

Abstract

A selective triazole-based COX-2 inhibitor, 4-(4-chlorophenyl)-3-[(4-fluorobenzyl)sulfanyl]-5-(thiophen-2-yl)-4H-1,2,4-triazole, C19H13ClFN3S2, has been synthesized, and its crystal structure was determined at 150 K. Single-crystal X-ray diffraction analysis revealed that the thiophene ring was disordered over two orientations. The crystal structure is stabilized by weak hydrogen and chalcogen bonds and unorthodox F···π and S···C(π) contacts. These noncovalent interactions cooperatively generate the supramolecular self-assembly in the crystalline state. The Hirshfeld surface and its associated two-dimensional (2D)-fingerprint plots were obtained to analyze the role of different noncovalent interactions in the crystal packing. Further, the enrichment ratio was obtained from different atom···atom pairs to calculate the propensity of these pairs to form noncovalent interactions. The strength of different dimeric motifs formed in the crystal structure and lattice energies was calculated by the PIXEL method. Furthermore, the topological analysis of the charge density of intermolecular interactions was described. A CSD survey of C–H···F hydrogen bond, C–S···Cl chalcogen bond, and unorthodox nonbonded contacts (F···π and S···C(π)) is presented. The title compound possesses selective inhibitory activity against human COX-2 enzyme rather than COX-1. The quantum mechanics (QM) polarized ligand docking analysis was used to predict the binding pose and study the title compound’s selectivity against COX-1/2 enzymes.
Introduction
1,2,4-Triazole heterocycles represent the essential core of clinically useful drugs.1 1,2,4-Triazole-based antifungal agents were early discovered as efficient drugs for treating topical and systemic fungal diseases. Fluconazole,2 Itraconazole,3 Terconazole,4 Voriconazole,5 Posaconazole,6 and Ravuconazole7 are the most frequently used antifungal drugs. The 1,2,4-triazole-based nucleosides Ribavirin and its derivatives Levovirin and Viramidine are clinically useful antiviral medications for treating chronic hepatitis C.8 1,2,4-Triazole derivatives were early recognized as anti-inflammatory agents.9 Besides, 1,2,4-triazole-based derivatives are clinically useful anticancer agents and the aromatase inhibitors Letrozole,10 Vorozole,11 and anastrozole12 are currently used for the treatment of breast cancer. Given the above medicinal importance, we synthesized the title compound, namely, 4-(4-chlorophenyl)-3-[(4-fluorobenzyl)sulfanyl]-5-(thiophen-2-yl)-4H-1,2,4-triazole.
Recently, the 1,2,4-triazole-based D−π–A system (4′-(4,5-diphenyl-4H-1,2,4-triazol-3-yl)-N,N-diphenyl- [1,1′-biphenyl]-4-amine) was designed, and it emitted true blue light with excellent electroluminescence performance.13 Zhou et al. described the crystal structures of a series of triaryl-1,2,4-triazoles in which benzimidazolyl, p-R-phenyl (R = H, Cl, Br, CH3, OCH3) and pyridyl substituents are present at 3, 4, and 5 positions, respectively. The intermolecular N–H···N, C–H···π, and C–H···Br/Cl interactions stabilize these structures.14 Loginova et al. showed that oxidation of polyfunctional sulfides with chlorine dioxide generates corresponding sulfoxides.15 Recently, Barman et al. designed a triazole derivative with pyrenes attached at 3 and 5 positions and characterized it as a molecular semiconductor.16 Some of the 1,2,4-triazole-3-thione- and 1,3,4-thiadiazole-based derivatives showed potential antibacterial activities.17
In our earlier study, we analyzed the crystal structure of a potential bioactive 1,2,4-triazole derivative and the molecular conformation of this molecule was locked by intramolecular C–S···F chalcogen bond and C–H···N interaction.18 The intermolecular interaction energies for dimers and charge density for these intermolecular interactions have been described in detail. Khan et al. investigated the role of electrostatic potential (ESP), deformation density, and energetics of hydrogen, chalcogen, and halogen bonds in crystalline triazolothiadiazole derivatives.19 Understanding the role of different noncovalent interactions such as hydrogen bonds of the types N/O–H···O/N,20 weak C–H···O/N/S/π interactions,21 halogen bonds,22 and chalcogen bonds23 in molecular solids is extremely important to design novel compounds with beneficial properties. The last two interactions belong to the attractive σ–hole interactions and received significant interest in different research areas, including structural chemistry, biology, and drug design.24
In this investigation, we analyzed the crystal structure of 4-(4-chlorophenyl)-3-[(4-fluorobenzyl)sulfanyl]-5-(thiophen-2-yl)-4H-1,2,4-triazole to understand the role of different noncovalent interactions in supramolecular self-assembly in the solid state. The energetics of the various motifs formed by hydrogen, chalcogen bonds, and other unorthodox nonbonded interactions in the title compound are presented based on Gavezzotti’s PIXEL energy.25 The Hirshfeld surface (HS) analysis26 was used to characterize the nature of the intermolecular interaction, and we also show the chemical species propensities in the title compound to form intermolecular interactions. The molecular electrostatic potential map was used to identify the complementary interacting sites in the title compound. Further, Bader’s quantum theory of atoms-in-molecules (QTAIM)27 approach was employed to study the nature and strength of intermolecular interactions formed in the crystal of the title compound. We also demonstrate the in vitro inhibitory potential of the title compound against COX-1/2 enzymes and its selectivity towards the COX-2 enzyme. The molecular docking analysis corroborates this experimental result.
Results and Discussion
Chemical Synthesis
The title compound 1 was prepared starting with thiophene-2-carbohydrazide A via reaction with 4-chlorophenyl isothiocyanate B, in ethanol to yield the corresponding 1,4-disubstituted thiosemicarbazide C, which was cyclized to the 1,2,4-triazole analogue D. Compound D was then reacted with 4-fluorobenzyl bromide E via stirring in N,N-dimethylformamide, in the presence of anhydrous potassium carbonate to yield the title compound 1 in 88% yield (Scheme 1). The structure confirmation of compound 1 was established based on 1H NMR, 13C NMR, and single-crystal X-ray diffraction data. The detailed reaction conditions, crystallization, and 1H and 13C NMR data in dimethylsulfoxide (DMSO)-d6 are shown in the Experimental Section.
Scheme 1. Synthetic Pathway for the Title Compound 1.

Crystal and Molecular Structures of 1
The title compound crystallizes in the orthorhombic crystal system with noncentrosymmetric space group P212121. The crystal data and refinement parameters of the title compound are summarized in Table 1. The title compound comprises two five-membered rings (thiophene and 1,2,4-triazole) and two aryl rings (4-chlorophenyl and 4-fluorophenyl). In the crystalline state, the thiophene ring was disordered over two orientations with the site-occupancy value of 0.832(5) for the major disordered component. The thermal ellipsoid plots for the major (Figure 1a) and minor disordered (Figure 1b) components of the title compound are illustrated. In the major disordered component, the central triazole ring is oriented at an angle of 31.14° with respect to the mean plane of the thiophene ring. The 4-chlorophenyl ring makes a right angle (88.79°) with respect to the central triazole ring. The dihedral angle between triazole and 4-fluorophenyl rings is 75.81°.
