

Abstract
The design, synthesis, and X-ray structural analysis of hybrid HIV-1 protease inhibitors (PIs) containing bis-tetrahydrofuran (bis-THF) in a pseudo-C2-symmetric dipeptide isostere are described. A series of PIs were synthesized by incorporating bis-THF of darunavir on either side of the Phe-Phe isostere of lopinavir in combination with hydrophobic amino acids on the opposite P2/P2′ position. Structure–activity relationship studies indicated that the bis-THF moiety can be attached at either the P2 or P2′ position without significantly affecting potency. However, the group on the opposite P2/P2′ position had a dramatic effect on potency depending on the size and shape of the side chains. Cocrystal structures of inhibitors with wild-type HIV-1 protease revealed that the bis-THF moiety retained similar interactions as observed in the darunavir-protease complex regardless of position on the Phe-Phe isostere. Analyses of cocrystal structures and molecular dynamics simulations provide insights for optimizing HIV-1 PIs containing bis-THF in non-sulfonamide dipeptide isosteres.
Graphical Abstract
INTRODUCTION
Combinations of drugs targeting HIV-1 enzymes essential for viral replication have been highly effective in reducing viral load in infected individuals and significantly increasing their life expectancy.1 HIV-1 protease inhibitors (PIs) have played an important role as a highly potent component of combination antiretroviral therapy (cART) for people living with HIV-1. There are nine HIV-1 PIs approved by the US FDA; however, clinical use of most of these has diminished over the years due to side effects, unfavorable pharmacokinetics, and drug resistance. Currently only three PIs, darunavir (DRV), lopinavir (LPV), and atazanavir (ATV) (Figure 1) – boosted with small doses of ritonavir (RTV) – are being used in cART as they have comparatively higher potency and better resistance profiles.2,3
Figure 1.

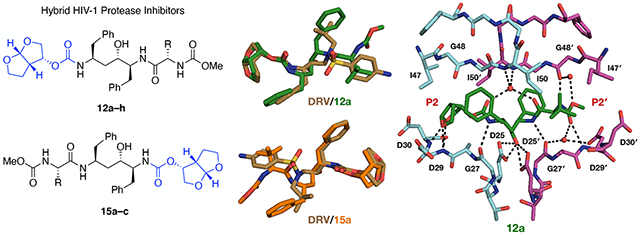
Structures of HIV-1 protease inhibitors darunavir (DRV) (1), lopinavir (LPV) (2), atazanavir (ATV) (3), previously reported hybrid compounds 4 and 5, and new hybrid compounds designed by incorporating the bis-THF moiety of DRV into the pseudo-symmetric Phe-Phe dipeptide isostere of LPV at the P2 (12a–h) or P2′ position (15a–c), in combination with an ATV- like hydrophobic amino acid moiety at the P2′ or P2 position, respectively. The canonical nomenclature is used to indicate the position of each inhibitor moiety.
Most HIV-1 PIs are substrate-based peptidomimetics containing a hydroxyl group as a transition state mimetic and diverse moieties that target the S1/S1′ and S2/S2′ subsites of the enzyme.4, 5 Among the PIs in clinical use, DRV is the most potent with the highest barrier to resistance and efficacy against multidrug-resistant HIV-1 strains.6 The excellent potency and resistance profiles of DRV are attributed to the bis-tetrahydrofuran (bis-THF) moiety, which forms strong hydrogen bonding interactions with the main-chain NH of Asp29 and Asp30 in the S2 subsite of the enzyme.7 Moreover, the extensive van der Waals (vdW) interactions of the bis-THF moiety, along with the sulfonamide-based Phe-Leu dipeptide isostere and the 4-aminobenzene group, also significantly contribute to the potency of DRV.8 Due to these unique binding characteristics the bis-THF moiety of DRV has emerged as a privileged ligand for targeting the S2 subsite of HIV-1 protease especially when incorporated into the (R)-(hydroxyethylamino)sulfonamide isostere.9
Efforts to improve potency, pharmacokinetics, and resistance profiles of HIV-1 PIs have led to the discovery of exceptionally potent compounds exhibiting a wide range of properties.5, 10-14 In recent years, these efforts have focused mainly on exploring DRV analogues with similar sulfonamide-based dipeptide isosteres and P2/P2′ moieties. Many analogues of DRV with modifications at P1/P1′ and P2/P2′ positions have been explored extensively,5, 15-18 including novel bis-THF analogues with improved hydrogen bonding and vdW interactions with the protease.19-22 Moreover, detailed structural analyses informing structure-based design of inhibitors with the DRV scaffold have been reported numerous times including recent structures determined by neutron crystallography.5, 7, 8, 23-25
However, very few inhibitors containing the bis-THF moiety in in non-sulfonamide dipeptide isosteres have been explored.26-28 Chen and colleagues at Abbott Laboratories explored PIs with bis-THF on either side of the pseudo-C2-symmetric Phe-Phe dipeptide isostere of LPV,26, 27 while Cannizzaro and coworkers at Gilead Sciences designed similar analogues using the aza-dipeptide isostere of ATV.28 Although some of the Phe-Phe isostere-based compounds, such as 4 and 5 (Figure 1), exhibited significant antiviral activity against wild-type HIV-1, in the absence of direct comparison, it is unclear if their potency is similar to that of DRV.26 Also, due to limited structure- activity relationship studies, the S2/S2′ subsite preference of the bis-THF moiety in the context of Phe-Phe isostere remains unclear,26, 27 as is the identity of an optimal moiety for targeting the opposite S2/S2′ subsite.26 Moreover, no cocrystal structures of such hybrid PIs have been reported, limiting opportunities for structure-guided design and optimization.
In this study, we investigate HIV-1 PIs containing the bis-THF moiety of DRV in the Phe-Phe dipeptide isostere of LPV with the aim to identify compounds with potency similar to that of DRV but with an alternate non-sulfonamide scaffold. Protease-inhibitor molecular interactions are characterized using X-ray crystallography and molecular dynamics (MD) simulations to aid structure-guided optimization of these hybrid PIs. In addition, interdependence of HIV-1 protease subsites was explored to provide insight into subsite preferences and influence of different inhibitor moieties on binding and potency.
The active site of HIV-1 protease exhibits subsite interdependence in substrate recognition and inhibitor binding,29, 30 but this feature of the protease has rarely been considered in inhibitor design. While identifying optimal moieties for targeting each of the subsites in the protease active site is important, understanding the subsite interdependence and selection of the best combination of moieties are also crucial to achieving higher potency especially against drug-resistant variants.19 The bis-THF moiety potentially can be incorporated in diverse dipeptide isosteres in combination with suitable ligands targeting the other subsites. Thus a series of hybrid HIV-1 PIs were synthesized by incorporating the bis-THF moiety of DRV on either side of the pseudo-C2- symmetric Phe-Phe dipeptide isostere of LPV in combination with ATV-like hydrophobic P2/P2′ ligands of varying size and shape (Figure 1). The hybrid PIs were tested for inhibitory activity against wild-type and two drug-resistant variants and for antiviral activity in cellular assays. We also determined high-resolution X-ray crystal structures of all designed hybrid compounds and previously reported PI 4 bound to wild-type HIV-1 protease and characterized the binding interactions of bis-THF in the context of Phe-Phe isostere. In addition, molecular dynamics simulations were utilized to assess the stability of protease-inhibitor interactions in solution. The comprehensive structural and dynamic analyses of hybrid PIs have provided insights into inhibitor binding and interdependence of protease subsites.
CHEMISTRY
The synthesis of hybrid PIs incorporating the bis-THF moiety at either the P2 or P2′ position is outlined in Scheme 1. The required Phe-Phe dipeptide isostere core intermediate 7 was synthesized from phenylalanine following previously reported methods with minor modifications (see Supporting Information for details) (Haight, OPRD, 1999).31 Reaction of the dipeptide isostere 7 with bis-THF activated carbonate 8 in the presence of DIEA provided intermediate 9. Debenzylation of 9 using ammonium formate and 10% palladium on activated carbon in methanol gave the corresponding deprotected amine 10, which was subsequently coupled with the amino acid derivatives 11a–h using HATU and DIEA to provide the target compounds 12a–h with bis-THF at the P2 position. While N-(methoxycarbonyl)-protected amino acid derivatives 11a and 11e were commercially available, intermediates 11b–d and 11f–h were prepared from the corresponding amino acids by reaction with methyl chloroformate under basic conditions following a reported procedure.32
Scheme 1.

Synthesis of Protease Inhibitors Incorporating Bis-Tetrahydrofurn in a Pseudo- Symmetric Phe-Phe Dipeptide Isostere
Reagents and conditions: (a) K2CO3, KOH, BnCl, H2O, 100 °C, 5 h; (b) NaNH2, CH3CN, MTBE, 0 °C, 2 h, then BnMgCl (2 M in THF), RT, 14 h; (c) NaBH4, MsOH, i-PrOH, EGDME, 0 °C, 12 h; (d) NaBH4, TEA, DMA, 0–15 °C, 2 h; (e) DIEA, CH3CN, RT, 24 h; (f) Pd/C, HCO2NH4, MeOH, 50 °C, 12 h; (g) N-(methoxycarbonyl)-capped amino acid, HATU, DIEA, DMF, RT, 4 h.
The PIs 15a–c with the bis-THF at the P2′ position were synthesized using a similar reaction sequence. Coupling of the dipeptide isostere 7 with acids 11a–c under HATU peptide coupling conditions gave the corresponding intermediates 13a–c. Catalytic debenzylation of compounds 13a–c followed by coupling with bis-THF activated carbonate 8 in the presence of DIEA provided the target compounds 15a–c. A pair of previously reported PIs 4 and 5 containing the dimethylphenoxy acetate and bis-THF moieties as the P2 or P2′ ligands in the Phe-Phe isostere were also synthesized using the same reaction sequences.
RESULTS AND DISCUSSION
We explored hybrid HIV-1 PIs containing the bis-THF moiety of DRV in the Phe-Phe isostere of LPV, with the goal to characterize the binding interactions of this P2 ligand in the context of a pseudo-C2-symmetric dipeptide isostere. The bis-THF moiety was used in combination with diverse N-(methoxycarbonyl)-capped amino acid derivatives similar to the P2/P2′ moiety of ATV to optimize hydrogen bonding and vdW interactions in both the S2 and S2′ subsites of HIV-1 protease. To explore subsite preference for the bis-THF moiety, pairs of compounds were synthesized with the bis-THF moiety attached at either the P2 (12a–c) or P2′ (15a–c) position with respect to the central hydroxyl group of the Phe-Phe isostere. Considering potential subsite interdependence, incorporating a relatively small and conformationally flexible moiety at the other P2/P2′ position was expected to allow the bis-THF moiety to optimally interact with the backbone atoms in the S2/S2′ subsite, likely improving overall inhibitor binding with the protease. Structure- activity relationships were explored by incorporating hydrophobic amino acids of varying size and shape to identify an optimal ligand for targeting the other S2/S2′ subsite.
Enzyme Inhibition and Antiviral Assays.
The potency of PIs was assessed using biochemical and antiviral assays. The enzyme inhibition constants (Ki) were determined against wild-type protease and two drug-resistant variants (I84V and I50V/A71V) using a highly sensitive fluorogenic assay (Table 1).33 Both I50V and I84V are major protease inhibitor resistance mutations that reduce susceptibility to LPV and DRV. For a subset of compounds, antiviral potencies (EC50) were determined against wild-type HIV-1 (NL4-3 strain) using a cell-based antiviral assay. DRV was used as a control in all assays.
Table 1.
