

Abstract
Single-cell genomic approaches have the potential to revolutionize the study of plant systems. Here, we highlight newly developed techniques to analyze transcriptomes at single-cell resolution. We focus on the rigorous standards necessary to generate and compare these data sets introducing analysis methods that can be applied to interpret their results. Lastly, we discuss the inherent limitations of single-cell studies and address future directions for plant single-cell genomics.
Bulk methods, those that average over many cells, can give us molecular insight into tissue, and/or timepoint/developmental groups. However, these methods are limited by the inherent biases introduced by averaging over distinct cell populations. In some instances, bulk averaging can even result in qualitatively incorrect conclusions, a phenomenon termed Simpson’s paradox [1]. Single cell genomic techniques are an incredibly powerful set of tools to uncover cellular heterogeneity, as well as the development and differentiation of cell types in complex tissues with high precision. These methods have primarily been applied to animal systems, however, recently several groups have applied high-throughput single-cell transcriptomics to plants [2••,3,4,5••,6,7•,8••,9••,10••]. The application of single-cell RNA-seq to plants brings the promise of comprehensively characterizing both common and rare cell types and cell states, identifying new cell types and bring about a dynamic understanding of how cell types relate to each other spatially and developmentally. Thus far, single-cell RNA-seq plant studies have focused mostly on the well-studied and understood Arabidopsis root system [2••,5••,8••,9••,10••]. Even in this highly tractable and well understood system, with many known marker genes, and cell-types these landmark studies revealed a plethora of unknown and more robust cell type marker genes and begun to define the transition states that give rise to developmental trajectories [2••,5••,8••,9••,10••].
Several methods exist to capture transcriptomic signatures of single cells isolated by mouth-pipetting [3,4] assaying hundreds of cells at very high resolution. Because of the limitation with regards to the numbers of cells analyzed, these methods have mostly been replaced by higher-throughput methods. Droplet based methods have become popular due to their simplicity and throughput [11,12]. The droplet based-method Drop-seq has been used successfully for Arabidopsis [9••]. Droplet-based single-cell genomic systems are also commercially available such as the 10x Chromium system from 10x Genomics which has been used in many of the recent Arabidopsis single-cell root RNA-seq reports [5••,8••,10••] (Jean-Baptiste, Zhang, Ryu). Other plate-based platforms have also been used successfully in plants including CEL-seq2 [13], a method that relies on in vitro transcription for library amplification, which has been used to assay maize pollen [7•,13]. Additionally, several other methods can be applied to perform single-cell RNA-seq in plants including plate-based method capable of profiling full length RNA (SMART-Seq2) [14] or the 3’ end of transcripts (SCRB-Seq) [15]; methods extending in vitro transcription based library construction with high-throughput unbiased indexed sorting (MARS-seq2.0) [16]; microwell techniques that generate single-cell RNA-seq libraries in situ after capture in nanoliter wells (Seq-Well) [17]; and single-cell combinatorial indexing RNA-seq methods (e.g. sci-RNA-seq, Split-seq) [18,19]. The latter of these having been demonstrated to scale to the level of millions of single-cell transcriptomes [20•].
Recent single-cell publications in Arabidopsis have highlighted the promise of single-cell in plants. All of these have used the well characterized Arabidopsis root, either using whole-roots [2••,5••,9••], or specifically enriching for root tips [8••,10••]. Because plants have a continuous body plan, we can capture cells of the same type at varying stages of development and age. Algorithms have been developed to reconstruct these continua within single-cell RNA-seq experiments [21,22], describing the sequence of molecular events that accompany these biological ‘trajectories’. Jean-Baptiste et al. identified strong hair cell trajectories, and less defined trajectories for both cortex and endodermis from whole seedling roots. Moreover, they were able to use the total amount of captured mRNA to gain insight into how total transcriptome size changes as cells progress along development. While endoreduplication is rampant in root hair cells, leading to as much as 32 copies per cell in older hair cells [23], they observed that the absolute amount of RNA is reduced with the exception of genes specific to hair cells, which increase as hair develops. This may give some indication of cells reaching terminal differentiation [5••]. They also applied a heat stress and found large changes in gene expression, that were more extreme on exterior tissues including the epidermis layers and the cortex [5••]. Shulse et al. used Drop-seq to assay 12 000 root cells, paying special attention to the endodermis trajectory [9••]. They also compared cell proportions of roots growth with or without sucrose, finding sucrose grown cells had a significantly higher proportion of hair cells, this may come as not a huge surprise as it has been demonstrated visually as well [24]. Zhang et al. isolated root tips, allowing them to draw trajectories from the meristem differentiation and root cap differentiation in great detail. They focus on the lateral root cap, and further explored the ARABIDOPSIS RESPONSE REGULATOR (arr) family, focusing on existing double and triple mutants, showing the arr1 arr10 arr12 triple mutant had reduced LRC cell layers [9••,10••]. Ryu et al. also used Arabidopsis root tips, identifying most major cell types, and further focusing on roots lacking non-hair cells (gl2) and lacking hair cells (rhd6), finding that corresponding mutants still maintain much of the transcriptional profile even with the observed phenotypic changes [8••,9••,10••]. These subtle changes that can give rise to drastic phenotypic differences have also been uncovered by single-cell RNA-seq in animal systems [25]. Overall, these studies had very similar outcomes, showing developmental trajectories of the major cell types (epidermis, endodermis, cortex, stele and root cap). No novel cell types were identified across all 5 studies, likely a result of both the simplicity of the Arabidopsis root architecture and how very well cell types have been characterized in the Arabidopsis model system.