Table 1. Crystal Data and Refinement Parameters of the Title Compound.
| crystal data | |
| empirical formula | C19H13ClFN3S2 |
| formula weight | 401.89 |
| crystal system, space group | orthorhombic, P212121 |
| temperature (K) | 150(2) |
| a, b, c (Å) | 5.2534 (3) |
| 12.4128 (5) | |
| 27.8469 (10) | |
| α, β, γ (deg) | 90, 90, 90 |
| V (Å3) | 1815.88 (14) |
| Z | 4 |
| radiation type | Cu Kα (λ = 1.54184) |
| μ (mm–1) | 4.17 |
| crystal size (mm3) | 0.31 × 0.08 × 0.06 |
| data collection | |
| diffractometer | Xcalibur, Ruby, Gemini diffractometer |
| absorption correction | multiscan |
| Tmin, Tmax | 0.704, 0.779 |
| no. of measured, independent, and observed [I > 2σ (I)] reflections | 10543, 3548, 3018 |
| Rint | 0.049 |
| (sin θ/λ)max (Å–1) | 0.628 |
| refinement | |
| R[F2 > 2σ(F2)], wR(F2), S | 0.043, 0.101, 1.03 |
| no. of reflections | 3548 |
| no. of parameters/restraints | 272/150 |
| H-atom treatment | H-atom parameters constrained |
| Δρmax, Δρmin (e Å–3) | 0.28, −0.22 |
| absolute structure | Flack x determined using 997 quotients28 |
| absolute structure parameter | –0.035 (16) |
| CCDC no. | 2050370 |
Figure 1.

Displacement ellipsoids are drawn at the 40% probability level for (a) major and (b) minor disordered components of the title compound and (c) structural overlay between major disordered component (X-ray: gray) and optimized structures of major (green) and minor (orange) disordered components.
Structural optimization was performed for both major and minor disordered components to identify the most stable thiophene ring orientation at the M062X-D3/cc-pVTZ level of theory. The vibrational frequency analysis of these optimized structures showed no imaginary frequency, confirming that they are minimum energy in their potential energy surface. The energy difference between these two conformers is as low as 0.4 kcal mol–1. Further, we superimposed optimized structures onto the X-ray conformation of the title compound (Figure 1c), and it reveals deviation observed in the substituted phenyl rings. This structural deviation could be correlated with the effect of crystal packing.
Hirshfeld Surface (HS) Analysis and Two-Dimensional (2D)-Fingerprint Plots
The major disordered component of the title compound was used for the HS analysis to characterize intermolecular interactions. The Hirshfeld surface (Figure 2a) and 2D-fingerprint plots (Figure 2b) for selected intercontacts are shown. The bright red spots appear for the short C–H···F, C–H···N, C–H···S, and C–H···π hydrogen bonds, C–S···Cl chalcogen bond, and an unorthodox S···C(π) contact.
Figure 2.

(a) Two different views of the Hirshfeld surfaces highlight the short intercontacts and (b) 2D-fingerprint plots for selected intercontacts observed in the title compound.
The decomposed 2D-fingerprint plots (2D-FP) and Table 2 show that the H···C (24.2%) and H···H (23.9) intercontacts contribute relatively higher compared to other interactions. It is evident from 2D-FP that the H···C contact, a representative of C–H···π interaction, is located around ∼2.6 Å (de + di). The next significant contribution arises from H···Cl contacts with 12.7% toward the crystal packing. The shortest intercontact distance for this pair located beyond 3 Å, which is longer than the sum of the vdW radii. This feature indicates that H···Cl contacts do not play an important role in the stabilization of the crystal structure.
Table 2. Hirshfeld Contact Surfaces, Derived Random Contacts, and Enrichment Ratios for the Title Compound.
| atoms | H | C | Cl | S | N | F |
|---|---|---|---|---|---|---|
| H | 23.9 | contact (%) | ||||
| C | 24.2 | 0.7 | ||||
| Cl | 12.7 | 0.4 | ||||
| S | 10.5 | 2.6 | 1 | |||
| N | 10.4 | 0.3 | 1.8 | |||
| F | 7.8 | 3.6 | ||||
| surface % | 56.7 | 16.3 | 7.0 | 8.0 | 6.3 | 5.7 |
| H | 32.1 | random contacts (%) | ||||
| C | 18.5 | 2.7 | ||||
| Cl | 7.9 | 2.3 | 0.5 | |||
| S | 9.1 | 2.6 | 1.1 | 0.6 | ||
| N | 7.1 | 2.1 | 0.9 | 1.0 | 0.4 | |
| F | 6.5 | 1.9 | 0.8 | 0.9 | 0.7 | 0.3 |
| H | 0.7 | enrichment ratio | ||||
| C | 1.3 | 0.3 | ||||
| Cl | 1.6 | 0.2 | ||||
| S | 1.2 | 1.0 | 0.9 | |||
| N | 1.5 | 0.1 | 1.8 | |||
| F | 1.2 | 1.9 | ||||
The intermolecular H···N/S interactions contribute nearly equally. The former contacts primarily represent C–H···N interactions and are located around 2.3 Å, while the latter contact, i.e., C–H···S interaction, is observed around 2.8 Å. These two contacts are less than the sum of the vdW radii of the interacting pair of atoms, and they are shown as red spots on the HS due to the short contact distances. The intermolecular H···F interactions appear as sharp double spikes with a de + di value of ∼2.2 Å. The chalcogen bond of the type S···Cl contributes about 1.0% to the total HS area. It is worthy to note that two unorthodox nonbonded F···C(π) and S···C(π) contacts contribute 3.6 and 2.6%, respectively, toward the crystal packing.
To identify the preferred contact partners, we have obtained the enrichment ratio from the decomposition of the crystal contacts surface between pairs of interacting atoms as described earlier.29 The enrichment ratio highlights the F···C(π) contact (1.9), which turns out to be favored in the crystal structure rather than the slightly enriched S···Cl chalcogen bond (0.9). All of the hydrogen bonds, including H···C/Cl/S/N/F contacts, show enrichment values larger than unity. In the case of S···C(π) contact, the enrichment value is unity. However, the S···N contact is found to be highly enriched with the value of 1.8. The existence of these noncovalent interactions was confirmed by the QTAIM analysis and discussed in detail in a later section.
Crystal Packing
The major disordered component of compound 1 was used for analysis. The molecules of 1 assemble as double layers projected onto the bc plane in the crystalline state. The PIXEL energy analysis suggests that there are six dimers (dimeric motifs: I–VI) found to be significant in the solid state, and these dimers are held together by different types of noncovalent interactions, including hydrogen bonds (C–H···N, C–H···S, C–H···π, and C–H···F), a chalcogen bond (C–S···Cl), and unorthodox short contacts such as S···C(π) and F···π contacts. The net interaction energy (Etot) for these dimers and its components (calculated by the PIXEL method with the MP2/6-31G** level of theory) along with geometrical parameters for intermolecular interactions observed in these dimers are summarized in Table 3. The basis set superposition error (BSSE)-corrected binding energy (ΔEcp) for the dimers calculated by the counterpoise method (with the M062X-D3/cc-pVTZ level of approximation) and the Etot values are comparable.
