

ABSTRACT
M2-like tumor-associated macrophages promote tumor progression by establishing an immunosuppressive tumor microenvironment. The phenotype and activity of immunosuppressive macrophages are related to their mitochondrial metabolism. Thus, we studied if drugs targeting mitochondrial metabolic pathways can repolarize macrophages from M2 into an M1-like phenotype or can prevent M0-to-M2 polarization. The drugs selected are clinically approved or in clinical trials and target M2-specific metabolic pathways: fatty acid oxidation (Perhexiline and Trimetazidine), glutaminolysis (CB-839), PPAR activation (HX531), and mitochondrial electron transport chain (VLX-600). Murine bone marrow-derived macrophages were either polarized to M2 using IL-4 in the presence of the drugs or polarized first into M2 and then treated with the drugs in presence of IFN-γ for re-polarization. Targeting both fatty acid oxidation with Perhexiline or the electron transport chain with VLX-600 in the presence of IFN-γ, impaired mitochondrial basal, and maximal respiration and resulted in M2 to M1-like re-polarization (increased iNOS expression, NO production, IL-23, IL-27, and TNF-α secretion), similar to LPS+IFN-γ re-polarization. Moreover, drug-induced macrophage re-polarization resulted in a strong tumor-cytotoxic activity. Furthermore, the polarization of M0- to M2-like macrophages was impaired by CB-839, Trimetazidine, HX531, and Perhexiline, while Hx531 and Perhexiline also reduced MCP-1 secretion. Our results show that by targeting cell metabolism, macrophages could be re-polarized from M2- into an anti-tumoral M1-like phenotype and that M0-to-M2 polarization could be prevented. Overall, this study provides rational for the use of clinically applicable drugs to change an immunosuppressive tumor environment into a pro-inflammatory tumor environment that could support cancer immunotherapies.
KEYWORDS: Macrophage polarization, macrophage re-polarization, immunometabolism, mitochondrial respiration
INTRODUCTION
The immunosuppressive tumor microenvironment (TME) is considered a major barrier for effective cancer immunotherapies based on checkpoint inhibitors.1 Besides tumor cell-induced immune suppressive mechanisms, tumor-associated macrophages (TAMs) have been shown to be involved in the establishment and maintenance of the immunosuppressive TME.2 In many tumors, TAMs display pro-tumoral activity, yet a whole spectrum from pro- to anti-tumoral macrophages can be present.3 TAMs have been classically categorized into a spectrum of functionally and phenotypically different subsets: in one extreme, macrophages with pro-inflammatory and anti-tumoral activity, which resemble the phenotype of in vitro LPS+IFN-γ stimulated macrophages (M1 from now on), and a second subset displaying anti-inflammatory and pro-tumoral activity, similar to in vitro IL-4 stimulated macrophages (M2).4 This complex mix makes it challenging to specifically target M2-like macrophages to prevent their activity or polarization and thus improve cancer immunotherapies.
To support tumor growth, tumor cells, and immune cells secrete chemokines (e.g., MCP-1) that promote the infiltration of monocytes and their polarization into macrophages.5 M1-like TAM activation can be induced by several pro-inflammatory signals (e.g., interferon-γ (IFN-γ) plus lipopolysaccharide (LPS)) which engage them to express co-stimulatory molecules (CD80 and CD86), secrete pro-inflammatory cytokines (IL-1β, IL-6, IL-27, and TNF-α), and produce effector molecules such as reactive nitrogen intermediates (e.g., nitric oxide), through inducible nitric oxide synthase (iNOS) increased expression.6 On the other hand, M2-like TAM polarization is driven by anti-inflammatory cytokines (e.g., IL-4, IL-10, and IL-13) and is associated with the expression of MHC-II and secretion of IL-10, TGF-β, and CCL2.6,7 Furthermore, in murine macrophages, increased Arginase 1 (Arg1) expression and low NO production are key Macrophage polarization involves changes in cell metabolism.8,9 LPS+IFN-γ polarized M1 macrophages show increased glycolysis and reduced mitochondrial respiration due to a broken tricarboxylic (TCA) cycle.10 On the other hand, IL-4-induced M2 macrophages mostly rely on fatty acid oxidation (FAO) and glutaminolysis to fuel the TCA cycle and to produce energy and metabolites required by oxidative phosphorylation (OXPHOS).11 The main controller of lipid metabolism upregulation is the peroxisome proliferator-activated receptor gamma (PPARγ) transcription factor, which is activated upon IL-4 stimulation12 and promotes an increase in oxygen consumption and mitochondrial biogenesis.11 PPARγ activity is modulated by the co-activators PGC-111 and RXR.13 Ultimately, IL-4 polarized macrophage will display an increased FAO activity, linked to an increase in fatty acid transport to the mitochondrial matrix by carnitine palmitoyl transferase (CPT) and the activity of enzymes responsible of β-oxidation, 11,14 whose products will fuel the TCA cycle and supply the electron transport chain (ETC). A functional TCA cycle is fundamental for M2 polarization10 and besides glycolysis and FAO, glutaminolysis replenishes it. It has been shown that glutamine deprivation prevents IL-4 differentiation, 3 supporting the importance of glutaminolysis metabolism for M2 polarization. In M2 macrophages, electron carriers NADPH and FADPH fuel the mitochondrial ETC, leading to ATP production; contrary to what is observed in M1 macrophages, in which the ETC is working in reverse to increase ROS production.15
Coordinating and fine-tuning of all metabolic changes is essential for IL-4 driven polarization. Disturbing these processes prevents functional polarization of macrophages and can lead to macrophage repolarization. Few studies have shown that IL-4 polarized macrophages can be re-polarized into a pro-inflammatory M1-like phenotype.16 Increasing glucose transporter 1 (GLUT-1) expression resulted in increased glycolysis and pentose phosphate pathway metabolites, reduced oxygen consumption, and enhanced LPS+IFN-γ polarization.17 Knock-down of pyruvate dehydrogenase kinase-1 -glycolysis regulatory enzyme- was shown to suppress LPS+IFN-γ polarization and improve IL-4-mediated M2 differentiation by enhancing mitochondrial respiration.18
Overall, preventing IL-4 mediated polarization or promoting re-polarization to a proinflammatory M1-like phenotype could be achieved by changing the metabolism of macrophages. While this has been mostly studied using reagents targeting specific pathways or by genetic modifications, 19,20 little is known whether clinically relevant drugs can reshape macrophage activity. Here we determined the activity of drugs (clinically approved or currently being evaluated in clinical trials) targeting mitochondrial metabolism, in driving M2-to-M1 re-polarization or preventing IL-4-induced M2 polarization.
MATERIALS AND METHODS
Media, cytokines, drugs, and reagents
RPMI-1640 with Glutamax, non-essential amino acids (NEAA), sodium pyruvate, and penicillin–streptomycin (P/S) were from Gibco (Paisley, Scotland, UK). Fetal bovine serum (FBS) was purchased from Life Science Production (Barnet, UK). Cytokines M-CSF, IL-4, and IFN-γ were purchased from Biolegend (San Diego, CA, USA). 2-Deoxy-D-glucose (2-DG), Oligomycin (Oligo), Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP), Rotenone (R/Rot,), and Antimycin A (AA) were purchased from Abcam (Cambridge, UK). Perhexiline (PerHx) and H531 (Hx) were purchased from Tocris (Bristol, UK). CB-839 was purchased from FOCUS Biomolecules (Plymouth Meeting, PA, USA). Trimetazidine (TMZ) was purchased from Sigma-Aldrich (St. Louis, MO, USA). VLX-600 (VLX) was purchased from Cayman Chemical (Ann Arbor, MI, USA). Lipopolysaccharide (LPS) from E. Coli was obtained from Sigma-Aldrich. Any other material used was of analytical grade.
Mice
Pathogen-free female C57BL/6 mice were used at 8–12 weeks of age. The animals were purchased from Envigo (The Netherlands) and kept according to UMCG institutional guidelines. All animal procedures were approved by the local Animal Experimentation Ethical Committee.
Bone marrow-derived macrophage culture and treatment
Bone marrow-derived macrophages (BM-M) were generated from flushed bone marrow suspension from freshly isolated bones (femurs, tibias, and hips). Cells were centrifuged for 5 min at 400xg, treated with ACK lysis buffer (NH4Cl 150 mM, KHCO3 10 mM, Na2EDTA 0.1 mM, pH 7.4) for 5 min, washed with PBS, centrifuged again, and then cultured for 6 days in a complete medium (RPMI-1640 with Glutamax supplemented with 10% FBS, 1% NEAA, 1% sodium pyruvate, and 1% P/S) supplemented with 20 ng/ml M-CSF. The medium was refreshed every 2–3 days. On day 6, BM-M were detached using citrate buffer (KCl 135 mM and Na₃C₆H₅O₇ 15 mM) for 15–20 min at 37°C and gentle resuspending. Cells were washed, counted, and cultured in a complete medium supplemented with M-CSF.