Very recent studies have begun to apply single-cell RNA-seq to other plant model systems besides Arabidopsis. A recent study assayed 144 early germinal cells in maize anthers, measuring in great detail the expression changes during mitosis and meiosis [7•]. Another used microcapillary manipulation to capture expression post leaf excision in physcomytrella finding several thousand differentially expressed genes as a function of excision [6]. This microcapillary manipulation technique is unique in that it retains position information for each cell. These studies highlight the potential of single-cell technologies to characterize plant systems. However, significant challenges exist in expanding single-cell genomics to less well studied and more complex systems in plants.
Methodology limitations
A potential limitation for plant systems may be the fact many/most of these studies rely on digestion of cell walls to dissociate tissues into single-cells, a process known as protoplasting [26,27]. While this method allows collection of many cells, there are obvious drawbacks. There are known stress-associated changes during this protoplasting protocol, including about 300 genes that are increased in expression in Arabidopsis root protoplasts [26]. Additionally there are clear biases in the capture of certain cell types, including difficulty in capturing vasculature, and a bias towards mesophyll cells in leaves (e.g. over representation of epidermis, under representation of stele) [5••]. We predict, much like in animal systems, the best, least biased method moving forward will be nuclei capture [28]. This has been done successfully for bulk-based assays using several methods, including Fluorescence Activated Nuclei Sorting, filtering crude plant tissue, or using the INTACT (isolation of nuclei tagged in specific cell types) method [29,30]. Each of these likely have unique drawbacks or advantages, but that has yet to be explored. Most platforms for single-cell genomics can also be used with nuclei, however to-date this has not been accomplished in plant systems. While nuclei only contain only a fraction of the RNA molecules whole cells do, they are enriched for nascent RNAs [31] which may contribute to better structure upon visualization [32]. Therefore, nuclei can result in similar cell type identification compared to whole cell preparations albeit with an increased gene dropout rates [33]. In animal systems, the power of these nuclei isolation techniques are highlighted by recent large scale single-nucleus RNA-seq studies of mice and human samples [20•,28,34]. An inherent limitation to the majority of single-cell RNA-seq methods is that they rely on 3’ end capture using an oligodT first strand synthesis primer, which carries the cell-specific barcode and a unique molecular identifier. This method relies on good annotations of plant 3’ UTRs and is largely incapable of determining changes in isoforms with the same 3’ end.
Analysis
The application of single-cell RNA-seq to plant systems has the potential to uncover previously unknown cell types, heterogeneity within known cell types, shed light on plant development from common cellular origins to diverse cell types and determine how each of those cell types respond to perturbation. Many algorithms have been developed with the goal of analyzing single-cell genomics data that aim to identify and characterize cell types and cell states. Here, we describe a general workflow and associated algorithms that can be readily applied to plant systems Figure 1. Initial processing steps such as the generation of count matrices, initial quality control and normalization are not covered in this review but we suggest [35–37] for further reading on the topics.
Figure 1.
