

Abstract
Alzheimer’s disease (AD) is a major neurological disorder impairing its carrier’s cognitive function, memory and lifespan. While the development of AD nanomedicine is still nascent, the field is evolving into a new scientific frontier driven by the diverse physicochemical properties and theranostic potential of nanomaterials and nanocomposites. Characteristic to the AD pathology is the deposition of amyloid plaques and tangles of amyloid beta (Aβ) and tau, whose aggregation kinetics may be curbed by nanoparticle inhibitors via sequence-specific targeting or nonspecific interactions with the amyloidogenic proteins. As literature implicates cell membrane as a culprit in AD pathogenesis, here we summarize the membrane axis of AD nanomedicine and present a new rationale that the field development may greatly benefit from harnessing our existing knowledge of Aβ-membrane interaction, nanoparticle-membrane interaction and Aβ-nanoparticle interaction.
Keywords: Aβ, AD, membrane, nanoparticle, nanomedicine
Graphical Abstract
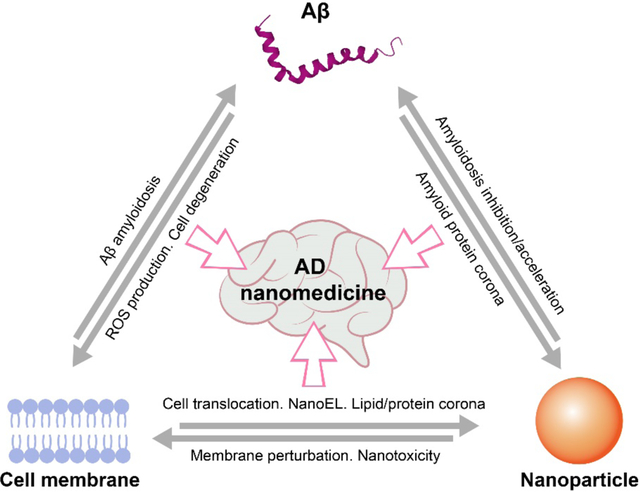
The development of AD nanomedicine, an emerging frontier in advanced nano-biomedical research, can benefit from our accumulating knowledge of Aβ-cell membrane, nanoparticle-cell membrane and Aβ-nanoparticle interactions.
1. Introduction
Alzheimer’s disease (AD) is the primary form of neurological disorder with a global burden of 50 million. Histopathologically, the AD brain post mortem is characterized by the presence of extracellular plaques comprised of amyloid beta (Aβ), lipids, metals (e.g., Zn2+, Cu2+ and Fe3+) as well as proteins.[1] Aβ, ranging from 36–43 in total number of residues, is a proteolytic product of transmembrane amyloid precursor protein (APP), cleaved off by β and γ secretases sequentially. The self-assembly of Aβ into cross-β fibrils, or Aβ amyloidosis in short, has been linked to neurodegeneration and the pathology of AD, as described by the amyloid cascade hypothesis.[2]
As Aβ plaques are primarily found in the extracellular space in the central nervous system (CNS), while the peptide itself is derived from APP expanding from extracellular to transmembrane domains (Scheme A), understanding Aβ amyloidosis in the presence of membrane mimetics (e.g., micelles, nanodisks, vesicles, liposomes, amphiphilic polymers and lipid bilayers), naturally, has been a major research undertaking over the past two decades, especially in the biophysical and biochemical communities.[3] Indeed, AD has been considered a membrane disorder, resulting from the interactions of Aβ with lipids, lipid rafts, transmembrane proteins and nicotinic acetylcholine receptors (nAChRs).[4] Furthermore, neurotoxicity in AD has been proposed to arise from the interaction of Aβ with membrane receptors[5] and the generation of membrane pores.[6–7] While membrane mimetics bear limited resemblance to cell membranes, fundamental research with these systems has nonetheless shed a crucial light on the structural transformation of Aβ in physiological environments as well as on their implications for neurotoxicity and AD pathology.
Scheme. Scope of the review.
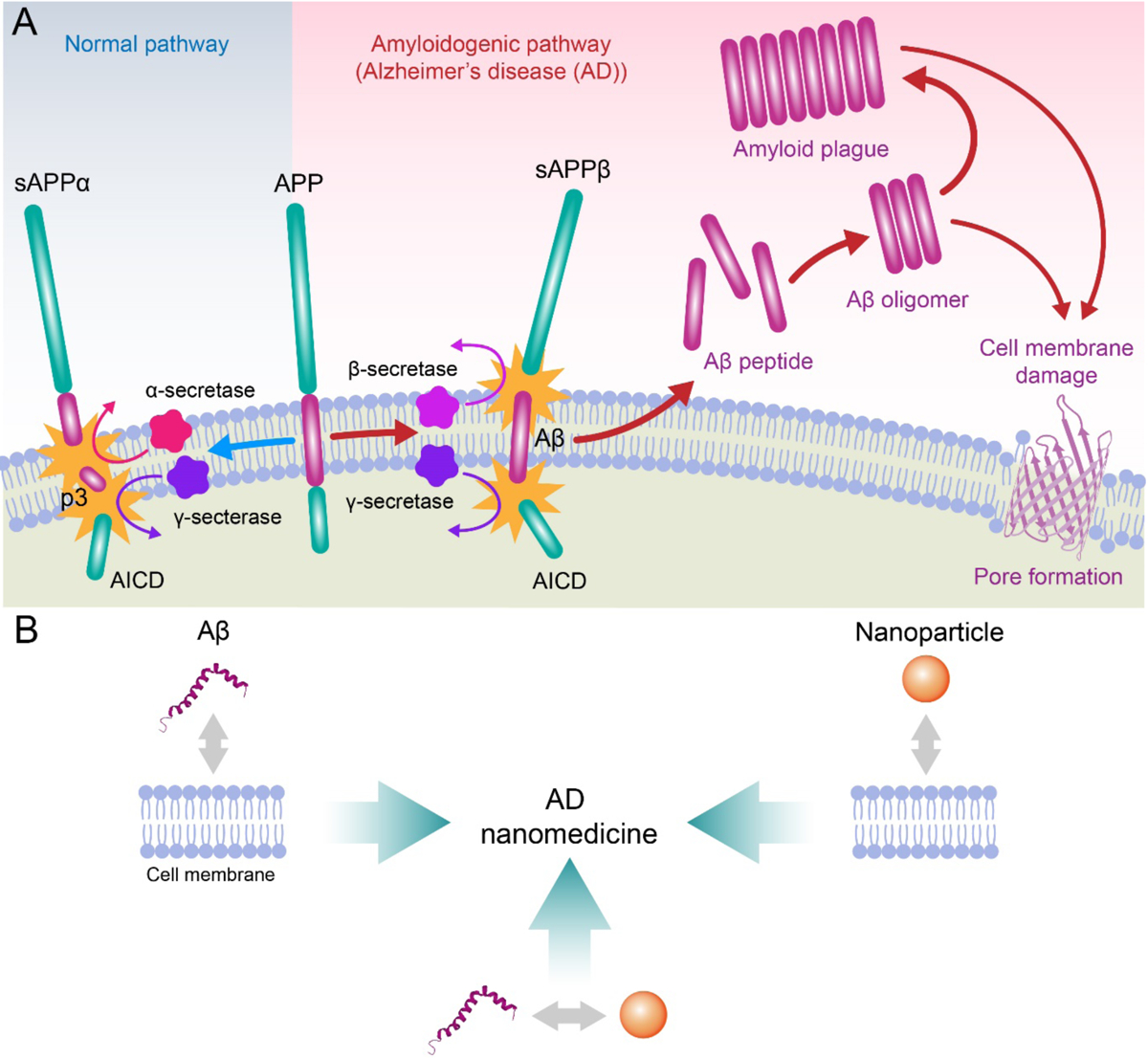
(A) Aβ is synthesized in situ by the consecutive cleavage of transmembrane amyloid precursor protein (APP) by β and γ secretases. Under aberrant conditions Aβ peptides self-assemble to render oligomeric species and amyloid fibrils, which may compromise membrane integrity via various mechanisms, such as poration. (B) The development of AD nanomedicines may benefit from better understanding of Aβ-membrane, nanoparticle-membrane and Aβ-nanoparticle interactions. AICD: APP intracellular domain; sAPPβ: β-secretase cleaved soluble APP N-terminal fragment; sAPPα: α-secretase cleaved soluble extracellular APP fragment; p3: 3-kDa peptide resulting from α- and γ-secretase cleavage of APP.
Towards the development of therapeutics against amyloid diseases, much research has been focused on the design and delivery of synthetic small molecules, natural polyphenols, peptidomimetics, monoclonal antibodies and, more recently, nanomaterials, in order to target or sequester the toxic oligomeric and fibrillar peptide species.[8–25] Especially with nanomaterials, their small size and diverse physicochemical properties have often entailed a remarkable inhibitory potential against the aggregation and toxicity of Aβ, alpha synuclein (αS) as well as human islet amyloid polypeptide (IAPP) associated with AD, Parkinson’s disease (PD) and type 2 diabetes (T2D), among others.[26]
In this review, we summarize the state-of-the-art of Aβ amyloidosis inhibition with nanomaterials by highlighting the central interactions between the peptide, the nanoparticle inhibitors, and the membrane substrates of various degrees of complexity. We present the rationale that, fundamentally, such application can greatly benefit from exploiting our prior and growing knowledge of a) Aβ-membrane interaction (section 2), b) nanoparticle-membrane interaction (section 3), and c) Aβ-nanoparticle interaction (section 4), for facilitating the development of potent AD nanomedicines (section 5) (Scheme B). An outlook on future AD nanomedicine is provided at the end of the article.
2. Aβ-membrane interaction
In this section, we summarize computational and experimental findings concerning Aβ-membrane interaction, the first of the three main constituents of this review that entail important implications for the development of AD nanomedicine.
2.1. Simulation studies
Although the interaction of Aβ with cellular membrane is likely a key factor in AD pathogenesis, the underlying molecular mechanisms remain largely unknown. While it is challenging to experimentally determine Aβ-membrane interaction with single-molecule resolution, computer simulations are ideally suited for delineating such interaction with molecular details and for providing new insights into the structure and function of complex biomolecular systems. It should be acknowledged that extensive computational studies have been conducted to date concerning the inner workings of Aβ-membrane binding. These studies can be categorized by the states of Aβ[27–30] (e.g., monomers, oligomers and fibrils), the positioning of Aβ with respect to the membrane[31–32] (e.g., adsorbed on or inserted into the membrane), and the computational methods adopted[33–34] (e.g., all-atom and coarse-grained/CG simulations), as discussed to various extents in previous reviews.[35–39] In this section, we summarize the critical findings revealed by simulation studies concerning the role of Aβ-membrane interaction in promoting the peptide aggregation and membrane damage.