Polarization and re-polarization
After being detached, BM-M were seeded in 24-well flat-bottom, non-treated plates from Corning (Corning, NY, USA) at a density of 2 × 105 cells/well. For M0-to-M2 polarization, cells were first treated with TMZ (100 µM), CB-839 (5 µM), HX531 (10 µM), PerHx (5 µM), or VLX (10 µM) for 1 h and then stimulated with IL-4 (20 ng/ml) for 24 h. For M2-to-M1 re-polarization studies, cells were first polarized to M2 using IL-4 and then washed and treated with the drugs. After a 1 h drug treatment, IFN-γ (20 ng/ml) was added for another 24 h. To induce M1 polarization (positive control) cells were stimulated with LPS (100 ng/ml) plus IFN-γ.
Flow cytometry
For surface markers, cells were stained for 30 min at 4°C in 50 μl of PBS-FBS 2% using anti-CD11b BV510-conjugated, anti-F/480 PerCP-Cy5.5-conjugated (Biolegend). Zombie Violet (BioLegend) was used to determine cell viability. For intracellular staining, cells were first fixed and permeabilized with the Fixation/Permeabilization Kit from BD Biosciences (San Jose, CA, USA) according to the manufacturer’s instructions. Briefly, cells were fixed (15 min), permeabilized (1 h), washed, and then stained for intracellular markers using anti-iNOS PE-Cy7-conjugated eBioscience (San Diego, CA, USA) and anti-Arginase-1 PE-conjugated from R&D systems (Minneapolis, MN, USA) for 30 min at 4°C in 50 μl of perm/wash buffer. After washing, the cells were resuspended in PBS-FBS 2% and analyzed by flow cytometry. 10,000 events were acquired within ZVlowCD11b+F4/80++ gate. Samples were assessed in a FACSVerse cytometer (BD Bioscience), and the data analyzed using FlowJo X (TreeStar Inc., Ashland, OR USA).
Cytokine bead array
BM-M (1x105 cells/well) were cultured in 96-well plates (flat bottom, cell culture-treated, from Corning) in 200 μL of complete media. Cells were polarized or re-polarized in the presence of drugs as described above. After 24 h of treatment, the supernatant was collected, centrifuged 600xg for 5 min to remove debris, and immediately analyzed using BioLegend’s LEGENDplex Mouse Inflammation Panel (13-plex) according to the manufacturer’s instructions. Samples were analyzed by flow cytometry in duplicate.
Extracellular bioenergetics-Seahorse analyses
Experiments were performed according to the manufacture’s recommendations (Agilent, Santa Clara, CA, USA). Briefly, 5 × 104 BM-M were seeded per well (6–8 replicates) of an XF96 culture plate (Seahorse XFe96 FluxPak from Agilent) for 1 h at room temperature and then incubated overnight at 37°C with 5% CO2. Cells were treated as described in the results section. Cells were washed with warm PBS and then assay medium (sodium bicarbonate- and glucose-free DMEM supplemented with sodium pyruvate 1 mM, pH 7.4) was added and the cells were kept for 1 h at 37°C without CO2. The background recording extracellular acidification rates (ECAR, milli pH/min) and oxygen consumption rates (OCR, pmol/min) were measured first. Glucose (25 mm; Sigma), Oligomycin (1,5 µM; Sigma), FCCP (1,5 µM; Sigma), and the mix of 2-deoxyglucose (50 mM), Antimycin A (1 µM), and Rotenone (1 µM) were injected when indicated. After lysis of the cells with RIPA buffer (Merck) with proteinase inhibitors (Roche complete Protease Inhibitor, Basel, Switzerland) protein concentrations were determined using BCA protein assay from Thermo-Fischer Scientific (Waltham, MA, USA). ECAR and OCR data were normalized to protein concentration and analyzed as described before.21
Measurement of NO production
The concentration of NO in culture supernatants was measured using Griess Reagent System from Promega (Madison, WI, USA). Macrophages were seeded in a 24-well, non-treated plate at a density of 1.2 × 105 per well and polarized or re-polarized as described in the results section. Supernatants were collected, mixed with Griess reagent (1:1), and incubated for 10 min at room temperature, avoiding direct light exposure. The samples were read at 545 nm in a plate reader (Synergy HT5, BioTek, Winooski, VT, USA) and NO concentration was calculated by referencing a standard curve generated from known concentrations of NaNO2.
Mitochondrial mass, potential, and ROS
BM-M (2x105 cells/well) were plated in a non-treated 24-well plate and polarized or re-polarized as described in the results section. BM-M were washed twice with PBS and then incubated with 500 μM of MitoSpy Green (Biolegend) and 500 μM of MitoTracker Red CMXRos (Thermo-Fischer Scientific) in PBS at 37 °C for 30 min, then washed once with PBS+FBS 10% and once with PBS. BM-M were detached using citrate buffer, washed, and analyzed by flow cytometry.
Co-culture and apoptosis determination
BM-M were harvested, counted, and plated (4.8x104/well) in 24-well, non-treated plates. After 18 h, macrophages were polarized to M2 and then re-polarized with the drugs plus IFN-γ, as described above. LPS+IFN-γ treatment was used as a positive control of re-polarization. TC-1 tumor cells were maintained and cultured in a complete RPMI medium. TC-1 cells were kindly donated by Dr. Cornelis J. Melief (Leiden University Medical Center, The Netherlands). The TC-1 cell line was generated from C57Bl/6 primary lung epithelial cells with a retroviral vector expressing HPV16 E6E7. 3 × 106 TC-1 cancer cells were incubated with 5 µM CFSE (Biolegend) for 15 min at 37°C. Unstained cells were kept as control and freshly stained cells were assessed by flow cytometry as input control. Macrophages were washed twice with warm PBS and CFSE-labeled TC-1 cells (2.4x104) were added to get a ratio of 2 to 1 (macrophages to TC-1). After 24 h, the cells were harvested and stained with anti-CD45 APC-Cy7-conjugated (Biolegend), CD11b, F4/80, and ZV, as described above. 10,000 events were acquired within the gate ZVlowCD45+.
The percentage of cytotoxicity was calculated as follows: % of cytotoxicity = 100x(1-X/Y), in which X is the calculated number of TC-1 cells in experimental co-cultures and Y is the calculated number of TC1 cells in the control co-cultures. The number of TC-1 cells was calculated based on the initial input of macrophages, the macrophage/TC-1 ratio after 24 h, and the assumption that the macrophages did not divide during the 24 h co-culture. For apoptosis determination, the co-cultured cells were stained with FITC-conjugated annexin V and propidium iodide (PI) following manufacturer’s guidelines (BD Biosciences).
Tumor cell proliferation
TC-1 (2x103/well) were seeded in a 96-well plate (flat bottom, cell culture treated) in three replicates. After 24 h, cells were treated with TMZ (100 µM), CB-839 (5 µM), HX531 (10 µM), PerHx (5 µM), and VLX (10 µM), leaving the drugs present during the whole experiment; or with the supernatant of drug-treated macrophages (1 h) after none, 1, 2 or 3 washes. Cell growth was monitored using the IncuCyte ZOOM Live Cell Imaging System (Essen Bioscience, Ann Arbor, MI, USA). Cell confluence (%) was calculated using IncuCyte ZOOM software.
Statistical analysis
Experimental data are presented as the mean ± SEM of the number of experiments indicated as “n”. For determination of significance, one-way analysis of variance was used (ANOVA) followed by Fisher’s LSD post hoc using Prism 6 (GraphPad. San Diego, CA, USA). A p value of 0.05 was considered a statistically significant difference between the data compared (* = p < .05, ** = p < .01 and *** = p < .001).
