

Abstract

Glycoconjugated chlorins represent a promising class of compounds that meet the requirements for the third-generation photosensitizer (PS) for photodynamic therapy (PDT). We have focused on the use of glucose (Glc) to improve the performance of the PS based on the Warburg effect—a phenomenon where tumors consume higher Glc levels than normal cells. However, as a matter of fact, Glc-conjugation has a poor efficacy in hydrophilic modification; thus, the resultant PS is not suitable for intravenous injection. In this study, a Glc-based oligosaccharide, such as maltotriose (Mal3), is conjugated to chlorin e6 (Ce6). The conjugation is assisted by two additional molecular tools, such as propargyl amine and a tetraethylene glycol (TEG) derivative. This route produced the target Mal3–Ce6 conjugate linked via the TEG spacer (Mal3–TEG–Ce6), which shows the required photoabsorption properties in the physiological media. The PDT test using canine mammary carcinoma (SNP) cells suggested that the antitumor activity of Mal3–TEG–Ce6 is extremely high. Furthermore, in vitro tests against mouse mammary carcinoma (EMT6) cells have been demonstrated, providing insights into the photocytotoxicity, subcellular localization, and analysis of cell death and reactive oxygen species (ROS) generation for the PDT system with Mal3–TEG–Ce6. Both apoptosis and necrosis of the EMT6 cells occur by ROS that is generated via the photochemical reaction between Mal3–TEG–Ce6 and molecular oxygen. Consequently, Mal3–TEG–Ce6 is shown to be a PS showing the currently desired properties.
Introduction
Photodynamic therapy (PDT) has increasingly attracted much attention as a minimally invasive cancer therapy.1−4 A brief overview of the therapy is that a photosensitizer (PS) is administered to the body by an intravenous injection and the cancer tissues are irradiated by visible red light. The photochemical reaction between PS and tissue oxygen (O2) occurs according to the mechanism described by the Jablonski diagram, eventually generating reactive oxygen species (ROS) that kill the target cell. Thus, the molecular properties of PS determine the performance of PDT, which require (a) strong photoabsorption of visible light in the longer wavelength region that is likely to penetrate deeper into tissues, (b) high ROS generation ability, (c) selective accumulation ability to the inside of the target tumor cells, (d) no toxicity in the dark, and (e) rapid clearance out of the body after the treatment. In clinical use, (f) compatibility with the physiological media (water-solubility) is added as a crucial property, which allows the administration of PS by intravenous injection without any toxic organic solvents and solubilizing agents that cause side effects.
Dehydroporphyrins, called chlorins, are a class of molecules satisfying the properties of the (a) strong photoabsorption and (b) high ROS generation ability. Representatively, 5,10,15,20-tetrakis(3-hydroxyphenyl)chlorin (Temoporfin, Foscan) is categorized as a chlorin-type PS, which is artificially synthesized from commercially available chemicals.5 Alternatively, mono-l-aspartyl chlorin e6 (NPe6, Talaporfin, Laserphyrin)6 is a PS derived from naturally occurring chlorophylls a. Pyropheophorbide-a analogues7 are also categorized as a PS that originate from natural dyes. A prime trend for the PS development is currently directed to utilize the ability of some other biologically active molecules to improve the requirement (c) selective accumulation ability to the inside of the target tumor cells. The light-sensitive drug in which the accumulation ability is intentionally enhanced by the incorporation of bioactive molecules is distinguished as “the third-generation PS”. A highlighted approach includes glycoconjugation.1,3 Saccharides are covalently bonded to the photoactive molecules derived from artificial chlorin dyes.8−22 As an example, a chlorin derivative possessing four perfluorinated aromatic rings encircled with four glucose (Glc) molecules has been synthesized. The product, called G–chlorin, shows a high photocytotoxicity with a half-maximal (50%) inhibitory concentration (IC50) value of less than 0.5 μM.14 The anticancer effects of G–chlorin for gastric and colon cancer have been reported to be high.16 These results strongly suggest that the Warburg effect23—a phenomenon where tumors consume higher Glc levels than normal cells—can be utilized in the conception to develop the third-generation PS. Naturally occurring chlorin derivatives are also targeted for glycoconjugation.24−30 Recently, chlorin e6 (Ce6) has been decorated with Glc to develop a new family of PS (Glc–Ce6), which shows an extremely high photocytotoxicity.27,28 Ce6 is a molecule that is derived from naturally occurring chlorophyll a; therefore, G–Ce6 is expected to show an improved biocompatibility and body clearance, meeting the requirements of both (d) no toxicity in the dark and (e) rapid clearance out of the body after the treatment. However, as a matter of fact, G–Ce6 is insoluble in water due to the strong hydrophobic property of the Ce6 unit. This result suggested that the strong hydrophobic property of the Ce6 unit dominates the solubility of G–Ce6. Another plausible reason is that the molecular size of Glc is small compared to that of Ce6 composed of the tetrapyrrole ring. Eventually, the Ce6 moiety is not sufficiently covered by hydrophilic moieties in the aqueous media, resulting in G–Ce6 forming precipitates. Thus, satisfying the requirement (f) compatibility with the physiological media (water-solubility) is the next challenge for the PDT with the glycoconjugated Ce6.
In this study, maltotriose (Mal3), a Glc-based trisaccharide, is conjugated to Ce6 to develop an advanced PS, showing both high performance and water solubility. Malto-oligosaccharide (Maln) is a general term for the oligosaccharides consisting of Glc as the repeating units, which are known to be found in a syrup as the main components. Maln exhibits not only the Warburg effect derived from the Glc unit but also the stronger and well-defined hydrophilic property due to possessing multiple and definite numbers of hydroxyl groups; thus, they are special molecules in bioconjugated chemistry and pharmacology. Indeed, we have previously reported that the Mal3-conjugation provides a very positive result for the preparation of the water-soluble PS.20,21Chart 1 depicts a synthetic conception in this study in which Mal3 is covalently linked to the Ce6 dimethyl ester (Ce6–DME) in which the connection is assisted by two other molecular tools such as propargyl amine (PA) and the tetraethylene glycol (TEG) derivative. The synthesis is achieved by three reaction steps staring from Mal3 without tedious protection/deprotection processes, which consist of (i) the direct N-glycosylation reaction, (ii) the copper-catalyzed azide/alkyne “click” (CuAAC) reaction, and (iii) the SN2 esterification reaction. The structure of the final product is assignable to the Mal3–Ce6 conjugate linked via the TEG spacer (Mal3–TEG–Ce6), which shows water solubility and characteristic photoabsorption properties at the longer wavelength in the visible-light area in aqueous physiological media. The performance of Mal3–TEG–Ce6 is clarified by the antitumor test against canine mammary carcinoma (SNP) cells, whose performance is compared to that of the previously reported glucose–Ce6 conjugate (G–Ce6). We also report the photocytotoxicity, subcellular localization, analysis of cell death, and ROS generation for the Mal3–TEG–Ce6 PDT system using mouse mammary carcinoma (EMT6) cells.
Chart 1. Synthetic Conception in Which Maltotriose (Mal3) Is Covalently Bonded with Chlorin e6 Dimethyl Ester (Ce6–DME) Assisted by Using Two Molecular Tools, Such as PA and the TEG Derivative, to Produce the Mal3–Ce6 Conjugate Linked with the TEG-Spacer (Mal3–TEG–Ce6).