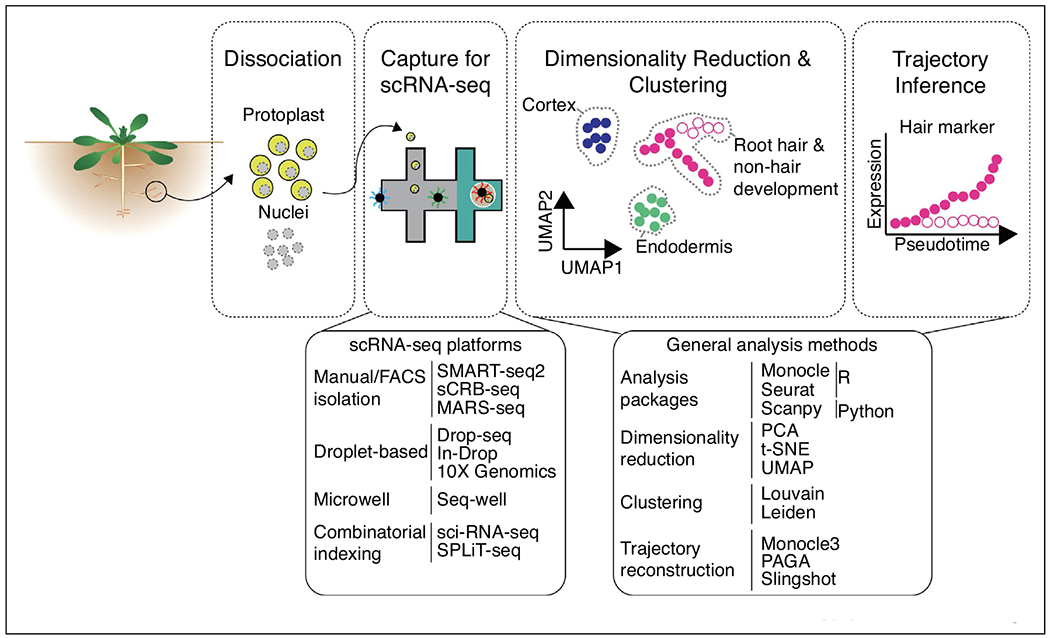
Overview of single-cell RNA-seq workflow and methods that can be applied to plants.
Dimensionality reduction techniques provide the ability to visualize the relationship between single-cell transcriptomes. These algorithms aim to embed cells that are transcriptionally similar, for example transcriptomes derived from the same cell type, in similar areas of a low dimensional representation, typically 2 or 3 dimensions. Popular techniques for this visualization are the t-distributed stochastic neighbor embedding (t-SNE) [38] and the uniform manifold approximation projection (UMAP) [39] algorithms, both of which are implemented in the single-cell analyses tool boxes Monocle3, Seurat and Scanpy [20•,40•,41,42]. We recommend the use of UMAP over t-SNE as it additionally aims to visualize large-scale distances, that is, the algorithm aims to maintain the relationships between different cell types [43]. Additional dimensionality reduction approaches, such as the potential of heat diffusion for affinity-based transition embedding (PHATE) algorithm, have been developed with the explicit goal of preserving both local and global distances from high-dimensional data such as that obtained from single-cell RNA seq [44]. Feature selection, that is performing dimensionality reduction on genes that are highly variable across the dataset [45] or expressed in at least a certain proportion of cells, can be used to aid in the interpretability of the resulting low dimensional embedding. However, we caution that feature selection may fail to capture genes that define rare cell types or key transition states and suggest that researchers compare to embeddings without feature selection.
After visualization, it is useful to cluster single-cell transcriptomes to create discrete groups of cells whose identity can then be closely examined. Many different algorithms have been applied to cluster single-cell RNA seq data including more traditional k-means and hierarchical clustering approaches as well as graph-based methods. For a detailed review of clustering methods in single-cell RNA-seq we point the reader to [46]. Graph-based Louvain community detection [47] has been applied to identify groups of cells from single-cell measurements [48]. Louvain identifies groups of cells across a k-nearest neighbor graph that have high similarity within a group and low similarity to other groups of cells in the embedding. Because of its ability to preserve relationships we suggest clustering cells in PCA space (for example on the top 50 PCA components). Alternatively, cells can be clustered in UMAP embeddings. Certain features that define similarity may be lost upon clustering in UMAP space but this approach offers considerable speed ups in the analysis of large datasets. Louvain community detection enables the identification of both distinct cell clusters as well as groups of clusters that may describe developmental processes [20•]. Recently, Leiden-based community detection has been introduced as an alternative to Louvain affording improvements in resolution and speed [49]. Cell identity of the resulting clusters/communities can be determined based on the expression of known marker genes or after determining differential gene expression between groups. As annotation of these datasets is laborious and time-consuming, tools that allow cell type annotations to be transferred across datasets have recently been developed [50,51].
Clustering of single-cell transcriptomes generally results in a collection of discrete cluster assignments that may not adequately represent instances where cells progress continually along a biological process or in response to a particular perturbation. In the context of single-cell RNA-seq in plant systems such instances are expected as cell types and tissues continually develop over the lifetime of a plant. Additionally, continua may also arise from small differences in the spatial distribution of a cell types across a tissue in the plant. Multiple algorithms have been developed and applied to single-cell RNA-seq to determine the location of a cell within these continua. These ‘trajectory inference’ methods aim to define new measures, ‘pseudotime’ [21,22] or ‘pseudospace’ (in the case of a spatial continuum) [52,53], along which cells progress along these biological continua. There are multiple trajectory inference methods to choose from [54], some of which learn tree topologies and allow for branching structures [55,56] whereas others, in addition, can learn disjointed trajectories and more complex topologies including cycles [20•,57].
