

Abstract
This Statement presents a proposal for harmonising the establishment of Health‐Based Guidance Values (HBGVs) for regulated products that are also nutrients. This is a recurrent issue for food additives and pesticides, and may occasionally occur for other regulated products. The Statement describes the specific considerations that should be followed for establishing the HBGVs during the assessment of a regulated product that is also a nutrient. It also addresses the elements to be considered in the intake assessment; and proposes a decision tree for ensuring a harmonised process for the risk characterisation of regulated products that are also nutrients. The Scientific Committee recommends the involvement of the relevant EFSA Panels and units, in order to ensure an integrated and harmonised approach for the hazard and risk characterisation of regulated products that are also nutrients, considering the intake from all relevant sources.
Keywords: nutrient, tolerable upper intake level (UL), Health‐Based Guidance Value (HBGV), acceptable daily intake (ADI), food additives, pesticides
Short abstract
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2021.EN-6480/full
1. Introduction
A Health‐Based Guidance Value (HBGV) is a science‐based recommendation for the maximum (oral) exposure to a substance that is not expected to result in an appreciable health risk, taking into account current safety data, uncertainties in these data, and the likely duration of consumption.
A nutrient is an element or compound needed for the normal growth, development and health maintenance of the organism. This includes vitamins, minerals and macronutrients. In this Statement, the term intake will be used to designate the dietary exposure to a nutrient.
Under the General Food Law (Regulation (EC) No 178/2002)1, the European Commission can ask the (European Food Safety Authority) EFSA to advise about HBGVs for nutrients through generic mandates, to support its legislative work in this field of nutrition (e.g. regarding the addition of vitamins and minerals to foods). These mandates are entrusted to the Panel on Nutrition, Novel Foods and Food Allergens (NDA Panel), which is responsible for establishing tolerable upper intake levels (ULs) as HBGVs for nutrients (see definition in Box 1).2
EFSA is also responsible for the evaluation of regulated products3 in food and feed that require a scientific risk assessment before their authorisation on the EU market. For food additives and pesticides, the relevant Panels and units regularly establish HBGVs, e.g. acceptable daily intake (ADI) (see definition in Box 1), as part of these assessments. For food additives and pesticides that are also nutrients, this can lead to a complex situation in which two assessments requiring the establishment of HBGVs for the same substance (i.e. a nutrient) are carried out under different regulatory frameworks, using similar scientific methodological approaches. Examples include the assessment of phosphates (EFSA FAF Panel, 2019a) and chlorides (EFSA FAF Panel, 2019b) as food additives, and copper used as a pesticide (EFSA, 2018a).
This Statement is meant to provide recommendations to address this particular situation in future EFSA assessments, with the view to ensure that there is an internal consistency when establishing HBGVs for regulated products (particularly for food additives and pesticides) that are also nutrients. It should be noted that for other regulated products that are also nutrients (e.g. feed additives, novel foods) the risk assessment is generally based on existing HBGVs (e.g. ULs).
Box 1. Definitions of ADI and UL .
1.
Acceptable daily intake (ADI): ‘an estimate of the amount of a substance in food or drinking water that can be consumed daily over a lifetime without presenting an appreciable risk to health. It is usually expressed as milligrams of the substance per kilogram of body weight per day and applies to chemical substances such as food additives, pesticide residues and veterinary drugs’ (EFSA Glossary4).
Tolerable upper intake level (UL): ‘the maximum level of total chronic daily intake of a nutrient (from all sources5) judged to be unlikely to pose a risk of adverse health effects to humans’ (SCF, 2000, 2000a, 2000b, 2000c, 2000d, 2000e, 2000f, 2000g). It is usually expressed as milligrams or micrograms of the nutrient per day for defined population groups (e.g., infants, children, adolescents, adults, pregnant women, lactating women).
The ADI and the UL are similar concepts. The concept of UL was introduced in the 1990s recognising that, like other chemicals, nutrients can produce adverse health effects at excessive intakes. In developing the methodology to establish ULs, it was recognised that the general principles of the risk assessment model developed for chemicals could be applied (IOM, 1997, 1998a,b, 2000, 2001; SCF, 2000, 2000a, 2000b, 2000c, 2000d, 2000e, 2000f, 2000g; WHO/IPCS, 2002; Renwick et al., 2004; WHO/FAO, 2006; Aggett, 2007). A fundamental difference though lies in the fact that nutrients are essential for human health within a certain range of intakes, i.e. intakes below the lower end of this range are associated with risk of nutrient deficiency. Another distinctive feature is that, for risk management purposes, ADIs are conventionally expressed relative to body weight (e.g. mg/kg body weight (bw) per day) and apply for the general population, while ULs are expressed in absolute amounts (e.g. mg/day) for defined life‐stage groups. Additional details are provided in Annex A.
1.1. Background and Terms of Reference as provided by EFSA
In November 2018, the FAF Panel consulted the Scientific Committee (SC) on the method to be used for setting HBGVs in the re‐evaluation of phosphoric acid, phosphates and polyphosphates as food additives. There are similar issues with other additives that are also nutrients, such as chlorides, but also with other regulated products, such as copper used as active substance in plant protection products.
The SC advised the FAF Panel to consider the total intake of phosphorus (including the intake from the diet) in the risk assessment. Having reviewed all the available scientific evidence from human and animal studies, the Panel identified a reference point for deriving an ADI from animal studies. In this specific case, however, with phosphorus being also a nutrient, the approach of the former Scientific Committee on Food (SCF) to derive a Tolerable Upper Intake Level (UL) could also be applied. The issue led to discussions within and between the Panels and units, and is just one of the several examples of similar situations still unresolved regarding the scientific methodology to be applied for additional exposure to nutrients through regulated products.
It is important to consider also the implications regarding the advice to risk managers, for their decision‐making on each regulated use, as well as for enforcement.
This is a recurrent issue, which has produced divergencies between different EFSA assessments, the most recent one being on copper.6 The FAF and NDA Panels, supported by the respective units, have suggested a clarification on the methodology to be used, and the approach for presenting the results in a way which is scientifically sound and fit for risk managers’ needs through a Statement from the Scientific Committee.
Terms of Reference
The SC is requested to provide scientific advice to EFSA Panels and units, taking into account risk managers’ needs, in line with the following terms of reference.
To review the background document produced by EFSA, describing current approaches for setting HBGV such as ADI and UL, and to define the general approach on how to estimate the risk to consumers regarding the exposure to additives and other substances in regulated products that are also nutrients.
To advise on the terms and definitions that should be used by EFSA in the hazard and risk characterisation in this type of assessments.
When setting this general approach, the SC should also consider how to present to risk managers information relevant for their decision making, covering the overall risk for consumers from all exposure sources, as well as the specific contribution to consumer's risk and health concerns from the exposure related to the regulated product, e.g. using the ‘total’ and ‘added’ risk concepts.
Where possible, to provide some recommendations for using and combining experimental animal studies and human nutrition information when setting HBGV for regulated substances that are also nutrients, accounting for the differences in background exposure levels between humans and experimental animals, as well as inter‐species differences in the physiological roles and homeostatic regulations between species and between nutrients.
The Scientific Committee is requested to consider also international approaches, including feedback obtained through ILMERAC (International Liaison Group for Methods of Risk Assessment for Chemicals in Food).
1.2. Interpretation of the Terms of Reference
This Statement provides recommendations for establishing the HBGVs to be used in dietary safety assessments of regulated products that are also nutrients. Therefore, the scope is limited to adverse effects resulting from high dietary intakes, as covered by the ULs and ADIs in the assessment of nutrients and regulated products, respectively. The assessment of dietary requirements for nutrients or deficiencies is outside the scope of this Statement.
In the present Statement, ‘all exposure sources’ refers to the total intake of nutrients from the diet. If information is available, other oral exposures to the nutrient (e.g. from drug adjuvants or consumer products) may be considered in order to provide comprehensive information to risk managers. Consequently, contributions related to occupational and non‐oral exposures are not covered in this Statement. Specific considerations regarding kinetics and homeostatic control are required when the assessment covers oral and non‐oral exposure routes; those considerations are also outside the scope of this Statement.
In line with the Terms of Reference, this document is a Statement, not a Guidance, i.e. it provides a set of recommendations for EFSA's units and Panels for facilitating the safety assessment of regulated products that are also nutrients. The details on how to conduct the hazard, exposure and risk assessments are provided in the relevant sectoral guidances.
In order to address the fourth ToR, a description of the biological‐based model (BBM) based on the IPCS/WHO proposal has been included. The interplay between HBGVs from the perspective of toxicological risk assessment and from the perspective of nutrition have been considered before in many papers, such as, but not limited to, Renwick et al. (2004), Verkaik‐Kloosterman et al. (2012), Bruins et al. (2015); EFSA, 2006; NASEM (2018). Moreover, HBGVs for micronutrients have been the underlying considerations for integrated risk‐benefit assessment for food and nutrition (Tijhuis et al., 2012a, 2012b; Boobis et al., 2013; Vidry et al., 2013). A detailed review of the different approaches proposed is outside the scope of this Statement.
2. Data and methodologies
This Statement is based on the guidance documents from the SCF and from different EFSA Panels and units, complemented with the assessment of approaches used by other organisations, for establishing HBGVs for nutrients and regulated products (focusing on food additives and pesticide residues). EFSA staff compiled information from previous assessments of nutrients as regulated products to map current practices and identify commonalities and differences in the approaches applied in the different areas, as a basis to formulate recommendations. A draft report was presented to the Scientific Committee and the revised version is included as Annex A.
The EFSA international network ILMERAC was sent a dedicated communication in order to identify recent or ongoing activities in this area and opportunities for collaboration, but no similar activities were identified. A draft Statement was published for public consultation. All received comments have been addressed in the complementary Technical Report (EFSA, 2021), and the Statement has been updated accordingly.
3. Assessment
3.1. Problem formulation and target population
The problem formulation is the first phase of a risk assessment. It includes: a) the clarification and acceptance of the mandate that takes place in dialogue with the requestor; and b) the translation of each mandate's terms of reference into one or more scientifically answerable assessment questions, to inform the definition of the related conceptual model and the selection of the overall approach for the assessment (EFSA, 2020).
Risk management options differ depending on whether the substance of interest is naturally present in foods, intentionally added (e.g. food additives), an unavoidable consequence of the intended use (e.g. residues of pesticides), or a contaminant. Risk managers frame the mandate to EFSA according to the regulatory actions, e.g. decision on product authorisation, management measures that must be taken.
For regulated products, the scope of the risk assessment is usually defined by the sectoral legislation and data requirements may differ across sectors (see Annex A).
The target population for the risk assessments of food additives, pesticides, and nutrients in the context of generic mandates, is the general population. The general population encompasses all age groups (i.e. infants, children and adolescents, adults, the elderly, pregnant and lactating women). The risk assessment process takes into account that some individuals may be more biologically sensitive than others. The HBGVs are usually based on protecting the most sensitive members of the general population, including young children, pregnant and lactating women, and elderly people. For some substances, there may be population subgroups who have distinct sensitivities that do not fall within the range of sensitivities expected for the general population, because of, e.g. specific genetic background, conditions or diseases. HBGVs may not apply to individuals under medical supervision. Considerations of susceptible groups of the population under the respective frameworks are described below and summarised in Table 1.
Table 1.
Target populations considered in deriving HBGVs and approaches regarding susceptible groups ‐ by sector
| Target population of the assessment | Consideration of susceptible groups | Refs | |
|---|---|---|---|
| Food additives |
General populationa Infants below 16 weeks of age are included in the assessment when the food additive(s) is/are authorised in food categories concerning this age group (e.g. infants formula, food for special medical purposes)b |
Covered by the HBGV case by case. Groups excluded are flagged in the risk characterisation for risk managers to take specific measures where appropriate |
EFSA ANS Panel (2012) and EFSA Scientific Committee (2017) |
| Pesticides |
General populationa Specific assessments for infants below 16 weeks of age address the presence of pesticide residues in food categories such as infant formula or baby foodb |
The HBGV should protect the whole population | Regulation 1107/2009, EFSA PPR Panel (2018) |
| Nutrients |
General populationa Exclude individuals receiving the nutrient under medical supervision Exclude individuals with extreme and distinct vulnerabilities due to genetic predisposition or other considerations |
Covered by the HBGV case by case. Groups excluded are flagged in the risk characterisation for risk managers to take specific measures where appropriate |
SCF (2000) |
HBGV: Health‐Based Guidance Value.
The general population encompasses all age groups (i.e. infants, children and adolescents, adults, the elderly, pregnant and lactating women).
Specific considerations are needed for determining whether the HBGV is applicable for infants below 16 weeks of age (EFSA Scientific Committee, 2017).
HBGVs for chemicals have traditionally not been considered applicable for infants below 16 weeks of age, and specific considerations are needed for determining whether an HBGV established for the general population is applicable for infants below 16 weeks of age (EFSA Scientific Committee, 2017).
For food additives, the aim of the HBGV is to be protective for the general population. However, in some instances susceptible subgroups may be identified and flagged as part of the risk characterisation so that risk managers can take specific measures for those groups, as appropriate. For instance, in relation to phosphate‐containing additives, the FAF Panel indicated that the ADI established was considered protective for healthy adults, but was not applicable to individuals with moderate to severe reduction in renal function (EFSA FAF Panel, 2019a). For pesticides, the HBGV should be protective for the general population including susceptible groups.
Regarding ULs for nutrients, the guidelines of the SCF state that ‘… adverse effects of nutrients are influenced by physiological changes and common conditions associated with growth and maturation that occur during an individual's lifespan. Therefore, where necessary, and to the extent possible, ULs are derived for each separate life‐stage group, e.g. infants, children, adults, the elderly, and women during pregnancy or lactation. Even within relatively homogenous life‐stage groups, there is a range of sensitivities to adverse effects, e.g. sensitivity is influenced by body weight and lean body mass’. The SCF's guidelines also state that ‘the derivation of ULs for the normal healthy population, divided into various life‐stage groups, accounts for normally expected variability in sensitivity, but it excludes sub‐populations with extreme and distinct vulnerabilities due to genetic predisposition or other considerations (including these would result in ULs which are significantly lower than are needed to protect most people against adverse effects of high intakes)’ (SCF, 2000). The guidelines indicate that ‘the extent to which a sub‐population becomes significant enough to be assumed to be representative of a general population is an area of judgement and of risk management and will be considered for individual nutrients’. Also, individuals receiving the nutrient under medical supervision are excluded (SCF, 2000).
3.2. Information used for the risk assessment
3.2.1. Assessment of regulated products
The safety assessment of regulated products is based on data required according to the relevant sectoral legislation or guidance.7 In practice, applications submitted to EFSA can relate to: the evaluation of products prior to their introduction on the EU market; the re‐evaluation of products due to the expiry of their authorisation; a re‐evaluation programme; a request for the extension of conditions of use or changes in technology; or development of new scientific knowledge.
