D (2019) An in vitro model maintaining taxon‐specific functional activities of the gut microbiome. Nat Commun 10: 4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Ning Z, Zhang X, Mayne J, Cheng K, Stintzi A, Figeys D (2020) RapidAIM: a culture‐ and metaproteomics‐based rapid assay of individual microbiome responses to drugs. Microbiome 8: 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindenbaum J, Rund DG, Butler VP, Tse‐Eng D, Saha JR (1981) Inactivation of digoxin by the gut flora: reversal by antibiotic therapy. N Engl J Med 305: 789–794 [DOI] [PubMed] [Google Scholar]
- Liu S, da Cunha AP, Rezende RM, Cialic R, Wei Z, Bry L, Comstock LE, Gandhi R, Weiner HL (2016) The host shapes the gut microbiota via fecal microRNA. Cell Host Microbe 19: 32–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukovac S, Belzer C, Pellis L, Keijser BJ, de Vos WM, Montijn RC, Roeselers G (2014) Differential modulation by akkermansia muciniphila and faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. MBio 5: e01438–e1514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn MA, Tumes DJ, Choo JM, Sribnaia A, Blake SJ, Leong LEX, Young GP, Marshall HS, Wesselingh SL, Rogers GB et al (2018) Early‐life antibiotic‐driven dysbiosis leads to dysregulated vaccine immune responses in mice. Cell Host Microbe 23: 653–660.e5 [DOI] [PubMed] [Google Scholar]
- Mager LF, Burkhard R, Pett N, Cooke NCA, Brown K, Ramay H, Paik S, Stagg J, Groves RA, Gallo M et al (2020) Microbiome‐derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 369: 1481–1489 [DOI] [PubMed] [Google Scholar]
- Maier L, Goemans CV, Pruteanu M, Wirbel J, Kuhn M, Cacace E, Banerjee T, Anderson EE, Milanese A, Löber U et al (2020) Dissecting the collateral damage of antibiotics on gut microbes microbiology. bioRxiv 10.1101/2020.01.09.893560 [PREPRINT] [DOI]
- Maier L, Pruteanu M, Kuhn M, Zeller G, Telzerow A, Anderson EE, Brochado AR, Fernandez KC, Dose H, Mori H et al (2018) Extensive impact of non‐antibiotic drugs on human gut bacteria. Nature 555: 623–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier L, Typas A (2017) Systematically investigating the impact of medication on the gut microbiome. Curr Opin Microbiol 39: 128–135 [DOI] [PubMed] [Google Scholar]
- Maini Rekdal V, Bess EN, Bisanz JE, Turnbaugh PJ, Balskus EP (2019) Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364: eaau6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malfatti MA, Kuhn EA, Murugesh DK, Mendez ME, Hum N, Thissen JB, Jaing CJ, Loots GG (2020) Manipulation of the gut microbiome alters acetaminophen biodisposition in mice. Sci Rep 10: 4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallory EK, Acharya A, Rensi SE, Turnbaugh PJ, Bright RA, Altman RB (2018) Chemical reaction vector embeddings: towards predicting drug metabolism in the human gut microbiome. In Biocomputing 2018 pp 56–67. Kohala Coast, Hawaii, USA: World Scientific; [PMC free article] [PubMed] [Google Scholar]
- Mateus A, Kurzawa N, Becher I, Sridharan S, Helm D, Stein F, Typas A, Savitski MM (2020) Thermal proteome profiling for interrogating protein interactions. Mol Syst Biol 16: e9232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurice CF, Haiser HJ, Turnbaugh PJ (2013) Xenobiotics shape the physiology and gene expression of the active human gut microbiome. Cell 152: 39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta RS, Abu‐Ali GS, Drew DA, Lloyd‐Price J, Subramanian A, Lochhead P, Joshi AD, Ivey KL, Khalili H, Brown GT et al (2018) Stability of the human faecal microbiome in a cohort of adult men. Nat Microbiol 3: 347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MetaCardis Consortium , Vieira‐Silva S, Falony G, Belda E, Nielsen T, Aron‐Wisnewsky J, Chakaroun R, Forslund SK, Assmann K, Valles‐Colomer M et al (2020) Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 581: 310–315 [DOI] [PubMed] [Google Scholar]