The resulting trajectories learned by trajectory inference methods are generally undirected, that is, it is not known a priori in which direction cells are progressing. This can be important in determining the validity or nature of a trajectory, for example whether cells progress to a more differentiated state. RNA velocity attempts to predict the future transcriptional state of a cell based on spliced and unspliced versions of transcripts found within single-cell transcriptomes, attempting to unbiasedly determine the direction of cells across single-cell trajectories [58]. Recently, the concepts introduced by RNA velocity have been extended to incorporate information from multimodal single-cell RNA-seq experiments [59], specifically protein levels via CITE-seq [60], as well as to build predictive models of past and future cell states [61]. Despite observing a low percentage of retained introns from A. thaliana single-cell RNA-seq (~4% informative for splicing), Jean-Baptiste et al. was able to use RNA velocity to corroborate the direction of their inferred pseudotime trajectory of root hair cell development [5••].
The application of single-cell RNA-seq technologies to plant biology will undoubtedly result in studies performed with many replicates and conditions each of which has the potential to introduce technical variation or ‘batch effects’ that can confound downstream analysis. Several approaches have been developed to correct for these batch effects. These include the use of linear regression to remove variation as a function of batch (limma, combat) [62,63], subtraction of background mRNA contamination [64,65] and mutual nearest neighbor approaches that attempt to align batches based on the similarity of cells across experiments [66,67]. The latter group of algorithms can also be used to perform comparative analyses between perturbations that may alter cell type identity. We recently applied a mutual nearest neighbor (MNN) alignment [66] to assign cell identity after heat-shock of Arabidopsis root cells [5••]. Heat-shock exposure led to large changes in gene expression that includes upregulation of heat-shock response genes with a concomitant global downregulaton of gene expression limiting cell type annotation via examination of established marker genes.
Dataset alignment [40•,41,66,67] also allows for the alignment of datasets across studies and platforms. To highlight the usefulness of this approach, we aligned all of the published Arabidopsis root cell single-cell RNA-seq datasets, visualizing approximately 50 000 cells (Figure 2a). By comparing to existing annotation information from Jean-Baptiste et al., we visually annotated groups of cells across all 5 data sets. Interestingly, although all cell types were present in each dataset there are substantial differences in the proportion of individual cell types. For example, 45% of cells in the Zhang et al. dataset were classified as stele, while on the other extreme the Shulse et al. only had 6%. This could be a result of differences in digestion time, where a long digestion (2 hours, Zhang et al.) may lead to better recovery of interior layers of the root and/or over digestion of exterior layers compared to shorter digestion times (1 hour, Shulse et al.). This combination of data sets can fill in many of the developmental trajectories within and across tissues, allowing for a more robust characterization of rare cell types in any one dataset. For example, after alignment we see clear focal accumulation of genes expressing the proposed quiescent center cell marker PLP6 (AT2G39220) identified by Ryu et al. on the basis of just two cellular transcriptomes [8••] (Figure 2b). Additional marker expression analysis finds that the placement of these cells in relation to other annotated cell types indeed supports their characterization as QC (Figure 2c). Multiple trajectories are apparent in this dataset including the development of root hair cells from the quiescent center, which is apparent by the expression of early (Figure 2d) and late (Figure 2e) root hair markers. Trajectory reconstruction using Monocle 3 (Figure 2f) results in the definition of a pseudotime measurement across root hair development through which the dynamics of gene expression can be examined (Figure 2g)
Figure 2.