Aβ has been shown to strongly interact with membranes in experiments. One of the main questions that computer simulations attempt at resolving is how the cell membrane modulates Aβ aggregation. Towards that goal, the adsorption of Aβ on the lipid bilayer and the early stage of the peptide oligomerization have been simulated. All-atom molecular dynamics (MD) simulations showed that Aβ was attracted from the solution to the surface of both zwitterionic dipalmitoylphosphatidylcholine (DPPC) and anionic dioleylphosphatidylserine (DOPS) bilayers driven by the electrostatic interactions between the charged peptide residues and the lipid headgroups, which accelerated Aβ aggregation by increasing the local peptide concentration and promoted Aβ diffusion by limiting their motion to the two dimensional surface compared with those in solution.[40–42] Once an Aβ monomer was bound to the membrane, their overall secondary structure did not change noticeably on the DPPC bilayer compared with their random coil structure in solution, whereas the DOPS bilayer strongly enhanced the helical structure, especially near the N-terminus of the peptide. No significant β-structure was observed except for a small, unstable β-hairpin formed on the anionic DOPS bilayer. However, Aβ bound to DOPS was able to adopt the conformation with a more exposed hydrophobic C-terminus that was more accessible to the adjacent Aβ peptides, which promoted protein-protein interaction and favored the early stage of Aβ aggregation. These results suggested that the structural conversions observed in experiments were not due to the ordering of monomeric Aβ on the bilayer surface but a result of protein-protein interaction that occurred during oligomerization, further enhanced by Aβ binding to the cell membrane.[43] Additional simulations considered the fact that the local pH near an anionic DOPS lipid bilayer was lower than the bulk, giving rise to transient stabilization of the peptide β-structure.[43] This observation implied that the Aβ-membrane interaction at the physiological pH can stabilize Aβ monomers in an aggregation-prone intermediate state. The hypotheses derived from simulations of Aβ monomers interacting with membranes have been validated by the dimerization of Aβ on the lipid membranes, where DOPS lipids promoted strong protein-protein interaction between Aβ monomers while weakened protein-lipid interaction during the dimerization.[44] For larger Aβ oligomers, the interaction of an Aβ tetramer with the membrane resulted in elongation of the tetramer in the presence of both pure 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and cholesterol-rich raft model membranes. Tetramer-raft interactions caused rearrangement of key hydrophobic regions in the tetramer and formation of a rod-like structure indicative of a fibril-seeding aggregate.[45] In addition, the chemical composition of the lipid membrane can also modulate the Aβ-membrane interaction.[46–47] Simulations showed that an increased cholesterol content significantly promoted the binding of Aβ monomers to a POPC lipid bilayer by increasing the bilayer thickness, hydrophobic chain order and surface hydrophobicity, while decreasing the lipid mobility.[48] In the presence of monosialotetrahexosylganglioside (GM1), which is enriched in the outer leaflet of the plasma cell membranes in the CNS, the GM1-Aβ interaction facilitated the formation of β-sheet structures.[49]
The molecular mechanism underpinning the toxicity of Aβ is another central topic in numerous simulation studies. It has been increasingly accepted that the cytotoxicity resulting from amyloidosis may be attributed to membrane disruption caused by amyloid aggregation, and the formation of membrane pores by amyloid proteins is regarded as a key event leading to membrane leakage and subsequent cell death.[50–51] Since it is computationally challenging to directly observe the spontaneous formation of amyloid pores by Aβ in membrane simulations, molecular modeling approaches combined with MD simulations are usually adopted. When inserted into the membrane, early-stage Aβ aggregates were postulated to adopt two models that mimicked the pores in membranes – one is the channel model where the peptides were inserted without inclination to the membrane normal axis, and the second one is the β-barrel model where the peptides were inclined with respect to the pore axis (Fig. 1A).[52–53] The dynamic properties of the two possible Aβ channel topologies were examined, as the CNpNC (C and N denote the C- and N-terminal β-strands, respectively, and p stands for the central pore), where the polar/charged N-terminal β-strand faced the water-filled pore and the hydrophobic C-terminal β-strand faced the bilayer, and the NCpCN, where the C-terminal β-strand faced the solvated pore (Fig. 1B). The CNpNC channel preserved the pore and conducted solvent, whereas the NCpCN channel failed to preserve the pore due to the hydrophobic collapse, in good agreement with the images acquired by atomic force microscopy (AFM).[54] Simulations of Aβ ion channels consisted of U-shaped β-strand-turn-β-strand motifs in the lipid bilayer showed that the optimized size of channels presenting a toxic ionic flux ranged between 16- to 24-mer with the subunit organization and dimensions being consistent with the experiments, whereas the smaller (12-mer) channel collapsed and the larger (36-mer) channel could not be supported by the bilayer.[55] Importantly, in agreement with AFM, the simulations indicated that the β-sheet channels broke into loosely associated mobile β-sheet subunits[56–57] (Fig. 1C). On the other hand, the β-barrels of Aβ(9–42) and Aβ(17–42) were also shown to preserve the U-shaped CNpNC topology with a similar subunit organization and dimensions as the channel models[58] (Fig. 1D). Moreover, the β-strands of the barrels adjusted the conformation by decreasing the tilt angle of the N-terminal β-strand and increasing the tilt angle of the C-terminal β-strand. As a result, the 12-mer Aβ barrel produced a well-defined pore, in contrast to the collapsed pore of the channel model, suggesting that the Aβ barrels provided a more optimized pore organization.[58] In addition to the N-terminal truncated Aβ, channels and barrels with L- and D-enantiomers Aβ[59] and Aβ mutants[60] (F19P, F20C and ΔE22) have also been identified in simulations. These models of Aβ channels and barrels in the bilayer have offered valuable insights into the molecular mechanisms of amyloid toxicity and revealed structural information pertinent to drug discovery efforts focused on inhibiting the formation of such toxic channels and pores in the cell membrane.
Figure 1. Aβ channels and β-barrels in lipid membranes.
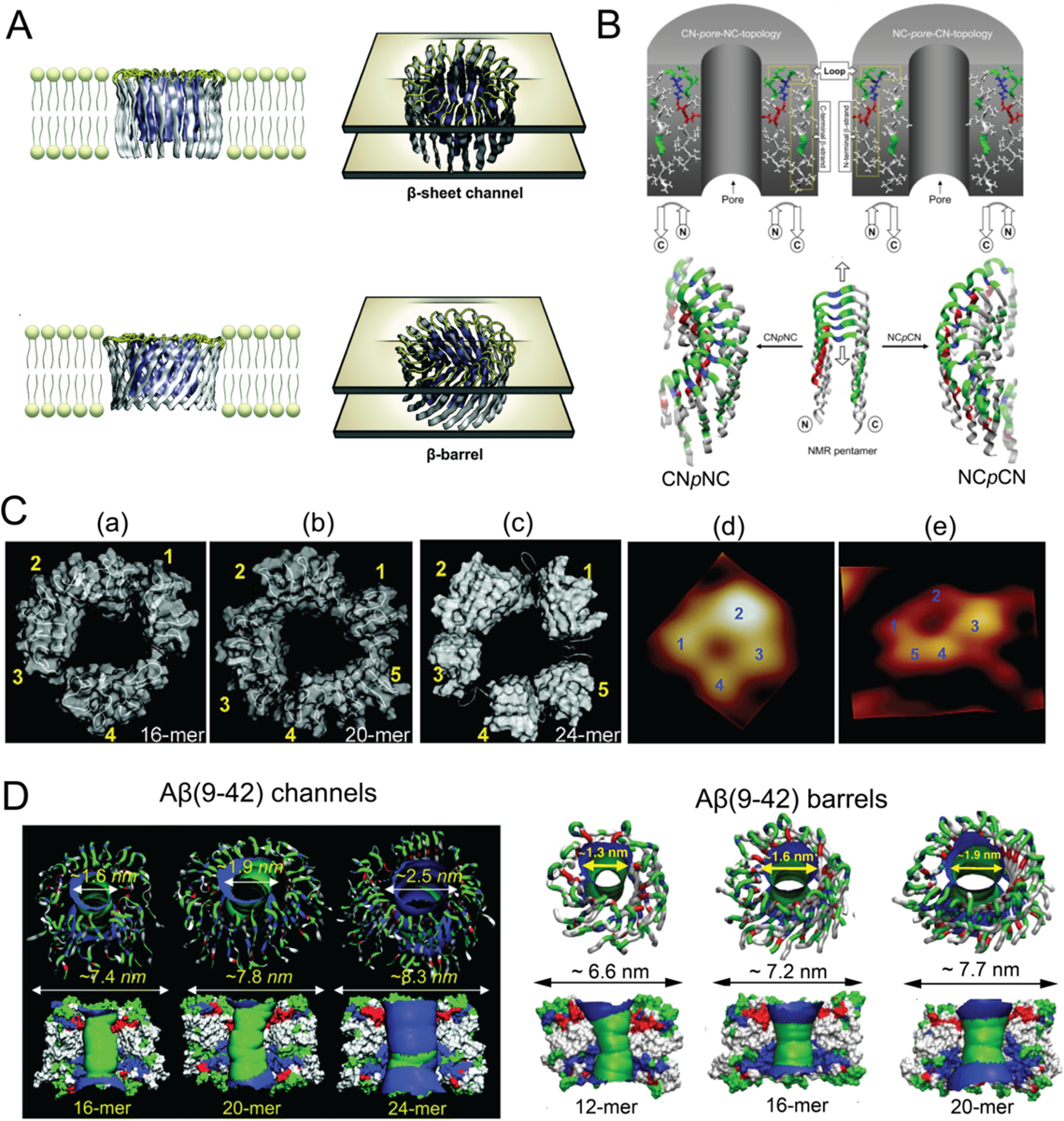
(A) Schematic illustrations of Aβ channels and β-barrels in lipid membranes. (B) Schematics of the CNpNC (left) and NCpCN (right) topologies of Aβ(17–42) channels. The Aβ monomers, which were taken from the NMR pentamer structure in the PDB databank (ID: 2BEG), exhibited the U-shaped strand-turn-strand conformation. Top panel: initial annular channel topologies shown as a cross-section of a hollowed cylinder in grey with a cut along the pore axis. The Aβ(17–42) peptide in ribbon representation was projected into the cross-section area. Middle panel: the topologies of Aβ peptides drawn by connected arrows. Bottom panel: ribbon representations of Aβ backbones. (C) Comparison between computed Aβ(17–42) channel structures and high-resolution AFM data. The simulated channel structures for (a) 16-mer, (b) 20-mer and (c) 24-mer showed four to five subunits, in agreement with the AFM images (d and e). (D) Aβ(9–42) channels (left) and β-barrels (right). Top panel: angle views of the pore structures, where pore structures were shown in ribbon representation. Bottom panel: lateral views of the pore structure, where the cross-sectioned pores were shown in surface representation with the degree of the pore diameter colored in the order of red < green < blue. Panels reproduced from Refs. 52, 54–55, 57–58. Copyrights 2014 The Royal Society of Chemistry, 2007 the Biophysical Society, 2010 American Chemical Society, 2010 National Academy of Sciences and 2010 Elsevier Ltd.
2.2. Experimental studies
Aβ peptides are produced from APP within the transmembrane region of neurons and synapses, mainly in the forms of Aβ40 and Aβ42, whose aggregation propensities are closely related to the occurrence and progression of AD. While Aβ generation is associated with the membrane lipid environment, Aβ-membrane interaction can induce membrane reorganization, deformation, pore formation, increased permeabilization, and lipid extraction, leading to intracellular ion dyshomeostasis, production of reactive oxygen species (ROS) with an excess of lipid peroxidation, increased lipid susceptibility to oxidative damage[61] and, eventually, neuronal death. As these processes are critical to the AD pathology, understanding Aβ-membrane interaction should directly benefit the development of new AD therapeutics.[62]
Accordingly, various types of membrane models with controlled characteristics, bilayer components, thickness and fluidity, have been designed to mimic the plasma membrane and screen their interactions with Aβ (Table 1). All Aβ species displayed a certain level of affinity for the anionic membrane,[63] with the oligomers displaying a greater membrane affinity than the monomers or fibrils.[64] Kayed et al. observed that synthetic Aβ oligomers, other than monomers or fibrils, could increase the conductance of the lipid bilayer and induce a sharp decrease in electron density throughout the lipid membrane, without generating discrete channel or pore formation or ion selectivity.[65] However, other studies revealed that Aβ fibrils could also be embedded in the upper leaflet of the bilayer.[66–67] The carpet model (Fig. 2) was proposed to explain the peripheral association of the bilayer and Aβ, which was accompanied by a general increase in membrane conductance either by membrane thinning or lateral spreading of lipid headgroups.[68–70] In addition, significant lipid extraction and disruption to bilayer were induced by oligomeric Aβ and were visualized through AFM and transmission electron microscopy (TEM) (Fig. 3A).[66] The phospholipid molecules extracted from both bilayer leaflets, or first from the outer leaflet and then the inner leaflet of the membrane, may contribute to the formation of Aβ-lipid complexes resulting in a detergent-like lipid removal action (Table 1, Fig. 2).[66, 68, 71] Thereafter, large pores spanning over the bilayer (~50 nm) were formed, and small Aβ oligomers could insert themselves into the pores that were expanding over time. Moreover, recent studies have revealed that this detergent-like activity of the peptide strongly depends on the structure and size of the Aβ oligomers.[63, 72] For example, globular nonfibrillar oligomers caused the largest reduction of lipid diffraction in anionic 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) lipid monolayers upon insertion, compared to short fibrillar oligomers and monomers.[63] Small-sized Aβ oligomers, corresponding to low molecular-weight Aβ oligomers, could destroy the brain total lipid extract (BTLE) membrane bilayer by lipid extraction. In contrast, the aggregation of large-sized Aβ oligomers was accelerated on the BTLE membrane surface without affecting the membrane integrity.[72] Small-sized Aβ oligomers and globular nonfibrillar oligomers have been considered as the more toxic species in causing neurodegeneration.[73] Alternatively, Aβ could induce channel-like perforation in neuronal cell membranes (Fig. 2), causing an increase in membrane conductance as well as intracellular calcium and ethidium bromide influxes in a concentration- and time-dependent manner.[74–75] Upon addition to large unilamellar vesicles (LUVs) soluble oligomers formed small cation-selective pores immediately, whereas large molecules were allowed to enter the cells with the membrane gradually losing its physical integrity over time.[76]
Table 1.
