

Abstract
Lower respiratory viral infections, such as influenza virus and severe acute respiratory syndrome coronavirus 2 infections, often cause severe viral pneumonia in aged individuals. Here, we report that influenza viral pneumonia leads to chronic nonresolving lung pathology and exacerbated accumulation of CD8+ tissue-resident memory T cells (TRM) in the respiratory tract of aged hosts. TRM cell accumulation relies on elevated TGF-β present in aged tissues. Further, we show that TRM cells isolated from aged lungs lack a subpopulation characterized by expression of molecules involved in TCR signaling and effector function. Consequently, TRM cells from aged lungs were insufficient to provide heterologous protective immunity. The depletion of CD8+ TRM cells dampens persistent chronic lung inflammation and ameliorates tissue fibrosis in aged, but not young, animals. Collectively, our data demonstrate that age-associated TRM cell malfunction supports chronic lung inflammatory and fibrotic sequelae after viral pneumonia.
INTRODUCTION
Aged individuals (65 and older) will comprise 20% of the population in developed countries in the coming decades (1). It is well established that the peripheral CD8+ T cell compartment that contributes to cellular immunity shrinks in number, diversity, and quality with age (2-4). As a consequence, memory T cells are enriched and T cell receptor (TCR) repertoires are narrowed during homeostasis (5). Subsequently, the magnitude and the quality of primary effector CD8+ T cell response often are compromised in aged hosts after pathogen challenge (6). Likewise, the development of CD8+ memory T cell responses in the circulation is often impaired during aging (7, 8). This age-related CD8+ T cell attrition in memory T cell differentiation and/or function has been linked to impaired host responses to pathogens after primary infection or vaccination in aged hosts (9).
Tissue-resident memory CD8+ T cells (TRM) with potent effector potential are mainly located in the peripheral tissue at portals of pathogen entry, whereas circulating central and effector memory T cells survey lymphoid organs and/or peripheral tissues (10, 11). CD8+ TRM cells can provide nearly sterilizing protection if present in sufficient numbers (12). Therefore, great interest has recently centered on enhancing local CD8+ TRM responses as a promising vaccination strategy to control mucosal infections including influenza virus infection (13). Much progress has been made regarding the cellular and molecular mechanisms regulating TRM cell development and maintenance (14, 15). However, less is known regarding the effects of aging in the development and function of TRM responses in mucosal tissues after primary infection. Of note, analysis of spatial and temporal maps of human T cell compartmentalization revealed that aged lungs harbor increased levels of CD8+ CD69+ TRM-like cells compared with young lungs (2). However, it is unclear whether these observations are due to a gradual buildup of memory T cells after frequent prior infections in the lungs or due to enhanced generation of CD8+ TRM cells after a single infection in the elderly. Therefore, the development and function of CD8+ TRM cells in aged hosts after primary infection remains to be examined.
Lower respiratory tract viral infections represent a major public health challenge and economic burden worldwide. In a matter of months, emerging respiratory viral diseases can alter social norms, stagnate economies, and overwhelm healthcare infrastructures across the globe, as demonstrated by the ongoing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) pandemic. Among respiratory viral pathogens, influenza virus can infect 5 to 10% of the population (16), resulting in up to 35.6 million illnesses and 56,000 deaths annually in the United States alone since 2010 (17). For reasons that are still not fully defined, many respiratory viruses including SARS-CoV-2 and influenza virus cause disproportionately severe morbidity in the elderly population (18, 19). For instance, the vast majority of the mortality associated with both influenza and SARS-CoV-2 infections occur in people that are 65 years and older. Influenza disease severity is elevated, prolonged, and linked with increased comorbidities and death in the elderly population (20). In addition to the mortality caused by respiratory viral infections, evidence has suggested that survivors of primary viral pneumonia may display persistent impairment of lung function because of lung fibrosis (21). Furthermore, it is predicted that a large proportion of elderly coronavirus disease 2019 (COVID-19) survivors will be prone to persistent impairment of lung function and pulmonary fibrosis, as was observed in survivors of SARS and Middle East respiratory syndrome (22). At this time, there are no preventative means or therapeutic interventions available to mitigate and/or reverse lung fibrosis development after viral pneumonia. Now, the cellular and molecular mechanisms regulating the development of chronic lung fibrotic sequelae after primary viral pneumonia are still largely unknown. This major knowledge gap likely represents a future bottleneck for development of successful therapeutics for patients with chronic lung fibrosis induced by SARS-CoV-2 infection.
Here, we report that influenza infection in aged mice leads to nonresolving inflammation and persistent chronic lung pathology. Transcriptional and flow cytometric analyses revealed that lungs from aged mice exhibited enhanced accumulation of CD8+ CD69+ memory T cells. Parabiosis experiments showed that these age-associated lung CD8+ CD69+ T cells were tissue resident. Furthermore, the excessive accumulation of lung CD8+ TRM cells likely depended on the aging-associated increase in environmental transforming growth factor-β (TGF-β). Single-cell (sc) RNA sequencing (RNA-seq) analysis demonstrated that TRM against a major influenza–protective epitope exhibited diminished effector function in response to TCR signaling. As a result, CD8+ TRM cells failed to provide protective immunity against a secondary heterologous virus infection in aged mice. We further showed that the depletion of CD8+ TRM cells resulted in decreased tissue inflammation and lowered lung collagen deposition. Thus, we have identified an age-associated paradox in CD8+ TRM cell responses. CD8+ TRM cell accumulation in aged hosts was not coupled to enhanced protective immunity but, rather, to chronic lung pathology and fibrotic sequelae after primary viral pneumonia.
RESULTS
Aged hosts develop nonresolving tissue inflammation and pathology after influenza virus infection
To investigate the short- and long-term effects of aging on influenza disease severity, young (2 months old) and aged (21 to 22 months old) C57BL/6 mice were infected with influenza A virus [A/PR8/34 strain (PR8); Fig. 1A]. Although 100% of young mice survived from intranasal infection with PR8, ~50% of aged mice succumbed to the infection by day 12 (Fig. 1B), which is consistent with previous reports that aged mice exhibit enhanced host mortality after primary influenza infection (23). Aged mice that survived exhibited prolonged weight recovery, although their weight almost completely recovered by 18 days post infection (d.p.i.) (Fig. 1C). These differences in acute morbidity and mortality were seen despite similar viral load at 9 d.p.i. and complete viral clearance in the airway in both groups by 15 d.p.i. (Fig. 1D) similar to previous reports (24).
Fig. 1. Aged lungs display nonresolving pulmonary immunopathology after influenza infection.

Young (2 month) or aged (22 month) C57BL/6 mice were infected or not with PR8 virus. (G and H) Anti-CD45 was injected intravenously to label circulating white blood cells before sacrifice. (A) Schematic of experimental procedure. (B) Percent of host survival after influenza infection in young or aged mice. (C) Percent of preinfection body weight after influenza infection in young or aged mice. (D) Airway viral titers were determined through pfu assay of bronchoalveolar lavage fluid at 9, 15, and 30 d.p.i. (E) Lung histopathology by H&E staining at 0, 9, 30, and 60 d.p.i. (F) Percent of left lung lobe parenchyma infiltrated by white blood cells quantitated by ImageJ analysis. (G and H) Lung cells were stained for monocytes (MNC; parent gates CD64− Siglec-F− CD11c− MHCII− CD11bhi) and neutrophils (Neu; parent gate Ly6G+), which were enumerated in the lung and divided into circulating (Cir) or parenchymal (Par) after intravenous labeling of immune cells at 9 (left) and 60 d.p.i. (right) (G) Representative plots. (H) After perfusion, cell numbers of lung-circulating and parenchymal monocytes and neutrophils at 9 and 60 d.p.i. (B) and (C) were n = 10 young and n = 14 aged mice (two experiments pooled). (D) was six BAL samples per group (two experiments pooled) for days 9 and 15 and three each for day 30. (E) and (G) were representatives of two to four experiments each; (H) is one to two independent replicates pooled. Scale bars (histology figures), 600 μm. Below limit of detection (<LOD). *P < 0.05 or not significant (ns) by Student’s two-tailed t test with unequal variance. Log-rank (Mantel-Cox) test was used for survival curve comparison in (B). ***P < 0.0005.
Recent clinical and experimental evidence suggests that severe acute influenza infection may cause persistent lung pathology, remodeling, and pulmonary dysfunction (21, 25). To determine whether these acute differences in host morbidity result in differential tissue pathological sequela, we examined the kinetics of histopathology in the lungs after acute PR8 infection in young and aged hosts. Although common features of pulmonary aging such as enlarged alveoli were observed before infection (0 d.p.i.), leukocytic infiltrates were not prominent in either aged or young naive lungs. At peak of host inflammatory responses and weight loss (9 d.p.i.), lung histopathology and tissue damage appeared comparable between young and aged mice (Fig. 1, E and F). However, at 60 d.p.i., lungs from aged mice showed a higher density of parenchymal disruption compared with lungs from young mice (Fig. 1, E and F). These data suggest that acute influenza virus infection in aged mice results in persistent nonresolving pathological pulmonary responses.
To confirm these lung histopathological data, we examined the presence of inflammatory monocytes and neutrophils, two major inflammatory cells that can cause pulmonary inflammation and tissue damage (23, 26). To discern the anatomical location of the cell infiltrates, we examined the vascular and parenchymal fractions of the lungs through the injection of intravenous CD45 antibody (Ab) 5 min before sacrifice (Fig. 1G) (27). In this setting, intravenous CD45− cells were within lung tissue, whereas intravenous CD45+ cells were in lung blood vessels. Neutrophil counts in the circulation exhibited a slight increase in aged relative to young mice at 9 d.p.i., as has been reported (23). Furthermore, there were more tissue-infiltrating neutrophils and monocytes in the aged group at 9 d.p.i. (Fig. 1H). By 60 d.p.i., the differences of neutrophil and monocyte numbers in the circulation were lost compared with 9 d.p.i., yet evidence of tissue-infiltrating neutrophils and monocytes persisted in aged lungs compared with young mice (Fig. 1H). Together, the data point to an infection-induced age-related nonresolving inflammatory response after primary influenza virus infection.