In the first case, a scientific dossier is submitted by an applicant, which contains the studies requested by the sectoral guidance with the aim of demonstrating the safety of the product. A set of toxicological studies is typically required, and this may vary depending on the sectors (see Annex A) and the nature and characteristics of the product. In most instances, an applicants’ dossier includes in vitro tests (e.g. genotoxicity tests) and in vivo animal studies (e.g. 90‐day subchronic toxicity study in rats) conducted according to standard protocols (e.g. OECD test guidelines). When available, human data are also submitted as part of the dossier.
In the event of a product review or re‐evaluation, the available data depend on the sector (see Annex A). For instance, in the case of the re‐evaluation of food additives, the assessment is based on the information available in the public domain or obtained after a call for data; while for the renewal process of active substances in plant protection products (PPPs), the applicant must submit a supplementary dossier with additional studies reflecting updated data requirements and guidance, when applicable, and a review of scientific peer‐reviewed publications.
3.2.2. Assessment of ULs for nutrients
The assessment of ULs for nutrients is conducted upon request from risk managers through a generic mandate. EFSA is responsible for collecting relevant information pertaining to the adverse effects of a given nutrient. The process used is the same as that for non‐nutrients. However, in general, the risk assessment for nutrients relies on the data published in the literature. There are few systematic toxicological data such as those generated for the risk assessment of additives (SCF, 2000). In its framework on developing ULs for vitamins and minerals, the SCF noted that the process depends on using all relevant human experimental and observational studies, as well as animal studies (SCF, 2000). It further noted that, given the limitations of available data, selecting an adverse event and assessing a tolerable intake involves ‘scientific judgement’ of the quality of evidence of adverse effects on humans, particularly the strength of causality and data on the underlying mechanisms. The consistency and completeness of the data overall should also be considered in informing hazard identification, setting of reference points and the application of uncertainty factors (UFs) for risk characterisation (SCF, 2000).
3.3. Hazard identification and characterisation
3.3.1. Hazard identification and characterisation of chemicals in food
The risk assessment of regulated products follows the classical approach applied to chemicals in food, which consists of 4 steps: hazard identification, hazard characterisation, exposure assessment and risk characterisation. Toxicological studies available for the assessment should allow the identification of potential adverse effects (hazard identification) and the assessment of dose–response relationships for the adverse effects (hazard characterisation). Data are evaluated to characterise the absorption, distribution, metabolism and excretion (ADME) of the product under evaluation, its general systemic toxicity as well as its potential genotoxicity. The conventional approach consists of assessing the dose–response relationships for the adverse effects and identifying a reference point (RP) (i.e. a no observed adverse effect level (NOAEL), a lowest observed adverse effect level (LOAEL) or the lower confidence limit of the calculated benchmark dose (BMDL) where applicable (see Annex A for details), based on the most sensitive endpoint relevant for humans. The RP is used to establish a HBGV – e.g. an ADI for chronic dietary exposure – by dividing the RP by UFs. A HBGV can be established for compounds for which thresholded mechanisms of toxicity can reasonably be expected based on the available data, and for which the safety‐related data are relatively complete. Alternatively, in particular when available data are not sufficient to establish a HBGV, the application of a margin of exposure (MoE) approach can be considered to conclude on the safety of the products, by considering the margin between the RP and the estimated exposure.
3.3.2. Conceptual and methodological specificities for nutrients
In general, the same principles of risk assessment also apply to nutrients and to other chemicals in food (Section 3.3.1). However, nutrients possess some distinguishing characteristics, which must be taken into account in the assessment. In brief, the underpinning assumption for the risk assessment of additives and pesticides is that they do not have a nutritional value or physiological role. In contrast, nutrients have distinctive biochemical and physiological roles, and specific and selective mechanisms for the regulation of the ADME of the nutrient or its metabolites, or both, within the body. These mechanisms are specific for each nutrient, and collectively they maintain the systemic homeostasis and body burden for the nutrient over a range of intakes (SCF, 2000; WHO/FAO, 2006).
The concept of acceptable range of oral intake (AROI)
A WHO/IPCS report of 2002 dealt with the principles and methods for the assessment of risk from essential trace elements (ETE) (WHO/IPCS, 2002). It considered the adverse effects of intakes below and above requirements and customary intakes. The report noted that uncritically applying usual toxicological approaches, involving the application of a (default) UF to a RP to establish a HBGV, was not appropriate for ETE. Indeed, while an ADI for food additives and pesticides assumes that zero exposure to the substance of concern is without risk, this assumption does not apply to ETEs. It was appreciated that the selection of appropriate UFs for ETEs must consider potential effects regarding both nutritional deficiency and toxicity. Establishing a HBGV for a nutrient below the reference dietary requirements would evidently be inappropriate in biological contexts and also for policy and practice in public and occupational health and in food safety (Mertz, 1993; WHO/IPCS, 2002).
The WHO/IPCS report adapted the concept of an AROI for essential trace elements, within which there is a small risk of toxicity or deficiency in a population, bounded by rising risks of either deficiency, as intake declines, or toxicity as intake increases, as illustrated in Figure 1. An important point is that these distributions represent the population heterogeneity in the rates at which deficiency and toxicity occur, and the report suggested that risk assessments for both deficiency and excess for ETEs should be based on biological endpoints.
Figure 1.
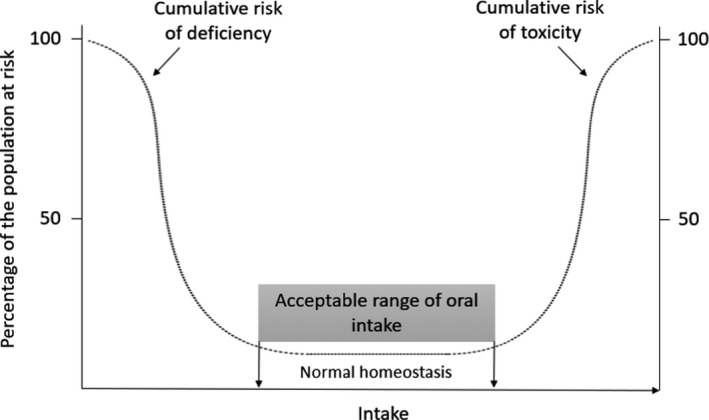
A theoretical representation of the percentage of the population at risk of deficiency and toxicity effects according to the dietary intake of a nutrient, adapted from WHO/IPCS (2002)
Thus, for risk assessment of excess intakes of ETEs, the WHO/IPCS report proposed that a ‘Biologically Based’ model or approach based on biological outcomes and mechanisms should be adopted, rather than using the customary toxicological approaches for establishing HBGVs (WHO/IPCS, 2002). In particular, it was appreciated that the sequence of accumulating events could be used to identify markers which could be characterised as endpoints of excess and of potential toxicity, rather than of overt toxicity.
Subsequently, a WHO/FAO exercise explored this approach to risk assessment for nutrients in general (WHO/FAO, 2006).
This framework was integrated by the NDA Panel in its principles for deriving dietary reference values (DRVs) for nutrients (EFSA NDA Panel, 2010a). At the lower bound of the range, dietary intakes necessary for meeting a population's nutritional requirements are described through the concepts of average requirements (ARs) and population reference intakes (PRIs) while, at the higher bound of the range, the UL is defined as ‘the maximum level of total chronic daily intake of a nutrient (from all sources) judged to be unlikely to pose a risk of adverse health effects to humans’ (SCF, 2000), which is similar to the definition of an ADI. The bounds of high and low exposures of an AROI are determined by the homeostatic and adaptive mechanisms.
A ‘Biological Based Model’ for nutrients
The IPCS/WHO working group ‘Biological Based Model’ for establishing HBGVs (ADIs/ULs) for ETEs, and the WHO/FAO working group on Nutrient Risk Assessment proposed the identification of critical endpoints from among the homeostatic and adaptive responses to excessive intakes of nutrients in nutrient risk assessment (WHO/IPCS, 2002; WHO/FAO, 2006). The following generalised description of responses to excess nutrient intake provides a background for identifying endpoints which could be candidates for critical endpoints. This is analogous to hazard identification and characterisation except that the endpoints identified would not be expected to be hazards or adverse events, i.e. they are predictive of adverse events that would occur if intake is not reduced. The identification and characterisation of critical endpoints depends on a sound understanding of the nutrient kinetics and dynamics of the nutrient of interest. It is noteworthy that the endogenous kinetics and homeostatic mechanisms of nutrients vary, and the risk assessment of endpoints related to excess intake may draw on an extensive resource of data. An illustration of the mechanisms applying to ETE is given in Appendix B.
The general features of the systemic responses to increasing intakes of nutrients are illustrated in Figure 2.
Figure 2.
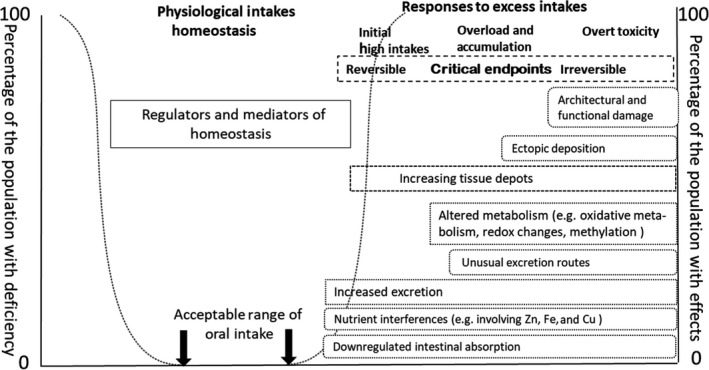
The generic chain of potential dose responses accompanying increasing intake and body burden of nutrients (see further explanation in text below and Appendix B)
The intakes indicated at the top of the Figure progress from deficiency to excessive intake. The text boxes describe potential physiological and pathological responses (i.e. endpoints) to increasing intakes, and, in this context of chronic excess and subsequent toxicity, the increasing body burden (i.e. the amount of a chemical accumulated in the body over time) of the nutrient being considered. The left‐hand margin of each box indicates the starting points for each response. These responses can involve, among others, reduced absorption, increased excretion, increased deposition of the nutrient in tissues, and/or increasing metabolism, to different extents depending on the nutrient. Prolonged excess intake leading to overload and systemic accumulation is indicative that physiological responses are being overwhelmed, and pathological events are developing. These are represented by the boxes positioned at those points of intake. Adverse biological changes (e.g. accumulation of a nutrient to a level that causes adverse effects) are likely to be reversible in response to subsequent reduced intake if they are under homeostatic control (e.g. nutrient ADME is regulated by its systemic levels). However, if high intake is maintained, phenomena arising from excess tissue deposition and ultimately ectopic deposition occur, with subsequent tissue and organ damage and failure. The periods over which the endpoints appear are highly variable; they can extend over decades, and often the events shown in Figure 2 occur concurrently.
The basis of using biological endpoints, as in Figure 2, is that these, as is stated above, can be used in risk assessment as discrete reliable endpoints that are predictive of impending overload and toxicity. These are more likely to be identified in the early stages of excess intake and impending toxicity. At these levels of intake, it should be possible, during the characterisation and validation of selected critical endpoints, to incorporate an assessment of the associated uncertainty in nutrient risk assessment.
The pathway of events from which biologically based critical endpoints can be identified extends from those based on homeostatic mechanisms to excess exposure that precedes cellular and tissue architectural and functional damage. Homeostatic mechanisms, and their markers, are reversible and not necessarily indicative of overt toxicity. However, at intakes higher than those responsive to homeostasis, the body's adaptive responses are less likely under chronic conditions to be reversible and are indicative of a higher probability of toxicity if intake (i.e. exposure) is not reduced. Critical endpoints would be those for which a mechanistic pathway can be discerned and which can be characterised and validated as indicative of probable toxicity (Aggett, 2007). Such endpoints can be regarded as predictive of toxicity and be used to establish HBGVs that are protective of human health.
Such biological and toxicological endpoints, have been ranked according to their potential value in risk assessment (Renwick et al., 2004) as follows:
1: Biochemical changes within the homeostatic range and without indication of adverse sequelae;
2: Biochemical changes outside the homeostatic range without known sequelae;
3: Biochemical changes outside the homeostatic range that represent a marker of potential adverse effects due to excess;
4: Clinical symptoms indicative of a minor but reversible change;
5: Clinical symptoms of significant but reversible effects;
6: Clinical signs indicative of significant but reversible organ damage; and
7: Clinical signs indicative of irreversible organ damage.
This ranking is useful in interpreting the events summarised in Figure 2. The markers of interest using a biologically based approach to the development of HBGVs are levels 1–3, and, possibly, level 4.
The advantages in a ‘BBM’ applied to nutrients are that it can use available information on ADME (i.e. nutrient kinetics) to interpret the mechanisms of observed effects in homeostasis, adaptation and initial dysfunction and morphological changes. It is also possible, using epidemiological techniques, to allow for a latency in the effect of long‐term intake and of the possible contribution to chronic disease. Exploration of the data on the mechanisms of homeostasis and adaptation, and the early pathogeneses of adverse effects, enables the identification of potential endpoints, which can be used for critical endpoint characterisation and risk assessment.
3.3.3. Evidence review and integration of lines of evidence for establishing HBGVs for nutrients
The BBM can use and integrate data from many sources. These have a commonality with the sources of evidence used in identifying environmental causes of disease including dietary and adventitious exposure to nutrients and other environmental chemicals (AMS, 2007). The BBM includes evidence from human studies such as randomised controlled trials, intervention studies in which experimental and reference groups have well characterised intakes, and relevant and validated endpoints, as well as observational studies in human populations. There are many experimental studies using animal models, but most of these studies have used excessive intakes of nutrients and are targeted at exploring the effects of high intakes on specific organs and functions; hence, they do not provide information on homeostatic and adaptive responses as body burden increases. Nonetheless such studies help to identify target organs and pathologies, and describe the sequential development of toxicological endpoints, which might enable the tracing of pathogenic events in the physio‐pathological pathway (Figure 2). Epidemiological studies in livestock, and reports including case reports of high intake and toxicities affecting humans and animals can also be helpful. Inborn errors of metabolism in humans and animals contribute to the understanding of underpinning genetic and consequent metabolic defects leading to toxicity. The quality of such data needs to be critically assessed for biologically based endpoints as would be the case for hazard identification and characterisation (SCF, 2000).
Nonetheless, the evidence available from studies in humans and animals to facilitate risk assessment is often limited both in quality and quantity. There is insufficient knowledge on the metabolism of many nutrients to enable the use and validation of markers at the threshold of developing potential adverse effects due to excess. It is possible, however, even if a critical endpoint cannot be identified, to use evidence derived from systematic studies of homeostasis and adaptation to high nutrient intakes in healthy individuals to identify predictive and therefore protective endpoints.
