

Abstract
SUMMARY
Many gene products exhibit great structural heterogeneity because of an array of modifications. These modifications are not directly encoded in the genomic template but often affect the functionality of proteins. Protein glycosylation plays a vital role in proper protein functions. However, the analysis of glycoproteins has been challenging compared with other protein modifications, such as phosphorylation. Here, we perform an integrated proteomic and glycoproteomic analysis of 83 prospectively collected high-grade serous ovarian carcinoma (HGSC) and 23 non-tumor tissues. Integration of the expression data from global proteomics and glycoproteomics reveals tumor-specific glycosylation, uncovers different glycosylation associated with three tumor clusters, and identifies glycosylation enzymes that were correlated with the altered glycosylation. In addition to providing a valuable resource, these results provide insights into the potential roles of glycosylation in the pathogenesis of HGSC, with the possibility of distinguishing pathological outcomes of ovarian tumors from non-tumors, as well as classifying tumor clusters.
In Brief
Hu et al. provide an integrated proteomic and glycoproteomic characterization of high-grade serous ovarian carcinomas and relevant non-tumor tissues, which reveals tumor-specific glycosylation, uncovers different glycosylation associated with three tumor clusters, and identifies glycosylation enzymes correlated with glycosylation alterations.
Graphical Abstract
INTRODUCTION
Ovarian cancer is the fifth leading cause of cancer death among women in the United States (Siegel et al., 2018; Torre et al., 2018). High-grade serous ovarian carcinomas (HGSCs) are the most common and lethal type of ovarian carcinoma responsible for the majority of ovarian cancer-related deaths (Siegel et al., 2018; Torre et al., 2018). The current standard of care is to perform an aggressive debulking surgery followed by platinum-taxane chemotherapy. However, the therapeutic approach is effective for only a small number of patients (Miller et al., 2009), and the 5-year survival rate is approximately 30% (Siegel et al., 2018; Torre et al., 2018). Results from the recent Prostate, Lung, Colorectal, and Ovarian (PLCO) Cancer Screening Trial, which combined transvaginal ultrasound and serum cancer antigen 125 (CA 125) levels for early detection, did not indicate a reduction in ovarian cancer mortality after 19 years of follow-up (Pinsky et al., 2016). Therefore, understanding the molecular mechanisms of ovarian cancer development, progression, and treatment susceptibility represents critical steps to further improve patient survival.
The molecular analysis of clinically annotated HGSCs using genomic, proteomic, and phosphoproteomic approaches highlighted by The Cancer Genome Atlas (TCGA; Cancer Genome Atlas Research Network, 2011) and Clinical Proteomic Tumor Analysis Consortium (CPTAC; Zhang et al., 2016) have provided an enhanced understanding of the impact of genomic alterations of HGSCs on protein networks and signaling pathways. TCGA identified a remarkable degree of genomic disarray in HGSC, including TP53 mutations in 96% of tumors and focal DNA copy number aberrations in 36% of cases; promoter methylation events involving 168 genes; and NF1, BRCA1, BRCA2, RB1, and CDK12 somatic mutations (Cancer Genome Atlas Research Network, 2011). CPTAC investigated the impact of genomic alterations on cancer biology at a functional level by comprehensively analyzing 169 HGSCs previously characterized by TCGA for proteomics and phosphoproteomics (Zhang et al., 2016). The study provided a number of important findings, such as the impact of copy number alterations on expression of proteins associated with chromosomal instability, protein acetylation associated with homologous recombination deficiency, and protein and phosphoprotein signaling pathways associated with cell survival (Coscia et al., 2018; Zhang et al., 2016). Although tumor tissues were extensively analyzed in these large-scale “omics” studies, strategies for the diagnosis and targeted therapy of HGSCs still need to be addressed.
In addition to genomic regulation, protein abundance and functions are further regulated by several factors, particularly proteinost-translational modifications (PTMs) (Vogel and Marcotte, 2012). Apart from phosphorylation, other protein modifications have not been investigated in large-scale proteomic studies (Mertins et al., 2016; Zhang et al., 2014). It is well-known that glycosylation plays a crucial role in cancer development processes, such as cell-cell adhesion, cell growth, ligand-receptor binding, and tumor metastasis (Hart and Copeland, 2010; Varki, 2017). Compared with other protein modifications, the analysis of glycoproteins has been limited because of the enormous complexity and heterogeneity of glycoprotein structures. Recent advances in glycoproteomic technologies have enabled the comprehensive analysis of complex glycoproteins (Narimatsu et al., 2018; Zielinska et al., 2010).
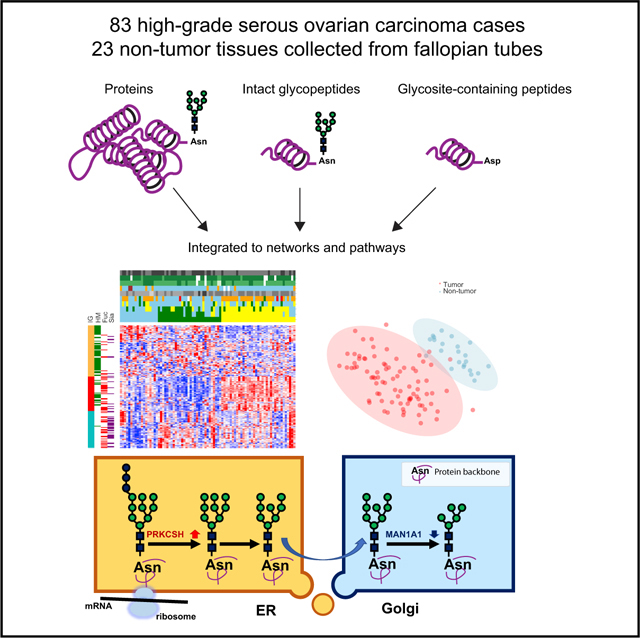
Here, we present the systematic proteomic and glycoproteomic analyses of 83 prospectively collected HGSC tissues and 23 relevant non-tumor tissues. The analysis allowed us to identify and quantify sites of N-linked glycosylation using a Solid-Phase Extraction of Glycosite-containing peptides (SPEG) approach (Zhang et al., 2003), as well as identification of the glycans occupying those N-linked sites using intact glycopeptides (IGPs) analysis (Khatri et al., 2016; Parker et al., 2013; Scott et al., 2011; Sun et al., 2016). This integrated approach provides a comprehensive look at the N-glycosylated proteins, their N-glycosylated sites, and the glycans occupying these sites in one study (Figure 1A). This is the first-of-its-kind large-scale omics analysis in clinical specimens with annotated clinical metadata. We found that glycoproteins in tumors compared with non-tumors and tumor clusters were regulated on multiple levels, including glycoprotein abundance, overall extent of glycosylation at specific glycosites identified, and type of glycosylation at the glycosites. Furthermore, using the integrated data from proteomics and glycoproteomics, we discovered that although the overall extent of glycosylation of each glycoprotein was strongly associated with its expression, glycans that modified glycoproteins had unique expression patterns that were correlated with glycosylation enzymes expressed in the tumors. These findings revealed the potential functions of protein glycosylation in ovarian cancer that have never been studied previously. Furthermore, altered glycoproteins from the extracellular space could provide a foundation for the development of diagnostic and/or therapeutic targets of HGSC.
Figure 1. The Workflow of the Integrated Glycoproteomic Strategy to Analyze HGSCs and Non-tumor Tissues.

(A) Proteins from 83 HGSC tumor tissues, 23 non-tumor tissues, pooled reference sample, and technical replicates of quality control sample were digested by trypsin to peptides, which were labeled by TMT and analyzed by global proteomic analysis (GLOBAL), as well as glycosite-containing peptides (SPEG), and intact glycopeptides (IGPs) analysis using LC-MS/MS.
(B) The clinical phenotypes and data profiling of proteomic (GLOBAL) and glycoproteomic (SPEG and IGP) data of 83 tumor and 23 non-tumor tissues.
RESULTS
The Landscape of Proteomic and Glycoproteomic Data
Proteins from tumors, non-tumors, and a quality control (QC) sample (separated into nine aliquots) were extracted, digested by trypsin, and labeled with tandem mass tags (TMTs) (Figure 1A). CPTAC prospectively collected 83 treatment-naive HGSC tumors and 23 relevant non-tumor tissues from normal fallopian tubes (FTs), including 13 paired FTs from the 83 HGSC patients. The median tumor cellularity of tumor samples is 75% (Table S1). The associated clinical data and metadata are provided in Table S1 and summarized in Figure 1B. The TMT-labeled peptides were divided into three aliquots: one aliquot for global proteomics (GLOBAL dataset) using liquid chromatography-tandem mass spectrometry (LC-MS/MS), one aliquot for N-linked glycosites identified by SPEG method (SPEG dataset), and one aliquot for enrichment of IGPs (IGP dataset) (Figure 1A). The global or non-modified proteomic measurements used TMT labeling in conjunction with offline basic reverse-phase liquid chromatography (bRPLC) fractionation and online LC-MS/MS to provide a broad coverage for peptide identification and quantification (STAR Methods; Figure 1A). The normalized relative abundance measurements (Figure S1A) were used to assess the analytical performance of each protein in all of the samples. We determined the reproducibility of the proteomic analysis using sample-sample correlation of the nine QC samples based on the absolute intensity measurements and the coefficient of variation (CV). As shown in Figure S1B, the median correlation of the quantified proteins in nine QC analyses was 0.90. The median CV of the quantified proteins in the nine QC analyses was 14% (Figure S1C). A total of 8,144 protein groups were identified with high confidence (protein-level false discovery rate [FDR] < 1%) in all of the tumor and non-tumor samples from the GLOBAL proteomic experiment as shown in Figure 1B, while there were 5,916 proteins identified crossing all the samples. The raw absolute intensity abundances of each protein in each tumor and non-tumor sample are given in Table S2.
The N-linked glycoproteomes of the two remaining aliquots of TMT-labeled peptides were analyzed for glycosites after releasing N-linked glycans using PNGase F (using SPEG methods; Zhang et al., 2003) and the enriched IGPs with associated glycans on specific glycosites (Sun et al., 2016; Zhang et al., 2003). Of the 8,144 protein groups identified from global proteomics, 1,690 N-linked glycosite-containing peptides and 3,202 intact N-linked glycopeptides were identified in the SPEG and IGP experiments, respectively, in which 5,916 protein groups, 490 glycosite-containing peptides, and 365 IGPs were identified from all samples (Tables S3 and S4; Figure 1B). Similar to the quality assessment for global proteomic data, we evaluated the reproducibility of the technical replicates from nine QC analyses in SPEG and IGP. The normalized data of all the samples were shown in Figures S1D and S1G. The median correlation of nine QC samples in the SPEG dataset was 0.88 (Figure S1E), while the median correlation value of the IGP QC samples was approximately 0.74 (Figure S1H). The median CV values were 22% for glycosites (Figure S1F) and 15% for IGP (Figure S1I).
According to the monosaccharide composition of N-linked glycans associated on the identified IGPs, three glycan types were defined and investigated in this study: oligomannose/high mannose (HM), sialylated glycans (Sia), and fucosylated glycans (Fuc). The HM glycans represent glycans containing two N-acetylhexosamine (N) and hexose (H) without additional N, fucose (F), or sialic acid (S). The Sia glycans represent any glycans containing S. The Fuc glycans represent any identified glycans containing F.