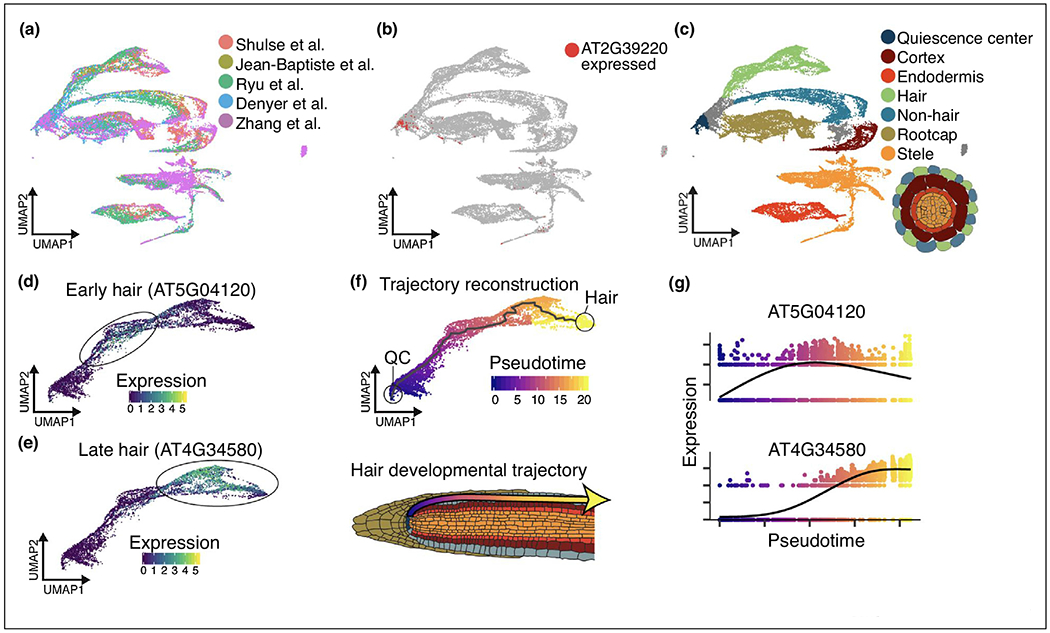
Alignment of publicly available A. thaliana single-cell RNA-seq datasets allows for the characterization of rare cell types and intermediate cell states. (a) UMAP embedding of publicly available A. thaliana single-cell RNA-seq datasets after mutual nearest neighbor alignment. Cells are colored by the study from which they originate. (b) UMAP embedding from (a) colored by whether a cell expresses (red) or doesn’t express (grey) the proposed [8••] QC marker PLP6. (c) UMAP embedding from (a) colored by cell type as determined via expression of cell type marker genes from [5••,8••]. (d-e) UMAP embedding of root hair cell development colored by the gene expression level of the early hair marker AT5G04120 (d) or the late hair marker AT4G34580 (e). Top: Trajectory reconstruction of root hair development using Monocle3. Line denotes the trajectory identified by Monocle 3. Cells are colored by progression along pseudotime. Bottom: Cartoon depicting the direction of root hair development in Arabidopsis roots. Arrow corresponds to the direction of inferred pseudotime using Monocle 3. (g) Expression levels of the early hair marker AT5G04120 (top) or the late hair marker AT4G34580 (bottom) over pseudotime.
Future direction for single-cell ‘omics in plants
Single-cell methods have immense potential to uncover mechanisms of gene regulation in plants. Additionally, resulting datasets can serve as a resource for the plant research community as a whole. As such, tools that allow exploration of single-cell gene expression data in Arabidopsis are already available (https://wwwdev.ebi.ac.uk/gxa/sc/experiments/). Single-cell genomic technologies could also play a major role in the development of crops. A major goal of crop genomics is to be able to manipulate gene expression in a single cell type and a specific time, or under specific conditions. Methods like laser capture microdissection and sorting based cell isolation of cells from reporter lines allow for specific cell type isolation and identification of associated expression patterns. However, these techniques are unable to simultaneously study hundreds or thousands of cell types in an efficient manner. Additionally, generation of reporter lines relies on the specificity of the chosen marker expression and necessitates creation of transgenic plants, a time intensive process for each cell type/tissue. Single cell genomics technologies constitute high-throughput platforms to help us understand tissue-level processes at the cell type and cell state level. Exciting future applications include unravelling the complex regulation of bundle sheath photosynthesis in grasses, and identifying approaches to manipulate root architecture with the goal of increasing uptake of fertilizers.
Combining this knowledge at single-cell resolution with new gene expression manipulation tools, like CRISPR/Cas technologies, would allow for specific changes to gene expression that improve agriculturally relevant traits and generate plants buffered against biotic and abiotic stresses. An exciting next step and an obvious extension of these profiling and gene manipulation tools will be expanding single-cell RNA-seq to crop species. Other single-cell genomic tools such as the single-cell assay for transposable accessible chromatin (scATAC) can also shed light on plant biology, enabling identification of accessible regions, a proxy for active regulatory elements, that are specific to cell-types or the response to abiotic stress. Plants species have a relatively low diversity of dynamic accessibility, where across tissues or conditions only about 5% of accessible regions change, this may be resolved if in fact this is due to heterogeneity across development of cell types. Combining scRNA-seq and scATAC-seq experiments could potentially give insight into which accessible regions are driving cell-type specific expression.
Single-cell genomic co-assays, those that simultaneously measure changes at multiple levels of gene and/or protein regulation will undoubtedly increase our understanding of unique molecular and cellular processes in plants. While the promise of single-cell technologies has massive potential for plants, caution must be taken when applying them to systems with fewer genetic tools and less ground-truth knowledge of cell types, subtissue architecture and tissue specific expression patterns. Annotation in these instances will continue to be a major limitation and one that needs to be addressed in the field using syntelogs and homologs across species resulting in a higher chance of identifying tissue specific clusters of cells in less well characterized plant species.