Recent advances in molecular biology and in computational modelling have enhanced the ability to use BBMs. Developments in bioinformatics have fostered Systems Biology which is being developed to enhance Toxicological Risk Assessment and Nutritional Science (Krewski et al., 2020) and which embraces and enables the integrated use of genomics, transcriptomics, proteomics and metabolomics to explore the dynamics and systemic kinetics of compounds. These platforms and subsidiary platforms focussing on epigenetic effects and e.g. nutrient metabolism, enable the intelligent integration of data sources. The use of such databases would enable deeper exploration of the interconnectivity at the biological levels involved in the reactions to intakes of nutrients above their physiological requirements. Such an exercise could contribute to identify knowledge gaps and research needed for further risk assessments of nutrients. Furthermore, systematic analysis to identify and characterise critical endpoints would inform the use of genomics, proteomics, and metabolomics either as markers themselves or as means to validate other markers, e.g. markers in tissues, that are more practical and ethical for risk assessment. This may also enable integration of the homeostatic and adaptive metabolomic data with emerging approaches to assessing environmental exposure and the human exposome, as well as epidemiological approaches to high dietary intakes and health outcomes.
It is noted that these approaches and data platforms are not unique to nutrients but part of ongoing developments in biology, toxicology, and exposure science, and are addressed in the EFSA Scientific Strategies (EFSA, 2016; Verhagen et al., 2019).
3.3.4. Hazard identification and characterisation of regulated products that are also nutrients
Minerals, vitamins and some fatty acids have been the subject of a risk assessment by the EFSA NDA Panel or SCF with a view of establishing ULs, whenever possible, based on available data in peer‐reviewed published papers (see Appendix A). In some cases, the safety of nutrients has also been assessed by other EFSA Panels in the context of the evaluation of regulated products (e.g. phosphate‐containing additives (EFSA FAF Panel, 2019a); copper used as feed additive (EFSA FEEDAP Panel, 2019) and active substance in PPPs (EFSA, 2018a)). When available, EFSA's existing Scientific Opinions should be used as a starting point for a hazard identification and characterisation. As described above, data available for EFSA's assessment may differ depending on the framework under which the assessment is conducted (Section 3.2). In addition, new scientific evidence may emerge. As a result, although concerning the same substance, the data available for the respective evaluations are likely to differ. For instance, a dossier submitted for a regulated product that is also a nutrient may contain information not available (e.g. unpublished proprietary studies) or not considered (e.g. new evidence) in the previous safety assessment of the nutrient. On the other hand, the dossier may contain the set of standard studies required by the sectoral guidance, while providing an incomplete or even no overview of the relevant data on the toxicity of the nutrient available in the literature.
In addition to the specificities of nutrient risk assessment discussed in Section 3.3.2, the hazard identification and characterisation should consider the following elements.
Human data provide the most relevant information for hazard identification. They are generally available for the risk assessment of nutrients, and, when they are of sufficient quality and extent, are given the greatest weight for the establishment of a HBGV. In such case, animal data (including data arising from the classical toxicity data set) may be used as supportive rather than as a primary source of evidence for the hazard identification and characterisation. Information from animal studies may also contribute to inform the biological‐based model described above (Sections 3.3.2 and 3.3.3), through the identification of homeostatic and pathological critical events in the context of nutrient kinetics and dynamics.
If animal studies are conducted or used for the purpose of demonstrating the safety of consumption of a regulated product which is also a nutrient, specific considerations are required, as follows.
Estimation of the dose of exposure to the nutrient should take into account the total amount, i.e. nutrient intake from the substance intentionally added to food and feed, and from the background diet. In case the study report does not include information about the nutrient content of the laboratory chow, the applicant should seek to obtain it from the entity that conducted the study or from veterinary guidelines valid at the time of the study. In case the intake from the background diet cannot be estimated, this uncertainty should be considered when characterising the dose–response relationships for the adverse effects.
Similarities and disparities between the animal species and humans regarding e.g. the nutrient homeostasis, the effect of nutrient interactions, etc. which may limit the external validity of the animal model, should be taken into account.
Mandates on regulated products that are also nutrients require collaboration among relevant EFSA Panels/units throughout the process. In the cases in which the hazard characterisation results in establishing a new HBGV for the regulated product that is also a nutrient, the scientific output should specify that the newly established HBGV replaces the previously established HBGV for that nutrient. A dialogue with the risk managers responsible for all relevant sectoral areas may also be needed to address possible impacts and needs for updating previous assessments of the nutrient.
For risk management purposes, ADIs are conventionally expressed relative to body weight (e.g. mg/kg body weight per day), which are applied to the general population, while ULs are traditionally expressed in absolute amounts (e.g. mg/day) for defined life‐stage groups (SCF, 2000; EFSA, 2006). When a new HBGV for a nutrient is established, it may be useful to express it in both manners, to facilitate the use by risk managers. This may require specific considerations regarding the extrapolation of values between different life‐stage groups, taking into account known differences in body size, physiology, metabolism, absorption and excretion of a nutrient (SCF, 2000) (see Annex A).
3.4. Exposure assessment
For nutrients, the exposure assessment focuses on the dietary intake, and combines data on concentrations of the nutrient present in foods and drinks with the quantity of those foods and drinks consumed. The choice of the method to estimate the intake and related degree of refinement of the intake estimates have to be tailored to the question addressed by the risk assessor. For regulated products, intake estimations addressing the intended uses are needed to support the decision‐making. Details on the data, tools and methods used by the different areas are available in the sectoral guidances.8
A comprehensive characterisation of risks associated with the dietary intake of a nutrient requires a complete intake assessment from all dietary sources, i.e. accounting for the natural nutrient content of foods as well as the additional contributions of regulated products. Total dietary intake of nutrients can be estimated by combining data from food composition databases and food consumption surveys. An accurate nutrient intake assessment requires to capture intake from intentional use in foods (e.g. as food additives), their migration into foods (e.g. from food contact materials), their use in food supplements or as nutrient sources (i.e. added to food) or their presence as residues (e.g. pesticides or feed additives).
Upon receipt of a new mandate/application for, or when conducting the re‐evaluation of a regulated product that is also a nutrient, previous EFSA assessments of the total intake of the nutrient by consumers should be considered.
When estimating total dietary intake of nutrients, potential sources of uncertainties regarding composition data include:
uncertainty regarding the extent to which the analytical values reported in the food composition databases include the contributions of nutrient‐containing regulated products (e.g. depending on the time of food sampling vs. the time of authorisation of the regulated uses, representativeness of the samples with regard to the authorisation conditions, and use patterns of the regulated products). This also includes uncertainties regarding nutrient contents in foods due to fortification. Also, composition data estimated from the ingredient list of foods require assumptions regarding the amounts of additives contained in foods (not reported on labels).
uncertainty related to speciation of the nutrient, i.e. distribution of its various chemical and physical forms, where applicable.
Chemical analyses of whole foods do not allow to distinguish between the naturally occurring fraction of the nutrient and the fraction contributed by regulated products (either intentionally added or present as residues). The relative contributions of regulated products to the overall nutrient intake have to be calculated from product‐specific concentration data, which may be inferred from e.g. authorised or reported use levels. For instance, two sets of concentration data can be used to estimate the intake of a nutrient from its food additive uses: (1) maximum permitted levels as set down in the EU legislation, and (2) use levels reported by food operators.
When available, biomarkers (e.g. blood levels, urinary excretion levels) may be useful to estimate overall nutrient intake. Biomarkers of exposure reflect the internal dose and exposure from all sources. However, reliable biomarkers of intake9 are available only for a limited number of nutrients. When these biomarkers are used, back‐calculation to dietary intake using kinetic modelling may be possible, but it triggers even more complex assessments to identify to which extent nutrients used as regulated products were included in the exposure assessment. In addition to model uncertainty, there can be uncertainty in identifying the contribution of a specific route of exposure (i.e. food) compared to other sources (e.g. inhalation).
Variability in the intake, as well as uncertainties regarding intake estimates should be presented in the scientific output.
3.5. Risk characterisation
In accordance with the principles of risk assessment of chemicals in food, the risk characterisation shall integrate the information from the hazard characterisation (Section 3.3) and the exposure assessment (Section 3.4) with the aim to provide practical scientific advice for risk managers.
Figure 3 presents the integrated approach proposed for the risk characterisation step of the safety assessment of regulated products that are also nutrients. The proposal addresses the need to ensure consistency across EFSA's assessments while providing the flexibility required by the specific regulatory frameworks.
Figure 3.
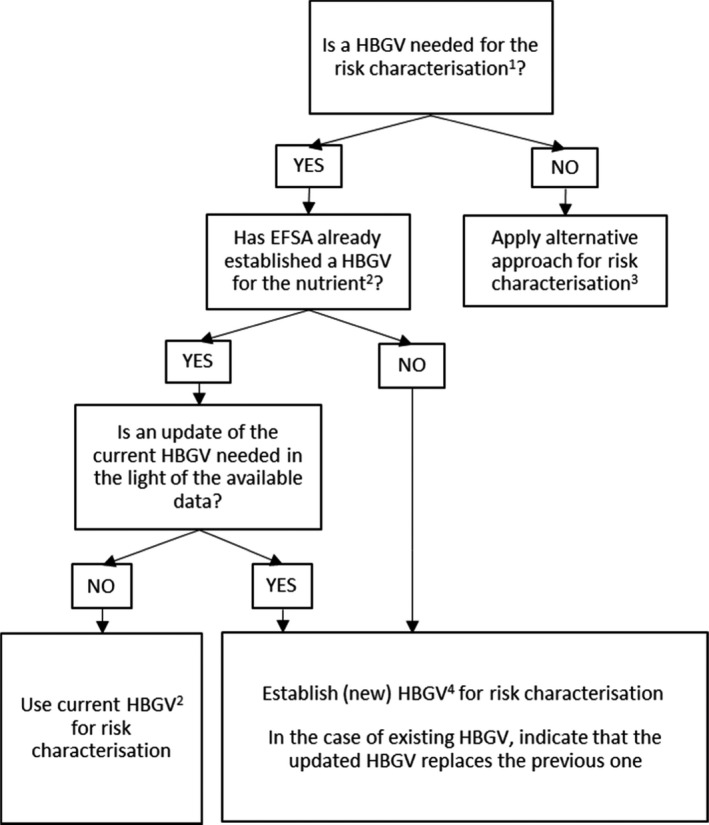
-
1The need for establishing a HBGV should be established according to the sectoral legislation.2This includes ULs established by the SCF/NDA Panel or other HBGVs for nutrients (e.g. ADI) established by other Panels in the context of previous assessments. Indications from the SCF/NDA Panel on the highest level of intake where there was reasonable confidence in data on the absence of adverse effects may also be considered.3Examples of alternative approaches include, for example, the calculation of a margin of exposure (MoE); comparative approaches in which the regulated product under evaluation can be considered ‘as safe as’ already authorised products; or estimations based on the relative contribution of the use as regulated product to total dietary intake.4When data are insufficient for establishing a HBGV, an indication on the highest level of intake where there is reasonable confidence in data on the absence of adverse effects may be provided.
In some circumstances, the establishment of a HBGV may not be necessary to provide the advice needed by risk managers. For instance, depending on the proposed uses and use levels of the regulated product, its contribution to the overall nutrient intake may be small and the risk associated with its consumption might be considered negligible. In some cases, it may be sufficient for risk assessors to comment on the MoE between the RP and the estimated human dietary intake, without establishing a HBGV. In other cases, risk assessors may base their conclusions on a comparative approach, i.e. the regulated product under evaluation is considered ‘as safe as’ already authorised products (e.g. based on their comparable composition and conditions of uses). In certain cases, the EFSA Panel or unit in charge of the assessment could justify that an overall risk assessment of the nutrient is not needed and proceed with the usual sectoral assessment.
When a HBGV is needed, the first step should be to consider whether EFSA has already established a HBGV for the nutrient, and in this case, to assess if the HBGV requires an update according to Section 3.3.4.
When a HBGV is established, the risk characterisation is based on the comparison with the estimated intake. The characterisation of the risk for consumers should include the total estimated intake of the nutrient, aggregating all sources of dietary intake. If the total estimated intake exceeds the HBGV, depending on the extent of this exceedance and the nature/severity of the potential adverse effects for the consumers, the scientific output should discuss its implications in order to inform risk managers’ decisions.
When the information is insufficient for establishing a HBGV, an indication may be given on the highest level of intake where there is reasonable confidence in data on the absence of adverse effects, in line with the approach previously applied by the SCF/NDA Panel (Appendix A). This is typically informed by human data about levels of nutrient intake significantly above those obtained from the diet (e.g. from food supplements), which have not been associated with adverse effects. Prevalence of adverse effects can be assumed to be low in populations with intakes below this value. However, characterisation of the risk is uncertain in populations with intakes above this value, because the relationship of such value to the toxicity for the nutrient is not known.
In addition, the relative contribution of the regulated use under assessment to the overall intake should be discussed as part of the risk characterisation. Whenever relevant, this information will be used by the relevant Panel or unit for proposing regulatory limits (e.g. maximum residue levels (MRLs) for pesticides in food) or for making specific recommendations for risk managers to consider, in accordance with EFSA's remit in the different sectors.
The risk characterisation should address all population groups included in the target population for the assessment (Section 3.1). If it is not possible for sub‐populations having distinct sensitivities to the adverse effects of the nutrient because of e.g. specific genetic background, conditions or diseases (‘susceptible groups’) to be covered by the HBGV, this should be indicated.
In line with EFSA's risk assessment principles, the risk characterisation should include a discussion and a characterisation of the uncertainties in the assessment.
4. Conclusions and recommendations
According to the General Food Law (Regulation (EC) No 178/2002)1, the EFSA Scientific Panels are responsible for providing scientific opinions within their own spheres of competence. A nutrient may be assessed under different regulatory frameworks by different Panels, i.e. following a generic mandate for establishing an UL (NDA Panel) or in the context of mandates addressing the safety of regulated products (by the respective Panel).
As the Scientific Committee is responsible for developing harmonised risk assessment methodologies and procedures relevant to cross‐cutting scientific matters, EFSA requested the Scientific Committee to provide recommendations to address such situation, with the view to ensure internal consistency regarding HBGVs for nutrients. This is particularly relevant for food additives and pesticides, for which HBGVs, e.g. ADIs, are regularly established.
To that end, the Scientific Committee has developed an approach, summarised in the decision tree shown in Figure 3, in order to guide the risk characterisation step of EFSA's assessments of regulated products that are also nutrients. In addition, the Scientific Committee formulates the following recommendations:
EFSA should use the integrated approach described herein for the assessment of applications for regulated products that are also nutrients, considering cross‐sectoral implications. The safety of these products should take into consideration the risk of adverse effects for the consumer associated with the total dietary intake of the nutrient.
The Panel/unit that has received the mandate should assess the need for establishing a HBGV for the nutrient in the context of such mandate.