Aged lungs fail to return to immune homeostasis after respiratory viral infection
The above data pointed to unresolved immune responses after influenza viral pneumonia in aged mice. To directly address this idea, we measured the expression of 560 immune-related genes in the lungs before (0 d.p.i.) or at 60 d.p.i. using NanoString. The immune gene profiles of the lungs from young or aged uninfected mice were quite similar except a few genes associated with “inflammaging,” which were modestly increased in aged lungs (Fig. 2A). Furthermore, the expression profile of the immune-related genes in the lungs from infected young mice (60 d.p.i.) was quite similar to those of lungs from uninfected mice, suggesting that lungs from infected young mice largely return to immune homeostasis at 60 d.p.i. However, the expression of most immune response–related genes were up-regulated in infected aged lungs relative to those of uninfected young or aged lungs or infected young lungs (Fig. 2A). The mRNA signal ratios of infected versus noninfected lungs in the young or aged groups identified 52 differentially expressed genes (DEGs) shared between young and aged groups; 12 were unique to young and 349 were unique to aged samples (Fig. 2A, top right). When comparing gene expression ratios of aged with young lungs, before and after infection, there were only 16 unique age-related DEGs before infection, whereas 32 were shared before and after infection between young and aged groups. In contrast, 348 unique DEGs were observed at day 60 after infection in aged groups (Fig. 2A, bottom right). Among those DEGs, influenza infection led to greatly increased expression of myeloid cell–associated genes, cytokine and chemokine genes, and genes involved in lymphocyte responses (fig. S1, A to C). Together, these data indicated that the immune landscape of aged and young lungs is grossly similar before infection, but aged lungs exhibit greatly exacerbated inflammatory gene expression and failed to return to immune homeostasis months after primary infection.
Fig. 2. Aged lungs fail to return to immune homeostasis after viral pneumonia.

(A) Five hundred sixty immune-associated transcripts were examined at the endogenous mRNA levels by NanoString from young (Y) or aged (A) lungs before (day 0) or 60 d.p.i. and displayed as a heatmap (purple, relatively low expression; orange, relatively high expression) after log2 transformation of raw expression data above the detection threshold. Venn diagrams displaying data in both number of genes and percent of total genes examined for the ratios infected versus noninfected by age (top right) or aged to young by d.p.i. (bottom right) from NanoString data. (B) Young and aged mice were infected with PR8. Bulk RNA-seq was performed on young or aged lungs at 60 d.p.i. Data is displayed as a heatmap (purple, relatively low expression; orange, relatively high expression) of 658 DEGs (549 up-regulated in aged relative to young and 109 up-regulated in young relative to aged) with P < 0.001. (C) GSEA for adaptive immune response genes with normalized enrichment score (NES) and false-discovery rate (FDR). (D) Ranked normalized gene set ESs from the C5 and Hallmark (HM) databases and associated FDR (q). (A) is representative of two independent repeats; (B) to (D) were performed once on triplicate samples. UV, ultraviolet.
To gain more insight into the lung global transcription profiles of influenza-infected young or aged mice, we performed bulk RNA-seq on lung tissues from infected young or aged mice at 60 d.p.i. There were around 660 genes differentially expressed between the lungs of young and aged mice (P < 0.001) (Fig. 2B). Among those 660 DEGs, aged lungs expressed higher levels of inflammatory cytokines and molecules associated with adaptive immune responses (fig. S1D). Gene set enrichment analysis (GSEA) found that young lungs were enriched with gene sets associated with tissue repair such as epithelial-mesenchymal transition (EMT) and apical junction (fig. S1E). In contrast, aged lungs were enriched with gene sets associated with inflammatory responses and adaptive immune responses (Fig. 2, C and D, and fig. S1F).
Aged hosts exhibit excessive accumulation of parenchymal CD8+ memory T cells
We were intrigued with the enrichment of adaptive immune responses in the aged lungs because previous reports described that influenza-infected aged mice had decreased levels of memory T cell responses in the circulation (28). Therefore, we performed flow cytometric analysis to measure splenic- and lung-circulating and parenchymal memory T cell responses after intravenous injection of CD45 Ab as above at 60 d.p.i. (fig. S2A). There were increased CD4+ and CD8+ T and B cell presence in aged lung parenchyma but not within the lung vasculature (circulation) (Fig. 3, A and B, and fig. S2, B and C). Lung CD8+ CD69+ CD103− or CD8+ CD69+ CD103+ TRM-like cells were also increased in aged lungs compared with those of young lungs (Fig. 3C). Consistent with previous reports, adaptive immune cells, particularly CD8+ T cells, were decreased in the spleens of aged mice (Fig. 3D) (29).
Fig. 3. Aged lungs exhibit enhanced local adaptive immune responses despite circulatory deficiencies in the memory phase.
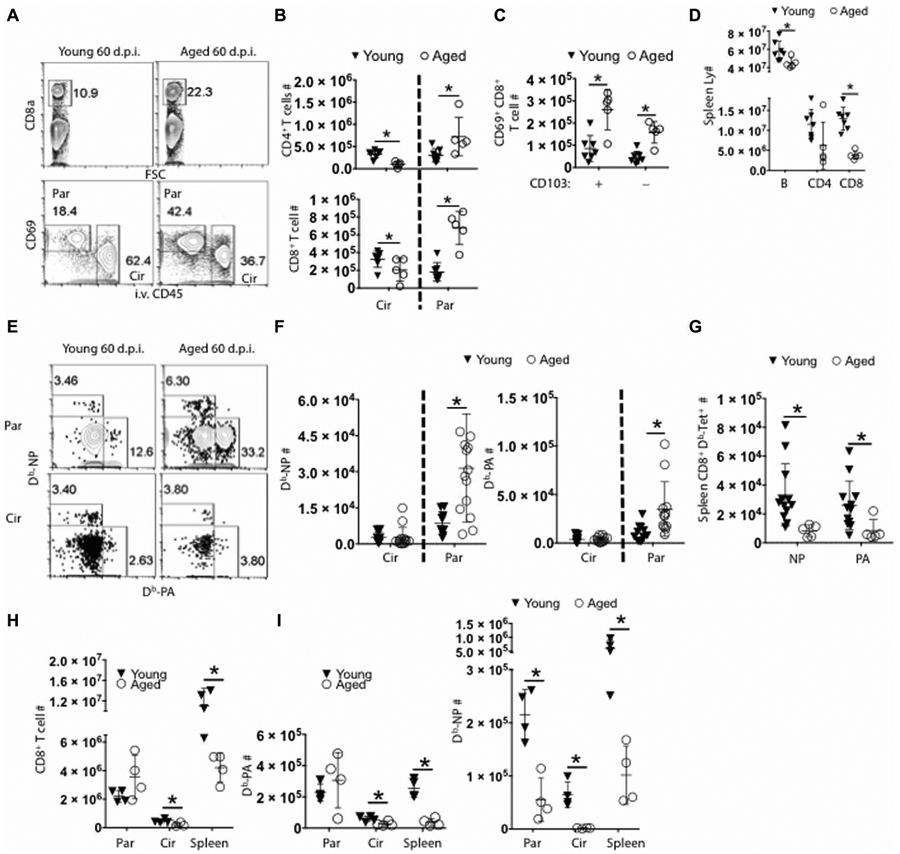
(A) Representative flow cytometry plot showing lung CD8+ T cells separated by CD69 and CD45 intravenous labeling, showing parenchymal (Par) and circulating (Cir) populations as indicated. (B) Quantification of CD4+ (top) and CD8+ T cells (bottom) in lung circulation (Cir) or parenchyma (Par) 60 d.p.i. (C) CD69+ parenchymal CD8+ T cells that either express CD103 (+) or not (−) were enumerated at 60 d.p.i. (D) T (CD4+ and CD8+) and B lymphocytes (B220+) were quantitated in spleens at 60 d.p.i. (Spleen Ly #). (E) Representative flow cytometry plots and (F) number of Db-NP (left) or Db-PA tetramer-positive (right) circulating (Circ) or parenchymal (Res) memory CD8+ T cells. (G) Number of Db-NP (NP) or Db-PA (PA) tetramer-positive CD8+ memory T cells in spleens. (H) Total CD8+, (I) CD8+ Db-PA (left), or CD8+ Db-NP (right) T cell numbers were quantitated in the lung parenchyma (Par), vasculature (Cir), or spleen (Spleen) at 10 d.p.i. (A) to (C), (E), and (H) were representatives of two to four experiments. (F) and (G) were pooled data from three independently significant experiments. *P < 0.05 Student’s two-tailed t test with unequal variance.
We next examined influenza-specific CD8+ memory T cells in the lungs and spleens of infected young or aged mice through the staining of H2Db/nucleoprotein (NP) peptide 366–374 tetramer (Db-NP) or polymerase (PA) peptide 224–233 (Db-PA) tetramers (Fig. 3E and fig. S2D). Aged lungs harbored significantly higher numbers of influenza-specific Db-NP and Db-PA memory T cells, selectively in the lung tissue, compared with young lungs (Fig. 3F). In contrast, there was a drastic reduction of influenza-specific CD8+ memory T cells in aged spleens (Fig. 3G and fig. S2D). Thus, we observed an unexpected increase of memory T cells present in aged lungs, despite the attrition of memory T cells in the spleen or circulation. Increased TRM responses in the lungs could be due to increased infiltration of effector T cells in aged mice. We, therefore, examined effector T cell responses in the lungs and spleens at 10 d.p.i. We found that although Db-PA responses appear to be equivalent in the lungs between young and aged mice, there was a deficit in the generation of Db-NP–specific CD8+ T cells in the lungs (Fig. 3, H and I). Furthermore, there were significantly lower levels of both Db-NP and Db-PA effector T cells in the spleens and lung circulation (Fig. 3I) of aged mice compared with controls as previously reported (30). Therefore, aged mice exhibit enhanced CD69+ parenchymal memory CD8+ T cell numbers despite having lower levels of effector T cell responses.
Influenza-specific lung memory CD8+ T cells of aged hosts are tissue resident
To verify whether the CD8+ CD69+ T cells in the lung that were protected from intravenous labeling were bona fide tissue-resident cells, we infected aged mice with influenza and performed parabiosis surgery to join the circulation of infected aged mice (CD45.2+) with a young naive congenic (CD45.1+) animal at 5 weeks after infection (Fig. 4A). Four weeks after parabiosis, we observed equilibration of CD8+ T cells from each parabiont in the blood and spleens (Fig. 4B and fig. S3, A and B), confirming the successful exchange of circulating immune cells between parabionts.
Fig. 4. CD8+ memory T cells in the lung parenchyma from aged mice are tissue resident.