Aβ-cell membrane interaction.
| Amyloid proteins | Membrane components | Interactants/binding sites | Toxicity | Mechanism | Year | Refs |
|---|---|---|---|---|---|---|
| Aβ40, Aβ42 | Bilayers formed from a mixture (1:1 by weight) of PC, PS, PE and cholesterol | Oligomers are only peripherally associated with the bilayer rather than stably inserted into the hydrocarbon core | Soluble oligomers eliciting cytotoxicity |
Carpet effect •Oligomers are only peripherally associated with the bilayer rather than stably inserted into the hydrocarbon core •Soluble oligomers increase membrane conductance in the absence of evident discrete ion channel or pore formation •Soluble oligomers Appear to facilitate ion transport through the lipid bilayer |
2004 | [65] |
| Aβ42 | Lipid vesicles, floating bilayer lipid membrane (fBLM) | Membrane hydrophobic core | Aβ perforation induces conformational and orientational changes of the lipid acyl chains, decreasing acyl chain mobility and altering the lipid packing unit cell | 2020 | [95] | |
| Aβ40 | Hippocampal and cortical neurons, HEK293 cell | •Aβ aggregates enhancing the release of synaptic vesicles from hippocampal neurons, engaging in synaptic dysfunction | •Aβ as a perforating toxic agent, rather than a classical pore-forming agent •Aβ excess disrupts membranes causing pore formation leading to alterations in ionic homeostasis •Perforating effects of Aβ are associated with microscopic structures resembling small fibrils, but not unstructured forms of Aβ |
2010 | [74] | |
| Aβ42, Aβ variants: L34T, G37C | DOPG, DOPC | DOPG | •G37C oligomer anchors on and reorganizes membrane, causing highest membrane perturbation and disruption •Aβ42 also perturbs liposome organization with membrane deformation rather than disruption |
2015 | [96] | |
| Aβ40, Aβ42 | OG, DM, DHPC, LDAO, DPC | •β-barrel pore-forming •Lipid-stabilized pores are mediated by the hydrophilic residues located on the core β-sheets edges of the oligomers |
2016, 2020 | [7, 50] | ||
| Aβ40, Aβ42 | HEK293 cells | Ile41-Ala42 residues in Aβ42 | Aβ42 more toxic | Channel formation: Aβ42 oligomer insert into the membrane to form membrane-spanning pores | 2017 | [75] |
| Aβ | Three zwitterionic lipid bilayers: DLPC, DOPC and POPC bilayers | •Thin bilayer-DLPC, 20.9 nm •Normal/thick bilayers: DOPC, 26.8 nm •Normal/thick bilayers: POPC, 27.1 nm |
•Aβ oligomers w/o DLPC reduce viability by 50% •With DLPC, nontoxic •Remodeled fibrils by DLPC inducing cellular death |
•Bilayer: alters membrane protein folding and modulates energy penalty due to hydrophobicity mismatch •Detergent effect: Aβ-DLPC interactions generate micelle-like lipid species that can prevent aggregation •Remodel preformed fibrils: (i)DLPC changes the monomer-fiber equilibrium and causes additional monomers to enter solution via dissociation from the ends of fibers, resulting in shorter aggregates (ii) DLPC LUVs alter the hydrogen bonding network found within the fibrils, generating distinct polymorphs of the aggregates that are less stable and can potentially become more similar to the immature protofibril structure (iii)DLPC LUVs rearrange the side chain packing of the mature fibrils and induce a partially unfolded state |
2017, 2018 | [69–70] |
| Aβ42, large-size Aβ oligomers, small-size Aβ oligomers | Porcine BTLE-supported lipid bilayer | phospholipids from bilayer leaflets | •Small-size Aβ oligomers more hydrophobic and toxic than large-size Aβ oligomers | •Pore formation: the pores expand over time, then small-sized Aβ oligomers insert themselves into these pores •Lipid extraction: extraction of phospholipid molecules from the membrane lead to the formation of Aβ-lipid complexes, via simultaneous involvement of phospholipids from both bilayer leaflets or, extraction phospholipids first from the outer and then, the inner leaflet |
2019 | [72] |
| Monomeric Aβ (Aβm), Fibrillar oligomers (FO), globular nonfibrillar oligomers (NFO) | Anionic DMPG lipid monolayers, POPC and POPG LUVs | •All Aβ species have affinity for the anionic membrane •Aβm and FO disrupt hexagonally packed lipid domains and result in membrane thinning and instability |
NFO the most toxic, followed by FO, and finally Aβm | •Detergent-like lipid removal mechanism: NFO-induced membrane destabilization and lipid extraction, potentially by forming stable protein/lipid complexes and detaching from membrane surface •Aβm and FO-induced reorganization of lipid packing and macroscale membrane deformation |
2019 | [63] |
| Aβ40, Aβ42 | Lipid bilayers contain PC, cholesterol, and GM1; LUVs | Detergent effect likely impacting cellular homeostasis and integrity, in line with the relative cytotoxicity of Aβ oligomers compared with fibrillar assembly states | •Only Aβo causes a detergent-like effect, extraction of lipids from the membrane •Fibers embed in the upper leaflet of the bilayer |
2019 | [66] | |
| Aβ40 | LUVs, BTLE | GM1 | •Soluble oligomers form small channel-like pores on the membrane •Aβ fibril elongation causes membrane fragmentation through a detergent-like mechanism •Membrane disruption by Aβ40 is transient and is abolished after fiber formation is complete, suggesting the pores are converted into fibers during membrane disruption that eventually detach from the membrane |
2012 | [76] | |
| Aβ40, Aβ42 | •GM1 clusters: GM1/SM/cholesterol (lipid raft) and GM1/PC •SH-SY5Y cells |
GM1/SM/cholesterol | •Aβ fibrils formed on ganglioside clusters cytotoxic, whereas fibrils formed in solution much less toxic | 2011 | [67] | |
| Aβ40, Aβ42 | J20 tg mice (hAPP transgenic mouse model), wild-type mice | GM1 and monosialotetrahexosyl head group of GM1 | Binding of Aβ to GM1 leading to downstream synaptotoxicity | Hydrophobic Aβ42 oligomers released into the extracellular fluid (ISF) become rapidly sequestered onto hydrophobic surfaces of adjacent lipid membranes | 2014 | [64] |
| Aβ42 oligomer, A+(toxic) and A−(benign) | SH-SY5Y neuroblastoma cells | GM1 | •Cytotoxicity of oligomers related to their membrane affinity •Disruption of lipid bilayers, alteration of their permeability and malfunction of raft associated Ca2+ channels, leading to Ca2+ influx into cells and toxicity |
2016 | [82] | |
| Aβ42 | Embryonic rat brain neuronal cell | PC | Neuronal death caused by Aβ42 completely preventable by pretreatment with PC in a dose-dependent manner | •PC modulates interactions between Aβ and membrane lipids, the phospholipid head group can stabilize Aβ to prevent fibrillogenesis •Decreased cholesterol-to-PC ratio attenuates aggregate formation of Aβ, probably due to enhanced Aβ insertion into the membrane |
2016 | [77] |
| Aβ42 | DMPC/cholesterol vesicles | Cholesterol | •Lipid membranes containing cholesterol promote Aβ42 aggregation by enhancing its heterogeneous primary nucleation rate by up to 20-fold through a heterogeneous nucleation pathway, while total load of toxic oligomers during the reaction is similar •Multiple cholesterol molecules cooperate with Aβ42 •Homogeneous pathway: new aggregates are formed solely from the interactions between soluble monomers •Heterogeneous pathway: generation of new aggregates depends on the concentrations of both monomeric Aβ42 and lipid vesicles |
2018 | [78] | |
| Aβ42 | •SCA-7 •Mouse embryonic day 17, wild-type cortical neurons •APPswe/PSen1-M146L mice |
PrPC | PrPC required by Aβ oligomers to induce Fyn activation and subsequent NR2B phosphorylation that is associated with transient increase in NR2B at the cell surface, with consequent excitotoxicity | PrPc, a cell surface glycoprotein that is associated with lipid rafts, as a receptor for Aβ oligomers | 2012 | [88] |
| Aβ | Human Brain Tissue; Wild type, APP/PSEN1, 5×FAD, tg2576, CRND8, 3×Tg mice | PrPC | PrPC-interacting Aβo species within human AD brain most critical for toxicity | Fraction of total Aβ oligomers interacting with PrPC vary considerably in different AD models and can determine the extent of contribution to AD-like symptoms of PrPC-dependent molecular mechanisms | 2015 | [89] |
| Aβ | APP23xTAU58 and APP51/16xTAU58 mice | PrPC | Mice overexpressing higher levels of soluble, presumably extracellular, APP-derived Aβ, show accelerated p-τ spreading accompanied by binding of Aβ and p-τ to PrPC | 2019 | [90] | |
| Aβ42 | BV2 microglia cells | TLR4, C and N-terminal region of Aβ42 interact with membrane | •Oligomeric aggregates causing induction of Ca2+ influx, activation of an innate immune response through TLR4 signaling •Monomeric or fibrillar forms of Aβ42 not causing inflammatory response •Antibody binding to the N-terminal region most effective in counteracting the toxicity of various Aβ42 aggregates |
•Synthetic Aβ aggregates exist in a range of different sizes and structures with the longer protofibrils being the inflammatory species and signal via TLR4 •Hydrophobic C-terminal region of these oligomeric species are responsible for observed disruption of the lipid bilayer •N-terminal region of these oligomeric species is important for inciting inflammation and toxicity |
2019 | [94] |
| Aβ42 | BV2 microglial cell, rat astrocytes | TLR4 | Soluble Aβ aggregates causing LTP deficits and neuronal death via an autocrine/paracrine mechanism due to TLR4 signaling | TLR4 mediates inflammatory response to aggregated Aβ42 | 2020 | [91] |
| Aβ40, Aβ42, 125I-Aβ40 | T2DM male db/db (BKS. Cg-m +/+ Leprdb/J) mice | RAGE | Inhibition of RAGE leading to inhibited neuronal Apoptosis, improved hippocampal plasticity, and ameliorated memory impairment | RAGE mediates Aβ accumulation, and implicates in inflammation, immunity, Apoptosis and cellular senescence through NF-κB signaling | 2018 | [92] |
| Aβ42, iso-Aβ (containing an isomerized Asp7 residue) | N2a, SH-SY5Y cells | α7nAChR | •Aβ-α7nAChR association disrupting the receptor’s function and causing neurotoxicity •Increased neurotoxicity Iso-Aβ in vitro, mediated by the α7nAChR |
•α7nAChR mediates cholinergic dysfunction in AD •Aβ binds to the α7nAChR, disrupting the receptor’s function and causing neurotoxicity •Effects of iso-Aβ and Aβ are α7nAChR-dependent |
2019 | [93] |
Figure 2. Hypothesized mechanisms of Aβ-membrane interaction.