Results and Discussion
Synthesis of the Maltotriose–Chlorin e6 Conjugate (Mal3–TEG–Ce6)
A synthetic protocol for the target PS is shown in Scheme 1. We performed the N-glycosylation reaction of PA to maltotriose (Mal3) according to a similar method described in the literature.31 The reaction produced Mal3 with an ethynyl group (Mal3–alkyne 1), which was then reacted with TEG with azido and tosylate groups32 (N3–TEG–OTs 2) using CuSO4 and sodium l-ascorbate to produce a product 3. It should be noted that this CuAAC reaction had to be performed before the conjugation with the chlorin derivative in order to avoid the contamination of the metal species for the final PS. Figure 1a shows the 1H NMR spectrum of 3 in D2O, exhibiting the signals due to the protons of the triazole ring (8.00 and 7.77 ppm), the aromatic ring (7.67 and 7.32 ppm), the Mal3 and TEG units (5.45–3.24 ppm), the methyl group (2.28 ppm), and the N-acetyl group (2.08 and 1.95 ppm). The sharp peak due to the azido group (2262 cm–1) is observed in the IR spectrum of 2 (Figure S1), while completely absent in that of 3 (Figure S2). Alternatively, the large peaks due to hydroxyl groups (3700–3000 cm–1) and acetyl group (1732 cm–1) are present in the IR spectrum of 3 (Figure S2). The signals observed for the 13C NMR spectrum were fully assigned (Figure S4). These results support that 3 is assignable to the target Mal3–TEG–OTs.
Scheme 1. Synthesis of the Mal3–Ce6 Conjugate Linked via the TEG Spacer (Mal3–TEG–Ce6).

Figure 1.

1H NMR spectra of (a) 3 in D2O and (b) Mal3–TEG–Ce6 in DMSO-d6 (the symbol * in the spectra corresponds to the protons due to the hydroxyl groups).
We finally performed the SN2 esterification reaction between 3 and the chlorin e6 dimethyl ester derivative (Ce6–DME 4) using Cs2CO3 in dry N,N-dimethylformamide at room temperature. The residue from the reaction mixture was purified by column chromatography to isolate the product. Figure 1b shows the 1H NMR spectrum of the product in DMSO-d6, displaying signals due to the protons derived from the 3 unit, such as the triazole ring (8.01 and 7.85 ppm), the Mal3–TEG unit (5.46–3.06 ppm), and the N-acetyl group (2.07 and 1.93 ppm). Additionally, the characteristic resonances due to the protons in the Ce6 unit (Hchls) distinctly appear, which include Hchl-10, Hchl-5, Hchl-20, Hchl-31, Hchl-32, Hchl-151, Hchl-18, Hchl-17, Hchl-81, Hchl-172, Hchl-171, Hchl-82, Hchl-181, Hchl-21, and Hchl-23. The area integration values for the respective signals are in good agreement with the target structures. The signals observed for the 13C NMR spectrum have been fully assigned (Figure S5). HR-ESI-MS exhibited the main peak at m/z 1431.60547, which agreed with the calculated [M + Na]+ ion peak value for the target compound of 1431.60713. These results indicated that the SN2 esterification reaction between the OT group of 3 and the COOH group of 4 proceeded to produce the target PS such as the Mal3–Ce6 conjugate linked via the TEG spacer (Mal3–TEG–Ce6).
Water Solubility
The desired result obtained was that Mal3–TEG–Ce6 readily dissolved in water, producing a green aqueous solution. The water solubility was determined to be >11.6 mg mL–1. We determined the partition coefficient (log P) to quantify the introduced hydrophilicity that is defined by eq 1
| 1 |
where [Coctanol] and [CPBS] denote the concentrations of PS being portioned into the 1-octanol phase and the PBS buffer phase, respectively. The log P value was determined to be −0.24 for Mal3–TEG–Ce6. We previously reported the synthesis of a fluorinated chlorin derivative that is encircled with four Mal3 molecules (Mal3–TFPC). Mal3–TFPC showed a high water solubility >37 mg mL–1 with the log P of −1.78.20 This result is simply attributable to the differences in the number of the introduced Mal3 molecules for Mal3–TEG–Ce6 (one Mal3) versus Mal3–TFPC (four Mal3’s). Consequently, the Mal3–TEG unit is shown to be a powerful tool to make the relatively large size of a hydrophobic molecule, such as Ce6, compatible in aqueous media.
Photoabsorption Property
Table 1 summarizes the UV–vis absorption property for Mal3–TEG–Ce6 together with that of Ce6–DME 4 as the control sample. Figure 2a displays the UV–vis spectra. In DMSO, Mal3–TEG–Ce6 shows an absorption due to the Soret band at the maximum absorption wavelength (λmax) of 404 nm with a molar absorption coefficient (ε) of 125,000 M–1 cm–1 (green solid line). Also, Mal3–TEG–Ce6 shows an absorption due to the Q band at λmax = 665 nm with ε = 37,000 M–1 cm–1. This absorption is important for PDT in which the photochemical property can be driven by the longer wavelengths regions that is favorable for the permeability of physiological tissues. Figure 2a also shows the UV–vis spectrum of 4 as a comparison, displaying the absorptions due to the Soret band at λmax = 404 nm with ε = 84,200 M–1 cm–1 and the Q band at λmax = 664 nm with ε = 25,700 M–1 cm–1. The ε values for 5 are 1.4–1.5 times higher than those of 4. A plausible explanation is that the intermolecular associations among the Ce6 moieties are suppressed by steric hindrance due to the introduced Mal3–TEG unit, eventually increasing the solubility of Mal3–TEG–Ce6 in DMSO. A more noteworthy result in this study is the photochemical properties in aqueous media. Figure 2b displays the UV–vis spectrum for Mal3–TEG–Ce6 in PBS, which exhibits absorptions due to the Soret band at λmax = 404 nm with ε = 52,300 M–1 cm–1 and the Q band at λmax = 664 nm with ε = 20,000 M–1 cm–1. The λmax values in PBS are consistent with those in DMSO for Mal3–TEG–Ce6. Although the ε values in PBS are decreased as compared to those in DMSO probably due to the intermolecular associations, Mal3–TEG–Ce6 shows photoabsorption properties derived from the Ce6 framework under the physiological conditions.
Table 1. Summary for Photoabsorption Properties of 4 in DMSO (Control) and Mal3–TEG–Ce6 in DMSO and PBS.
| λ/nm (ε/M–1 cm–1) |
||||||
|---|---|---|---|---|---|---|
| sample | solvent | Soret | Q bands | |||
| 4 | DMSO | 404 (84,200) | 502 (7450) | 531 (2680) | 609 (2630) | 664 (25,700) |
| Mal3–TEG–Ce6 | DMSO | 404 (125,000) | 502 (11,900) | 530 (5580) | 609 (5690) | 665 (37,000) |
| Mal3–TEG–Ce6 | PBS | 404 (52,300) | 502 (6850) | 531 (3800) | 609 (3050) | 664 (20,000) |
Figure 2.

UV–vis spectra of (a) Mal3–TEG–Ce6 (solid line) and 4 (dashed line) in DMSO and (b) Mal3–TEG–Ce6 in PBS.