Proteomic and Glycoproteomic Tumor Sample Clusters
To investigate the cancer heterogeneity of HGSC, we used the Z score transformed log2 ratio expression of the top 50% most variable proteins, N-linked glycosite-containing peptides, and intact N-linked glycopeptides for GLOBAL, SPEG, and IGP tumor sample clustering analysis, respectively. The consensus clustering result illustrated that three tumor clusters could be distinguished (see Figure 2A; Table S5). The three clusters were conservative for GLOBAL, SPEG, and IGP clusters, especially for cluster 3 (Figure 2A). Using the correlation to compare the three clustering results from IGP with GLOBAL and SPEG showed that IGP cluster 3 was the most conserved (Figures 2B and 2C).
Figure 2. The Proteomic and Glycoproteomic Investigation in Tumor Clusters.

(A) Hierarchical clustering of tumor samples based on their Z score transformed abundance of IGPs from the IGP dataset. The clinical phenotypes of all 83 tumor samples, including tumor cellularity, tumor grade, tumor stage, participant race, participant age, anatomic site, origin site, and the labeled tumor clusters classified from GLOBAL, SPEG, and IGP datasets, were shown in the top rows of the clustered heatmap. The left columns showed the overrepresented pathways in the three IGP groups (IGs) and the associated glycan types on the IGPs. HM, high-mannose glycans; Fuc, fucosylated glycans; Sia, sialylated glycans.
(B) The pairwise correlation values between the three tumor clusters based on GLOBAL and IGP datasets, respectively.
(C) The pairwise correlation values between the three tumor clusters based on SPEG and IGP datasets, respectively.
(D) The pairwise correlation values between the three clinical phenotypes (tumor cellularity, anatomic site, and origin site) and the three tumor clusters in the IGP dataset.
(E) The abundance comparison of three IGP groups (IGs) grouped by three tumor clusters in the IGP dataset.
Fuc, fucose; HM, high mannose; Sia, Sialic acid. See also Table S5 and Figure S2.
To investigate whether clinical phenotypes were associated with tumor clusters, we calculated the correlation of IGP clusters to clinical phenotypes, and the result showed that the IGP cluster 3 reversely correlated with tumor cellularity and correlated with anatomic site of omentum (−0.45 and 0.45 for IGP cluster 3, respectively), but did not significantly correlate with other clinical phenotypes, such as tumor origin site of FT (Figure 2D). Most of the samples in cluster 3 were from omentum and have relatively lower tumor cellularity (Figure 2A).
The IGP clustering showed three IGP groups (IGs; Figure 2A). The Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway (Kanehisa, 2019; Kanehisa and Goto, 2000; Kanehisa et al., 2019) analysis using DAVID 6.8 (Huang et al., 2009a, 2009b) indicated that different pathways were significantly enriched in each IG. Lysosome was enriched in the IG1; the phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway, focal adhesion, and extracellular matrix (ECM)-receptor interaction were enriched in IG2; and complement and coagulation cascades were enriched in IG3 (Table S5).
The result also showed the three IG-associated glycans, HM glycans in IG1, HM and Fuc glycans in IG2, and Fuc and Sia glycans in IG3 (Figure 2A). The relationship of the tumor clusters and IGs was revealed using glycans determined by IPGs from each tumor cluster. As shown in Figure 2E, the relative abundance of IG2 was observed to be decreased in tumor cluster IGP2 but increased in cluster IGP3 (Figure 2E). IG3 was decreased in tumor cluster IGP1 but increased in cluster IGP2. There is no significant difference for IG1 levels among the three tumor clusters.
To determine the relationship of IGP tumor clusters with tumor subtypes, the identified 44 GLOBAL protein expression out of 100 subtype signature genes derived by Verhaak et al. (2013) were used to associate with the IGP tumor clusters and showed that the tumor clustering result from the IGP dataset was relevant to the four historical subtypes: differentiated, immunoreactive, mesenchymal, and proliferative (Figure S2A). The signature proteins of immunoreactive subtype were elevated in IGP cluster 1, while the signature proteins of mesenchymal were decreased in IGP2 and elevated in IGP3 (Figure S2B). According to the signature comparison, this observation indicated that IGP cluster 1 is relevant to immunoreactive subtype and IGP cluster 3 is relevant to mesenchymal subtype. We also applied ESTIMATE (Yoshihara et al., 2013) on the protein expression of 5,916 proteins identified crossing all tumor samples in the GLOBAL dataset to estimate the stromal cell and immune cell influence on the clustering result (Figures S2C–S2E). The IGP1 cluster seemed not to be influenced by the tumor purity or stromal score. IGP2 had relatively higher tumor purity and lower stromal and immune scores. IGP3 had lower tumor purity and higher stromal and immune scores.
Proteomic and Glycoproteomic Analyses of HGSC Tumor and Non-tumor Tissues
In the previous retrospective study (Zhang et al., 2016), only tumor samples were considered. Here, we included relevant “non-tumor” samples to investigate HGSC in a more comprehensive approach. The “non-tumor” samples were from normal FT (STAR Methods), which is an adjacent critical organ considered as the start point of genetic alterations in HGSC development (Labidi-Galy et al., 2017; Ducie et al., 2017). The comparison between the tumor and non-tumor tissues could lead to the discovery of specific protein changes for HGSC. The relative abundance of each protein, glycosite-containing peptide, or IGP was determined by the log2 ratio of each protein level to the abundance of the reference sample from each TMT-10 plex (Tables S2, S3, and S4). The reference sample was pooled from all samples and serves as a common denominator for normalization of each sample for quantification in several multiplexed proteomic experiments.
In the IGP dataset, the principal-component analysis (PCA) of log2 ratio of tumors and non-tumors illustrated the formation of distinct clusters of the tumors and non-tumors (Figure 3A). All 83 tumors were assembled and basically differentiated from the non-tumor samples. To determine the differential protein expression in the tumor and non-tumor samples, we applied a t test to the 365 intact N-linked glycopeptides expressed in all of the samples (Figure 3B). Filtering by permutation corrected FDR = 0.01 using Perseus (Tyanova and Cox, 2018; Tyanova et al., 2016), we identified 142 differentially expressed IGPs. Among them, 48 IGPs were significantly upregulated in tumors compared with non-tumor samples, while 94 IGPs were significantly downregulated (Figure 3B; Table S6).
Figure 3. Proteomic and Glycoproteomic Analyses of 83 Ovarian Tumors and 23 Non-tumors Revealed Alterations of Proteins and Glycoproteins in Ovarian Tumors.

(A) Principal-component analysis (PCA) based on the abundance of IGPs from the IGP dataset to reveal the difference between 83 tumor and 23 non-tumor samples.
(B) Volcano plot of IGPs of 83 tumor and 23 non-tumor samples to reveal the significantly upregulated and downregulated IGPs.
(C) Receiver operating characteristic (ROC) curves of selected IGPs: HYOU1_931_N2H8 (AEPPLNASASDQGEK), FKBP10 _70_N2H8 (YHYNGTFEDGK), PSAP_80_ N2H3F1S0G0 (DNATEEEILVYLEK), and PPT1_212_N2H7 (GINESYK).The format is GeneName_Glycosite_GlycanComposition (PeptideSequence). In the glycan composition, N = HexNAc and H = Hex.
(D) Overrepresentation analysis (ORA) of significantly upregulated and downregulated IGPs using DAVID 6.8 referring to the KEGG pathway database.
(E) The relative abundances of IGPs in tumor and non-tumor samples.
(F) The enriched pathways from the gene sets obtained from the identified IGPs under three different glycosylation types (HM, Fuc, and Sia).
Two-sample t tests were also applied to the proteins (5,916 proteins) expressed in all of the samples, as well as glycosite-containing peptides (490 peptides), to determine the differentially expressed proteins and N-linked glycosite-containing peptides in the tumor and non-tumor samples. Similar to the IGP result, the PCA of log2 relative abundance of glycosite-containing peptides and proteins illustrated the formation of distinct clusters of the tumors and non-tumors (Figures S3A and S3B). Filtering by permutation corrected FDR = 0.01 using Perseus (Tyanova and Cox, 2018; Tyanova et al., 2016), we identified 1,232 proteins and 173 glycosite-containing peptides differentially expressed in tumors compared with non-tumors (Table S6). Among them, 645 proteins and 59 glycosite-containing peptides were significantly upregulated, while 587 proteins and 114 glycosite-containing peptides were significantly downregulated (Figures S3C and S3D).
The significantly altered proteins or glycoproteins between tumor and non-tumor samples could be potentially useful for the diagnosis of ovarian cancer. We used CombiROC (Mazzara et al., 2017) to select signatures of IGPs to distinguish tumor and non-tumor samples (STAR Methods). As shown in Figure 3C, receiver operating characteristic (ROC) curves were presented for four selected IGPs from HYOU1, FKBP10, PSAP, and PPT1 to classify tumor and non-tumor tissues.
The KEGG pathway using DAVID 6.8 (Huang et al., 2009a, 2009b) was applied on the significantly positive and negative regulated proteins, glycosite-containing peptides, and IGPs in tumors based on their corresponding genes. The KEGG pathway analysis on IGPs revealed that lysosome was the overrepresented pathway (Benjamini-adjusted p < 0.05) for the significantly upregulated IGPs in the tumor samples, while complement and coagulation cascades pathways, ECM-receptor interaction, PI3K-Akt signaling pathway, focal adhesion, and protein digestion and absorption were the top overrepresented (Benjamini-adjusted p < 0.05) pathways among the significantly downregulated IGPs in the tumor samples (Table S6; Figure 3D). Some pathways such as focal adhesion, ECM-receptor interaction, complement and coagulation cascades, and PI3K-Akt signaling pathway showed consistent overrepresented results from all GLOBAL, SPEG, and IGP datasets (Figures S3E and S3F).
Comparing the relative abundances of IGPs between tumor and non-tumor samples, we observed that the IGPs with HM type of glycan were in high abundance in tumors, while Fucand Sia-containing IGPs were in low abundance in tumors (Figure 3E). The pathways involved in the HM-, Fuc-, or Sia-containing IGPs showed that the lysosome pathway was the top enriched pathway in HM-containing IGPs. The ECM-receptor interaction was enriched in Fuc-IGPs, and component and coagulation cascades were enriched in Sia-IGPs (Figure 3F).
The clustering heatmap of all the abundances of IGPs and their corresponding SPEG peptides and global proteins illustrated that most of the upregulated IGPs were associated with HM-containing IGPs, in which the lysosome pathway was overrepresented (Figure S3G). Several lysosomal proteins were identified as upregulated in both protein and IGP levels in tumor samples, including proteases (CTSC, CTSD, CTSL, and LGMN), glycosidases (GAA and HEXA), sulfatase (GNS), phosphatase (ACP2), ceramidase (ASAH1), and other lysosomal enzymes and associated activators (PPT1 and PSAP).