Enzyme Inhibitory Activity and Antiviral Potency of Hybrid HIV-1 Protease Inhibitors
| Inhibitor | Structure | Ki (nM)a | Ec50 (nM)b | ||
|---|---|---|---|---|---|
| WT | I84V | I50V/A71V | WT | ||
| 12a |  |
0.018 ± 0.003 | 0.208 ± 0.014 | 0.273 ± 0.043 | 11 |
| 15a |  |
0.013 ± 0.001 | 0.372 ± 0.021 | 0.072 ± 0.005 | 39 |
| 12b |  |
46 ± 3 | 236 ± 39 | 484 ± 70 | NT |
| 15b |  |
7.3 ± 0.5 | 191 ± 30 | 40 ± 3 | NT |
| 12c |  |
0.235 ± 0.016 | 0.71 ± 0.10 | 2.35 ± 0.39 | NT |
| 15c |  |
0.161 ± 0.011 | 1.18 ± 0.26 | 1.39 ± 0.12 | NT |
| 12d |  |
0.019 ± 0.004 | 0.069 ± 0.013 | 0.073 ± 0.003 | 66 |
| 12e |  |
0.074 ± 0.006 | 0.302 ± 0.022 | 0.295 ± 0.082 | 55 |
| 12f |  |
0.571 ± 0.076 | 1.82 ± 1.50 | 1.66 ± 0.48 | NT |
| 12g | 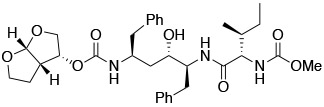 |
0.055 ± 0.004 | 0.198 ± 0.014 | 0.560 ± 0.094 | 70 |
| 12h |  |
0.027 ± 0.004 | 0.091 ± 0.013 | 0.029 ± 0.003 | 240 |
| 4 |  |
0.017 ± 0.002 | 0.069 ± 0.005 | 0.072 ± 0.002 | 38 |
| 5 |  |
0.037 ± 0.005 | 0.259 ± 0.055 | NT | 16 |
| LPV |  |
< 0.005 | 0.051 ± 0.004 | 0.061 ± 0.011 | 12 |
| DRV |  |
< 0.005 | 0.025 ± 0.006 | 0.075 ± 0.006 | 5 |
Enzyme inhibition constants against wild-type HIV-1 protease and drug resistant variants
Antiviral potency against wild-type HIV-1; NT = Not Tested
Compounds 12a and 15a, with the bis-THF moiety at the P2 and P2′ position, respectively, and the N-(methoxycarbonyl)-tert-leucine at the opposite P2/P2′ position, showed excellent inhibitory potencies against wild-type protease and the drug resistant variants. Compared to DRV and LPV, both compounds were less active, particularly against the I84V variant. Interestingly, compounds 12a and 15a exhibited similar Ki values, though the latter was 4-fold more active against the I50V/A71V variant, indicating no clear S2/S2′ subsite preference for the bis-THF moiety. However, in antiviral assays compound 12a was 3-fold more potent than 15a against wild-type HIV-1. The compound 12a, with the bis-THF moiety at the P2 position, exhibited excellent antiviral potency (EC50 = 11 nM) comparable to that of LPV (EC50 = 12 nM) and DRV (EC50 = 5 nM). A similar potency profile was observed for previously reported compounds 4 and 5 containing the bis-THF and the dimethylphenoxy acetate moieties as the P2 and P2′ ligands. Compound 5 exhibited 2-fold better antiviral potency than 4, indicating a reverse subsite preference for the bis-THF moiety relative to the 12a/15a pair. The small variation in the antiviral potencies of compounds 12a/15a and 4/5 indicate that the position of the bis-THF moiety with respect to the central hydroxyl group of the Phe-Phe isostere, though not critical for binding, may affect potency in cellular assays.
Although the potency of the hybrid compounds was not particularly sensitive to the position of the bis-THF moiety, compounds with different hydrophobic amino acid derivatives at the opposite P2/P2′ position showed markedly different inhibitory potencies. The compounds with the cycloleucine moiety, 12b and 15b, were significantly less active (>2500-fold and 560-fold, respectively) than the corresponding tert-leucine analogues 12a and 15a. The dramatic loss of potency is likely due to the altered shape and flexibility of the hydrophobic amino acid moiety. This was further supported by the fact that the corresponding cyclopentylglycine analogues 12c and 15c, though also an order of magnitude less active than 12a and 15a, still exhibited sub- nanomolar to low nM potency against wild-type protease and drug-resistant variants. Together these data confirm that the hydrophobic amino acid moiety at the opposite P2/P2′ position significantly influence potency in both biochemical and antiviral assays.
Since potency of the compounds with bis-THF moiety attached to the Phe-Phe isostere largely depended on the hydrophobic amino acid moiety at the opposite side of the core scaffold, we investigated size and shape requirements for optimally targeting the S2′ subsite. Analogues of compound 12a were synthesized with diverse hydrophobic amino acid derivatives as P2′ ligands while keeping the bis-THF moiety at the P2 position. The alanine analogue 12d exhibited excellent inhibitory potency with Ki values against resistant variants comparable to that of LPV and DRV. The corresponding valine analogue 12e was 4-fold less active against wild-type protease but maintained similar inhibitory potency as 12a against the resistant variants. However, compared to 12a compounds 12d and 12e were 6- and 5-fold less active in antiviral assays, respectively, indicating that factors other than binding affinity are responsible for lower antiviral potency. The cyclopropyl analogue 12f was considerably less active than 12a, with Ki values similar to the cyclopentyl analogue 12c, strongly suggesting that cyclic moieties at the P2/P2′ position are unfavorable. Replacement of tert-leucine with an isoleucine (compound 12g) or alloisoleucine (compound 12h) moiety at the P2′ position also resulted in slightly lower activity against wild- type protease but similar or better potency against resistant variants. This further confirmed earlier findings that the P2/P2′ group opposite to the bis-THF moiety on the Phe-Phe core has a significant impact on potency. These results are in contrast to the sulfonamide-based dipeptide isostere of DRV, where diverse P2′ modifications result in relatively minor changes in potency.6 The identity of the P2/P2′ moiety opposite to the bis-THF on the Phe-Phe core considerably impacts potency in both biochemical and antiviral assays.
Analysis of Protease-Inhibitor Complexes.
To characterize protease-inhibitor molecular interactions, twelve high-resolution (1.8–2.0 Å) crystal structures of hybrid PIs and compound 4 bound to wild-type HIV-1 protease of the NL4-3 strain were determined. A cocrystal structure of LPV was also determined bound to the same protease enzyme for direct comparison. The crystallographic data collection and refinement statistics are summarized in Table S1. Following convention, protease chains were assigned chain A (non-prime) or chain B (prime) depending on the interactions between the central hydroxyl group of the Phe-Phe isostere and the catalytic aspartates Asp25/Asp25′.
LPV and DRV achieve low picomolar affinity to wild-type HIV-1 protease by making a number of hydrogen bonds and vdW interactions with the active site residues (Figure S1). The interactions of the transition state mimetic secondary hydroxyl group with the catalytic aspartates, together with the vdW interactions of the adjacent P1/P1′ moieties, are critical for overall inhibitor binding. Together these interactions determine the positions of the P2/P2′ moieties in the active site, which in turn determine the patterns of hydrogen bonding and vdW interactions of these moieties with the protease. DRV contains a sulfonamide-based Phe-Leu dipeptide isostere core with the (R)- configuration of the secondary hydroxyl group, whereas LPV has a pseudo-C2-symmetric Phe-Phe dipeptide isostere with the (S)-configuration of the hydroxyl group. Though chemically quite distinct, the core scaffolds of LPV and DRV make largely similar hydrogen bonding and vdW interactions with the protease. These inhibitors, however, differ in their interactions with the protease in the S2 and S2′ subsites.
The bis-THF moiety of DRV forms direct hydrogen bonds with the main-chain NH of Asp29 and Asp30 in the S2 subsite, while the 4-aminobenzene makes a direct hydrogen bond with the main-chain carbonyl of Asp30′ as well as a water-mediated interactions with the side-chain carboxylate of the same residue in the S2′ subsite (Figure S1). The cyclic urea moiety of LPV makes direct hydrogen bonds with the main-chain NH and side-chain carboxylate of Asp29. The same moiety also makes water-mediated interactions with the main-chain carbonyl of Gly27 and the side-chain carboxylate of Asp29 in the S2 subsite. However, in contrast to the P2′ moiety of DRV, the dimethylphenoxy acetate moiety of LPV only makes hydrophobic interactions in the S2′ subsite (Figure S1). The hybrid compounds combine the pseudo-C2-symmetric Phe-Phe isostere of LPV with the bis-THF moiety of DRV and an ATV-like amino acid moiety to enhance direct hydrogen bonding and vdW interactions in the S2 and S2′ subsites.
The cocrystal structures of hybrid compounds revealed similar binding conformations of the Phe-Phe isostere and the bis-THF moiety as observed in the protease complexes with LPV and DRV, respectively (Figure 2). Importantly, the bis-THF moiety is positioned to interact with the main-chain NH of residues Asp29 and Asp30, mimicking the binding interactions of DRV (Figure 3). This binding conformation is maintained in all hybrid compounds including 4 regardless of the position of the bis-THF moiety. As a result, within each compound series (with the bis-THF moiety at the P2 and P2′ position respectively), the structures aligned well with variations occurring only at the hydrophobic amino acid moiety (Figure 4).
Figure 2.

Overlays of protease cocrystal structures bound to LPV (PDB 6PJB) (grey, top) and DRV (6DGX) (brown, bottom) with compounds 12a (bis-THF at P2; PDB 6PJD) (green), 15a (bis-THF at P2′; PDB 6PJM) (orange), and 4 (PDB 6PJC) (salmon) revealed similar binding conformations of the Phe-Phe isostere and the bis-THF moiety as observed in the protease complexes with LPV and DRV, respectively. Note that DRV is flipped 180° to compare with bis-THF of 15a at the P2′ position.
Figure 3.

Crystal structures of wild-type HIV-1 protease in complex with LPV (PDB 6PJB) (A) and hybrid compounds 4 (PDB 6PJC) (B), 12a (PDB 6PJD) (C), and 15a (PDB 6PJM) (D). The bis-THF moiety maintains the hydrogen bonding interactions with the main-chain NH of Asp29/Asp29′ and Asp30/Asp30′ in the S2/S2′ subsites regardless of the its position on the Phe- Phe core. Hydrogen bonds and water-mediated interactions as determined by PyMol are shown as black dashed lines with waters shown as red spheres.
Figure 4.

Superposition of HIV-1 Protease complexes with compounds 12a–12h (A) and compounds 15a–15c (B), showing similar binding conformations of the bis-THF moiety regardless of the position and minor differences at the Phe-Phe isostere, but major differences at the P2/P2′ amino acid moiety. The protease active site is shown in a ribbon representation, with bound inhibitors depicted as sticks.
In all complexes, the central hydroxyl group of the Phe-Phe core maintains the key hydrogen bonds with the side-chain carboxylates of the catalytic residues Asp25/Asp25′. Nevertheless, subtle differences in hydrogen bonding and vdW interactions were observed between the Phe-Phe cores in LPV and the hybrid compounds. Notably, one of the four water-mediated interactions with the flap residue Ile50 is consistently longer (2.9–3.4 Å) than what is observed in the LPV structure (2.7 Å) (Table S2). The two amide NH groups on each side of the Phe-Phe core could potentially interact with the backbone carbonyl of Gly27 and Gly27′, but in the LPV structure only the Gly27 is close enough to form a hydrogen bond (2.9 Å versus 3.9 Å) (Figure 3). This phenomenon is also observed in the protease complex with compound 4 (2.9 Å versus 3.7 Å) indicating that the large hydrophobic P2′ moiety prevents the additional hydrogen bond from occurring with Gly27′. In contrast, in all hybrid compound structures the core NH groups are within 3.5 Å from the backbone carbonyls of Gly27 and Gly27′ (Table S2), although simultaneous interactions with both Gly27/Gly27′ may not be stable. Nevertheless, the combination of bis-THF and a flexible, hydrophobic amino acid moiety allow both core NH groups to interact with the main-chain carbonyls of residues Gly27/Gly27′.
The Phe-Phe isostere in hybrid compounds, in addition to maintaining key hydrogen bonding interactions with the protease, makes a number of favorable vdW interactions with the hydrophobic residues that make up the S1/S1′ subsites. Despite the same phenylalanine side chains at the P1 and P1′ positions, there are a few notable differences in vdW contacts with the protease (Figure S2). Compared to LPV, the P1/P1′ phenyl group adjacent to the amino acid moiety is shifted towards Val82 resulting in improved vdW contacts with that residue, particularly for compounds with larger P2/P2′ amino acid moieties. These localized gains in vdW contacts are slightly lower for compounds 15a–c with bis-THF at the P2′ position than their corresponding analogues 12a–c. The shift of the P1/P1′ phenyl group towards Val82, creating more vdW contacts, resulted in a nearly equal and opposite loss of contacts with Ile50 in the same subsite. The packing of the phenyl group adjacent to the bis-THF moiety resulted in a small increase in vdW contacts with the 80’s loop (residues 81, 82 and 84) but decrease in contacts with flap residue Gly49. For compounds 15a–c, gains in vdW contacts are smaller and the losses are greater. Overall, despite localized losses and gains in vdW interactions for both series of compounds, the packing of the P1 and P1′ groups remain generally similar to LPV.
The cocrystal structures of hybrid compounds (and 4) also revealed similar binding interactions of the bis-THF moiety as in DRV-protease complex regardless of the position, with the two ring oxygen atoms positioned to form hydrogen bonds with the main-chain NH of Asp29 and Asp30. However, there are subtle differences in the position and puckering of the bis-THF rings, causing the carbamate-linked THF ring oxygen atom to slightly shift away from the backbone NH of Asp30 compared to DRV (3.1–3.4 Å versus 2.9 Å) (Figure 5). Whereas the oxygen atom of the other THF ring maintains a similar distance to the backbone NH of Asp29 (2.7–2.9 Å versus 2.8 Å) (Table S2). For compounds 12a–c, these subtle changes in the bis-THF moiety arise from the differences in the binding of core isosteres, where the P1 benzyl group and the carbamate are shifted towards the B-chain compared to their position in DRV. This is likely due to the additional carbon in the core or to accommodate the larger P1′ group. In compounds 15a–c, the core scaffold from the central hydroxyl to the bis-THF moiety is identical. However, the opposite stereochemistry of the central hydroxyl causes a subtle shift preventing the bis-THF moiety from optimally interacting with Asp30′ (Figure 5). Despite these minor differences in the position of the bis-THF moiety, the hybrid compounds make similar hydrogen bonding interactions with the protease in the S2/S2′ subsite compared to DRV.
Figure 5.