In Vitro PDT Test against SNP Cells
Osaki et al. reported the photocytotoxicity of G–Ce6 against canine mammary carcinoma (SNP) cells.28 The performance of G–Ce6 has been roughly estimated to be 30-fold higher than that of NPe6, a clinically approved PDT drug in Japan. Very recently, Shinoda et al. reported that the anticancer effect of G–Ce6 was 1000-fold higher than that of TS for the system using the human glioblastoma U251 cells.30 Thus, the extremely high performance of G–Ce6 has already been proven. We now clarified the performance of Mal3–TEG–Ce6 using the SNP cell system for the first time, and the result is compared to that of G–Ce6. The SNP cells were treated with different Mal3–TEG–Ce6 concentrations (0.16, 0.80, 4.0, and 20 μM) and exposed to 671 nm red light (fluence rate = 7.3 mW/cm2, light dose = 5 J/cm2). Using the WST-8 assay, the cell viabilities for the photoirradiated and photounirradiated groups were measured and the ratio (%) compared to the untreated cells were determined. For the injection of G–Ce6, the use of a surfactant or organic solvent, such as DMSO, is necessary. However, Mal3–TEG–Ce6 is a molecule that readily dissolves in water, which allows direct injection into the cell media. Figure 3a displays a plot of the cell viability as a function of the Mal3–TEG–Ce6 concentrations. For the photounirradiated group (0 J/cm2), no cell is killed at the Mal3–TEG–Ce6 concentrations ranging from 0.16 to 20 μM. Hence, Mal3 is a powerful molecular tool to introduce biocompatibility. On the other hand, for the photoirradiated group (5 J/cm2), a very high photocytotoxicity was observed. For example, the cell viability reduces to 50% for the group using 0.16 μM Mal3–TEG–Ce6. Furthermore, almost all the cells are killed in the group using 8.0 μM Mal3–TEG–Ce6. Figure 3b shows the result of the G–Ce6 groups as a comparison. The half maximal (50%) inhibitory concentration (IC50) values are approximately determined to be 0.26 μM for G–Ce6 and 0.15 μM for Mal3–TEG–Ce6, indicating that Mal3–TEG–Ce6 possesses a very high PDT activity comparable to that of G–Ce6. Thus, the availability of Mal3 to endow both a high water solubility and high tumor cell accumulation ability to Ce6 has been proven. As for the effect of Mal3, Nishie et al. has already reported that a fluorinated chlorin derivative encircled with four Mal3 molecules (Mal3–TFPC) exhibited a high accumulation ability to both HKN45 human gastric cancer cells and HT29 colon cancer cells.21 We have not obtained a clear answer for the mechanism of how the Mal3 and/or Mal3-conjugates incorporate into the cancer cells. The facts that upregulation of glucose transporters (GLUTs) has been reported in numerous cancer types33 suggest the possibility that the GLUTs are related to the cellar uptake for the Mal3-system. The PDT test in the presence of the GLUT inhibitors would provide some insights into the mechanism that we would like to try in the near future.
Figure 3.

Photodynamic cytotoxicity of (a) Mal3–TEG–Ce6 and (b) G–Ce6 in SNP cells. The cell viabilities are plotted as a function of the concentrations of the PSs for the photoirradiated system (671 nm) (green circles) and photounirradiated system (black circles). Each value represents the mean ± SD. (n = 6).
In Vitro PDT Test against EMT6 Cells
The efficacy of Mal3–TEG–Ce6 has been confirmed using mouse mammary carcinoma (EMT6) cells. The cells were treated with diverse Mal3–TEG–Ce6 concentrations (0.032, 0.16, 0.80, 4.0, and 20 μM) and exposed to 671 nm red light (fluence rate = 8.3 mW/cm2). We have selected the light doses of 0, 1, 5, and 15 J/cm2 to obtain the optimal light condition. The viabilities of EMT6 cells for the respective groups were determined after 4 h. Figure 4 displays a plot of the cell viability as a function of the Mal3–TEG–Ce6 concentrations. Cell death is not observed for any photounirradiated groups at the Mal3–TEG–Ce6 concentrations ranging from 0.032 to 20 μM (0 J/cm2). On the other hand, for the photoirradiated groups (1, 5, and 15 J/cm2), the cells were effectively killed. Even for the group using a light dose (1 J/cm2), the photocytotoxicity was clearly observed and almost all the cells were killed in the 4.0 μM Mal3–TEG–Ce6 group. The cell viability is dependent on the Mal3–TEG–Ce6 concentrations, roughly providing IC50 = 0.80 μM. A more significant effect is observed for the group using the increased light dose (5 J/cm2). Cell viability reaches <5% when the Mal3–TEG–Ce6 concentration has a low value of 0.80 μM. The tendency in the cell viability for the 15 J/cm2 group is almost the same as that for the 5 J/cm2 group. Consequently, the PDT using Mal3–TEG–Ce6 induces cell death in the EMT6 cells in a manner dependent on the PS dose and the light dose.
Figure 4.
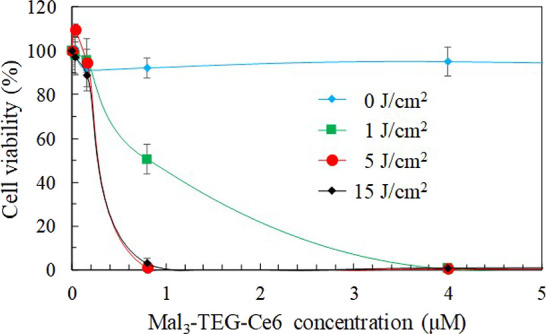
Photodynamic cytotoxicity of Mal3–TEG–Ce6 in EMT6 cells (0.032, 0.16, 0.8, 4.0, and 20 μM). Each value represents the mean ± SD. (n = 6).
Subcellular Localization in EMT6 Cells
Figure 5a shows fluorescence micrographs of Mal3–TEG–Ce6 taken in EMT6 which are stained with the lysosome, mitochondrial, and endoplasmic reticulum (ER) probes. The overlaid images have yellow-green fluorescent spots, indicating an overlap of Mal3–TEG–Ce6 (red) and the respective probes (green). We ascertain that Mal3–TEG–Ce6 primarily accumulates in the lysosomes. This tendency is similar to other systems with the Glc-conjugate (G–Ce6) and Mal3–conjugate (Mal3–TFPC). The three PSs are similar in that they are nonionic compounds.
Figure 5.
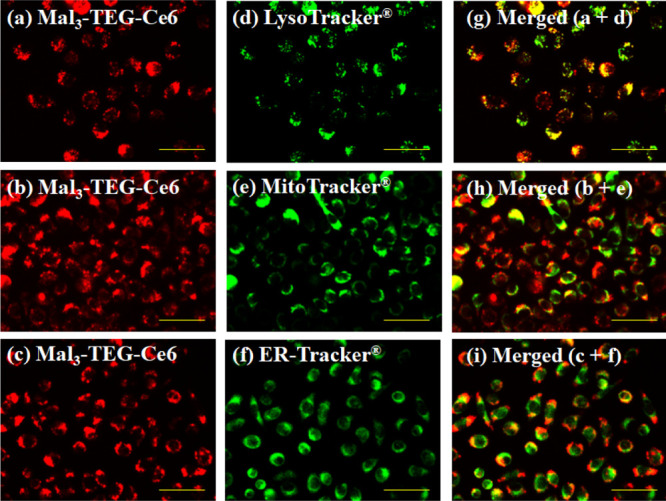
Subcellular localization of Mal3–TEG–Ce6 in the EMT6 cells. The images (a–c) show the red fluorescences of Mal3–TEG–Ce6. The images (d–f) show the green fluorescences of the probe-stained lysosome, mitochondria, and ER, respectively, which are in the same views as (a–c). The images (g–i) are the merged images of the left and middle ones. Scale bar, 50 μm.