Footnotes
Conflict of interest statement
Nothing declared.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Trapnell C: Defining cell types and states with single-cell genomics. Genome Res 2015, 25:1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.••.Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K, Timmermans MCP: Spatiotemporal developmental trajectories in the arabidopsis root revealed using high-throughput single-cell RNA sequencing. Dev Cell 2019, 48:840–852.e5 [DOI] [PubMed] [Google Scholar]; Denyer et al. used the 10x Genomics platform to assay transcriptomes for 4700 Arabidopsis root cells, paying special attention to the Quiescent center and several developmental trajectories. Amongst the most recent Arabidopsis root single-cell studies, they are the only group to validate the expression using promoter reports fluorescence fusions.
- 3.Efroni I, Ip P-L, Nawy T, Mello A, Birnbaum KD: Quantification of cell identity from single-cell gene expression profiles. Genome Biol 2015, 16:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Efroni I, Mello A, Nawy T, Ip P-L, Rahni R, DelRose N, Powers A, Satija R, Birnbaum KD: Root regeneration triggers an embryo-like sequence guided by hormonal interactions. Cell 2016, 165:1721–1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.••.Jean-Baptiste K, McFaline-Figueroa JL, Alexandre CM, Dorrity MW, Saunders L, Bubb KL, Trapnell C, Fields S, Queitsch C, Cuperus JT: Dynamics of gene expression in single root cells of A. thaliana. Plant Cell 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors assayed single cell transcriptomes of thousands of Arabidopsis roots using the 10x Genomics platform, drawing developmental trajectories for several cell types. They also applied a heat shock resulting in large transcriptional changes for several cell-types.
- 6.Kubo M, Nishiyama T, Tamada Y, Sano R, Ishikawa M, Murata T, Imai A, Lang D, Demura T, Reski R et al. : Single-cell transcriptome analysis of Physcomitrella leaf cells during reprogramming using microcapillary manipulation. Nucleic Acids Res 2019, 47:4539–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.•.Nelms B, Walbot V: Defining the developmental program leading to meiosis in maize. Science 2019, 364:52–56 [DOI] [PubMed] [Google Scholar]; Nelms and Walbot used size differences between somatic and developing pollen cells in the anthers of maize, isolating cells during pollen development. They used single-cell RNA-seq of 144 of these cells to identify changes during and throughout meiosis.
- 8.••.Ryu KH, Huang L, Kang HM, Schiefelbein J: Single-cell rna sequencing resolves molecular relationships among individual plant cells. Plant Physiol 2019, 179:1444–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors used Arabidopsis root tips isolating several thousand cells for single-cell transcriptomic profiling using the 10x Genomics platform. They focused on roots lacking non-hair cells (gl2) and lacking hair cells (rhd6), finding that corresponding mutants still maintain much of the transcriptional profile even with the observed phenotypic changes.
- 9.••.Shulse CN, Cole BJ, Ciobanu D, Lin J, Yoshinaga Y, Gouran M, Turco GM, Zhu Y, O’Malley RC, Brady SM et al. : High-throughput single-cell transcriptome profiling of plant cell types. Cell Rep 2019, 27:2241–2247.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]; Authors used Drop-seq to assay 12 000 root cells, drawing a developmental trajectory for the endodermis cell type. They compared roots growth with or without sucrose, finding sucrose grown cells had a significantly higher proportion of hair cells.
- 10.••.Zhang T-Q, Xu Z-G, Shang G-D, Wang J-W: A single-cell RNA sequencing profiles the developmental landscape of Arabidopsis root. Mol Plant 2019, 12:648–660 [DOI] [PubMed] [Google Scholar]; Using isolated root tips, the authors collected thousands of single-cells using the 10x genomics platform. They focus on the root cap, and further explored the ARABIDOPSIS RESPONSE REGULATOR (arr) family, focusing on existing double and triple mutants, showing the arr1 arr10 arr12 triple mutant had reduced root cap cell layers.