Aged mice (CD45.2+) were infected with PR8 and parabiosed with young naive (CD45.1+) mice at 5 weeks after infection. Four weeks later, CD45+ white blood cells were intravenously labeled in each host before sacrifice. (A) Schematic of experimental procedure. (B) Each animal was bled at time of sacrifice, and CD8+ T cells were examined by flow cytometery for origin (representative flow plot). (C) Representative flow plot of parenchymal Db-NP tetramer+ CD8+ T cells (CD69+CD8+ T cells protected from intravenous CD45 labeling) derived from host or donor. (D) Enumeration of Db-NP+ cells that are resident (left; TRM) or circulating (right). (E) Representative flow plot of parenchymal Db-PA tetramer+ CD8+ T cells (CD69+CD8+ T cells protected from intravenous CD45 labeling) derived from host or donor. (F) Enumeration of Db-PA+ cells that are resident (left; TRM) or circulating (right). (G to K) Young (CD45.1+) or aged mice were infected and parabiosed at 5 weeks after infection and examined 4 weeks later. (H) Each animal was bled at time of sacrifice and frequencies of CD8+ T cells from young (CD45.1+) or aged (CD45.1−) origin in each parabionts were examined. (I) Frequencies of splenic Db-NP− (top) or Db-PA–specific CD8+ T cells (bottom) from young or aged origin in each parabiont. (J) Frequencies of lung parenchymal Db-NP− or (K) Db-PA–specific CD69+ CD8+ T cells from young or aged origin in each parabionts. (B) to (F) were repeated twice with three pairs each and data were pooled. (G) to (K) were repeated twice with a total of five pairs and pooled. *P < 0.05 or not significant (ns) by Student’s two-tailed t test with unequal variance.
The parenchymal compartment of the aged infected host lungs had a significantly larger number of CD8+ CD69+ T cells than the uninfected parabiont, of which ~95% were from the aged parabiont, despite indistinguishable numbers of CD8+ T cells in both the lung vasculature and spleens between the young and aged parabionts (fig. S3, C and D). About 99% of total parenchymal CD8+ CD69+ Db-NP+ cells in the lungs of two parabionts were found in aged lungs. In contrast, there were comparable intravenous CD45+ CD8+Db-NP+ cells that were found in the lung circulation between young and aged parabionts (Fig. 4, C and D, and fig. S3E). Likewise, we observed similar patterns on the distribution of the Db-PA–specific CD8+ T cells in lung parenchyma and circulation (Fig. 4, E and F, and fig. S3F). There were no major differences in CD8+ splenic memory T cells between the two parabionts for the same antigen specificities (fig. S3, G and H).
It remains possible that the resident status of the CD8+ T cells in the aged host would be influenced if the parabiont had a prior influenza infection. To address this possibility, we joined aged mice with infection-matched young congenic (CD45.1+) mice 5 weeks after infection. Four weeks later, the circulating and parenchymal compartments were examined as above (Fig. 4G). Four weeks after parabiosis, we observed equilibration of total CD8+ T cells in the blood or lung circulation (Fig. 4H, and fig. S3, I and J). Furthermore, antigen-specific memory CD8+ T cells from each parabiont in the spleens were roughly equilibrated (Fig. 4I). These data suggest that the aged and young parabionts had a successful exchange of circulating memory CD8+ T cells. In contrast, CD69+ parenchymal total or antigen-specific CD8+ T cells from the aged host greatly outnumbered those from the young host in the aged lung tissue, and CD69+ parenchymal total or antigen-specific CD8+ T cells from the young host outnumbered those from the aged host in the young lung tissue (fig. S3, K to M). Furthermore, more than 95% of parenchymal Db-NP–specific memory T cells were derived from the host lung (Fig. 4J). In addition, the distribution of memory Db-PA–specific CD8+ T cells found in the parenchyma of each host was similar to Db-NP memory T cells (Fig. 4K). Together, these data indicate that circulating memory T cells contribute limitedly to the local memory CD8+ T cell pool and that lung parenchymal CD8+ CD69+ influenza-specific memory T cells observed in aged lungs are overwhelmingly tissue resident.
Enhanced TRM cell accumulation during aging is associated with increased environmental TGF-β expression
Resident memory CD8+ T cells in the lungs are relatively short lived after primary respiratory virus exposure (15, 31). Yet, in humans, more pulmonary-resident CD8+ T cells are seen in advanced age (32). It is now unclear whether this is the result of accumulation of memory T cells over a lifetime and/or a difference in the permissivity between young and aged tissue environments. We examined parenchymal CD8+ T cell numbers in influenza-naive aged and young hosts kept in specific pathogen–free environments (Fig. 5A). There was a 40-fold increase in parenchymal CD69+ CD8+ T cells in aged over young lungs (Fig. 5B), indicating that aged hosts may provide a niche for enhanced TRM accumulation in the tissue relative to young hosts. To further examine whether the excessive accumulation of TRM cells in aged lungs is due to the aged environment or intrinsic differences in T cell development between young and aged mice, we adoptively transferred CD8+ ovalbumin (OVA)–TCR transgenic OT-I T cells from young naive donors to young or aged hosts. We then infected the mice with recombinant influenza virus expressing cognate OVA peptide (SIINFEKL) (PR8-OVA) (33) and evaluated memory T cell responses at 7 weeks after infection (Fig. 5C). There were increased numbers of OT-I TRM cells present in aged lung tissue versus young (Fig. 5D), whereas equivalent numbers of lung-circulating or splenic memory OT-I cells were observed between young and aged mice (Fig. 5E and fig. S4). Together, these data indicated that the aged microenvironment facilitates lung TRM cell accumulation after influenza virus infection although the responding cells were from the same young donor.
Fig. 5. Age-associated CD8+ TRM cell accumulation is dependent on TGF-βR signaling.

(A and B) influenza-naive young and aged mice were intravenously labeled with CD45 Ab. (A) Representative flow cytometry plot showing frequency of circulating and parenchymal CD8+ T cells. (B) CD69+ CD8+ ivCD45− T cells were enumerated in influenza-naive (day 0) young and aged mice. (C to E) OT-i T cells (CD90.1+) from young donors were adoptively transferred into young or aged C57BL/6 mice 1 day before infection with PR8-OVA virus. (C) Schematic of experimental design. Donor OT-I cells were enumerated in young and aged hosts in the lung vasculature (Cir) or parenchyma (Par) (D) or in spleens (E) at 50 d.p.i. (F) Relative transcripts (relative to Hprt) of II-15, Tnf, and Tgfb1 (left to right) from total lung samples obtained from aged and young animals at 60 d.p.i. was evaluated by qPCR. (G and H) WT (CD45.1+) or TGFbR2fl/fl dLck-Cre OT-I cells (KO, CD45.1+) were adoptively transferred from young donors into separate young or aged hosts (CD45.2+) 1 day before PR8-OVA infection. Donor OT-I cells were enumerated in young and aged hosts in the lung vasculature (Cir) or parenchyma (Par) (G) or in spleens (H) at 50 d.p.i. (B) is an independent repeat of two experiments. (D) is pooled data of two independent experiments; (E) is an individual experiment. (F) is representative of two repeats. (G) and (H) are pooled from two repeats *P < 0.05 or not significant (ns) by Student’s two-tailed t test with unequal variance.
To explore the potential mechanisms regulating CD8+ TRM cell accumulation during aging, we performed quantitative reverse transcription polymerase chain reaction (qRT-PCR) analysis on infected lungs to examine the expression of Il15, Tnf, and Tgfb1, which are important in TRM cell development and/or maintenance (34-38). We found that Tgfb1 transcript levels were elevated in infected aged lungs compared with those of infected young lungs (Fig. 5F), which is consistent with previous reports that aged tissues express higher levels of TGF-β than young tissues (7, 8). Previously, TGF-βR signaling was shown to be critical for the development of both CD69+ CD103+ and CD69+ CD103− lung TRM cells (39). Therefore, we examined whether the excessive accumulation of CD8+ TRM cells in aged lungs is dependent on TGF-βR signaling. To this end, we adoptively transferred young wild-type (WT) or TGFβRII-deficient [TGFβRII knockout (KO), dlLck-Cre Tgfbr2fl/fl] OT-I cells into young or aged hosts and then infected the mice with PR8-OVA. TGFβRII deficiency not only diminished OT-I TRM development in young mice but also abolished the excessive accumulation of OT-I TRM in aged mice (Fig. 5G), whereas TGFβRII deficiency minimally affected splenic memory T cell responses in young or aged mice (Fig. 5H). Thus, these data suggest that excessive accumulation of CD8+ TRM cells in aged lungs is associated with enhanced TGF-β signals in aged tissue environment.
Age-associated TRM cells against a major protective epitope are dysfunctional
TRM cells are vital for heterologous influenza virus reinfection (40). Because aged lungs accumulated significantly more CD8+ TRM cells, it is plausible that aged mice may exhibit enhanced protective heterologous immunity against viral reinfection. To this end, we used a heterologous infection model in which we infected young or aged mice with PR8 virus and then rechallenged the mice with a lethal dose of X31 virus in the presence of FTY720, which blocks the contribution of protective circulating memory T cells (Fig. 6A) (13). PR8 and X31 viruses differ in viral surface proteins but share internal viral proteins such as NP, which are cross-recognized by memory CD8+ T cells (41, 42). We confirmed that FTY720 treatment abolished T cell migration (fig. S5A). We then followed host morbidity and mortality daily after X31 challenge. All naive young and aged mice succumbed to X31 infection (fig. S5B), whereas PR8-infected young mice were fully protected from lethal X31 infection in the presence of FTY720 (Fig. 6A). Seventy-five percent of PR8-infected aged animals succumbed to secondary X31 rechallenge, which was similar to mortality in primary X31 infection (Fig. 6A and fig. S5B). Thus, the increased density of CD8+ TRM cells in aged lungs failed to provide heterologous protection against influenza reinfection. Most previously infected mice survived reinfection in both young and aged groups in the absence of FTY720 treatment (fig. S5C), indicating that circulating memory CD8+ T cells can provide significant heterosubtypic protection in aged hosts. However, consistent with a previous study (9), aged mice had enhanced weight loss after a secondary viral challenge compared with those of young mice, suggesting that aged mice have impaired protective heterologous immunity in both the circulating and resident memory T cell compartments.
Fig. 6. Db-NP TRM cells from aged lungs are dysfunctional in protective immunity.