Analysis of Annexin V (+) Cells and ROS (+) Cells
The EMT6 cells were incubated with the 0.8 μM Mal3–TEG–Ce6 and exposed to 671 nm red light (fluence rate = 8.3 mW/cm2, 0, 1, 5, or 15 J/cm2). The PDT-treated cells were stained using the Apoptotic/Necrotic Cells Detection Kit (Takara Bio, Inc., Japan). Figure 6 shows an image of the resultant cells, which are divided into the following groups; control (no treatment), laser (irradiated with a light dose of 15 J/cm2), Mal3–TEG–Ce6 (treated with 0.8 M Mal3–TEG–Ce6), and PDT (treated with 0.8 μM Mal3–TEG–Ce6 and then irradiated with a light dose of 1, 5, or 15 J/cm2). The images include the ones for the dead cells stained by ethidium homodimer III (EthD-III) (red) and annexin V-fluorescein isothiocyanate (annexin V) (green). The cells are rarely stained in the control, laser, and Mal3–TEG–Ce6 groups, whereas a large number of cells are positively stained by EthD-III or annexin V in the PDT groups (1 and 5 J/cm2). This indicated the occurrence of both the phosphatidylserine translocation and the loss of the plasma membrane integrity, implying that the cells are either in late apoptotic or early necrotic stages. Another result to be noted is that the number of cells is extremely low in the 15 J/cm2 group. A possible explanation for this result is that the photocytotoxicity is so high for the 15 J/cm2 group that the cells are destroyed and disappear.
Figure 6.

Representative images of EMT6 cells stained with EthD-III (red) and annexin V (green) for the PDT groups using the conditions of the Mal3–TEG–Ce6 concentration and the light dose of (a) 0 μM and 0 J/cm2, (b) 0 μM and 15 J/cm2, (c) 0.8 μM and 0 J/cm2, (d) 0.8 μM and 1 J/cm2, (e) 0.8 μM and 5 J/cm2, and (f) 0.8 μM and 15 J/cm2, respectively. Scale bar, 500 μm.
Apoptosis has been assessed for the PDT-treated cells using the Muse Annexin V and Dead Cell Assay kit. Figure 7a shows the percentage of annexin V positive (+) cells. The apoptotic rates are 15.1, 55.0, and 42.6% for the cells in the 1, 5, and 15 J/cm2 PDT groups, respectively. Thus, the apoptotic rates of the PDT group are clearly higher than that in the control and laser groups (all p < 0.05). Notably, the high apoptotic rate of 55.0% was observed in the 5 J/cm2 group. The rate in the 15 J/cm2 group is lower than that in the 5 J/cm2 group. As already mentioned, there is a possibility that cells treated at 15 J/cm2 are severely destroyed, providing inaccurate measurements as for the percentage for the annexin V (+) cells. This result suggested that the Mal3–TEG–Ce6 PDT increased the annexin V (+) cells in a light dose-dependent manner. Finally, the generation for ROS has been assessed using the Muse Oxidative Stress kit. Figure 7b shows the percentage of the ROS positive (+) cells. A high value of 56.7% was observed for the 5 J/cm2 group. The percentage of ROS positive (+) cells of the 5 J/cm2 PDT group was significantly higher than that for the laser group (p < 0.05). The percentage of ROS (+) cells fairly agrees with that of the annexin V (+) cells, suggesting that apoptosis of the EMT6 cells is due to the ROS generated as a result of the photoirradiation of Mal3–TEG–Ce6 in the presence of molecular oxygen.
Figure 7.

Percentages of (a) annexin V (+) cells and (b) ROS (+) cells. Data were analyzed using Dunn’s multiple comparison test [(a) *p < 0.05; control vs 5 J/cm2, laser vs 5 J/cm2 and (b) *p < 0.05; laser vs 5 J/cm2]. The results are presented as the mean ± standard deviation.
Conclusions
Mal3, an oligosaccharide in which three glucose molecules are coupled together, has been conjugated to Ce6 by using two additional molecular tools such as PA and the TEG derivative. This allowed the production of the Mal3–Ce6 conjugate linked via the TEG-spacer, Mal3–TEG–Ce6. A remarkable result is that Mal3–TEG–Ce6 acquires water solubility, while its original photoabsorption properties due to Ce6 remain intact even in physiological media, which has not been achieved by the monosaccharide conjugation. In vitro tests against SNP cells have indicated that Mal3–TEG–Ce6 possesses an extremely high PDT activity comparable to that of the previously reported monosaccharide-based PS such as G–Ce6. In vitro tests against EMT6 cells provided further insights into the Mal3–TEG–Ce6 PDT system that includes both apoptosis and necrosis of the EMT6 cells is due to the ROS generated as a result of the photochemical reaction between Mal3–TEG–Ce6 and tissue molecular oxygen. We have experimentally proven that Mal3–TEG–Ce6 has a high potential as an advanced PS showing the currently desired properties.
Experimental Section
Materials
N3–TEG–OTs 2(32) and chlorin e6–DME 4(34) were prepared according to the literature. Mal3 (Sigma-Aldrich, >90%), PA (Tokyo Chemical Industry Co., Japan, >95.0%), CuSO4 (Wako Pure Chemical Industries, Japan, 97.5%), sodium L-ascorbate (Wako Pure Chemical Industries, Japan, 98.0%), Cs2CO3 (Tokyo Chemical Industry Co., Japan, >98%), tert-butyl alcohol (Tokyo Chemical Industry Co., Japan, >99.0%), and dry N,N-dimethylformamide (Wako Pure Chemical Industries, Japan, 99.5%) were used as received. All other materials were obtained from commercial sources and used as received, unless otherwise stated.
Instruments
The 1H and 13C NMR spectra were recorded using JEOL JNM-ECX400 and JNM-ECZ600R instruments. The infrared (IR) spectra were recorded using a Horiba FT-720 spectrometer. The UV–vis spectra were recorded by a JASCO V-500 spectrophotometer. The high-resolution mass spectra were recorded by a JEOL AccuTOF JMS-T100LC (ESI-MS). Aqueous preparative size-exclusion chromatography (SEC) was performed using ChromNAV software, a JASCO LC-NetII/ADC interface box, a JASCO FC-2088-30 fraction collector controller, a JASCO PU-2086 Plus pump, a JASCO UV-2075 Plus detector, an Advantec CHF 122 SC fraction collector, and a Shodex OH pak SB-2002.5 column (20 × 300 mm, average bead size: 10 μm, exclusion limit: 1 × 104) using water as the eluent at a flow rate of 2.5 mL min–1 and room temperature.