- Mills RH, Wozniak JM, Vrbanac A, Campeau A, Chassaing B, Gewirtz A, Knight R, Gonzalez DJ (2020) Organ‐level protein networks as a reference for the host effects of the microbiome. Genome Res 30: 276–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito T, Mulet C, De Castro C, Molinaro A, Saffarian A, Nigro G, Bérard M, Clerc M, Pedersen AB, Sansonetti PJ et al (2017) Lipopolysaccharide from crypt‐specific core microbiota modulates the colonic epithelial proliferation‐to‐differentiation balance. MBio 8: e01680–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama H, Kinouchi T, Kataoka K, Akimoto S, Matsuda Y, Ohnishi Y (1997) Intestinal anaerobic bacteria hydrolyse sorivudine, producing the high blood concentration of 5‐(E)‐(2‐bromovinyl)uracil that increases the level and toxicity of 5‐fluorouracil. Pharmacogenetics 7: 35–43 [DOI] [PubMed] [Google Scholar]
- Napolitano A, Miller S, Nicholls AW, Baker D, Van Horn S, Thomas E, Rajpal D, Spivak A, Brown JR, Nunez DJ (2014) Novel Gut‐based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS One 9: e100778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Shi ZJ, Seshadri R, Pollard KS, Kyrpides NC (2019) New insights from uncultivated genomes of the global human gut microbiome. Nature 568: 505–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KM, Aranda‐Díaz A, Tropini C, Frankel MR, Van Treuren W, O’Laughlin CT, Merrill BD, Yu FB, Pruss KM, Oliveira RA et al (2019) Recovery of the Gut microbiota after antibiotics depends on host diet, community context, and environmental reservoirs. Cell Host Microbe 26: 650–665.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TLA, Vieira‐Silva S, Liston A, Raes J (2015) How informative is the mouse for human gut microbiota research? Dis Model Mech 8: 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro G, Rossi R, Commere P‐H, Jay P, Sansonetti PJ (2014) The cytosolic bacterial peptidoglycan sensor Nod2 affords stem cell protection and links microbes to gut epithelial regeneration. Cell Host Microbe 15: 792–798 [DOI] [PubMed] [Google Scholar]
- Nobel YR, Cox LM, Kirigin FF, Bokulich NA, Yamanishi S, Teitler I, Chung J, Sohn J, Barber CM, Goldfarb DS et al (2015) Metabolic and metagenomic outcomes from early‐life pulsed antibiotic treatment. Nat Commun 6: 7486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norvaisas P, Cabreiro F (2018) Pharmacology in the age of the holobiont. Curr Opin Syst Biol 10: 34–42 [Google Scholar]
- O’Reilly LP, Luke CJ, Perlmutter DH, Silverman GA, Pak SC (2014) C. elegans in high‐throughput drug discovery. Adv Drug Deliv Rev 69–70: 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole PW, Marchesi JR, Hill C (2017) Next‐generation probiotics: the spectrum from probiotics to live biotherapeutics. Nat Microbiol 2: 17057 [DOI] [PubMed] [Google Scholar]
- Pál C, Papp B, Lázár V (2015) Collateral sensitivity of antibiotic‐resistant microbes. Trends Microbiol 23: 401–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palleja A, Mikkelsen KH, Forslund SK, Kashani A, Allin KH, Nielsen T, Hansen TH, Liang S, Feng Q, Zhang C et al (2018) Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat Microbiol 3: 1255–1265 [DOI] [PubMed] [Google Scholar]