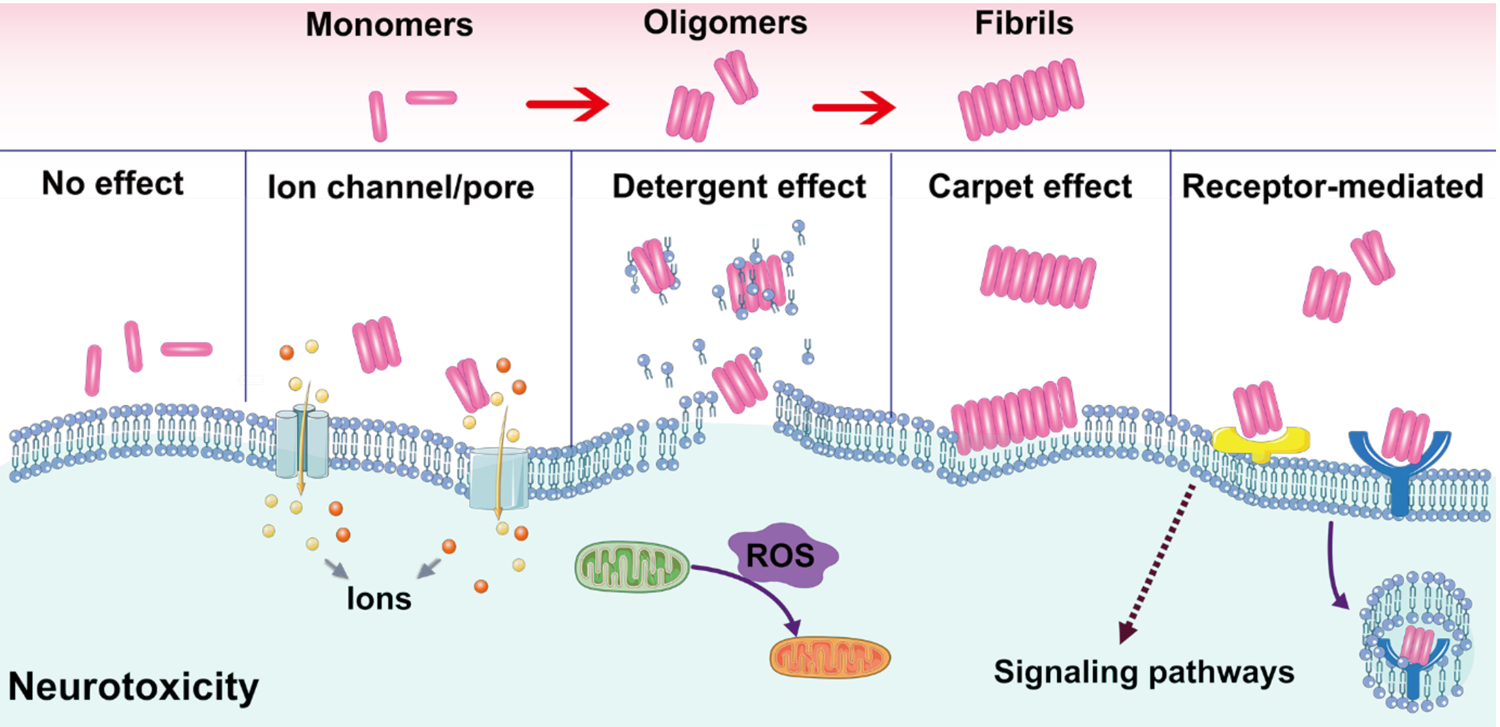
Carpet effect, ion channel, pore formation, detergent effect and receptor-mediated interaction are the most accepted modes of interaction.
Figure 3. Aβ-membrane interaction and subsequent biological effects.
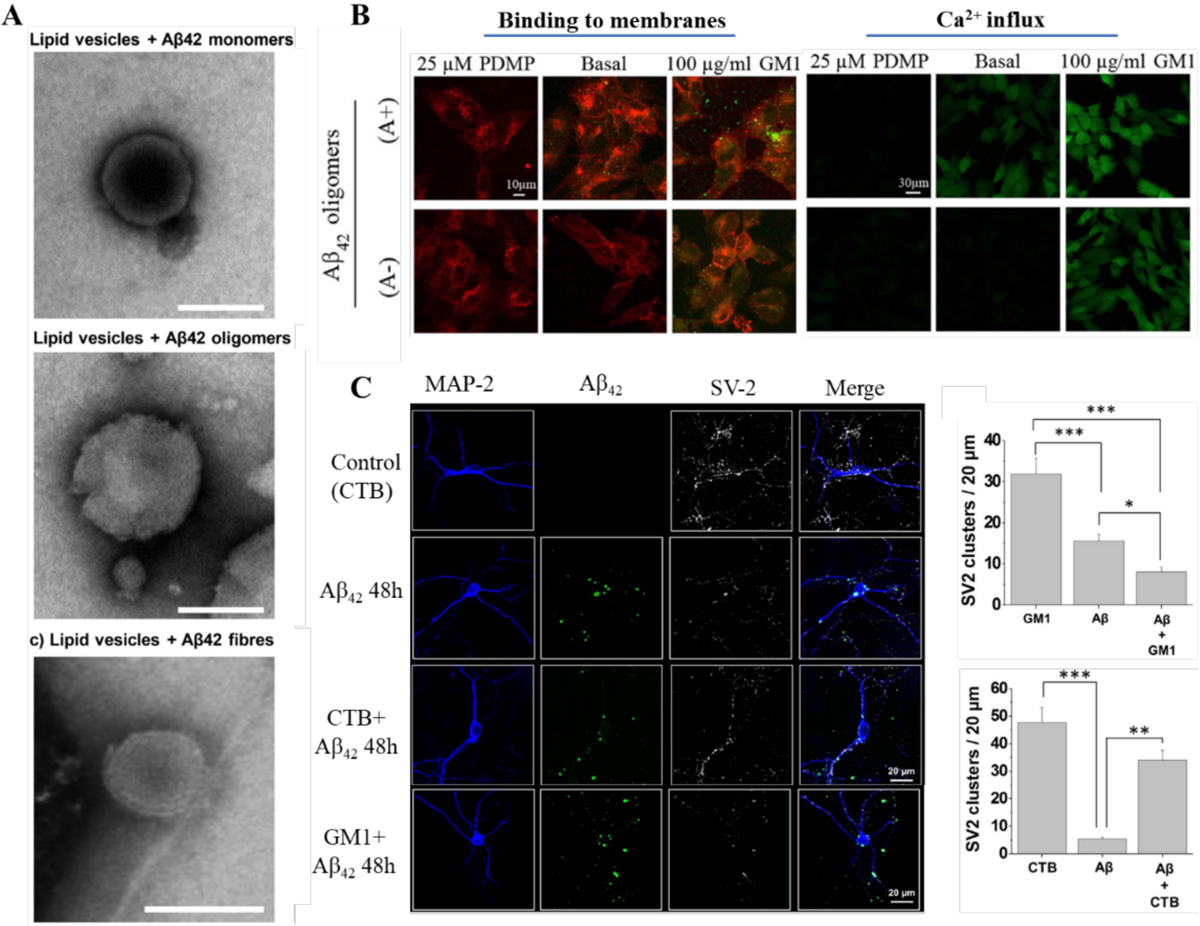
(A) Disruption of lipid vesicles by Aβ42 species. (B) Binding of Aβ42 oligomers to cells with different GM1 contents and ensuing Ca2+ influxes. GM1-depleted (25 μM D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol, PDMP), basal and GM1 enriched (100 μ g/mL GM1) cells treated for 1 h with 10 μM A+ (toxic, obtained by 1 day incubation) or A− (benign, obtained by 4 day incubation) Aβ42 oligomers. Red and green fluorescence emissions were from the cell membranes and Aβ42 oligomers, respectively. (C) Confocal micrographs and quantification of fluorescent Aβ clusters showing the distribution of Aβ clusters on the cell membrane surface along with the synaptic vesicle protein 2 (SV2) immunoreactivity in hippocampal neurons treated with Aβ-FAM. Cholera Toxin Subunit-B could block the interaction of Aβ with GM1. Panels reproduced from Refs. 66, 82–83. Copyrights 2019 The American Society for Biochemistry and Molecular Biology, Inc., 2016 Springer Nature, 2017 Elsevier B.V.
The interaction sites for Aβ peptides within the membrane were localized at the lipid rafts enriched in GM1, cholesterol and phosphatidylcholine (Table 1).[64, 76–78] The increment of GM1 distributed in the membrane was reported in the temporal and frontal cortices of the brain suffering from AD, and a specific form of Aβ bound to GM1 may serve as a seed for the formation of toxic amyloid aggregates.[64, 79–81] The more hydrophobic Aβ42 oligomers were rapidly sequestered from the brain interstitial fluid in vivo.[64] Compared to the non-toxic monomers, the Aβ42 oligomers could strongly bind to the GM1 on neuronal membranes to induce disruption of lipid bilayers via pore formation and malfunction of raft-associated Ca2+ channels, leading to Ca2+ influx into cells and downstream synaptotoxic effects (Fig. 3)[82–83] Hence, blocking the interaction between GM1 and Aβ peptides has been suggested as a strategy for AD drug discovery.[74, 84] Cholesterol, as a principal constituent in lipid rafts, not only facilitates Aβ binding to GM1 but also interacts with Aβ directly.[4, 67, 78] Cholesterol-containing lipid vesicles could promote Aβ42 aggregation, as evidenced by the enhanced primary nucleation rate of Aβ42 by up to 20-fold through a heterogeneous nucleation pathway. GM1 and cholesterol cooperation has been proposed to expedite amyloid pore formation in the cell membrane: amyloid peptides first bind to the negative charged sialic acid in GM1 via electrostatic interaction, then cholesterol interacts with the inserted peptides to finalize the pore formation process.[4, 85] A membrane therapy, targeting either cholesterol or GM1 or both, was successfully demonstrated by effectively disrupting the interactions of Aβ with the two types of membrane lipids.[85]
In addition to membrane lipids, other cell surface molecules have been considered as possible receptors of Aβ peptides, including cell-prion peptide (PrPC) and Toll-like receptor 4 (TLR4), the receptor for advanced glycosylation end products (RAGE) and nAChRs (Table 1).[86–93] Aβ oligomers were found to bind with PrPC specifically and the binding complex was related to synaptotoxicity, inhibition of long-term potentiation (LTP), memory impairment, and decreased survival in AD mice models.[88–89] PrPC-binding Aβ oligomers, and even their reactions with phosphorylated τ-protein (p-τ) in neurofibrillary tangles were also found in the human brain.[88, 90] Concomitant hippocampal Aβ burden and RAGE upregulation were discovered in mice, and the inhibition of RAGE ameliorated memory impairment.[92] The LTP deficits and neuronal death caused by Aβ were also found through an autocrine/paracrine mechanism due to TLR4 signaling.[91] Longer Aβ protofibrils caused inflammation and increased production of TNF-α and IL-β via TLR4.[94] Therefore, characterization of the binding of membrane surface receptors and Aβ will undoubtedly facilitate the development of novel AD therapies. Furthermore, alternations to lipid rafts and perturbations to membrane signaling may also be considered as promising molecular targets for AD.
3. Nanoparticle-membrane interaction
In this section, we highlight computational and experimental findings of nanoparticle-membrane interactions, which provide a valuable basis for describing the behavior and fate of AD nanomedicines in cellular environments.
3.1. In silico studies
Theoretical studies of nanoparticle-membrane interaction first began in 2007, when Qiao et al. examined a DPPC bilayer exposed to a pristine fullerene C60 and its hydroxylated counterpart fullerol C60(OH)20 (Fig. 4A).[97] Using atomistic molecular dynamics simulations the researchers found that the potential of mean force (PMF) favored uptake of the hydrophobic C60, while the amphiphilic C60(OH)20 was excluded by the lipid bilayer. However, the hydroxyl groups of the fullerol enabled their H-bonding with the head groups of the DPPC bilayer, rendering their close association with the top leaflet of the artificial membrane. This study provided the first theoretical support to the experimentally observed phenomenon that the toxicity of nanoparticles usually increases with their increasing hydrophobicity.[98–99]
Figure 4. Nanoparticle-membrane interactions in silico.