Comparison of the binding conformations of the bis-THF moiety and protease-inhibitor intramolecular distances with the main-chain NH of Asp29 and Asp30 in the S2/S2′ subsite of HIV-1 protease. As observed in the DRV-protease complex (PDB 6DGX), the bis-THF moiety retained similar interactions with the protease in complexes with compound 4 (PDB 6PJC) (top), the most potent hybrid compounds 12a (PDB 6PJD) and 15a (PBD 6PJM) (middle), and the least potent hybrid compounds 12b (PDB 6PJE) and 15b (PDB 6PJN) (bottom), irrespective of position on the Phe-Phe core.
The bis-THF moiety of DRV is critical to its potency, not only for the hydrogen bonding interactions with Asp29 and Asp30, but also vdW packing with residues in the S2 subsite, particularly Val32 and Ile47. In hybrid compounds, the minor change in the position and puckering of the bis-THF moiety causes reduced vdW interactions with Ala28 and Asp30, but increased interactions with Ile47 and Gly48 (Figure S3). The main-chain carbonyl of Gly48 is oriented towards the hydrophobic region of bis-THF, so enhanced contacts with this residue may be unfavorable. In contrast, compounds 4, 15a and 15b do not make these unfavorable contacts with Gly48. With the exception of enhanced interactions with Ile47, the bis-THF moiety in most hybrid compounds make less optimal vdW interactions with the protease compared to those of DRV.
The major difference between the hybrid compounds and the previously reported Phe-Phe core- based bis-THF containing PIs 4 and 5 is the introduction of an ATV-like amino acid moiety to target the opposite S2/S2′ subsite. The amino acid side chain interacts with hydrophobic protease residues, while the N-(methoxycarbonyl)-capping group allows for additional hydrogen bonding interactions with the protease. As already noted, LPV’s P2′ moiety does not form any hydrogen bonds, and DRV’s P2′ moiety makes one direct and one water-mediated interactions in the S2′ subsite (Figure S1). In most hybrid compounds, the carbonyl oxygen of the carbamate makes one direct hydrogen bond with the backbone NH of Asp29′ (Asp29), and the carbamate NH makes another direct hydrogen bond with the backbone carbonyl of Gly48′ (Gly48) in the flaps (Figure 6). There are also a number of coordinated water molecules that allow for additional water- mediated interactions. The carbonyl oxygen of the carbamate coordinates the same water-mediated interactions as the cyclic urea moiety of LPV, with the backbone carbonyl of Gly27′ (Gly27) and the side-chain carboxylate of Asp29′ (Asp29) (Figure 6). This network of polar interactions is maintained by all amino acid moieties in both the S2′ and S2 subsites except the cycloleucine in compound 12b.
Figure 6.

Comparison of protease-inhibitor direct hydrogen bonds and water-mediated interactions in the S2/S2′ subsites for the cyclic urea moiety of LPV (PDB 6PJB) (A), N- (methoxycarbonyl)-tert-leucine of ATV (PDB 3EKY) (B), and the N-(methoxycarbonyl)-capped amino acid moieties of hybrid compound 15a (PDB 6PJM) (C), 12a (PDB 6PJD) (D), 15b (PDB 6PJN) (E), and 12b (PDB 6PJE) (F). Polar interactions are shown as black dashed lines (as determined by PyMol) with waters shown as red spheres.
The carbamate of compound 12b is flipped relative to all other inhibitors losing the direct hydrogen bond to flap residue Gly48′ (Figure 6F). This flip positions the carbonyl oxygen of the carbamate close to the carbonyl oxygen of Gly48′ (3.3 Å), creating a very unfavorable interaction and preventing compound 12b from utilizing the same water-mediated interactions as LPV and most hybrid compounds. Instead 12b forms a hydrogen bond with a “backside” water in the S2′ subsite, creating a water-mediated interaction with the main-chain carbonyl and NH of Asp30′ (Figure 6F). The marked changes in the hydrogen bonding interactions in the S2/S2′ subsite likely underlie the greatly reduced potency of 12b compared to all other hybrid compounds. Unlike 12b, the P2 carbamate group in compound 15b maintained a similar conformation as in the tert-leucine analogue 15a but with a slight change in the position of the carbonyl oxygen away from Asp29. This shift resulted in increased hydrogen bond distance between the P2 carbamate carbonyl and the main-chain NH of Asp29 compared to 15a (3.3 Å versus 2.9 Å). However, the reduced potency of 15b could not result from minor differences in hydrogen bonding interactions alone, highlighting the significance of non-polar interactions for potency.
The protease-inhibitor vdW interactions in the S2/S2′ subsite varied significantly depending on the size and shape of the hydrophobic amino acid moiety (Figure S4). Compounds 12a and 15a with the tert-leucine make highly distributed vdW contacts with a number of residues. In contrast, compared to tert-leucine, the cycloleucine side chain in 12b predominantly interacts with Ile50 and I84V, while losing significant contacts with residues 28–30 (Figure 7). The cycloleucine side chain of 15b packs in the same hydrophobic area but makes less vdW contacts than 12b. Compound 12c with cyclopentyl glycine also has enhanced vdW contacts with Ile50 but without experiencing losses at other resides as observed for 12b. In general, the cyclic hydrophobic amino acid derivatives make more localized vdW interactions in the S2/S2′ subsite while the acyclic moieties make more distributed contacts. Thus, in addition to altered polar interactions, the differences in vdW contacts due to the size and shape of the hydrophobic amino acid moiety are likely responsible for the varied inhibitor potencies.
Figure 7.

Comparison of binding conformation (A) and van der Waals packing with residues in the S2′ subsite for the most and least potent inhibitors 12a (PDB 6PJD) (B, green sticks) and 12b (PDB 6PJE) (C, blue sticks). Compound 12a makes more extensive contacts with the protease as estimated by relative vdW contact energy compared to 12b (−91 versus −86 kcal/mol), with major difference occurring at the S2′ subsite (−27 versus −23 kcal/mol). Protease residues are colored red to blue for highest to lowest vdW interactions with the inhibitor.
Molecular Dynamics (MD) Simulations.
Starting from the cocrystal structures, molecular dynamics (MD) simulations were utilized to interrogate the stability of protease-inhibitor interactions in the dynamic ensemble of the complexes. Three replicates of fully hydrated 100 ns MD simulations were performed on each protease-inhibitor complex. All simulations reached convergence (Figure S5).
The protease active site and flaps dynamics were analyzed by measuring Cα distances between specific residues. The analysis focused on the most and least potent pairs of compounds containing bis-THF at either the P2 or P2′ position (12a/15a and 12b/15b) compared to LPV. The cocrystal structures suggested the flaps and the 80’s loops surrounding the inhibitor may be perturbed when bound to different compounds. To monitor dynamics of the flaps, distance distributions between Cα atoms of Ile50–Ile84′, Ile50′–Ile84 and Ile50–Ile50′ were calculated during the MD trajectories. Distances for Ile84–Ile84′ and Pro81–Pro81′ were used to probe the expansion or narrowing of the “lower” and “upper” side wall of the active site, respectively (Figure S6). In protease complexes with hybrid compounds, the active site remained largely unchanged in the “lower” part of the active site as indicated by a single, narrow distribution of Ile84–Ile84′ distance centered around 15.2 Å. However, compared to LPV the shift in the Pro81–Pro81′ distance distribution with compounds 12a, 15a, 12b, and 15b indicated the active site was narrower in the “upper” portion close to the flaps. In addition, the active site was asymmetrically longer (Ile50′–Ile84 or Ile50–Ile84′), sampling bimodal distance distributions, suggesting the sampling of a semi-open conformation of the flaps. This was supported by the Ile50–Ile50′ distance distribution, with one major peak around 5.3 Å (closed flaps) and another ~10 Å corresponding to semi-open flaps with compounds 15a and 15b (Figure S6). Increased distance between residues Ile50 and Ile50′ in the relatively short time (nanoseconds) during MD simulations has been previously suggested to indicate flap opening.34, 35 Thus, perturbed flaps dynamics and narrowing of the active site may indicate unfavorable binding of these compounds compared to LPV, which is supported by the Ki data.
Next, the stability of inhibitor binding was analyzed by calculating root-mean-square fluctuations (RMSF) of each atom (Figure 8). In all complexes, the bis-THF moiety often displayed the lowest RMSF amongst all moieties, regardless of its position on the Phe-Phe core. Notably, even in the least active compounds with nanomolar potency (12b and 15b), the bis-THF moiety showed fluctuations similar to that observed in 12a and 15a. Moreover, the phenylalanine side chain adjacent to the bis-THF moiety displayed lower RMSF compared to the distal phenylalanine side chain, despite the pseudo-symmetric nature of the Phe-Phe core. The P2/P2′ amino acid moieties showed varying RMSF profiles, independent of the size or shape of the hydrophobic side chain. The phenylalanine adjacent to the amino acid moiety consistently displayed greater RMSF, with the exception of 12e. The varying fluctuations of the P1/P1′ moiety depending on the size and shape of the P2/P2′ moiety indicate interdependence between the S1/S1′ and S2/S2′ subsites. The P2/P2′ amino acid moiety also affected the fluctuations of the bis-THF moiety but to a much lesser extent. This data suggests that in hybrid compounds the bis-THF moiety and the adjacent P1/P1′ moiety remain relatively stable in the protease active site regardless of the position on the Phe-Phe core. In contrast, the fluctuations of the amino acid and the adjacent P1/P1′ moieties appear to destabilize overall inhibitor binding. Thus, due to interdependence between the S1/S1′ and S2/S2′ subsites, the optimization of the bis-THF containing hybrid PIs, would require modification of the amino acid moiety together with the adjacent P1/P1′ group.
Figure 8.

Inhibitor root-mean-square fluctuation (RMSF) of each non-hydrogen atom of DRV, LPV, and hybrid compounds mapped onto their crystal structure. Warmer colors indicate larger fluctuations.
The relationship between inhibitor and protease conformational dynamics was examined via simultaneous monitoring of inhibitor bond rotations and protease residue distances. The dihedral angles of all inhibitor rotatable bonds were calculated throughout the MD simulations. Given the bimodal sampling of the flap distances (Ile50–Ile50′, Ile50–Ile84′ and Ile50′–Ile84), we examined whether the semi-open conformation of flaps was associated with certain inhibitor bond rotations. In protease complexes with compounds 12a and 15a, the two most potent inhibitors, the separation of the flap tips (Ile50–Ile50′) was associated with the conformational sampling of the dihedral controlling the amino acid moiety (Figure 9). In this semi-open flap conformation, the Ile50/Ile50′ residues at the top of the flaps also lost intra-protease vdW contacts with residues 32, 47–49 and 84 (Figure S7). Loss of intra-protease vdW interactions could result in a less stable protease- inhibitor complex, leading to an overall decrease in inhibitor binding affinity.
Figure 9.

The dihedral angle (ϕ5) responsible for conformational sampling of the tert-leucine moiety on 12a (A) and 15a (B) is associated with increased Ile50–Ile50′ distance sampling.
In addition, protein dynamics were compared by calculating the root-mean-square fluctuations (RMSF) of Cα atoms. Consistent with our observations from the distance distributions, there were significant increases in fluctuations at the flaps and 80’s loop (Figure S8). In complex with LPV, the flaps and active site show little fluctuations, comparable to DRV. However, compounds containing bis-THF at the P2′ position showed increased fluctuations at both flaps, with fluctuations asymmetrically greater at the B chain flap, contacting the amino acid moiety. The increased protein fluctuation could impact the stability of protease-inhibitor hydrogen bonding interactions.
In the cocrystal structures, the hybrid compounds make a number of direct and water-mediated interactions with the protease active site, including the central hydroxyl with catalytic residues, coordinated water with Ile50/50′, the Phe-Phe core nitrogen atoms with Gly27, bis-THF with Asp29 and Asp30, carbamate with Asp29 and Gly48, and the coordinated S3′ water with Gly27′ and Asp29’. To assess if the increased protein and ligand fluctuations impact hydrogen bonding stability, the patterns and frequencies of these hydrogen bonds were monitored throughout the MD simulations (Figure 10, Figure S9). In MD simulations, LPV and the hybrid compounds did not maintain all hydrogen bonds observed in the cocrystal structures. LPV and all compounds with bis-THF at the P2 position (4 and 12a–h) maintained a hydrogen bond with Asp25′ (66–99% frequency) but did not form a hydrogen bond with Asp25. Whereas compounds 15a and 15b containing bis-THF at the P2 position formed the hydrogen bonds with Asp25 and Asp25′ at roughly 100% and 50% frequency, respectively. In contrast, the hydrogen bond between compound 15c and Asp25 was unstable, while the hydrogen bond with Asp25′ was highly stable (92% frequency). Hybrid compounds maintained the network of water-mediated interactions with the main-chain NH of Ile50 and Ile50′ at varying stability. The interactions with Ile50 were observed to be less stable compared to the ones with Ile50′ (21–65% versus 49–78%). For most of the hybrid compounds, the hydrogen bonds between the Phe-Phe core NH groups and the main- chain carbonyls of Gly27/Gly27′ were either not observed or were very unstable.
Figure 10.

Protease-inhibitor hydrogen bond frequencies from MD simulations mapped onto the cocrystal structures of compounds 12a (A), 12b (B), 15a (C), and 15b (D). The protease side chains and inhibitors are shown as sticks; chains A and B are colored cyan and magenta, respectively.