- Pandey UB, Nichols CD (2011) Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol Rev 63: 411–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, Usui Y, Hatano N, Shinohara M, Saito Y et al (2016) Promotion of intestinal epithelial cell turnover by commensal bacteria: role of short‐chain fatty acids. PLoS One 11: e0156334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasolli E, Asnicar F, Manara S, Zolfo M, Karcher N, Armanini F, Beghini F, Manghi P, Tett A, Ghensi P et al (2019) Extensive unexplored human microbiome diversity revealed by over 150,000 genomes from metagenomes spanning age, geography, and lifestyle. Cell 176: 649–662.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce SC, Al‐Jawadi A, Kishida K, Yu S, Hu M, Fritzky LF, Edelblum KL, Gao N, Ferraris RP (2018) Marked differences in tight junction composition and macromolecular permeability among different intestinal cell types. BMC Biol 16: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppercorn MA, Goldman P (1972) The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther 181: 555–562 [PubMed] [Google Scholar]
- Pleguezuelos‐Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, Gurjao C, Manders F, Dalmasso G, Stege PB et al (2020) Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli . Nature 580: 269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollet RM, D’Agostino EH, Walton WG, Xu Y, Little MS, Biernat KA, Pellock SJ, Patterson LM, Creekmore BC, Isenberg HN et al (2017) An atlas of β‐glucuronidases in the human intestinal microbiome. Structure 25: 967–977.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poyet M, Groussin M, Gibbons SM, Avila‐Pacheco J, Jiang X, Kearney SM, Perrotta AR, Berdy B, Zhao S, Lieberman TD et al (2019) A library of human gut bacterial isolates paired with longitudinal multiomics data enables mechanistic microbiome research. Nat Med 25: 1442–1452 [DOI] [PubMed] [Google Scholar]
- Pryor R, Martinez‐Martinez D, Quintaneiro L, Cabreiro F (2020) The role of the microbiome in drug response. Annu Rev Pharmacol Toxicol 60: 417–435 [DOI] [PubMed] [Google Scholar]
- Pryor R, Norvaisas P, Marinos G, Best L, Thingholm LB, Quintaneiro LM, De Haes W, Esser D, Waschina S, Lujan C et al (2019) Host‐microbe‐drug‐nutrient screen identifies bacterial effectors of metformin therapy. Cell 178: 1299–1312.e29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rashid M‐U, Zaura E, Buijs MJ, Keijser BJF, Crielaard W, Nord CE, Weintraub A (2015) Determining the long‐term effect of antibiotic administration on the human normal intestinal microbiota using culture and pyrosequencing methods. Clin Infect Dis 60: S77–S84 [DOI] [PubMed] [Google Scholar]
- Rask‐Andersen M, Almén MS, Schiöth HB (2011) Trends in the exploitation of novel drug targets. Nat Rev Drug Discov 10: 579–590 [DOI] [PubMed] [Google Scholar]
- Ratzke C, Gore J (2018) Modifying and reacting to the environmental pH can drive bacterial interactions. PLOS Biol 16: e2004248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roemhild R, Linkevicius M, Andersson DI (2020) Molecular mechanisms of collateral sensitivity to the antibiotic nitrofurantoin. PLOS Biol 18: e3000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N et al (2018) Environment dominates over host genetics in shaping human gut microbiota. Nature 555: 210–215 [DOI] [PubMed] [Google Scholar]
- Ruiz VE, Battaglia T, Kurtz ZD, Bijnens L, Ou A, Engstrand I, Zheng X, Iizumi T, Mullins BJ, Müller CL et al (2017) A single early‐in‐life macrolide course has lasting effects on murine microbial network topology and immunity. Nat Commun 8: 518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ et al (2009) Single Lgr5 stem cells build crypt‐villus structures in vitro without a mesenchymal niche. Nature 459: 262–265 [DOI] [PubMed] [Google Scholar]