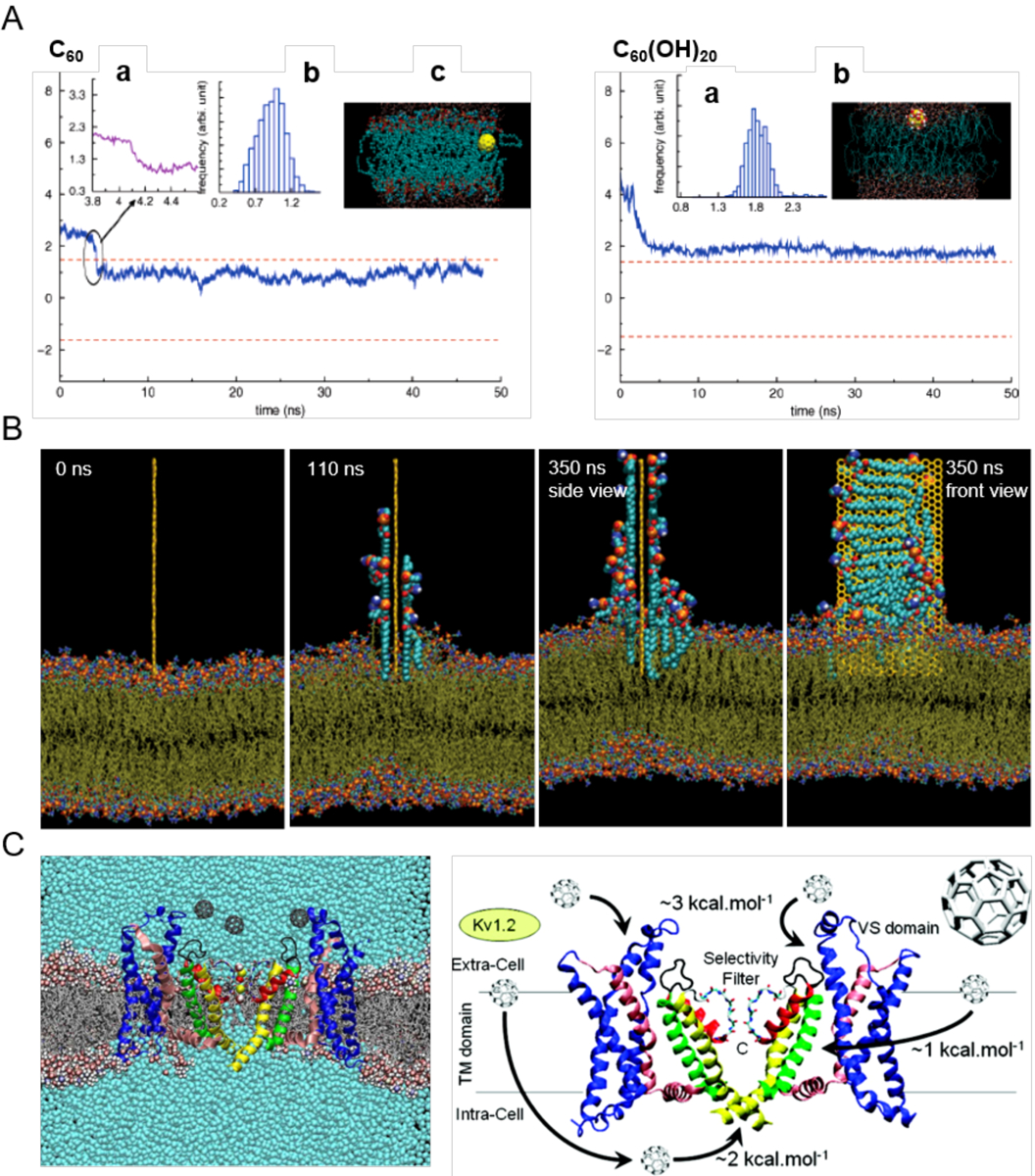
(A) Uptake of C60 (left) and C60(OH)20 (right) in the transmembrane (z) direction. The two dashed lines denote the upper and lower leaflets of the DPPC bilayer. Top left panel: (a) Zoomed trajectory at t = 4.09 ns. (b) Center-of-mass (COM) of C60 after it enters the bilayer (t > 4.2 ns). (c) Side view of the simulation system at t = 34.5 ns. The yellow ball denotes the C60 particle, cyan dots denote the lipid tail groups, and the red and blue dots represent the lipid head groups. Top right panel: (a) COM of C60(OH)20 during simulation. (b) Side view of the simulation system. Yellow balls and the attached large red and white dots denote the C60(OH)20 particle, cyan dots denote the tail groups of the DPPC lipids, and the small red and blue dots denote the lipid head groups.[97] (B) Lipid extraction by graphene in docking simulations over time. The restrained graphene nanosheet was docked at the surface of the outer POPE membrane of E. coli.[100] (C) Affinity of C60 with membrane potassium channels. (Left) A Kv1.2 channel embedded in a POPC lipid bilayer in the presence of C60 nanoparticles and their associated energetics.[101] Panels reproduced from Refs. 97, 100, 101. Copyrights 2007 American Chemical Society, 2013 Springer Nature and 2010 American Chemical Society.
Many computational works have ensued, offering new insights into nanoparticle-cell membrane interactions and their biological implications. Using CG simulations Wong-ekkabut et al. examined the translocation of many C60 nanoparticles across a dioleoylphosphatidylcholine (DOPC) lipid bilayer, which was shown to be thermodynamically driven and occur on the microsecond timescale.[102] In addition, high concentrations of the fullerene nanoparticles induced changes in both the structure and elasticity of the lipid bilayer. Using dissipative particle dynamics (DPD), Yang and Ma found that the membrane-penetrating capabilities of nanoparticles possessing the shapes of spheres, ellipsoids, rods, discs and pushpin-like were determined by the contact area between the nanoparticles and the lipid bilayer, as well as the local curvature of the nanoparticles at the contact points.[103] Using CG simulations, Shi et al. noted that the cell entry of anisotropic one-dimensional carbon nanotubes favored tip recognition and was accompanied by rotation.[104] In addition, the insertion and lipid extraction capacities of two-dimensional graphene nanosheets interacting with a lipid bilayer of Escherichia coli (E. coli) (outer layer: palmitoyloleoylphosphatidylethanolamine or POPE; inner layer: both POPE and palmitoyloleoylphosphatidylglycerol or POPG) were reported by Tu et al. employing CG MD simulations (Fig. 4B).[100] Utilizing MD simulations, Kraszewski et al. examined the association of C60 with potassium ion channels in a DOPC lipid bilayer, where the free energies of the binding between the nanoparticle and the KcsA, MthK, and Kv1.2 protein domains of the potassium channels ranged from 0.7–7.6 kcal/mol to offer a quantitative baseline for nanoparticle-ion channel association (Fig. 4C).[101]
3.2. Experimental studies
Several experimental studies have reported the dependence of cell/lipid membrane interactions with 0D-2D nanomaterials on the nanoparticle size, hydrophobicity, morphology, charge as well as surface modification. With carbon-based nanomaterials (Table 2), Chen et al.[105] reported the plasma membrane translocation of C70 assembled with natural organic matter (NOM) led to their increased cellular uptake in mammalian cells. This effect was not found with plant cells, however, due to the size-exclusion effect afforded by the plant cell wall. With carbon nanotubes (CNTs), their cell internalization was highly dependent on their physical size. Unlike multi-walled CNTs, single-walled CNTs exhibited high cellular uptake and internalization via the energy-dependent/independent endocytotic pathways based on their length (100–200 nm: clathrin dependent; 50–100 nm: clathrin and caveolae dependent, <50 nm: energy independent) (Table 2).[106] In vitro study on the intracellular transport of graphene quantum dots (GQDs) in Madin-Darby Canine Kidney (MDCK) monolayer cells showed a lipid-raft mediated transcytosis of GQDs across the plasma membrane and the cytoplasm, which was inhibited by a lipid-raft inhibitor methyl-β-cyclodextrin.[107] For graphene oxide (GO) nanosheets, quartz crystal microbalance with dissipation monitoring (QCM-D) sensing showed the rupture of positively charged POPC/POEPC (3:1) liposomes mainly driven by electrostatic interactions.[108] In a cellular membrane environment, exposure to GO nanosheets induced pore formation in both viable and non-viable A549 and Raw264.7 cells initiated by lipid extraction.[109] Using a 2D sandwiched graphene-membrane superstructure, Chen et al.[110] reported the Brownian diffusion pattern of GO across cell membranes. Taking advantage of the formed pores the researchers demonstrated GO as efficient drug nanocarriers against breast cancer cells (Table 2). Exposing large and small GO sheets to neutrophils, furthermore, elicited membrane stripping, prompted the formation of neutrophil extracellular traps (NET), and elevated the production of carbon radicals and oxidized cholesterol species, in a GO-size dependent manner.[111]
Table 2.
Nanoparticle-membrane interaction.
| Nanomaterial class | Nanomaterial | Cell lines/ lipid membranes | Material properties | Biological effect | Interactions/Mediated pathway | Year | Ref. |
|---|---|---|---|---|---|---|---|
| Carbon based | C60(OH)20 | Allium cepa cells | Concentration: 10–110 mg L−1 Solubility: water soluble Size: (1–24 nm) Surface charge: −43 mV |
Plant cell wall permeation, Plasma membrane exclusion, cell damage | Hydrophilicity, electrostatic repulsion, H-bonding, Van der Waals | ||
| HT-29 cells | Low affinity for cell membrane | ||||||
| C70–NOM | Allium cepa cells | Concentration: 10–110 mg L−1
Solubility: hydrophobic and clustered in high concentrations (10–100 nm) Surface charge: −34 mV |
Plant cell wall protection | Hydrophobic interactions, non-covalent assembly | 2010 | [105] | |
| HT-29 cells | Cell lysis | ||||||
| S/L-SWNTs | Size (diameter): (1–3 nm) Size (length): S-SWNTs (50–100 nm) |
Cell nucleus localization | Nuclear envelope transport, energy independent pathway, clathrin-dependent endocytosis, caveolae-dependent endocytosis | 2010 | [106] | ||
| Hep G2 cells | Size (diameter): (1–3 nm) Size (length): L-SWNTs (100–200 nm) |
Cytoplasm localization | Clathrin-dependent endocytosis | ||||
| S/L-MWNTs | Size (diameter): (10–30 nm) Size (length): S-MWNTs (0.5–1 μm) Size (diameter): (10–30 nm) Size (length): L-MWNTs (1–2 μm) |
FA receptor mediated endocytosis | Receptor binding | ||||
| GQDs | MDCK monolayer cells | Size: 3 nm, 12 nm Surface charge: −1 mV (3 nm), −15 mV (12 nm) |
Time, concentration (~300 mg L−1) and size dependent cell membrane permeability. Noninduced cell lysis and plasma membrane penetration | Lipid raft-mediated transcytosis | 2015 | [107] | |
| GO-OH/COOH | POPC/POEPC, POPC/POPS (3:1) liposomes | Surface charge: 22 mV (POPC/POEPC (3:1) liposomes), −26 mV (POPC/POPS (3:1) liposomes), −56 mV (GO, pH 4) | Rupture of pre-adsorbed positively charged liposomes (QCM-D frequency shift), multilayered structure formation | Electrostatic interactions, H-bonding | 2012 | [108] | |
| GO (pristine/oxidized) | A549 and Raw264.7 cells | Layer: Single (thickness ~1 nm) Size: (200–700 nm) Raman bands: D (~1350 cm−1) and G (~1598 cm−1) Oxidization degree: 26.1% |
Cell death and pore hole formation (non- autophagy) induced at concentrations 50 to 200 μg/mL. | Phospholipid extraction on GO surface (unstable Stage I) (↓Van der Waals energy), balance between internal membrane tension and graphene-mediated dispersion force (metastable Stage II), membrane tension, lipid extraction and pore formation (↓ Van der Waals energy) (stable Stage III) | 2017 | [109] | |
| DOPC lipid bilayer sandwiched GO | J774A (macrophage) and 4T1 (breast cancer) cells | Size: ~40 nm (GO) Height: Less than 2 nm Layer: Single or double |
Transport inside of cell membrane (Lévy and directional dynamics), pore hole formation | 2019 | [110] | ||
| S/L-GO sheets | Primary human neutrophils | Layer: Single or double (thickness: 1–2 nm) Size: GO-S (50–300 nm), GO-L (10–40 μm) Surface charge: −55 mV, −37 mV in cell culture medium |
Dose-dependent loss of cell viability, membrane stripping, size-dependent (GO-L) NET induced production (Ca+2, ROS dependent), elevation of oxidized cholesterol species | Potential interaction with positively charged histones (NET), lipid oxidation | 2018 | [111] | |
| Metal based | TiO2NPs | HMVEC monolayer and MDA-MB-231 cells | Size: 57 nm (in cell culture medium) Surface charge: −24 mV (in cell culture medium) | Dose (10–1250 μM) and size (nanoscale) dependent endothelial cell leakiness (NanoEL) due to cell junction (VE-cadherin) disruption, further promoting breast cancer cell intravasation | Electrostatic interactions between negatively charged TiO2 NPs and positively charged VE-cadherin side chains. Homophilic loss of cell-cell interaction | 2013, 2019 | [112– 113] |