When an existing HBGV for the nutrient established by EFSA is available, it should be used as the HBGV for the risk characterisation of regulated products that are also nutrients. The Panel/unit that has received the mandate should assess, in consultation with the NDA Panel and other relevant EFSA Panels/units, whether the data available for the assessment of the regulated product are consistent with the existing value or whether an update of the HBGV is needed.
When an existing HBGV (e.g. UL) requires an update, or there is no existing HBGV, the hazard identification and hazard characterisation steps should take into account the specificities pertaining to nutrient risk assessment, e.g. consideration of their biological role, homeostatic mechanisms and their regulation, and requirement, as described in Sections 3.3.2 and 3.3.4 of this Statement. The assessment should be conducted in consultation with the NDA Panel and other relevant EFSA Panels/units throughout the process.
When an update of the existing HBGV (e.g. UL) for a nutrient is required, the scientific output should clearly state that the new value replaces the previous one. The Scientific Committee recommends that EFSA maintains a centralised database of HBGVs for nutrients.
To establish a HBGV for regulated products that are nutrients, EFSA should ensure an integrated and harmonised hazard characterisation across EFSA's sectors. The Scientific Committee should be engaged whenever, during the assessment of a regulated product, a Panel or unit finds evidence that a pre‐existing HBGV for a nutrient (e.g. UL) needs to be updated (see point 2), or a new HBGV for the nutrient is needed (see point 3), because of the multidisciplinary nature of such assessment and the need to evaluate cross‐sectoral implications.
The HBGVs for regulated products that are also nutrients should be expressed relative to body weight (e.g. mg/kg body weight per day) and also in absolute amounts (mg/day). Specific values for particular subpopulations may be derived, where appropriate. The lowest value expressed on a per kg bw basis could be used by risk managers as equivalent to the ADI mentioned in the sectoral legislation. For practical reasons and to facilitate the use by risk managers, the Scientific Committee recommends that the values are also expressed in absolute amounts by life‐stage groups, in line with the guiding principles for establishing ULs (SCF, 2000).
When a quantitative risk characterisation is conducted, the HBGV should be compared with consumers’ total intake of the nutrient from all dietary sources (from natural occurrence, from contaminants and regulated uses).
In order to inform risk managers, the dietary intake assessment should, as much as possible, characterise the contribution from the regulated use relative to the intake from other dietary sources.
An active dialogue with risk managers should occur throughout the assessment process to discuss the potential regulatory implications that the assessment may have beyond the sectoral legislation under which the application was submitted.
Abbreviations
- ADI
acceptable daily intake
- ADME
absorption, distribution, metabolism, and excretion
- ANS
EFSA Panel on Panel on Food Additives and Nutrient Sources Added to Food
- AR
average requirement
- ARfD
acute reference dose
- AROI
acceptable range of oral intake
- BBM
biological‐based model
- BMDL
lower confidence limit of the benchmark dose
- bw
body weight
- DRV(s)
dietary reference value(s)
- ETE(s)
essential trace element(s)
- FAF
EFSA Panel on Food Additives and Flavourings
- FAO
Food and Agriculture Organization
- FEEDAP
EFSA Panel on Additives and Products or Substances used in Animal Feed
- HBGV(s)
health‐based guidance value(s)
- IAEA
International Atomic Energy Agency
- ILMERAC
International Liaison Group for Methods on Risk Assessment of Chemicals in Food
- IPCS
International Programme on Chemical Safety
- LOAEL
lowest observed adverse effect level
- MoE
margin of exposure
- MRL(s)
maximum residue level(s)
- NDA
EFSA Panel on Nutrition, Novel Foods and Food Allergens
- NOAEL
no observed adverse effect level
- OECD
Organisation for Economic Co‐operation and Development
- PoD
point of departure (used as equivalent to the RP in some jurisdictions)
- PPP(s)
plant protection product(s)
- PPR
EFSA Panel on Plant Protection Products and their Residues
- PRI(s)
population reference intake(s)
- RP
reference point
- SC
Scientific Committee
- SCF
Scientific Committee on Food
- TDI
tolerable daily intake
- ToR
terms of reference
- UF
uncertainty factor
- UL
tolerable upper intake level
- WHO
World Health Organization
Glossary
The terms described below have been used in this document according to the meaning described in this glossary.
- Essential Trace Element (ETE)
The term is used in line with the definition proposed by the Joint FAO/IAEA/WHO Expert Consultation on Trace Elements in Human Nutrition ‘arbitrarily, the term “trace” has been applied to concentrations of element not exceeding 250 pg per g of matrix. An element is considered essential to an organism when reduction of its exposure below a certain limit results consistently in a reduction in a physiologically important function, or when the element is an integral part of an organic structure performing a vital function in that organism’ (WHO/IAEA/FAO, 1996).
- Exposure
Amount of a particular substance that is taken in a specific frequency over a certain amount of time. Dietary exposure assessment is the qualitative and/or quantitative evaluation of the likely intake of a substance from the diet by an individual or population.
- Health‐Based Guidance Value (HBGV)
HBGV is an umbrella term for values that are established as the result of the risk assessment of chemical substances and provides guidance on safe consumption of substances, taking into account current safety data, uncertainties in these data, and the likely duration of consumption (EFSA Glossary10). Depending on their nature and applications, a HBGV for oral exposure may be termed tolerable upper intake level (UL) (nutrients), acceptable daily intake (ADI) (food additives, pesticides), tolerable daily intake (TDI) (contaminants) or acute reference dose (ARfD).
- Nutrient
An element or compound needed for the normal growth, development and health maintenance of the organism. This includes vitamins, minerals and macronutrients.
- Regulated product
Food‐ and feed‐related products that require a scientific assessment by EFSA to evaluate their safety for supporting marketing authorisation decisions by risk managers. Regulated products include substances used in food and feed (such as additives, enzymes, flavourings, and nutrient sources), food contact materials and pesticides, genetically modified organisms, novel foods, food‐related processes and processing aids. This Statement only covers regulated products that are also nutrients.
- Regulated products that are also nutrients
Regulated products that, aside from their regulated use, are, by nature, forms of a nutrient (see definition above).
- Regulated use
The uses of regulated products requiring a scientific assessment by EFSA. Depending on the scope of the mandate, the risk assessment may be extended to regulated uses falling outside of EFSA's remit
Appendix A – Overview of EFSA's evaluations of tolerable upper intake levels (ULs)
1.
The SCF and, subsequently, EFSA received the mandate to assess the tolerable upper intake levels of the substances listed below.
The SCF/EFSA NDA Panel established ULs11 for:
boron (sodium borate and boric acid) (EFSA, 2004a)
copper (SCF, 2003e)
fluoride (EFSA, 2005a)
folate (synthetic folic acid) (SCF, 2000f)
iodine (SCF, 2002a)
magnesium (SCF, 2001c)
molybdenum (SCF, 2000)
nicotinic acid and nicotinamide (niacin) (SCF, 2002b)
selenium (SCF, 2000a)
preformed vitamin A (retinol and retinyl esters) (SCF, 2002c)
vitamin B6 (SCF, 2000g)
vitamin E (SCF, 2003c)
zinc (SCF, 2003d)
At the time of the assessment, data were insufficient to establish ULs for any population group for:
β‐carotene (SCF, 2000d)
biotin (SCF, 2001b)
chromium (trivalent) (SCF, 2003b)
folate (natural) (SCF, 2000f)
iron (EFSA, 2004b)
manganese (SCF, 2000e)
nickel (EFSA, 2005c)
pantothenic acid (SCF, 2002d)
phosphorus (EFSA, 2005d)
potassium (EFSA, 2005e)
silicon (EFSA, 2004c)
sodium15 (EFSA, 2005f)
tin (EFSA, 2005g)
vanadium (EFSA, 2004d)
vitamin B1 (SCF, 2001a)
vitamin B2 (SCF, 2000b)
vitamin B12 (SCF, 2000c)
vitamin C (L‐ascorbic acid, its calcium, potassium and sodium salts and L‐ascorbyl‐6-palmitate) (EFSA, 2004e)
vitamin K (SCF, 2003a)
In those cases, the SCF/EFSA NDA Panel provided advice on the highest level of intake where there is reasonable confidence in data on the absence of adverse effects.
Appendix B – Illustrations of the endogenous kinetics and homeostatic mechanisms of essential trace elements
1.
Inorganic micronutrients such as sodium, potassium, chloride, sulfur, phosphate, iodine and selenium are soluble at physiological pH and are absorbed easily. Then, they are systemically distributed either as ions or associated loosely with low molecular weight ligands (e.g. amino acids, polypeptides, organic acids) or with albumin. Their systemic burdens are controlled principally by renal excretion. In the case of magnesium and calcium their absorption is regulated, they exist as ions or protein bound states in the systemic circulation, and their excretion is via renal and intestinal excretion.
Metals such as zinc, copper, iron, manganese are poorly soluble at physiological pH and need specific ligands to protect tissues from the damage their free ions would cause and to support the metals’ kinetics and dynamics. Thus, each of these elements has specific carriers to facilitate its absorption, distribution (organ uptake), excretion and deposition, as well as its cellular biochemical function. Their homeostasis varies. For example, zinc homeostasis is regulated by control of its absorption and its excretion via gastrointestinal (including pancreatic) secretion and renal excretion; excessive systemic accumulation of zinc is countered by intracellular sequestration by hepatic metallothionein. The systemic burden of copper at high intakes is limited initially by down regulation of its intestinal uptake and by hepatobiliary excretion. At high intakes, copper is stored in metallothionein pools in the liver and gut mucosa, simultaneously and, perhaps in advance of this, there is an accumulation of copper in integuments and copper appears in the urine. The latter phenomena are regarded as early evidence of failed copper homeostasis and excessive internal copper burden.
In the case of iron, there is no excretory route to reduce its systemic burden. The only physiological means of doing this is by preventing the acquisition of iron. This is achieved by the down regulation of intestinal uptake of iron by a direct effect on enterocytic uptake mechanisms through modulation of expression of transfer mechanisms for iron. A further control on transfer of iron to the portal circulation is achieved by induction of enterocytic apoferritin which sequesters iron in ferritin inside the enterocytes. Subsequently, the unabsorbed iron in the ferritin is lost in the faeces when the enterocytes are shed. Excessive systemic iron burden and toxicity is countered by systemic apoferritins which sequester iron, these depots also provide a reserve of iron at time of deficient intake. The homeostasis of iron is sensitive also to systemic responses to inflammation and hypoxia, which demonstrate the subtlety of iron homeostasis in the context of these conditions. It is noteworthy that at high dietary intakes and intraluminal contents of iron, physiological barriers to iron absorption are ineffective because iron transfer across the intestinal mucosa occurs passively along a paracellular transepithelial concentration gradient bypassing the enterocytes. A similar phenomenon occurs with high intakes of other trace metals and dietary cations.
Interferences may occur between iron, copper and zinc which are thought to arise from competition for similar carriers in their chain of carriers. It is possible that these may affect their kinetics and dynamics resulting in altered absorption and physiological effectiveness. These interactions affecting absorption may vary according to the dietary milieu in which they are consumed.
Annex A – Review of current EFSA approaches for establishing HBGVs for nutrients used as regulated products
A.1. Introduction
Several EFSA Panels and Units establish Health‐Based Guidance Values (HBGV) as part of the hazard characterisation process. A HBGV is a science‐based recommendation for the maximum (oral) exposure to a substance that is not expected to result in an appreciable health risk, taking into account current data, uncertainties in these data, and the likely duration of consumption. The HBGV represents the highest exposure level that is considered of presenting no health concerns based on all the known facts at the time of the evaluation (FAO/WHO, 2009).
The terminology and the methodology for establishing HBGVs have evolved with time in the different sectors covered by EFSA. The term acceptable daily intake (ADI) was introduced in 1957 by the Council of Europe (Chemicals Regulation Directorate and Safety Executive, 2013). The ADI is generally used for substances intentionally added to food, such as additives, or to the residues in food following intended uses of the substance in the process for food production, such as residues of pesticides and feed additives in foods, including not only the active substance but also the relevant metabolites. In the US, the equivalent general concept of oral reference dose (RfD) or reference level (RfL) uses different terms as HBGV in different frameworks (US EPA, 2002).
Most chemicals for which an ADI has been established do not have human or animal physiological requirements, thus levels of exposure from zero up to the ADI are considered acceptable.
For vitamins and minerals, with particular physiological functions in the human body, the situation is different. Minimum intakes are required in order to fulfil physiological requirements, while excess intakes may lead to adverse health effects. Thus, a set of reference values, the Dietary Reference Values (DRVs), are typically derived for nutrients (EFSA, 2017). On one side, the average requirement (AR) and the population reference intake (PRI) or the adequate intake (AI) if a PRI cannot be established, and the reference intake (RI) range for nutrients provide guidance on the amount of a nutrient needed to maintain health in a healthy group of people. On the other side, the tolerable upper intake level (UL) represents the maximum level of total chronic daily intake of a nutrient (from all sources) judged to be unlikely to pose a risk of adverse health effects to humans (SCF, 2000).
A particularly complex situation arises when the substances intentionally added to food as additives, or the residues in food (and drinks) resulting from regulated uses of a substance such as a feed additive or pesticide, are also nutrients. This is not unusual: phosphates, chlorides, vitamin C or copper are just some examples that have received recent EFSA attention, leading to assessments of the same substance under different scientific methodological approaches and regulatory frames. This Annex summarises the current practices for establishing ADIs and ULs by EFSA Panels and Units.
A.2. Generic methodology for establishing HBGVs
Establishing a HBGV is the key step in the hazard characterisation process for consumer risk assessments. The basic principles and concepts were already defined in the 1950s, as part of the foundation for supporting chemical control through science‐based assessments. The basic concepts for establishing HBGVs have been reviewed by several authors and by WHO (e.g. Herrman and Younes, 1999; Speijers, 1999; Dybing et al., 2002; FAO/WHO, 2009).
Basically, during the hazard identification step of the risk assessment, all available information on the effects of the substance is assessed. Then, the relevant effects and their dose–response relationships are assessed in order to propose a level of exposure without observable adverse health effects. The process for the establishment of the HBGV includes the selection of a dose that can be used as a starting point for risk assessment as the ‘Reference Point’ (RP), also named ‘Point of Departure’ (PoD), followed by the selection of uncertainty factors (UFs) or safety factors, which are applied to the RP to ensure a sufficient level of protection for humans.
During the hazard identification, all available information, e.g. laboratory toxicity studies in animals and human evidence, such as data from experimental and observational studies and case reports, are evaluated in order to identify critical endpoints representing potential concerns for human health. For regulated products, the applicant is usually requested to submit a dossier containing a set of mandatory safety studies and a compilation of additional information, such as a review of published studies and previous assessments from other regulatory agencies. The data requirements depend on the sectoral legislation and are described in the relevant guidance documents.