- Sausset R, Petit MA, Gaboriau‐Routhiau V, De Paepe M (2020) New insights into intestinal phages. Mucosal Immunol 13: 205–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schopper S, Kahraman A, Leuenberger P, Feng Y, Piazza I, Müller O, Boersema PJ, Picotti P (2017) Measuring protein structural changes on a proteome‐wide scale using limited proteolysis‐coupled mass spectrometry. Nat Protoc 12: 2391–2410 [DOI] [PubMed] [Google Scholar]
- Scott TA, Quintaneiro LM, Norvaisas P, Lui PP, Wilson MP, Leung K‐Y, Herrera‐Dominguez L, Sudiwala S, Pessia A, Clayton PT et al (2017) Host‐microbe co‐metabolism dictates cancer drug efficacy in C. elegans . Cell 169: 442–456.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selwyn FP, Cheng SL, Klaassen CD, Cui JY (2016) Regulation of hepatic drug‐metabolizing enzymes in germ‐free mice by conventionalization and probiotics. Drug Metab Dispos 44: 262–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shetty SA, Smidt H, de Vos WM (2019) Reconstructing functional networks in the human intestinal tract using synthetic microbiomes. Curr Opin Biotechnol 58: 146–154 [DOI] [PubMed] [Google Scholar]
- Shin N‐R, Lee J‐C, Lee H‐Y, Kim M‐S, Whon TW, Lee M‐S, Bae J‐W (2014) An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet‐induced obese mice. Gut 63: 727–735 [DOI] [PubMed] [Google Scholar]
- Sivan A, Corrales L, Hubert N, Williams JB, Aquino‐Michaels K, Earley ZM, Benyamin FW, Man Lei Y, Jabri B, Alegre M‐L et al (2015) Commensal Bifidobacterium promotes antitumor immunity and facilitates anti‐PD‐L1 efficacy. Science 350: 1084–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F, Anderson JM, Bharti R, Raes J, Rosenstiel P (2017) The resilience of the intestinal microbiota influences health and disease. Nat Rev Microbiol 15: 630–638 [DOI] [PubMed] [Google Scholar]
- Sousa T, Yadav V, Zann V, Borde A, Abrahamsson B, Basit AW (2014) On the colonic bacterial metabolism of azo‐bonded prodrugsof 5‐aminosalicylic acid. J Pharm Sci 103: 3171–3175 [DOI] [PubMed] [Google Scholar]
- Spanogiannopoulos P, Bradley PH, Melamed J, Malig YNA, Lam KN, Gerona RR, Pollard KS, Turnbaugh PJ (2019) Drug resistant gut bacteria mimic a host mechanism for anticancer drug clearance. bioRxiv 10.1101/820084 [PREPRINT] [DOI]
- van de Steeg E, Schuren FHJ, Obach RS, van Woudenbergh C, Walker GS, Heerikhuisen M, Nooijen IHG, Vaes WHJ (2018) An ex vivo fermentation screening platform to study drug metabolism by human gut microbiota. Drug Metab Dispos 46: 1596–1607 [DOI] [PubMed] [Google Scholar]
- Suez J, Zmora N, Zilberman‐Schapira G, Mor U, Dori‐Bachash M, Bashiardes S, Zur M, Regev‐Lehavi D, Ben‐Zeev Brik R, Federici S et al (2018) Post‐antibiotic gut mucosal microbiome reconstitution is impaired by probiotics and improved by autologous FMT. Cell 174: 1406–1423.e16 [DOI] [PubMed] [Google Scholar]
- Suez J, Zmora N, Segal E, Elinav E (2019) The pros, cons, and many unknowns of probiotics. Nat Med 25: 716–729 [DOI] [PubMed] [Google Scholar]
- Sun L, Xie C, Wang G, Wu Y, Wu Q, Wang X, Liu J, Deng Y, Xia J, Chen B et al (2018) Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat Med 24: 1919–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanoue T, Morita S, Plichta DR, Skelly AN, Suda W, Sugiura Y, Narushima S, Vlamakis H, Motoo I, Sugita K et al (2019) A defined commensal consortium elicits CD8 T cells and anti‐cancer immunity. Nature 565: 600–605 [DOI] [PubMed] [Google Scholar]