Table 3. Intermolecular Interaction Energies for Dimers along with Interaction Geometrical Parameters and Lattice Energy for the Crystal Structure of Compound 1a.
| PIXEL
method at MP2/6-31G** |
DFT at M062X-D3/cc-PVTZ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| motif | CD | symmetry | important interactions | geometryb H···A (Å), ∠D–H···A (deg) | ECoul | Epol | Edisp | Erep | Etot | ΔEcp |
| compound 1 (major disordered component) | ||||||||||
| I | 5.253 | x – 1, y, z | C8–H8···Cg1 | 2.31, 174 | –7.8 | –5.2 | –19.5 | 18.2 | –14.3 | –15.5 |
| S1A···C1(π) | 3.396 (2) | |||||||||
| II | 7.977 | x – 1/2, −y + 1/2, −z + 1 | C2–S2···Cl1 | 3.403 (2), 171.05 (2) | –3.1 | –1.8 | –10.3 | 7.4 | –7.8 | –5.9 |
| III | 10.408 | –x + 2, y – 1/2, −z + 1/2 | C6A–H6A···N2 | 2.34, 156 | –5.0 | –2.7 | –7.0 | 8.2 | –6.5 | –5.1 |
| C6A–H6A···N1 | 2.41, 125 | |||||||||
| C5A–H5A···S1A | 3.00, 150 | |||||||||
| IV | 9.243 | –x + 1, y – 1/2, −z + 1/2 | C19–H19···Cg1 | 2.85, 169 | –2.7 | –1.7 | –6.3 | 4.7 | –6.0 | –5.2 |
| C13–H13B···C6A | 2.79, 157 | |||||||||
| V | 12.413 | x, y – 1, z | F1···Cg2 | 3.676 (2) | –0.5 | –0.4 | –4.5 | 1.4 | –3.9 | –3.2 |
| VI | 13.479 | –x + 1, −y – 1, −z + 1 | C9–H9···F1 | 2.23, 169 | –1.7 | –0.4 | –1.4 | 1.6 | –1.8 | –1.7 |
| lattice energy | –21.8 | –11.5 | –52.1 | 41.7 | –43.7 | |||||
All of the energies are expressed in kcal mol–1.
Neutron values are given for all D–H···A interactions. CD: centroid-to-centroid distance of the molecular pair. Cg1 and Cg2 are the centroids of the thiophene and 4-chlorophenyl rings, respectively.
As shown in Figure 3a, the most stabilized molecular dimer (motif I, Etot: −14.3 kcal mol–1) is formed by a short C–H···π interaction (involving centroid of the thiophene ring and CH group of the 4-chlorophenyl ring) and also supported by an unorthodox contact of the type S···C(π). These interactions link the molecules into a chain that runs parallel to the crystallographic a axis. The electrostatic energy contributes about 40% toward the stabilization of this dimer. To gain more insights into the nature of the noncovalent bond, the molecular electrostatic potential (ESP) was mapped on the Hirshfeld surface. The ESP map reveals that the negative electrostatic potential region over the thiophene S1A atom interacts with the positive electrostatic potential region on the C1 atom of the adjacent triazole ring (Figure 3b).
Figure 3.

Dimers observed in compound 1. (a) Dimer I, (b) decomposition of Hirshfeld surface mapped over electrostatic potential showing the complementary region of S1A and C1 atoms, (c) dimer II, (d) electrostatic potential map showing σ-hole around S2 atom (positive region) and the negative belt around Cl atom in dimer II, and (e) deformation density map showing charge concentration (CC) (blue) region near Cl atom and charge depletion (red) region near S atom in dimer II. The electrostatic potential and deformation density were calculated at the HF/6-311G(d,p) level of theory.
The dimer II stabilizes by a chalcogen bond C–S···Cl (involving S2 and Cl atoms; Etot: −7.8 kcal mol–1; Figure 3c). The molecule in one layer interacts with the molecule in an adjacent layer via this chalcogen bond. The dispersion energy contributes (68%) more than 2-fold than electrostatic energy (32%) toward stabilizing this dimer. The ESP map indicates the presence of a σ-hole and the positive electrostatic potential observed on the S2 atom with the value of +0.038 au. The positive region interacts with one of the negative belt sites around the Cl atom with the negative ESP value −0.012 au (Figure 3d). To gain further insights into this chalcogen bond’s nature, we computed deformation density for the title molecule. This plot reveals the charge depletion (CD) region of the S2 atom that is directed toward the charge concentration (CC) region around the Cl1 atom, facilitating the formation of a C–S···Cl chalcogen bond (Figure 3e).
Two intermolecular C–H···N (involving one of the CH of the thiophene ring and N1/N2 of triazole ring) interactions and an intermolecular C–H···S interaction (involving H5A and S1A atoms; Figure 4a) produce dimer III. The contribution of electrostatic energy is about 52% toward the stabilization of this dimer. We note that these three interactions link the molecules into a zigzag chain in which molecules in one layer interlinked with molecules in an adjacent layer. These chains run parallel to the b axis. The negative ESP values for N1 and N2 atoms are −0.075 and −0.065 au, respectively, and these atoms interact with the most positive region of the thiophene ring.
Figure 4.

(a) Dimer III stabilized by bifurcated C–H···N and C–H···S interactions, (b) dimer IV forms by C–H···π interactions and decomposed H···C contacts showing complementary regions near H and thiophene ring, and (c) F···π contact stabilizes dimer IV and the ESP mapped over Hirshfeld surface (0.007 au (blue); 0 au (white); and −0.007 au (red)).
Dimer IV (Etot: −6.0 kcal mol–1) is stabilized by two C–H···π interactions, in which CH of the phenyl and methylene groups are involved as donors and the thiophene ring acts as a π-acceptor. The role of these interactions primarily is very similar to that of the interactions observed in dimer III. The dispersion energy contributes about 59% to the stabilization of dimer IV. The thiophene ring’s negative region interacts with the complementary positive region on the CH groups of methylene and 4-chlorophenyl ring (Figure 4b).
The organic fluorine has involved in an unorthodox nonbonded contact with the π-hole of the 4-chlorophenyl ring. This F···π contact with a separation of 3.676 (2) Å forms the dimer V with the net intermolecular interaction energy of −3.9 kcal mol–1 (Figure 4c). This noncovalent interaction links the molecules in the same layer. Furthermore, this dimer is predominantly dispersive, with an 83% contribution to the stabilization. The ESP map reveals a σ-hole with the most positive ESP value of −0.039 au on the C–F bond’s outer surface and the negative belt around the F atom with the negative ESP value −0.041 au. In dimer V, the π-hole at the center of one side of the 4-chlorophenyl ring with a relatively small positive ESP value of 0.0037 au interacts with the F atom’s negative region. These features support the dispersive origin of the formation of dimer V. The directional C(sp3)–F···π type of interaction was observed in substituted benzanilides containing fluorine and trifluoromethyl group.30 This interaction was either formed solely or presented along with other interactions.
The least dimer (VI; Etot: −1.8 kcal mol–1; 60% of electrostatic energy contributes towards stabilization) in this structure is formed by an intermolecular C–H···F hydrogen bond [2.23 Å and 169°], which links the molecules into a chain forming a layer. This layer interconnects with an adjacent layer by a chalcogen bond (motif II). The resulting supramolecular architecture generates a double layer. Overall, the hydrogen and chalcogen bonds and unorthodox nonbonded motifs are cooperatively involved in the generation of supramolecular architecture for compound 1 in the solid state (Figure 5). As shown in Table 3, the lattice energy calculation using the PIXEL method reveals that dispersion and electrostatic energies contribute 61 and 39% to the stabilization of the crystal structure, respectively.
Figure 5.

Crystal structure of compound 1 viewed down the a axis. The hydrogen and chalcogen bonds and other unorthodox noncovalent interactions cooperatively generate supramolecular sheet. Dimers are labeled, and small red and blue spheres correspond to the centroid of the respective ring.