PR8 virus–infected young (filled triangles) or aged (open circles) mice were treated with FTY720 daily starting at 60 d.p.i. per Materials and Methods. Then, mice were infected with a lethal dose of X31 at 61 d.p.i. (A) Schematic of experimental procedure. Survival (middle) and weight loss (right) as percent of pre-secondary infection weight that were determined daily (d.p.2°i.). (B) Db-NP–specific TRM cells (CD8+CD44HiCD69+i.v.C D90−Db-NP tetramer+) from PR8-infected young (Y; n = 18) or aged (A; n = 11) mice that were pooled and sorted for scRNA-seq analysis. GSEA of NF-κB signaling (top) and inflammatory response (bottom) with associated NES and FDR. (C) Seurat UMAP plots of clusters 0 to 3 (left) separated by age (right) from scRNA-seq data. (D) Color-coded composition of aged or young TRM cell numbers per cluster from (C). (E) Cluster and age-differentiated heatmap of indicated genes. (F) UMAP feature plots for Bhlhe40, Junb, Tnf, and Ifng. (G) Representative flow cytometry plots of the production of IFN-γ and TNF in lung-resident CD8+ T cells after NP 366–374 peptide stimulation (1 ng/ml). (H) Percent of IFN-γ+–resident CD8+ T cells after stimulation with increasing amount of NP 366–374 peptide (0.01 to 10 ng/ml). (I) Geometric mean fluorescence intensity (gMFI) IFN-γ levels after stimulation with increasing amount of NP 366–374 peptide. (J) Percent of IFN-γ+ TNF+-resident CD8+ T cells after stimulation with increasing amount of NP 366–374 peptide (0.01 to 10 ng/ml). (K) Percent of IFN-γ+-resident CD8+ T cells after stimulation with increasing amount of PA 224–233 peptide (0.01 to 10 ng/ml). (L) WT OT-I were adoptively transferred into aged or young animals as per Fig. 3. Fifty d.p.i., percent of IFN-γ+ TNF+-resident (left) or splenic (right) OT-I cells after stimulation with increasing amount of SIINFEKL peptide (0.01 to 20 ng/ml). (A) was pooled data from two experiments. (G) to (K) are representatives of four experiments. (L) was pooled from two to three experiments. *P < 0.05 or not significant by Student’s two-tailed t test with unequal variance or log-rank (Mantel-Cox) test for survival curve comparison (A).
Among influenza-specific CD8+ T cells, Db-NP–specific T cells appear to be the dominant protective epitope against secondary heterologous viruses (43). To examine the mechanisms underlying the impaired protective function of TRM cells in aged lungs, we sorted polyclonal Db-NP–specific TRM from infected young or aged lungs and performed scRNA-seq at 60 d.p.i. (fig. S5D). Compared with Db-NP TRM cells from aged mice, Db-NP TRM from young mice were enriched in gene sets associated with nuclear factor κB (NF-κB) signaling and inflammatory immune responses (Fig. 6B). We performed shared nearest neighbor clustering algorithm and visualized TRM cells in two-dimensional (2D) space with uniform manifold approximation and projection (UMAP).
We found that polyclonal Db-NP TRM cells from young and aged mice can be divided into four clusters (Fig. 6C and fig. S5E). Cluster 0 cells were mainly composed of TRM cells from young mice, and clusters 2 and 3 cells consisted mainly of TRM cells from aged mice, whereas cluster 1 cells were equally divided by TRM cells from young and aged mice (Fig. 6, C and D). Compared with the rest of the clusters, cells in cluster 0 express high levels of genes associated with classical TRM signature (such as Bhleh40, Cd69, and Jun), molecules associated with stronger TCR signaling (such as Lck, Cd3e, and Cd28), and genes associated with CD8+ T cell effector function (such as Ccl4 and Tnf) (Fig. 6, E and F, and fig. S5, E and F). Thus, age-associated lung TRM cells lose a functional subpopulation that is characterized with robust expression of CD8+ T cell effector and functional molecules, which likely explain the lack of protection against secondary viral rechallenge.
To further test the capacity of TRM cells to elicit a protective response, we stimulated lung cells from infected young or aged mice with NP 366–374 peptide and then measured interferon-γ (IFN-γ) and tumor necrosis factor (TNF) production by CD8+ TRM cells through intracellular staining. Consistent with the scRNA-seq data, Db-NP–specific TRM cells from aged mice exhibited diminished production of IFN-γ compared with young mice, particularly when IFN-γ levels were evaluated on a per cell basis (Fig. 6, G to I). Moreover, Db-NP CD8+ TRM cells from aged lungs were defective in simultaneously producing both IFN-γ and TNF after peptide restimulation (Fig. 6J). To exclude the possibility that the diminished cytokine production by aged Db-NP CD8+ TRM cells was due to the impaired lung antigen-presenting cell (APC) function in the aged lungs during in vitro restimulation, we labeled infected young lung cells with ef670 dye then mixed the labeled young lung cells with unlabeled aged lung cells (1:1) and measured IFN-γ and TNF production by young and aged TRM cells in the same sample. We found that the aged TRM cells were still dysfunctional for production of TNF and IFN-γ in response to NP peptide stimulation relative to young TRM cells despite the same APCs present in the culture (fig. S5G).
In contrast to their diminished cytokine production after peptide stimulation (TCR signaling), total CD8+ or Db-NP–specific TRM cells were capable of coproducing IFN-γ and TNF after phorbol 12-myristate 13-acetate (PMA)/ionomycin restimulation (fig. S5, H and I). These data suggest that Db-NP TRM cells exhibit impaired production of effector molecules specifically after TCR stimulation. After ex vivo stimulation of Db-PA–specific TRM cells by PA 224–233 peptide, we found that Db-PA TRM cells from aged and young hosts had similar ability to produce IFN-γ and TNF (Fig. 6K). Collectively, these data suggest that the impaired production of effector molecules by Db-NP–specific TRM cells is likely not due to the aged environment but their developmental intrinsic defects as demonstrated before (28). Consistent with this idea, young OT-I TRM cells in aged hosts did not exhibit functional defects in TCR-mediated cytokine production compared with young hosts (Fig. 6L and fig. S5J).
Resident CD8+ T cells in aged hosts drive persistent pulmonary inflammation and lung fibrosis
Excessive accumulation of TRM cells after programmed death-ligand 1 (PD-L1) blockade in influenza-infected mice leads to persistent inflammatory and fibrotic responses in the lungs at the memory stage (39). Because aged lungs harbored enhanced TRM cells and persistent lung pathology, we hypothesized that excessive accumulation of CD8+ TRM cells in aged lungs may cause persistent inflammation and fibrotic sequela. To test this hypothesis, we infected aged mice with PR8 virus and then injected the mice with high or low doses of CD8-depleting Ab (α-CD8) at 21 d.p.i. to deplete CD8+ T cells systemically in lymphoid and peripheral tissues (high dose) or only in lymphoid organs and the blood (low dose) (Fig. 7A) (38, 44, 45). Both low- and high-dose CD8 Ab treatment largely ablated splenic CD8+ T cells; however, only high-dose CD8 Ab injection caused significant ablation of CD8+ T cells in the lung parenchyma (Fig. 7B and fig. S6A). Neither high nor low dose of CD8 Ab treatment caused significant reduction of CD4+ T or B cell compartments in the lungs (fig. S6B). Lung histopathological analysis revealed marked reduction of inflammatory lesions after high dose but not low dose of CD8 Ab treatment particularly in aged mice after infection (Fig. 7, C to E), indicating that lung-resident CD8+ T cells drive chronic lung pathology at the memory phase. Consistent with the histology data, high dose, but not low dose, of CD8 Ab treatment caused significant reduction of Ly6C+ monocyte and neutrophil infiltration in aged but not young lung tissues (Fig. 7F). Thus, resident CD8+ T cells are responsible for the constant recruitment of monocytes and neutrophils to the tissue at the memory phase. To further explore the roles of resident CD8+ T cells in promoting tissue inflammation at the memory stage, we measured immune-associated genes in the lungs by NanoString. There was a marked decrease in the expression of multiple cytokines and particular chemokines in the lungs of mice that received high CD8 Ab treatment compared with those that received control Ab or low dose of CD8 Ab at 60 d.p.i. (Fig. 7G and fig. S6, C and D), likely explaining the diminished monocyte and neutrophil infiltration in the tissue after resident CD8+ T cell depletion.
Fig. 7. Resident CD8+ T cells support chronic parenchymal inflammation and fibrosis in aged hosts.

Aged or young WT mice were Infected with PR8 virus. (A) Experimental procedure of high (500 μg) or low (20 μg) dose of CD8 Ab (αCD8) treatment starting at 21 d.p.i. (B) Lung-resident CD8+ T cell numbers in aged (left) or young (right) mice after CD8 Ab treatment. (C and D) H&E staining of whole left lung lobes from young (C) or aged mice (D) treated with indicated Abs. (E) Percent of left lung parenchyma infiltrated by white blood cells via ImageJ analysis in aged mice from (D). (F) Quantitation of resident monocytes (Ly6CHi; ivCD45− CD64− CD11c>Lo Siglec-F− CD11bHi Ly6CHi) and neutrophils (Neu; ivCD45− Ly6GHi CD11bHi) in IgG or high-dose CD8-depleted young (left) or aged (right) mice treated with indicated Ab. (G) Heatmap for select DEGs of the lungs from aged mice treated with IgG (in triplicate), high dose (αCD8Hi; in triplicate), or low dose (αCD8Lo; pooled n = 3) of CD8 Ab as analyzed by NanoString. Orange is relatively high expression and purple is relatively low expression. (H) Representative 200× micrographs of Masson’s trichromatic stain on young lung and sections at 60 d.p.i. after treating with IgG or high-dose CD8-depleting Ab (αCD8Hi). (I) Hydroxyproline (reflective of collagen content) assay on young lung samples treated as in (H). (J) Masson’s trichromatic stain or (K) hydroxyproline assay on aged lung samples from mice treated with IgG, low (αCD8Lo), or high (αCD8Hi) dose of CD8-depleting Ab. Scale bars, 600 μm (C and D) and 100 μm for (H and J). (C), (D), (H), and (J) were representative of at least two experiments. (B), (F), (I), and (K) were two pooled experiments. (E) and (G) were single replicates. *P < 0.05, **P < 0.005, or not significant by Student’s two-tailed t test with unequal variance (I) or for ANOVA with correction for multiple tests (B, E, F, and K).
Acute influenza virus infection can cause persistent lung remodeling and collagen deposition (25). We, therefore, examined whether CD8+ T cell depletion at 3 weeks after infection could affect lung fibrosis at the memory stage. CD8+ T cell depletion did not affect lung collagen content in infected young mice revealed by both Masson’s trichrome staining and hydroxyproline assay (Fig. 7, H and I), which measures total collagen protein (46). These data suggest that normal CD8+ TRM cell responses in young mice contribute minimally to lung fibrotic responses after acute influenza virus infection, which is in contrast to the excessive CD8+ TRM cell–induced fibrotic sequelae after PD-L1 blockade (39). In contrast to young mice, aged mice exhibited increased lung collagen deposition, particularly in the lung parenchyma (Fig. 7, H to K). Mice that received high CD8 Ab treatment had markedly reduced lung fibrosis as revealed by trichrome staining and hydroxyproline assay compared with lungs of mice that received control Ab or low dose of CD8 Ab (Fig. 7, J and K). We previously reported that PD-1 expression in resident CD8+ T cells suppressed TRM cell fibrogenic potential (39). Examination of PD-1 expression on CD8+ T cells in young and aged lungs indicated higher expression of PD-1 in aged CD8+ T cells in both the circulation and CD69+ parenchymal compartments (fig. S6E). Although Db-PA–specific resident memory T cells also had higher PD-1 expression in aged lungs, Db-NP specific did not (fig. S6F). Thus, the development of age-associated lung fibrosis is not due to the lack of PD-1 on CD8+ TRM cells.