- 11.Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, Kirschner MW: Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015, 161:1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM et al. : Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015, 161:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hashimshony T, Senderovich N, Avital G, Klochendler A, de Leeuw Y, Anavy L, Gennert D, Li S, Livak KJ, Rozenblatt-Rosen O et al. : CEL-Seq2: sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol 2016, 17:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, Sandberg R: Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc 2014, 9:171–181. [DOI] [PubMed] [Google Scholar]
- 15.Soumillon M, Cacchiarelli D, Semrau S, van Oudenaarden A, Mikkelsen TS: Characterization of directed differentiation by high-throughput single-cell RNA-Seq. bioRxiv 2020. [Google Scholar]
- 16.Keren-Shaul H, Kenigsberg E, Jaitin DA, David E, Paul F, Tanay A, Amit I: MARS-seq2.0: an experimental and analytical pipeline for indexed sorting combined with single-cell RNA sequencing. Nat Protoc 2019, 14:1841–1862. [DOI] [PubMed] [Google Scholar]
- 17.Gierahn TM, Wadsworth MH 2nd, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, Shalek AK: Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods 2017, 14:395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao J, Packer JS, Ramani V, Cusanovich DA, Huynh C, Daza R, Qiu X, Lee C, Furlan SN, Steemers FJ et al. : Comprehensive single-cell transcriptional profiling of a multicellular organism. Science 2017, 357:661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Graybuck LT, Peeler DJ, Mukherjee S, Chen W et al. : Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 2018, 360:176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.•.Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, Zhang F, Mundlos S, Christiansen L, Steemers FJ et al. : The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019, 566:496–502 [DOI] [PMC free article] [PubMed] [Google Scholar]; Cao et al. apply a novel single-cell combinatorial indexing RNA-seq method, that allows for the profiling of millions of cellular transcriptomes, to create an atlas of mammalian development. This study introduces the newest iteration of Monocle, Monocle3, a scRNA-seq toolbox that scales to the analysis of millions of cells to jointly identify many cell types and cellular trajectories.
- 21.Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Simonds EF, Chen TJ, Shenfeld DK, Nolan GP, Pe’er D: Single-Cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell 2014, 157:714–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL: The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Blotechnol 2014, 32:381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bhosale R, Boudolf V, Cuevas F, Lu R, Eekhout T, Hu Z, Van Isterdael G, Lambert GM, Xu F, Nowack MK et al. : A spatiotemporal DNA endoploidy map of the arabidopsis root reveals roles for the endocycle in root development and stress adaptation. Plant Cell 2018, 30:2330–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jain A, Poling MD, Karthikeyan AS, Blakeslee JJ, Peer WA, Titapiwatanakun B, Murphy AS, Raghothama KG: Differential effects of sucrose and auxin on localized phosphate deficiency-induced modulation of different traits of root system architecture in Arabidopsis. Plant Physlol 2007, 144:232–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saunders LM, Mishra AK, Aman AJ, Lewis VM, Toomey MB, Packer JS, Qiu X, McFaline-Figueroa JL, Corbo JC, Trapnell C et al. : Thyroid hormone regulates distinct paths to maturation in pigment cell lineages. eLlfe 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN: A gene expression map of the Arabidopsis root. Science 2003, 302:1956–1960. [DOI] [PubMed] [Google Scholar]
- 27.Yoo S-D, Cho Y-H, Sheen J: Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2007, 2:1565–1572. [DOI] [PubMed] [Google Scholar]
- 28.Habib N, Avraham-Davidi I, Basu A, Burks T, Shekhar K, Hofree M, Choudhury SR, Aguet F, Gelfand E, Ardlie K et al. : Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat Methods 2017, 14:955–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deal RB, Henikoff S: The INTACT method for cell type-specific gene expression and chromatin profiling in Arabidopsis thaliana. Nat Protoc 2011, 6:56–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slane D, Kong J, Berendzen KW, Kilian J, Henschen A, Kolb M, Schmid M, Harter K, Mayer U, De Smet I et al. : Cell type-specific transcriptome analysis in the early Arabidopsis thaliana embryo. Development 2014, 141:4831–4840. [DOI] [PubMed] [Google Scholar]
- 31.Lake BB, Ai R, Kaeser GE, Salathia NS, Yung YC, Liu R, Wildberg A, Gao D, Fung H-L, Chen S et al. : Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352:1586–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lake BB, Codeluppi S, Yung YC, Gao D, Chun J, Kharchenko PV, Linnarsson S, Zhang K: A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci Rep 2017, 7:6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bakken TE, Hodge RD, Miller JA, Yao Z, Nguyen TN, Aevermann B, Barkan E, Bertagnolli D, Casper T, Dee N et al. : Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One 2018, 13:e0209648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sathyamurthy A, Johnson KR, Matson KJE, Dobrott CI, Li L, Ryba AR, Bergman TB, Kelly MC, Kelley MW, Levine AJ: Massively parallel single nucleus transcriptional profiling defines spinal cord neurons and their activity during behavior. Cell Rep 2018, 22:2216–2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.AlJanahi AA, Danielsen M, Dunbar CE: An introduction to the analysis of single-cell RNA-sequencing data. Mol Ther Methods Clin Dev 2018, 10:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hwang B, Lee JH, Bang D: Single-cell RNA sequencing technologies and bioinformatics pipelines. Exp Mol Med 2018, 50:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stegle O, Teichmann SA, Marioni JC: Computational and analytical challenges in single-cell transcriptomics. Nat Rev Genet 2015, 16:133–145. [DOI] [PubMed] [Google Scholar]
- 38.Lvander Maaten, Hinton G: Visualizing data using t-SNE. J Mach Learn Res 2008, 9:2579–2605. [Google Scholar]
- 39.McInnes L, Healy J, Saul N, Großberger L: UMAP: uniform manifold approximation and projection. J Open Source Softw 2018, 3:861. [Google Scholar]
- 40.•.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R: Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018, 36:411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]; Butler et al. present a dataset alignment and integration functionality for the single-cell analysis toolbox, Seurat, based on canonical correlation analysis and dynamic time warping. This study presents various applications for dataset alignment likely to be useful in the analysis of plant scRNA-seq, including integration of datasets from different species and different scRNA-seq platforms.