Topological Analysis of Electron Density of Noncovalent Interactions
We calculated the topological parameters for all six dimers to
delineate the nature and strength of noncovalent interactions in compound 1. Table 4 summarizes
selected the topological parameters for different noncovalent interactions
observed in dimers, and the molecular graphs for these dimers are
shown (see Figure S1, Supporting Information,
SI). All of the interactions observed in this structure belong to
closed-shell interactions as evident from the value of 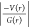 < 1, the positive
value of the Laplacian
of electron density (∇2ρ(r) > 0), and the total electronic energy density, H(r) > 1.31
< 1, the positive
value of the Laplacian
of electron density (∇2ρ(r) > 0), and the total electronic energy density, H(r) > 1.31
Table 4. Topological Parameters for Intermolecular Interactions (in Different Dimers of Compound 1) at Their (3, −1) BCPsa.
| interaction | Rij | ρ(r) | ∇2ρ(r) | V(r) | G(r) | H(r) | 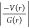 |
De |
|---|---|---|---|---|---|---|---|---|
| I | ||||||||
| C8–H8···C3A | 2.928 | 0.079 | 0.981 | –18.8 | 22.8 | 4.0 | 0.83 | 2.3 |
| S1A···C1 | 3.414 | 0.053 | 0.613 | –11.4 | 14.1 | 2.6 | 0.81 | 1.4 |
| II | ||||||||
| S2···Cl1 | 3.421 | 0.059 | 0.737 | –12.3 | 16.2 | 3.9 | 0.76 | 1.5 |
| III | ||||||||
| C6A–H6A···N2 | 2.362 | 0.079 | 1.163 | –20.0 | 25.8 | 5.9 | 0.77 | 2.4 |
| C6A–H6A···N1 | 2.451 | 0.073 | 1.167 | –18.8 | 25.3 | 6.5 | 0.74 | 2.3 |
| C5A–H5A···S1A | 3.021 | 0.035 | 0.408 | –6.1 | 8.6 | 2.5 | 0.71 | 0.7 |
| IV | ||||||||
| C19–H19···S1A | 2.818 | 0.060 | 0.645 | –12.0 | 14.8 | 2.8 | 0.81 | 1.4 |
| C13–H13B···C6A | 2.810 | 0.042 | 0.437 | –7.4 | 9.7 | 2.2 | 0.77 | 0.9 |
| V | ||||||||
| F1···C10(π) | 3.839 | 0.019 | 0.261 | –3.9 | 5.5 | 1.6 | 0.70 | 0.5 |
| VI | ||||||||
| C9–H9···F1 | 2.249 | 0.068 | 1.149 | –19.1 | 25.2 | 6.1 | 0.76 | 2.3 |
Definitions: Rij, bond path (Å); ρ(r), electron density (e Å–3); ∇2ρ(r), Laplacian of electron density (e Å–5); V(r), potential electron density (kJ mol–1 br–3); G(r), kinetic electron density (kJ mol–1 br–3); H(r), total electronic energy density (kJ mol–1 br–3); De, dissociation energy (kcal mol–1).
The dissociation energies (De) for C8–H8···C3A, C6A–H6A···N2/N1, and C9–H9···F1 interactions are nearly the same and strong among other interactions. The bond path (Rij) for H···F is shorter with a distance of 2.249 Å in compound 1 compared to the corresponding contact (2.655 Å) in one of the 1,2,4-triazole derivatives, namely, 6-(2-chloro-4,5-difluorophenyl)-3-(pyridin-3-yl)[1,2,4]-triazolo[3,4-b][1,3,4]thiadiazole.19 The dissociation energy for the H···F interaction (2.4 kcal mol–1) in compound 1 is more than 2-fold compared to the related reported compound (1.1 kcal mol–1). The strength of the S···C(π) contact observed in dimer I is equal to the chalcogen bond S···Cl observed in dimer II and a C–H···S interaction in dimer IV. We also note that the F···C(π) contact is 3-fold weaker than the S···Cl chalcogen bond and also weaker than the F···C(π) interactions reported earlier.30
CSD Survey of Noncovalent Interactions
To understand the existence and the geometrical preferences of some of the noncovalent interactions observed in compound 1, we performed a CSD search using the fragments illustrated in Scheme 2. A CSD search resulted in 5145 hits, leading to 9587 C–H···F hydrogen-bonded motifs when X was H (case 1). We also found that there were 1901 (2733 C–H···F contacts) and 102 hits (114 C–H···F contacts) identified from the CSD for X = any halogen (case 2) and Cl (case 3), respectively. The significant reduction in the number of hits in the third case (X = Cl) suggests that the C–H···F hydrogen-bonded motif observed in the title compound has packing constraints in the solid state in comparison to the other two cases (X = H and any halogen). The scatter plots showing the distribution of H···F distance and C–H···F angle are given for all three cases in Figure S2 (SI). The scatter plots reveal that the H···F contacts well distributed in the range of 2.4–2.65 Å and the C–H···F angle is distributed over 120°.
Scheme 2. Different Fragments Showing Different Motifs Observed in Compound 1 Used for the CSD Survey.

The CSD survey of C–F···π contact shorter than 3.8 Å yielded 5426 hits providing 15137 contacts. The scatter plot shows that more points localized between 3.1 and 3.8 Å and angle range from 70 to 140° (Figure S3, SI). This contact (3.676 (2) Å) observed in compound 1 lies within this limit. A survey of a chalcogen contact, namely, C–S···Cl from the CSD, resulted in 98 hits with 109 contacts, and more contacts are located above 3.40 Å. However, the C–Cl···S angle is distributed in three different regions (< 120, 120–140, and > 150°) as shown in Figure S4 (SI). It is to be noted that only 27 contacts (∼25%) show directional (> 150°) among 109 contacts, and it indicates that this one of the rare chalcogen bonds. In compound 1, the C–S···Cl-type chalcogen bond is highly directional. The S···C(π) contact is observed in four structures (csd refcodes: TOSYEI, PACVUL, QIQGOP, and VUYPIO)32 with a distance range of 3.371–3.488 Å.
In Vitro COX Inhibition Assay and Quantum Mechanics (QM) Polarized Ligand Docking Analysis
It has been reported that nonsteroidal anti-inflammatory drugs were commonly used to treat inflammation, pain, and fever. These drugs suppress the activities of two distinct isoforms of cyclooxygenase, namely, COX-1 and COX-2 enzymes. Moreover, the nonselective COX inhibitors show severe side effects, including stomach bleeding and gastric ulcers. Therefore, the development of selective COX inhibitors with low side effects is an important medicinal chemistry problem. As mentioned earlier, the 1,2,4-triazole-containing compounds are recognized as anti-inflammatory agents. In this study, we performed in vitro COX inhibition assay for the title compound, which is also a nonsteroidal compound. Table 5 summarizes the in vitro activity of the title compound against two isoforms of COX and the selectivity index (SI). The assay results suggest that compound 1 exhibits marked COX inhibitory activity with almost double selectivity toward COX-2 (SI = 1.89), compared to the selective COX-2 inhibitor celecoxib (SI = 308.57) and the nonselective COX inhibitor diclofenac (SI = 0.90).
Table 5. In Vitro COX-1 and COX-2 Inhibitory Activities and the COX-2 Selectivity Index.
| IC50 (μM)a |
|||
|---|---|---|---|
| COX-1 | COX-2 | SIb | |
| compound 1 | 8.04 | 4.26 | 1.89 |
| celecoxib | 21.60 | 0.07 | 308.57 |
| diclofenac | 2.72 | 3.02 | 0.90 |
IC50 value is the compound concentration required to produce 50% inhibition of COX-1 or COX-2 for means of three determinations.
Selectivity index (IC50 COX-1/IC50 COX-2).