The identification of the adverse effects produced by the substance should consider the biological relevance for humans, integrate different sets of data through a weight of evidence approach, and consider the uncertainties. The EFSA Scientific Committee has developed specific guidances for covering these critical steps: assessment of biological relevance (EFSA Scientific Committee, 2017c), weighing and integrating the different lines of evidence (EFSA Scientific Committee, 2017b); and assessing the uncertainty of the available data and scientific knowledge (EFSA Scientific Committee, 2018a,b).
Following the identification of hazards, the hazard characterisation step considers the exposure (e.g. the dose in experimental toxicity studies) at which critical effects are observed. The no observed adverse effect level (NOAEL) has been historically used as the RP for animal studies. At present EFSA considers that the benchmark dose (BMD) approach is scientifically more advanced and should be preferred, when possible, over the NOAEL approach for deriving human (health‐based) guidance values (EFSA Scientific Committee, 2017d).
Currently the NOAEL and the lowest observed adverse effect level (LOAEL) are still the most frequently used RPs in the existing EFSA assessments. Both are derived from tested doses and selected in line with a statistical analysis. The NOAEL is the highest level of a test substance that does not cause any observed and statistically significant adverse effect compared with the controls. Similarly, the LOAEL is the lowest dose where there is a statistically significant adverse effect compared with the controls. The NOAEL and LOAEL are consecutive dose levels within a study and should be considered in combination. The effects observed at the LOAEL in the different studies, and the progression to adversity, are used for identifying critical effects associated to expected adverse effects in humans, and then the lowest relevant NOAEL is used as RP. If statistically significant effects are observed at the lowest tested dose, only a LOAEL can be identified, and under certain circumstances this LOAEL can be used as RP. In some cases, the consideration of adversity is not evident, and the study reports N/LOELs (no/lowest observed effect levels), those can be also used as RP under certain conditions.
Regarding the UF to be applied to the RP for establishing a HBGV, EFSA has adopted the standard approaches developed during the last decades by different bodies. In its guidance on default values (EFSA Scientific Committee, 2012), EFSA proposed the default UF of 100, introduced in 1954 (Lehman and Fitzhugh, 1954) and adopted by JECFA in 1958; as well as the further division of UF into inter‐ and intra‐species subfactors as proposed by WHO/IPCS (2005). The default value of 100 is composed by a factor of 10 to account for interspecies differences and a factor of 10 for intraspecies (human interindividual) differences, and the two factors each consist of toxicokinetic and toxicodynamic subfactors of 4 and 2.5 for interspecies toxicokinetic and toxicodynamic differences, respectively; and 3.16 (100.5) each for human interindividual toxicokinetic and toxicodynamic differences. These and additional recommendations are summarised in Table A.1.
Table A.1.
Default uncertainty factors proposed by EFSA to be considered when setting HBGVs from animal studies to humans (EFSA Scientific Committee, 2012)
| Source of uncertainty | EFSA recommended default value | Comments | Reference provided in (EFSA Scientific Committee, 2012) |
|---|---|---|---|
| Inter‐species toxicokinetic variation | 4.0a | Combined inter‐species variation: 10 | WHO/IPCS, 2005 |
| Inter‐species toxicodynamic variation | 2.5a | WHO/IPCS, 2005 | |
| Human interindividual toxicokinetic variation | 3.16a | Combined human interindividual variation: 10 | WHO/IPCS, 2005 |
| Human interindividual toxicodynamic variation | 3.16a | WHO/IPCS, 2005 | |
| Subchronic (90‐day study) to chronic | 2 | If similar parameters investigated as usually done in chronic studies | (ECHA, 2010) (Zarn et al., 2011, 2015) |
| Subacute to chronic | Case by case | ||
| LOAEL as replacement for NOAEL | Case by case | ||
| Severity of the effect | Case by case |
If relevant chemical‐specific data on kinetics and/or dynamics are available, the relevant subfactor should be replaced by actual data.
Establishing a HBGV is not considered appropriate for substances with genotoxicity concerns. However, Chapter 8.1 of the Scientific Committee Opinion on genotoxicity testing strategies (EFSA Scientific Committee, 2011) describes some circumstances under which genotoxicity might occur only at doses resulting in saturation of detoxification pathways or in cases of substances that interact with molecular targets other than DNA (e.g. DNA polymerases, topoisomerases and spindle proteins). In such cases, provided robust data on the underlying mode of action are available and taking into account all other relevant information, establishing a HBGV might be possible.
A.3. Establishing ULs for nutrients
Nutrients may have adverse health effects if consumed in excessive amounts. HBGVs for nutrients are referred to as tolerable upper intake levels (UL). Guidelines for the development of ULs for vitamins and minerals were developed by the Scientific Committee on Food (SCF) in 2000 and subsequently applied by the NDA Panel (EFSA, 2006; EFSA NDA Panel, 2012a,b, 2018). The concept of UL also applies to other nutrients (EFSA NDA Panel, 2012c; EFSA, 2018b).
The UL is not a recommended level of intake. It is an estimate of the highest level of intake which carries no appreciable risk of adverse health effects (SCF, 2000). Thus, it has a similar meaning to the ADI. To establish whether an exposed population is at risk requires a risk assessment to determine what is the fraction (if any) of the population whose intake exceeds the UL and the magnitude and duration of the excessive intake. By definition, ULs are derived for the normal healthy population but excludes sub‐populations with extreme and distinct vulnerabilities due to genetic predisposition or other considerations (SCF, 2000). Sub‐populations needing special protection are better served through the use of public health screening, health care providers, product labelling, or other individualised strategies. The extent to which a sub‐population becomes significant enough to be assumed to be representative of a general population is an area of judgement and of risk management and is considered for individual nutrients (SCF, 2000).
Committees in charge of setting ULs for nutrients used the classical risk assessment framework of chemical substances: identifying potential hazards associated with high intake of the nutrient, characterising those hazards on the basis of dose–response analyses, and establishing an UL, where possible (IOM, 1998; SCF, 2000; WHO, 2002; EVM, 2003; EFSA, 2006; WHO, 2006; IOM, 2007).
Nutrient risk assessment is associated with specific challenges in relation to: the nature of the available evidence; the interpretation of observed effects in the context of the normal physiology of the nutrient; and ultimately, the establishment of a HBGV at a level, which cannot be the same or less than the nutrient adequacy level. In other words: there can be adverse health effects resulting from intakes that are either too low or too high. The acceptable range of intake should prevent deficiencies and toxicities. Consequently, the following elements require special attention for the establishment of HBGVs for nutrients:
Nutrients are elements or compounds needed for the normal growth, development and health maintenance of the organism i.e. are required from the diet to satisfy nutritional needs.
There is a long history of safe consumption of nutrients at the levels found in human diets; because nutrients are often subject to homeostatic regulation, which provides a measure of protection against excessive intakes.
Data on adverse effects are available from studies in humans (in particular experimental studies in which the nutrient was used as food supplement or as drug, as well as case reports). On the other hand, human intervention studies are generally not designed for evaluating adverse effects or toxicities but rather to evaluate beneficial or metabolic effects of nutrients.
For many nutrients experimental studies in animals aimed at detecting adverse effects are often not available.
These elements are not relevant for additives and other regulated products, unless they are also nutrients.
A methodology for establishing ULs for nutrients and related substances was proposed by IPCS (2002) and reviewed at a joint FAO/WHO workshop in 2005 (WHO, 2006). The report proposed a model for nutrient risk assessment highlighting the differences with the general assessment of chemicals that are non‐nutrients. Accounting for uncertainties in the evidence base is an important step in establishing ULs. If available data allow, a quantitative adjustment for uncertainties may be applied to the value derived from the intake–response assessment. Generally, however, adjustments for uncertainty must make use of UF. The FAO/WHO model suggests the use of a single composite factor rather than applying individual UFs and advised uncertainty considerations to be checked against the level of recommended intake relative to biological essentiality or the levels of intake associated with the demonstrated impact on health.
ULs are derived for different life‐stage groups using relevant data (SCF, 2000). For a specific life‐stage group for which insufficient or no data is available, extrapolations may be made from the UL for other groups on the basis of known differences in weight, body size, physiology or metabolism, absorption and excretion of a nutrient (SCF, 2000). For instance, values for specific groups may be established by extrapolating values for adults on the basis of body weight or relative energy expenditure, depending on the nutrient. In practice, the SCF and EFSA NDA Panel scaled down values for adults to children and adolescents for a number of vitamins and minerals, based on relative body weights (e.g. vitamin B6, folate, nicotinic acid, nicotinamide, molybdenum, copper, fluoride) or body weight to account for difference in basal metabolic rate (e.g. vitamin A, vitamin E, iodine, zinc, calcium), using reference weights (EFSA, 2006).
A.4. Establishing ADIs for additives and pesticides
For food additives and pesticide residues, the long‐term oral HBGV is expressed as the ADI. For short‐term oral exposures to pesticide residues, the ADI may be complemented with an acute reference dose (ARfD) as guidance for a maximum short‐term ingestion during a single day or single meal. The regulatory framework and the specific considerations used by the EFSA Panels and units when setting the ADI are summarised below. Table A.2 compares the different risk assessment steps for the assessment of nutrients with those for food additives and pesticide residues.
A.4.1. Food additives
Regulation (EC) No 1331/200818 of the European Parliament and Council establishing a common authorisation procedure for food additives, food enzymes and food flavourings lays down a common procedure for the assessment and authorisation of food additives, food enzymes and food flavourings in view of updating the Community lists of permitted substances defined in the relevant sectoral food laws.
The risk assessment process for food additives follows the standard 4 steps: hazard identification, hazard characterisation, exposure assessment and risk characterisation. For food additives, the HBGV is the ADI and is applicable for the general population, except for infants below 16 weeks of age. For compounds with (or presumed to have) a common mode of action, a group ADI may be set, which is applicable to the sum of the compounds in the group.
The data requirements for the risk assessment of food additives are described in the Guidance for submission for food additive evaluations (EFSA ANS Panel, 2012) as follows: ‘This guidance describes a tiered approach which balances data requirements against other considerations such as use and animal welfare. The tiered approach initially uses less complex tests to obtain hazard data; these are then evaluated to determine if they are sufficient for risk assessment or, if not, to design studies at higher tiers. The intention is that in developing their dossier, applicants will be able to more readily identify relevant data needs which will allow adequate assessment of risks to humans from the intended use whilst strengthening the scientific basis for the assessment. In addition, this approach takes into consideration animal welfare by adopting animal testing strategies in line with the 3 Rs (replacement, refinement, reduction). The Panel recommends that an integrated testing strategy, which may include alternative approaches, should be used to further support the risk assessment. The Panel has sought to provide an overall concept with clear information on a tiered approach for risk assessment. Using this tiered approach, a minimal dataset applicable to all compounds has been developed under Tier 1. Compounds which are systemically absorbed or for which toxic or genotoxic effects are found in Tier 1 will require Tier 2 testing to generate more extensive data. Tier 3 defines detailed testing for specific endpoints, for which Tier 2 testing results raised concerns, and is performed on a case‐by‐case basis’. The guidance uses the term Margin of Safety (MoS) to represent the margin between the NOAEL or BMDL and the estimated exposure. It should be noted that according to EFSA harmonised terminology the margin between the NOAEL or BMDL and the estimated exposure should be named margin of exposure (MoE) instead of MoS.
This guidance is complemented by a statement on the conceptual framework for the risk assessment of certain food additives re‐evaluation (EFSA ANS Panel, 2014) to facilitate the risk assessment process.
An ADI is established for compounds for which thresholded mechanisms of toxicity can be reasonably expected based on the available data. Where the available data are limited, the application of the MoE approach can be considered.
During the evaluation of food additives, the EFSA Panels have already conducted several assessments for nutrients used as additives. The establishment of the HBGV in those cases has been adapted to each case and available information. For example, an ADI was established during the re‐evaluation of carotenes (EFSA ANS Panel, 2012b) and phosphates (EFSA FAF Panel, 2019a); while a risk characterisation based on lack of safety concern at the estimated exposure levels, without establishing a HBGV due to lack of data, was the approach used for Vitamin C (EFSA ANS Panel, 2015) and chlorides (EFSA FAF Panel, 2019b).
A.4.2. Pesticide active substances in Plant Protection Products
Regulation EC 1107/200919 concerning the placing of plant protection products on the market replaced Directive 91/414/EEC and is complemented with specific provisions on data requirements (Regulations EU 283/201320 and 284/201321).
The use of the ADI as HBGV for pesticides was included in Directive 91/414/EEC and confirmed as a legally binding value in Regulation (EC) No 1107/2009, and is complemented with an ARfD for assessing acute (one meal or one day) exposures and an acceptable operator exposure level (AOEL) (and an acute‐AOEL when relevant) for non‐dietary exposures. The legislation also set a minimum UF (safety margin), at least 100, to be used when establishing a HBGV: ‘Where relevant, an ADI, AOEL and ARfD shall be established. When establishing such values an appropriate safety margin of at least 100 shall be ensured taking into account the type and severity of effects and the vulnerability of specific groups of the population. When the critical effect is judged of particular significance, such as developmental neurotoxic or immunotoxic effects, an increased margin of safety shall be considered, and applied if necessary’ (Regulation (EC) No 1107/2009).
Following the evaluation of the submitted dossier by the rapporteur Member State (RMS) and the peer review by EFSA, the proposed values are discussed by risk managers, included in the review report prepared by the European Commission, and following the risk managers agreement, become mandatory and included in the EU pesticides database.22
A specific guidance, defined as a working document of the European Commission services, for setting ARfDs has been prepared for pesticides in the EU context,23 whereas no specific guidance for setting an ADI for pesticides at the European level is available, with the exception of the legal requirements for a UF of at least 100, and that data collected on humans shall not be used to lower the UF resulting from tests or studies on animals.
Based on these legal principles, the derivation of ADIs through the peer‐review process in the EFSA Conclusions on Pesticides (EFSA Scientific Committee, 2012) is based on the standard default approach and a safety margin of 100 is applied to the selected RP.
The RP is usually the NOAEL for the critical effect observed in the animal studies. Although the use of the BMD approach for selecting the RP has been discussed and proposed as a scientifically justified improvement (Chemicals Regulation Directorate and Safety Executive, 2013; EFSA, 2014), it has not been used by EFSA in the regulatory assessments of pesticides yet, and the NOAEL, and alternatively the LOAEL with an additional safety factor, are still the RP currently used for pesticides.
The UF may be increased in case of incomplete datasets, uncertainties, or concerns related to the severity of the observed effects. This is in line with the international provisions in the area of pesticides (WHO, 2015). At the international level, there are no legal limitations for using human data for reducing the UF. JMPR also uses the standard justification for the default UF of 100 and the IPCS (WHO/IPCS, 2005) recommendation for subdividing the two 10‐fold factors into toxicokinetic and toxicodynamic subfactors (WHO/IPCS, 2015). There are also numerical recommendations for the ‘extra’ UF to be used to account for the use of a LOAEL instead of a NOAEL, use the NOAEL from short‐term toxicity studies in order to account for the short duration of the study and for covering deficiencies in the database.