Synthesis
N-Acetyl-N-ethynyl-[(O-α-d-glucopyranosyl)-(1 → 4)]2-β-d-glucopyranoside (Mal3–Alkyne 1)
The synthesis of Mal3–alkyne 1 has already been reported elsewhere.35 The mixture of Mal3 (500 mg, 991 μmol) and PA (900 μL, 14.1 mmol) was stirred at room temperature. The consumption of Mal3 was monitored by TLC (silica gel 60 F254, CH3CN/H2O = 2:1). After 72 h, dry MeOH (1.00 mL) was added and the mixture was poured into dry CH2Cl2 (150 mL). The formed precipitates were collected by filtration using a 1.0 μm pore-sized PTFE membrane filter and washed with a mixture of 1:3 dry MeOH/dry CH2Cl2 (8.00 mL). A solution of Ac2O (3.56 g, 34.9 mmol) in dry MeOH (66.0 mL) was added to the obtained solid, and the resulting mixture was stirred overnight at room temperature and then evaporated. A mixture of 1:1 MeOH/toluene (10.0 mL) was added, and the mixture was evaporated. This procedure was repeated until Ac2O and acetic acid were removed from the mixture. The residue was redissolved in H2O (5 mL) and freeze-dried to give the target 1 as a white solid (360 mg, 62.2%). Rf = 0.53 (CH3CN/H2O = 2/1).
2-(2-(2-(2-[[(4-Methylphenyl)sulfonyl]oxy]ethoxy)ethoxy)ethoxy)ethyl-1H-1,2,3-triazol-1-yl-methyl O-(α-d-glucopyranosyl)-(1 → 4)-(α-d-glucopyranosyl)-(1 → 4)-N-acetyl-N-β-d-glucopyranoside (Mal3–TEG–OTs 3)
A mixture of Mal3–alkyne 1 (193 mg, 331 μmol) and N3–TEG–OTs 2 (124 mg, 331 μmol) was stirred in a mixture of 1:1 tert-butyl alcohol/H2O (33.0 mL). CuSO4 (52.8 mg, 331 μmol) and sodium l-ascorbate (197 mg, 993 μmol) were added to the mixture, which was stirred at room temperature. After 24 h, the mixture was evaporated to dryness and the residue was purified by aqueous preparative SEC. The target product was redissolved in H2O (5.00 mL) and freeze-dried to give 3 as a white solid (126 mg, 39.7%). Rf = 0.33 (CHCl3/CH3OH = 3/1). 1H NMR (400 MHz, D2O): δ (ppm) = 8.00 and 7.77 (1H, 2 × s, minor and major rotamers, respectively, =CHN), 7.67 (2H, d, J = 8.2 Hz, ArH), 7.32 (2H, d, J = 8.2 Hz, ArH), 5.45 and 4.97 (1H, 2 × d, minor and major rotamers, J = 9.1 Hz and J = 8.2 Hz, respectively, H-1Mal1), 5.25–5.21 (2H, m, H-1Mal2–Mal3), 4.45 (2H, s, N(COCH3)CH2), 4.40 (2H, t, J = 10 Hz, NCH2), 4.11 (2H, t, J = 8.6 Hz, CH2OTs), 3.83–3.24 (30H, m, H-2Mal1–Mal3, H-3Mal1–Mal3, H-4Mal1–Mal3, H-5Mal1–Mal3, H-6Mal1–Mal3, NCH2CH2(OCH2CH2)2OCH2CH2OTs), 2.28 (3H, s, ArCH3), 2.08 and 1,95 (3H, 2 × s, major and minor rotamers, respectively, NCOCH3). 13C NMR (100 MHz, D2O): δ (ppm) = 174.9 (COCH3), 146.5 (Ar), 146.5 (NC=CHN), 130.9 (Ar), 130.2 (Ar), 127.8 (Ar), 124.9 (NC=CHN), 100.0 (C-1Mal2–Mal3), 87.0 (C-1Mal1), 76.6, 73.4, 72.7, 71.8, 71.5, 71.2 (C-2Mal1–Mal3, C-3Mal1–Mal3, C-4Mal1–Mal3, C-5Mal1–Mal3), 70.2, 70.1, 69.7, 69.5, 69.3, 68.8, 68.0 (NCH2CH2O, OCH2CH2O), 60.6 (C-6Mal1–Mal3), 50.1 (N(COCH3)CH2), 21.2 (ArCH3), 20.9 (NCOCH3). FT-IR (KBr): ν (cm–1) 3394, 2923, 1732, 1450, 1356, 1034, 924, 816, 771, 698, 561. HR-ESI-MS: [M + Na]+ ion peak at m/z 979.33019 corresponding to C38H60N4O22SNa (calcd. 979.33176).
131-(2-(2-(2-(2-[1H-1,2,3-Triazol-1-yl-methyl-O-(α-d-glucopyranosyl)-(1 → 4)-(α-d-glucopyranosyl)-(1 → 4)-N-acetyl-N-β-d-glucopyranosyl]ethoxy)ethoxy)ethoxy)ethoxycarbonyl) Chlorin e6 Dimethyl Ester (Mal3–TEG–Ce6)
The mixture of Mal3–TEG–OTs 3 (23.8 mg, 24.9 μmol), chlorin e6-DME 4 (15.6 mg, 24.9 μmol), and Cs2CO3 (8.10 mg, 24.9 μmol) in dry N,N-dimethylformamide (500 μL) was stirred at room temperature for 48 h. The mixture was evaporated to dryness, and the residue was purified by silica gel column chromatography (CHCl3/MeOH = 4/1 Rf = 0.11) to give the target Mal3–TEG–Ce6 as a green solid (22.1 mg, 63.1%). Data for Mal3–TEG–Ce6: Rf = 0.32 (CHCl3/CH3OH = 3/1). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 9.78 (1H, s, Hchl-10), 9.67 (1H, s, Hchl-5), 9.07 (1H, s, Hchl-20), 8.29 (1H, dd, J = 11, J = 12 Hz, Hchl-31), 8.01 and 7.85 (1H, 2 × s, minor and major rotamers, respectively, =CHN), 6.46 and 6.20 (2H, d, J = 19 and 12 Hz, respectively, Hchl-32), 5.46 and 4.85 (1H, 2 × d, minor and major rotamers, J = 4.8 Hz and J = 9.0 Hz, respectively, H-1Mal1), 5.31 (1H, br, Hchl-151), 5.03–5.00 (2H, m, H-1Mal2–Mal3), 4.80–4.74 (1H, m, CH2OC=O), 4.60–4.56 (1H, m, Hchl-18), 4.56–4.53 (2H, m, N(COCH3)CH2), 4.45 (1H, m, Hchl-17), 4.45–4.38 (2H, m, NCH2CH2), 3.97 (2H, t, J = 12 Hz, OCH2CH2OC=O), 3.81–3.77 (2H, q, Hchl-81), 3.75–3.06 (44H, m, H-2Mal1–Mal3, H-3Mal1–Mal3, H-4Mal1–Mal3, H-5Mal1–Mal3, H-6Mal1–Mal3, NCH2CH2(OCH2CH2)2O, Hchl-21, Hchl-71, Hchl-121, Hchl-154, Hchl-175), 2.70 and 2.09 (1H, m, Hchl-172), 2.39 and 1.59 (1H, m, Hchl-171), 2.07 and 1.93 (3H, 2 × s, major and minor rotamers, respectively, NCOCH3), 1.65–1.67 (6H, m, Hchl-82, Hchl-181), −1.50 (1H, s, Hchl-21), −1.70 (1H, s, Hchl-23). 13C NMR (150 MHz, DMSO-d6): δ (ppm) = 173.8 (Cchl-152), 173.1 (Cchl-173), 171.3 (NCOCH3), 171.1 (Cchl-131), 168.7 (Cchl-19), 168.1 (Cchl-16), 154.6 (Cchl-6), 148.8 (Cchl-9), 145.5 (Cchl-8) 145.4 (NC=CHN), 139.4 (Cchl-2), 136.7 (Cchl-4), 136.5 (Cchl-7), 135.3 (Cchl-12), 134.9 (Cchl-14), 134.7 (Cchl-3), 131.4 (Cchl-1, Cchl-11), 129.6 (Cchl-31), 129.0 (Cchl-13), 124.3 (NC=CHN), 122.8 (Cchl-32), 102.9 (Cchl-15), 102.5 (Cchl-10), 101.4 (C-1Mal2,3), 98.8 (Cchl-5), 94.7 (Cchl-20), 87.4 (C-1Mal1), 80.1, 74.0, 73.8, 73.1, 72.5, 72.3 (C-2Mal1–3, C-3Mal1–3, C-4Mal1–3, C-5Mal1–3), 70.7, 70.3, 70.2, 70.1, 69.1, 68.9, 65.6 (NCH2CH2O, OCH2CH2O), 61.3 (C-6Mal1–3), 52.9 (Cchl-17), 52.5 (Cchl-154), 51.9 (Cchl-175), 49.8 (N(COCH3)CH2), 48.7 (Cchl-18), 38.1 (Cchl-151), 31.0 (Cchl-172), 29.9 (Cchl-171), 23.3 (Cchl-181), 22.3 (NCOCH3), 19.3 (Cchl-81), 18.3 (Cchl-82), 12.5 (Cchl-21), 12.3 (Cchl-121), 11.4 (Cchl-71). FT-IR (KBr): ν (cm–1) 3411, 2922, 2868, 2571, 1795, 1723, 1643, 1539, 1437, 1344, 1147, 1066, 1030, 843, 798, 766, 723, 620. HR-ESI-MS: [M + Na]+ ion peak at m/z 1431.60547 corresponding to C67H92N8O25Na (calcd. 1431.60713). UV–vis (c 10.0 μM, DMSO, path length = 1 cm, 25 °C): λ/nm (ε × 10–3/M–1 cm–1) = 404 (124), 502 (11.3), 530 (4.98), 609 (5.23), 665 (36.7).