Integrated Glycoproteomic Analyses Revealed Changes in Glycosylation Sites and Glycans
Comparing differential expressed global proteins (GLOBAL), glycosite-containing peptides, and IGPs in tumor and non-tumor samples, we observed overlapping protein and glycoprotein changes; however, the glycosite-containing peptides and IGPs showed distinct levels of regulation in tumors (Figures 4A and 4B). The t test-based comparative analysis was performed on the glycoproteins quantified by GLOBAL proteomic dataset and SPEG dataset to investigate whether the tumor-specific changes in glycosites were also present in global protein abundance of the glycoproteins (Figure 4A). The correlation of the two t tests’ statistical significance scores from proteins in GLOBAL and glycosite-containing peptides in SPEG was 0.84 (R2 = 0.52), indicating that glycoproteins could be regulated by glycosylation occupancy, as well as global protein expression. Interestingly, although most of the differential abundance changes of glycosite-containing peptides were still positively correlated with the corresponding global protein expression, the abundance changes of glycosites of certain glycoproteins could exhibit distinguishable expression patterns from their global levels (Figure 4A). MUC16 (also named as CA125), for example, was previously reported as a tumor biomarker of ovarian cancer (Bast et al., 1983, 2005). MUC16 showed no significant abundance change in the global protein expression level (p = 0.70; Figure 4C). However, this glycoprotein showed significantly differential levels in two glycosites, MUC16_12272 (p < 0.05; Figure 4D) and MUC16_12586 (p < 0.05; Figure 4E), between the tumors and non-tumors. These results suggested that simple measurement of protein abundance and subsequent protein-based clustering might be insufficient in comprehensively understanding tumor biology, and that clinical assays to measure CA125 glycosylation levels in multiple glycosites could be more informative than measuring only CA125 protein abundance in diagnosis and monitoring of ovarian cancer.
Figure 4. Proteomic and Glycoproteomic Analyses of 83 Ovarian Tumors and 23 Non-tumors Reveal Alterations of Proteins and Glycoproteins in Ovarian Tumors.

(A) A comparative analysis of the differential abundance changes of glycosite-containing peptides and their corresponding proteins in tumors comparing with non-tumor samples from SPEG glycoproteomic data and GLOBAL proteomic data, respectively.
(B) A comparative analysis of the differential abundance changes of IGPs and glycosite-containing peptides in tumors comparing with non-tumors from intact glycoproteomic data and SPEG glycoproteomic data, respectively. The attached glycans were classified and highlighted by three groups (HM, Fuc, and Sia) according to their identified glycan compositions.
(C) The abundance changes of global protein expression of CA125 (MUC16), an ovarian cancer biomarker, in the tumor and non-tumor samples.
(D) The abundance changes of glycosite-containing peptides NTSVGPLYSGCR of protein CA125 (MUC16) in the comparison between tumors and non-tumors. The identifier of each glycosite-containing peptide was presented using the specific format: MUC16(gene name)_12272(start position of the peptide) _NTSVGPLYSGCR(peptide sequence)_1(number of glycosites)_12272(glycosite position(s)).
(E) The abundance changes of glycosite-containing peptides NTSVGLLYSGCR of protein CA125 (MUC16) in the comparison between tumors and non-tumors. (F–H) Micro-heterogeneity of glycosylation expression on the same IGPs of translocon-associated protein subunit beta (SSR2). The identifier of IGPs was presented using the format: SSR2(gene name) IAPASNVSHTVVLRPK(peptide sequence)+N2H8F0S0G0(glycan composition), in which N2H8F0S0G0 represents the glycan composition of HexNAc/N:2, Hexose/H:8, Fucose/F:0, Neu5Ac/S:0, and Neu5Gc/G:0.
To further investigate whether the alteration of glycosites was also reflected in the IGP analysis, we used the similar approach of t tests to compare the glycoproteins quantified by the SPEG and IGP datasets (Figure 4B). The comparison of t test scores from SPEG and IGP (correlation = 0.69, R2 = 0.56) and the analysis of associated glycan types on the IGPs indicated the abundance changes of IPGs in tumors comparing with non-tumors were not only regulated by the extent of glycosylation at each glycosite but also influenced by glycans that modify the glycosite. As shown in Figure 4B, the IGPs containing HM glycans were mostly overexpressed in tumors according to their quantitative values in the SPEG and IGP experiments, while the abundance changes of IGPs containing other types of glycans-containing IGPs were various. The heterogeneity of glycosylation on the same glycosite was also observed in the IGP analysis. An example of differential regulation of glycosylation at the same glycosite showed that glycosite-containing peptide, IAPASNVSHTVVLRPLK from the signal sequence receptor (SSR2), was modified by three different glycans, including Man 8 (N2H8), Man 9 (N2H9), and complex glycan (N4H7F1) (Figures 4F–4H). Peptides carrying the HM type of glycan were elevated in tumors (Figures 4F and 4G), while glycopeptides with complex glycan showed no significant difference between the tumor and non-tumor samples (Figure 4H). SSR2 is a glycosylated endoplasmic reticulum (ER) membrane receptor that functions to translocate proteins from the ribosome across the ER membrane (Wiedmann et al., 1987). Because the synthesized glycoproteins are modified by HM in ER, the elevated levels of HM modified SSR2 may represent the elevated levels of newly synthesized SSR in ER and play the translocation function of signal sequences of newly synthesized proteins to ER. SSR2 was reported to play a pro-survival role in human melanoma cells (Garg et al., 2016).
Altered Glycosylation Biosynthesis in HGSC
To investigate the regulation of glycan expression, we correlated the abundance of IGPs from each tumor and non-tumor sample in the IGP dataset with the protein abundance of the glycosylation enzymes that were identified and quantified from the GLOBAL proteomic dataset (Figure 5A). We found that the IGPs with glycosylation of HM glycans were positively correlated with the expression of Glucosidase 2 subunit beta (PRKCSH), but negatively correlated with several other glycosylation enzymes, including the expression of Mannosyl-oligosaccharide 1, 2-alpha-mannosidase IA (MAN1A1). Among all of the identified glycosylation enzymes, only PRKCSH was found to be significantly upregulated in tumors, while most of the other glycosylation enzymes were downregulated in tumors (Figure 5B). The correlations of the expression of IGPs and protein expression of Alpha-(1, 3)-fucosyltransferase 11 (FUT11), PRKCSH, and MAN1A1 in the 83 tumor and 23 non-tumor samples were shown in Figures 5C–5E, respectively. We observed statistical significantly positive correlations of FUT11 with IGPs modified by Fuc glycans (Figure 5C), as well as PRKCSH with IGPs modified by HM glycans (Figure 5D), but a negative correlation of MAN1A1 with IGPs modified by HM (Figure 5E). The quantitative measurement of FUT11, PRKCSH, and MAN1A1 showed no significant differential expression of FUT11, but a significantly increased level of PRKCSH and decreased level of MAN1A1 in the tumor samples comparing with non-tumors (Figures 5F–5H). These results were consistent with the observation that the IGPs modified by HM glycans were increased in the tumor samples (Figure 3E).
Figure 5. Association of IGP Abundance and Protein Levels of Glycosylation Enzymes in 83 Tumors and 23 Non-tumors.

(A) The hierarchal-clustered correlation matrix of IGPs and glycosylation enzymes. The glycan types were highlighted in the top rows.
(B) The bar chart log2 fold change (FC) ratio values of glycosylation enzymes between tumor and non-tumor samples from the GLOBAL dataset.
(C) Correlation between FUT11 and IGPs with/without Fuc glycans (Fuc and non-Fuc).
(D) Correlation between PRKCSH and IGPs with/without HM (HM and non-HM).
(E) Correlation between MAN1A1 and IGPs with/without HM (HM and non-HM).
(F) The abundances of FUT11 in tumors and non-tumors.
(G) The abundances of PRKCSH in tumors and non-tumors.
(H) The abundances of MAN1A1 in tumors and non-tumors.
To determine the potential roles of HM modifications to glycoproteins, the partial glycosylation biosynthetic pathway was analyzed for the synthesis of HM with the functions of key glycosylation enzymes (Figure 6A). The increased expression of PRKCSH and decreased level of MAN1A1 in tumor cells could result in elevated glycoproteins with HM glycosylation, thus preventing further detailed complex carbohydrate synthesis. The function of the HM in cancer is not clear. This increment of HM glycan modifications could be critical for glycoproteins that are synthesized in large quantities for tumor growth. Investigation of the network of glycoproteins that are modified by HM in cancer cells might be helpful to identify the glycoproteins required for fast cell growth; we applied protein-protein interaction analysis of STRING 10.5 (Szklarczyk et al., 2017) on the proteins with HM IGPs upregulated in tumors, and we found that they were involved in a network mainly related to lysosome, collagen metabolic process, and endomembrane system (Figure 6B). Previous studies done by looking at only glycans found that HM glycans were elevated in several cancer types, including breast cancer (de Leoz et al., 2011), cholangiocarcinoma (Talabnin et al., 2018), ovarian cancer (Chen et al., 2017), colorectal cancer (Balog et al., 2012; Sethi et al., 2014), and prostate cancer (PCa) sera (Tabarés et al., 2006). By analyzing the IGPs modified by HM glycans, this study identified the glycoproteins as the potential targets required for cancer growth.
Figure 6. The Synthesis Pathway and Protein-Protein Interaction (PPI) Network of the Elevated IGPs Modified by HM Glycans in Tumor.

(A) The possible mechanism of glycan biosynthesis with the elevated HM glycosylation in ovarian cancer.
(B) The PPI network of significantly upregulated HM IGPs in tumors. The annotations were also marked by different colors on the nodes of the involved genes.
DISCUSSION
The integrated glycoproteomic analysis on the proteins, glycosite-containing peptides, and IGPs illustrated the reliable power of the MS-based proteomic and glycoproteomic methods on the molecular profiling of HGSCs and non-tumor tissues (Figure 1; Figure S1). The statistics and evaluations demonstrated these datasets (GLOBAL, SPEG, and IGP) provide an integrated proteomic and glycoproteomic data resource for HGSC study.
The hierarchical clustering method suggested that ovarian tumors could be separated into three different clusters (Figure 2A). The tumor clustering analysis at the IGP level showed apparent consistency with the clustering results of GLOBAL and SPEG datasets (Figures 2B and 2C), and further revealed that the clusters could be correlated with the clinical phenotypes of tumor cellularity and anatomic site (Figures 2A and 2D). Overall, glycans are differentially presented in three tumor clusters. The tumor cluster IGP1 has the lowest level of IG3, which is dominated by the IGPs modified by complex glycans containing Fuc and/or Sia types of glycans from complement and coagulation cascades pathway. The tumor cluster IGP2 has the lowest level of IG2, which contains the IGPs modified mainly by HM or Fuc type of glycans from the ECM-receptor interaction pathway. Meanwhile, the tumor cluster IGP3 has the highest level of IG2 (Figure 2E). This result suggests that multi-omics data should be considered to guide the classification of ovarian cancers into molecular clusters, which is helpful to understand the relationship between the molecular alteration and the clinical phenotypes.