- Taur Y, Coyte K, Schluter J, Robilotti E, Figueroa C, Gjonbalaj M, Littmann ER, Ling L, Miller L, Gyaltshen Y et al (2018) Reconstitution of the gut microbiota of antibiotic‐treated patients by autologous fecal microbiota transplant. Sci Transl Med 10: eaap9489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MR, Flannigan KL, Rahim H, Mohamud A, Lewis IA, Hirota SA, Greenway SC (2019) Vancomycin relieves mycophenolate mofetil–induced gastrointestinal toxicity by eliminating gut bacterial β‐glucuronidase activity. Sci Adv 5: eaax2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Integrative HMP (iHMP) Research Network Consortium (2019) The integrative human microbiome project. Nature 569: 641–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ticinesi A, Milani C, Lauretani F, Nouvenne A, Mancabelli L, Lugli GA, Turroni F, Duranti S, Mangifesta M, Viappiani A et al (2017) Gut microbiota composition is associated with polypharmacy in elderly hospitalized patients. Sci Rep 7: 11102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tramontano M, Andrejev S, Pruteanu M, Klünemann M, Kuhn M, Galardini M, Jouhten P, Zelezniak A, Zeller G, Bork P et al (2018) Nutritional preferences of human gut bacteria reveal their metabolic idiosyncrasies. Nat Microbiol 3: 514–522 [DOI] [PubMed] [Google Scholar]
- Trinder M, Daisley BA, Dube JS, Reid G (2017) Drosophila melanogaster as a high‐throughput model for host‐microbiota interactions. Front Microbiol 8: 751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchimura Y, Fuhrer T, Li H, Lawson MA, Zimmermann M, Yilmaz B, Zindel J, Ronchi F, Sorribas M, Hapfelmeier S et al (2018) Antibodies set boundaries limiting microbial metabolite penetration and the resultant mammalian host response. Immunity 49: 545–559.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underhill GH, Khetani SR (2018) Advances in engineered human liver platforms for drug metabolism studies. Drug Metab Dispos 46: 1626–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega NM, Gore J (2014) Collective antibiotic resistance: mechanisms and implications. Curr Opin Microbiol 21: 28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venturelli OS, Carr AV, Fisher G, Hsu RH, Lau R, Bowen BP, Hromada S, Northen T, Arkin AP (2018) Deciphering microbial interactions in synthetic human gut microbiome communities. Mol Syst Biol 14: e8157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vétizou M, Pitt JM, Daillère R, Lepage P, Waldschmitt N, Flament C, Rusakiewicz S, Routy B, Roberti MP, Duong CPM et al (2015) Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science 350: 1079–1084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viaud S, Saccheri F, Mignot G, Yamazaki T, Daillere R, Hannani D, Enot DP, Pfirschke C, Engblom C, Pittet MJ et al (2013) The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science 342: 971–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vich Vila A, Collij V, Sanna S, Sinha T, Imhann F, Bourgonje AR, Mujagic Z, Jonkers DMAE, Masclee AAM, Fu J et al (2020) Impact of commonly used drugs on the composition and metabolic function of the gut microbiota. Nat Commun 11: 362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace BD, Wang H, Lane KT, Scott JE, Orans J, Koo JS, Venkatesh M, Jobin C, Yeh L‐A, Mani S et al (2010) Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330: 831–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter J, Armet AM, Finlay BB, Shanahan F (2020) Establishing or exaggerating causality for the gut microbiome: lessons from human microbiota‐associated rodents. Cell 180: 221–232 [DOI] [PubMed] [Google Scholar]