According to a review conducted in 2013, in the EFSA assessments of pesticides the default value of 100 has been applied in 187 out of 213 (88%) cases; additional factors, ranging from 2 to 20, have been added, mostly related to the use of a LOAEL instead of a NOAEL or to the severity of the observed effects (Chemicals Regulation Directorate and Safety Executive, 2013). The review also indicated that for 128 compounds (57%) the original proposal by the RMS was in agreement with the final ADI value; and that for the other cases the changes were justified by the use of different RP or a different UF, in addition to the consideration of new data submitted during the EFSA procedure (Chemicals Regulation Directorate and Safety Executive, 2013).
A case of particular interest is copper, also used as pesticidal active substance, for which the ADI is based on human data for infants (adults: 0.2 mg Cu/kg bw per day and infants: 0.15 mg Cu/kg bw per day). The ADI is supported by animal data (90‐day rat study) with a NOAEL of 16 mg Cu/kg bw per day (EFSA, 2018a). It should be noted that the EFSA Conclusion on copper used the term ADI as this is the term included in the sectorial legislation and in the EU pesticides database. Nevertheless, it is important to mention that as stated in the EFSA Peer Review Report ‘it was felt by the experts that the term ‘ADI’ was not fully adequate to copper as a micronutrient essential for life; the term ‘upper limit’ used in the nutrient area would be more appropriate; therefore, in the specific case of copper, the ADI is considered equivalent to an UL’.
In addition to the pesticide active substance, the EFSA assessments include the evaluation of metabolites observed in food commodities of plant and animal origin. The first step is the identification of the metabolites that may be present in the different food commodities according to the intended uses and metabolisms studies. Then the assessment focused on whether the metabolites are of higher, equal or lower toxicity than the parent compound. The metabolic pathway of the parent and specific toxicity data for the metabolite, including genotoxicity, guide how the decision is taken. The conclusion that the metabolite is of equal or lower toxicity than the parent implies that HBGV values of the parent could apply to the metabolite for the consumer's risk assessment. If the toxicological profile of the metabolite is qualitatively different from that of the parent or if the metabolite is quantitatively of higher toxicity than the parent, specific HBGVs should be established for the metabolite (EFSA, 2016). Although not yet implemented as mandatory guidance in the regulatory context, EFSA has updated the scientific methodology for assessing the metabolism studies, deciding on further testing, and establishing the residue definition (EFSA PPR Panel, 2016).
Table A.2.
Comparison of the HBGV and consumer risk assessment frameworks applicable to nutrients, food additives and pesticides according to current sectoral guidance documents
| Nutrient | Food Additives | Pesticides | |
|---|---|---|---|
| Scope of the RA | Risk associated to the intake of the nutrient from all foods and drinks | Risk associated to the intake of the additive from its proposed uses and use levels | Risk associated to the presence of residues of the pesticides or their metabolites in food |
| Term used for the oral HBGV | Tolerable Upper Intake Level (UL) | Acceptable daily intake (ADI) |
Acceptable daily intake (ADI) Acute reference dose (ARfD) for short‐term exposures |
| Definition | Maximum level of total chronic daily intake of a nutrient (from all sources) judged to be unlikely to pose a risk of adverse health effects to humans. |
Estimate of the amount of a substance in food or drinking water that can be consumed daily over a lifetime without presenting an appreciable risk to health NB: For compounds with (or presumed to have) a common mode of action, group ADIs may be set which apply to the sum of the compounds in the group. |
ADI: Estimate of the amount of a substance in food or drinking water that can be consumed daily over a lifetime without presenting an appreciable risk to health ARfD: Estimated intake of a chemical substance in food, expressed on a body weight basis, that can be ingested over a short period of time, usually during one meal or one day, without posing a health risk. NB: Metabolites may be covered by the assessment of the active substance or require specific ADIs, ARfDs |
| Expression |
Absolute daily amount (e.g. mg/day) Specific values derived for various life‐stage groups in the population, e.g. infants, children, adults, the elderly, and women during pregnancy or lactation. |
Daily amount per kg body weight (e.g. mg/kg bw per day) | Daily amount per kg body weight (e.g. mg/kg bw per day) |
| Establishing HBGV and approach taken in case a HBGV cannot be established |
|
|
ADI/ARfD are established for active substances and relevant metabolites for which there is sufficient information and no concerns on genotoxicity have been identified. If concerns on genotoxicity are identified, no ADI/ARfD are proposed and the risk assessment indicates concerns for consumers (if genotoxicity is confirmed) or is not finalised pending submission of additional information for clarifying the genotoxicity potential. |
| Target population and consideration of susceptible groups |
Normal healthy population, divided into various life‐stage groups to account for normally expected variability in sensitivity. Excludes sub‐populations with extreme and distinct vulnerabilities due to e.g. genetic predisposition or other considerations (e.g. certain disease states)a. Groups excluded are flagged in the risk characterisation for risk managers to take specific measures where appropriate. |
General population; specific assessments may be conducted for infants below 16 weeks. Susceptible groups covered by the HBGV case by case. Groups excluded are flagged in the risk characterisation for risk managers to take specific measures where appropriate |
General population; specific assessments may be conducted for infants below 16 weeks. The HBGV should protect the whole population. |
| Regulatory implications | The UL is not a regulatory limit; it is a value meant to support decision‐making. | The maximum use levels for food additives are established taking into consideration that the intake of an additive from all its uses should not exceed its ADI. | The ADI/ARfD proposed by EFSA are discussed by risk managers. If agreed, they become mandatory and are published by European Commission. |
| Evidence base |
|
|
Regulatory GLP animal toxicity studies covering the active substance and if needed relevant metabolites The review of scientific literature provided by the applicant in the dossier (mandatory requirement but not always covered at the levels required by the EFSA guidance) Additional information provided during the Member State or EFSA process, including the public and expert consultations Human data even if available cannot be used for increasing the ADI/ARfD according to the legislation |
| Toxicokinetics (ADME) |
|
|
The dossier should contain sufficient information on metabolism in animals, plants and degradation products in the environment for the identification of relevant metabolites, those not covered by the active substances, and for setting the residue definitions applicable to plant and animal commodities |
| Hazard identification |
|
|
An extensive set of toxicological studies is mandatory and specifically mention in the regulatory framework (regulations on data requirements for active substances and for Plant Protection Products). The information is compared with the criteria for classification and labelling established under the CLP Regulation. The RMS is expected to submit a proposal for classification and labelling to ECHA. |
| Hazard characterisation |
|
|
An extensive set of toxicological studies is mandatory and specifically mention in the regulatory framework (regulations on data requirements for active substances and for Plant Protection Products). The information is checked for completeness and used for setting the RPs. A minimum UF of 100 is established in the legislation, and additional factors can be added in case of non‐standard uncertainties in the data set. |
| Exposure assessment |
|
|
Occurrence in plants, expected levels of residues in plant commodities is assessed through supervised field trials. Occurrence in animal commodities following the consumption of feed containing residues is assessed through agreed exposure models. Consumer exposure is quantified according to the Pesticides Residues Intake Model (PRIMo) developed by EFSA based on internationally agreed methodologies and covering different national and general diets. |
| Risk characterisation |
|
|
For the chronic assessment, the higher level of exposure (considering all crops with MRLs) should not exceed 100% of the ADI. For the acute exposure, the higher level of exposure for each commodity should not exceed 100% of the ARfD. |
The extent to which a sub‐population becomes significant enough to be assumed to be representative of a general population is an area of judgement and of risk management and is considered for individual nutrients.
For additives the term MoS has been used as representation of the margin between the NOAEL or BMDL and the anticipated exposure (EFSA ANS Panel, 2012a), however according to EFSA terminology this comparison should be named as Margin of Exposure (MoE).
References
Chemicals Regulation Directorate, Health & Safety Executive, UK, 2013. Investigation of the state of the art on identification of appropriate reference points for the derivation of health‐based guidance values (ADI, AOEL and AAOEL) for pesticides and on the derivation of uncertainty factors to be used in human risk assessment. Supporting Publications 2013;10(4):EN‐413, 169 pp. https://doi.org/10.2903/sp.efsa.2013.EN‐413
Dybing E, Doe J, Groten J, Kleiner J, O'Brien J, Renwick AG, Schlatter J, Steinberg P, Tritscher A, Walker R and Younes M, 2002. Hazard characterisation of chemicals in food and diet: dose response, mechanisms and extrapolation issues. Food and Chemical Toxicology, 40, 237–282. https://doi.org/10.1016/s0278-6915(01)00115-6
ECHA (European Chemicals Agency), 2010. Guidance on information requirements and chemical safety assessment. Chapter R.8: Characterisation of dose[concentration]‐response for human health. Guidance for implementation of REACH.
EFSA (European Food Safety Authority), 2005. Opinion of the Scientific Committee on a request from EFSA related to A Harmonised Approach for Risk Assessment of Substances Which are both Genotoxic and Carcinogenic. EFSA Journal 2005;3(10):282, 33 pp. https://doi.org/10.2903/j.efsa.2005.282
EFSA (European Food Safety Authority), 2006. Tolerable upper intake levels for vitamins and minerals. Available online: http://www.efsa.europa.eu/sites/default/files/efsa_rep/blobserver_assets/ndatolerableuil.pdf
EFSA (European Food Safety Authority), 2014. Conclusion on the peer review of the pesticide human health risk assessment of the active substance chlorpyrifos. EFSA Journal 2014;12(4):3640, 34 pp. https://doi.org/10.2903/j.efsa.2014.3640
EFSA (European Food Safety Authority), 2016. Outcome of the pesticides peer review meeting on general recurring issues in mammalian toxicology. EFSA Journal 2016;13(8):1074E, 24 pp. https://doi.org/10.2903/sp.efsa.2016.EN-1074
EFSA (European Food Safety Authority), 2017. Dietary reference values for nutrients: Summary report. EFSA supporting publication 2017;14(12):15121, 98 pp. https://doi.org/10.2903/sp.efsa.2017.15121
EFSA (European Food Safety Authority), 2018a. Peer review of the pesticide risk assessment of the active substance copper compounds copper(I), copper(II) variants namely copper hydroxide, copper oxychloride, tribasic copper sulfate, copper(I) oxide, Bordeaux mixture. EFSA Journal 2018;16(1):5152, 25 pp. https://doi.org/10.2903/j.efsa.2018.5152
EFSA (European Food Safety Authority), 2018b. Protocol for the scientific opinion on the Tolerable Upper Intake Level of dietary sugars. EFSA Journal 2018;16(8):5393, 47 pp. https://doi.org/10.2903/j.efsa.2018.5389
EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources Added to Food), 2012a. Guidance for submission for food additive evaluations. EFSA Journal 2012, 10(7):2760, 60 pp. https://doi.org/10.2903/j.efsa.2012.2760
EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), 2012b. Scientific Opinion on the re‐evaluation of Mixed Carotenes (E 160a (i)) and beta‐Carotene (E 160a (ii)) as a food additive. EFSA Journal 2012;10(3):2593, 67 pp. https://doi.org/10.2903/j.efsa.2012.2593
EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources Added to Food), 2014. Statement on a conceptual framework for the risk assessment of certain food additives re‐evaluated under Commission Regulation (EU) No 257/2010. EFSA Journal 2014;12(6):3697, 11 pp. https://doi.org/10.2903/j.efsa.2014.3697
EFSA ANS Panel (EFSA Panel on Food Additives and Nutrient Sources added to Food), 2015. Scientific Opinion on the re‐evaluation of ascorbic acid (E 300), sodium ascorbate (E 301) and calcium ascorbate (E 302) as food additives. EFSA Journal 2015;13(5):4087, 124 pp. https://doi.org/10.2903/j.efsa.2015.4087
EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings), 2019a. Scientific Opinion on the re‐evaluation of phosphoric acid–phosphates – di‐, tri‐ and polyphosphates (E 338–341, E 343, E 450–452) as food additives and the safety of proposed extension of use. EFSA Journal 2019;17(6):5674, 156 pp. https://doi.org/10.2903/j.efsa.2019.5674
EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings), 2019b. Scientific Opinion on the re‐evaluation of hydrochloric acid (E 507), potassium chloride (E 508), calcium chloride (E 509) and magnesium chloride (E 511) as food additives. EFSA Journal 2019;17(7):5751, 51 pp. https://doi.org/10.2903/j.efsa.2019.5751
EFSA NDA Panel (Panel on Dietetic Products, Nutrition and Allergies), 2012a. Scientific Opinion on the Tolerable Upper Intake Level of vitamin D. EFSA Journal 2012;10(7):2813, 45 pp. https://doi.org/10.2903/j.efsa.2012.2813
EFSA NDA Panel (Panel on Dietetic Products, Nutrition, and Allergies), 2012b. Scientific Opinion on the Tolerable Upper Intake Level of calcium. EFSA Journal 2012;10(7):2814, 44 pp. https://doi.org/10.2903/j.efsa.2012.2814.
EFSA NDA Panel (Panel on Dietetic Products, Nutrition and Allergies), 2012c. Scientific Opinion related to the Tolerable Upper Intake Level of eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and docosapentaenoic acid (DPA). EFSA Journal 2012;10(7):2815, 48 pp. https://doi.org/10.2903/j.efsa.2012.2815
EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2018. Scientific opinion on the update of the tolerable upper intake level for vitamin D for infants. EFSA Journal 2018;16(8):5365, 118 pp. https://doi.org/10.2903/j.efsa.2018.5365
EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues), 2016. Guidance on the establishment of the residue definition for dietary risk assessment. EFSA Journal 2016;14(12):4549, 129 pp. https://doi.org/10.2903/j.efsa.2016.4549
EFSA Scientific Committee, 2011. Scientific opinion on genotoxicity testing strategies applicable to food and feed safety assessment. EFSA Journal 2011;9(9):2379, 69 pp. https://efsa.onlinelibrary.wiley.com/doi/abs/10.2903/j.efsa.2011.2379
EFSA Scientific Committee, 2012. Guidance on selected default values to be used by the EFSA Scientific Committee, Scientific Panels and Units in the absence of actual measured data. EFSA Journal 2012;10(3):2579, 32 pp. https://doi.org/10.2903/j.efsa.2012.2579
EFSA Scientific Committee, 2017a. Clarification of some aspects related to genotoxicity assessment. EFSA Journal 2017;15(12):e05113, 25 pp. https://doi.org/10.2903/j.efsa.2017.5113
EFSA Scientific Committee, 2017b. Guidance on the use of the weight of evidence approach in scientific assessments. EFSA Journal 2017;15(8):e04971, 69 pp. https://doi.org/10.2903/j.efsa.2017.4971
EFSA Scientific Committee, 2017c. Guidance on the assessment of the biological relevance of data in scientific assessments. EFSA Journal 2017;15(8):4970, 73 pp. https://doi.org/10.2903/j.efsa.2017.4970
EFSA Scientific Committee, 2017d. Update: use of the benchmark dose approach in risk assessment. EFSA Journal 2017;15(1):e04658, 41 pp. https://doi.org/10.2903/j.efsa.2017.4658
EFSA Scientific Committee, 2018a. The principles and methods behind EFSA's Guidance on Uncertainty Analysis in Scientific Assessment. EFSA Journal 2018;16(1):5122, 235 pp. https://doi.org/10.2903/j.efsa.2018.5122
EFSA Scientific Committee, 2018b. Guidance on Uncertainty Analysis in Scientific Assessments. EFSA Journal 2018;16(1);5123, 39 pp. https://doi.org/10.2903/j.efsa.2018.5123
EVM (Expert Group on Vitamins and Minerals), 2003. Safe Upper Levels for Vitamins and Minerals.