| AuNPs | HMVEC, HMMEC and HUVEC monolayer | Size: 10–30 nm Surface charge: ~−8 mV (in cell culture medium) SPR peak: 518 nm (Au10), 523 nm (Au30) |
NanoEL | Disruption of VE-cadherin-VE-cadherin interactions | 2017 | [114] | |
| MSNs (MCM-41 and SBA-15) | RBC membranes | Size: ~600 nm (SBA-15), ~100 nm (MCM-41) | Size- (larger-> higher hemolytic activity) and surface (large surface->larger binding energy)- dependent interaction and engulfment between MSN and RBC membranes | Binding of silanol-rich surface of MSNs with phosphatidyl choline-rich RBC membrane | 2011 | [115] | |
| Surface modified (Au based) | AuNPs | CP70, ASM, A2780, BEC cells | Plasma membrane charge: between −75 and −55 mV Size: 2 nm (AuNP core) |
Surface charge (+AuNPs) → rapid plasma membrane depolarization, intracellular uptake and [Ca2+]i elevation. Concentration dependant (0–1.2 mM) | Electrostatic interactions | 2010 | [116] |
| PVA-AuNPs | A549 and J774A.1 cells | Surface charge: Range of −3 mV to −14 mV in cell culture medium Functional groups: Primary, secondary, tertiary amino groups | Increased NP-cell membrane association for primary amines with high amine density (A549) combined with high protein adsorption (serum albumin, alpha-2-HS-glycoprotein) irrespective of amine density | Salt bridge formation, hydrophobicity, conformational effect on PVA coating and accessibility toward polar amino acids of serum proteins | 2018 | [117] | |
| Ligand-AuNPs | POPC, DOPC/DOPS (−), DOPC/DOEPC (+) A549 cells | Size: 13 nm Surface ligands: MW, charge, and bonding categorization, small molecules, biomacromolecules |
Ligand’s size and adsorption affinity-two main factors for ligand exchange with lipid molecules Influence on endocytic pathways, uptake efficiency, cell membrane integrity |
Electrostatic, hydrophobic and Van der Waals interactions | 2019 | [118] | |
| Ligand-coated AuNRs | SOPC monolayer, THP-1 cells | Diameter, length: CTAB/PDC-AuNRs (~15, ~60 nm) Surface charge: CTAB/PDC-AuNRs (30–40 mV in H2O, −13 mV in serum and H2O) Ligand-NR stability: (PDC>CTAB) |
Weaker ligand stability and further detachment lead to membrane thickness decrease (SOPC), lysosomal membrane penetration, concentration dependent cytotoxicity and inflammation | CTAB-AuNRs (Van der Waals), PDCAuNRs (Hydrophobic and electrostatic interactions). CTAB-AuNRs-SOPC (↓Van der Waals, electrostatic interactions) | 2019 | [119] | |
| Protein corona | BSA-PMASH NPPs | THP-1 monocytic and dTHP-1 macrophage cells | Surface charge: −25 mV in BSA medium | Reduced cellular association and internalization by protein-NPP complexes than NPPs in monocyte cells. Differential cellular association in dTHP-1 cells (SR-A-mediated phagocytosis) | Structural conformation of BSA upon protein corona formation influences binding with SR-A | 2013 | [121] |
| BSA/HSA/HDL-AgNPs | RLE and RAEC cells | Size: 19 nm (AgNPs), 70 nm (HSA-AgNPs), 31 nm (BSA-AgNPs), 62 nm (HDL-AgNPs) Surface charge: −35 mV (AgNPs), −25 mV (HSA-AgNPs), −18 mV (BSA-AgNPs), −8 mV (HDL-AgNPs) |
Reduced cytotoxicity of protein corona AgNPs in both cells lines at high concentrations (50 μg/ml) at 3 h and 6 h. Increased inflammatory response for HDL-AgNPs in RLE cells IL-6 mRNA expression: AgNPs (control, RLE, RAEC cells), HSA-AgNPs (↓-RLE, RAEC cells) BSA-AgNPs (↓-RLE, RAEC cells), HDL-AgNPs (↑-RLE cells, ↓-RAEC cells), Samples in the presence of Blt2 inhibitor (↓-RLE, RAEC cells) |
Cell surface receptor-mediated signaling. Intracellular Ag+ release. Loss of protein corona upon internalization | 2014 | [122] | |
| HSA (modified)- DHLA-QDs | HeLa cells | HSA modified species: succinylated HSA (HSAsuc), aminated HSA (HSAam) Surface charge: −11 mV (HSA-QDs), −19 mV (HSAsuc-QDs), −6 mV (HSAam-QDs) |
Enhanced membrane binding and cell internalization of HSAsuc-coated-QDs and respective suppression by HSAam-coated-QDs due to modified charge distributions | Active pinocytic (clathrin-mediated) pathway to endosomes and lysosomes | 2014 | [123] | |
| Deglycosylated protein corona SiO2NPs | dTHP-1 (M1, M2 macrophages) cells | Size: 126–139 nm Surface charge: ~9 mV |
Enhanced cell membrane adhesion, nanoparticle uptake and stimulation of pro-inflammatory responses | Influence of glycosylation on immune activation | 2015 | [124] | |
| Hard/soft corona (human serum) PS-NPs (PS, PS-COOH, PS-NH2 | Raw264.7 cells | Size (NP soft corona): 140 nm (PS), 178 nm (PS-COOH), 188 nm (PS-NH2) Size (NP hard corona):92 nm (PS), 80 nm (PS-COOH), 88 nm (PS-NH2) Hard corona thickness: 15 nm Surface charge: −4 mV (PS), −1 (PS-COOH), 1 mV (PS-NH2) |
Differential cellular uptake between soft corona or Apo-A1 PS-NPs (inhibition) and hard corona PS-NPs. Significant enhanced uptake for hard corona PS-COOH-NPs | CLIC/GEEC endocytosis pathway for bare PS-NPs. Similar structural properties (large corona diameter ~100 nm and large number of Apo-A1 proteins) for soft corona and Apo-A1 PS-NPs lead to uptake inhibition | 2017 | [125] |
In addition to bare nanomaterials, negatively charged inorganic TiO2, SiO2, gold nanoparticles (AuNPs) have been reported to interact and disrupt VE-cadherin-VE-cadherin homophilic interactions in endothelial cells, causing leakiness (termed as “NanoEL”) in a dose- and size-dependent manner (Table 2).[112–114] This transient cellular phenomenon provides both a new mechanism for understanding nanotoxicity and a strategy for devising new nanoparticle-based cancer drug delivery and therapies. On the other hand, as with any viable nanomedicine, their prolonged blood circulation is essential for desired drug delivery and efficacy. Zhao et al.[115] reported that the adsorption of large mesoporous silica nanoparticles (600 nm) to silanol rich red blood cell (RBC) membranes induced disruption, nanoparticle internalization and hemolysis, compared with functionalized small mesoporous silica nanoparticles (~100 nm) (Table 2). Surface-modified AuNPs/AuNRs (gold nanorods) have been studied for their interactions with cell membranes. The electrostatic interactions between positively charged AuNPs and cell membranes exerted a significant effect on the integrity of plasma membranes causing rapid membrane depolarization and intracellular uptake of the nanoparticles.[116] Apart from charge, poly(vinyl alcohol-co-N-vinylamine) (PVA)-coated AuNPs displayed an increased effect of their backbone-dense primary amines on cellular association and the susceptibility of primary amines to protein corona formation.[117] Applying a library of ligand-coated AuNPs (Table 2) to supported lipid bilayers, Wang et al.[118] identified size and adsorption affinity as two key parameters for ligand exchange at the nanoparticle-cell membrane interface, and determined the role of ligand stability on membrane integrity in a separate study.[119]
A wealth of literature over the past decade[120] has implicated the central role of the protein corona for determining the biological behavior and fate of nanoparticles, including the cellular association and subsequent translocation of nanoparticles. The conformational changes of proteins upon binding with nanoparticles have been shown to trigger the activation of macrophage class A scavenger receptor (SR-A)-mediated phagocytosis of the nanoparticles in macrophage cells, compared to the reduced nanoparticle internalization in monocytic cells.[121] On the other hand, incubation of silver nanoparticles with human serum and cell culture medium and subsequent treatment of the nanoparticle-protein complexes with the scavenger receptor BI decreased their toxicity and IL-6 mRNA expression in rat lung epithelial and rat aortic endothelial cells.[122] By modifying protein charge distribution[123] or deglycosylating the coronal proteins,[124] other studies have revealed the importance of Coulombic interactions and immune activation for the cellular recognition and uptake of nanoparticles. In addition, surface chemistry of polystyrene nanoparticles has been found crucial for the rendition of a hard protein corona, thereby influencing the cellular uptake and endocytotic pathways of carboxylated vs. bare polystyrene nanoparticles.[125]
4. Aβ-nanoparticle interaction
The interactions between amyloidogenic human β2-microglobulin and a host of nanoparticles (copolymer particles, cerium oxide particles, quantum dots, and CNTs) were first examined by Linse et al. in 2007,[126] around the same time as the protein corona concept was put forth by Dawson and Linse et al.[127] Since then, amyloidosis inhibition with nanomaterials has evolved into a major effort concerning the aggregation, toxicity and mitigation of a range of amyloid proteins and peptides. In the following section, we highlight recent computational and experimental findings of Aβ-nanoparticle interactions, a foundation for the development of AD nanomedicines.