The integrated glycoproteomic analysis of HGSCs and non-tumor samples demonstrated that there was a distinct expression pattern of proteins and glycoproteins in the tumors and non-tumors (Figures 3A and 3B), which can potentially be used as targets for the diagnosis and/or treatment of HGSCs, especially those glycoproteins or glycopeptides that are preferentially expressed on the cell surface or secreted in extracellular space with the likelihood of releasing into body fluids (Figure 3C). Using DAVID 6.8 on the significantly upregulated and downregulated gene names of identified proteins, glycosite-containing peptides, and IGPs compared with the KEGG pathway database, we observed lysosome was an enriched pathway in upregulated glycopeptides and focal adhesion, PI3-Akt signaling pathway, ECM-receptor interaction, and complement and coagulation cascades in downregulated glycopeptides (Figure 3D). The upregulation of proteins in the ECM-receptor interaction pathway was observed in a clear cell renal cell carcinoma (ccRCC) study (Clark et al., 2019), but the expression of proteins in the ECM-receptor interaction pathway was significantly downregulated in HSGC tumors in protein, glycosite-containing peptides, and IGPs. The significant changed abundance of the relevant proteins and glycosylation could be also regarded as informative indicators of the development of HGSC. Another interesting find is that the significantly upregulated IGPs in the lysosome pathway were dominantly occupied by HM type of glycans (Figure 3E), which was further confirmed by the enriched pathway comparison in tumor and non-tumor tissues shown in Figure 3F. The lysosomes are the recycling centers in cells, where organelles and proteins are degraded during autophagy and micropinocytosis (Towers and Thorburn, 2017), which are also critical components for tumor cell resistance to stress and to survival and growth. On the other hand, the exocytosis of acid hydrolases inside lysosomes could cause the ECM degradation (Kallunki et al., 2013), which has been reported as important in invasion and metastasis of tumor cells (Guan, 2015; Jiang et al., 2015). The inhibition of multiple lysosomal activities is one of the important directions of cancer therapy methods, but rarely considered is the inhibition of glycosylation on lysosomes, which could be a new direction for future investigation.
Although our integrated glycoproteomic analysis of global proteins, glycosite-containing peptides, and IGPs illustrated the formation of distinct clusters of the tumors and non-tumors, and that the trends of changes in proteins, glycosite-containing peptides, and IGPs (T scores) of the tumors compared with the non-tumors were mainly positively correlated, some glycosites were differentially regulated as compared with their global expression levels (Figure 4A). This finding was also observed in the comparative analysis of the IGPs and glycosite-containing peptides from IGP and SPEG datasets (Figure 4B). CA125 (MUC16) was developed for monitoring treatment response of ovarian cancer, distinguishing malignant from benign pelvic masses, assessment of prognosis, prediction of response to drugs, and detection of primary cancer at its early stage (Bast et al., 2005). An elevated serum CA125 level (>35 U/mL) by a monoclonal antibody CA125 assay was found in patients with a variety of cancers, particularly in ovarian cancer (Fedele et al., 2010). However, elevated serum CA125 levels were also found in patients with benign conditions, such as endometriosis, menstruation, and pregnancy, as well as in patients with non-ovarian malignancy. Furthermore, CA125 is not detected in 20% of ovarian tumor tissue sections (Bast et al., 2005; Ooms et al., 2015). The limitation of a lack of sensitivity and specificity of current CA125 testing in clinical practice precipitates the urgency for the development of an alternative testing strategy. Our proteomic and glycoproteomic analysis showed that there was no evidence indicating that CA125 was differentially expressed between the tumor and non-tumor samples according to its global expression measurement (Figure 4C). However, as shown in Figures 4D and 4E, the glycosite-containing peptides of CA125 detected in all ovarian tumors demonstrated differential expression between the tumor and non-tumor samples. Our unique findings indicated that the analysis of glycosites of CA125 protein could be used for the detection of ovarian tumors. Although the protein expression of CA125 was not different in the tumors and non-tumors, the degree of protein glycosylation at specific glycosites could be influenced by the pathological status of the tissues. This observation suggests that glycosylation changes may occur independently of protein expression. Thus, both the measurement of protein expression and the glycosylation are critical to characterize tumor-specific changes. Several studies have employed MS to independently verify antibody-based CA125 detection (Swiatly et al., 2018; Weiland et al., 2012). However, currently published mass spectrometric data based on global proteomic experiments may be insufficient to satisfy complete profiling of CA125 and identify alterations on glycosylation level. Our finding highlights the need for high-quality mass spectrometric data to enable comprehensive CA125 analysis at each glycosite. Ultimately, the improved knowledge about the nature of CA125 may lead to the development of assays for quantification of CA125 glycosites specific to ovarian tumors, which can potentially increase current specificities of CA125 for ovarian cancer diagnosis and monitoring.
Differential protein expression was observed not only on the global proteins or glycosites but also on the specific N-linked glycans that modified the glycoproteins in the tumors. As shown in Figure 4B, elevated levels of HM IGPs could be observed between tumors and non-tumors. The elevated HM type of glycans was previously observed in breast cancer progression (de Leoz et al., 2011) and also observed by glycomics analysis in epithelial ovarian cancer progression (Chen et al., 2017). HM glycans are not commonly detected in normal serum or presented as cell surface proteins because of extensive glycan processing within the Golgi, which yields highly processed complex and hybrid glycans on the mature proteins. The presence of increased levels of HM glycans in cancer represents an aberrant biosynthetic pathway of protein glycosylation in cancer cells. Indeed, HM-reactive antibodies have been isolated from patients with late-stage PCa (Wang et al., 2013), suggesting that glycan biosynthesis is dysregulated at the HM stage within the glycan processing pathway.
The expression levels of glycosylation-related enzymes are known to be disrupted during tumorigenesis (Meany and Chan, 2011; Stowell et al., 2015). Indeed, aberrant glycosylation in cancer progression can be used to distinguish cancerous cells from healthy cells and is one of the few distinctive details that can be used to distinguish between self-derived antigens (Gilgunn et al., 2013; Padler-Karavani, 2014). As a non-template-mediated PTM, glycans are not regulated by the genetic code; however, the pattern of glycosylation is controlled by the expression levels of glycosyltransferase and exo-/endo-glycosidase enzymes. In addition, glycosylation-related enzymes have been demonstrated to be pleiotropic drivers of the epithelial-mesenchymal transition (EMT) process, particularly the influential and oncogenic fucosylation modification (Chen et al., 2013; Wang et al., 2014). Given the importance of aberrant glycosylation in cancer progression, a number of glycogenes have been analyzed to discover their cancer-associated functions. The ability of glycosylation-related enzymes to alter cancer-associated processes like migration demonstrates the multifaceted role of glycosylation enzymes (Wang et al., 2014). The observation that HM glycan abundance is increased in ovarian tumors compared with non-tumors suggests that there is dysregulation of the enzymes responsible for trimming mannose during glycan biosynthesis. The pathological dysregulation of oligomannose-trimming enzymes could be related to the enhancement of tumorigenesis. Most of the glycosyltransferases were identified at lower expression levels in tumor tissues compared with non-tumors, except FUT11 and PRKCSH (Figure 5A). PRKCSH is a critical component of the glycan biosynthesis pathway and a positive regulator of Wnt/beta-catenin signaling and autophagy and apoptosis (Khaodee et al., 2017; Rauscher et al., 2018). PRKCSH was found to play an important role in tumorigenesis by selectively boosting the IRE1 signaling pathway (Shin et al., 2019). The upregulation of PRKCSH and the downregulation of most of the downstream glycosylation enzymes, such as MAN1A1 in tumors, actively promote the expression of the N-linked glycoproteins carrying HM glycans (Figure 6A). The HM type of glycans might occupy more glycosylation sites associated with the peptides in the tumors. As shown in Figures 3E and 4B, most of the HM type of glycan were upregulated in the tumor tissues, while the downstream hybrid or complex glycans were downregulated in the tumors. According to the observations above, we hypothesize that the glycosylation biosynthesis pathway may be partially disabled in tumor tissues because of the downregulation of a series of glycosylation enzymes starting after the truncation of glucose in the ER (Figure 6A). Many protein glycosylation events could be terminated after exiting the ER with a premature glycosylation without further decoration on glycan structures in Golgi. This could be an energy-saving mechanism in tumor to more efficiently manufacture glycoproteins or adapt to environmental stress. Moreover, the overactivation of the lysosome pathway might release overexpressed HM glycosylated acid hydrolases via exocytosis to cause ECM degradation, including changes of stiffness, elasticity, and remodeling of ECM, and consequently contribute to tissue fibrosis and tumor metastasis. Due to the HM type of glycan’s protection, these acid hydrolases could be more difficult to degrade and result in more tissue damage.
In this study, the integrated multi-omics analysis, including proteomics and glycoproteomics analysis of HGSC, demonstrated the linkage of glycosylation to ovarian cancer. By applying the differential expression of multi-omics data between tumors and non-tumors, we identified several potential tumor-specific proteins, glycoproteins, and glycans. Further investigation showed that the differential glycoprotein expression in tumors could be shown as differential extent of glycosylation at glycosites, as well as types of glycan on the glycosites. The glycosylation biosynthetic pathways of tumors differ from those of non-tumors. Due to the upregulation of PRKCSH and the downregulation of MAN1A1, the N-linked glycoproteins could carry more HM glycans but fewer hybrid or less complex glycans in tumors as compared with non-tumors. This could be a common mechanism regulated by PRKCSH in tumors for efficient glycoprotein production, resistance to environmental stress, and overactivation of lysosomes. Finally, the comprehensive proteomic and glycoproteomic measurements for the HGSC tumor samples provide a valuable public resource. The glycoproteomic data linking glycoproteins with their extent of glycosylation, glycan modifications, and the glycosylation enzymes will improve our understanding of the molecular basis of ovarian cancer.
STAR+METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hui Zhang (huizhang@jhu.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets generated during this study are available at CPTAC data portal and publicly available (https://cptac-data-portal.georgetown.edu/study-summary/S038). The codes supporting the current study are publicly available and listed in the Key Resources Table.
KEY RESOURCES TABLE
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Specimen acquisition
The ovarian tumor and non-tumor tissue samples used in this study were acquired from the prospective project of Clinical Proteomic Tumor Analysis Consortium (CPTAC). Biospecimens were collected from 83 patients who were recently diagnosed with high-grade serous ovarian adenocarcinoma, underwent surgical resection and did not receive any prior treatment for their disease, including chemotherapy or radiotherapy. For each patient, up to 3 individual fimbria from each normal FT were collected as non-tumor tissue control. Twenty-three relevant non-tumor tissues from FTs included 13 paired non-tumor samples from the 83 patients. Ten patients provided FTs only as the matched tumor tissues from these 10 cases failed molecular qualification (McDermott et al., 2020). There were 83 tumor and 23 non-tumor tissue samples were applied in this study. All cases were required to be of serous histology but were collected regardless of surgical stage or histologic grade. Cases were staged according to the 1988 International Federation of Gynecology and Obstetrics (FIGO) staging system.
Each specimen endured cold ischemia for ≤ 30 minutes prior to freezing in liquid nitrogen. The specimens were used for the global proteomics (Global) and glycoproteomics studies including solid phase extraction of N-linked glycosite-containing peptide (SPEG) and intact N-linked glycopeptide (IGP) analyses. Each specimen was embedded in optimal cutting temperature (OCT) medium, and histologic sections were obtained from the top and bottom portions for pathology review. Each case was reviewed by a board-certified pathologist to confirm the assigned pathology. For inclusion in this study, the top and bottom sections were required to contain 60% tumor cell nuclei with < 20% necrosis. The specimens were serially curled at the Biospecimen Core Resource, and the curled sections were then transferred into pre-cooled cryovials (Corning).