To find a binding mode of compound 1 at the active site of the COX-1 and COX-2 enzymes and the selectivity, we have performed a flexible ligand docking simulation with glide XP scoring scheme, followed by a QM polarized ligand docking (QPLD) approach. The glide XP scores for compound 1 with COX-1 and COX-2 enzymes are −2.796 and −7.317 kcal mol–1, respectively. This glide XP score suggests that compound 1 is selective against the COX-2 enzyme rather than COX-1. As shown in Figure 6, compound 1 makes a cation−π stacking interaction, in which Arg 120 residue of COX-1 and thiophene ring of compound 1 have participated. In COX-2, the active site residue Tyr 355 makes two π-stacking interactions with the 4-chlorophenyl ring and the thiophene ring of compound 1.
Figure 6.

Predicted binding modes of the title compound at the active sites of (a) COX-1 and (b) COX-2.
Our previous study reported the glide scores for control inhibitors with COX-1/2 enzymes,18 i.e., mefanamic acid with COX-2: −3.813 kcal mol–1 and ibuprofen with COX-1: −8.525 kcal mol–1. Overall, the title compound shows a relatively better binding potential against COX-2 than the control inhibitor mefenamic acid and displays selectivity against the COX-2 enzyme. The in silico molecular docking analysis is in good agreement with the experimental in vitro data.
Conclusions
A promising triazole-based COX-2 inhibitor, namely, 4-(4-chlorophenyl)-3-[(4-fluorobenzyl)sulfanyl]-5-(thiophen-2-yl)-4H-1,2,4-triazole, has been synthesized, and its crystal structure has been analyzed in detail. The Hirshfeld surface analysis revealed the nature of the C–H···F, C–H···N, C–H···S, and C–H···π hydrogen bonds, a rare C–S···Cl chalcogen bond, and an unorthodox S···C(π) contact. The C–H···π hydrogen bonds contribute more in comparison to the other interactions toward the crystal packing. Though F···C(π) and S···Cl contacts contribute only 3.6 and 1.0% to the total HS area, their enrichment ratio was ≥0.9, suggesting that the former contact is more favorable than the latter contact. We also found that the enrichment ratio for F···C(π) was found to be 1.9 and highest among other contacts observed in compound 1. Further, the enrichment ratio for the unorthodox S···C(π) contact is also likely to form. All of the possible hydrogen bonds, such as C–H···N/S/F/Cl/C bonds, have an enrichment ratio of more than 1, indicating their possibility of making these contacts in the solid state. The electrostatic potential map revealed σ-hole on the halogens (Cl and F) and the S atoms. The self-assembly of compound 1 mediated by different types of hydrogen bonds, a rare chalcogen bond, and unorthodox S···C(π) and F···C(π) contacts acted together to generate a supramolecular sheet. The PIXEL energy analysis showed that the strong dimer was formed by a C–H···π interaction with further support from a S···C(π) contact. The topological analysis revealed that all of the intermolecular interactions observed in this structure belong to closed-shell interactions. The strength of the C–H···F/N/C interactions was found to be similar. We found that a chalcogen bond and a S···C(π) contact provided significant stability to motifs formed in the solid state. The experimental and theoretical studies on the inhibitory activity of the COX-2 enzyme agreed well.
Experimental Section
Instruments and Materials
Melting point (°C, uncorrected) was measured in open glass capillaries using a Branstead 9100 melting point apparatus. NMR spectra were recorded at room temperature on a Jeol ECA 500 III NMR spectrometer at 500.16 MHz for 1H and 125.77 MHz for 13C, using DMSO-d6 as a solvent. Monitoring the reactions and checking the purity of the final product were carried out by thin-layer chromatography (TLC) using silica-gel-precoated aluminum sheets (60 F254, Merck) and visualization with ultraviolet light (UV) at 365 and 254 nm. All chemicals and solvents were used as purchased without further purification. The standard COX inhibitors celecoxib (CAS #169590-42-5) and diclofenac sodium (CAS #15307-79-6) were purchased from Sigma-Aldrich Co.
Synthesis and Crystallization
A mixture of 4-(4-chlorophenyl)-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-thiol D (2.96 g, 0.01 mol), 4-fluorobenzyl bromide E (1.89 g, 0.01 mol), and anhydrous potassium carbonate (1.38 g, 0.01 mol), in N,N-dimethylformamide (10 mL), was stirred at room temperature for 24 h. Water (15 mL) was gradually added, and the reaction mixture was stirred for further 1 h. The precipitated crude product was filtered, washed with water, dried, and crystallized from aqueous ethanol to yield 3.54 g (88%) of the title compound 1. Mp 231–233 °C. 1H NMR (DMSO-d6, 500.16 MHz): δ 4.49 (s, 2H, CH2), 6.98–7.17 (m, 2H, Ar-H), 7.15 (d, 2H, Ar-H, J = 8.0 Hz), 7.33–7.36 (m, 2H, Ar-H), 7.41 (d, 1H, Thiophene-H, J = 4.5 Hz), 7.51–7.55 (m, 4H, 2 Ar-H & 2 thiophene-H). 13C NMR (DMSO-d6, 125.77 MHz): δ 39.50 (SCH2), 118.62, 124.82, 125.55, 128.84, 128.57, 129.0, 130.01, 131.03, 131.07, 131.10, 143.20, 159.08 (Ar-C & Thiophene-C), 137.51, 154.32 (Triazole C).
Single-Crystal X-ray Diffraction
A suitable single crystal of the title compound was chosen for X-ray intensity data collected at 150 K on an Xcalibur, Ruby, Gemini diffractometer. Before experiment, data collection, data reduction, and analytical absorption correction33 were performed with the program suite CrysAlisPro (Rigaku Oxford Diffraction). The structure was solved with the SHEXT-2014/5 program,34 and the refinement was performed with the SHELXL 2018/3 program.35 The thiophene ring was disordered over two orientations with a refined occupancy ratio of 0.832(5):0.168(5). The EADP and EXYZ constraints were applied to the disordered thiophene ring, and the similar atomic displacement parameter (ADP) restraint SIMU was also used to make the ADP values of the disordered atoms more reasonable. Further, all of the H atoms were placed in geometrically idealized positions (C–H = 0.95–0.99 Å) and were constrained to ride on their parent atoms with Uiso(H) = 1.2Ueq(C). During the final refinement, the reflections 6 3 2 and 6 4 1 were omitted due to [Error/esd] > 10.
The crystal packing and dimeric structures were produced using the program MERCURY.36 The specific motifs were searched in the Cambridge Structural Database (CSD version 5.41, updated March 2020) using the following criteria: (i) three-dimensional (3D)-coordinates determined, (ii) nondisordered, (iii) no errors, (iv) not polymeric, (v) no ions, (vi) single-crystal structures, and (vii) only organics.37
Computational Details
Intermolecular interactions present in the title compound were analyzed using the Hirshfeld surface,38 and 2D-fingerprint plots39 were obtained with the CrystalExplorer-17.5 package.40 For the analysis, we considered only the title compound’s major disordered component with its occupancy. Further, H atom’s distances moved to their typical neutron diffraction values (C–H = 1.083 Å). The electrostatic potential map and deformation density map were obtained at the HF/6-311G(d,p) level of theory using CrystalExplorer program.41
The CLP program25,42 was used to calculate the lattice energy for the title compound’s crystal structure. We also obtained intermolecular interaction energies (Etot) for various dimers observed in the crystal structure using the CLP program’s PIXELC module. This calculation required electron density for the title compound, and it was obtained at the MP2/6-31G** level of theory using the Gaussian 09 package.43 The sum of the Coulombic (ECoul), polarization (Epol), dispersion (Edisp), and repulsion (Erep) energies gave the lattice energy for the crystal structure and the net intermolecular interaction energy for dimers. The binding energies for dimers observed in the crystal structure were also calculated using the counterpoise method44 with the M062X/cc-pVTZ level of theory,45 and Grimme’s empirical dispersion correction (D3) was incorporated.46 The binding energies (ΔEcp) were also corrected for basis set superposition error (BSSE).