The bis-THF moiety in all hybrid compounds maintained two hydrogen bonds with the main- chain NH of Asp29/Asp29′ and Asp30/Asp30′ at high frequency, regardless of the position on the Phe-Phe isostere (Figure 10, Figure S9). In contrast, the P2/P2′ amino acid moiety had varying effects on hydrogen bonding interactions in the S2/S2′ subsite. The carbamate nitrogen of hybrid compounds formed a hydrogen bond with the backbone carbonyl of Gly48′ at moderate to high frequency, but this interaction was not observed for the least potent compound 12b and was less stable for 15b. The carbonyl oxygen of the P2′ amino acid moiety in compounds 12a and 12c formed relatively stable water-mediated interactions with the sidechain of Asp30′. In contrast, the carbonyl oxygen of the P2 moiety in compounds 15a and 15c formed three low-frequency (14–39%) water-mediated interactions, bridging the main-chain carbonyl of Gly27 and the side-chain carboxylate of Asp29. These data suggest that for most hybrid compounds the key hydrogen bonding interactions of the bis-THF moiety and the Phe-Phe isostere with protease remain relatively stable. In addition, the size and shape of the P2/P2′ amino acid moiety not only influences the interactions in the S2/S2′ subsites but can also propagate changes in the water-mediate interactions between the inhibitors and Ile50/Ile50′.
CONCLUSIONS
We have explored hybrid HIV-1 PIs containing the bis-THF moiety of DRV on either side of the Phe-Phe dipeptide isostere of LPV to ascertain whether this moiety could maintain key interactions with the protease and improve potency against wild-type and primary drug-resistant variants of the enzyme. Most of the hybrid compounds retained picomolar biochemical potency irrespective of the position of bis-THF relative to the central hydroxyl group, likely due to the pseudo-C2-symmetric nature of the Phe-Phe isostere. But in both compound series the identity of the other P2/P2′ moiety significantly affected potency, favoring relatively flexible, hydrophobic moieties. The cocrystal structures of hybrid compounds revealed that the bis-THF moiety maintains the key hydrogen bonding interactions with the protease in the S2/S2′ subsite as observed in the DRV-protease complex regardless of its position on the Phe-Phe isostere. In contrast, the binding of the hydrophobic amino acid moiety in the other S2/S2′ subsite was greatly influenced by the size, shape, and flexibility of the hydrophobic group, which affected hydrogen bonding and vdW interactions with the protease. Moreover, this moiety appears to influence binding of the adjacent P1/P1′ group as well as the water-mediated interactions between the inhibitor and flap residues, indicating dynamic interdependence between the protease subsites. This is further supported by the relatively stable binding interactions of the bis-THF moiety and the adjacent P1/P1′ group. The dynamic interdependence between subsites in the protease active site can be exploited to optimize inhibitor potency against drug-resistant protease variants. The detailed structural characterization of hybrid HIV-1 PIs containing bis-THF in non-sulfonamide dipeptide isosteres offers opportunities for structure-guided optimization of these promising inhibitors.
EXPERIMENTAL SECTION
General.
All reactions were performed in oven-dried round-bottom flasks fitted with rubber septa under argon atmosphere unless otherwise noted. All reagents and solvents, including anhydrous solvents, were purchased from commercial sources and used as received. Flash column chromatography was performed on an automated Teledyne ISCO CombiFlash Rf+ system equipped with a UV-vis detector using disposable Redisep Gold high performance silica gel columns or was performed manually using silica gel (230–400 mesh, EMD Millipore). Thin- layer chromatography (TLC) was performed using silica gel (60 F254) coated aluminum plates (EMD Millipore), and spots were visualized by exposure to ultraviolet light (UV), exposure to iodine adsorbed on silica gel, and/or staining with alcohol solutions of phosphomolybdic acid (PMA) and ninhydrin followed by brief heating. 1H NMR and 13C NMR spectra were acquired on Varian Mercury 400 MHz and Bruker Avance III HD 500 MHz NMR instruments. Chemical shifts are reported in ppm (δ scale) with the residual solvent signal used as a reference and coupling constant (J) values are reported in hertz (Hz). Data are presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublet, dd = doublet of triplet, t = triplet, m = multiplet, br s = broad singlet), coupling constant in Hz, and integration. High-resolution mass spectra (HRMS) were recorded on a Thermo Scientific Orbitrap Velos Pro mass spectrometer coupled with a Thermo Scientific Accela 1250 UPLC and an autosampler using electrospray ionization (ESI) in the positive mode. The purity of final compounds was determined by analytical HPLC and was found to be ≥95% pure. HPLC was performed on a Agilent 1200 system equipped with a multiple wavelength detector and a manual injector under the following conditions: column, Phenomenex Hypersil-BDS-5u-C18 (5 μm, 4.6 mm × 250 mm, 130 Å); solvent A, H2O containing 0.1% trifluoroacetic acid (TFA); solvent B, CH3CN containing 0.1% TFA; gradient, 20% B to 100% B over 15 min followed by 100% B over 5 min; injection volume, 20 μL; flow rate, 1 mL/min. The wavelengths of detection were 254 nm and 280 nm. Retention times and purity data for each target compound are provided in the Experimental Section.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-5-(dibenzylamino)-4-hydroxy-1,6-diphenylhexan-2-yl)carbamate (9).
A solution of the Phe-Phe core intermediate 7 (3.00 g, 6.64 mmol) in anhydrous CH3CN (50 mL) was cooled to 0 °C and treated with diisopropylethylamine (DIEA) (3.37 mL, 19.35 mmol) followed by bis-THF activated carbonate 8 (1.93 g, 7.10 mmol). The reaction mixture was warmed to room temperature and stirred for 24 h, concentrated under reduced pressure and dried under high vacuum. The residue was purified by flash column chromatography (RediSep Gold, 80 g, gradient elution with 0–100% ethyl acetate/hexanes) to provide the intermediate 9 (4.01 g, 100%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.34–7.17 (m, 16 H), 7.10 (d, J = 7.5 Hz, 2 H), 7.01 (d, J = 7.0 Hz, 2 H), 5.69 (d, J = 5.0 Hz, 1 H), 5.29 (d, J = 6.0 Hz, 1 H), 5.12 (q, J = 6.5 Hz, 1 H), 4.52 (br s, 1 H), 4.02 (dd, J = 9.0, 7.0 Hz, 1 H), 3.95–3.73 (m, 6 H), 3.65–3.58 (m, 1 H), 3.38 (d, J = 13.0 Hz, 2 H), 3.10 (dd, J = 14.0, 5.5 Hz, 1 H), 3.04–2.97 (m, 1 H), 2.85–2.76 (m, 2 H), 2.66 (dd, J = 13.0, 6.5 Hz, 1 H), 2.54 (dd, J = 14.5, 7.0 Hz, 1 H), 1.87–1.75 (m, 2 H), 1.52 (dd, J = 14.5, 3.5 Hz, 1 H), 1.11 (dt, J = 14.5, 8.5 Hz, 1 H) ppm; 13C NMR (125 MHz, CDCl3) δ 155.41, 140.15, 138.74, 138.18, 129.61, 129.15, 128.86, 128.69, 128.40, 127.53, 126.47, 126.40, 109.48, 73.00, 71.03, 69.77, 69.74, 64.31, 53.95, 52.28, 45.49, 41.35, 38.15, 32.15, 26.09 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C39H45N2O5, 621.3323; found 621.3306.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-4-hydroxy-5-((S)-2-((methoxycarbonyl)amino)-3,3-dimethylbutanamido)-1,6-diphenylhexan-2-yl)carbamate (12a).
A solution of compound 9 (0.50 g, 0.81 mmol) in anhydrous MeOH (10 mL) was treated with ammonium formate (0.30 g, 4.83 mmol) and 10% Pd/C (0.1 g) and the reaction mixture was stirred at 50 °C for 14 h. The reaction mixture was filtered through a pad of Celite and the filtrate was concentrated under reduced pressure. The resulting deprotected amine 10 was dissolved in anhydrous CH2Cl2 (10 mL) and the solution was cooled to 0 °C. N-(methoxycarbonyl)-L-tert- leucine 11a (0.18 g, 0.97 mmol), DIEA (0.280 ml, 1.61 mmol) and HATU (0.48 g, 1.26 mmol) were added. The reaction mixture was stirred at room temperature for 16 h, concentrated under reduced pressure, and dried under high vacuum. The residue was purified by flash column chromatography (RediSep Gold, 24 g, gradient elution with 0–15% methanol/dichloromethane) to provide the target compound 12a (0.27 g, 55%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.25–7.13 (m, 8 H), 7.07 (d, J = 7.0 Hz, 2 H), 6.57 (d, J = 9.0 Hz, 1 H), 5.65 (d, J = 5.0 Hz, 1 H), (d, J = 9.0 Hz, 1 H), 5.12 (d, J = 8.0 Hz, 1 H), 5.04 (q, J = 6.5 Hz, 1 H), 4.15 (q, J = 7.0 Hz, 1 H), 4.06–3.96 (m, 1 H), 3.93 (dd, J = 9.5, 6.5 Hz, 1 H), 3.87–3.63 (m, 4 H), 3.66 (s, 3 H, overlapping), 2.96–2.81 (m, 3 H), 2.80 (s, 2 H), 2.66 (dd, J = 14.0, 7.5 Hz, 1 H), 1.74–1.55 (m, 4 H), 0.93 (s, 9 H) ppm; 13C NMR (125 MHz, CDCl3) δ 170.93, 157.18, 155.58, 137.95, 137.39, 129.39, 128.67, 128.60, 126.82, 126.60, 109.44, 73.47, 70.97, 69.74, 69.67, 63.44, 54.83, 52.59, 50.95, 45.51, 41.72, 40.19, 38.35, 34.21, 26.71, 26.02 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H46N3O8, 612.3280; found 612.3276; Anal. HPLC: tR 10.86 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-4-hydroxy-5-(1-((methoxycarbonyl)amino)cyclopentane-1-carboxamido)-1,6-diphenylhexan-2-yl)carbamate (12b).
The same procedure was used as described above for compound 12a. Compound 9 (1.00 g, 1.61 mmol) was treated with ammonium formate (0.61 g, 9.66 mmol) and 10% Pd/C (0.25 g) to give the corresponding deprotected amine 10, which was coupled with N-(methoxycarbonyl)-cycloleucine 11b (0.36 g, 1.93 mmol) using DIEA (1.12 ml, 6.44 mmol) and HATU (0.96 g, 2.51 mmol) to provide the target compound 12b (0.66 g, 67%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.28–7.22 (m, 4 H), 7.21–7.16 (m, 4 H), 7.14 (d, J = 7.0 Hz, 2 H), 6.61 (d, J = 8.5 Hz, 1 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.07 (app q, J = 6.5 Hz, 2 H), 4.96 (br s, 1 H), 4.07 (q, J = 7.5 Hz, 1 H), 4.02–3.93 (m, 2 H), 3.90–3.84 (m, 1 H), 3.76–3.65 (m, 3 H), 3.63 (s, 3 H), 3.35 (br s, 1 H), 2.99–2.93 (m, 1 H), 2.90–2.84 (m, 2 H), 2.83 (dd, J = 14.5, 7.0 Hz, 1 H, overlapping), 2.75 (dd, J = 12.5, 7.0 Hz, 1 H), 2.27–2.18 (m, 1 H), 2.00–1.91 (m, 1 H), 1.86–1.59 (m, 10 H) ppm; 13C NMR (125 MHz, CDCl3) δ 174.05, 156.34, 155.58, 138.29, 137.85, 129.53, 129.34, 128.57, 126.64, 126.55, 109.45, 73.31, 70.91, 69.74, 67.49, 55.07, 52.53, 51.16, 45.50, 41.84, 39.19, 37.80, 36.60, 26.00, 24.11 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H44N3O8, 610.3123; found 610.3117; Anal. HPLC: tR 9.74 min, purity 96%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-5-((S)-2-cyclopentyl-2-((methoxycarbonyl)amino)acetamido)-4-hydroxy-1,6-diphenylhexan-2-yl)carbamate (12c).
The same procedure was used as described above for compound 12a. Compound 9 (0.50 g, 0.81 mmol) was treated with ammonium formate (0.30 g, 4.83 mmol) and 10% Pd/C (0.13 g) to give the corresponding deprotected amine 10, which was coupled with N-(methoxycarbonyl)-L-cyclopentylglycine 11c (0.19 g, 0.97 mmol) using DIEA (0.566 ml, 3.22 mmol) and HATU (0.48 g, 1.26 mmol) to provide the target compound 12c (0.32 g, 64%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.22 (m, 4 H), 7.21–7.16 (m, 4 H), 7.11 (d, J = 7.5 Hz, 2 H), 6.35 (d, J = 6.5 Hz, 1 H), 5.67 (d, J = 5.0 Hz, 1 H), 5.12 (d, J = 7.5 Hz, 1 H), 5.07 (q, J = 6.5 Hz, 1 H), 4.94 (d, J = 8.5 Hz, 1 H), 4.11 (app q, J = 8.5 Hz, 1 H), 4.03–3.93 (m, 2 H), 3.92–3.83 (m, 2 H), 3.77–3.66 (m, 3 H), 3.67 (s, 3 H, overlapping), 3.30 (br s, 1 H), 2.99–2.93 (m, 1 H), 2.91–2.85 (m, 2 H), 2.82 (dd, J = 14.5, 6.5 Hz, 1 H), 2.70 (dd, J = 13.5, 7.5 Hz, 1 H), 2.22–2.11 (m, 1 H), 1.76–1.46 (m, 10 H), 1.27–1.14 (m, 2 H) ppm; 13C NMR (125 MHz, CDCl3) δ 172.09, 157.16, 155.62, 138.05, 137.55, 129.43, 129.36, 128.66, 128.62, 126.80, 126.62, 109.46, 73.47, 70.93, 70.15, 69.73, 59.47, 54.91, 52.63, 51.02, 45.53, 41.94, 41.81, 39.98, 38.17, 29.59, 28.73, 26.00, 25.51, 25.28 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H46N3O8, 624.3280; found 624.3273; Anal. HPLC: tR 10.98 min, purity 96%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-4-hydroxy-5-((S)-2-((methoxycarbonyl)amino)propanamido)-1,6-diphenylhexan-2-yl)carbamate (12d).