- 41.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R: Comprehensive integration of single-cell data. Cell 2019, 177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wolf FA, Angerer P, Theis FJ: SCANPY: large-scale single-cell gene expression data analysis. Genome Biol 2018, 19:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, Newell EW: Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol 2018. [DOI] [PubMed] [Google Scholar]
- 44.Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, van den Elzen A, Hirn MJ, Coifman RR et al. : Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol 2019, 37:1482–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC et al. : Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods 2013, 10:1093–1095. [DOI] [PubMed] [Google Scholar]
- 46.Kiselev VY, Andrews TS, Hemberg M: Challenges in unsupervised clustering of single-cell RNA-seq data. Nat Rev Genet 2019, 20:273–282. [DOI] [PubMed] [Google Scholar]
- 47.Blondel VD, Guillaume J-L, Lambiotte R, Lefebvre E: Fast unfolding of communities in large networks. J Stat Mech: Theory Exp 2008, 2008:P10008. [Google Scholar]
- 48.Levine JH, Simonds EF, Bendall SC, Davis KL, Amir E-AD, Tadmor MD, Litvin O, Fienberg HG, Jager A, Zunder ER et al. : Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell 2015, 162:184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Traag VA, Waltman L, van Eck NJ: From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 2019, 9:5233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pliner HA, Shendure J, Trapnell C: Supervised classification enables rapid annotation of cell atlases. Nat Methods 2019, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang AW, O’Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, Wiens M, Walters P, Chan T, Hewitson B et al. : Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods 2019, 16:1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joost S, Zeisel A, Jacob T, Sun X, La Manno G, Lönnerberg P, Linnarsson S, Kasper M: Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst 2016, 3:221–237.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scialdone A, Tanaka Y, Jawaid W, Moignard V, Wilson NK, Macaulay IC, Marioni JC, Göttgens B: Resolving early mesoderm diversification through single-cell expression profiling. Nature 2016, 535:289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Saelens W, Cannoodt R, Todorov H, Saeys Y: A comparison of single-cell trajectory inference methods. Nat Biotechnol 2019, 37:547–554. [DOI] [PubMed] [Google Scholar]
- 55.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, Trapnell C: Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 2017, 14:979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N, Purdom E, Dudoit S: Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics 2018, 19:477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wolf FA, Hamey FK, Plass M, Solana J, Dahlin JS, Göttgens B, Rajewsky N, Simon L, Theis FJ: PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol 2019, 20:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A et al. : RNA velocity of single cells. Nature 2018, 560:494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gorin G, Svensson V, Pachter L: RNA velocity and protein acceleration from single-cell multiomics experiments. Genome Biol 2020, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, Satija R, Smibert P: Simultaneous epitope and transcriptome measurement in single cells. Nat Methods 2017, 14:865–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Qiu X, Zhang Y, Yang D, Hosseinzadeh S, Wang L, Yuan R, Xu S, Ma Y, Replogle J, Darmanis S et al. : Mapping vector field of single cells. bioRxiv 2020. [Google Scholar]
- 62.Johnson WE, Li C, Rabinovic A: Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8:118–127. [DOI] [PubMed] [Google Scholar]
- 63.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK: Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015, 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Packer JS, Zhu Q, Huynh C, Sivaramakrishnan P, Preston E, Dueck H, Stefanik D, Tan K, Trapnell C, Kim J et al. : A lineage-resolved molecular atlas of C. elegans embryogenesis at single cell resolution. Science 2019, 365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Young MD, Behjati S: SoupX removes ambient RNA contamination from droplet based single cell RNA sequencing data. bioRxiv 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Haghverdi L, Lun ATL, Morgan MD, Marioni JC: Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol 2018, 36:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hie B, Bryson B, Berger B: Efficient integration of heterogeneous single-cell transcriptomes using Scanorama. Nat Biotechnol 2019, 37:685–691. [DOI] [PMC free article] [PubMed] [Google Scholar]