Nevertheless, our data suggest that resident but not circulating CD8+ T cells are important in promoting chronic lung collagen deposition after the resolution of primary influenza infection. In summary, our data indicate that excessive accumulation of lung-resident CD8+ T cells does not lead to enhanced protective immunity in aged mice but, rather, drives age-associated persistent lung inflammation and chronic fibrosis after viral pneumonia.
DISCUSSION
Here, we demonstrate that acute influenza infection leads to persistent chronic pulmonary inflammation and fibrosis, which are largely propagated by an exaggerated CD8+ TRM response in aged hosts. Somewhat counterintuitively, elevated CD8+ TRM cells in the aged lung fail to provide the protective heterologous immunity observed in young mice, likely because of a selective dysfunction in a protective CD8+ TRM cell population (i.e., Db-NP CD8+ T cells) (47, 48).
Previous reports suggested that both T cell–intrinsic and –extrinsic environmental factors could contribute to the suboptimal effector and memory CD8+ T cell responses during aging (9, 49, 50). To our surprise, we observed enhanced TRM cell accumulation, despite diminished lung effector T cell responses and lower levels of splenic memory T cells observed in aged hosts. Our data further indicate that the aged environment, likely through elevated tissue TGF-β, promotes TRM cell generation and/or maintenance in a T cell–extrinsic fashion during aging. Of note, aging has been linked to altered naive T cell repertoires and enhanced representation of senescent virtual memory T cells (51). Whether these T cell–intrinsic changes in aged hosts, in addition to environmental factors, contribute to an enhanced TRM generation requires further investigation. Despite a numeric increase in TRM cells, protective heterologous immunity is impaired in aged hosts because of a qualitative defect in TRM cells not present in young mice. The protective function of TRM cells is usually associated with their effector activities at the site of pathogen reentry (52). For instance, it has been shown that IFN-γ is important for TRM cell protective immunity against secondary heterologous influenza virus infection (40). Consistent with this notion, scRNA-seq analysis found that TRM cells against a major protective CD8 epitope (Db-NP) from aged mice lose a functional TRM subset observed in young hosts; this subset was characterized by robust expression of molecules associated with TCR signaling and effector molecules. As a result, Db-NP–specific TRM cells are less sensitive to TCR stimulation for the simultaneous production of IFN-γ and TNF. Thus, defective TCR-mediated effector cytokine production could underlie the impaired protection against secondary influenza virus infection. TRM cells can undergo in situ proliferation and generate secondary effector T cells (39, 53, 54). Previously, it has been shown that memory T cells in aged mice are senescent in proliferation during secondary expansion (9). Therefore, it is also possible that impaired TRM proliferation and secondary effector T cell expansion could, together, contribute to the impaired TRM protective function.
Impaired effector cytokine production by Db-NP TRM cells was not observed in Db-PA or transferred young OT-I TRM cells, suggesting that this functional defect in Db-NP T cells is likely not caused by the aged environment. It has been reported previously that influenza-specific Db-NP, but not Db-PA, T cells exhibited altered TCR-β usage during aging (7, 28, 55). Further, the narrowed TCR repertoire specifically in Db-NP T cells correlated with the age-associated loss in the responsiveness in expansion during infection (28). On the basis of these previous data and our data presented here, it is reasonable that there is a developmental defect in Db-NP, but not Db-PA, CD8+ T cells during aging, which leads to decreased responsiveness of Db-NP–specific CD8+ T cells to TCR simulation but not PMA-ionomycin stimulation. Thus, intrinsic developmental defects are likely the cause of the functional decline in Db-NP TRM cells (28). Our data highlight the importance of Db-NP TRM cells in protective immunity as functional impairment specifically in Db-NP cells that could translate into differences in the secondary protective immunity. This notion is consistent with previous reports, which have pinpointed the critical function of Db-NP TRM cells in the protection against heterologous influenza reinfection and the robust expansion of Db-NP memory T cells over Db-PA memory T cells in the secondary effector responses after heterologous viral challenge (39, 43, 56).
In experimental models, both CD4+ and CD8+ TRM cells have been implicated in causing tissue immunopathology ranging from persistent allergic inflammation to inflammatory bowel disease (57, 58). Previous reports have demonstrated that influenza infection in young mice leads to long-lasting inflammation and collagen deposition (25). Resident CD8+ T cell depletion only moderately affected pulmonary inflammation and lung collagen deposition in young mice, although exaggerated CD8+ TRM responses after PD-1 blockade can lead to inflammatory and fibrotic sequelae (39). These data suggest that influenza-infected young lungs are able to tolerate “normal levels” of TRM cells, likely due to the expression of various inhibitory molecules expressed by TRM cells (39). Although influenza infection during infancy leads to poor viral-specific TRM cell accumulation (59), during aging, influenza virus infection leads to excessive accumulation of TRM cells, which exceeds the ability of the aged tissue to “counterbalance” the pathogenic activities of TRM cells, driving enhanced pulmonary inflammation and parenchymal fibrosis. The cellular and molecular mechanisms underlying TRM-mediated lung pathology and fibrosis during aging are unclear currently. However, aging is associated with the impaired capability of tissue repair (60). It is possible that epithelial injury caused by persistent low-level release of cytopathic molecules by TRM cells surpasses the limited recovery capability of the aged tissue. To this end, although Db-NP TRM cells have diminished TCR signaling for IFN-γ and TNF production, their responses to PMA and ionomycin are intact. Therefore, they may remain capable of producing low levels of effector molecules in response to environmental inflammatory factors that are constitutively observed in aged tissues (23, 60). Alternatively, it is possible that enhanced accumulation of those TRM cells that exhibit no defects for effector molecule expression after TCR stimulation, such as Db-PA TRM cells, may be responsible for the persistent tissue inflammation and fibrosis in aged hosts. In addition, TRM cells in aged mice may have different traits than TRM cells in young mice (61, 62), thereby causing increased lung inflammation and tissue fibrosis.
Aging is associated with enhanced development of a number of chronic lung diseases including chronic obstructive pulmonary diseases and pulmonary fibrosis (63). Aging is also associated with increased susceptibility of acute lung injury and severe disease development after respiratory viral infections such as influenza virus and SARS-CoV-2 (18, 64, 65). Emerging evidence has also suggested that COVID-19 survivors will likely exhibit persistent impairment of lung function and the development of pulmonary fibrosis (22, 66-70). Our data here provide a potential mechanistic explanation and point out future directions for therapeutics to those aged patients with COVID-19 that would suffer persistent impairment of lung function. To this end, recent data on scRNA-seq analyses have suggested that pulmonary T cells from patients with COVID-19 are enriched with TRM signature and CD8+ T cell clonal expansion in the airways (71). Thus, strategies aimed at decreasing exuberant TRM responses and/or inhibiting the pathogenic potential of TRM cells may be promising in treating chronic lung diseases after viral pneumonia in the elderly, including COVID-19 survivors.
In summary, we have unveiled an age-associated paradox in CD8+ TRM cell responses. The excessive accumulation of TRM cells is not protective but rather drives inflammatory and fibrotic sequelae after primary respiratory viral infection. Moving forward, it is crucial to better understand the mechanisms of age-associated TRM cell malfunction so that we can selectively restore TRM cell protective function while minimizing their pathogenic potential in the elderly.
MATERIALS AND METHODS
Study design
The aim of the study was to determine the protective and/or pathogenic function of CD8+ TRM cells in aged hosts after respiratory viral infection. We conducted a kinetic analysis on tissue pathology and lung CD8+ T cell responses after influenza virus infection in young or aged mice. Adoptive transfer of WT or TGFβRII-deficient T cells into aged animals was used to determine the roles of environmental TGF-β in CD8+ TRM cell accumulation in aged hosts. A heterotypic influenza virus reinfection model was used to examine the protective function of CD8+ TRM cells in aged hosts. We performed scRNA-seq and intracellular cytokine staining to evaluate the protective potential of CD8+ TRM cells. Last, we conducted histopathology, NanoString, and hydroxyproline analysis to determine lung inflammatory and fibrotic responses after CD8+ TRM cell depletion. Experiments were conducted in replicates as indicated in figure legends. No outliers were removed.
Mice, infections, and adoptive transfers
Female C57BL/6 were originally purchased from the Jackson Laboratory (Harbor, ME) and mostly bred in house. Female aged mice were received at 20 to 21 months of age from the National Institutes of Aging and maintained in the same specific pathogen-free conditions for at least 1 month before infection. In most of cases, aged and young subject bedding was cross-contaminated weekly to control for different microbial environments. Young OT-I and dLck-Cre Tgfbr2fl/fl OT-I lymphoid tissues (KO, CD45.1+) were shipped from N.Z. (University of Texas, Health Science Center at San Antonio). All mice were housed in a specific pathogen–free environment and used under conditions fully reviewed and approved by the institutional animal care and use committee guidelines at the Mayo Clinic (Rochester, MN). For primary influenza virus infection, influenza A/PR8/34 strain [~100 plaque-forming units (pfu) per mouse] was diluted in fetal bovine serum (FBS)–free Dulbecco’s modified Eagle’s medium (DMEM) (Corning) on ice and inoculated in anesthetized mice through intranasal route as described before (72). X-31 strain was prepared identically, and 2.8 × 105 pfu was administered per mouse for secondary challenge. During the secondary challenge phase, experimental mice were treated with FTY720 (1 μg/g) daily starting 1 day before rechallenge. For adoptive transfers, 1 × 104 OT-I cells from lymph nodes of 8- to 10-week-old females were transferred intravenously, and mice were infected with PR8-SIINFEKL (33) (150 pfu) 1 day later.
Cell depletions
For depletion of CD8+ T cells, starting at 21 d.p.i., mice were given 20 μg (low dose) or 500 μg of anti-CD8 Ab (53 to 6.7) or control rat immunoglobulin G (IgG) (500 μg) in 200 μl of phosphate-buffered saline (PBS) through once a week of intraperitoneal injection. Mice were sacrificed 3 days after the last treatment.