RESULTS
Selection of drugs
Drugs were selected based on their mechanism of action, i.e., targeting fundamental metabolic pathways. From these, we considered the ones that are clinically available or are undergoing clinical evaluations and that have proven in vitro efficacy in other cells than macrophages. Overall, we selected 5 drugs to be tested. To impair fatty acid transport into mitochondria through CPT-1 inhibition, Perhexiline (PerHx) was selected.22 PerHx is used as a prophylactic antianginal agent.23 To decrease FAO, we targeted 3-ketoacylthiolase (3-KAT) -which catalyzes the final step in FAO- by using Trimetazidine (TMZ), an FDA-approved drug for the treatment of angina pectoris.24 To inhibit glutaminolysis, we targeted the first enzyme of the pathway, glutaminase 1 (GLS) using CB-839 (Telaglenastat), 25 a promising anti-cancer drug currently in clinical phase 2 trials (NCT03875313). VLX600 (VLX), a novel iron chelator that interferes with intracellular iron metabolism was used to impair mitochondrial respiration.26 VLX has shown promising results preventing tumor cell proliferation in vitro27 and displayed a high-safety profile in clinical studies.28 Finally, to decrease the activity of PPARγ, we selected HX531, a compound that blocks the activity of RXR (part of the heterodimer PPARγ/RXR).29 Although HX531 is not yet FDA-approved, it has shown promising results for diabetes type 2 treatment.30
Mitochondrial respiration is impaired upon LPS+IFN-γ treatment of IL-4-polarized macrophage
Considering that most TAMs are anti-inflammatory M2-like macrophages, re-polarizing them into a pro-inflammatory M1-like phenotype would induce an inflammatory milieu, which could support cancer immunotherapies.20 We first analyzed the extent of M2-to-M1 re-polarization that can be achieved in vitro upon LPS+IFN-γ stimulation. For this, mouse bone marrow-derived macrophages (BM-M) were first polarized using IL-4 for 24 hours, washed, and then treated with LPS+IFN-γ for another 24 hours (gating strategy Sup. Figure 1a). From the CD11b+F4/80+ population, we assessed iNOS and Arg1 expression as M1 and M2 markers, respectively. As shown in Figure 1a (upper, left panel), IL-4-induced polarization increased Arg1 but not iNOS expression. Opposite, LPS+IFN-γ polarized macrophages highly increased iNOS. Re-polarization of IL-4-polarized macrophages with LPS+IFN-γ (from now on referred to as IL-4→LPS+IFN-γ) increased iNOS expression to the same extent as achieved in LPS+IFN-γ polarized macrophages. Yet, Arg1 expression did not change significantly as compared to IL-4 polarized macrophages.
Figure 1.
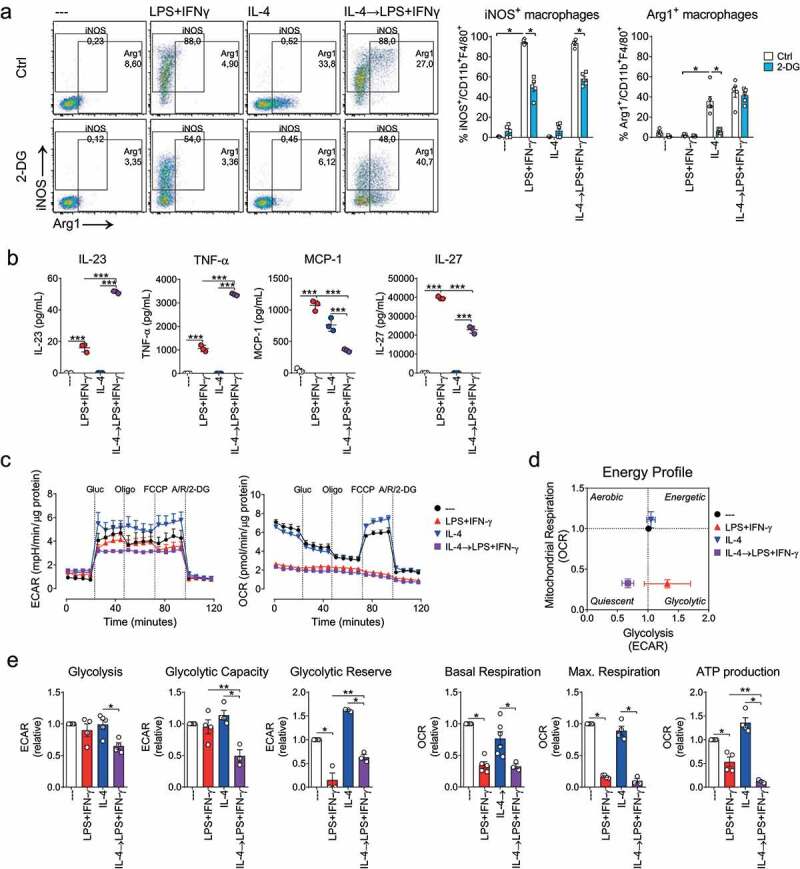
Macrophage repolarization depends on glycolysis and impairs mitochondrial respiration. Re-polarization scheme. BM-M were polarized using LPS+INF-γ (100 and 20 ng/ml, respectively) or IL-4 (20 ng/ml) (24 h). IL-4 macrophages were further re-polarized with LPS+IFN-γ (24 h). A, macrophages were (re)polarized in the presence or absence of 2-deoxiglucose (2-DG, 5 mM). iNOS and Arg1 expression was assessed by flow cytometry. Left, dot plots depicting gating strategy and population percentages of a typical experiment; right, iNOS+ or Arg1+ macrophage percentage (n = 5, two-way ANOVA followed by Fisher’s LSD post hoc). B, cytokine secretion in supernatant (cytokine bead array) (n = 3). C, metabolic profiles as determined by extracellular flux analysis (Seahorse XF). Extracellular acidification rate (ECAR, left) and mitochondrial oxygen consumption rates (OCR, right) were determined following the addition of glucose (Glu, 25 mM), oligomycin (Oligo, 1,5 µM), FCCP (1,5 µM) and Rotenone (Rot, 100 nM), antimycin A (AA,1 µM) and 2-DG (50 mg/ml). D, energy profile in the presence of glucose. E, calculated levels of glycolysis, glycolytic capacity and reserve, mitochondrial basal and maximal respiration, and ATP production (mean ± SEM, n = 3, paired one-way ANOVA followed by Fisher’s LSD test) (p values: * = p < .05, ** = p < .01 and *** = p < .001)
It has been proposed that both LPS+IFN-γ- and IL-4-mediated polarization depend on glycolysis.31 To assess whether glycolysis contributes to M2-to-M1 macrophage re-polarization, BM-M were re-polarized in the presence of the glycolysis inhibitor 2-deoxiglucose (2-DG). The increase in iNOS expression induced upon IL-4→LPS+IFN-γ re-polarization was partially blocked by 2-DG, similarly to the 2-DG effect on LPS+IFN-γ polarization (Figure 1a, lower panel dot plots). Arg1 expression was strongly impaired when 2-DG was used during IL-4 promoted M2 polarization (Figure 1a), while it was not affected by 2-DG during IL-4→LPS+IFN-γ re-polarization, suggesting that maintenance of Arg1 expression in previously IL4-polarized macrophages does not require glucose. These results support the role of glycolysis in macrophage re-polarization but also point toward a contribution of other metabolic pathways.
To gain insight into the functional activity after IL-4→LPS+IFN-γ re-polarization, we determined cytokine secretion. As expected, re-polarization resulted in an increased secretion of pro-inflammatory cytokines (IL-23, TNF-α, and IL-27) comparable to LPS+IFN-γ polarized macrophages (Figure 1b and Sup. Figure 1b). MCP-1 secretion was lower in re-polarized macrophages than in IL-4 polarized macrophages.Next, we assessed the metabolic rewiring after LPS+IFN-γ-induced re-polarization as real-time changes in the extracellular acidification rate (ECAR, for aerobic glycolysis) and the oxygen consumption rate (OCR, an OXPHOS indicator) (Figure 1c). The overall energy profile was determined (Figure 1d). The energy profile of IL-4-treated macrophages resembled that of the untreated control M0 macrophages whilst IL-4→LPS+IFN-γ re-polarized macrophage displayed a decreased glycolysis and impaired mitochondrial respiration, the latter was comparable to the response of LPS+IFN-γ M1 macrophages. A detailed analysis (Figure 1e) revealed that IL-4→LPS+IFN-γ re-polarization reduced glycolysis, glycolytic capacity, and glycolytic reserve and that mitochondrial respiration parameters (basal respiration, maximal respiration, and ATP production) were as impaired as seen after LPS+IFN-γ polarization, suggesting a metabolic re-polarization.
Metabolic drugs PerHx and VXL promote M2 to M1-like re-polarization
As LPS+IFN-γ re-polarization impaired mitochondrial respiration, we next studied if selected metabolic drugs could also induce M2-to-M1 re-polarization. Treatment of M2-polarized macrophages with the drugs PerHx, VLX, CB-839, TMZ, and HX531 for 24 h did not significantly change Arg1 or iNOS expression (Sup. Figure 2a), suggesting that an additional signal is needed for macrophage re-polarization. Since in a tumor microenvironment, numerous cells of innate and adaptive immunity produce IFN-γ, 32 we explored the effect of the metabolic drugs in the presence of IFN-γ. As a control, IL-4-polarized macrophages were incubated with IFN-γ for 24 hours (Figure 2a). This IFN-γ treatment alone induced a small, yet significant increase in the percentage of cells expressing iNOS (7,7% versus 1,3%) which however is much lower than the ~90% that is obtained upon IL-4→LPS+IFN-γ re-polarization (Figure 1a). Notably, treatment of IL4-polarized macrophages with VLX and PerHx in combination with IFN-γ increased the frequency of iNOS-expressing macrophages to 55% and 87%, respectively. The population of Arg1 expressing macrophages did not change significantly remaining near 40% for both conditions. TMZ, CB-839, or HX531 did not induce re-polarization (Sup. Figure 2a). Cell viability was not affected by any of the drugs (Sup. Figure 2b).