- Wampach L, Heintz‐Buschart A, Hogan A, Muller EEL, Narayanasamy S, Laczny CC, Hugerth LW, Bindl L, Bottu J, Andersson AF et al (2017) Colonization and succession within the human gut microbiome by archaea, bacteria, and microeukaryotes during the first year of life. Front Microbiol 8: 738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegh CAM, Geerlings SY, Knol J, Roeselers G, Belzer C (2019) Postbiotics and their potential applications in early life nutrition and beyond. Int J Mol Sci 20: 4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson B, Wilson C, Dagnell G, Riley RJ (2017) Harmonised high throughput microsomal stability assay. J Pharmacol Toxicol Methods 84: 31–36 [DOI] [PubMed] [Google Scholar]
- Willing BP, Russell SL, Finlay BB (2011) Shifting the balance: antibiotic effects on host–microbiota mutualism. Nat Rev Microbiol 9: 233–243 [DOI] [PubMed] [Google Scholar]
- Wilson ID, Nicholson JK (2017) Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res 179: 204–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wlodarska M, Luo C, Kolde R, d’Hennezel E, Annand JW, Heim CE, Krastel P, Schmitt EK, Omar AS, Creasey EA et al (2017) Indoleacrylic acid produced by commensal peptostreptococcus species suppresses inflammation. Cell Host Microbe 22: 25–37.e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås‐Holm L, Ståhlman M, Olsson LM, Serino M, Planas‐Fèlix M et al (2017) Metformin alters the gut microbiome of individuals with treatment‐naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 23: 850–858 [DOI] [PubMed] [Google Scholar]
- Yin X, Farin HF, van Es JH, Clevers H, Langer R, Karp JM (2014) Niche‐independent high‐purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat Methods 11: 106–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zampieri M, Szappanos B, Buchieri MV, Trauner A, Piazza I, Picotti P, Gagneux S, Borrell S, Gicquel B, Lelievre J et al (2018) High‐throughput metabolomic analysis predicts mode of action of uncharacterized antimicrobial compounds. Sci Transl Med 10: eaal3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D‐W, Dong X, Pan P, Chen K‐W, Fan J‐X, Cheng S‐X, Zhang X‐Z (2019) Phage‐guided modulation of the gut microbiota of mouse models of colorectal cancer augments their responses to chemotherapy. Nat Biomed Eng 3: 717–728 [DOI] [PubMed] [Google Scholar]
- Zimmermann J, Obeng N, Yang W, Pees B, Petersen C, Waschina S, Kissoyan KA, Aidley J, Hoeppner MP, Bunk B et al (2020) The functional repertoire contained within the native microbiota of the model nematode Caenorhabditis elegans . ISME J 14: 26–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Zimmermann‐Kogadeeva M, Wegmann R, Goodman AL (2019a) Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science 363: eaat9931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Zimmermann‐Kogadeeva M, Wegmann R, Goodman AL (2019b) Mapping human microbiome drug metabolism by gut bacteria and their genes. Nature 570: 462–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann‐Kogadeeva M, Zimmermann M, Goodman AL (2020) Insights from pharmacokinetic models of host‐microbiome drug metabolism. Gut Microbes 11: 587–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Ma Y, Raoult D, Kroemer G, Gajewski TF (2018) The microbiome in cancer immunotherapy: diagnostic tools and therapeutic strategies. Science 359: 1366–1370 [DOI] [PubMed] [Google Scholar]
- Zmora N, Zilberman‐Schapira G, Suez J, Mor U, Dori‐Bachash M, Bashiardes S, Kotler E, Zur M, Regev‐Lehavi D, Brik RB‐Z et al (2018) Personalized gut mucosal colonization resistance to empiric probiotics is associated with unique host and microbiome features. Cell 174: 1388–1405.e21 [DOI] [PubMed] [Google Scholar]
- Zou Y, Xue W, Luo G, Deng Z, Qin P, Guo R, Sun H, Xia Y, Liang S, Dai Y et al (2019) 1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses. Nat Biotechnol 37: 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]