Cell Culture
Canine mammary carcinoma (SNP) cells were established by the author.36 The mouse mammary carcinoma (EMT6) cells were supplied by Professor Yoshihiro Uto of Tokushima University (Tokushima, Japan). The SNP and EMT6 cells were cultured in a 250 mL tissue culture flask (Corning Incorporated, Corning, NY, USA) containing RPMI 1640 medium (Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10% heat-inactivated fetal bovine serum (Nichirei Biosciences, Tokyo, Japan) and PSN (5 mg/mL penicillin, 5 mg/mL streptomycin, and 10 mg/mL neomycin) solution (Invitrogen), then incubated in 5% CO2 at 37 °C. The cells were washed with phosphate-buffered saline for subculturing and then harvested from near-confluent cultures via a brief exposure to a solution containing 0.25% trypsin and 1 mmol/L tetrasodium ethylenediaminetetraacetic acid with phenol red (Invitrogen). Trypsinization was terminated using RPMI 1640 medium containing 10% fetal bovine serum. The trypsinized cells were transferred to a new tissue culture flask.
In Vitro PDT Test
We seeded 1 × 104 SNP cells or EMT6 cells into each well of 96-well plates (Corning, Inc., New York, NY, USA) followed by overnight incubation. The cells were then incubated with various Mal3–TEG–Ce6 concentrations for 24 h at 37 °C. After washing with fresh media, the cells were irradiated with 671 nm light emitted by a semiconductor laser (Osada Electric Co., Ltd., Tokyo, Japan), using an optical fiber with a microlens delivery attachment (Pioneer Optics, Inc., Windsor Lock, CT, USA). PDT was performed at 7.3 mW/cm2 in cells exposed to four concentrations of Mal3–TEG–Ce6 (0, 0.16, 0.8, 4, and 20 μM) using a light dose of 5 J/cm2. The cells were then incubated for 24 h in the dark prior to examining the cell viability using the Cell Counting Kit-8 (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions.
Subcellular Localization
We cultured 1 × 105 EMT6 cells in a 35-mm Petri dish (Thermo Fisher Scientific, Waltham, MA, USA). The EMT6 cells were then incubated with Mal3–TEG–Ce6 at a final concentration of 20 μM in complete cell culture medium for 6 h, followed by coincubation with 50 nM LysoTracker Yellow HCK-123 (Invitrogen), 50 nM MitoTracker Green FM (Invitrogen), and 50 nM ER-Tracker Green (Invitrogen) and for an additional 30 min at 23 °C in the culture medium before fluorescence microscopy. The fluorescence of Mal3–TEG–Ce6 was detected with a filter (excitation, 405 nm; emission, 640 nm) using an all-in-one fluorescence microscope (BZ-X800, Keyence Co., Osaka, Japan). A BZ-X filter GFP (excitation, 470 nm; emission, 525 nm) was used to observe the mitochondria and lysosome; the green and red images were combined to form an overlay image.
Analysis of Apoptosis and ROS
The EMT6 cells were seeded at a density of 1.0 × 105 cells/well in 35 mm Petri dishes containing 2 mL of culture medium. Following 24 h of incubation, the cells were divided into the following groups: control (no treatment); laser (irradiated with a light dose of 15 J/cm2); Mal3–TEG–Ce6 (treated with 0.8 M Mal3–TEG–Ce6); and PDT (treated with 0.8 μM Mal3–TEG–Ce6 and then irradiated with a light dose of 1, 5, or 15 J/cm2). The cells were incubated with 0.8 μM Mal3–TEG–Ce6 for 4 h. After washing with fresh media, the cells were irradiated by a 671 nm laser light (8.3 mW/cm2; 1, 5, and 15 J/cm2) emitted by a DPPS laser (HangZhou NaKu Technology Co., Ltd., China, Zhejiang) using an optical fiber with a microlens delivery attachment. Apoptosis was assessed 4 h after the laser irradiation using the Muse Annexin V and Dead Cell Assay Kit (Merk Millipore, Germany) according to the manufacturer’s protocols. Annexin V was used to detect phosphatidylserine on the external membrane of the apoptotic cells. The ROS generation was assessed 4 h after laser irradiation using the Muse Oxidative Stress Kit (Merk Millipore, Germany) according to the manufacturer’s protocols; this kit determines the percentage of cells that are negative (healthy cells) and positive for ROS (cells containing ROS). Single-cell suspensions were then loaded onto the Muse Cell Analyzer (EMD Millipore Co.).
Acknowledgments
The author thanks the Yamagata University YU-COE(C) project.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c06316.
IR spectra of 2, 3, and Mal3–TEG–Ce6 and the 13C NMR spectra of 3 and Mal3–TEG–Ce6 (PDF)
Author Contributions
The manuscript was written through contributions of all the authors. All the authors have given approval to the final version of the manuscript.