Figure 2.
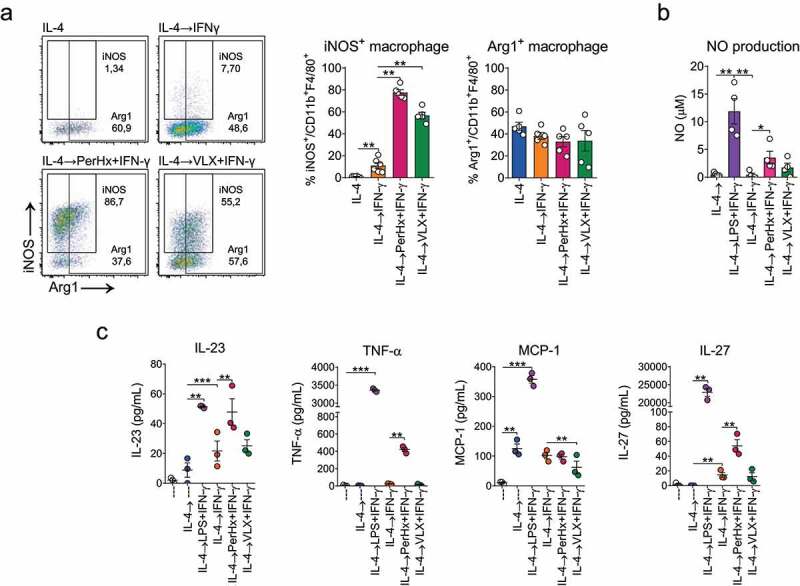
Metabolic drugs increase IFN-γ-mediated macrophage re-polarization. BM-M were polarized with IL-4 (20 ng/ml, 24 h), washed and treated with PerHx (5 µM) or VLX (10 µM) and after 1 h IFN-γ (20 ng/ml) was added for another 24 h. Cells were analyzed by flow cytometry and the supernatants were collected to assess NO production and cytokine secretion. A, iNOS and Arg1 expression. Left, representative dot plots; right, percentages of iNOS+ and Arg1+ macrophages (n = 5). B, NO production as determined by Griess reaction (n = 3). C, cytokines secreted in supernatants (cytokine beads array) (n = 3). Bars represent the mean ± SEM of 3–5 experiments, analyzed by paired one-way ANOVA followed by Fisher’s LSD test (p values: * = p < .05, ** = p < .01 and *** = p < .001)
Since PerHx and VLX in the presence of IFN-γ stimulated macrophage re-polarization, we further characterized the changes elicited with these drugs. As in pro-inflammatory macrophages NO production is increased, we evaluated NO production upon drug-induced re-polarization. As shown in Figure 2b, IL-4→LPS+IFN-γ re-polarization increased NO up to ~12 µM, compared with 1 µM in IL-4 polarized macrophage. IFN-γ re-polarization did not increase NO production, yet PerHx in the presence of IFN-γ (PerHx+IFN-γ) significantly enhanced NO production (~3 µM) and also VLX+IFN-γ resulted in a low, but not significant, enhanced NO production.
We next determined cytokine secretion during the drug-mediated re-polarization (Figure 2c). While IFN-γ alone had no significant effect on the level of cytokine secretion, PerHx+IFN-γ significantly increased TNF-α, IL-23, and IL-27 secretion. VLX+IFN-γ treatment however did not change the levels of TNF-α, IL-23, and IL-27 secretion but did suppress MCP-1 secretion compared to IFN-γ treatment alone. Thus, although PerHx and VLX seem to result in different re-polarization pathways, these results support our hypothesis that targeting mitochondrial metabolic pathways can induce macrophage re-polarization.
Drugs promoting macrophage re-polarization impair OXPHOS
As LPS+IFN-γ-mediated re-polarization impaired OXPHOS, we determined whether VLX and PerHx impair mitochondrial respiration. IFN-γ treatment of IL-4-polarized macrophages did not induce changes in the energy profile (Figure 3a). However, upon VLX+IFN-γ and PerHx+IFN-γ treatment, oxygen consumption was abrogated, resembling the effect of LPS+IFN-γ during re-polarization (Figure 1c). The ECAR profile slightly differed between the conditions. A further analysis revealed that under VLX+IFN-γ re-polarization glycolysis was reduced while PerHx+IFN-γ re-polarization did not alter glycolysis, as compared to IFN-γ alone (Figure 3c). The maximal respiration and glycolytic reserve were decreased by both drugs and comparable to the responses seen upon LPS+IFN-γ re-polarization (Figure 3c and Figure 1c-E). Furthermore, both drugs abrogated the basal and maximal respiration, as well as ATP production (Figure 3c). Combined, these results suggest that PerHx and VLX impair mitochondrial respiration and thus promote M2 to M1-like re-polarization. Since impaired mitochondrial respiration has been associated with damaged mitochondria, we assessed mitochondrial mass and membrane potential (ψ) (Figure 3d and E) to measure mitochondrial healthiness. VLX+IFN-γ treatment decreased mitochondrial mass and reduced membrane potential although the ratio membrane potential: mitochondrial mass remained unchanged (data not shown), while PerHx+IFN-γ treatment had no effect on mitochondrial mass or membrane potential.
Figure 3.
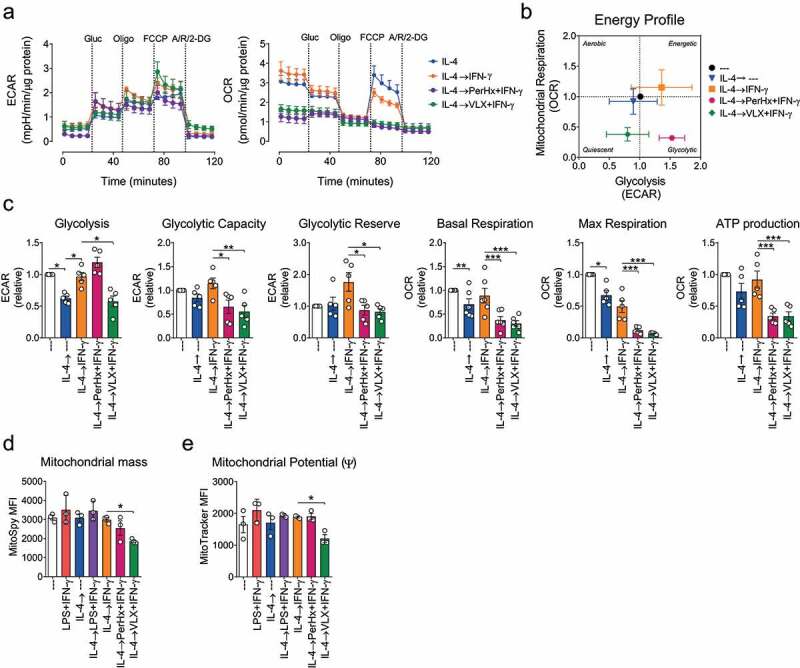
Drug-mediated macrophage re-polarization impairs OXPHOS. BM-M were polarized with IL-4 (20 ng/ml, 24 h), washed and treated with PerHx (5 µM) or VLX (10 µM) after 1 h IFN-γ (20 ng/ml) was added for another 24 h. A, metabolic profile as determined by extracellular flux analysis. ECAR (left) and OCR (right) were determined following the addition of glucose (Glu, 25 mM), oligomycin (Oligo, 1,5 µM), FCCP (1,5 µM) and Rotenone (Rot, 100 nM), antimycin A (AA,1 µM) and 2-DG (50 mg/ml). B, energy profile in the presence of glucose. C, calculated levels of glycolysis, glycolytic capacity and reserve, mitochondrial basal and maximal respiration, and ATP production (n = 5). D, mitochondrial mass and E mitochondrial potential as determined by flow cytometry using MitoSpy Green FM and MitoTracker Red CMXRos, respectively (measured MFI is shown, n = 3). Bars represent the mean ± SEM. Data were analyzed by paired one-way ANOVA followed by Fisher’s LSD test (p values: * = p < .05, ** = p < .01 and *** = p < .001)
Macrophage metabolic re-polarization induces antitumor cytotoxicity
We hypothesized that re-polarizing anti-inflammatory macrophages into pro-inflammatory cells would also unleash macrophage cytotoxic activity against tumor cells. In order to test whether the drug-promoted re-polarization increases macrophage-mediated cytotoxicity, re-polarized macrophages were co-cultured with CFSE-stained TC-1 tumor cell in media without drugs or cytokines (Figure 4a). After 24 hours of co-culture total cells were analyzed by flow cytometry, based on CD45 expression and CFSE staining (Figure 4b, gating strategy Sup. Figure 3a). As shown in Figure 4c, the percentage of TC-1 cells (CD45-) obtained after the coculture was lower when cocultured with M1-like re-polarized macrophages. As expected, both LPS+IFN-γ polarization and IL-4→LPS+IFN-γ re-polarization decreased TC-1 cells, while IL-4→IFN-γ re-polarized macrophages had no effect on the number of TC-1 recovered. PerHx+IFN-γ slightly decreased the percentage of TC-1 cells, yet VLX+IFN-γ re-polarization induced a similar effect as LPS+IFN-γ re-polarization. Since the number of macrophages did not change over time (Sup. Figure 3b) the cytotoxic activity of macrophages toward TC-1 (Figure 4c) could be determined based on the percentage of TC-1 cells after the co-culture (Sup. Figure 3c). The response of untreated macrophages was defined as no cytotoxicity. IL-4 polarized macrophage promoted TC-1 survival (negative cytotoxicity) and IL-4→IFN-γ re-polarized macrophages exhibited a slight, yet not significant, increase in cytotoxicity. LPS+IFN-γ polarized and IL-4→LPS+IFN-γ re-polarized macrophages induced 75% cytotoxicity. Notably, PerHx+IFN-γ treatment resulted in approximately 40% and VLX+IFN-γ treatment up to 75% of cytotoxicity, which was comparable to the response seen upon LPS+IFN-γ re-polarization. To further confirm the cytotoxic effect of re-polarized macrophages, we determined whether tumor cells undergo apoptosis. As shown in Figure 4d (and supplementary figure 3f-G), IL-4→LPS+IFN-γ re-polarized macrophages induced a strong increase in Annexin V surface expression up to 56%. Similarly, VLX+IFN-γ re-polarization induced apoptosis in up to 50% of the cell. PerHx+IFN-γ re-polarization was less effective in promoting apoptosis (30% of annexin V+ cells). Combined, our data suggest that M1-like macrophages promote tumor cell cytotoxicity by triggering apoptosis.