4.1. Simulation studies
Due to their versatile physiochemical properties, nanoparticles are promising amyloidosis inhibitors.[26, 128] Accurate and efficient modelling of Aβ-nanoparticle interaction can significantly contribute to our understanding of the bio-nano interfacial phenomena and provide a guidance to the design of anti-amyloid nanomedicines. Extensive simulations have been conducted to date to characterize the interactions between Aβ and various types of nanoparticles, including soft polymeric nanoparticles (e.g., dendrimers and hydrogels), carbon-based nanoparticles (e.g., fullerenes, carbon nanotubes, graphene oxides and carbon quantum dots), and metallic nanoparticles (e.g., gold nanoparticles), among others.[129–130]
Aβ showed a strong affinity for PEGylated nanoparticles due to the combination of both hydrophilic and hydrophobic interactions between the PEG shell and the peptide, which enabled the nanoparticle to encapsulate Aβ monomers and oligomers.[131] Carbon-based nanoparticles have been widely used in various biomedical applications such as biosensing, imaging and drug delivery.[132–133] C60 disrupted Aβ fibrillization by destructing the β-helical twist, destabilizing the D23–K28 salt bridge and hydrophobic interactions between the Aβ peptides.[134] 1,2-(dimethoxymethano) fullerene, a water-soluble fullerene derivative, inhibited the dimerization of Aβ by binding with the peptide aromatic residues (F4 and Y10), the central hydrophobic region (L17–A21) and the C-terminal hydrophobic regions (I31–V40).[135] Aβ peptide was found to adsorb on the surface of single-walled CNT rapidly, during which the hydrophobic core of Aβ initiated the adsorption process and subsequently brought the peptide N-terminal region in contact with the CNT. Once adsorbed onto the CNT, the peptide propensity of collapsing between the central hydrophobic core region and the hydrophobic patches near the peptide C-terminus was reduced significantly, indicating that CNT could disrupt Aβ aggregation.[136] Driven by the strong hydrophobic interaction between Aβ and CNTs, Aβ(25–35) peptides with mixed antiparallel-parallel β-strands assembled into a β-barrel wrapping around the CNTs.[137] Similarly, carbon quantum dots (CQDs) exhibited strong binding with Aβ, especially with the peptide N-terminal region. Upon adsorption onto the CQD surface, Aβ peptides preferred to bind with CQDs instead of other peptides, resulting in a reduction of the inter-peptide contacts and subsequent fibrillization inhibition.[138] In addition, graphene nanosheets were shown to effectively break and clear the pre-formed fibrillar structures of Aβ by extraction of the peptides, which was attributed to the exceptionally strong dispersion interactions between the peptides and graphene that were further enhanced by their strong π–π stacking.[139] AuNPs are often employed as theranostic agents in drug delivery and photothermal therapy due to their relatively low toxicity and surface chemistry for functionalization.[140] By parameterizing interactions between AuNPs and proteins in MD simulations using density functional theory (DFT) calculations,[141] bare AuNPs facilitated the adsorbed Aβ to adopt the fibril-prone conformations, suggesting that bare AuNPs could promote the peptide fibrillization.[142–143] Other simulations with negatively charged AuNPs showed that the fibrillization process of Aβ was actually inhibited due to the electrostatic interactions between Lys 16 and Lys 28 of the peptide with the nanoparticle surface charge.[144]
Based on the experimental and simulation studies, it can be concluded that nanoparticles can exhibit both promotion and inhibition effects on the aggregation of Aβ under different conditions.[145] To understand these seemingly conflicting effects of nanoparticles on Aβ aggregation, CG molecular dynamics simulations have been conducted to probe the effects of the strength of nonspecific nanoparticle-protein attraction and the relative concentration between protein and nanoparticles (Fig. 5).[146] It was revealed that the aggregation of Aβ on the nanoparticle surface was initially promoted with increasing nanoparticle-peptide attraction due to increased local peptide concentration on the nanoparticle surface and destabilization of the peptide folded state (Fig. 5A&C). However, further increase of nanoparticle-peptide attraction decreased the stability of amyloid fibrils and reduced their lateral diffusion on the nanoparticle surface necessary for peptide conformational changes and self-association, thus prohibiting amyloid aggregation (Fig. 5B&D). Moreover, it was found that the amyloid aggregation was also regulated by the relative concentration between protein and nanoparticles (Fig. 5E&F). Specifically, with a high nanoparticle/protein ratio, nanoparticles with intrinsic promotion effects may inhibit amyloid aggregation by depleting the proteins in solution while having a low concentration of the proteins on each nanoparticle’s surface. These results offered a molecular basis for delineating the contrasting and seemingly conflicting effects of nanoparticles on amyloid aggregation,[26] which can be utilized for tailoring the design of AD nanomedicines.[146]
Figure 5. Coarse-grained simulations of Aβ-nanoparticle interactions.
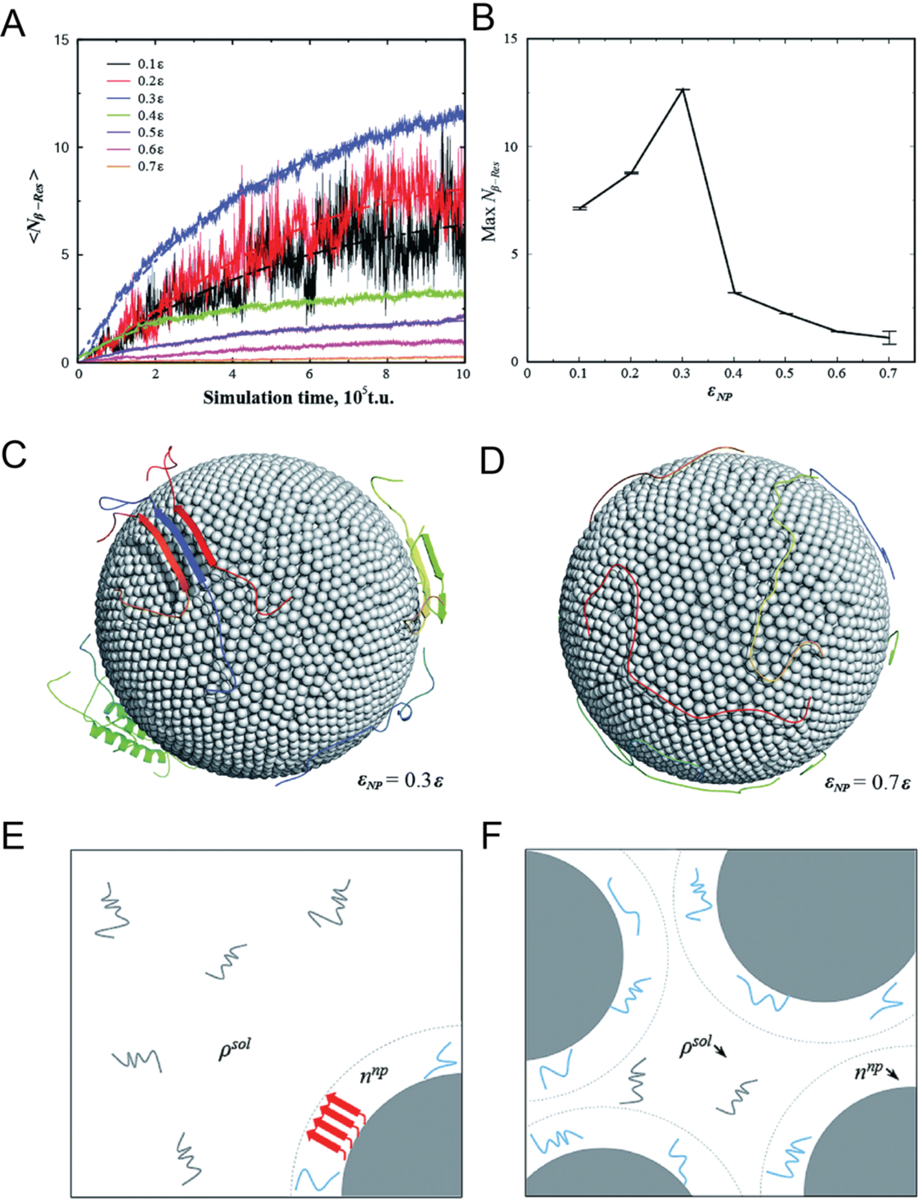
Dependence of Aβ aggregation on nanoparticle–protein interaction strength. (A) Average number of residues per chain that formed inter-peptide beta-sheets, Nβ–Res as a function of simulation time. (B) Maximum Nβ–Res as a function of nanoparticle-protein interaction strength εNP. Representative conformations with (C) εNP = 0.3ε (D) εNP = 0.74ε, where ε is the protein contact energy. (E) With a high protein/nanoparticle ratio, the nanoparticles (depicted as gray solid spheres) may promote the formation of amyloid fibrils (represented by red arrows) by increasing the local concentration of Aβ peptides on the nanoparticle surface (denoted as lines in cyan). (F) With a low protein/nanoparticle ratio, the nanoparticles displayed an inhibitive effect on the Aβ aggregation by depleting the peptides in solution but effectively having a low peptides concentration on nanoparticle surface. Reproduced with permission from Ref. 146. Copyrights 2015 The Royal Society of Chemistry.
Most of the existing simulations so far have only considered pristine or bare nanoparticles. However, upon entry into biological systems such as the plasma, the nanoparticle surfaces will be covered by various sugars, lipids and proteins, which leads to the formation the bio-corona.[147] As a result, it is the corona-coated nanoparticles rather than the pristine or bare nanoparticles that interact with Aβ in vivo. Therefore, it is of practical importance to investigate the effects of the protein corona on Aβ-nanoparticle interaction in the future. In a recent work by Javed et al., the interaction between Aβ and casein coated-gold nanoparticles (βCas AuNPs) was examined.[24] The results showed that Aβ monomer became more helical and an Aβ oligomer could not grow into an extended β-sheet structure upon binding with the intrinsically disordered βCas with high conformational flexibility. Moreover, there was no overlapping between the binding sites of βCas with Aβ and those with the AuNPs, suggesting that the chaperone-like βCas in the AuNP corona could trap Aβ monomers in the aggregation-incompetent forms and cap Aβ fibrils from elongation.
4.2. Experimental studies
The oligomers are the major toxic species of Aβ amyloidogenesis, inducing disruptions to membrane structure and cellular ion homeostasis as well as inflammation, immunity and apoptosis due to their interactions with cell membranes, proteins, chaperones and small ligands.[26, 148] The assembly of Aβ into pathological seeds, including the oligomers and subsequent tertiary aggregates such as protofibrils and amyloids, constitutes the primary neuropathological hallmark of AD. Thus, the design of AD therapeutics is usually focused on inhibiting and disrupting the Aβ aggregation pathway or accelerating Aβ elimination in the patient brain. To achieve this, a range of inorganic, polymeric, carbon-based and biomimetic nanoparticles and nanocomposites have been developed for potential AD treatment (Table 3, Fig. 6).
Table 3.
Aβ-nanoparticle interaction.