Specimens were shipped overnight from the Tissue Source Sites to the Proteome Characterization Center located at Johns Hopkins University (JHU) in Baltimore, MD using a cryoport that maintained an average temperature of < −150°C. All procedures were carried out on dry ice to maintain the tissue in a frozen state and processed for mass spectrometric (MS) analysis at JHU.
Clinical data annotation
Clinical data were obtained from Tissue Source Sites and aggregated by the Biospecimen Core Resource. Data forms were stored as Microsoft Excel files (.xlsx). Clinical data can be accessed and downloaded from the CPTAC Data Portal (https://cptac-data-portal.georgetown.edu/cptac/documents/CPTAC_S038_ovarian_cancer_clinical_data_r1.xlsx). Demographics, histopathologic information, and treatment details were collected. Supplemental clinical data were collected directly from the original file, and the updated clinical data are provided in Table S1. As shown in Table S1, the characteristics of the CPTAC Prospective specimens reflect the general population of women with advanced ovarian cancer. The average age at diagnosis was 59.94 years, all cases were of serous histology. Most cases were at late stage, with 76% (63 of 83) of cases at FIGO stage III and 18% (15 of 83) at FIGO stage IV. The ‘SPL’ column was used to indicate the internal sample index for simplifying the sample name.
METHOD DETAILS
Protein extraction and tryptic digestion
The experimental design is shown in Figure 1A. Approximately 30–200 mg of each of the sectioned ovarian tumor tissues or non-tumor tissues were homogenized separately in lysis buffer (8 M urea, 1.0 M NH4HCO3, pH 8.0) by sonication (Branson Sonifier 250, 15 s cycles with 1 min cool down, 4 times, 20% output). Lysates were precleared by centrifugation at 16,500 g for 15 min at 4°C and protein concentrations were determined by BCA assay (Pierce). Proteins (2mg/mL) were reduced with 10 mM tris (2-carboxyethyl) phosphine (TCEP) for 1 h at 37°C, and subsequently alkylated with 15mM iodoacetamide for 1 h at room temperature (RT) in the dark. Samples were diluted 1:5 with deionized water and digested with sequencing grade modified trypsin (Promega) at a 1:50 enzyme-to-substrate ratio. After overnight digestion at 37°C, another aliquot of the same amount of trypsin was added to the samples and further incubated at 37°C overnight. The digested samples were then acidified with 50% trifluoroacetic acid (TFA, Sigma) to ~pH 2. Tryptic peptides were desalted on reversed phase C18 SPE columns (Waters) and dried using a Speed-Vac (Thermo Scientific).
TMT labeling of peptides
Desalted peptides from each sample were labeled with 10-plex TMT (Tandem Mass Tag) reagents (Thermo Fisher Scientific). Peptides (300 μg) from each of the prospective ovarian samples were dissolved in 55 μL of 0.5 M triethylammonium bicarbonate (TEAB), pH 8.5 solution, and mixed with 3 units of TMT reagent that was freshly dissolved in 130 μL of ethanol. After 1h incubation at RT, the reaction was quenched by acidification with 50% TFA to pH < 3. A reference sample was created by pooling an aliquot of peptides from each individual tumor and non-tumor sample, and TMT Channel 126 was used to label the pooled reference sample throughout the proteomic analysis. A single HGSOC tumor sample previously used as an internal quality control (QC) for the analysis of the prospectively-collected tumors (Zhang et al., 2016) was prepared and repeatedly analyzed in the same manner in the current study. A total of 83 prospectively-collected tumors and 23 non-tumor samples together with 9 QC aliquots were co-randomized to 13 TMT sets. The sample-to-TMT channel mapping is shown in the “Experiment Design” sheet of Table S1. After labeling, in each TMT set, peptides labeled by different TMT reagents were mixed and desalted on C18 SPE columns. After desalting, the peptides from each sample (3 mg) were divided to 4 groups: 200 μg for proteomic analysis, 400 μg for SPEG analysis, 1.1 mg for intact glycopeptide analysis, and 1.3 mg for additional analysis, if needed.
Peptide fractionation by basic reversed-phase liquid chromatography (bRPLC)
Extensive fractionation was performed by bRPLC to reduce sample complexity and thus reduce the likelihood of peptides being co-isolated and co-fragmented. This approach has been well-documented to reduce isobaric (i.e., iTRAQ, TMT) reporter ion ratio distortion effects (Bantscheff et al., 2008) and it was applied in this study.
The samples were fractionated using bRPLC. Approximately 200 μg of 10-plex TMT labeled sample was first purified on strong cation exchange columns (Glygen), and then separated on a reversed phase Zorbax extend-C-18 column (4.6 × 100 mm column containing 1.8-um particles; Agilent) using an Agilent 1200 Infinity HPLC System. The solvent A consisted of 10 mM ammonium formate, pH 10.0. Solvent B consisted of 10 mM ammonium formate, pH 10, 90% acetonitrile as mobile phase. The separation gradient was set as follows: 2% B for 10 min, from 2 to 15% B for 5 min, from 15 to 45% B for 85 min, from 45 to 95% B for 5 min, and 95% B for 15 min. A total of 96 fractions were collected into a 96 well plate in a time-based mode. These fractions were then concatenated into 24 fractions by combining 4 fractions that are 24 fractions apart (i.e., combining fractions #1, #25, #49, and #73; #2, #26, #50, and #74; and so on). Each concatenated fraction was dried down in a Speed-Vac and re-suspended in 2% acetonitrile, 0.1% formic acid for LC-MS/MS analysis.
Enrichment of intact glycopeptides by Retain AX cartridges (RAX)
A total of 1.1 mg TMT labeled peptides from each set were adjusted to 95% ACN (v/v), 1% TFA (v/v) for intact glycopeptide enrichment using Retain AX Cartridges (RAX) (particle size 30–50 μm, 30 mg sorbent per cartridge, Thermo Fisher Scientific) (Yang et al., 2017). The RAX columns were equilibrated three times with 1 mL of ACN, three times with 100 mM triethylammonium acetate, three times with water, and finally three times with 95% ACN (v/v), 1% TFA (v/v). The samples were loaded on to RAX columns and washed four times with 1 mL of 95% ACN, 1% TFA. Finally, bound intact glycopeptides were eluted in 400 μL of 50% ACN (v/v), 0.1% TFA (v/v). The intact glycopeptides were then dried in a Speed-Vac and stored in −80°C prior to LC–MS/MS analysis.
Solid phase extraction of N-linked glycosite-containing peptides (SPEG)
N-linked glycopeptides were captured by solid phase extraction of N-linked glycosite-containing peptides (SPEG) as described previously (Zhang et al., 2003). Briefly, 400 μg TMT-labeled peptides (in C18 elution buffer: 60% ACN, 0.1%TFA) of each TMT set were oxidized by 10 mM of sodium periodate at room temperature for 1 h in the dark. After oxidation, samples were desalted on C18 SPE columns to remove sodium periodate. Then the sample was conjugated to 40μl hydrazide resin (Bio-Rad) in the presence of 1% Aniline at room temperature overnight by gentle shaking. Non-glycopeptides were removed by centrifugation at 6000 rpm for 1 min. Then the resin was intensively washed sequentially with 1) 50% ACN/50% deionized water (v/v), 2) 1.5M NaCl, 3) deionized water and 4) 25mM NH4HCO3, three times for each wash step, by vortexing and centrifugation. After the last wash, the hydrazide resin was reconstituted in 200μL 25mM NH4HCO3. The N-linked glycopeptides were released from the resin by incubation with 2μL PNGase F (New England Biolabs Inc) at 37°C overnight with gentle shaking. The released de-glycopeptides were dried and stored in −80°C prior to LC-MS/MS analysis.
LC-MS/MS for global proteomic analysis
The global proteome fractions were separated on a Dionex Ultimate 3000 RSLC nano system (Thermo Scientific) with a 75 μm × 50 cm PepMap RSLC C18 Easy-Spray column (Thermo Scientific) protected by a 100 μm × 2 cm Acclaim PepMap 100 guard column (Thermo Scientific). The mobile phase flow rate was 450 nL/min and consisted of 0.1% formic acid in water (A) and 0.1% formic acid/95% acetonitrile (B). The sample injected (6 μL) was trapped using 100% mobile phase A for 13 min at a flow rate of 5 μL/min before being placed in-line with the analytical column and subjected to a gradient profile which was set as follows: 2%–4% B for 10 min, 4%–24% B for 80 min, 24%–33% B for 22 min, 33%–95% B for 3 min, 95% B for 10 min at a flow rate of 320 nL/min. MS analysis was performed using a Q-Exactive mass spectrometer (Thermo Scientific). The Q-Exactive mass spectrometer parameters were as follows: electrospray voltage was 2.2 kV; following a 20 min delay from the end of sample trapping, Orbitrap precursor spectra (AGC 3×106) were collected from 400–1800 m/z for 110 minutes at a resolution of 70K along with the top 12 data dependent Orbitrap HCD MS/MS spectra at a resolution of 35K (AGC 2×105) and max ion time of 120 msec; ions selected for MS/MS were isolated at a width of 1.4 m/z and fragmented using a normalized collision energy of 31%; peptide match was set to ‘Preferred’; exclude isotopes was set to ‘on’; and charge state screening was enabled to reject unassigned 1+, and > 8+ ions with a dynamic exclusion time of 30 s to discriminate against previously analyzed ions.
LC-MS/MS for glycoproteomic analysis
The de-glycosylated glycosite-containing peptides isolated by SPEG were separated on a Dionex Ultimate 3000 RSLC nano system (Thermo Scientific) with a 75 um × 50 cm Acclaim PepMap RSLC C18 Easy-Spray column (Thermo Scientific) protected by a 100um × 2 cm Acclaim PepMap 100 guard column (Thermo Scientific). The mobile phase flow rate in the analytical column was 320 nL/min and consisted of 0.1% formic acid in water (A) and 0.1% formic acid/95% acetonitrile (B). The sample injected (6 μL) was trapped using 100% mobile phase A for 13 min at a flow rate of 5 μL/min before being placed in-line with the analytical column and subjected to the gradient profile which was set as follows: 2%–7% B for 10 min, 7%–27% B for 80 min, 27%–34% B for 22 min, 34%–95% B for 3 min, 95% B for 10 min. MS analysis was performed using a Q-Exactive mass spectrometer (Thermo Scientific). The Q-Exactive mass spectrometer parameters were as follows: electrospray voltage was 2.2 kV; following a 20 min delay from the end of sample trapping, Orbitrap precursor spectra (AGC 3×106) were collected from 400–1800 m/z for 110 minutes at a resolution of 70K along with the top 12 data dependent Orbitrap HCD MS/MS spectra at a resolution of 35K (AGC 2×105) and max ion time of 120 msec; ions selected for MS/MS were isolated at a width of 1.4 m/z and fragmented using a normalized collision energy of 31%; peptide match was set to ‘Preferred’; exclude isotopes was set to ‘on’; and charge state screening was enabled to reject unassigned 1+, and > 8+ ions with a dynamic exclusion time of 30 s to discriminate against previously analyzed ions. Each sample was analyzed by LC-MS/MS in triplicate.