Figure 4.
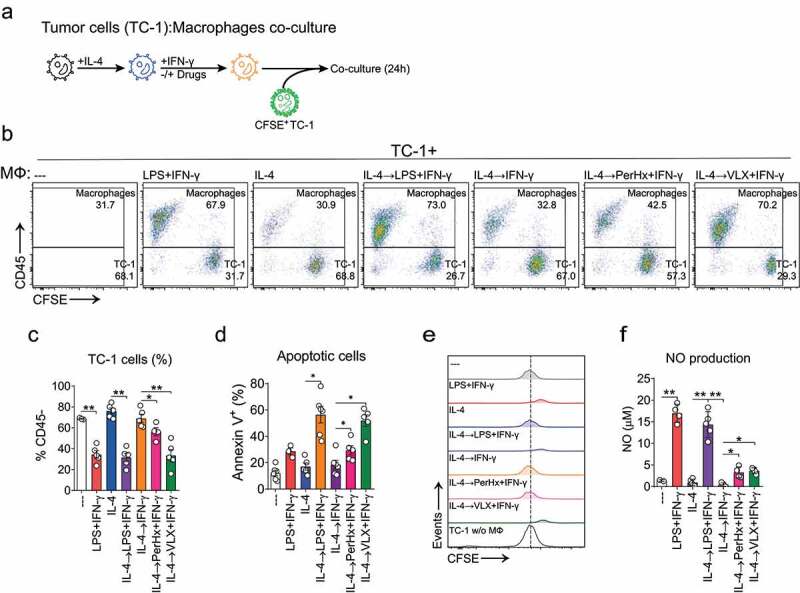
Macrophage metabolic re-polarization improves antitumoral cytotoxicity. BM-M were polarized with IL-4 (20 ng/ml, 24 h), washed and treated with PerHx (5 µM) or VLX (10 µM), after 1 h IFN-γ (20 ng/ml) was added. After another 24 h the wells were washed and CFSE-stained TC-1 tumor cells were added at a macrophage to TC-1 cell ratio of 2 to 1. A, protocol summary. B, after 24 h of co-culture, cells were harvested and analyzed by flow cytometry. Macrophage frequency was based on the percentage of CD45+ cells as TC-1 cells are CD45-. Numbers indicate percentages after co-culture (n = 4). C, percentage of TC-1 (CD45-) cells recovered after co-culture. D, apoptosis induce by macrophage was determined as annexin V+ cell (%) (n = 3–6). E, CFSE dilution in TC-1 cells. CFSE histograms of TC-1 cells (gated from living cells CD45-). A representative figure is shown (n = 5–6). F, NO production as determined by Griess reaction (n = 4–5). Bars represent the mean ± SEM. Data were analyzed by paired one-way ANOVA followed by Fisher’s LSD test (p values: * = p < .05, ** = p < .01 and *** = p < .001)
We next evaluated the dilution of CFSE stain as an indicator of tumor cell proliferation (gating strategy Sup. Figure 3a). As depicted in Figure 4d, TC-1 cells cultured without macrophages, with untreated macrophages or with IL-4 polarized, IFN-γ or PerHx+IFN-γ treated macrophages displayed the same level of CFSE dilution. This suggests that surviving TC-1 cells proliferated to the same extent. Opposite, TC-1 cultured with LPS+IFN-γ polarized, IL-4→LPS+IFN-γ or VLX+IFN-γ treated macrophages retained higher levels of CFSE (Figure 4d, supplementary Figure 3b), suggesting inhibition of proliferation.
To exclude that the TC-1 cytotoxicity was merely due to direct toxicity of drug remaining after washing, TC-1 cells were cultured in the presence of supernatants harvested from drug-repolarized macrophages that had previously been washed 1–3 times after the 1 h-incubation with the drugs. As shown in supplementary Figure 3e, the culture supernatant of drug-treated macrophages already after 1 wash did not affect TC-1 proliferation, supporting that the cytotoxic effect is macrophage-mediated. It should be noted that when incubating TC-1 directly with VLX, cell survival was strongly impaired (Sup. Figure 3d). On the other hand, PerHx did not affect TC-1 cell proliferation.
Next, NO production during the co-culture was determined. As shown in figure 4f, IL-4→LPS+IFN-γ re-polarized macrophages increased NO production up to 15 µM, as compared to IL-4 polarized cells (around 1 µM). In line with the experiments depicted in Figure 2c, IL-4→IFN-γ re-polarization did not induce NO production. Yet, PerHx or VLX in the presence of IFN-γ significantly increased NO production (both appr. 4 µM). Combined, our results suggest that drug-induced macrophage re-polarization unleashes anti-tumor cell responses.
Metabolic drugs can prevent mouse macrophage polarization
As monocytes infiltrating the TME will primarily differentiate into M2 macrophages, preventing M2 polarization could decrease anti-inflammatory responses. Thus, we evaluated whether the selected drugs can also prevent IL-4-mediated M2 polarization. M0 macrophages were pre-treated with the drugs PerHx, VLX, CB-839, HX531, or TMZ for 1 hour and then cultured for 24 hours with IL-4 (Figure 5a). While in M0 macrophages approximately 3% of the cells expressed Arg1, this level increased up to 30% in IL-4 polarized M2 macrophages. TMZ and CB-839 treatment partially prevented Arg1 expression (reaching 20% and 17%, respectively). HX531 had a more pronounced effect, reducing Arg1 expression to 10%, and PerHx almost completely impaired its expression (5%). Moreover, PerHx slightly increased the number of cells expressing iNOS (~2% vs 1% in IL-4-polarized cells). Unexpectedly, VLX increased IL-4-promoted Arg1 expression up to 40%. None of the drugs affected cell viability or induced macrophage polarization (Sup. Figure 4a).
Figure 5.