The same procedure was used as described above for compound 12a. Compound 9 (0.45 g, 0.73 mmol) was treated with ammonium formate (0.28 g, 4.36 mmol) and 10% Pd/C (0.11 g) to give the corresponding deprotected amine 10, which was coupled with N-(methoxycarbonyl)-L-alanine 11d (0.14 g, 0.95 mmol) using DIEA (0.377 ml, 2.18 mmol) and HATU (0.47 g, 1.23 mmol) to provide the target compound 12d (0.20 g, 47%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.28–7.22 (m, 4 H), 7.21–7.16 (m, 4 H), 7.11 (d, J = 7.5 Hz, 2 H), 6.39 (d, J = 9.0 Hz, 1 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.17–5.09 (m, 1 H), 5.06 (q, J = 6.0 Hz, 1 H), 4.99 (d, J = 8.0 Hz, 1 H), 4.16–4.03 (m, 2 H), 3.99–3.91 (m, 2 H), 3.85 (td, J = 8.0, 2.0 Hz, 1 H), 3.77–3.65 (m, 3 H), 3.67 (s, 3 H, overlapping), 2.97–2.91 (m, 1 H), 2.88 (d, J = 7.5 Hz, 2 H), 2.81 (dd, J = 13.5, 5.5 Hz, 1 H), 2.71 (dd, J = 14.0, 8.0 Hz, 1 H), 1.75–1.58 (m, 4 H), 1.25 (d, J = 7.0 Hz, 3 H) ppm; 13C NMR (125 MHz, CDCl3) δ 172.73, 156.74, 155.66, 138.05, 137.61, 129.44, 129.36, 128.65, 126.79, 126.66, 109.45, 73.48, 70.96, 70.03, 69.73, 54.92, 52.61, 51.01, 45.54, 41.82, 39.87, 37.99, 26.00, 18.76 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C30H40N3O8, 570.2810; found 570.2811.; Anal. HPLC: tR 9.14 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-4-hydroxy-5-((S)-2-((methoxycarbonyl)amino)-3-methylbutanamido)-1,6-diphenylhexan-2-yl)carbamate (12e).
The same procedure was used as described above for compound 12a. Compound 9 (0.44 g, 0.70 mmol) was treated with ammonium formate (0.27 g, 4.22 mmol) and 10% Pd/C (0.11 g) to give the corresponding deprotected amine 10, which was coupled with N-(methoxycarbonyl)-L-valine 11e (0.15 g, 0.84 mmol) using DIEA (0.364 ml, 2.11 mmol) and HATU (0.42 g, 1.10 mmol) to provide the target compound 12e (0.29 g, 70%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.23 (m, 4 H), 7.22–7.17 (m, 4 H), 7.10 (d, J = 7.5 Hz, 2 H), 6.30 (d, J = 9.0 Hz, 1 H), 5.67 (d, J = 5.0 Hz, 1 H), 5.12–5.04 (m, 2 H), 4.93 (d, J = 8.5 Hz, 1 H), 4.12 (q, J = 7.5 Hz, 1 H), 4.01–3.93 (m, 2 H), 3.92–3.83 (m, 2 H), 3.75–3.67 (m, 3 H), 3.68 (s, 3 H, overlapping), 3.26 (br s, 1 H), 2.99–2.93 (m, 1 H), 2.91–2.84 (m, 2 H), 2.81 (dd, J = 14.5, 6.5 Hz, 1 H), 2.70 (dd, J = 13.5, 7.5 Hz, 1 H), 2.15–2.05 (m, 1 H), 1.77–1.57 (m, 4 H), 0.90 (d, J = 7.0 Hz, 3 H), 0.78 (d, J = 6.5 Hz, 3 H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.58, 157.28, 155.67, 138.00, 137.49, 129.42, 129.35, 128.67, 126.82, 126.66, 109.45, 73.50, 70.93, 70.12, 69.72, 61.01, 54.88, 52.67, 51.09, 45.52, 41.86, 40.04, 38.26, 30.50, 26.01, 19.54, 17.54 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C32H44N3O8, 598.3123; found 598.3122; Anal. HPLC: tR 10.23 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-5-((S)-2-cyclopropyl-2-((methoxycarbonyl)amino)acetamido)-4-hydroxy-1,6-diphenylhexan-2-yl)carbamate (12f).
The same procedure was used as described above for compound 12a. Compound 9 (0.46 g, 0.75 mmol) was treated with ammonium formate (0.28 g, 4.49 mmol) and 10% Pd/C (0.12 g) to give the corresponding deprotected amine 10, which was coupled with N-(methoxycarbonyl)-L-cyclopropylglycine 11f (0.18 g, 1.05 mmol) using DIEA (0.390 ml, 2.25 mmol) and HATU (0.42 g, 1.05 mmol) to provide the target compound 12f (0.27 g, 61%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.22 (m, 4 H), 7.21–7.15 (m, 4 H, overlapping), 7.10 (d, J = 7.5 Hz, 2 H), (d, J = 7.5 Hz, 1 H), 5.66 (d, J = 5.0 Hz, 1 H), 5.49 (d, J = 6.5 Hz, 1 H), 5.10–5.02 (m, 2 H), 4.10 (app, q, J = 8.0 Hz, 1 H), 4.02–3.91 (m, 2 H), 3.89–3.82 (m, 1 H), 3.78–3.64 (m, 3 H), 3.65 (s, 3 H, overlapping), 3.54–3.47 (br s, 1 H), 3.46–3.39 (m, 1 H), 2.98–2.91 (m, 1 H), 2.89 (d, J = 7.5Hz, 2 H), 2.81 (dd, J = 14.0, 6.0 Hz, 1 H), 2.70 (dd, J = 13.5, 7.5 Hz, 1 H), 1.75–1.57 (m, 4 H), 0.96 (br s, 1 H), 0.55–0.44 (m, 2 H), 0.43–0.33 (m, 2 H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.65, 156.96, 155.58, 138.08, 137.65, 129.44, 129.36, 128.62, 128.59, 126.72, 126.62, 109.43, 73.41, 70.94, 69.70, 59.32, 54.96, 52.56, 50.82, 45.54, 41.63, 39.78, 37.95, 25.98, 14.59, 3.68, 3.40 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C32H42N3O8, 596.2967; found 596.2957; Anal. HPLC: tR 10.54 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-4-hydroxy-5-((2S,3S)-2-((methoxycarbonyl)amino)-3-methylpentanamido)-1,6-diphenylhexan-2-yl)carbamate (12g).
The same procedure was used as described above for compound 12a. Compound 9 (0.44 g, 0.70 mmol) was treated with ammonium formate (0.27 g, 4.22 mmol) and 10% Pd/C (0.11 g) to give the corresponding deprotected amine 10, which was coupled with N-(methoxycarbonyl)-L-isoleucine 11g (0.15 g, 0.84 mmol) using DIEA (0.364 ml, 2.11 mmol) and HATU (0.42 g, 1.10 mmol) to provide the target compound 12g (0.29 g, 40%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.23 (m, 4 H), 7.22–7.17 (m, 4 H), 7.11 (d, J = 7.5 Hz, 2 H), 6.26 (d, J = 9.0 Hz, 1 H), 5.67 (d, J = 5.5 Hz, 1 H), 5.07 (q, J = 6.5 Hz, 1 H), 5.02 (d, J = 8.0 Hz, 1 H), 4.89 (d, J = 8.0 Hz, 1 H), 4.13 (q, J = 7.0 Hz, 1 H), 4.00–3.90 (m, 3 H), 3.89–3.84 (m, 1 H), 3.75–3.67 (m, 3 H), 3.68 (s, 3 H, overlapping), 3.16 (br s, 1 H), 2.99–2.93 (m, 1 H), 2.91–2.84 (m, 2 H), 2.81 (dd, J = 15.5, 7.5 Hz, 1 H), 2.71 (dd, J = 14.0, 8.0 Hz, 1 H), 1.90–1.80 (m, 1 H), 1.77–1.56 (m, 4 H), 1.33–22 (m, 1 H), 1.03–0.93 (m, 1 H), 0.87 (d, J = 7.0 Hz, 3 H), 0.84 (t, J = 7.5 Hz, 3 H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.58, 157.21, 155.66, 137.99, 137.51, 129.42, 129.36, 128.68, 128.64, 126.82, 126.64, 109.45, 73.49, 70.91, 70.24, 69.73, 60.40, 56.02, 54.80, 52.67, 51.10, 45.52, 43.96, 41.90, 40.06, 38.25, 36.82, 26.00, 24.48, 18.78, 17.40, 15.85, 12.65, 11.57 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H46N3O8, 612.3280; found 612.3275; Anal. HPLC: tR 10.82 min, purity 99%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,4S,5S)-4-hydroxy-5-((2S,3R)-2-((methoxycarbonyl)amino)-3-methylpentanamido)-1,6-diphenylhexan-2-yl)carbamate (12h).
The same procedure was used as described above for compound 12a. Compound 9 (0.53 g, 0.86 mmol) was treated with ammonium formate (0.33 g, 5.17 mmol) and 10% Pd/C (0.13 g) to give the corresponding deprotected amine 10, which was coupled with N-(methoxycarbonyl)-L-alloisoleucine 11h (0.15 g, 0.84 mmol) using DIEA (0.450 ml, 2.59 mmol) and HATU (0.59 g, 1.55 mmol) to provide the target compound 12h (0.30 g, 58%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.23 (m, 4 H), 7.22–7.17 (m, 4 H), 7.11 (d, J = 7.0 Hz, 2 H), 6.28 (d, J = 9.0 Hz, 1 H), 5.67 (d, J = 5.5 Hz, 1 H), 5.07 (q, J = 6.5 Hz, 1 H), 4.96 (d, J = 7.5 Hz, 1 H), 4.93 (d, J = 8.0 Hz, 1 H), 4.16–4.09 (m, 1 H), 4.07 (dd, J = 7.0, 4.5 Hz, 1 H), 3.97 (dd, J = 9.5, 6.5 Hz, 2 H), 3.90–3.84 (m, 1 H), 3.75–3.67 (m, 3 H), 3.69 (s, 3 H, overlapping), 2.99–2.93 (m, 1 H), 2.90–2.84 (m, 2 H), 2.81 (dd, J = 15.5, 7.5 Hz, 1 H), 2.72 (dd, J = 14.0, 8.0 Hz, 1 H), 1.96–1.88 (m, 1 H), 1.76–1.57 (m, 4 H), 1.40–1.30 (m, 1 H), 1.21–1.11 (m, 1 H), 0.90 (t, J = 7.0 Hz, 3 H), 0.67 (d, J = 6.5 Hz, 3 H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.84, 157.29, 155.66, 138.02, 137.54, 129.42, 129.34, 128.67, 126.81, 126.67, 109.45, 73.48, 70.89, 70.36, 69.73, 59.12, 54.88, 52.71, 51.16, 45.52, 41.96, 39.95, 38.22, 36.82, 26.52, 26.00, 14.17, 11.80 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H46N3O8, 612.3280; found 612.3277; Anal. HPLC: tR 10.87 min, purity 95%.
Methyl ((S)-1-(((2S,4S,5S)-5-(dibenzylamino)-4-hydroxy-1,6-diphenylhexan-2-yl)amino)-3,3-dimethyl-1-oxobutan-2-yl)carbamate (13a).