The intact glycopeptides were analyzed on the Orbitrap Fusion Lumos system (Thermo Scientific). The intact glycopeptides were separated using an Easy nLC 1200 UPLC system (Thermo Scientific) on an in-house packed 20 cm × 75 mm diameter C18 column (1.9 mm Reprosil-Pur C18-AQ beads, Dr. Maisch GmbH); Picofrit 10 mm opening (New Objective). The column was heated to 50°C using a column heater (Phoenix-ST). The flow rate was 200 nL/min with 0.1% formic acid and 2% acetonitrile in water (A) and 0.1% formic acid/90% acetonitrile (B). Injected peptides were subjected to the following gradient: 2%–6% B for 1 min, 6%–30% B for 84 min, 30%–60% B for 9 min, 60%–90% B for 1 min, 90% B for 5 min and then back to 50% B for 10 min. The Fusion Lumos mass spectrometer parameters were as follows: electrospray voltage was 1.8 kV; the ion transfer tube temperature was at 250°C; Orbitrap precursor spectra (AGC 4×105) were collected from 350–1800 m/z for 110 min at a resolution of 60K along with data dependent Orbitrap HCD MS/MS spectra (centroided) at a resolution of 50K (AGC 2×105) and max ion time of 105 msec for a total duty cycle of 2 s; masses selected for MS/MS were isolated (quadrupole) at a width of 0.7 m/z and fragmented using a high energy collision dissociation of 38%; peptide charge state screening was enabled to reject unassigned 1+, 7+, 8+, and > 8+ ions with a dynamic exclusion time of 45 s to discriminate against previously analyzed ions between ± 10 ppm. Each sample was analyzed by LC-MS/MS in triplicate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Identification and quantification of global proteins
LC-MS/MS analysis of the TMT-labeled, bRPLC fractionated samples generated a total of 312 global proteomics data files. The Thermo RAW files were processed with ProteoWizard 3.0(Chambers et al., 2012) using ‘peak-picking’ for MS1 and MS2 spectra and converted to ‘.mzML’ format, and protein identification was conducted using MS-PyCloud (Chen et al., 2018). MS-GF+v9881 (Kim et al., 2008; Kim and Pevzner, 2014) was the default search engine in MS-PyCloud applied to match against the RefSeq human protein sequence database, released on May 02, 2016 (101,661 proteins). The partially tryptic search used a ± 10 ppm parent ion tolerance, 0.5 m/z fragment ion tolerance, allowed for isotopic error in precursor ion selection [−1,2], and searched a decoy database composed of the forward and reversed protein sequences. MS-GF+ settings included static carbamidomethylation (+57.0215 Da) on Cys residues, TMT modification (+229.1629 Da) on the peptide N terminus and Lys residues, and dynamic oxidation (+15.9949 Da) on Met residues for searching the global proteome data. Peptide identification stringency was tuned to not exceed a false discovery rate (FDR) of 1% at the peptide-spectrum match (PSM) level. In the protein inference conducted by MS-PyCloud, a minimum of 3 PSMs per peptide and 2 unique peptides per protein were required for achieving FDR < 1% at the protein level within the full dataset. Inference of parsimonious protein set resulted in a total of 8,144 common protein groups among all the tumor, non-tumor, pooled reference, and QC samples (Table S2).
The intensities of all ten TMT reporter ions in each MS/MS spectrum were extracted using MS-PyCloud. Next, PSMs were linked to the extracted reporter ion intensities by scan number. The relative protein abundance was calculated using the ‘log2-median-median’ strategy. The pooled reference sample was labeled with TMT 126 reagent, allowing comparison of relative abundances across the normalized intensity values of the remaining 9 channels of the TMT 10-plexes on the PSM level. The median value of the log2-transformed relative abundances from different scans and different bRPLC fractions corresponding to the same peptide were used as the relative abundance of the peptide. The final relative protein abundance was calculated as the median value of the log2-transformed relative abundance from each protein’s constituent peptides. Small differences in sample handling can result in detectable systematic, sample-specific bias in the quantification of protein levels. In order to mitigate these effects, we computed the median, log2 relative protein abundance over all identified proteins for each sample followed by re-centering to achieve a common median of 0 (see Figure S1A).
Identification and quantification of glycosite-containing peptides isolated with SPEG
The glycosite-containing peptide identification for the 39 SPEG data files (each set has 3 replicated runs) were performed as described above (e.g., peptide level FDR < 1%), with an additional dynamic deamidation (+0.984016 Da) modification on Asn and Gln residues. For SPEG datasets, the TMT-10 quantitative data was summarized at the glycosite-containing peptide level (Table S3). All the peptides (glycosite-containing peptides and global peptides) were labeled with TMT-10 reagent simultaneously. SPEG and intact glycopeptide analyses were performed after the TMT labeling. Thus, all the biases upstream of labeling are assumed to be identical between the global proteomics and glycoproteomics samples isolated by SPEG and intact glycopeptide enrichment. Therefore, to account for sample-specific biases in the glycosite-containing peptide analysis we normalized the relative abundance of the glycosite-containing peptides by subtracting the median values of log2-transformated relative abundance of glycoproteins in each sample (see Figure S1D).
Identification and quantification of intact N-linked glycopeptides
The intact N-linked glycopeptides were identified using GPQuest 2.1 software (Hu et al., 2018; Mertins et al., 2018). Prior to database search, ProteoWizard 3.0 was used to convert the .RAW files to .mzML files with the “centroid all scans” option selected. GPQuest 2.1 was applied to identify intact glycopeptides to MS/MS spectra using two approaches: searching spectra containing oxonium ions (‘oxo-spectra’) and identifying intact N-linked glycopeptides. The oxonium ions were used as the signature features of the glycopeptides from the MS/MS spectra, which were caused by the fragmentation of glycans attached to intact glycopeptides in the mass spectrometer. In this study, the MS/MS spectra containing the oxonium ions (m/z 204.0966) in the top 10 abundant peaks after removing TMT reporter ions were considered as the potential glycopeptide candidates. The intact N-linked glycopeptides were identified by using GPQuest 2.1 to search against the database of unique deglycosylated peptide sequences identified from the SPEG method and a database containing 178 N-linked glycan compositions. The glycan database was collected from the public database of GlycomeDB (Ranzinger et al., 2011) (http://www.glycome-db.org). Each tandem mass spectrum was first processed in a series of preprocessing procedures, including removing reporter ions, spectrum de-noising, intensity square root transformation (Liu et al., 2007), oxonium ions evaluation and glycan type prediction (Toghi Eshghi et al., 2016). The top 100 peaks in each preprocessed spectrum were matched to the fragment ion index generated from a peptide sequence database to identify all the candidate peptides. All the qualified (> 6 fragment ions matchings) candidate peptides were compared with the spectrum again to calculate the Morpheus scores (Wenger and Coon, 2013) by considering all the peptide fragments, glycopeptide fragments, and their isotope peaks. The peptide having the highest Morpheus score was then assigned to the spectrum. The mass gap between the assigned peptide and the precursor mass was searched in the glycan database to find the associated glycan. The best hits of all ‘oxo-spectra’ were ranked by the Morpheus score in descending order, in which those with FDR < 1% and covering > 10% total intensity of each tandem spectrum were reserved as qualified identifications. The precursor mass tolerance was set as 10ppm, and the fragment mass tolerance was 20 ppm.
Similar to the process described for the analysis of glycosite-containing peptides in SPEG, the quantification of the intact glycopeptides was also conducted at the peptide level. The median log2 ratio value of all the PSMs of an identical intact glycopeptide was used as the relative abundance of the intact glycopeptide. The relative abundances of intact glycopeptides of samples were also normalized by subtracting the median value of glycoproteins in each corresponding sample expressed in the global datasets (See Figure S1G and Table S4).
Quality control assessment
The sample correlation was the indicator of the similarity of the expression values of the samples. To eliminate the influence of the pooled reference channel, the absolute intensity matrix was applied in the sample correlation procedure. Instead of using a ‘log2-median-median’ strategy, the ‘sum-of-intensity’ approach was used to generate the intensity matrix of protein expression. The median (MD) strategy is 1) calculate median log2 value of the ith sample (mi = median(yij; where j = 1…p; i = 1...n). Here, p is the total protein or peptide identification number, and n is the total sample number. 2) record m0 = median (mi; where i = 1…n). 3) center the data of each sample by subtracting median from each value . The sum of intensity of all the reporter ions of the PSMs from all the fractions assigned to the same peptide was used as the absolute abundance of the peptide. The sum of peptide intensity values of the same protein was regarded as the absolute abundance of the protein. A Spearman’s rank correlation value was calculated between the two samples using their shared proteins (See Figure S1B). As the correlation is a rank-based correlation, no normalization is required before the calculation. The sample correlation was also applied on the ‘sum-of-intensity’ peptide matrices of all the quality control samples of the SPEG dataset and the intact N-linked glycopeptide dataset (See Figures S1E and S1H). The coefficient of variation (CV) values of the relative abundance (ratio values) of proteins or peptides of the QC samples were also calculated to evaluate the stability of the reproducibility of proteins or peptides expressed in the 9 QC samples (See Figures S1C, S1F, and S1I).
Proteomic and glycoproteomic clustering analysis
The top 50% of most variable global proteins (2,958) without missing values were analyzed by CancerSubtypes (Xu et al., 2017) for consensus clustering (Monti et al., 2003) of tumor subtypes. For the glycosite-containing peptide and IGP data, an identical approach was applied on the 50% most variable glycosite-containing peptides and IGPs. Specifically, 80% of the original sample pool was randomly subsampled without replacement and partitioned into three major clusters using hierarchical clustering, which was repeated 500 times (Wilkerson and Hayes, 2010). The expression values were transformed into Z scores using the built-in standardization function of R. The sample clustering result was reported in Table S5. For the IGP clustering, the corresponding glycan types were also listed on the left side of the heatmap of the clustered expression matrix to illustrate the possible relationship between tumor clusters and the associated glycan types (Figure 2A). The preferential glycan types and enriched pathways of different intact glycopeptides were grouped and shown in the left side columns of Figure 2A. The Z-score transformed the abundance of intact glycopeptides were grouped by the IG types in each IGP cluster to show the preferential glycosylation in each tumor cluster (Figure 2E).
Correlation between tumor clusters and clinical phenotype associations
The abundance levels of GLOBAL, SPEG, and IGP were transformed to binary vectors. The spearman’s rank correlation coefficient values of each pair of binary vectors were calculated by using Python SciPy package. The results were visualized in the Figures 2B and 2C for GLOBAL and SPEG comparing to IGP respectively. The categorical clinical phenotypes, such as tumor grade, tumor stage, participant race, anatomic site, origin site were also transformed to binary vectors for each class of the corresponding clinical phenotype and then correlated with the tumor clusters of IGP datasets. The numeric clinical phenotypes, such as tumor cellularity and participant age were directly correlated with the tumor clusters using spearman’s rank correlation method. The result was shown in Figure 2D.
Principal component analysis of tumor and non-tumor samples
The principal component analysis (PCA) function under OmicsOne (Hu et al., 2019) using scikit-learn package (Pedregosa et al., 2011) was implemented to conduct the unsupervised clustering analysis with the parameter ‘n_components = 2′ on the expression matrix of global proteomic data, in which there are 106 samples (observations) and 365 intact glycopeptides (features). The 95% confidence coverage was represented by an ellipse for each group, which was calculated based on the mean and covariance of points in that group (see Figure 3A). A similar approach was also applied on the GLOBAL proteomic and SPEG glycoproteomic datasets (see Figures S3A and S3B).