FAO (Food and Agriculture Organization of the United Nations), WHO (World Health Organization), 2009. Principles and Methods for the Risk Assessment of Chemicals in Food. Environmental health criteria, 240.
Herrman JL and Younes M, 1999. Background to the ADI/TDI/PTWI. Regulatory Toxicology and Pharmacology, 30, S109–S113. https://doi.org/10.1006/rtph.1999.1335
IOM (Institute of Medicine), 1998. Dietary reference intakes. A risk assessment model for establishing upper intake levels for nutrients. National Academies Press, Washington, DC.
IOM (Institute of Medicine), 2007. Nutritional Risk Assessment: Perspectives, Methods, and Data Challenges: Workshop Summary. National Academies Press, Washington, DC.
Krewski D, Andersen ME, Tyshenko MG, Krishnan K, Hartung T, Boekelheide K, Wambaugh JF, Jones D, Whelan M, Thomas R, Yauk C, Barton‐Maclaren T and Cote I, 2020. Toxicity testing in the 21st century: progress in the past decade and future perspectives. Archives of Toxicology, 94, 1‐58. https://doi.org/10.1007/s00204-019-02613-4
Lehman AJ and Fitzhugh OG, 1954. 100‐Fold Margin of Safety. Assoc. Food Drug Off. U.S.Q. Bulletin, 13, 51–58.
SCF (Scientific Committee on Food), 2000. Guidelines of the Scientific Committee on Food for the development of tolerable upper intake levels for vitamins and minerals.
Speijers GJ, 1999. Precision of estimates of an ADI (or TDI or PTWI). Regulatory Toxicology and Pharmacology, 30, S87–S93. https://doi.org/10.1006/rtph.1999.1331
U.S. EPA (United States Environmental Protection Agency), 2002. A Review of the Reference Dose and Reference Concentration Processes. U.S. Environmental Protection Agency, Risk Assessment Forum, Washington, DC, EPA/630/P‐02/002F.
WHO (World Health Organization), IPCS (International Programme on Chemical Safety), 2002. Principles and methods for the assessment of risk from essential trace elements. Available online: https://apps.who.int/iris/handle/10665/42416
WHO (World Health Organization), IPCS (International Programme on Chemical Safety), 2005. Chemical‐specific adjustment factors for interspecies differences and human variability : guidance document for use of data in dose/concentration‐response assessment. World Health Organization.
WHO (World Health Organization), FAO (Food and Agriculture Organization of the United Nations) and IPCS (International Programme on Chemical Safety), 2006. A model for establishing upper levels of intake for nutrients and related substances: report of a Joint FAO/WHO Technical Workshop on Food Nutrient Risk Assessment, WHO Headquarters, Geneva, Switzerland, 2‐6 May 2005. World Health Organization. https://apps.who.int/iris/handle/10665/43451
WHO (World Health Organization), 2015. Pesticide residues in food: WHO core assessment group on Pesticide Residues: guidance document for WHO monographers and reviewers.
Zarn JA, Engeli BE and Schlatter JR, 2011. Study parameters influencing NOAEL and LOAEL in toxicity feeding studies for pesticides: Exposure duration versus dose decrement, dose spacing, group size and chemical class. Regulatory Toxicology and Pharmacology, 61, 243–250. https://doi.org/10.1016/j.yrtph.2011.08.004
Zarn JA, Hanggi E and Engeli BE, 2015. Impact of study design and database parameters on NOAEL distributions used for toxicological concern (TTC) values. Regulatory Toxicology and Pharmacology, 72, 491–500. https://doi.org/10.1016/j.yrtph.2015.05.015
Abbreviations
- 3Rs
replacement, refinement, reduction
- ADI
acceptable daily intake
- AI
adequate intake
- AOEL
acceptable operator exposure levels
- AR
average requirement
- ARfD
acute reference dose
- AROI
acceptable range of oral intake
- BMD
benchmark dose
- BMDL
lower confidence limit of the benchmark dose
- CLP
Classification, Labelling and Packaging
- DRV(s)
dietary reference value(s)
- ECHA
European Chemicals Agency
- FAF
EFSA Panel on Food Additives and Flavourings
- FAO
Food and Agriculture Organization
- FEEDAP
EFSA Panel on Additives and Products or Substances used in Animal Feed
- GLP
good laboratory practice
- HBGV(s)
health‐based guidance value(s)
- IOM
US Institute of Medicine
- IPCS
International Programme on Chemical Safety
- JECFA
Joint FAO/WHO Expert Committee on Food Additives
- LOAEL
lowest observed adverse effect level
- MoE
margin of exposure (Ratio of the no‐observed‐adverse‐effect level or benchmark dose lower confidence limit for the critical effect to the theoretical, predicted or estimated exposure dose or concentration).
- MoS
margin of safety (The margin between the health‐based guidance value (reference dose) and the actual or estimated exposure dose or concentration. Used for referring to the MoE in the EFSA ANS Panel 2012 guidance)
- MRL(s)
maximum residue level(s)
- NDA
EFSA Panel on Nutrition, Novel Foods and Food Allergens
- NOAEL
no observed adverse effect level
- OECD
Organisation for Economic Co‐operation and Development
- PoD
point of departure (used as equivalent to the RP in some jurisdictions)
- PPP(s)
plant protection product(s)
- PPR
EFSA Panel on Plant Protection Products and their Residues
- PRI(s)
population reference intake(s)
- PRIMO
Pesticides Residues Intake Model
- RfD
oral reference dose (used as equivalent to the ADI in some jurisdictions)
- RfL
reference level (used as equivalent to the HBGV in some jurisdictions)
- RI
reference intake
- RMS
rapporteur member state
- RP
reference point
- SC
Scientific Committee
- SCF
Scientific Committee on Food
- TDI
tolerable daily intake
- UF
uncertainty factor
- UL
tolerable upper intake level
- US EPA
United States Environmental Protection Agency
- WHO
World Health Organization
Suggested citation: EFSA Scientific Committee , More S, Bampidis V, Benford D, Bragard C, Halldorsson T, Hougaard Bennekou S, Koutsoumanis K, Machera K, Naegeli H, Nielsen S, Schlatter J, Schrenk D, Silano V, Turck D, Younes M, Aggett P, Castenmiller J, Giarola A, de Sesmaisons‐Lecarré A, Tarazona J, Verhagen H and Hernández‐Jerez A, 2021. Statement on the derivation of Health‐Based Guidance Values (HBGVs) for regulated products that are also nutrients. EFSA Journal 2021;19(3):6479, 39 pp. 10.2903/j.efsa.2021.6479
Requestor: EFSA
Question number: EFSA‐Q‐2019‐00505
Panel members: Vasileos Bambidis, Diane Benford, Claude Bragard, Thorhallur I Halldorsson, Antonio Hernandez‐Jerez, Susanne Hougaard Bennekou, Kostas Koutsoumanis, Kyriaki Machera, Simon More, Hanspeter Naegeli, Søren Nielsen, Josef R Schlatter, Dieter Schrenk, Vittorio Silano†, Dominique Turck and Maged Younes.
Declarations of interest: The declarations of interest of all scientific experts active in EFSA's work are available at https://ess.efsa.europa.eu/doi/doiweb/doisearch.
Acknowledgments: The Scientific Committee wishes to thank the EFSA trainee Maria Chiara Astuto for the support provided to this scientific output.
Adopted: 17 February 2021
This publication is linked to the following EFSA Supporting Publications article: http://onlinelibrary.wiley.com/doi/10.2903/sp.efsa.2021.EN-6480/full
Notes
Regulation (EC) No 178/2002 of the European Parliament and of the Council of 28 January 2002 laying down the general principles and requirements of food law, establishing the European Food Safety Authority and laying down procedures in matters of food safety. OJ L 31, 1.2.2002, p. 1–24.
Before the establishment of EFSA in 2002, EU risk managers had commissioned this task to the Scientific Committee on Food (SCF). In 2006, EFSA published a report gathering the scientific opinions of the SCF and the NDA Panel on the ULs for vitamins and minerals. Since then, EFSA continues to receive requests from the European Commission to update ULs for specific nutrients.
Regulated products include substances used in feed and food (such as additives, enzymes, flavourings, nutrient sources), novel foods, infant formulae, food contact materials, pesticides, genetically modified organisms (GMOs). They are submitted to EFSA under a sectoral legislation and/or under the general food law (Regulation (EC) No 178/2002). An overview of all regulatory framework of regulated products evaluated by EFSA is available here: http://www.efsa.europa.eu/en/applications
EFSA glossary: https://www.efsa.europa.eu/en/glossary/acceptable-daily-intake
All dietary sources.
The HBGV for copper is currently under revision by the Scientific Committee (EFSA‐Q‐2020‐00399).
An overview of all regulatory framework of regulated products evaluated by EFSA is available here: https://www.efsa.europa.eu/en/applications
EFSA's sectoral guidance documents are available at: https://www.efsa.europa.eu/en/applications/pesticides/regulationsandguidance http://www.efsa.europa.eu/en/applications/foodingredients/regulationsandguidance
The term ‘biomarker of intake’ refers to biomarkers of exposure that specifically reflect the exposure to the nutrient through the diet (i.e. its dietary intake).
For quick reference, an overview table of all UL values is available at: http://www.efsa.europa.eu/sites/default/files/assets/UL_Summary_tables.pdf.
This opinion reviews the tolerable upper intake level of calcium for the general population; it supersedes the opinion from the SCF published in 2003 (EFSA, 2006).
This opinion reviews the tolerable upper intake level of vitamin D for the general population; it supersedes the opinion from the SCF published in 2002 (EFSA, 2006).
This opinion reviews the tolerable upper intake level of vitamin D for infants; it supersedes the NDA Panel opinion published in 2012 for this age group (EFSA NDA Panel, 2012a).
For sodium and chloride, safe and adequate intakes have been established by the NDA Panel (EFSA NDA Panel, 2019a,b).
The safety of individual amino acids was not reviewed in that opinion.
At the time of this Statement, a review was on‐going (EFSA, 2018b).
Regulation (EC) No 1331/2008 of the European Parliament and of the Council of 16 December 2008 establishing a common authorisation procedure for food additives, food enzymes and food flavourings. OJ L 354, 31.12.2008, p. 1–6.
Regulation (EC) No 1107/2009 of the European Parliament and of the Council of 21 October 2009 concerning the placing of plant protection products on the market and repealing Council Directives 79/117/EEC and 91/414/EEC. OJ L 309, 24.11.2009, p. 1–50.
Commission Regulation (EU) No 283/2013 of 1 March 2013 setting out the data requirements for active substances, in accordance with Regulation (EC) No 1107/2009 of the European Parliament and of the Council concerning the placing of plant protection products on the market. OJ L 93, 3.4.2013, p. 1–84.
Commission Regulation (EU) No 284/2013 of 1 March 2013 setting out the data requirements for plant protection products, in accordance with Regulation (EC) No 1107/2009 of the European Parliament and of the Council concerning the placing of plant protection products on the market. OJ L 93, 3.4.2013, p. 85–152.
EU pesticides database, available at: http://ec.europa.eu/food/plant/pesticides/eu-pesticides-database/public/?event=homepage&language=EN
EC Guidance for the setting of an Acute Reference Dose (ARfD), available at: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_ppp_app-proc_guide_tox_acute-ref-dose.pdf
References
- Aggett PJ, 2007. Nutrient risk assessment: setting upper levels and an opportunity for harmonization. Food and Nutrition Bulletin, 28, S27–S37. 10.1177/15648265070281s104 [DOI] [PubMed] [Google Scholar]
- AMS (Academy of Medical Sciences), 2007. Identifying the environmental causes of disease: how should we decide what to believe and when to take action? Available online: https://acmedsci.ac.uk/file-download/34590-IECDWebD.pdf
- Boobis A, Chiodini A, Hoekstra J, Lagiou P, Przyrembel H, Schlatter J, Schutte K, Verhagen H and Watzl B, 2013. Critical appraisal of the assessment of benefits and risks for foods, ‘BRAFO Consensus Working Group’. Food and Chemical Toxicology, 55, 659–675. 10.1016/j.fct.2012.10.028 [DOI] [PubMed] [Google Scholar]
- Bruins MJ, Mugambi G, Verkaik‐Kloosterman J, Hoekstra J, Kraemer K, Osendarp S, Melse‐Boonstra A, Gallagher AM and Verhagen H, 2015. Addressing the risk of inadequate and excessive micronutrient intakes: traditional versus new approaches to setting adequate and safe micronutrient levels in foods. Food & Nutrition Research, 59, 26020. 10.3402/fnr.v59.26020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2004a. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] related to the Tolerable Upper Intake Level of Boron (Sodium Borate and Boric Acid). EFSA Journal 2004;2(8):80, 22 pp. 10.2903/j.efsa.2004.80 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2004b. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] related to the Tolerable Upper Intake Level of Iron. EFSA Journal 2004;2(11):125, 34 pp. 10.2903/j.efsa.2004.125 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2004c. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] related to the Tolerable Upper Intake Level of Silicon. EFSA Journal 2004;2(5):60, 11 pp. 10.2903/j.efsa.2004.60 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2004d. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] related to the Tolerable Upper Intake Level of Vanadium. EFSA Journal 2004;2(3):33, 22 pp. 10.2903/j.efsa.2004.33 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2004e. Opinion of the Scientific Panel on Dietetic Products, Nutrition and Allergies (NDA) a request from the Commission related to the Tolerable Upper Intake Level of Vitamin C (L‐Ascorbic acid, its calcium, potassium and sodium salts and L‐ascorbyl‐6‐palmitate). EFSA Journal 2004;2(5):59, 20 pp. 10.2903/j.efsa.2004.59 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2005a. Opinion of the Scientific Committee on a request from EFSA related to A Harmonised Approach for Risk Assessment of Substances Which are both Genotoxic and Carcinogenic. EFSA Journal 2005;3(10):282, 33 pp. 10.2903/j.efsa.2005.282 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2005b. Opinion of the Scientific Panel on Dietetic Products, Nutrition and Allergies (NDA) on a request from the Commission related to the Tolerable Upper Intake Level of Fluoride. EFSA Journal 2005;3(3):192, 40 pp. 10.2903/j.efsa.2005.192 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2005c. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] on a request from the Commission related to the Tolerable Upper Intake Level of Chloride. EFSA Journal 2005;3(6):210, 9 pp. 10.2903/j.efsa.2005.210 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2005d. Opinion of the Scientific Panel on Dietetic Products, Nutrition and Allergies (NDA) on a request from the Commission related to the Tolerable Upper Intake Level of Nickel. EFSA Journal 2005;3(2):146, 21 pp. 10.2903/j.efsa.2005.146 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2005e. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] related to the tolerable upper intake level of phosphorus. EFSA Journal 2005;3(1):233, 19 pp. 10.2903/j.efsa.2005.233 [DOI]
- EFSA (European Food Safety Authority), 2005f. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] on a request from the Commission related to the Tolerable Upper Intake Level of Potassium. EFSA Journal 2005;3(2):193, 19 pp. 10.2903/j.efsa.2005.193 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2005g. Opinion of the Scientific Panel on Dietetic products, nutrition and allergies [NDA] related to the Tolerable Upper Intake Level of Sodium. EFSA Journal 2005;3(6):209, 26 pp. 10.2903/j.efsa.2005.209 [DOI] [Google Scholar]
- EFSA (European Food Safety Authority), 2006. Tolerable upper intake levels for vitamins and minerals. Scientific Committee on Food, Scientific Panel on Dietetic Products, Nutrition and Allergies. EFSA Journal 2006;480 pp.