The theoretical charge density analysis was performed for the dimers obtained using the crystal structure geometry with normalized H-positions with the AIMALL package.47 The wave functions were generated for these dimers at the M062X-D3/cc-pVTZ level of theory. Selected topological parameters such as electron density (ρ(r)), Laplacian of the electron density (∇2ρ(r)), potential energy density (V(r)), kinetic energy density (G(r)), and total electronic energy density (H(r) = V(r) + G(r)) were used at their bond critical points (BCPs) to characterize the nature and strength of intermolecular interactions. The dissociation energy for interaction was calculated using the EML empirical scheme, i.e., De = −0.5 × V(r) to assess the strength of the interaction.48
In Vitro COX Inhibition Assay
The in vitro inhibitory activity of compound 1 against cyclooxygenases COX-1 and COX-2 was evaluated using COX inhibitor screening assay kit of Cayman Chemical, Ann Arbor, MI (Catalog No. 560131). The preparation of the reagents and the testing procedure was performed according to the manufacturer recommendations using various concentrations (0.01–100 μM) of compound 1, celecoxib (selective COX-2 inhibitor), and diclofenac (nonselective COX inhibitor) in dimethylsulfoxide (DMSO). The concentration that causes 50% enzyme inhibition (IC50) was calculated from the concentration inhibition response curve, and the selectivity index (SI) was calculated by dividing IC50 COX-1 by IC50 COX-2.
Molecular Docking
The anti-inflammatory drug targets such as COX-1 and COX-2 structures were used for molecular docking analysis to assess the inhibitory potential and its selectivity. The 3D structures of human COX-1 (pdb id: 6Y3C) and human COX-2 (pdb id: 5IKR) were retrieved from the Protein Data Bank (www.rcsb.org). The molecular docking simulation was performed using Schrödinger suite 2019-4 (Schrödinger, LLC, New York, NY, 2019). The proteins and title ligands were prepared for the docking analysis by modules of protein prepwizard and ligprep, respectively. The OPLS-3e force field was used for both protein and ligand molecules.49 After model preparations, the initial molecular docking was performed with the glide XP approach.50 The predicted poses were further subjected to QM polarized ligand docking (QPLD) analysis to get an accurate docking score and pose.51
Acknowledgments
This research was funded by the Deanship of Scientific Research at Princess Nourah bint Abdulrahman University through the Research Groups Program (grant no. RGP-1442-0010-4). S.T. and M.J.P. thank Laboratorio Nacional de Supercomputo del Sureste (LNS-BUAP) for computational resources.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c06287.
Molecular graphs of dimers showing bond critical points for intermolecular interactions and distribution of distance and angles for C–H···F, C–F···Cg(π), and C–S···Cl contacts (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Aggarwal R.; Sumran G. An insight on medicinal attributes of 1,2,4-triazoles. Eur. J. Med. Chem. 2020, 205, 112652 10.1016/j.ejmech.2020.112652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debruyne D.; Ryckelynck J. P. Clinical pharmacokinetics of fluconazole. Clin. Pharmacokinet. 1993, 24, 10–27. 10.2165/00003088-199324010-00002. [DOI] [PubMed] [Google Scholar]
- Piérard G. E.; Arrese J. E.; Piérard-Franchimont C. Itraconazole. Expert Opin. Pharmacother. 2000, 1, 287–304. 10.1517/14656566.1.2.287. [DOI] [PubMed] [Google Scholar]
- Tolman E. L.; Isaacson D. M.; Rosenthale M. E.; McGuire J. L.; Van Cutsem J.; Borgers M.; Van den Bossche H. Anticandidal activities of terconazole, a broad-spectrum antimycotic. Antimicrob. Agents Chemother. 1986, 29, 986–991. 10.1128/AAC.29.6.986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbrecht R.; Denning D. W.; Patterson T. F.; Bennett J. E.; Greene R. E.; Oestmann J.-W.; Kern W. V.; Marr K. A.; Ribaud P.; Lortholary O.; Sylvester R.; Rubin R. H.; Wingard J. R.; Stark P.; Durand C.; Caillot D.; Thiel E.; Chandrasekar P. H.; Hodges M. R.; Schlamm H. T.; Troke P. F.; de Pauw B. Voriconazole versus Amphotericin B for Primary Therapy of Invasive Aspergillosis. N. Engl. J. Med. 2002, 347, 408–415. 10.1056/NEJMoa020191. [DOI] [PubMed] [Google Scholar]
- Schiller D. S.; Fung H. B. Posaconazole: An extended-spectrum triazole antifungal agent. Clin. Ther. 2007, 29, 1862–1886. 10.1016/j.clinthera.2007.09.015. [DOI] [PubMed] [Google Scholar]
- Pfaller M. A.; Messer S. A.; Hollis R. J.; Jones R. N.; Diekema D. J. In Vitro Activities of Ravuconazole and Voriconazole Compared with Those of Four Approved Systemic Antifungal Agents against 6,970 Clinical Isolates of Candia spp. Antimicrob. Agents Chemother. 2002, 46, 1723–2002. 10.1128/AAC.46.6.1723-1727.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Reddy K. R.; Nelson D. R.; Zeuzem S. Ribavirin: Current role in the optimal clinical management of chronic hepatitis C. J. Hepatol. 2009, 50, 402–411. 10.1016/j.jhep.2008.11.006. [DOI] [PubMed] [Google Scholar]; b Watson J. Prospects for hepatitis C virus therapeutics: levovirin and viramidine as improved derivatives of ribavirin. Curr. Opin. Invest. Drugs 2002, 3, 680–683. [PubMed] [Google Scholar]
- a Wade P. C.; Vogt B. R.; Kissick T. P.; Simpkins L. M.; Palmer D. M.; Millonig R. C. 1-Acyltriazoles as antiinflammatory agents. J. Med. Chem. 1982, 25, 331–333. 10.1021/jm00345a021. [DOI] [PubMed] [Google Scholar]; b Maxwell J. R.; Wasdahl D. A.; Wolfson A. C.; Stenberg V. I. Synthesis of 5-aryl-2H-tetrazoles, 5-aryl-2H-tetrazole-2-acetic acids, and [(4-phenyl-5-aryl-4H-1,2,4-triazol-3-yl)thio]acetic acids as possible superoxide scavengers and antiinflammatory agents. J. Med. Chem. 1984, 27, 1565–1570. 10.1021/jm00378a007. [DOI] [PubMed] [Google Scholar]; c Jiang B.; Huang X.; Yao H.; Jiang J.; Wu X.; Jiang S.; Wang Q.; Lu T.; Xu J. Discovery of potential anti-inflammatory drugs: diaryl-1,2,4-triazoles bearing N-hydroxyurea moiety as dual inhibitors of cyclooxygenase-2 and 5-lipoxygenase. Org. Biomol. Chem. 2014, 12, 2114–2127. 10.1039/c3ob41936c. [DOI] [PubMed] [Google Scholar]; d Navidpour L.; Shafaroodi H.; Abdi K.; Amini M.; Ghahremani M. H.; Dehpour A. R.; Shafiee A. Design, synthesis, and biological evaluation of substituted 3-alkylthio-4,5-diaryl-4H-1,2,4-triazoles as selective COX-2 inhibitors. Bioorg. Med. Chem. 2006, 14, 2507–2517. 10.1016/j.bmc.2005.11.029. [DOI] [PubMed] [Google Scholar]; e Al-Abdullah E. S.; Asiri H. H.; Lahsasni S.; Habib E.; Ibrahim T.; El-Emam A. A. Synthesis, antimicrobial, and anti-inflammatory activity, of novel S-substituted and N-substituted 5-(1-adamantyl)-1,2,4-triazole-3-thiols. Drug Des., Dev. Ther. 2014, 8, 505–518. 10.2147/DDDT.S62465. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Alam M. M.; Nazreen S.; Haider S.; Shafi S.; Yar M. S.; Hamid H.; Alam M. S. Synthesis of Some New S-Alkylated 1,2,4-Triazoles, Their Mannich Bases and Their Biological Activities. Arch. Pharm. 2012, 345, 203–214. 10.1002/ardp.201100128. [DOI] [PubMed] [Google Scholar]