A solution of the Phe-Phe core intermediate 7 (1.00 g, 2.16 mmol) in DMF (15 mL) was cooled to 0 °C and treated with N-(methoxycarbonyl)-L-tert-leucine 11a (0.48 g, 2.58 mmol), DIEA (1.1 ml, 6.30 mmol) and HATU (1.28 g, 3.36 mmol). The resulting reaction mixture was stirred at room temperature for 4 h, concentrated under reduced pressure, and dried under high vacuum. The residue was purified by flash column chromatography (RediSep Gold, 24 g, gradient elution with 0–10% methanol/dichloromethane) to give the intermediate 13a (1.20 g, 87%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.34–7.29 (m, 4 H), 7.28–7.23 (m, 4 H), 7.22–7.14 (m, 8 H), 7.06 (d, J = 7.5 Hz, 2 H), 7.00 (d, J = 6.5 Hz, 2 H), (d, J = 6.0 Hz, 1 H), 5.42 (d, J = 9.0 Hz, 1 H), 4.65 (br s, 1 H), 4.06–3.97 (m, 1 H), 3.91–3.79 (m, 3 H), 3.66 (s, 3 H), 3.53 (t, J = 8.5 Hz, 1 H), 3.36 (d, J = 13.5 Hz, 2 H), 3.06 (dd, J = 14.5, 5.5 Hz, 1 H), 2.92 (dd, J = 13.5, 5.0 Hz, 1 H), 2.76 (q, J = 7.0 Hz, 1 H), 2.59–2.46 (m, 2 H), 1.50 (dd, J = 14.5, 5.0 Hz, 1 H), 1.17–1.08 (m, 1 H), 0.99 (s, 9 H) ppm; 13C NMR (125 MHz, CDCl3) δ 170.57, 157.02, 140.03, 138.77, 138.10, 129.54, 129.15, 129.06, 128.80, 128.64, 128.36, 127.49, 126.40, 126.29, 69.83, 64.22, 63.31, 53.90, 52.34, 51.54, 41.12, 37.42, 34.78, 32.01, 26.60 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C40H50N3O4, 636.3796; found 636.3793.
Methyl (1-(((2S,4S,5S)-5-(dibenzylamino)-4-hydroxy-1,6-diphenylhexan-2-yl)carbamoyl)cyclopentyl)carbamate (13b).
The same procedure was used as described above for 13a. The Phe-Phe core intermediate 7 (1.00 g, 2.16 mmol) was coupled with N-(methoxycarbonyl)-cycloleucine 11b (0.47 g, 2.50 mmol) using DIEA (1.1 ml, 6.30 mmol) and HATU (1.21 g, 3.18 mmol) to provide 13b (1.30 g, 95%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.33–7.27 (m, 4 H), 7.26–7.15 (m, 12 H), 7.11 (d, J = 7.5 Hz, 2 H), 7.04 (d, J = 7.0 Hz, 2 H), 6.80 (br s, 1 H), 5.00 (br s, 1 H), 4.32 (br s, 1 H), 4.04 (m, 1 H), 3.93 (d, J = 13.0 Hz, 2 H), 3.55 (s, 4 H), 3.39 (d, J = 13.0 Hz, 2 H), 3.05 (dd, J = 13.5, 5.0 Hz, 1 H), 2.81 (dd, J = 13.5, 5.0 Hz 1 H), 2.74 (app q, J = 7.5 Hz, 1 H), 2.70–2.59 (m, 2 H), 2.28–2.17 (m, 1 H), 2.12–2.04 (m, 1 H), 1.93–1.78 (m, 1 H), 1.77–1.65 (m, 4 H), 1.51 (dd, J = 14.5, 3.5 Hz, 1 H), 1.35–1.24 (m, 1 H) ppm; 13C NMR (125 MHz, CDCl3) δ 173.64, 156.09, 140.40, 139.32, 138.38, 129.69, 129.27, 129.10, 128.74, 128.57, 128.30, 127.33, 126.28, 70.03, 67.31, 64.31, 54.27, 52.20, 50.93, 41.33, 38.17, 37.23, 36.65, 31.65, 24.30, 24.26 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C40H48N3O4, 634.3640; found 634.3615.
Methyl((S)-1-cyclopentyl-2-(((2S,4S,5S)-5-(dibenzylamino)-4-hydroxy-1,6-diphenylhexan-2-yl)amino)-2-oxoethyl)carbamate (13c).
The same procedure was used as described above for 13a. The Phe-Phe core intermediate 7 (1.04 g, 2.23 mmol) was coupled with N-(methoxycarbonyl)-cyclopentylglycine 11c (0.45 g, 2.23 mmol) using DIEA (1.17 ml, 6.69 mmol) and HATU (1.02 g, 2.68 mmol) to give 13c (1.30 g, 90%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.34–7.28 (m, 4 H), 7.27–7.22 (m, 4 H), 7.21–7.14 (m, 8 H), 7.06 (d, J = 7.0 Hz, 2 H), 7.00 (d, J = 6.5 Hz, 2 H), 6.57 (d, J = 5.5 Hz, 1 H), 5.22 (d, J = 8.0 Hz, 1 H), 4.66 (br s, 1 H), 4.06–3.98 (m, 1 H), 3.97–3.90 (m, 1 H), 3.86 (d, J = 13.0 Hz, 2 H), 3.64 (s, 3 H), 3.53 (t, J = 8.5 Hz, 1 H), 3.36 (d, J = 13.0 Hz, 2 H), 3.05 (dd, J = 14.5, 5.5 Hz, 1 H), 2.88 (dd, J = 13.5, 5.0 Hz, 1 H), 2.76 (q, J = 6.5 Hz, 1 H), 2.59–2.47 (m, 2 H), 2.23 (app q, J = 8.0 Hz, 1 H), 1.74–1.54 (m, 4 H), 1.53–1.43 (m, 3 H), 1.40–1.22 (m, 2 H), 1.15–1.06 (m, 1 H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.64, 157.03, 140.10, 138.79, 138.14, 129.56, 129.16, 129.11, 128.83, 128.66, 128.38, 127.51, 126.42, 126.33, 70.00, 64.20, 58.96, 53.88, 52.38, 51.53, 42.75, 41.40, 37.59, 32.05, 29.29, 28.55, 25.46, 25.20 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C41H50N3O4, 648.3796; found 648.3795.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3S,5S)-3-hydroxy-5-((S)-2-((methoxycarbonyl)amino)-3,3-dimethylbutanamido)-1,6-diphenylhexan-2-yl)carbamate (15a).
A solution of intermediate 13a (1.20 g, 1.89 mmol) in anhydrous MeOH (15 mL) was treated with ammonium formate (0.71 g, 11.32 mmol) and 10% Pd/C (0.3 g). The resulting reaction mixture was stirred at 50 °C for 15 h, cooled to room temperature, filtered through a pad of Celite, and the filtrate was concentrated under reduced pressure. The resulting deprotected amine was dissolved in acetonitrile (10 mL) and the solution was cooled to 0 °C. DIEA (0.98 ml, 5.66 mmol) was added followed by bis-THF activated carbonate 8 (0.56 g, 2.08 mmol). The reaction mixture was warmed up to room temperature and stirred for 24 h. The solvents were removed under reduced pressure, and the residue was purified by automated flash chromatography using a silica gel column (RediSep Gold, 40 g, gradient elution with 0–15% methanol/dichloromethane) to provide the target compound 15a (0.36 g, 31%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.22 (m, 4 H), 7.21–7.16 (m, 4 H), 7.10 (d, J = 7.0 Hz, 2 H), 6.07 (d, J = 6.0 Hz, 1 H), 5.67 (d, J = 5.0 Hz, 1 H), 5.36–5.22 (m, 1 H), 5.10 (d, J = 9.5 Hz, 1 H), 5.05 (q, J = 6.5 Hz, 1 H), 4.22–4.13 (m, 1 H), 3.98 (dd, J = 9.5, 6.5 Hz, 1 H), 3.87 (td, J = 8.5, 3.0 Hz, 1 H), 3.82–3.58 (m, 6 H), 3.67 (s, 3 H, overlapping), 2.98–2.90 (m, 1 H), 2.88–2.73 (m, 4 H), 1.73–1.57 (m, 4 H), 0.91 (s, 9 H) ppm; 13C NMR (125 MHz, CDCl3) δ 171.09, 157.19, 155.83, 138.13, 137.12, 129.41, 129.32, 128.73, 128.64, 126.91, 126.61, 109.47, 73.39, 70.98, 70.35, 69.78, 63.35, 56.60, 52.65, 49.38, 45.49, 41.43, 40.37, 38.87, 34.35, 26.65, 25.98 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H46N3O8, 612.3280; found 612.3278; Anal. HPLC: tR 10.70 min, purity 97%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3S,5S)-3-hydroxy-5-(1-((methoxycarbonyl)amino)cyclopentane-1-carboxamido)-1,6-diphenylhexan-2-yl)carbamate (15b).
The same procedure was used as described above for 15a. Compound 13b (0.70 g, 1.10 mmol) was treated with ammonium formate (0.42 g, 6.63 mmol) and 10% Pd/C (0.17 g) to give the corresponding deprotected amine, which was subsequently treated with DIEA (0.380 ml, 2.20 mmol) and bis-THF activated carbonate 8 (0.33 g, 1.21 mmol) to provide the target compound 15b (0.23 g, 35%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.22 (m, 4 H), 7.21–7.16 (m, 4 H), 7.11 (d, J = 7.0 Hz, 2 H), 6.49 (d, J = 7.0 Hz, 1 H), 5.67 (d, J = 5.5 Hz, 1 H), 5.14 (d, J = 9.5 Hz, 1 H), 5.07 (q, J = 6.5 Hz, 1 H), 4.97 (s, 1 H), 4.23 (br s, 1 H), 3.99 (dd, J = 9.5, 6.5 Hz, 1 H ), 3.94 (br s, 1 H), 3.88 (td, J = 8.0, 2.0 Hz, 1 H ), 3.85–3.69 (m, 4 H), 3.65 (s, 3 H), 2.98–2.91 (m, 1 H), 2.89 (dd, J = 14.0, 7.5 Hz, 1 H, overlapping ), 2.83 (dd, J = 15.5, 8.0 Hz, 2 H ), 2.71 (dd, J = 13.5, 7.5 Hz, 1 H ), 2.19–2.11 (m, 1 H), 1.94–1.59 (m, 11 H) ppm; 13C NMR (125 MHz, CDCl3) δ 173.71, 156.67, 155.77, 138.34, 137.62, 129.52, 129.40, 128.56, 126.73, 126.49, 109.51, 73.29, 71.11, 70.40, 69.81, 67.39, 57.24, 52.62, 49.96, 45.50, 41.98, 40.64, 39.01, 37.14, 36.65, 26.03, 24.01, 23.81 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C33H44N3O8, 610.3123; found 610.3118; Anal. HPLC: tR 7.73 min, purity 95%.
(3R,3aS,6aR)-Hexahydrofuro[2,3-b]furan-3-yl ((2S,3S,5S)-5-((S)-2-cyclopentyl-2-((methoxycarbonyl)amino)acetamido)-3-hydroxy-1,6-diphenylhexan-2-yl)carbamate (15c).
The same procedure was used as described above for 15a. Compound 13c (0.70 g, 1.08 mmol) was treated with ammonium formate (0.41 g, 6.48 mmol) and 10% Pd/C (0.17 g) to give the corresponding deprotected amine, which was subsequently treated with DIEA (0.570 ml, 3.24 mmol) and bis-THF activated carbonate 8 (0.32 g, 1.19 mmol) to provide the target compound 15c (0.26 g, 39%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.29–7.22 (m, 4 H), 7.21–7.16 (m, 4 H), 7.11 (d, J = 7.5 Hz, 2 H), 6.29 (br s, 1 H), 5.67 (d, J = 5.0 Hz, 1 H), 5.19–5.07 (m, 2 H), 5.05 (q, J = 6.0 Hz, 1 H, overlapping), 4.24–4.15 (m, 1 H), 3.98 (dd, J = 9.5, 6.0 Hz, 1 H), 3.87 (td, J = 8.0, 2.0 Hz, 1 H), 3.85–3.78 (m, 2 H, overlapping), 3.77–3.63 (m, 3 H), 3.67 (s, 3 H, overlapping), 2.99–2.91 (m, 1 H), 2.90–2.71 (m, 4 H), 2.22–2.09 (m, 1 H), 1.83–1.45 (m, 10 H), 1.22–1.07 (m, 2 H) ppm; 13C NMR (125 MHz, CDCl3) δ 172.11, 157.29, 155.83, 138.24, 137.34, 129.45, 129.34, 128.63, 128.60, 126.80, 126.56, 109.48, 73.36, 71.00, 70.33, 69.79, 59.46, 56.68, 52.71, 49.33, 45.54, 41.70, 41.46, 40.51, 38.92, 29.48, 28.71, 25.97, 25.47, 25.22 ppm; HRMS (ESI) m/z: [M + H]+ calcd for C34H46N3O8, 624.3279; found 624.3282; Anal. HPLC: tR 10.83 min, purity 99%.
Protease Gene Construction.
Protease gene construction was carried out as previously described.36, 37 The NL4-3 strain has four naturally occurring polymorphisms in the protease relative to the SF2 strain. In short, the protease variant genes (I84V, I50V/A71V) were constructed using QuikChange site-directed mutagenesis (Genewiz) onto NL4-3 wild-type protease on a pET11a plasmid with codon optimization for protein expression in Escherichia coli. A Q7K mutation was included to prevent autoproteolysis.38
Protein Expression and Purification.
The expression, isolation, and purification of WT and mutant HIV-1 proteases used for the kinetic assays and crystallization were carried out as previously described.36, 37 Briefly, the gene encoding the HIV protease was subcloned into the heat-inducible pXC35 expression vector (ATCC) and transformed into E. coli TAP-106 cells. Cells grown in 6 L of Terrific Broth were lysed with a cell disruptor and the protein was purified from inclusion bodies.39 The inclusion body centrifugation pellet was dissolved in 50% acetic acid followed by another round of centrifugation to remove impurities. Size exclusion chromatography was used to separate high molecular weight proteins from the desired protease. This was carried out on a 2.1 L Sephadex G-75 superfine column (Millipore Sigma) equilibrated with 50% acetic acid. The cleanest fractions of HIV protease were refolded into a 10-fold dilution of 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Folded protein was concentrated down to 1–2 mg/mL and stored. This stored protease was used in Ki assays. For crystallography, a final purification was performed with a Pharmacia Superdex 75 FPLC column equilibrated with 0.05 M sodium acetate at pH 5.5, 5% ethylene glycol, 10% glycerol, and 5 mM DTT. Protease fractions purified from the size exclusion column were concentrated to 1–2 mg/mL using an Amicon Ultra-15 10-kDa device (Millipore) for crystallization.