Parabiosis surgery
To examine tissue residency of lung CD69+ parenchymal CD8+ T cells, parabiotic surgery was performed. CD45.1+ young naive or CD45.1+ influenza-infected mice (at 35 d.p.i.) were paired with infected aged CD45.2+ congenic mice (also at 35 d.p.i.). Briefly, mice were anesthetized with ketamine and xylazine. After disinfection, incisions were made in the shaved skin area, and then the olecranon of the knee joints of each mouse was joined. Opposing incisions were closed with a continuous suture on the dorsal and ventral sides. Parabionts were then allowed to rest for 4 weeks before sacrifice and examining the lung vasculature, parenchyma, and spleen immunoprofiles. Equilibration of white blood cells between parabionts was confirmed in the peripheral blood before tissue analysis by flow cytometry.
Tissue processing, cellular isolation, stimulations, and plaque assays
Animals were injected intravenously with 4 μg of CD45 or 2 μg of CD90.2 Ab labeled with various fluorochromes. Two minutes after injection, animals were euthanized with an overdose of ketamine/ xylazine and processed 3 min later. After euthanasia, spleens were removed, and the right ventrical of the heart was gently perfused with PBS (10 ml). Lungs were instilled with either 1 ml of 10% formalin for histology studies [hematoxylin and eosin (H&E) or Masson’s trichrome stains performed by the Mayo Clinic Pathology Research core] or 1 ml of digestion buffer [90% DMEM and 10% PBS and calcium with type 2 collagenase (180 U/ml) (Worthington) and DNase (15 μg/ml) (Sigma-Aldrich) additives]. Tissue was processed on a gentleMACS tissue disrupter (Miltenyi) for 40 min at 37°C followed by hypotonic lysis of red blood cells in ammonium-chloride-potassium buffer and filtering through a 70-μm mesh. FC-γ receptors were blocked with anti-CD16/32 (2.4G2). Cells were washed twice in PBS and stained with near-infrared Zombie viability dye per the manufacturers protocol (BioLegend). Cell surfaces were immunostained with the following cocktails of fluorochrome-conjugated Abs (BioLegend). Immunostaining was performed at 4°C for 30 min. Ab clones are as follows: Siglec-F brilliant violet 421 (E50-2440), CD11c (N418), CD11b (M1/70), CD64-R-phycoerythrin (X54-4/7.1), Ly6G brilliant violet 711 (1A8), I-A/I-E-allophycocyanin (M5/114.15.2), Ly6C (HK1.4), CD8α (53-6.7), CD69 brilliant violet 421 or FITC (H1.2F3), CD44 (IM7), CD103 (2E7), and PD-1 brilliant violet 711 (29F.1A12). For above clones where not noted otherwise, fluorophores varied between experiments. Stains with H2Db tetramers for PA224-233 (allophycocyanin) or NP366-374 (R-phycoerythrin) were incubated for 1 hour with above surface stains. Cells were washed twice with fluorescence-activated cell sorting buffer (PBS, 2 mM EDTA, 2% FBS, and 0.09% sodium azide) before fixation and ran on an Attune NxT autosampler (Life Technologies). Fluorescence correlation spectroscopy files were analyzed with FlowJo 10.0 (Tree Star). For functional assays, following digestion protocols above, cells were resuspended at 3 × 106/ml in RPMI with 10% fetal calf serum and stimulated with an indicated amount of peptide or PMA (100 ng/ml) and ionomycin (1 μg/ml) for 3 hours before adding monensin (2 nM) for the last 2 hours of culture. For mixed young and aged cell cultures, young lung cells were labeled with 1 μM ef670 dye (eBioscience) and then 1:1 mixed with unlabeled aged lung cells. Peptide stimulation and cytokine staining were performed as above. Plaque assays were carried out as previously described from bronchial alveolar lavage samples (73).
Hydroxyproline assay
Fifty milligrams of lung tissue was hydrolyzed in 1 ml of 6 M HCl at 95°C overnight. Hydroxyproline standard solution was purchased from Sigma-Aldrich. Hydroxyproline concentration is determined by the reaction of oxidized hydroxyproline with 4-(dimethylamino) benzaldehyde. The product was read at 560-nm wavelength in a Thermax plate reader as described (39, 74).
Quantification of histopathology
H&E-stained formalin-fixed lung sections were scanned using an Aperio image scanner (Leica). Tiff files were converted to 16-bit black and white, and two threshold measurements were taken using the ImageJ software [National Institutes of Health (NIH)]. One measured the total parenchymal area and the other the inflamed area of the tissue. The caveat of the method is that bronchiole epithelium is thresholded in both measurements. To account for this, values from a group of age-matched naive mice were subtracted from the above ratios to yield percent of inflamed or infiltrated parenchyma.
Quantitative RT-PCR
Total RNA was extracted from lung tissue from TRIzol preparations. Random primers (Invitrogen) and MMLV reverse transcriptase (Invitrogen) were used to synthesize first-strand complementary DNAs (cDNAs) from equivalent amounts of RNA from each sample. These cDNA were subjected to real-time PCR with Fast SYBR Green PCR Master Mix (Applied Biosystems). Real-time PCR was conducted on QuantStudio 3 (Applied Bioscience). Data were generated with the comparative threshold cycle (ΔCt) method by normalizing to hypoxanthine-guanine phosphoribosyltransferase transcripts in each sample as reported previously (73).
NanoString analysis
Total RNA was extracted from bulk lung samples in TRIzol at 0 or 60 d.p.i. One hundred nanograms of RNA were either pooled between groups or used from individual samples as indicated. Library hybridization was established by following the instructions of the manufacturer. Aliquots of Reporter CodeSet and Capture ProbeSet were thawed at room temperature. Then, a master mix was created by adding 70 μl of hybridization buffer to the tube containing the Reporter CodeSet. Eight microliters of this master mix was added to each of the tubes for different samples; 5 μl of the total RNA sample was added into each tube. Then, 2 μl of the well-mixed Capture ProbeSet was added to each tube and placed in the preheated 65°C thermal cycler. All the sample mixes were incubated for 18 hours at 65°C for completion of hybridization. The samples were then loaded into the cartridge and loaded into the NanoString nCounter SPRINT Profiler machine (NanoString). When the corresponding Reporter Library File running was finished, the raw data were downloaded and analyzed with the NanoString Software nSolver 3.0 (NanoString). mRNA counts were processed to account for hybridization efficiency, background noise, and sample content and were normalized using the geometric mean of housekeeping genes. Heatmaps were generated by an assortment of R language packages using log2 transformed ratios of greater than 1.5-fold differences between pooled samples. For fig. S1 (A to C), gene groups were manually selected from NanoString data that represented surface molecules associated with myeloid or lymphoid cells or mRNA for intracellular cytokines and chemokines.
Single-cell RNA sequencing
Sorted (CD8+CD44HiCD69+i.v.CD90−Db-NP tetramer PE/APC-double fluorochrome positive) T cells from pooled lung cells of mice (10 to 18 mice) infected with influenza virus (60 d.p.i.) were loaded on the Chromium Controller (10x Genomics). Single-cell libraries were prepared using the Chromium Single Cell 3′ Reagent Kit (10x Genomics) following the manufacturer’s instruction. Paired-end sequencing was performed, and 10x Genomics Cell Ranger Single Cell Software Suite (v2.0.2) was used to demultiplex raw base call files into FASTQ files, align reads to the reference genome (mm10), filter, and generate barcode and unique molecular identifier (UMI) counts. All scRNA-seq analyses were performed in R using the Seurat package (version 3.0). Cells that had 200 to 4500 genes, which included less than 5% mitochondrial genes, were included in further analysis with Seurat. The same number of cells from each group was used for log normalization, followed by identifying DEGs using variance-stabilizing transformation (vst). The processed data were scaled, and the heterogeneity introduced by mitochondria contamination was regressed out. Clustering was performed using the shared nearest neighbor clustering algorithm with the Louvain method for modularity optimization. UMAP dimensional reduction was performed to visualize data in 2D space. DEGs between clusters were determined using a Wilcoxon rank sum test.
Total RNA-sequencing
RNA was extracted from bulk lung samples with TRIzol. After quality control, high-quality [Agilent Bioanalyzer, RIN (RNA integrity number) >7.0] total RNA was used to generate the RNA-seq library. cDNA synthesis, end repair, A-base addition, and ligation of the Illumina indexed adapters were performed according to the TruSeq RNA Sample Prep Kit v2 (Illumina). The concentration and size distribution of the completed libraries were determined using an Agilent Bioanalyzer DNA 1000 chip and Qubit fluorometry (Invitrogen). Paired-end libraries were sequenced on an Illumina HiSeq 4000 following Illumina’s standard protocol using the Illumina cBot and HiSeq 3000/4000 PE Cluster Kit. Base calling was performed using Illumina’s RTA software (version 2.5.2). Paired-end RNA-seq reads were aligned to the mouse reference genome (GRCm38/mm10) using RNA-seq spliced read mapper TopHat2 (version 2.1.1). Pre- and post-alignment quality controls, gene level raw read count, and normalized read count [i.e., fragments per kilobase million (FPKM)] were performed using RSeQC package (version 2.3.6) with National Center for Biotechnology Information mouse RefSeq gene model. For functional analysis, GSEA was used to identify enriched gene sets, from the Hallmark and C5 databases of MSigDB, having up- and down-regulated genes, using a weighted enrichment statistic and a log2 ratio metric for ranking genes. DEGs were considered to have a 1.5-fold change with a false discovery rate (FDR) of <0.01.
Statistical analysis
Quantitative data are presented as means ± SD. Unpaired two-tailed Student’s t test (two-tailed, unequal variance) was used to determine statistical significance with Prism software (GraphPad). Where appropriate, analysis of variance (ANOVA) corrected for multiple comparisons was used (GraphPad). Log-rank (Mantel-Cox) test was used for survival curve comparison. We considered P < 0.05 as significant in all statistical tests and denoted within figures as a * for each order of magnitude P value.
Data and materials availability:
The data for this study have been deposited in the GEO database (GSE159065 and GSE159066). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials.
Supplementary Material
Fig. S1. Aged lungs exhibit persistent inflammatory responses after influenza infection.
Fig. S2. Enhanced presence of adaptive immune cells in aged lung parenchyma.
Fig. S3. Aged parenchymal CD8+ CD69+ memory T cells are tissue resident.
Fig. S4. The accumulation of CD8+ TRM in aged lungs is dependent on TGF-βR signaling.