- Rugo H. S.; Finn R. S.; Diéras V.; Ettl J.; Lipatov O.; Joy A. A.; Harbeck N.; Castrellon A.; Iyer S.; Lu D. R.; Mori A.; Gauthier E. R.; Bartlett C. H.; Gelmon K. A.; Slamon D. J. Palbociclib plus letrozole as first-line therapy in estrogen receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer with extended follow-up. Breast Cancer Res. Treat. 2019, 174, 719–729. 10.1007/s10549-018-05125-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott K. K.; McGinley J. N.; Lubet R. A.; Steele V. E.; Thompson H. J. Effect of the aromatase inhibitor vorozole on estrogen and progesterone receptor content of rat mammary carcinomas induced by 1-methyl-1-nitrosourea. Breast Cancer Res. Treat. 2001, 70, 171–183. 10.1023/A:1013051107535. [DOI] [PubMed] [Google Scholar]
- Cuzick J.; Sestak I.; Forbes J. F.; Dowsett M.; Cawthorn S.; Mansel R. E.; Loibl S.; Bonanni B.; Evans D. G.; Howell A. Use of anastrozole for breast cancer prevention (IBIS-II): long-term results of a randomised controlled trial. Lancet 2020, 395, 117–122. 10.1016/S0140-6736(19)32955-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Ding Q.; Xu L.; Lv X.; Liu Z.; Sun Q.; Pan Y.; Xue S.; Yang W. A simple D−π–A hybrid mode for highly efficient non-doped true blue OLEDs with CIEy < 0.05 and EQE up to 6%. J. Mater. Chem. C 2018, 6, 11063–11070. 10.1039/C8TC03777A. [DOI] [Google Scholar]
- Zhou Y.-F.; Zhang S.-P.; Feng Z.; Shen X.; Zhu D.-R. Syntheses, Crystal Structures, and Spectral Characterization of Six Novel Benzimidazolyl Substituted Triaryltriazoles. J. Heterocycl. Chem. 2017, 54, 2773–2780. 10.1002/jhet.2880. [DOI] [Google Scholar]
- Loginova I. V.; Rodygin K. S.; Rubtsova S. A.; Slepukhin P. A.; Kuchin A. V.; Polukeev V. A. Oxidation of polyfunctional sulfides with chlorine dioxide. Russ. J. Org. Chem. 2011, 47, 124–130. 10.1134/S1070428011010167. [DOI] [Google Scholar]
- Barman B. K.; Guru M. M.; Panda G. K.; Maji B.; Vijayaraghavan R. K. Pyrene-affixed triazoles: a new class of molecular semiconductors for robust, non-volatile resistive memory devices. Chem. Commun. 2019, 55, 4643–4646. 10.1039/C8CC10185J. [DOI] [PubMed] [Google Scholar]
- Popiołek Ł.; Kosikowska U.; Mazur L.; Dobosz M.; Malm A. Synthesis and antimicrobial evaluation of some novel 1,2,4-triazole and 1,3,4-thiadiazole derivatives. Med. Chem. Res. 2013, 22, 3134–3147. 10.1007/s00044-012-0302-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wahaibi L. H.; Akilandeswari G.; Anusha R.; Al-Shaalan N. H.; Alkmali O. M.; El-Emam A. A.; Percino J. M.; Thamotharan S. Insights into the nature of weak noncovalent interactions in 3-(4-fluorophenyl)-6-(2-fluorophenyl)-1,2,4-triazolo[3,4-b][1,3,4]thiadiazole, a potential bioactive agent: X-ray, QTAIM and molecular docking analysis. J. Mol. Struct. 2019, 1183, 331–341. 10.1016/j.molstruc.2019.01.106. [DOI] [Google Scholar]
- Khan I.; Panini P.; Khan S. U.-D.; Rana U. A.; Andleeb H.; Chopra D.; Hameed S.; Simpson J. Exploiting the Role of Molecular Electrostatic Potential, Deformation Density, Topology, and Energetics in the Characterization of S···N and Cl···N Supramolecular Motifs in Crystalline Triazolothiadiazoles. Cryst. Growth Des. 2016, 16, 1371–1386. 10.1021/acs.cgd.5b01499. [DOI] [Google Scholar]
- Desiraju G. R. A Bond by Any Other Name. Angew. Chem., Int. Ed. 2011, 50, 52–59. 10.1002/anie.201002960. [DOI] [PubMed] [Google Scholar]
- a Desiraju G. R.; Steiner T.. The Weak Hydrogen Bond: In Structural Chemistry and Biology; Oxford University Press, 1999. [Google Scholar]; b Takahashi O.; Kohno Y.; Nishio M. Relevance of Weak Hydrogen Bonds in the Conformation of Organic Compounds and Bioconjugates: Evidence from Recent Experimental Data and High-Level ab Initio MO Calculations. Chem. Rev. 2010, 110, 6049–6076. 10.1021/cr100072x. [DOI] [PubMed] [Google Scholar]; c Desiraju G. R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. 10.1021/ja403264c. [DOI] [PubMed] [Google Scholar]; d Nangia A.; Desiraju Gautam R.. Design of Organic Solids; Weber E., Ed.; Springer-Verlag: Berlin, 1998. [Google Scholar]
- a Metrangolo P.; Resnati G.; Pilati T.; Biella S.. Halogen Bonding: Fundamentals and Applications; Metrangolo P.; Resnati G., Eds.; Springer-Verlag: Berlin, 2008. [Google Scholar]; b Metrangolo P.; Resnati G.. Halogen Bonding II: Impact on Materials Chemistry and Life Sciences; Springer International Publishing: Switzerland, 2015. [Google Scholar]
- Vogel L.; Wonner P.; Huber S. M. Chalcogen Bonding: An Overview. Angew. Chem., Int. Ed. 2019, 58, 1880–1891. 10.1002/anie.201809432. [DOI] [PubMed] [Google Scholar]
- a Mahmudov K. T.; Kopylovich M. N.; Guedes da Silva M. F. C.; Pombeiro A. J. L. Chalcogen bonding in synthesis, catalysis and design of materials. Dalton Trans. 2017, 46, 10121–10138. 10.1039/C7DT01685A. [DOI] [PubMed] [Google Scholar]; b Lu Y.; Liu Y.; Xu Z.; Li H.; Liu H.; Zhu W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discovery 2012, 7, 375–383. 10.1517/17460441.2012.678829. [DOI] [PubMed] [Google Scholar]; c Kříž K.; Fanfrlík J.; Lepšík M. Chalcogen Bonding in Protein–Ligand Complexes: PDB Survey and Quantum Mechanical Calculations. ChemPhysChem 2018, 19, 2540–2548. 10.1002/cphc.201800409. [DOI] [PubMed] [Google Scholar]
- Gavezzotti A. Calculation of lattice energies of organic crystals: the PIXEL integration method in comparison with more traditional methods. Z. Kristallogr. - Cryst. Mater. 2005, 220, 499–510. 10.1524/zkri.220.5.499.65063. [DOI] [Google Scholar]
- McKinnon J. J.; Jayatilaka D.; Spackman M. A. Towards quantitative analysis of intermolecular interactions with Hirshfeld surfaces. Chem. Commun. 2007, 3814–3816. 10.1039/b704980c. [DOI] [PubMed] [Google Scholar]
- Bader R.Atoms in Molecules: A Quantum Theory; Oxford University Press: USA, 1994. [Google Scholar]