This research was partly supported by JSPS KAKENHI grant numbers 25288028 and 18K05161.
The authors declare no competing financial interest.
Supplementary Material
References
- Yano S.; Hirohara S.; Obata M.; Hagiya Y.; Ogura S.-i.; Ikeda A.; Kataoka H.; Tanaka M.; Joh T. Current States and Future Views in Photodynamic Therapy. J. Photochem. Photobiol., C 2011, 12, 46–67. 10.1016/j.jphotochemrev.2011.06.001. [DOI] [Google Scholar]
- Ethirajan M.; Chen Y.; Joshi P.; Pandey R. K. The Role of Porphyrin Chemistry in Tumor Imaging and Photodynamic Therapy. Chem. Soc. Rev. 2011, 40, 340–362. 10.1039/b915149b. [DOI] [PubMed] [Google Scholar]
- Singh S.; Aggarwal A.; Bhupathiraju N. V. S. D. K.; Arianna G.; Tiwari K.; Drain C. M. Glycosylated Porphyrins, Phthalocyanines, and Other Porphyrinoids for Diagnostics and Therapeutics. Chem. Rev. 2015, 115, 10261–10306. 10.1021/acs.chemrev.5b00244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhupathiraju N. V. S. D. K.; Rizvi W.; Batteas J. D.; Drain C. M. Fluorinated Porphyrinoids as Efficient Platforms for New Photonic Materials, Sensors, and Therapeutics. Org. Biomol. Chem. 2016, 14, 389–408. 10.1039/c5ob01839k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnett R.; White R. D.; Winfield U. J.; Berenbaum M. C. Hydroporphyrins of the Meso-Tetra(Hydroxyphenyl)Porphyrin Series as Tumor Photosensitizers. Biochem. J. 1989, 261, 277–280. 10.1042/bj2610277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spikes J. D.; Bommer J. C. Photosensitizing Properties of Mono-L-Aspartyl Chlorin e6 (NPe6) - a Candidate Sensitizer for the Photodynamic Therapy of Tumors. J. Photochem. Photobiol., B 1993, 17, 135–143. 10.1016/1011-1344(93)80006-u. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Zheng X.; Dobhal M. P.; Gryshuk A.; Morgan J.; Dougherty T. J.; Oseroff A.; Pandey R. K. Methyl Pyropheophorbide-a Analogues: Potential Fluorescent Probes for the Peripheral-Type Benzodiazepine Receptor. Effect of Central Metal in Photosensitizing Efficacy. J. Med. Chem. 2005, 48, 3692–3695. 10.1021/jm050039k. [DOI] [PubMed] [Google Scholar]
- Sol V.; Blais J. C.; Bolbach G.; Carré V.; Granet R.; Guilloton M.; Spiro M.; Krausz P. Toward Glycosylated Peptidic Porphyrins: a New Strategy for PDT?. Tetrahedron Lett. 1997, 38, 6391–6394. 10.1016/s0040-4039(97)01489-5. [DOI] [Google Scholar]
- Maillard P.; Hery C.; Momenteau M. Synthesis, Characterization and Photocytotoxicity of a Glycoconjugated meso-Monoarylbenzochlorin. Tetrahedron Lett. 1997, 38, 3731–3734. 10.1016/s0040-4039(97)00711-9. [DOI] [Google Scholar]
- Mikata Y.; Onchi Y.; Shibata M.; Kakuchi T.; Ono H.; Ogura S.-i.; Okura I.; Yano S. Synthesis and Phototoxic Property of tetra- and octa-Glycoconjugated Tetraphenylchlorins. Bioorg. Med. Chem. Lett. 1998, 8, 3543–3548. 10.1016/s0960-894x(98)00645-3. [DOI] [PubMed] [Google Scholar]
- Sol V.; Blais J. C.; Carré V.; Granet R.; Guilloton M.; Spiro M.; Krausz P. Synthesis, Spectroscopy, and Photocytotoxicity of Glycosylated Amino Acid Porphyrin Derivatives as Promising Molecules for Cancer Phototherapy. J. Org. Chem. 1999, 64, 4431–4444. 10.1021/jo982499+. [DOI] [Google Scholar]
- Silva A. M. G.; Tomé A. C.; Neves M. G. P. M. S.; Silva A. M. S.; Cavaleiro J. A. S.; Perrone D.; Dondoni A. Porphyrins in 1,3-Dipolar Cycloaddition Reactions with Sugar Nitrones. Synthesis of Glycoconjugated Isoxazolidine-Fused Chlorins and Bacteriochlorins. Tetrahedron Lett. 2002, 43, 603–605. 10.1016/s0040-4039(01)02243-2. [DOI] [Google Scholar]
- Hirohara S.; Obata M.; Alitomo H.; Sharyo K.; Ogata S.-i.; Ohtsuki C.; Yano S.; Ando T.; Tanihara M. Structure-Photodynamic Effect Relationships of 24 Glycoconjugated Photosensitizers in HeLa Cells. Biol. Pharm. Bull. 2008, 31, 2265–2272. 10.1248/bpb.31.2265. [DOI] [PubMed] [Google Scholar]
- Hirohara S.; Obata M.; Alitomo H.; Sharyo K.; Ando T.; Tanihara M.; Yano S. Synthesis, Photophysical Properties and Sugar-Dependent in Vitro Photocytotoxicity of Pyrrolidine-Fused Chlorins bearing S-Glycosides. J. Photochem. Photobiol., B 2009, 97, 22–33. 10.1016/j.jphotobiol.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Singh S.; Aggarwal A.; Thompson S.; Tomé J. P. C.; Zhu X.; Samaroo D.; Vinodu M.; Gao R.; Drain C. M. Synthesis and Photophysical Properties of Thioglycosylated Chlorins, Isobacteriochlorins, and Bacteriochlorins for Bioimaging and Diagnostics. Bioconjugate Chem. 2010, 21, 2136–2146. 10.1021/bc100356z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M.; Kataoka H.; Mabuchi M.; Sakuma S.; Takahashi S.; Tujii R.; Akashi H.; Ohi H.; Yano S.; Morita A.; Joh T. Anticancer Effects of Novel Photodynamic Therapy with Glycoconjugated Chlorin for Gastric and Colon Cancer. Anticancer Res. 2011, 31, 763. [PubMed] [Google Scholar]
- Aggarwal A.; Thompson S.; Singh S.; Newton B.; Moore A.; Gao R.; Gu X.; Mukherjee S.; Drain C. M. Photophysics of Glycosylated Derivatives of a Chlorin, Isobacteriochlorin and Bacteriochlorin for Photodynamic Theragnostics: Discovery of a Two-photon-absorbing Photosensitizer. Photochem. Photobiol. 2014, 90, 419–430. 10.1111/php.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M.; Kataoka H.; Yano S.; Ohi H.; Moriwaki K.; Akashi H.; Taguchi T.; Hayashi N.; Hamano S.; Mori Y.; Kubota E.; Tanida S.; Joh T. Antitumor Effects in Gastrointestinal Stromal Tumors Using Photodynamic Therapy with a Novel Glucose-Conjugated Chlorin. Mol. Cancer Ther. 2014, 13, 767–775. 10.1158/1535-7163.mct-13-0393. [DOI] [PubMed] [Google Scholar]