Enzyme Inhibition Assays.
The enzyme inhibition assays were carried out as previously described.33, 40 To determine the enzyme inhibition constant (Ki), in a 96-well plate, each inhibitor was serially diluted, including a no drug control, and incubated with 0.35 nM protein for 1 hour. A 10-amino acid substrate containing an optimized protease cleavage site with an EDANS/DABCYL FRET pair was dissolved in 4% DMSO at 120 μM. Using the Envision plate reader, 5 μL of the 120 μM substrate was added to the 96-well plate to a final concentration of 10 μM. The fluorescence was observed with an excitation at 340 nm and emission at 492 nm and monitored for 200 counts, for approximately 60 min. Data was analyzed with Prism7. DRV was used as a control in all assays.
Antiviral Assays.
293T and TZM-BL41 cells (NIH AIDS Research and Reference Reagent Program) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum in the presence of penicillin and streptomycin at 37 °C with 5% CO2. To determine the concentration of drugs achieving 50% inhibition of infection compared with the drug-free control, 4.5×106 293T cells were seeded onto a 10-cm plate 24 h before transfection. Cells were transfected with 8 μg of either the wild-type plasmid, infectious molecular clone pNL-CH derived from the pNL4-3 clone of HIV-1 using FuGENE 6 transfection reagent (Roche). The culture supernatant of 293T cells transfected with wild-type or Pi-resistant HIV-1 variant was removed 18 h after transfection and the cells were washed with 1 × PBS. The 293T cells were collected and transferred to wells of a 24-well plate. Briefly, each drug was serially diluted in the culture medium and the dilutions were added to the wells of a 24-well plate. The 293T cells (0.5 × 106 per well) collected from the transfection were added to wells containing various concentrations of drug. The culture supernatant containing virus particles was harvested 18 h after the 293T cells were reseeded in the presence of drug. This supernatant was filtered through a 0.45-μm-pore-size membrane (Millipore) to remove cell debris then used to infect 2 × 104 TZM-BL cells in a 96-well plate following a procedure previously described.42 The culture supernatant was removed from each well 48 h post-infection, and the cells were washed with 1 × PBS. For the luciferase assay, infected TZM-BL cells were lysed in 1× reporter lysis buffer (Promega) and the cells were kept at −80 °C. After one freeze-thaw cycle, the cell lysates were transferred into a 96-well assay plate (Costar), and luciferase activity was measured using a luminometer (Promega). The culture supernatant harvested from 293T cells reseeded in the absence of drugs was used as a drug-free control. EC50 was determined based on a dose-response curve generated using GraphPad Prism (version 7.0).
Protein Crystallization.
The condition reliably producing cocrystals of NL4-3 WT protease bound to PIs was discovered and optimized as previously described.40, 43 Briefly, all cocrystals were grown at room temperature by hanging drop vapor diffusion method in a 24-well VDX hanging-drop trays (Hampton Research) with a protease concentration of 1.4–1.7 mg/mL with 3-fold molar excess of inhibitors and mixed with the precipitant solution at a 1:2 ratio. The reservoir solution was 23–27% (w/v) ammonium sulfate with 0.1 M bis-Tris-methane buffer at pH 5.5, and the crystallization drops were set with 2 μL of well solution and 1 μL of protein-inhibitor solution and micro-seeded with a cat whisker. Diffraction quality crystals were obtained within 1 week. As data were collected at 100 K, cryogenic conditions contained the precipitant solution supplemented with 25% glycerol.
X-Ray Data Collection and Structure Solution.
X-ray diffraction data were collected and solved as previously described.36, 40, 43 Diffraction quality crystals were flash frozen under a cryostream when mounting the crystals either at our in-house Rigaku_Saturn944 X-ray system or the Chicago APS Synchrotron Beamline 23-1D-D. The cocrystal diffraction intensities from the Rigaku system were indexed, integrated, and scaled using HKL3000.44 Structures were solved using molecular replacement with PHASER.45 Model building and refinement were performed using Coot46 and Phenix.47 Ligands were designed in Maestro and the output sdf files were used in the Phenix program eLBOW48 to generate cif files containing atomic positions and constraints necessary for ligand refinement. Iterative rounds of crystallographic refinement were carried out until convergence was achieved. To limit bias throughout the refinement process, five percent of the data were reserved for the free R-value calculation.49 MolProbity50 was applied to evaluate the final structures before deposition in the PDB. Structure analysis, superposition and figure generation was done using PyMOL.51 X-ray data collection and crystallographic refinement statistics are presented in the Supporting Information (Table S1).
The cocrystal structures of all new hybrid compounds were solved in the P212121 space group with one protease homodimer in the asymmetric unit and only one orientation of the bound inhibitor in the active site, which was crucial for direct comparison of inhibitor structures. The cocrystal structure of compound 4 was solved in the P21 space group with two protease homodimers in the asymmetric unit, and one inhibitor bound to each dimer in one orientation, allowing one P21 dimer to be directly compared to the crystal structures solved in the P212121 space group. Three structures (10a, 10b, 13c) had significant electron density at the flap tips (residues 50–51) indicating that the backbone atoms interacted in two conformations, as both hydrogen bond donors and acceptors. Figure generation and structural analysis calculations (distance differences, vdW, hydrogen bonds) were performed with the flap tips modeled in one conformation while the other conformation was excluded. Compound 5 formed cocrystals but were inadequate for X-ray data collection.
Intermolecular vdW Contact Analysis of Crystal Structures.
To calculate the intermolecular vdW interaction energies the crystal structures were prepared using the Schrödinger Protein Preparation Wizard.52 Hydrogen atoms were added, protonation states were determined, and the structures were minimized. The protease active site was monoprotonated at Asp25. Subsequently, force field parameters were assigned using the OPLS3 force field.53 Interaction energies between the inhibitor and protease were estimated using a simplified Lennard-Jones potential, as previously described in detail.54 Briefly, the vdW energy was calculated for pairwise interactions depending on the types of atoms interacting and the distance between them. For each protease residue, the change in vdW interactions relative to a reference complex in the same space group was also calculated for each variant structure.
Molecular Dynamics Simulations
System Preparation.
High resolution crystal structures of LPV and the designed inhibitors bound to WT protease were prepared using the Protein Preparation Wizard using Maestro within the Schrodinger Suite52 as previously described.55, 56 Briefly, missing atoms were added using Prime57 and PROPKA58 was used to determine the protonation state of the side chains at pH 7.0. The catalytic aspartic acid with a pKa higher than 7.0 was protonated whereas the one with a pKa less than 7.0 was unprotonated. Co-crystallized fragments such as phosphate ions were removed. Lastly, the structure was minimized to a convergence criterion of 0.3 Å using Impref.59
Molecular Dynamics Simulations.
The prepared systems were placed in a cubic TIP3P implicit water box measuring 12Å on each side. Molecular dynamics simulations were carried out as previously described55 using Desmond within Schrödinger Suite.52 Briefly, chloride ions were used to neutralize the system and 0.15 M salt were added using sodium and chloride ions. The OPLS3 force field was used to parameterize the ligand and protein. Prior to starting the 100 ns MD simulations, the solvated system was minimized using the stepwise procedure described previously.55 Triplicates of 100 ns simulations for LPV and the designed inhibitors in complex with WT protease each with a randomized velocity were started using a protocol previously developed.55, 56 The root-mean-square deviation (RMSD) and root-mean-square fluctuation (RMSF) were calculated using tools within the Schrodinger Suite.
Supplementary Material
ACKNOWLEDGMENT
This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. GM/CA@APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). The Eiger 16M detector was funded by an NIH–Office of Research Infrastructure Programs, High-End Instrumentation Grant (1S10OD012289-01A1). We thank the beamline specialists at 23-ID-D for their help in data collection. We thank Ellen A. Nalivaika for providing the HIV-1 protease variants, the AIDS Research and Reference Reagent Program (NIAD, NIH) for reference protease inhibitors, and Drs. William Royer, Brian Kelch and members of the Schiffer and Miller laboratories for helpful discussions. This work was supported by a grant from the National Institute of General Medical Sciences of the NIH (P01 GM109767).
ABBREVIATIONS
- ATV
atazanavir
- HIV-1
human immunodeficiency virus type 1
- bis-THF
bis-tetrahydrofuran
- Boc
tert-butoxycarbonyl
- DIEA
N,N-diisopropylethylamine
- DRV
darunavir
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
- FRET
fluorescence resonance energy transfer
- LPV
lopinavir
- MDR
multidrug-resistant
- PIs
protease inhibitors
- SAR
structure-activity relationships
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
Footnotes
Supporting Information
Synthesis details and analytical data of Phe-Phe isostere 7, amino acid derivatives 11a–h, and inhibitors 4–5; Figures S1-S9; Tables S1-S3 (PDF)
Molecular formula strings (CSV)
Accessions Codes
The PDB accession codes for X-ray cocrystal structures of wild-type HIV-1 protease with DRV, LPV, 4, 12a, 12b, 12c, 12d, 12e, 12f, 12g, 12h, 15a, 15b, and 15c are 6DGX, 6PJB, 6PJC, 6PJD, 6PJE, 6PJG, 6PJH, 6PJI, 6PJK, 6PJL, 6PJM, 6PJN, and 6PJO, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
The authors declare no competing financial interest.