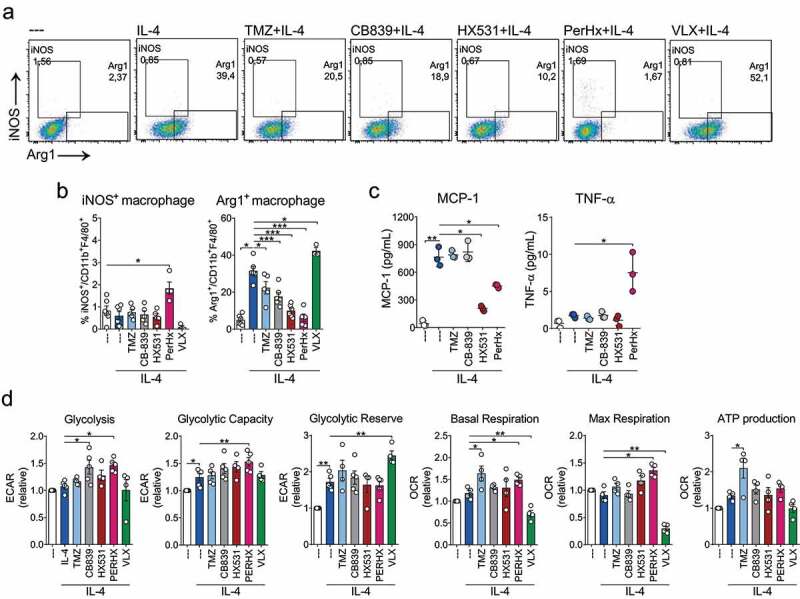
Metabolic drugs prevent IL-4-mediated polarization. BM-M were treated with TMZ (100 µM), CB-839 (5 µM), HX531 (10 µM), PerHx (5 µM) and VLX (10 µM) for 1 h and then IL-4 (20 ng/ml) was added for 24 h. A, iNOS and Arg1 expression was assessed by flow cytometry. Dot plots depicting gating strategy and populations percentages. B, iNOS+ and Arg1+ macrophage percentages (n = 5). C, cytokines were evaluated from the supernatant by flow cytometry (beads array) (n = 3). D, metabolic profile as determined by extracellular flux analysis (Seahorse XF). Calculated levels of glycolysis, glycolytic capacity and reserve, mitochondrial basal and maximal respiration, and ATP production (n = 5) mean ± SEM, paired one-way ANOVA followed by Fisher’s LSD test, p values: * = p < .05, ** = p < .01 and *** = p < .001)
To assess the functional changes induced by the drugs, we measured cytokine secretion, focusing only on those drugs that impaired IL-4 polarization. Interestingly, neither TMZ nor CB-839 altered cytokine secretion (Figure 5c). IL-4-induced MCP-1 secretion was strongly impaired by HX531 and PerHx (Figure 5c) and PerHx treatment resulted in a minor increase in TNF-α secretion.
Next, we determined metabolic changes. Non-polarized and IL-4 polarized macrophages displayed similar metabolic profiles. Interestingly, PerHx treatment increased glycolysis and maximal glycolytic capacity. Moreover, PerHx did not impair mitochondrial respiration as it did during IFN-γ+ PerHx re-polarization; instead, it increased both basal and maximal mitochondrial respiration. On the other hand, VLX increased glycolytic reserve while decreasing basal and maximal respiration. HX531 had no effect on cell metabolism, CB-839 increased glycolysis and TMZ increased basal respiration and ATP production.
Combined our data suggest that IL-4 polarization can be prevented by the interference of metabolic pathways, which differentially impacts macrophage metabolism. Moreover, IL-4 polarization can be prevented without impairing mitochondrial respiration.
DISCUSSION
It is generally assumed that the effectiveness of cancer immunotherapies can be enhanced with strategies that change the immunosuppressive tumor environment. In this study, we, therefore, focused on a strategy to employ metabolic drugs to modify immunosuppressive M2-like macrophages into antitumoral M1-like macrophages and to prevent the differentiation of M0 macrophages into M2-like macrophages. Here, we demonstrate that the drugs PerHx and VLX, in the presence of IFN-γ, can indeed change immunosuppressive M2 macrophages into macrophages with M1-like features, including tumor cytotoxic activity, by impairing mitochondrial respiration. This study furthermore demonstrates that the metabolic drugs PerHx, CB-839, Hx-531, and TMZ can prevent IL-4-driven M2 polarization. These are encouraging data that suggest that drugs targeting mitochondrial metabolism can promote a pro-inflammatory microenvironment and prevent a macrophage-mediated immunosuppressive response.
In this study, LPS+IFN-γ induced M2-to-M1 re-polarization increased iNOS expression, NO production, and pro-inflammatory cytokines secretion, in line with a previous report that showed up-regulation of CD80 and CD86 expression after re-polarization.16 Here we also show that LPS+IFN-γ re-polarization did not change the expression of Arg1. Apparently, LPS+IFN-γ driven induction of M1 features in previously M2 polarized macrophages does not abrogate all M2 features. Similarly, Vidyarthi et al.33 showed that TLR-3 ligation to skew M2 into an M1 phenotype resulted in a pronounced increase of M1 markers (CD86, CD80, CD40, iNOS, and pro-inflammatory cytokine secretion), yet only in a slight decrease of M2 markers (CD206 and Tim-3). Surprisingly, TNF-α and IL-23 secretion were higher in IL-4→LPS+IFN-γ re-polarized macrophages than in LPS+IFN-γ polarized. It has been reported that IL-4 potentiated the secretion of proinflammatory cytokines upon additional bacterial stimulation (a kind of M1-like polarization) in a mechanism dependent of the MyD88 signaling pathway.7 Thus, IL-4 stimulation might activate signaling pathways involved in protein synthesis and secretion that are enhanced by LPS+IFN-γ re-polarization.
The metabolic drugs PerHx or VLX in the presence of IFN-γ promoted macrophage re-polarization. In the absence of IFN-gamma, the drugs did not induce significant changes. However, considering that IFN-γ is produced by both innate and adaptive immune cells, it will be present in the tumor microenvironment. And notably, anti-cancer treatments often result in increased intratumoral IFN-γ levels.
M2 macrophages treated with VLX+IFN-γ displayed a strong anti-tumoral response, which was comparable to the antitumor activity observed upon treatment with LPS+IFN-γ. VLX+IFN-γ treatment also moderately increased iNOS expression and NO production but did not significantly change pro-inflammatory cytokine secretion. The effect of VLX in the tumor cytotoxicity assay can be fully ascribed to its effect on macrophages as direct toxicity of VLX on TC-1 cells in this assay could be excluded. Notably, VLX-induced reduction of MCP-1 secretion may prevent the infiltration and development of immunosuppressive macrophages in vivo.34 Macrophage-mediated cytotoxicity elicited upon PerHx+IFN-γ treatment of M2 macrophages was moderately increased and accompanied by moderate NO production and TNF-α secretion. Yet, increased secretion of IL-23 and IL-27 elicited upon PerHx+IFN-γ re-polarization could enhance the anti-tumor immune response in vivo.35 Based on our results we will next study the in vivo effect of PerHx and VLX. We anticipate that metabolic re-polarization of macrophages 1) may induce a direct anti-tumoral effect (as shown in this report) and/or 2) create a more pro-inflammatory tumor milieu thereby increasing the efficacy of current immunotherapies.36 For instance, re-programming macrophages into a pro-inflammatory immune cell improved the efficacy of anti-PD-1 therapy.37 In addition, Palmieri E. et al. showed that the deletion of glutamine synthase in macrophages skews M2-polarized macrophages toward M1-like, promoting the accumulation of cytotoxic T cells and inhibition of metastasis in tumor-bearing mice.38
Little is known about metabolic changes during M2 to M1-like re-polarization. Our data show that glycolysis contributed to IL-4→LPS+IFN-γ re-polarization; while impaired mitochondrial respiration is a feature of M1 re-polarized macrophages, similar to what has been described for M1 polarization.17 By selecting drugs targeting mitochondrial metabolism, we sought to impair mitochondrial respiration and thus shift cell metabolism into a glycolysis to mimic the metabolic response upon LPS+IFN-γ polarization. Our data point to increased glycolytic activity and impaired OXPHOS after LPS+IFN-γ polarization (Figure 1d) is in line with previous reports;8 however, after re-polarization using LPS+IFN-γ (Figure 1d) or VLX+IFN-γ (Figure 3b) cells displayed an impaired OXPHOS but also a slightly reduction in glycolysis. Recently, Palmieri E. et al. showed that nitric oxide (NO) produced upon LPS+IFN-γ treatment targets pyruvate dehydrogenase (PDH), decreasing glucose-derived carbon influx and thus induces uptake of glutamine and its utilization as a carbon source.9 Since it is known that glutaminolysis-related genes are increased in IL-4-induced M2 macrophages,10 we hypothesize that the increase in glutamine metabolism during M2 polarization provides M1-like re-polarized macrophages of the machinery to use glutamine as a carbon source instead of glucose. In our experimental setting, PerHx+IFN-γ and VLX+IFN-γ abrogated mitochondrial respiration. A reduced activity of the electron transport chain (ETC) would be the most likely explanation, as it has been described that LPS+IFN-γ treatment impairs the activity of complexes I and II of the ETC.16 Likewise, it has been proposed that VLX impairs ETC activity and mitochondrial respiration by decreasing complex IV activity.27 However, PerHx does not target the ETC directly; it impairs CPT-1/2 activity and thus decreases the availability of fatty acids to be oxidized.22,39 Yet recently it has also been demonstrated that in the cell lines MCF-7 and T47D, PerHx did not directly impair FAO.40 Even more, the same mechanism was described for Etomoxir (another CPT-1 inhibitor), which impaired IL-4 mediated polarization independently of CPT-1 inhibition by an off-target inhibition of the Adenine Nucleotide Translocator (ANT) and depletion of cytoplasmic CoA.41 Further studies are needed to unravel the effect of PerHx on mitochondrial respiration and immune cells, considering that PerHx dosage may have to be adjusted per patient to prevent side effects.42