- Hayashi N.; Kataoka H.; Yano S.; Tanaka M.; Moriwaki K.; Akashi H.; Suzuki S.; Mori Y.; Kubota E.; Tanida S.; Takahashi S.; Joh T. A Novel Photodynamic Therapy Targeting Cancer Cells and Tumor-Associated Macrophages. Mol. Cancer Ther. 2015, 14, 452–460. 10.1158/1535-7163.mct-14-0348. [DOI] [PubMed] [Google Scholar]
- Narumi A.; Tsuji T.; Shinohara K.; Yamazaki H.; Kikuchi M.; Kawaguchi S.; Mae T.; Ikeda A.; Sakai Y.; Kataoka H.; Inoue M.; Nomoto A.; Kikuchi J.-i.; Yano S. Maltotriose-Conjugation to a Fluorinated Chlorin Derivative Generating a PDT Photosensitizer with Improved Water-Solubility. Org. Biomol. Chem. 2016, 14, 3608–3613. 10.1039/c6ob00276e. [DOI] [PubMed] [Google Scholar]
- Nishie H.; Kataoka H.; Yano S.; Kikuchi J.-i.; Hayashi N.; Narumi A.; Nomoto A.; Kubota E.; Joh T. A Next-Generation Bifunctional Photosensitizer with Improved Water-Solubility for Photodynamic Therapy and Diagnosis. Oncotarget 2016, 7, 74259–74268. 10.18632/oncotarget.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda Y.; Takahashi T.; Akimoto J.; Ichikawa M.; Yamazaki H.; Narumi A.; Yano S.; Fujiwara Y. Comparative Photodynamic Therapy Cytotoxicity of Mannose-Conjugated Chlorin and Talaporfin Sodium in Cultured Human and Rat Cells. J. Toxicol. Sci. 2017, 42, 111–119. 10.2131/jts.42.111. [DOI] [PubMed] [Google Scholar]
- Warburg O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Zheng G.; Graham A.; Shibata M.; Missert J. R.; Oseroff A. R.; Dougherty T. J.; Pandey R. K. Synthesis of β-Galactose-Conjugated Chlorins Derived by Enyne Metathesis as Galectin-Specific Photosensitizers for Photodynamic Therapy. J. Org. Chem. 2001, 66, 8709–8716. 10.1021/jo0105080. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Zhang Z.; Blessington D.; Li H.; Busch T. M.; Madrak V.; Miles J.; Chance B.; Glickson J. D.; Zheng G. Pyropheophorbide 2-Deoxyglucosamide: A New Photosensitizer Targeting Glucose Transporters. Bioconjugate Chem. 2003, 14, 709–714. 10.1021/bc034038n. [DOI] [PubMed] [Google Scholar]
- Cieckiewicz E.; Mathieu V.; Angenot L.; Gras T.; Dejaegher B.; de Tullio P.; Pirotte B.; Frédérich M. Semisynthesis and in Vitro Photodynamic Activity Evaluations of Halogenated and Glycosylated Derivatives of Pheophorbide a. Eur. J. Org. Chem. 2015, 2015, 6061–6074. 10.1002/ejoc.201500387. [DOI] [Google Scholar]
- Nishie H.; Kataoka H.; Yano S.; Yamaguchi H.; Nomoto A.; Tanaka M.; Kato A.; Shimura T.; Mizoshita T.; Kubota E.; Tanida S.; Joh T. Excellent Antitumor Effects for Gastrointestinal Cancers Using Photodynamic Therapy with a Novel Glucose Conjugated Chlorin e6. Biochem. Biophys. Res. Commun. 2018, 496, 1204–1209. 10.1016/j.bbrc.2018.01.171. [DOI] [PubMed] [Google Scholar]
- Osaki T.; Hibino S.; Yokoe I.; Yamaguchi H.; Nomoto A.; Yano S.; Mikata Y.; Tanaka M.; Kataoka H.; Okamoto Y. A Basic Study of Photodynamic Therapy with Glucose-Conjugated Chlorin e6 Using Mammary Carcinoma Xenografts. Cancers 2019, 11, 636. 10.3390/cancers11050636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvagin V. F.; Kuzmina N. S.; Krylova L. V.; Volovetsky A. B.; Nyuchev A. V.; Gavryushin A. E.; Meshkov I. N.; Gorbunova Y. G.; Romanenko Y. V.; Koifman O. I.; Balalaeva I. V.; Fedorov A. Y. Water-Soluble Chlorin/Arylaminoquinazoline Conjugate for Photodynamic and Targeted Therapy. J. Med. Chem. 2019, 62, 11182–11193. 10.1021/acs.jmedchem.9b01294. [DOI] [PubMed] [Google Scholar]
- Shinoda Y.; Kujirai K.; Aoki K.; Morita M.; Masuda M.; Zhang L.; Kaixin Z.; Nomoto A.; Takahashi T.; Tsuneoka Y.; Akimoto J.; Kataoka H.; Rachi R.; Narumi A.; Yoshimura T.; Yano S.; Fujiwara Y. Novel Photosensitizer β-Mannose-Conjugated Chlorin e6 as a Potent Anticancer Agent for Human Glioblastoma U251 Cells. Pharmaceuticals 2020, 13, 316. 10.3390/ph13100316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togashi D.; Otsuka I.; Borsali R.; Takeda K.; Enomoto K.; Kawaguchi S.; Narumi A. Maltopentaose-Conjugated CTA for RAFT Polymerization Generating Nanostructured Bioresource-Block Copolymer. Biomacromolecules 2014, 15, 4509–4519. 10.1021/bm501314f. [DOI] [PubMed] [Google Scholar]
- Yue X.; Feng Y.; Yu Y. B. Synthesis and Characterization of Fluorinated Conjugates of Albumin. J. Fluorine Chem. 2013, 152, 173–181. 10.1016/j.jfluchem.2013.01.026. [DOI] [Google Scholar]
- Ancey P. B.; Contat C.; Meylan E. Glucose Transporters in Cancer - from Tumor Cells to the Tumor Microenvironment. FEBS J. 2018, 285, 2926–2943. 10.1111/febs.14577. [DOI] [PubMed] [Google Scholar]
- Jinadasa R. G. W.; Hu X.; Vicente M. G. H.; Smith K. M. Syntheses and Cellular Investigations of 173-, 152-, and 131-Amino Acid Derivatives of Chlorin e6. J. Med. Chem. 2011, 54, 7464–7476. 10.1021/jm2005139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K.; Tanaka S.; Yamamoto T.; Tajima K.; Borsali R.; Isono T.; Satoh T. Chain-End Functionalization with a Saccharide for 10 nm Microphase Separation: ″Classical″ PS-b-PMMA versus PS-b-PMMA-Saccharide. Macromolecules 2018, 51, 8870–8877. 10.1021/acs.macromol.8b02069. [DOI] [Google Scholar]
- Osaki T.; Sunden Y.; Sugiyama A.; Azuma K.; Murahata Y.; Tsuka T.; Ito N.; Imagawa T.; Okamoto Y. Establishment of a Canine Mammary Gland Tumor Cell Line and Characterization of Its miRNA Expression. J. Vet. Sci. 2016, 17, 385–390. 10.4142/jvs.2016.17.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.