- EFSA (European Food Safety Authority), 2016. EFSA Strategy 2020, Trusted science for safe food, Protecting consumers’ health with independent scientific advice on the food chain.
- EFSA (European Food Safety Authority), 2018a. Conclusion on the peer review of the pesticide risk assessment of the active substance copper compounds copper(I), copper(II) variants namely copper hydroxide, copper oxychloride, tribasic copper sulfate, copper(I) oxide, Bordeaux mixture. EFSA Journal 2018;16(1):5152, 25 pp. 10.2903/j.efsa.2018.5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2018b. Protocol for the scientific opinion on the Tolerable Upper Intake Level of dietary sugars. EFSA Journal 2018;16(8):5393, 47 pp. 10.2903/j.efsa.2018.5389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA (European Food Safety Authority), 2020. Draft framework for protocol development for EFSA's scientific assessments. EFSA supporting publication 2020;17(4):EN‐1843, 46 pp. 10.2903/sp.efsa.2020.EN-1843 [DOI]
- EFSA (European Food Safety Authority), 2021. Outcome of the public consultation on draft EFSA Statement on the derivation of Health‐Based Guidance Values (HBGVs) for regulated products that are also nutrients. EFSA supporting publication 2021:EN‐6480. 64 pp. 10.2903/sp.efsa.2021.EN-6480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA ANS Panel (Panel on Food Additives and Nutrient Sources Added to Food), 2012. Guidance for submission for food additive evaluations. EFSA Journal 2012;10(7):2760, 60 pp. 10.2903/j.efsa.2012.2760 [DOI] [Google Scholar]
- EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings), 2019a. Scientific Opinion on the re‐evaluation of phosphoric acid–phosphates – di‐, tri‐ and polyphosphates (E 338–341, E 343, E 450–452) as food additives and the safety of proposed extension of use. EFSA Journal 2019;17(6):5674, 156 pp. 10.2903/j.efsa.2019.5674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA FAF Panel (EFSA Panel on Food Additives and Flavourings), 2019b. Scientific Opinion on the re‐evaluation of hydrochloric acid (E 507), potassium chloride (E 508), calcium chloride (E 509) and magnesium chloride (E 511) as food additives. EFSA Journal 2019;17(7):5751, 51 pp. 10.2903/j.efsa.2019.5751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA FEEDAP Panel (EFSA Panel on Additives and Products or Substances used in Animal Feed), 2019. Scientific Opinion on the safety and efficacy of copper chelates of lysine and glutamic acid as a feed additive for all animal species. EFSA Journal 2019;17(6):5728, 14 pp. 10.2903/j.efsa.2019.5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic products, Nutrition, and Allergies), 2010a. Scientific Opinion on principles for deriving and applying Dietary Reference Values. EFSA Journal 2010;8(3):1458, 30 pp. 10.2903/j.efsa.2010.1458 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition, and Allergies), 2010b. Scientific Opinion on Dietary Reference Values for fats, including saturated fatty acids, polyunsaturated fatty acids, monounsaturated fatty acids, trans fatty acids, and cholesterol. EFSA Journal 2010;8(3):1461, 107 pp. 10.2903/j.efsa.2010.1461 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition, and Allergies), 2010c. Scientific Opinion on Dietary Reference Values for carbohydrates and dietary fibre. EFSA Journal 2010;8(3):1462, 77 pp. 10.2903/j.efsa.2010.1462 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2012a. Scientific Opinion on the Tolerable Upper Intake Level of vitamin D. EFSA Journal 2012;10(7):2813, 45 pp. 10.2903/j.efsa.2012.2813 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition, and Allergies), 2012b. Scientific Opinion on the Tolerable Upper Intake Level of calcium. EFSA Journal 2012;10(7):2814, 44 pp. 10.2903/j.efsa.2012.2814 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2012c. Scientific Opinion related to the Tolerable Upper Intake Level of eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA) and docosapentaenoic acid (DPA). EFSA Journal 2012;10(7):2815, 48 pp. 10.2903/j.efsa.2012.2815 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2012d. Scientific Opinion on Dietary Reference Values for protein. EFSA Journal 2012;10(2):2557, 66 pp. 10.2903/j.efsa.2012.2557 [DOI] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Dietetic Products, Nutrition and Allergies), 2018. Scientific opinion on the update of the tolerable upper intake level for vitamin D for infants. EFSA Journal 2018;16(8):5365, 118 pp. 10.2903/j.efsa.2018.5365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens), 2019a. Scientific Opinion on dietary reference values for chloride. EFSA Journal 2019;17(9):5779, 24 pp. 10.2903/j.efsa.2019.5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA NDA Panel (EFSA Panel on Nutrition, Novel Foods and Food Allergens), 2019b. Scientific Opinion on the dietary reference values for sodium. EFSA Journal 2019;17(9):5778, 191 pp. 10.2903/j.efsa.2019.5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA PPR Panel (EFSA Panel on Plant Protection Products and their Residues), 2018. Scientific opinion on pesticides in foods for infants and young children. EFSA Journal 2018;16(6):5286, 75 pp. 10.2903/j.efsa.2018.5286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- EFSA Scientific Committee , 2017. Guidance on the risk assessment of substances present in food intended for infants below 16 weeks of age. EFSA Journal 2017;15(5):4849, 58 pp. 10.2903/j.efsa.2017.4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- IOM (Institute of Medicine), 1997. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. National Academy Press, Washington, DC. [PubMed] [Google Scholar]
- IOM (Institute of Medicine), 1998a. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. National Academy Press, Washington, DC. [PubMed] [Google Scholar]
- IOM (Institute of Medicine), 1998b. Dietary Reference Intakes: a Risk Assessment Model for Establishing Upper Intake Levels for Nutrients. Washington, DC. National Academies Press. [PubMed] [Google Scholar]
- IOM (Institute of Medicine), 2000. Dietary Reference Intakes for vitamin C, vitamin E, selenium, and carotenoids. Washington, DC. National Academy Press. [PubMed]
- IOM (Institute of Medicine), 2001. Dietary Reference Intakes for vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. Washington, DC. National Academy Press. [PubMed] [Google Scholar]
- Krewski D, Andersen ME, Tyshenko MG, Krishnan K, Hartung T, Boekelheide K, Wambaugh JF, Jones D, Whelan M, Thomas R, Yauk C, Barton‐Maclaren T and Cote I, 2020. Toxicity testing in the 21st century: progress in the past decade and future perspectives. Archives of Toxicology, 94, 1–58. 10.1007/s00204-019-02613-4 [DOI] [PubMed] [Google Scholar]
- Mertz W, 1993. Essential trace metals: new definitions based on new paradigms. Nutrition Reviews, 51, 287–295. 10.1111/j.1753-4887.1993.tb03057.x [DOI] [PubMed] [Google Scholar]
- NASEM (National Academies of Sciences, Engineering, and Medicine), 2018. Global harmonization of methodological approaches to nutrient intake recommendations: proceedings of a workshop—in brief. The National Academies Press, Washington, DC. 10.17226/24989 [DOI]
- Renwick AG, Flynn A, Fletcher RJ, Muller DJG, Tuijtelaars S and Verhagen H, 2004. Risk‐benefit analysis of micronutrients. Food and Chemical Toxicology, 42, 1903–1922. 10.1016/j.fct.2004.07.013 [DOI] [PubMed] [Google Scholar]
- SCF (Scientific Committee on Food), 2000. Guidelines of the Scientific Committee on Food for the development of tolerable upper intake levels for vitamins and minerals. SCF/CS/NUT/UPPLEV/11 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80a_en.pdf
- SCF (Scientific Committee on Food), 2000a. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Selenium. SCF/CS/NUT/UPPLEV/25 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80g_en.pdf
- SCF (Scientific Committee on Food), 2000b. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Vitamin B2. SCF/CS/NUT/UPPLEV/33 final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80i_en.pdf
- SCF (Scientific Committee on Food), 2000c. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Vitamin B12. SCF/CS/NUT/UPPLEV/42 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80d_en.pdf
- SCF (Scientific Committee on Food), 2000d. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Beta Carotene. SCF/CS/NUT/UPPLEV/37 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80b_en.pdf
- SCF (Scientific Committee on Food), 2000e. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Manganese. SCF/CS/NUT/UPPLEV/21 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80f_en.pdf
- SCF (Scientific Committee on Food), 2000f. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Folate. SCF/CS/NUT/UPPLEV/18 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80e_en.pdf
- SCF (Scientific Committee on Food), 2000g. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Vitamin B6. SCF/CS/NUT/UPPLEV/16 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80c_en.pdf
- SCF (Scientific Committee on Food), 2001a. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Vitamin B1. SCF/CS/NUT/UPPLEV/46 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out93_en.pdf
- SCF (Scientific Committee on Food), 2001b. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Biotin. SCF/CS/NUT/UPPLEV/55 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out106_en.pdf
- SCF (Scientific Committee on Food), 2001c. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Magnesium. SCF/CS/NUT/UPPLEV/54 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out105_en.pdf
- SCF (Scientific Committee on Food), 2002a. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Iodine. SCF/CS/NUT/UPPLEV/26 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out146_en.pdf
- SCF (Scientific Committee on Food), 2002b. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Levels of Nicotinic Acid and Nicotinamide (Niacin). SCF/CS/NUT/UPPLEV/39 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80j_en.pdf
- SCF (Scientific Committee on Food), 2002c. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Preformed Vitamin A (retinol and retinyl esters). SCF/CS/NUT/UPPLEV/24 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out145_en.pdf
- SCF (Scientific Committee on Food), 2002d. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Pantothenic Acid. SCF/CS/NUT/UPPLEV/61 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out80k_en.pdf
- SCF (Scientific Committee on Food), 2003a. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Vitamin K. SCF/CS/NUT/UPPLEV/32 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out196_en.pdf
- SCF (Scientific Committee on Food), 2003b. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Trivalent Chromium. SCF/CS/NUT/UPPLEV/67 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out197_en.pdf
- SCF (Scientific Committee on Food), 2003c. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Vitamin E. SCF/CS/NUT/UPPLEV/31 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out195_en.pdf
- SCF (Scientific Committee on Food), 2003d. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Zinc. SCF/CS/NUT/UPPLEV/62 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out177_en.pdf
- SCF (Scientific Committee on Food), 2003e. Opinion of the Scientific Committee on Food on the Tolerable Upper Intake Level of Copper. SCF/CS/NUT/UPPLEV/57 Final. Available online: https://ec.europa.eu/food/sites/food/files/safety/docs/sci-com_scf_out176_en.pdf
- Tijhuis MJ, de Jong N, Pohjola MV, Gunnlaugsdottir H, Hendriksen M, Hoekstra J, Holm F, Kalogeras N, Leino O, van Leeuwen FX, Luteijn JM, Magnusson SH, Odekerken G, Rompelberg C, Tuomisto JT, Ueland O, White BC and Verhagen H, 2012a. State of the art in benefit‐risk analysis: food and nutrition. Food and Chemical Toxicology, 50, 5–25. 10.1016/j.fct.2011.06.010. [DOI] [PubMed] [Google Scholar]
- Tijhuis MJ, Pohjola MV, Gunnlaugsdottir H, Kalogeras N, Leino O, Luteijn JM, Magnusson SH, Odekerken‐Schroder G, Poto M, Tuomisto JT, Ueland O, White BC, Holm F and Verhagen H, 2012b. Looking beyond borders: integrating best practices in benefit‐risk analysis into the field of food and nutrition. Food and Chemical Toxicology, 50, 77–93. 10.1016/j.fct.2011.11.044 [DOI] [PubMed] [Google Scholar]
- Verhagen H, Robinson T, Gallani B, Hugas M, Kleiner J, Hardy A and Devos Y, 2019. EFSA's third Scientific Conference ‘Science, Food, Society’: concluding remarks. EFSA Journal, 17, e170723. 10.2903/j.efsa.2019.e170723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkaik‐Kloosterman J, McCann MT, Hoekstra J and Verhagen H, 2012. Vitamins and minerals: issues associated with too low and too high population intakes. Food & Nutrition Research, 56. 10.3402/fnr.v3456i3400.5728.10.3402/fnr.v56i0.5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidry S, Hoekstra J, Hart A, Watzl B, Verhagen H, Schütte K, Boobis A and Chiodini A, 2013. Benefit‐Risk Analysis for Foods (BRAFO)‐ Executive Project Summary. European Journal of Nutrition & Food Safety, 3, 146–153. 10.9734/ejnfs/2013/7007 [DOI] [Google Scholar]
- WHO/FAO (World Health Organization, Food and Agriculture Organization of the United Nations) and International Programme on Chemical Safety, 2006. A model for establishing upper levels of intake for nutrients and related substances: report of a Joint FAO/WHO Technical Workshop on Food Nutrient Risk Assessment, WHO Headquarters, Geneva, Switzerland, 2‐6 May 2005. World Health Organization. Available online: https://apps.who.int/iris/handle/10665/43451
- WHO/IAEA/FAO (World Health Organization, International Atomic Energy Agency, Food and Agriculture Organization of the United Nations, 1996. Trace elements in human nutrition and health. World Health Organization. Available online: https://apps.who.int/iris/handle/10665/37931
- WHO/IPCS (World Health Organization) and International Programme on Chemical Safety), 2002. Principles and methods for the assessment of risk from essential trace elements. Available online: https://apps.who.int/iris/handle/10665/42416