Preventing monocytes to become M2-like macrophages could also decrease immunosuppression in the TME We found that four out of five drugs tested prevented IL-4-mediated M2 polarization. The effect of PerHx and TMZ suggests that an impaired FAO prevents the acquisition of an M2 phenotype. PerHx strongly suppressed the expression of Arg1 and even slightly increased iNOS expression and TNF-α secretion, while preventing MCP-1 release. Intriguingly, PerHx increased both mitochondrial respiration and glycolysis but did not impair mitochondrial activity which contrasts with its effect on re-polarization (Figure 3). This response is likely a consequence of metabolic compensation, as it has been described that macrophages can adjust their metabolism to cope with stress.43 On the other hand, TMZ prevented M2 polarization less efficiently. Although it has been shown that TMZ targets 3-ketoacyl-CoA thiolase (3-KAT),44 it has been suggested that it does not affect cardiac FAO,45 while it can support ATP synthesis by elevating mitochondrial Ca2+ level,46 in line with our results. As expected, the glutaminolysis inhibitor CB-839 impaired M2 polarization, since glutamine metabolism is fundamental for M2 polarization,10 likely reducing α-ketoglutarate synthesis. α-Ketoglutarate, besides its functions in cell metabolism, is an anti-inflammatory metabolite that promotes M2 polarization.47 Interestingly, HX531 strongly prevented M2 polarization while it had no impact on cell metabolism, probably since HX531 antagonizes the nuclear receptor RXR, which in turn controls other transcriptional factors (e.g., PPARs, retinoic acid receptor, vitamin D receptor).48 PPARγ−/- macrophages display a reduced MCP-1 expression,49 in line with the effect observed with Hx531 (Figure 5c). To our surprise, VLX increased Arg1 expression upon IL-4 polarization while strongly impairing mitochondrial metabolism. We hypothesize that VLX-induced OXPHOS impairment (Figure 5d) decreases metabolite production, which is critical for M2 phenotype polarization, leading to the increase in Arg1 expression as a coping mechanism. For instance, an impaired OXPHOS could lead to a decrease production of metabolites like proline, citrulline, and ornithine,50 which are essential for IL-4-polarized M2 macrophage wound-healing functions.51 Nonetheless, further studies are required to determine if VLX indeed can enhance M2 polarization and not only Arg1 expression. Despite the fact that we focused on a mouse model, we believe that this knowledge could be translated to human monocyte-to-macrophage differentiation. Further experiments are being conducted to demonstrate the effect of these metabolic drugs on human monocyte/macrophage polarization.
In conclusion, this study shows that macrophage re-polarization from M2 into an M1-like phenotype as well as prevention of M2 polarization was achieved by targeting mitochondrial metabolism. We showed that impaired mitochondrial respiration correlates with enhanced re-polarization into M1-like phenotype and anti-tumoral functions. Moreover, we have established a new platform to test whether drugs clinically available can promote macrophage re-polarization into a pro-inflammatory phenotype, which opens the window for new research lines and treatments aiming for functional re-polarization of macrophages.
Supplementary Material
Acknowledgments
The authors acknowledge the graduate school GSMS of the UMCG for a scholarship for AVC and the Chinese Scholarship Council (CSC) for a scholarship for SC.
Funding Statement
This research was funded by the Dutch Cancer Society grant 10330.
DECLARATIONS
Ethics approval and consent to participate
The mice were kept according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals guidelines and all experiments were approved by the Animal Ethical Committee of the University of Groningen.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Competing interests
Not applicable.
Authors’ contributions
All authors made substantial contributions to the manuscript. Conception and design: CO, TD. Supervision and funding acquisition: CO, TD. Collection and assembly of data: CO, AVC, SC, AB. Data analysis and interpretation: CO, AVC, SC, AB, TD. Manuscript writing: CO, TD. All authors reviewed iterations of the report and approved the final version for submission.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, Coussens LM, Gabrilovich DI, Ostrand-Rosenberg S, Hedrick CC, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24(5):541–13. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caux C, Ramos RN, Prendergast GC, Bendriss-Vermare N, Menetrier-Caux C.. A Milestone Review on How Macrophages Affect Tumor Growth. Cancer Res. 2016;76(22):6439–6442. doi: 10.1158/0008-5472.CAN-16-2631. [DOI] [PubMed] [Google Scholar]
- 3.Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26(1):78. doi: 10.1186/s12929-019-0568-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. Journal of Immunology. 2000;164(12):6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 5.Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage Polarization: different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front Immunol. 2019;10:1084. doi: 10.3389/fimmu.2019.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Galvan-Pena S, O’Neill LA. Metabolic reprograming in macrophage polarization. Front Immunol. 2014;5:420. doi: 10.3389/fimmu.2014.00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viola A, Munari F, Sanchez-Rodriguez R, Scolaro T, Castegna A. The Metabolic Signature of Macrophage Responses. Front Immunol. 2019;10:1462. doi: 10.3389/fimmu.2019.01462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jha AK, Huang SC, Sergushichev A, Lampropoulou V, Ivanova Y, Loginicheva E, Chmielewski K, Stewart KM, Ashall J, Everts B, et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity. 2015;42(3):419–430. doi: 10.1016/j.immuni.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Vats D, Mukundan L, Odegaard JI, Zhang L, Smith KL, Morel CR, Wagner RA, Greaves DR, Murray PJ, Chawla A. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 2006;4(1):13–24. doi: 10.1016/j.cmet.2006.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. 2007;447(7148):1116–1120. doi: 10.1038/nature05894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, Rastinejad F. Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature. 2008;456(7220):350–356. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nomura M, Liu J, Rovira II, Gonzalez-Hurtado E, Lee J, Wolfgang MJ, Finkel T. Fatty acid oxidation in macrophage polarization. Nat Immunol. 2016;17(3):216–217. doi: 10.1038/ni.3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mills EL, Kelly B, Logan A, Costa ASH, Varma M, Bryant CE, Tourlomousis P, Dabritz JHM, Gottlieb E, Latorre I, et al. Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell. 2016;167(2):457–70 e13. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Van Den Bossche J, Baardman J, Otto NA, Van Der Velden S, Neele AE, Van Den Berg SM, Luque-Martin R, Chen HJ, Boshuizen MC, Ahmed M, et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016;17(3):684–696. doi: 10.1016/j.celrep.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 17.Freemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, Macintyre AN, Goraksha-Hicks P, Rathmell JC, Makowski L. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. 2014;289(11):7884–7896. doi: 10.1074/jbc.M113.522037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan Z, Xie N, Cui H, Moellering DR, Abraham E, Thannickal VJ, Liu G. Pyruvate dehydrogenase kinase 1 participates in macrophage polarization via regulating glucose metabolism. Journal of Immunology. 2015;194(12):6082–6089. doi: 10.4049/jimmunol.1402469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mitchem JB, Brennan DJ, Knolhoff BL, Belt BA, Zhu Y, Sanford DE, Belaygorod L, Carpenter D, Collins L, Piwnica-Worms D, et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells, relieves immunosuppression, and improves chemotherapeutic responses. Cancer Res. 2013;73(3):1128–1141. doi: 10.1158/0008-5472.CAN-12-2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang F, Parayath NN, Ene CI, Stephan SB, Koehne AL, Coon ME, Holland EC, Stephan MT, et al. Genetic programming of macrophages to perform anti-tumor functions using targeted mRNA nanocarriers. Nat Commun. 2019;10(1):3974. doi: 10.1038/s41467-019-11911-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Den Bossche J, Baardman J, De Winther MP. Metabolic Characterization of Polarized M1 and M2 Bone Marrow-derived Macrophages Using Real-time Extracellular Flux Analysis. Journal of Visualized Experiments: JoVE. 2015;105:105. doi: 10.3791/53424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol. 1996;52(2):273–280. doi: 10.1016/0006-2952(96)00204-3. [DOI] [PubMed] [Google Scholar]