REFERENCES
- 1.Cihlar T; Fordyce M Current status and prospects of HIV treatment. Curr. Opin. Virol 2016, 18, 50–56. [DOI] [PubMed] [Google Scholar]
- 2.Saag MS; Benson CA; Gandhi RT; Hoy JF; Landovitz RJ; Mugavero MJ; Sax PE; Smith DM; Thompson MA; Buchbinder SP; Del Rio C; Eron JJ Jr.; Fatkenheuer G; Gunthard HF; Molina JM; Jacobsen DM; Volberding PA Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2018 recommendations of the international antiviral society-USA panel. JAMA 2018, 320, 379–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV; Department of Health and Human Services. Available at https://files.aidsinfo.nih.gov/contentfiles/lvguidelines/AdultandAdolescentGL.pdf. (Accessed February 15, 2020). [Google Scholar]
- 4.Ali A; Bandaranayake RM; Cai Y; King NM; Kolli M; Mittal S; Murzycki JF; Nalam MN; Nalivaika EA; Ozen A; Prabu-Jeyabalan MM; Thayer K; Schiffer CA Molecular basis for drug resistance in HIV-1 protease. Viruses 2010, 2, 2509–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ghosh AK; Osswald HL; Prato G Recent progress in the development of HIV-1 protease inhibitors for the treatment of HIV/AIDS. J. Med. Chem 2016, 59, 5172–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Surleraux DL; Tahri A; Verschueren WG; Pille GM; de Kock HA; Jonckers TH; Peeters A; De Meyer S; Azijn H; Pauwels R; de Bethune MP; King NM; Prabu- Jeyabalan M; Schiffer CA; Wigerinck PB Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor. J. Med. Chem 2005, 48, 1813–1822. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh AK; Chapsal BD; Weber IT; Mitsuya H Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res 2008, 41, 78–86. [DOI] [PubMed] [Google Scholar]
- 8.Tie Y; Boross PI; Wang YF; Gaddis L; Hussain AK; Leshchenko S; Ghosh AK; Louis JM; Harrison RW; Weber IT High resolution crystal structures of HIV-1 protease with a potent non-peptide inhibitor (UIC-94017) active against multi-drug-resistant clinical strains. J. Mol. Biol 2004, 338, 341–352. [DOI] [PubMed] [Google Scholar]
- 9.Ghosh AK; Ramu Sridhar P; Kumaragurubaran N; Koh Y; Weber IT; Mitsuya H Bis-tetrahydrofuran: a privileged ligand for darunavir and a new generation of hiv protease inhibitors that combat drug resistance. ChemMedChem 2006, 1, 939–50. [DOI] [PubMed] [Google Scholar]
- 10.Hazen R; Harvey R; Ferris R; Craig C; Yates P; Griffin P; Miller J; Kaldor I; Ray J; Samano V; Furfine E; Spaltenstein A; Hale M; Tung R; St Clair M; Hanlon M; Boone L In vitro antiviral activity of the novel, tyrosyl-based human immunodeficiency virus (HIV) type 1 protease inhibitor brecanavir (GW640385) in combination with other antiretrovirals and against a panel of protease inhibitor-resistant HIV. Antimicrob. Agents Chemother 2007, 51, 3147–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cihlar T; He GX; Liu X; Chen JM; Hatada M; Swaminathan S; McDermott MJ; Yang ZY; Mulato AS; Chen X; Leavitt SA; Stray KM; Lee WA Suppression of HIV-1 protease inhibitor resistance by phosphonate-mediated solvent anchoring. J. Mol. Biol 2006, 363, 635–647. [DOI] [PubMed] [Google Scholar]
- 12.Dandache S; Sevigny G; Y elle J; Stranix BR; Parkin N; Schapiro JM; Wainberg MA; Wu JJ In vitro antiviral activity and cross-resistance profile of PL-100, a novel protease inhibitor of human immunodeficiency virus type 1. Antimicrob. Agents Chemother 2007, 51, 4036–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kesteleyn B; Amssoms K; Schepens W; Hache G; Verschueren W; Van De Vreken W; Rombauts K; Meurs G; Sterkens P; Stoops B; Baert L; Austin N; Wegner J; Masungi C; Dierynck I; Lundgren S; Jonsson D; Parkes K; Kalayanov G; Wallberg H; Rosenquist A; Samuelsson B; Van Emelen K; Thuring JW Design and synthesis of HIV-1 protease inhibitors for a long-acting injectable drug application. Bioorg. Med. Chem. Lett 2013, 23, 310–317. [DOI] [PubMed] [Google Scholar]
- 14.Bungard CJ; Williams PD; Schulz J; Wiscount CM; Holloway MK; Loughran HM; Manikowski JJ; Su HP; Bennett DJ; Chang L; Chu XJ; Crespo A; Dwyer MP; Keertikar K; Morriello GJ; Stamford AW; Waddell ST; Zhong B; Hu B; Ji T; Diamond TL; Bahnck-Teets C; Carroll SS; Fay JF; Min X; Morris W; Ballard JE; Miller MD; McCauley JA Design and synthesis of piperazine sulfonamide cores leading to highly potent HIV-1 protease inhibitors. ACS Med. Chem. Lett 2017, 8, 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller JF; Andrews CW; Brieger M; Furfine ES; Hale MR; Hanlon MH; Hazen RJ; Kaldor I; McLean EW; Reynolds D; Sammond DM; Spaltenstein A; Tung R; Turner EM; Xu RX; Sherrill RG Ultra-potent P1 modified arylsulfonamide HIV protease inhibitors: the discovery of GW0385. Bioorg. Med. Chem. Lett 2006, 16, 1788–1794. [DOI] [PubMed] [Google Scholar]
- 16.He G-X; Yang Z-Y; Williams M; Callebaut C; Cihlar T; Murray BP; Yang C; Mitchell ML; Liu H; Wang J; Arimilli M; Eisenberg E; Stray KM; Tsai LK; Hatada M; Chen X; Chen JM; Wang Y; Lee MS; Strickley RG; Iwata Q; Zheng X; Kim CU; Swaminathan S; Desai MC; Lee WA; Xu L Discovery of GS-8374, a potent human immunodeficiency virus type 1 protease inhibitor with a superior resistance profile. MedChemComm 2011, 2, 1093–1098. [Google Scholar]
- 17.Windsor IW; Palte MJ; Lukesh JC 3rd; Gold B; Forest KT; Raines RT Sub- picomolar inhibition of HIV-1 protease with a boronic acid. J. Am. Chem. Soc 2018, 140, 14015–14018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nalam MN; Ali A; Reddy GS; Cao H; Anjum SG; Altman MD; Yilmaz NK; Tidor B; Rana TM; Schiffer CA Substrate envelope-designed potent HIV-1 protease inhibitors to avoid drug resistance. Chem. Biol 2013, 20, 1116–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aoki M; Hayashi H; Rao KV; Das D; Higashi-Kuwata N; Bulut H; Aoki-Ogata H; Takamatsu Y; Yedidi RS; Davis DA; Hattori SI; Nishida N; Hasegawa K; Takamune N; Nyalapatla PR; Osswald HL; Jono H; Saito H; Yarchoan R; Misumi S; Ghosh AK; Mitsuya H A novel central nervous system-penetrating protease inhibitor overcomes human immunodeficiency virus 1 resistance with unprecedented aM to pM potency. Elife 2017, 6, e28020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh AK; P RN; Kovela S; Rao KV; Brindisi M; Osswald HL; Amano M; Aoki M; Agniswamy J; Wang YF; Weber IT; Mitsuya H Design and synthesis of highly potent HIV-1 protease inhibitors containing tricyclic fused ring systems as novel P2 ligands: structure-activity studies, biological and X-ray structural analysis. J. Med. Chem 2018, 61, 4561–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh AK; Rao KV; Nyalapatla PR; Osswald HL; Martyr CD; Aoki M; Hayashi H; Agniswamy J; Wang YF; Bulut H; Das D; Weber IT; Mitsuya H Design and development of highly potent HIV-1 protease inhibitors with a crown-like oxotricyclic core as the P2-ligand to combat multidrug-resistant HIV variants. J. Med. Chem 2017, 60, 4267–4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghosh AK; Chapsal BD; Baldridge A; Steffey MP; Walters DE; Koh Y; Amano M; Mitsuya H Design and synthesis of potent HIV-1 protease inhibitors incorporating hexahydrofuropyranol-derived high affinity P2 ligands: structure-activity studies and biological evaluation. J. Med. Chem 2011, 54, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.King NM; Prabu-Jeyabalan M; Nalivaika EA; Schiffer CA Combating susceptibility to drug resistance: lessons from HIV-1 protease. Chem. Biol 2004, 11, 1333–1338. [DOI] [PubMed] [Google Scholar]
- 24.Gerlits O; Wymore T; Das A; Shen CH; Parks JM; Smith JC; Weiss KL; Keen DA; Blakeley MP; Louis JM; Langan P; Weber IT; Kovalevsky A Long-range electrostatics-induced two-proton transfer captured by neutron crystallography in an enzyme catalytic site. Angew. Chem. Int. Ed. Engl 2016, 55, 4924–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kovalevsky A; Gerlits O; Beltran K; Weiss KL; Keen DA; Blakeley MP; Louis JM; Weber IT Proton transfer and drug binding details revealed in neutron diffraction studies of wild-type and drug resistant HIV-1 protease. Methods Enzymol. 2020, 634, 257–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X; Kempf DJ; Li L; Sham HL; Vasavanonda S; Wideburg NE; Saldivar A; Marsh KC; McDonald E; Norbeck DW Synthesis and SAR studies of potent HIV protease inhibitors containing novel dimethylphenoxyl acetates as P2 ligands. Bioorg. Med. Chem. Lett 2003, 13, 3657–3660. [DOI] [PubMed] [Google Scholar]
- 27.Chen X; Li L; Kempf DJ; Sham H; Wideburg NE; Saldivar A; Vasavanonda S; Marsh KC; McDonald E; Norbeck DW Evaluation of furofuran as a P2 ligand for symmetry- based HIV protease inhibitors. Bioorg. Med. Chem. Lett 1996, 6, 2847–2852. [Google Scholar]
- 28.Cannizzaro CE; Yang Z-Y; Desai MC; Mitchell ML; Xu L; Swaminathan S; Chen JM Antiviral protease inhibitors. PCT Int. App. WO 2008/011117, 2008. [Google Scholar]
- 29.Ozen A; Haliloglu T; Schiffer CA Dynamics of preferential substrate recognition in HIV-1 protease: redefining the substrate envelope. J. Mol. Biol 2011, 410, 726–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paulsen JL; Leidner F; Ragland DA; Kurt Yilmaz N; Schiffer CA Interdependence of inhibitor recognition in HIV-1 protease. J. Chem. Theory Comput 2017, 13, 2300–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haight AR; Stuk TL; Allen MS; Bhagavatula L; Fitzgerald M; Hannick SM; Kerdesky FAJ; Menzia JA; Parekh SI; Robbins TA; Scarpetti D; Tien J-HJ Reduction of an enaminone: synthesis of the diamino alcohol core of ritonavir. Org. Process Res. Dev 1999, 3, 94–100. [Google Scholar]
- 32.Engle KM; Wang DH; Yu JQ Ligand-accelerated C-H activation reactions: evidence for a switch of mechanism. J. Am. Chem. Soc 2010, 132, 14137–14151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Windsor IW; Raines RT Fluorogenic Assay for Inhibitors of HIV-1 Protease with Sub-picomolar Affinity. Sci. Rep 2015, 5, 11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scott WR; Schiffer CA Curling of flap tips in HIV-1 protease as a mechanism for substrate entry and tolerance of drug resistance. Structure 2000, 8, 1259–1265. [DOI] [PubMed] [Google Scholar]
- 35.Yu Y; Wang J; Chen Z; Wang G; Shao Q; Shi J; Zhu W Structural insights into HIV-1 protease flap opening processes and key intermediates. RSC Adv. 2017, 7, 45121–45128. [Google Scholar]
- 36.Ozen A; Lin KH; Kurt Yilmaz N; Schiffer CA Structural basis and distal effects of Gag substrate coevolution in drug resistance to HIV-1 protease. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 15993–15998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.King NM; Melnick L; Prabu-Jeyabalan M; Nalivaika EA; Yang SS; Gao Y; Nie X; Zepp C; Heefner DL; Schiffer CA Lack of synergy for inhibitors targeting a multi- drug-resistant HIV-1 protease. Protein Sci. 2002, 11, 418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rose JR; Salto R; Craik CS Regulation of autoproteolysis of the HIV-1 and HIV-2 proteases with engineered amino acid substitutions. J. Biol. Chem 1993, 268, 11939–11945. [PubMed] [Google Scholar]
- 39.Hui JO; Tomasselli AG; Reardon IM; Lull JM; Brunner DP; Tomich CS; Heinrikson RL Large scale purification and refolding of HIV-1 protease from Escherichia coli inclusion bodies. J. Protein Chem 1993, 12, 323–327. [DOI] [PubMed] [Google Scholar]
- 40.Rusere LN; Lockbaum GJ; Lee SK; Henes M; Kosovrasti K; Spielvogel E; Nalivaika EA; Swanstrom R; Yilmaz NK; Schiffer CA; Ali A HIV-1 protease inhibitors incorporating stereochemically defined P2' ligands to optimize hydrogen bonding in the substrate envelope. J. Med. Chem 2019, 62, 8062–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wei X; Decker JM; Liu H; Zhang Z; Arani RB; Kilby JM; Saag MS; Wu X; Shaw GM; Kappes JC Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother 2002, 46, 1896–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee SK; Harris J; Swanstrom R A strongly transdominant mutation in the human immunodeficiency virus type 1 gag gene defines an Achilles heel in the virus life cycle. J. Virol 2009, 83, 8536–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lockbaum GJ; Leidner F; Rusere LN; Henes M; Kosovrasti K; Nachum GS; Nalivaika EA; Ali A; Yilmaz NK; Schiffer CA Structural Adaptation of Darunavir Analogues against Primary Mutations in HIV-1 Protease. ACS Infect. Dis 2019, 5, 316–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Otwinowski Z; Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 45.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emsley P; Cowtan K Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 47.Adams PD; Afonine PV; Bunkoczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung LW; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moriarty NW; Grosse-Kunstleve RW; Adams PD electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr., Sect. D: Biol. Crystallogr 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brunger AT Free R value: a novel statistical quantity for assessing the accuracy of crystal structures. Nature 1992, 355, 472–475. [DOI] [PubMed] [Google Scholar]
- 50.Davis IW; Leaver-Fay A; Chen VB; Block JN; Kapral GJ; Wang X; Murray LW; Arendall WB 3rd; Snoeyink J; Richardson JS; Richardson DC MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.PyMOL. The PyMOL Molecular Graphics System, Version 2.3; Schrödinger, LLC. [Google Scholar]
- 52.Schrödinger. Schrödinger Release 2019-2; Schrödinger, LLC, New York, NY, United States, 2019. [Google Scholar]
- 53.Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; Kaus JW; Cerutti DS; Krilov G; Jorgensen WL; Abel R; Friesner RA OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- 54.Nalam MN; Ali A; Altman MD; Reddy GS; Chellappan S; Kairys V; Ozen A; Cao H; Gilson MK; Tidor B; Rana TM; Schiffer CA Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance. J. Virol 2010, 84, 5368–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Leidner F; Kurt Yilmaz N; Paulsen J; Muller YA; Schiffer CA Hydration structure and dynamics of inhibitor-bound HIV-1 protease. J. Chem. Theory Comput 2018, 14, 2784–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Henes M; Lockbaum GJ; Kosovrasti K; Leidner F; Nachum GS; Nalivaika EA; Lee SK; Spielvogel E; Zhou S; Swanstrom R; Bolon DNA; Kurt Yilmaz N; Schiffer CA Picomolar to micromolar: elucidating the role of distal mutations in HIV-1 protease in conferring drug resistance. ACS Chem. Biol 2019, 14, 2441–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jacobson MP; Friesner RA; Xiang Z; Honig B On the role of the crystal environment in determining protein side-chain conformations. J. Mol. Biol 2002, 320, 597–608. [DOI] [PubMed] [Google Scholar]
- 58.Olsson MH; Sondergaard CR; Rostkowski M; Jensen JH PROPKA3: consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput 2011, 7, 525–537. [DOI] [PubMed] [Google Scholar]
- 59.Banks JL; Beard HS; Cao Y; Cho AE; Damm W; Farid R; Felts AK; Halgren TA; Mainz DT; Maple JR; Murphy R; Philipp DM; Repasky MP; Zhang LY; Berne BJ; Friesner RA; Gallicchio E; Levy RM Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem 2005, 26, 1752–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.