Tumor and non-tumor differential expression
To uncover discriminating features between tumors and non-tumors, we performed the t test analysis on the global proteomic dataset of 5916 global proteins expressed on tumor and non-tumor samples. The permutation corrected p values were calculated using Perseus with setting the FDR = 0.01 to identify the significant alternations. A total of 645 significantly upregulated and 587 significantly downregulated proteins were observed in the filtered results (see Figure S3C). A similar approach was also applied to the SPEG and IGP glycoproteomic data (See Figures S3D and 3B and Table S6).
CombiROC is an interactive web tool for selecting accurate marker combinations of omics data (Mazzara et al., 2017). It was applied to plot the Receiver operating characteristic (ROC) curves for the differential intact glycopeptides in tumor and non-tumor samples, in which both signal cutoff and minimum features were set to 1 to plot the results. The result was shown in Figure 3C.
DAVID 6.8 was applied on the 48 significantly upregulated and 94 significantly downregulated intact glycopeptides to perform gene-annotation enrichment analysis and shown in Figure 3D. The 365 identified glycopeptides were classified to HM, Fuc, and Sia types based on their glycan compositions, and separately plotted according to their median log2 ratio values in tumor and non-tumor sample groups as shown in Figure 3E. DAVID 6.8 was also applied on the gene name list of intact glycopeptides associated with HM, Fuc, and Sia glycan types for enriched pathways (Figure 3F).
Integrated proteomic and glycoproteomic analysis
The t tests were applied to the common global proteins, glycosite-containing peptides, and intact glycopeptides respectively to determine their differential expression in the tumor and non-tumor tissues (Figures 4A and 4B). The glycosylation sites of CA125 (MUC16) and its identified glycosite-containing peptides (SPEG) were highlighted in Figures 4C–4E to indicate the differential expression of global protein and the three glycosite-containing peptides. For further comparison, their corresponding expression values across all samples are shown as four boxplots representing expression in tumors and non-tumors (Figures 4D and 4E). The heterogenous glycosylation events on the identical glycosite of SSR2 were plotted in Figures 4F–4H.
Glycosylation biosynthetic pathway analysis
The intact glycopeptide expression was hypothesized to be influenced at least by the expression of substrate glycoproteins and glycosylation enzymes. The log2 ratio values of intact glycopeptides were correlated with the 22 glycosylation enzymes identified from the global proteomic data in this study. The correlation matrix was further arranged by hierarchical clustering on glycopeptides (columns) and glycosylation enzymes (rows) and visualized in Figure 5A. The glycan compositions were linked to the intact glycopeptides. The intact glycopeptides were classified as different groups for two comparisons based on the glycan structure they carry: one comparison is whether glycopeptides contained HM glycans (Figures 5D and 5E); the other is whether glycopeptides contained Fuc glycans (Figure 5C). For each comparison, the correlations between the IPGs and specific glycosylation enzyme (FUT11, PRKCSH, or MAN1A1) that correlated with the IGPs across all samples were calculated and shown in a boxplot. The hypothesis of tumor-specific glycosylation mechanism was shown in Figure 6A.
The gene names of significantly elevated intact glycopeptides modified by HM glycans in tumors were submitted in STRING 10.5 (Szklarczyk et al., 2017). The minimum required interaction score was set to 0.7. The protein-protein interaction network was shown in Figure 6B by disabling structure previews inside network bubbles, hiding disconnected nodes and small groups in the network.
Supplementary Material
Highlights.
Proteomics and glycoproteomics of 83 ovarian cancer and 23 relevant non-tumor tissues
Glycosylation is associated with three tumor clusters
Tumor-specific changes of glycoproteins and glycosites are apparent
Enzymes responsible for the glycosylation alterations are identified
ACKNOWLEDGMENTS
This work was supported by the National Cancer Institute (NCI) Clinical Proteomic Tumor Analysis Consortium (CPTAC; grants U24CA160036 and U24CA210985).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108276.
CONSORTIA
The members of the National Cancer Institute Clinical Proteomic Tumor Analysis Consortium are Yingwei Hu, Jianbo Pan, Punit Shah, Minghui Ao, Stefani N. Thomas, Yang Liu, Lijun Chen, Michael Schnaubelt, David J. Clark, Qing Kay Li, Jiang Qian, Matthew J.C. Ellis, Zhiao Shi, Bing Zhang, Jasmin Bavarva, Melissa Borucki, Kimberly Elburn, Linda Hannick, Mathangi Thiagarajan, Negin Vatanian, Samuel H. Payne, Steven A. Carr, Karl R. Clauser, Michael A. Gillette, Eric Kuhn, D.R. Mani, Shuang Cai, Karen A. Ketchum, Ratna R. Thangudu, Gordon A. Whiteley, Amanda Paulovich, Jeffrey Whiteaker, Nathan J. Edwards, Subha Madhavan, Peter B. McGarvey, Daniel W. Chan, Ie-Ming Shih, Hui Zhang, Zhen Zhang, Heng Zhu, Steven J. Skates, Forest M. White, Philip Mertins, Akhilesh Pandey, Robert J.C. Slebos, Emily S. Boja, Tara Hiltke, Christopher R. Kinsinger, Mehdi Mesri, Robert C. Rivers, Henry Rodriguez, Stephen E. Stein, David Fenyo, Kelly Ruggles, Douglas A. Levine, Mauricio Oberti, Tao Liu, Jason E. McDermott, Karin D. Rodland, Richard D. Smith, Lisa J. Zimmerman, Paul A. Rudnick, Michael Snyder, David L. Tabb, Yingming Zhao, Xian Chen, David F. Ransohoff, Andrew Hoofnagle, Daniel C. Liebler, Melinda E. Sanders, Yue Wang, Sherri R. Davies, Li Ding, R. Reid Townsend, Mark Watson, and Ana I. Robles.
REFERENCES
- Balog CI, Stavenhagen K, Fung WL, Koeleman CA, McDonnell LA, Verhoeven A, Mesker WE, Tollenaar RA, Deelder AM, and Wuhrer M (2012). N-glycosylation of colorectal cancer tissues: a liquid chromatography and mass spectrometry-based investigation. Mol. Cell. Proteomics 11, 571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantscheff M, Boesche M, Eberhard D, Matthieson T, Sweetman G, and Kuster B (2008). Robust and Sensitive iTRAQ Quantification on an LTQ Orbitrap Mass Spectrometer. Mol. Cell Proteomics 7, 1702–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bast RC Jr., Klug TL, St John E, Jenison E, Niloff JM, Lazarus H, Berkowitz RS, Leavitt T, Griffiths CT, Parker L, et al. (1983). A radioimmuno-assay using a monoclonal antibody to monitor the course of epithelial ovarian cancer. N. Engl. J. Med 309, 883–887. [DOI] [PubMed] [Google Scholar]
- Bast RC Jr., Badgwell D, Lu Z, Marquez R, Rosen D, Liu J, Baggerly KA, Atkinson EN, Skates S, Zhang Z, et al. (2005). New tumor markers: CA125 and beyond. Int. J. Gynecol. Cancer 15 (Suppl 3), 274–281. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011). Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers MC, Maclean B, Burke R, Amodei D, Ruderman DL, Neumann S, Gatto L, Fischer B, Pratt B, Egertson J, et al. (2012). A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol 30, 918–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CY, Jan YH, Juan YH, Yang CJ, Huang MS, Yu CJ, Yang PC, Hsiao M, Hsu TL, and Wong CH (2013). Fucosyltransferase 8 as a functional regulator of nonsmall cell lung cancer. Proc. Natl. Acad. Sci. USA 110, 630–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Deng Z, Huang C, Wu H, Zhao X, and Li Y (2017). Mass spectrometric profiling reveals association of N-glycan patterns with epithelial ovarian cancer progression. Tumour Biol. 39, 1010428317716249. [DOI] [PubMed] [Google Scholar]
- Chen L, Zhang B, Schnaubelt M, Shah P, Aiyetan P, Chan D, Zhang H, and Zhang Z (2018). MS-PyCloud: an open-source, cloud computing-based pipeline for LC-MS/MS data analysis. bioRxiv. 10.1101/320887. [DOI] [Google Scholar]
- Clark DJ, Dhanasekaran SM, Petralia F, Pan J, Song X, Hu Y, da Veiga Leprevost F, Reva B, Lih TM, Chang HY, et al. ; Clinical Proteomic Tumor Analysis Consortium (2019). Integrated Proteogenomic Characterization of Clear Cell Renal Cell Carcinoma. Cell 179, 964–983.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coscia F, Lengyel E, Duraiswamy J, Ashcroft B, Bassani-Sternberg M, Wierer M, Johnson A, Wroblewski K, Montag A, Yamada SD, et al. (2018). Multi-level proteomics identifies CT45 as a chemosensitivity mediator and immunotherapy target in ovarian cancer. Cell 175, 159–170.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leoz ML, Young LJ, An HJ, Kronewitter SR, Kim J, Miyamoto S, Borowsky AD, Chew HK, and Lebrilla CB (2011). High-mannose glycans are elevated during breast cancer progression. Mol. Cell. Proteomics 10, M110.002717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, Shih I-M, Soslow RA, Cope L, and Levine DA (2017). Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun 8, 990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedele CG, Ooms LM, Ho M, Vieusseux J, O’Toole SA, Millar EK, Lopez-Knowles E, Sriratana A, Gurung R, Baglietto L, et al. (2010). Inositol polyphosphate 4-phosphatase II regulates PI3K/Akt signaling and is lost in human basal-like breast cancers. Proc. Natl. Acad. Sci. USA 107, 22231–22236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg B, Pathria G, Wagner C, Maurer M, and Wagner SN (2016). Signal Sequence Receptor 2 is required for survival of human melanoma cells as part of an unfolded protein response to endoplasmic reticulum stress. Mutagenesis 31, 573–582. [DOI] [PubMed] [Google Scholar]
- Gilgunn S, Conroy PJ, Saldova R, Rudd PM, and O’Kennedy RJ (2013). Aberrant PSA glycosylation—a sweet predictor of prostate cancer. Nat. Rev. Urol 10, 99–107. [DOI] [PubMed] [Google Scholar]
- Guan X (2015). Cancer metastases: challenges and opportunities. Acta Pharm. Sin. B 5, 402–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart GW, and Copeland RJ (2010). Glycomics hits the big time. Cell 143, 672–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Shah P, Clark DJ, Ao M, and Zhang H (2018). Reanalysis of Global Proteomic and Phosphoproteomic Data Identified a Large Number of Glycopeptides. Anal. Chem 90, 8065–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Ao M, and Zhang H (2019). OmicsOne: Associate Omics Data with Phenotypes in One-Click. bioRxiv. 10.1101/756544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jiang WG, Sanders AJ, Katoh M, Ungefroren H, Gieseler F, Prince M, Thompson SK, Zollo M, Spano D, Dhawan P, et al. (2015). Tissue invasion and metastasis: Molecular, biological and clinical perspectives. Semin. Cancer Biol 35 (Suppl), S244–S275. [DOI] [PubMed] [Google Scholar]
- Kallunki T, Olsen OD, and Jäättelä M (2013). Cancer-associated lysosomal changes: friends or foes? Oncogene 32, 1995–2004. [DOI] [PubMed] [Google Scholar]