Fig. S5. scRNA-seq on Db-NP TRM from young or aged lungs.
Fig. S6. High dose of CD8 Ab treatment diminishes lung inflammatory responses.
Acknowledgments:
We thank the NIH tetramer core for tetramers and the Mayo flow cytometry core and genomic core for technical assistance.
Funding: This study was funded by the U.S. National Institutes of Health grants AI112844, AI147394, AG069264, and AG047156 to J.S.; NS103212 to A.J.J.; AI125701 and AI139721 to N.Z.; HL52160 to A.H.L.; and T32AG04967 to N.P.G.; Mayo Clinic CBD Research Fund to J.S., E.N.C., and R.V.; Cancer Research Institute CLIP program and American Cancer Society grant RSG-18-222-01-LIB to N.Z.; and Three Lakes Partners Foundation to A.H.L.
Footnotes
Competing interests: R.V. receives funding from Pfizer, Bristol-Myers-Squibb, and Sun Pharmaceuticals. J.S. laboratory receives funding from Evive Biotech. E.N.C. is a shareholder and consultant for TeneoBio. E.N.C. is also a consultant for Calico, Mitobridge, Cytokinetics, and Nestle Health. E.N.C. has sponsor research agreements with Calico. E.N.C. is an inventor on patent application 15/590,107 held/submitted by Mayo Clinic and Harvard that covers the use of CD38 inhibitors for age-realted diseases and metabolic syndromes.
REFERENCES AND NOTES
- 1.Scardamalia R, Aging in America, County and city extra series (Bernan Press, 2014), 416 p. [Google Scholar]
- 2.Thome JJC, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, Farber DL, Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159, 814–828 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akondy RS, Fitch M, Edupuganti S, Yang S, Kissick HT, Li KW, Youngblood BA, Abdelsamed HA, Mc Guire DJ, Cohen KW, Alexe G, Nagar S, McCausland MM, Gupta S, Tata P, Haining WN, McElrath MJ, Zhang D, Hu B, Greenleaf WJ, Goronzy JJ, Mulligan MJ, Hellerstein M, Ahmed R, Origin and differentiation of human memory CD8 T cells after vaccination. Nature 552, 362–367 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goronzy JJ, Fang F, Cavanagh MM, Qi Q, Weyand CM, Naive T cell maintenance and function in human aging. J. Immunol 194, 4073–4080 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sant S, Grzelak L, Wang Z, Pizzolla A, Koutsakos M, Crowe J, Loudovaris T, Mannering SI, Westall GP, Wakim LM, Rossjohn J, Gras S, Richards M, Xu J, Thomas PG, Loh L, Nguyen THO, Kedzierska K, Single-cell approach to influenza-specific CD8+ T cell receptor repertoires across different age groups, tissues, and following influenza virus infection. Front. Immunol 9, 1453 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Po JLZ, Gardner EM, Anaraki F, Katsikis PD, Murasko DM, Age-associated decrease in virus-specific CD8+ T lymphocytes during primary influenza infection. Mech. Ageing Dev 123, 1167–1181 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Valkenburg SA, Venturi V, Dang THY, Bird NL, Doherty PC, Turner SJ, Davenport MP, Kedzierska K, Early priming minimizes the age-related immune compromise of CD8+ T cell diversity and function. PLOS Pathog. 8, e1002544 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nikolich-Žugich J, The twilight of immunity: Emerging concepts in aging of the immune system. Nat. Immunol 19, 10–19 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Decman V, Laidlaw BJ, Dimenna LJ, Abdulla S, Mozdzanowska K, Erikson J, Ertl CJ, Wherry EJ, Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J. Immunol 184, 5151–5159 (2010). [DOI] [PubMed] [Google Scholar]
- 10.Davies B, Prier JE, Jones CM, Gebhardt T, Carbone FR, Mackay LK, Cutting Edge: Tissue-resident memory T cells generated by multiple immunizations or localized deposition provide enhanced immunity. J. Immunol 198, 2233–2237 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Steinert EM, Schenkel JM, Fraser KA, Beura LK, Manlove LS, Igyártó BZ, Southern PJ, Masopust D, Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161, 737–749 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Z, Kedzierski L, Nuessing S, Chua BY, Quiñones-Parra SM, Huber VC, Jackson DC, Thomas PG, Kedzierska K, Establishment of memory CD8+ T cells with live attenuated influenza virus across different vaccination doses. J. Gen. Virol 97, 3205–3214 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Wu T, Hu Y, Lee Y-T, Bouchard KR, Benechet A, Khanna K, Cauley LS, Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol 95, 215–224 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Zhu B, Son YM, Wang Z, Jiang L, Xiang M, Ye Z, Beckermann KE, Wu Y, Jenkins JW, Siska PJ, Vincent BG, Prakash YS, Peikert T, Edelson BT, Taneja R, Kaplan MH, Rathmell JC, Dong H, Hitosugi T, Sun J, The transcription factor bhlhe40 programs mitochondrial regulation of resident CD8+ T cell fitness and functionality. Immunity 51, 491–507.e7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayward SL, Scharer CD, Cartwright EK, Takamura S, Li Z-RT, Boss JM, Kohlmeier JE, Environmental cues regulate epigenetic reprogramming of airway-resident memory CD8+ T cells. Nat. Immunol 21, 309–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruf BR, Knuf M, The burden of seasonal and pandemic influenza in infants and children. Eur. J. Pediatr 173, 265–276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rolfes MA, Foppa IM, Garg S, Flannery B, Brammer L, Singleton JA, Burns E, Jernigan, Olsen SJ, Bresee J, Reed C, Annual estimates of the burden of seasonal influenza in the United States: A tool for strengthening influenza surveillance and preparedness. Influenza Other Respi. Viruses 12, 132–137 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W; China Novel Coronavirus Investigating and Research Team, A novel coronavirus from patients with pneumonia in china, 2019. N. Engl. J. Med 382, 727–733 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casas-Aparicio GA, León-Rodríguez I, de Jesús Hernández-Zenteno R, Castillejos-López M, la Barrera C. A.-d., Ormsby CE, Reyes-Terán G, Aggressive fluid accumulation is associated with acute kidney injury and mortality in a cohort of patients with severe pneumonia caused by influenza A H1N1 virus. PLOS ONE 13, e0192592 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keilich SR, Bartley JM, Haynes L, Diminished immune responses with aging predispose older adults to common and uncommon influenza complications. Cell. Immunol 345, 103992 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, Wu J, Hao S, Yang M, Lu X, Chen X, Li L, Long term outcomes in survivors of epidemic Influenza A (H7N9) virus infection. Sci. Rep 7, 17275 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hosseiny M, Kooraki S, Gholamrezanezhad A, Reddy S, Myers L, Radiology perspective of coronavirus disease 2019 (COVID-19): Lessons from severe acute respiratory syndrome and Middle East respiratory syndrome. AJR Am. J. Roentgenol 214, 1078–1082 (2020). [DOI] [PubMed] [Google Scholar]
- 23.Kulkarni U, Zemans RL, Smith CA, Wood SC, Deng JC, Goldstein DR, Excessive neutrophil levels in the lung underlie the age-associated increase in influenza mortality. Mucosal Immunol. 12, 545–554 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lanzer KG, Cookenham T, Reiley WW, Blackman MA, Virtual memory cells make a major contribution to the response of aged influenza-naïve mice to influenza virus infection. Immun. Ageing 15, 17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keeler SP, Agapov EV, Hinojosa ME, Letvin AN, Wu K, Holtzman MJ, Influenza a virus infection causes chronic lung disease linked to sites of active viral RNA remnants. J. Immunol 201, 2354–2368 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, Chen C-I, Anekalla KR, Joshi N, Williams KJN, Abdala-Valencia H, Yacoub TJ, Chi M, Chiu S, Gonzalez-Gonzalez FJ, Gates K, Lam AP, Nicholson TT, Homan PJ, Soberanes S, Dominguez S, Morgan VK, Saber R, Shaffer A, Hinchcliff M, Marshall SA, Bharat A, Berdnikovs S, Bhorade SM, Bartom ET, Morimoto RI, Balch WE, Sznajder JI, Chandel NS, Mutlu GM, Jain M, Gottardi CJ, Singer BD, Ridge KM, Bagheri N, Shilatifard A, Budinger GRS, Perlman H, Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J. Exp. Med 214, 2387–2404 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson KG, Sung H, Skon CN, Lefrancois L, Deisinger A, Vezys V, Masopust D, Cutting edge: Intravascular staining redefines lung CD8 T cell responses. J. Immunol 189, 2702–2706 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yager EJ, Ahmed M, Lanzer K, Randall TD, Woodland DL, Blackman MA, Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J. Exp. Med 205, 711–723 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tishon A, Borrow P, Evans C, Oldstone MB, Virus-induced immunosuppression. 1. Age at infection relates to a selective or generalized defect. Virology 195, 397–405 (1993). [DOI] [PubMed] [Google Scholar]
- 30.Ely KH, Roberts AD, Kohlmeier JE, Blackman MA, Woodland DL, Aging and CD8+ T cell immunity to respiratory virus infections. Exp. Gerontol 42, 427–431 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Van Braeckel-Budimir N, Varga SM, Badovinac VP, Harty JT, Repeated antigen exposure extends the durability of influenza-specific lung-resident memory CD8+ T cells and heterosubtypic immunity. Cell Rep. 24, 3374–3382.e3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar BV, Connors TJ, Farber DL, Human T cell development, localization, and function throughout life. Immunity 48, 202–213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jenkins MR, Webby R, Doherty PC, Turner SJ, Addition of a prominent epitope affects influenza A virus-specific CD8+ T cell immunodominance hierarchies when antigen is limiting. J. Immunol 177, 2917–2925 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Adachi T, Kobayashi T, Sugihara E, Yamada T, Ikuta K, Pittaluga S, Saya H, Amagai M, Nagao K, Hair follicle-derived IL-7 and IL-15 mediate skin-resident memory T cell homeostasis and lymphoma. Nat. Med 21, 1272–1279 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nath AP, Braun A, Ritchie SC, Carbone FR, Mackay LK, Gebhardt T, Inouye M, Comparative analysis reveals a role for TGF-β in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLOS ONE 14, e0210495 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang N, Bevan MJ, Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skon CN, Lee J-Y, Anderson KG, Masopust D, Hogquist KA, Jameson