- Parsons S.; Flack H. D.; Wagner T. Use of intensity quotients and differences in absolute structure refinement. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2013, 69, 249–259. 10.1107/S2052519213010014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jelsch C.; Soudani S.; Ben Nasr C. Likelihood of atom-atom contacts in crystal structures of halogenated organic compounds. IUCrJ 2015, 2, 327–340. 10.1107/S2052252515003255. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jelsch C.; Ejsmont K.; Huder L. The enrichment ratio of atomic contacts in crystals, an indicator derived from the Hirshfeld surface analysis. IUCrJ 2014, 1, 119–128. 10.1107/S2052252514003327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panini P.; Gonnade R. G.; Chopra D. Experimental and computational analysis of supramolecular motifs involving Csp2(aromatic)–F and CF3 groups in organic solids. New J. Chem. 2016, 40, 4981–5001. 10.1039/C5NJ03211C. [DOI] [Google Scholar]
- Gatti C. Chemical bonding in crystals: new directions. Z. Kristallogr. - Cryst. Mater. 2005, 220, 399–457. 10.1524/zkri.220.5.399.65073. [DOI] [Google Scholar]
- a Aggarwal R.; Sharma S.; Hooda M.; Sanz D.; Claramunt R. M.; Twamley B.; Rozas I. Visible-light mediated regioselective approach towards synthesis of 7-aroyl-6-methyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Tetrahedron 2019, 75, 130728 10.1016/j.tet.2019.130728. [DOI] [Google Scholar]; b Marubayashi N.; Ogawa T.; Moriwaki M.; Haratake M. Atropisomerism in 4-(2-Thienyl)-4H-1, 2, 4-triazole Derivatives. Chem. Pharm. Bull. 1992, 40, 220–223. 10.1248/cpb.40.220. [DOI] [Google Scholar]; c Kemmish H. J.; Hamor T. A. Structure of 2-bromo-4-(2-chlorophenyl)-6H-thieno[3,2-f][1,2,4]triazolo[4,3-a][1,4]diazepine (I) and of its 9-cyclohexyl derivative (II). Acta Crystallogr., Sect. C: Cryst. Struct. Commun 1988, 44, 1653–1656. 10.1107/S0108270188005773. [DOI] [Google Scholar]; d Sirakanyan S. N.; Spinelli D.; Geronikaki A.; Kartsev V. G.; Hakobyan E. K.; Hovakimyan A. A. Synthesis of New Heterocyclic Systems: Pyrido[3′,2′:4,5]thieno(furo)[2,3-e][1,2,4]triazolopyrimidines and an Unusual ANRORC Rearrangement in the Fused Pyrimidine Series. ChemistrySelect 2018, 3, 10938–10942. 10.1002/slct.201802221. [DOI] [Google Scholar]
- Clark R. C.; Reid J. S. The analytical calculation of absorption in multifaceted crystal. Acta Crystallogr., Sect. A: Found. Adv. 1995, 51, 887–897. 10.1107/S0108767395007367. [DOI] [Google Scholar]
- Burla M. C.; Caliandro R.; Camalli M.; Carrozzini B.; Cascarano G. L.; Giacovazzo C.; Mallamo M.; Mazzone A.; Polidori G.; Spagna R. SIR2011: a new package for crystal structure determination and refinement. J. Appl. Crystallogr. 2012, 45, 357–361. 10.1107/S0021889812001124. [DOI] [Google Scholar]
- Sheldrick G. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez-Monge L.; Taylor R.; van de Streek J.; Wood P. A. Mercury CSD 2.0—new features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/S0021889807067908. [DOI] [Google Scholar]
- Groom C. R.; Bruno I. J.; Lightfoot M. P.; Ward S. C. The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, 72, 171–179. 10.1107/S2052520616003954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spackman M. A.; Jayatilaka D. Hirshfeld surface analysis. CrystEngComm 2009, 11, 19–32. 10.1039/B818330A. [DOI] [Google Scholar]
- Spackman M. A.; McKinnon J. J. Fingerprinting intermolecular interactions in molecular crystals. CrystEngComm 2002, 4, 378–392. 10.1039/B203191B. [DOI] [Google Scholar]
- Turner M. J.; McKinnon J. J.; Wolff S. K.; Grimwood D. J.; Spackman P. R.; Jayatilaka D.; Spackman M. A.. CrystalExplorer17; University of Western Australia, 2017. [Google Scholar]
- Spackman M. A.; McKinnon J. J.; Jayatilaka D. Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals. CrystEngComm 2008, 10, 377–388. 10.1039/b715227b. [DOI] [Google Scholar]
- a Gavezzotti A. Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. I. Electrostatic and Polarization Energies in Molecular Crystals. J. Phys. Chem. B 2002, 106, 4145–4154. 10.1021/jp0144202. [DOI] [Google Scholar]; b Gavezzotti A. Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities. 2. An Improved Polarization Model and the Evaluation of Dispersion and Repulsion Energies. J. Phys. Chem. B 2003, 107, 2344–2353. 10.1021/jp022288f. [DOI] [Google Scholar]; c Gavezzotti A. Efficient computer modeling of organic materials. The atom–atom, Coulomb–London–Pauli (AA-CLP) model for intermolecular electrostatic-polarization, dispersion and repulsion energies. New J. Chem. 2011, 35, 1360–1368. 10.1039/c0nj00982b. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.; et al. Gaussian 09, revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013.
- Boys S. F.; Bernardi F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. 10.1080/00268977000101561. [DOI] [Google Scholar]
- a Zhao Y.; Truhlar D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]; b Dunning T. H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. 10.1063/1.456153. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Keith T. A.AIMAll, version 19.02.13; TK Gristmill Software: Overland Park KS, USA, 2019.
- Espinosa E.; Molins E.; Lecomte C. Hydrogen bond strengths revealed by topological analyses of experimentally observed electron densities. Chem. Phys. Lett. 1998, 285, 170–173. 10.1016/S0009-2614(98)00036-0. [DOI] [Google Scholar]
- Harder E.; Damm W.; Maple J.; Wu C.; Reboul M.; Xiang J. Y.; Wang L.; Lupyan D.; Dahlgren M. K.; Knight J. L.; Kaus J. W.; Cerutti D. S.; Krilov G.; Jorgensen W. L.; Abel R.; Friesner R. A. OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J. Chem. Theory Comput. 2016, 12, 281–296. 10.1021/acs.jctc.5b00864. [DOI] [PubMed] [Google Scholar]
- Friesner R. A.; Murphy R. B.; Repasky M. P.; Frye L. L.; Greenwood J. R.; Halgren T. A.; Sanschagrin P. C.; Mainz D. T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein–Ligand Complexes. J. Med. Chem. 2006, 49, 6177–6196. 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- a QM-Polarized Ligand Docking Protocol; Schrödinger Release 2019-4; Glide, Schrödinger, LLC: New York, NY, 2019.; b Schrödinger Release 2019-4; Jaguar, Schrödinger, LLC: New York, NY, 2019.; c Schrödinger Release 2019-4; QSite, Schrödinger, LLC: New York, NY, 2019.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.