- 23.Sallustio BC, Westley IS, Morris RG. Pharmacokinetics of the antianginal agent perhexiline: relationship between metabolic ratio and steady-state dose. Br J Clin Pharmacol. 2002;54(2):107–114. doi: 10.1046/j.1365-2125.2002.01618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lopaschuk GD, Barr R, Thomas PD, Dyck JR. Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long-chain 3-ketoacyl coenzyme a thiolase. Circ Res. 2003;93(3):e33–7. doi: 10.1161/01.RES.0000086964.07404.A5. [DOI] [PubMed] [Google Scholar]
- 25.Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther. 2014;13(4):890–901. doi: 10.1158/1535-7163.MCT-13-0870. [DOI] [PubMed] [Google Scholar]
- 26.Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, Gogvadze V, Dang L, Pahlman S, Schughart LA, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun. 2014;5:3295. doi: 10.1038/ncomms4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fryknas M, Zhang X, Bremberg U, Senkowski W, Olofsson MH, Brandt P, Persson I, D'Arcy P, Gullbo J, Nygren P, et al. Iron chelators target both proliferating and quiescent cancer cells. Sci Rep. 2016;6:38343. doi: 10.1038/srep38343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mody K, Mansfield AS, Vemireddy L, Nygren P, Gulbo J, Borad M. A phase I study of the safety and tolerability of VLX600, an Iron Chelator, in patients with refractory advanced solid tumors. Invest New Drugs. 2019;37(4):684–692. doi: 10.1007/s10637-018-0703-9. [DOI] [PubMed] [Google Scholar]
- 29.Kanayasu-Toyoda T, Fujino T, Oshizawa T, Suzuki T, Nishimaki-Mogami T, Sato Y, Sawada J, Inoue K, Shudo K, Ohno Y, et al. HX531, a retinoid X receptor antagonist, inhibited the 9-cis retinoic acid-induced binding with steroid receptor coactivator-1 as detected by surface plasmon resonance. J Steroid Biochem Mol Biol. 2005;94(4):303–309. doi: 10.1016/j.jsbmb.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 30.Yamauchi T, Waki H, Kamon J, Murakami K, Motojima K, Komeda K, Miki H, Kubota N, Terauchi Y, Tsuchida A, et al. Inhibition of RXR and PPARgamma ameliorates diet-induced obesity and type 2 diabetes. J Clin Invest. 2001;108(7):1001–1013. doi: 10.1172/JCI12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao Q, Chu Z, Zhu L, Yang T, Wang P, Liu F, Huang Y, Zhang F, Zhang X, Ding W, et al. 2-Deoxy-d-Glucose Treatment Decreases Anti-inflammatory M2 Macrophage Polarization in Mice with Tumor and Allergic Airway Inflammation. Front Immunol. 2017;8:637. doi: 10.3389/fimmu.2017.00637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mojic M, Takeda K, Hayakawa Y. The Dark Side of IFN-gamma: its Role in Promoting Cancer Immunoevasion. Int J Mol Sci. 2017;19(1):1. doi: 10.3390/ijms19010089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vidyarthi A, Khan N, Agnihotri T, Negi S, Das DK, Aqdas M, Chatterjee D, Colegio OR, Tewari MK, Agrewala JN. TLR-3 Stimulation Skews M2 Macrophages to M1 Through IFN-alphabeta Signaling and Restricts Tumor Progression. Front Immunol. 2018;9:1650. doi: 10.3389/fimmu.2018.01650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, Lopez-Bravo M, Joven J, Ardavin C, Rodriguez-Fernandez JL, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. Journal of Immunology. 2014;192(8):3858–3867. doi: 10.4049/jimmunol.1302821. [DOI] [PubMed] [Google Scholar]
- 35.Cocco C, Di Carlo E, Zupo S, Canale S, Zorzoli A, Ribatti D, Morandi F, Ognio E, Airoldi I. Complementary IL-23 and IL-27 anti-tumor activities cause strong inhibition of human follicular and diffuse large B-cell lymphoma growth in vivo. Leukemia. 2012;26(6):1365–1374. doi: 10.1038/leu.2011.363. [DOI] [PubMed] [Google Scholar]
- 36.Duan Q, Zhang H, Zheng J, Zhang L. Turning Cold into Hot: firing up the Tumor Microenvironment. Trends Cancer. 2020;6(7):605–618. doi: 10.1016/j.trecan.2020.02.022. [DOI] [PubMed] [Google Scholar]
- 37.De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, Budhu S, Ghosh A, Pink M, Tchaicha J, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature. 2016;539(7629):443–447. doi: 10.1038/nature20554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palmieri EM, Menga A, Martin-Perez R, Quinto A, Riera-Domingo C, De Tullio G, Hooper DC, Lamers WH, Ghesquiere B, McVicar DW, et al. Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep. 2017;20(7):1654–1666. doi: 10.1016/j.celrep.2017.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kennedy JA, Kiosoglous AJ, Murphy GA, Pelle MA, Horowitz JD. Effect of perhexiline and oxfenicine on myocardial function and metabolism during low-flow ischemia/reperfusion in the isolated rat heart. J Cardiovasc Pharmacol. 2000;36(6):794–801. doi: 10.1097/00005344-200012000-00016. [DOI] [PubMed] [Google Scholar]
- 40.Ma Y, Wang W, Devarakonda T, Zhou H, Wang XY, Salloum FN, Spiegel S, Fang X. Functional analysis of molecular and pharmacological modulators of mitochondrial fatty acid oxidation. Sci Rep. 2020;10(1):1450. doi: 10.1038/s41598-020-58334-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Divakaruni AS, Hsieh WY, Minarrieta L, Duong TN, Kim KKO, Desousa BR, Andreyev AY, Bowman CE, Caradonna K, Dranka BP, et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018;28(3):490–503 e7. doi: 10.1016/j.cmet.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Horowitz JD, Sia ST, Macdonald PS, Goble AJ, Louis WJ. Perhexiline maleate treatment for severe angina pectoris–correlations with pharmacokinetics. Int J Cardiol. 1986;13(2):219–229. doi: 10.1016/0167-5273(86)90146-4. [DOI] [PubMed] [Google Scholar]
- 43.Raulien N, Friedrich K, Strobel S, Rubner S, Baumann S, Von Bergen M, Korner A, Krueger M, Rossol M, Wagner U. Fatty Acid Oxidation Compensates for Lipopolysaccharide-Induced Warburg Effect in Glucose-Deprived Monocytes. Front Immunol. 2017;8:609. doi: 10.3389/fimmu.2017.00609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kantor PF, Lucien A, Kozak R, Lopaschuk GD. The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long-chain 3-ketoacyl coenzyme A thiolase. Circ Res. 2000;86(5):580–588. doi: 10.1161/01.RES.86.5.580. [DOI] [PubMed] [Google Scholar]
- 45.Cavar M, Ljubkovic M, Bulat C, Bakovic D, Fabijanic D, Kraljevic J, Karanovic N, Dujic Z, Lavie CJ, Wisloff U, et al. Trimetazidine does not alter metabolic substrate oxidation in cardiac mitochondria of target patient population. Br J Pharmacol. 2016;173(9):1529–1540. doi: 10.1111/bph.13454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guarnieri C, Finelli C, Zini M, Muscari C. Effects of trimetazidine on the calcium transport and oxidative phosphorylation of isolated rat heart mitochondria. Basic Res Cardiol. 1997;92(2):90–95. doi: 10.1007/BF00805569. [DOI] [PubMed] [Google Scholar]
- 47.Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al. alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat Immunol. 2017;18(9):985–994. doi: 10.1038/ni.3796. [DOI] [PubMed] [Google Scholar]
- 48.Roszer T, Menendez-Gutierrez MP, Cedenilla M, Retinoid RM. X receptors in macrophage biology. Trends Endocrinol Metab. 2013;24(9):460–468. doi: 10.1016/j.tem.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 49.Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25(8):1647–1653. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- 50.Chen Q, Kirk K, Shurubor YI, Zhao D, Arreguin AJ, Shahi I, Valsecchi F, Primiano G, Calder EL, Carelli V, et al. Rewiring of Glutamine Metabolism Is a Bioenergetic Adaptation of Human Cells with Mitochondrial DNA Mutations. Cell Metab. 2018;27(5):1007–25 e5. doi: 10.1016/j.cmet.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Campbell L, Saville CR, Murray PJ, Cruickshank SM, Hardman MJ. Local arginase 1 activity is required for cutaneous wound healing. J Invest Dermatol. 2013;133(10):2461–2470. doi: 10.1038/jid.2013.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.