SC, Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat. Immunol 14, 1285–1293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Slutter B, Van Braeckel-Budimir N, Abboud G, Varga SM, Salek-Ardakani S, Harty JT, Dynamics of influenza-induced lung-resident memory T cells underlie waning heterosubtypic immunity. Sci. Immunol 2, eaag2031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, Huang S, Shan J, Ma C, Ye Z, Xiang M, Limper AH, Porquera E-C, Kohlmeier JE, Kaplan MH, Zhang N, Johnson AJ, Vassallo R, Sun J, PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci. Immunol 4, eaaw1217 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McMaster SR, Wilson JJ, Wang H, Kohlmeier JE, Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-γ production. J. Immunol 195, 203–209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haanen JBAG, Wolkers MC, Kruisbeek AM, Schumacher TNM, Selective expansion of cross-reactive CD8+ memory T cells by viral variants. J. Exp. Med 190, 1319–1328 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo H, Topham DJ, Multiple distinct forms of CD8+ T cell cross-reactivity and specificities revealed after 2009 H1N1 influenza A virus infection in mice. PLOS ONE 7, e46166 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ballesteros-Tato A, Leon B, Lee BO, Lund FE, Randall TD, Epitope-specific regulation of memory programming by differential duration of antigen presentation to influenza-specific CD8+ T cells. Immunity 41, 127–140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo H, Baker SF, Martínez-Sobrido L, Topham DJ, Induction of CD8 T cell heterologous protection by a single dose of single-cycle infectious influenza virus. J. Virol 88, 12006–12016 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richmond JM, Strassner JP, Rashighi M, Agarwal P, Garg M, Essien KI, Pell LS, Harris JE, Resident memory and recirculating memory T cells cooperate to maintain disease in a mouse model of vitiligo. J. Invest. Dermatol 139, 769–778 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hausmann E, Neuman WF, Conversion of proline to hydroxyproline and its incorporation into collagen. J. Biol. Chem 236, 149–152 (1961). [PubMed] [Google Scholar]
- 47.Nguyen THO, Sant S, Bird NL, Grant EJ, Clemens EB, Koutsakos M, Valkenburg SA, Gras S, Lappas M, Jaworowski A, Crowe J, Loh L, Kedzierska K, Perturbed CD8+ T cell immunity across universal influenza epitopes in the elderly. J. Leukoc. Biol 103, 321–339 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Crowe SR, Turner SJ, Miller SC, Roberts AD, Rappolo RA, Doherty PC, Ely KH, Woodland DL, Differential antigen presentation regulates the changing patterns of CD8+ T cell immunodominance in primary and secondary influenza virus infections. J. Exp. Med 198, 399–410 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quinn KM, Kan W-T, Watson KA, Liddicoat BJ, Swan NG, Quilten HM, Denton AE, Li J, Chen W, Brown LE, Jackson DC, Reading PC, Doherty PC, Kedzierska K, Kedzierski L, Turner SJ, La Gruta NL, Extrinsically derived TNF is primarily responsible for limiting antiviral CD8+ T cell response magnitude. PLOS ONE 12, e0184732 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jergović M, Smithey MJ, Nikolich-Žugich J, Intrinsic and extrinsic contributors to defective CD8+ T cell responses with aging. Exp. Gerontol 105, 140–145 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Hussain T, Quinn KM, Similar but different: Virtual memory CD8 T cells as a memory-like cell population. Immunol. Cell Biol 97, 675–684 (2019). [DOI] [PubMed] [Google Scholar]
- 52.Schenkel JM, Fraser KA, Vezys V, Masopust D, Sensing and alarm function of resident memory CD8+ T cells. Nat. Immunol 14, 509–513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, Alexandre YO, Gregory JL, Russell TA, Gebhardt T, Carbone FR, Tscharke DC, Heath WR, Mueller SN, Mackay l. K., Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat. Immunol 19, 183–191 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Fonseca R, Beura LK, Quarnstrom CF, Ghoneim HE, Fan Y, Zebley CC, Scott MC, Fares-Frederickson NJ, Wijeyesinghe S, Thompson EA, da Silva HB, Vezys V, Youngblood A, Masopust D, Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol 21, 412–421 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.La Gruta NL, Rothwell WT, Cukalac T, Swan NG, Valkenburg SA, Kedzierska K, Thomas PG, Doherty PC, Turner SJ, Primary CTL response magnitude in mice is determined by the extent of naive T cell recruitment and subsequent clonal expansion. J. Clin. Invest 120, 1885–1894 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grant E, Wu C, Chan K-F, Eckle S, Bharadwaj M, Zou QM, Kedzierska K, Chen W, Nucleoprotein of influenza A virus is a major target of immunodominant CD8+ T-cell responses. Immunol. Cell Biol 91, 184–194 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Ichikawa T, Hirahara K, Kokubo K, Kiuchi M, Aoki A, Morimoto Y, Kumagai J, Onodera A, Mato N, Tumes DJ, Goto Y, Hagiwara K, Inagaki Y, Sparwasser T, Tobe K, Nakayama T, CD103hi Treg cells constrain lung fibrosis induced by CD103lo tissue-resident pathogenic CD4 T cells. Nat. Immunol 20, 1469–1480 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Jabri B, Abadie V, IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol 15, 771–783 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zens KD, Chen JK, Guyer RS, Wu FL, Cvetkovski F, Miron M, Farber DL, Reduced generation of lung tissue-resident memory T cells during infancy. J. Exp. Med 214, 2915–2932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sueblinvong V, Neujahr DC, Mills ST, Roser-Page S, Ritzenthaler JD, Guidot D, Rojas M, Roman J, Predisposition for disrepair in the aged lung. Am. J. Med. Sci 344, 41–51 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Quinn KM, Fox A, Harland KL, Russ BE, Li J, Nguyen THO, Loh L, Olshanksy M, Naeem H, Tsyganov K, Wiede F, Webster R, Blyth C, Sng XYX, Tiganis T, Powell D, Doherty PC, Turner SJ, Kedzierska K, La Gruta NL, Age-related decline in primary CD8+ T cell responses is associated with the development of senescence in virtual memory CD8+ T cells. Cell Rep. 23, 3512–3524 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Quinn KM, Zaloumis SG, Cukalac T, Kan W-T, Sng XYX, Mirams M, Watson KA, McCaw JM, Doherty PC, Thomas PG, Handel A, La Gruta NL, Heightened self-reactivity associated with selective survival, but not expansion, of naive virus-specific CD8+ T cells in aged mice. Proc. Natl. Acad. Sci. U.S.A 113, 1333–1338 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, Selman M, Pardo A, White ES, Levy BD, Busse PJ, Tuder RM, Antony VB, Sznajder JI, Budinger GRS, Blue journal conference. Aging and susceptibility to lung disease. Am. J. Respir. Crit. Care Med 191, 261–269 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, Hu Y, Tao Z-W, Tian J-H, Pei Y-Y, Yuan M-L, Zhang Y-L, Dai F-H, Liu Y, Wang Q-M, Zheng J-J, Xu L, Holmes EC, Zhang Y-Z, A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Acosta E, Hallman SA, Dillon LY, Ouellette N, Bourbeau R, Herring DA, Inwood K, Earn DJD, Madrenas J, Miller MS, Gagnon A, Determinants of Influenza Mortality Trends: Age-period-cohort analysis of influenza mortality in the United States, 1959-2016. Demography 56, 1723–1746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Han R, Huang L, Jiang H, Dong J, Peng H, Zhang D, Early clinical and CT manifestations of coronavirus disease 2019 (COVID-19) Pneumonia. AJR Am. J. Roentgenol 215, 338–343 (2020). [DOI] [PubMed] [Google Scholar]
- 67.Xu Y-H, Dong J-H, An W-M, Lv X-Y, Yin X-P, Zhang J-Z, Dong L, Ma X, Zhang H-J, Gao A-L, Clinical and computed tomographic imaging features of novel coronavirus pneumonia caused by SARS-CoV-2. J. Infect 80, 394–400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chan JF-W, Yuan S, Kok K-H, To KK-W, Chu H, Yang J, Xing F, Liu J, Yip CC-Y, Poon RW-S, Tsoi H-W, Lo SK-F, Chan K-H, Poon VK-M, Chan W-M, Ip JD, Cai J-P, Cheng VC-C, Chen H, Hui CK-M, Yuen K-Y, A familial cluster of pneumonia associated with the 2019 novel coronavirus indicating person-to-person transmission: A study of a family cluster. Lancet 395, 514–523 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chan JF-W, Yuan S, Zhang AJ, Poon VK-M, Chan CC-S, Lee AC-Y, Fan Z, Li A, Liang R, Cao J, Tang K, Luo C, Cheng VC-C, Cai J-P, Chu H, Chan K-H, To KK-W, Sridhar S, Yuen K-Y, Simulation of the clinical and pathological manifestations of Coronavirus Disease 2019 (COVID-19) in golden Syrian hamster model: Implications for disease pathogenesis and transmissibility. Clin. Infect. Dis, ciaa644 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen J, Qi T, Liu L, Ling Y, Qian Z, Li T, Li F, Xu Q, Zhang Y, Xu S, Song Z, Zeng Y, Shen Y, Shi Y, Zhu T, Lu H, Clinical progression of patients with COVID-19 in Shanghai, China. J. Infect 80, e1–e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, Cheng L, Li J, Wang X, Wang F, Liu L, Amit H, Zhang S, Zhang Z, Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med 26, 842–844 (2020). [DOI] [PubMed] [Google Scholar]
- 72.Goplen NP, Huang S, Zhu B, Cheon IS, Son YM, Wang Z, Li C, Dai Q, Jiang L, Sun J, Tissue-resident macrophages limit pulmonary CD8 resident memory T cell establishment. Front. Immunol 10, 2332 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Huang S, Zhu B, Cheon IS, Goplen NP, Jiang L, Zhang R, Peebles RS, Mack M, Kaplan MH, Limper AH, Sun J, PPAR-γ in macrophages limits pulmonary inflammation and promotes host recovery following respiratory viral infection. J. Virol 93, e00030–19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Huang S, Goplen NP, Zhu B, Cheon IS, Son Y, Wang Z, Li C, Dai Q, Jiang L, Xiang M, Carmona EM, Vassallo R, Limper AH, Sun J, Macrophage PPAR-γ suppresses long-term lung fibrotic sequelae following acute influenza infection. PLOS ONE 14, e0223430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Aged lungs exhibit persistent inflammatory responses after influenza infection.
Fig. S2. Enhanced presence of adaptive immune cells in aged lung parenchyma.
Fig. S3. Aged parenchymal CD8+ CD69+ memory T cells are tissue resident.
Fig. S4. The accumulation of CD8+ TRM in aged lungs is dependent on TGF-βR signaling.
Fig. S5. scRNA-seq on Db-NP TRM from young or aged lungs.
Fig. S6. High dose of CD8 Ab treatment diminishes lung inflammatory responses.