- Kanehisa M (2019). Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Sato Y, Furumichi M, Morishima K, and Tanabe M (2019). New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47 (D1), D590–D595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaodee W, Inboot N, Udomsom S, Kumsaiyai W, and Cressey R (2017). Glucosidase II beta subunit (GluIIb) plays a role in autophagy and apoptosis regulation in lung carcinoma cells in a p53-dependent manner. Cell Oncol. (Dordr.) 40, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri K, Klein JA, White MR, Grant OC, Leymarie N, Woods RJ, Hartshorn KL, and Zaia J (2016). Integrated Omics and Computational Glycobiology Reveal Structural Basis for Influenza A Virus Glycan Micro-heterogeneity and Host Interactions. Mol. Cell. Proteomics 15, 1895–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, and Pevzner PA (2014). MS-GF+ makes progress towards a universal database search tool for proteomics. Nat. Commun 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Gupta N, and Pevzner PA (2008). Spectral probabilities and generating functions of tandem mass spectra: a strike against decoy databases. J. Proteome Res 7, 3354–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S, Phallen J, et al. (2017). High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun 8, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Bell AW, Bergeron JJ, Yanofsky CM, Carrillo B, Beaudrie CE, and Kearney RE (2007). Methods for peptide identification by spectral comparison. Proteome Sci. 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzara S, Rossi RL, Grifantini R, Donizetti S, Abrignani S, and Bombaci M (2017). CombiROC: an interactive web tool for selecting accurate marker combinations of omics data. Sci. Rep 7, 45477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDermott JE, Arshad OA, Petyuk VA, Fu Y, Gritsenko MA, Clauss TR, Moore RJ, Schepmoes AA, Zhao R, Monroe ME, et al. ; Clinical Tumor Analysis Consortium (2020). Proteogenomic Characterization of Ovarian HGSC Implicates Mitotic Kinases, Replication Stress in Observed Chromosomal Instability. Cell Rep Med 1, 100004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meany DL, and Chan DW (2011). Aberrant glycosylation associated with enzymes as cancer biomarkers. Clin. Proteomics 8, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, Wang X, Qiao JW, Cao S, Petralia F, et al. ; NCI CPTAC (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, et al. (2018). Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc 13, 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller DS, Blessing JA, Krasner CN, Mannel RS, Hanjani P, Pearl ML, Waggoner SE, and Boardman CH (2009). Phase II evaluation of pemetrexed in the treatment of recurrent or persistent platinum-resistant ovarian or primary peritoneal carcinoma: a study of the Gynecologic Oncology Group. J. Clin. Oncol 27, 2686–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti S, Tamayo P, Mesirov J, and Golub T (2003). Consensus Clustering: A Resampling-Based Method for Class Discovery and Visualization of Gene Expression Microarray Data. Machine Learning 52, 91–118. [Google Scholar]
- Narimatsu H, Kaji H, Vakhrushev SY, Clausen H, Zhang H, Noro E, Togayachi A, Nagai-Okatani C, Kuno A, Zou X, et al. (2018). Current Technologies for Complex Glycoproteomics and Their Applications to Biology/Disease-Driven Glycoproteomics. J. Proteome Res 17, 4097–4112. [DOI] [PubMed] [Google Scholar]
- Ooms LM, Binge LC, Davies EM, Rahman P, Conway JR, Gurung R, Ferguson DT, Papa A, Fedele CG, Vieusseux JL, et al. (2015). The Inositol Polyphosphate 5-Phosphatase PIPP Regulates AKT1-Dependent Breast Cancer Growth and Metastasis. Cancer Cell 28, 155–169. [DOI] [PubMed] [Google Scholar]
- Padler-Karavani V (2014). Aiming at the sweet side of cancer: aberrant glycosylation as possible target for personalized-medicine. Cancer Lett. 352, 102–112. [DOI] [PubMed] [Google Scholar]
- Parker BL, Thaysen-Andersen M, Solis N, Scott NE, Larsen MR, Graham ME, Packer NH, and Cordwell SJ (2013). Site-specific glycanpeptide analysis for determination of N-glycoproteome heterogeneity. J. Proteome Res 12, 5791–5800. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn Res 12, 2825–2830. [Google Scholar]
- Pinsky PF, Yu K, Kramer BS, Black A, Buys SS, Partridge E, Gohagan J, Berg CD, and Prorok PC (2016). Extended mortality results for ovarian cancer screening in the PLCO trial with median 15years follow-up. Gynecol. Oncol 143, 270–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzinger R, Herget S, von der Lieth CW, and Frank M (2011). GlycomeDB—a unified database for carbohydrate structures. Nucleic Acids Res. 39, D373–D376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauscher B, Heigwer F, Henkel L, Hielscher T, Voloshanenko O, and Boutros M (2018). Toward an integrated map of genetic interactions in cancer cells. Mol. Syst. Biol 14, e7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott NE, Parker BL, Connolly AM, Paulech J, Edwards AV, Crossett B, Falconer L, Kolarich D, Djordjevic SP, Højrup P, et al. (2011). Simultaneous glycan-peptide characterization using hydrophilic interaction chromatography and parallel fragmentation by CID, higher energy collisional dissociation, and electron transfer dissociation MS applied to the N-linked glycoproteome of Campylobacter jejuni. Mol. Cell. Proteomics 10, M000031MCP201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi MK, Thaysen-Andersen M, Smith JT, Baker MS, Packer NH, Hancock WS, and Fanayan S (2014). Comparative N-glycan profiling of colorectal cancer cell lines reveals unique bisecting GlcNAc and α−2,3-linked sialic acid determinants are associated with membrane proteins of the more metastatic/aggressive cell lines. J. Proteome Res 13, 277–288. [DOI] [PubMed] [Google Scholar]
- Shin G-C, Moon SU, Kang HS, Choi H-S, Han HD, and Kim K-H (2019). PRKCSH contributes to tumorigenesis by selective boosting of IRE1 signaling pathway. Nat. Commun 10, 3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2018). Cancer statistics, 2018. CA Cancer J. Clin 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Stowell SR, Ju T, and Cummings RD (2015). Protein glycosylation in cancer. Annu. Rev. Pathol 10, 473–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Shah P, Eshghi ST, Yang W, Trikannad N, Yang S, Chen L, Aiyetan P, Hoöti N, Zhang Z, et al. (2016). Comprehensive analysis of protein glycosylation by solid-phase extraction of N-linked glycans and glycosite-containing peptides. Nat. Biotechnol 34, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swiatly A, Horala A, Matysiak J, Hajduk J, Nowak-Markwitz E, and Kokot ZJ (2018). Understanding Ovarian Cancer: iTRAQ-Based Proteomics for Biomarker Discovery. Int. J. Mol. Sci 19, 2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45 (D1), D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabarés G, Radcliffe CM, Barrabés S, Ramírez M, Aleixandre RN, Hoesel W, Dwek RA, Rudd PM, Peracaula R, and de Llorens R (2006). Different glycan structures in prostate-specific antigen from prostate cancer sera in relation to seminal plasma PSA. Glycobiology 16, 132–145. [DOI] [PubMed] [Google Scholar]
- Talabnin K, Talabnin C, Ishihara M, and Azadi P (2018). Increased expression of the high-mannose M6N2 and NeuAc3H3N3M3N2F tri-antennary N-glycans in cholangiocarcinoma. Oncol. Lett 15, 1030–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toghi Eshghi S, Yang W, Hu Y, Shah P, Sun S, Li X, and Zhang H (2016). Classification of Tandem Mass Spectra for Identification of N- and O-linked Glycopeptides. Sci. Rep 6, 37189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, Gaudet MM, Jemal A, and Siegel RL (2018). Ovarian cancer statistics, 2018. CA Cancer J. Clin 68, 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towers CG, and Thorburn A (2017). Targeting the Lysosome for Cancer Therapy. Cancer Discov. 7, 1218–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, and Cox J (2018). Perseus: a bioinformatics platform for integrative analysis of proteomics data in cancer research. In Cancer Systems Biology: Methods and Protocols, von Stechow L, ed. (Springer; New York: ), pp. 133–148. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, and Cox J (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. [DOI] [PubMed] [Google Scholar]
- Varki A (2017). Biological roles of glycans. Glycobiology 27, 3–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Tamayo P, Yang JY, Hubbard D, Zhang H, Creighton CJ, Fereday S, Lawrence M, Carter SL, Mermel CH, et al. ; Cancer Genome Atlas Research Network (2013). Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Invest 123, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, and Marcotte EM (2012). Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13, 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Dafik L, Nolley R, Huang W, Wolfinger RD, Wang L-X, and Peehl DM (2013). Anti-Oligomannose Antibodies as Potential Serum Biomarkers of Aggressive Prostate Cancer. Drug Dev. Res 74, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Chen J, Li QK, Peskoe SB, Zhang B, Choi C, Platz EA, and Zhang H (2014). Overexpression of α (1,6) fucosyltransferase associated with aggressive prostate cancer. Glycobiology 24, 935–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland F, Fritz K, Oehler MK, and Hoffmann P (2012). Methods for identification of CA125 from ovarian cancer ascites by high resolution mass spectrometry. Int. J. Mol. Sci 13, 9942–9958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger CD, and Coon JJ (2013). A proteomics search algorithm specifically designed for high-resolution tandem mass spectra. J. Proteome Res 12, 1377–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedmann M, Kurzchalia TV, Hartmann E, and Rapoport TA (1987). A signal sequence receptor in the endoplasmic reticulum membrane. Nature 328, 830–833. [DOI] [PubMed] [Google Scholar]
- Wilkerson MD, and Hayes DN (2010). ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 26, 1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Le TD, Liu L, Su N, Wang R, Sun B, Colaprico A, Bontempi G, and Li J (2017). CancerSubtypes: an R/Bioconductor package for molecular cancer subtype identification, validation and visualization. Bioinformatics 33, 3131–3133. [DOI] [PubMed] [Google Scholar]
- Yang W, Shah P, Hu Y, Toghi Eshghi S, Sun S, Liu Y, and Zhang H (2017). Comparison of Enrichment Methods for Intact N- and O-Linked Glycopeptides Using Strong Anion Exchange and Hydrophilic Interaction Liquid Chromatography. Anal. Chem 89, 11193–11197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li XJ, Martin DB, and Aebersold R (2003). Identification and quantification of N-linked glycoproteins using hydrazide chemistry, stable isotope labeling and mass spectrometry. Nat. Biotechnol 21, 660–666. [DOI] [PubMed] [Google Scholar]
- Zhang B, Wang J, Wang X, Zhu J, Liu Q, Shi Z, Chambers MC, Zimmerman LJ, Shaddox KF, Kim S, et al. ; NCI CPTAC (2014). Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu T, Zhang Z, Payne SH, Zhang B, McDermott JE, Zhou JY, Petyuk VA, Chen L, Ray D, et al. ; CPTAC Investigators (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinska DF, Gnad F, Wiśniewski JR, and Mann M (2010). Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study are available at CPTAC data portal and publicly available (https://cptac-data-portal.georgetown.edu/study-summary/S038). The codes supporting the current study are publicly available and listed in the Key Resources Table.
KEY RESOURCES TABLE