| Nanomaterials class | Nanomaterials name | Nanomaterials properties | Peptide | Biological effect | Effect on amyloidosis | Mode of interaction | Year | Ref. |
|---|---|---|---|---|---|---|---|---|
| Inorganic nanomaterials | Nanoceria | Size: 3–8 nm | Aβ25–35 | •Accumulate at mitochondrial outer membrane and plasma membrane •Reduce mitochondrial fragmentation •Reduce neuronal cell death |
Block Aβ-mediated mitochondrial fragmentation via the reduction of DRP1 S616 hyperphosphorylation | 2014 | [157] | |
| SPIONs-PEG-NH2 | Size: 20 nm Surface charge: 17.4 ± 2.5 mV |
Aβ42 | Dual effects on Aβ fibrillization: •High concentrations accelerate fibrillization under magnetic field •Lower concentrations inhibit fibrillization under magnetic field |
Magnetic field on the size and surface charge of SPIONs, thereby impacting Aβ fibrillization | 2015 | [156] | ||
| AuNPs | Size: 20, 50 and 80 nm | Aβ | Aβ oligomers more toxic than Aβ fibrils or plaques in inducing acute cell death | •Aβ aggregates on nanoparticle surfaces •Larger particles induce more Aβ aggregation on particle surfaces with a shortened lag phase |
2015 | [149] | ||
| Surface charge: positive (amine-AuNPs), negative (citrate-AuNPs) | Aβ | •Amine-AuNPs are more strongly attracted to Aβ, forming smaller aggregates, not protofibrils •Citrate-AuNPs act as nucleation seeds to accelerate fibrillization |
•Electrostatic interactions •Replacement of citrate on AuNPs with Aβ and direct attachment of AuNPs to Aβ |
|||||
| Shape: Spherical AuNPs, nanorods (AuNRs), and nanocubes (AuNCs) | Aβ | Nanostructure-dependent cytotoxicity on neuroblastoma cells | •AuNCs interact with Aβ to produce fibril networks •AuNRs inhibit Aβ aggregation |
•AuNCs possess a larger effective surface area and are more isotropic than AuNRs •Larger aggregates form on AuNCs | ||||
| CeONP@POMs | Size: ~5 nm Surface charge: −48.2 mV |
Aβ40 | •Reduce intracellular ROS •Promote PC12 cell proliferation •Cross the BBB •Inhibit Aβ-induced BV2 microglial cell activation |
•Inhibit Aβ fibrillization •Disaggregate peptide fibrils •Hydrolyze peptide monomers |
Hydrolytic effect | 2016 | [151] | |
| MoS2 NPs | Size: ~100 nm FTIR peaks: 1630, 1420, and 1280 cm−1 |
Aβ42 | •Scavenge ROS •Block formation of Ca2+ channel in cell membrane •Alleviate cell toxicity |
•Inhibit Aβ fibrillization •Destabilize Aβ fibrils |
Electrostatic attraction and high surface ratio effects | 2017 | [150] | |
| CGA@SeNPs | Size: ~100 nm | Aβ40 | •Reduce ROS generation •Inhibit neurotoxicity of Aβ40 |
Inhibit Aβ aggregation | Aβ binds on SeNPs via N-donors containing side chains of amino acids to form an Se–N bond, blocking direct contact between peptide monomers | 2018 | [155] | |
| βCas AuNPs | Size: 7.5 ± 2.6 nm Surface charge: −11.7 ± 1.8 mV |
Aβ42 | •No lethality but reduced locomotion, nonresponsive mobility and a loss of balance in larvae upon Aβ injection •βCas AuNPs recover the mobility and cognitive function of adult zebrafish |
•No such mitigation is obtained with caseins alone •βCas promote fast Aβ nucleation |
Sequester toxic Aβ42 through a nonspecific, chaperone-like manner | 2019 | [24] | |
| SiO2–cyclen | Size: 65.2 ± 4.9 nm | Aβ40 | •Cross the BBB •Reduce cytotoxicity and ROS |
Inhibit metal-induced aggregation (Zn2+, Cu2+) | •Metal-chelation •Crossing the BBB via adsorptive or receptor-mediated transportation |
2019 | [154] | |
| BP@BTA | Height: 3–5 nm UV–vis: 345 nm (BTA, covalent interaction with BP) |
Aβ42 | •Reduce the peptide cytotoxicity •Attenuate peptide neurotoxicity to CL2006, extend the lifespan of worms •BP@BTA and its degradation products are nontoxic and biocompatible |
•Inhibit Aβ aggregation under NIR, no effect under dark •Oxygenate Aβ |
High affinity for Aβ due to specific amyloid selectivity of BTA | 2019 | [153] | |
| AgTNPs | Edge length: 70 ± 8 nm Surface charge: −41 ± 1.4 mV |
Aβ40 | Increase cell viability | •Prevent formation of Aβ fibrils •Dissolve mature Aβ fibrils |
•AgTNPs selective bind the positively charged amyloidogenic sequence of Aβ monomer •AgTNPs dissolve mature Aβ fibrils via plasmonic photothermal property |
2019 | [152] | |
| Polymeric nanomaterials | PMA-nanodiscs | Size: ~10 nm | Aβ40 | Aβ40 oligomers incubated with PMA-nanodiscs exhibit relatively less neurotoxic and neuronal damage | •Lipid concentration and composition are important to regulate Aβ fibrillization •Form low-ordered Aβ aggregates in the presence of nanodiscs |
Amide (H–N) and side chain protons of Aβ show a correlation with PMA functional groups, hydrophobic chain and quaternary ammonium group | 2018 | [159] |
| NC-KLVFF | Size: 14 ± 4 nm | Aβ42 | •Attenuate neuron damage •Regain endocranial microglia’s capability to phagocytose Aβ •Protect hippocampal neurons against Apoptosis |
•Inhibit self-aggregation of Aβ •Dissociate Aβ fibrils |
•Polymeric surface property impacts peptide binding affinity •Block interaction between Aβ oligomers and cell membranes |
2019 | [22] | |
| CPNPs | Size: ~4.7 nm Surface charge: ~ 30 mV |
Aβ40 | Inhibit Aβ fibrillization | Binding to the termini of seed fibrils can effectively inhibit fibrillization | 2019 | [165] | ||
| G5-PAMAM G6-PAMAM | NMR: 7.05–7.25 ppm UV peak: 260 nm |
Aβ42 | Phenyl derivatives of high-generation dendrimers (G5-P and G6-P) significantly inhibit Aβ42 aggregation and alter ultrastructure of Aβ42 aggregates | Hydrophobic binding-electrostatic repulsion theory | 2019 | [160] | ||
| Carbon-based nanomaterials | GQDs | Size: ~8 nm Surface charge: negative |
Aβ42 | •Increase survival rate •Great biocompatibility |
Inhibit Aβ fibrillization | Hydrophobic and electrostatic interactions | 2015 | [166] |
| C60(OH)16 | Aβ40 | Biocompatible materials | •Reduce the formation of amyloid fibrils | •Electrostatic interactions •Binding with hydrophobic Aβ C-terminus |
2016 | [167] | ||
| SWNT-OH | Size: ~8 nm | Aβ42 | Cytoprotective effects against Aβ42 fibrillization-induced cytotoxicity | •Inhibit Aβ42 fibrillization •Disaggregate preformed amyloid fibrils |
Nonpolar interactions, especially van der Waals forces | 2019 | [168] | |
| CQDs | Size: ~2.8 nm Surface charge: ~−44.6 mV |
Aβ42 | Restore embryo survival rate by 32% Decrease ROS production | Inhibit Aβ fibrillization | Hydrophobic interaction and H-bonding | 2020 | [138] | |
| Biologically inspired nanomaterials | ApoE3-rHDL nanodiscs | Size: 27.9 ± 8.9 nm Surface charge: −4.07 ± 0.83 mV |
Aβ40, Aβ42 | •Accelerate microglial, astroglial, and liver cell degradation of Aβ by facilitating lysosomal transport •Cross the BBB |
Inhibit Aβ aggregation | •Receptor-mediated endocytosis •High binding affinity for Aβ monomers and oligomers |
2014 | [162] |
| αNAP-GM1-rHDL | Size: 25.42 ± 1.18 nm Surface charge: −15.70 ± 0.93 mV |
Aβ42 | •Decrease cell toxicity in vitro •Reduce Aβ deposition, ameliorate neurologic changes, and rescue memory loss in vivo |
•Inhibit Aβ aggregation •High Aβ binding affinity and clearance activity |
•Aβ targeting: GM1 •ApoE-concentration-dependent binding synergetic effect of DMPC, GM1 and ApoE |
2015 | [163] | |
| ANC-α-M | Size: 35.95 ± 9.05 nm Surface charge: negative |
Aβ42 | •Accelerate Aβ42 degradation •Facilitate microglia-mediated uptake in vitro •Decrease amyloid deposition, attenuate microgliosis, and rescue memory deficit in AD mice |
Block formation of both Aβ oligomers and fibrils, disturb preformed fibrils | •High affinity for Aβ monomers and oligomers •ApoE-dependent cellular uptake |
2016 | [161] | |
| dcHGT NPs | Size: ~15 nm | Aβ42 | •Relieve inflammation and protect primary neurons from Aβ oligomer-induced neurotoxicity in vitro •Reduce Aβ deposition, ameliorate neuron morphological changes, rescue memory deficits, and improve acetylcholine regulation ability in vivo |
Inhibit and eliminate Aβ aggregation | •Aβ targeting-GM 1 •Metal-ion chelation and inhibition of AChE activity |
2020 | [164] |
Figure 6. Modes of action for nanoparticle inhibitors against Aβ amyloidosis.
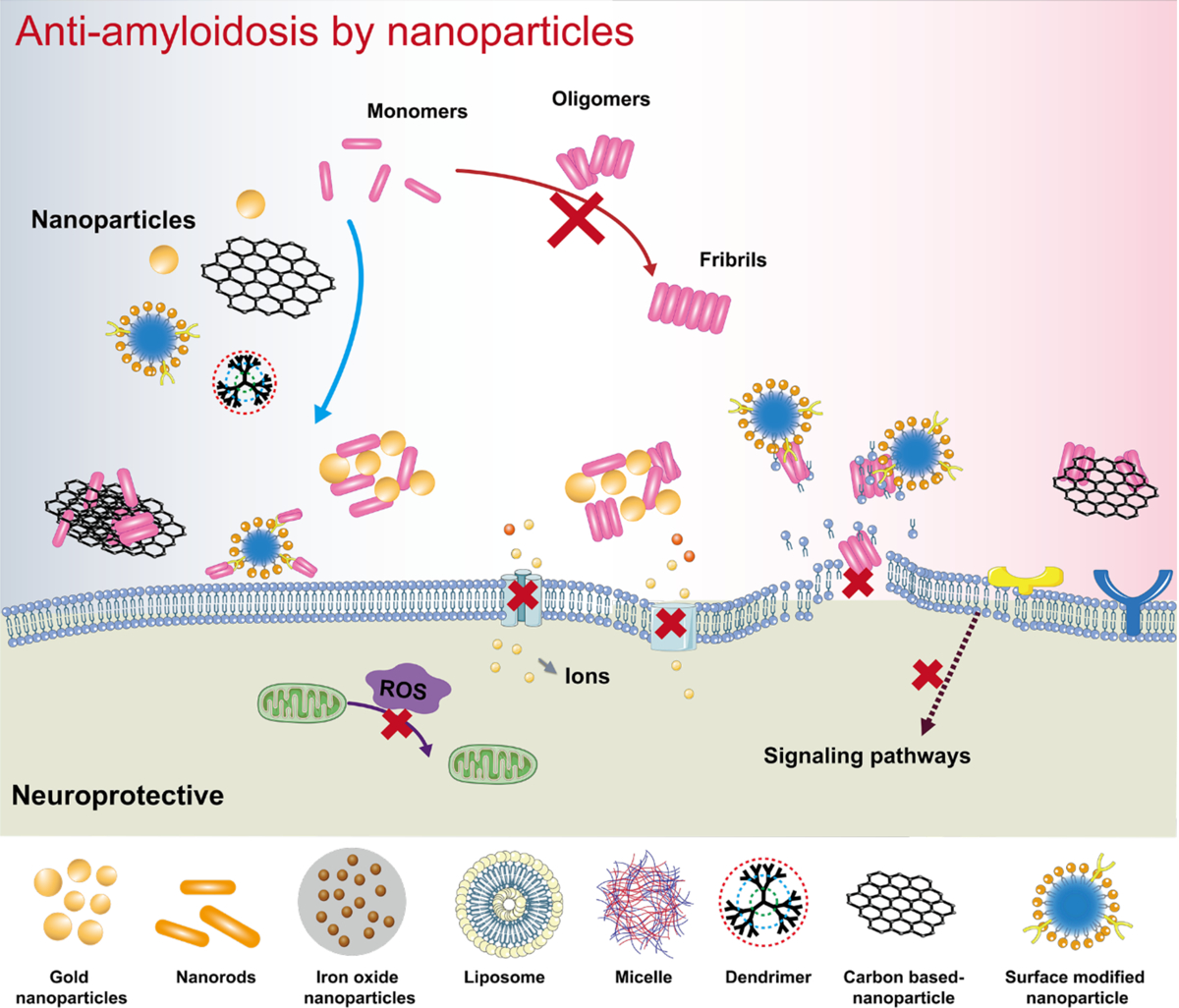
AuNPs have been recognized as an excellent anti-amyloid agent. A systematic study on the effects of AuNP size, shape and surface charge on Aβ aggregation was performed on a total brain lipid-based supported lipid bilayer.[149] Larger AuNPs induced large amorphous Aβ aggregates on the lipid bilayer with a shortened lag phase of aggregation, whereas smaller AuNPs rendered protofibrillar Aβ structures. Naturally, positively charged AuNPs were more strongly attracted to the anionic Aβ, giving rise to the formation of small amorphous aggregates of fewer β-sheets and more random coil structures, which also elicited significant toxicity to SH-SY5Y neuroblastoma cells. In contrast, negatively charged AuNPs acted as nucleation seeds to accelerate the formation of larger Aβ aggregates by gathering more peptides and AuNPs together. Hence, anionic or neutrally charged nanoparticles appeared to be more effective candidates against the amyloidosis of Aβ, whereas cationic nanoparticles were more prone to protein fouling and membrane damage. Furthermore, a comparison of different shaped AuNPs revealed that the longer gold nanorods elicited a greater affinity for Aβ to inhibit its fibrillization, whereas all the facets of gold nanocubes were accessible for interacting with Aβ to produce a fibrillar network.[149] The mitigation potential of βCas AuNPs against Aβ toxicity and Alzheimer’s-like symptoms was demonstrated in vitro and in a zebrafish model.[24] βCas, a milk protein adsorbed onto the surface of AuNPs, facilitated the binding of the nanoparticles with Aβ monomers/oligomers. The small βCas AuNPs (5–8 nm size) administrated to the bloodstream were able to cross the blood-brain barrier (BBB) and subsequently sequester toxic Aβ42 in the brain of larval and adult zebrafish through a nonspecific, chaperone-like mechanism (Fig. 7A&B). The mobility and cognitive function of adult zebrafish were recovered after the treatment of βCas AuNPs (Fig. 7C&D). In addition to AuNPs, nanoceria, iron oxide nanoparticles, silicon dioxide nanoparticles, silver nanoparticles as well as transition-metal nanomaterials such as molybdenum disulfide and black phosphorus have been shown as promising biocompatible nanomedicines for sequestering toxic amyloid species and alleviating their cell toxicity (Table 3).[150–157]
Figure 7. Mitigation of Aβ toxicity and Alzheimer’s-like symptoms in adult zebrafish with βCas AuNPs.[24].
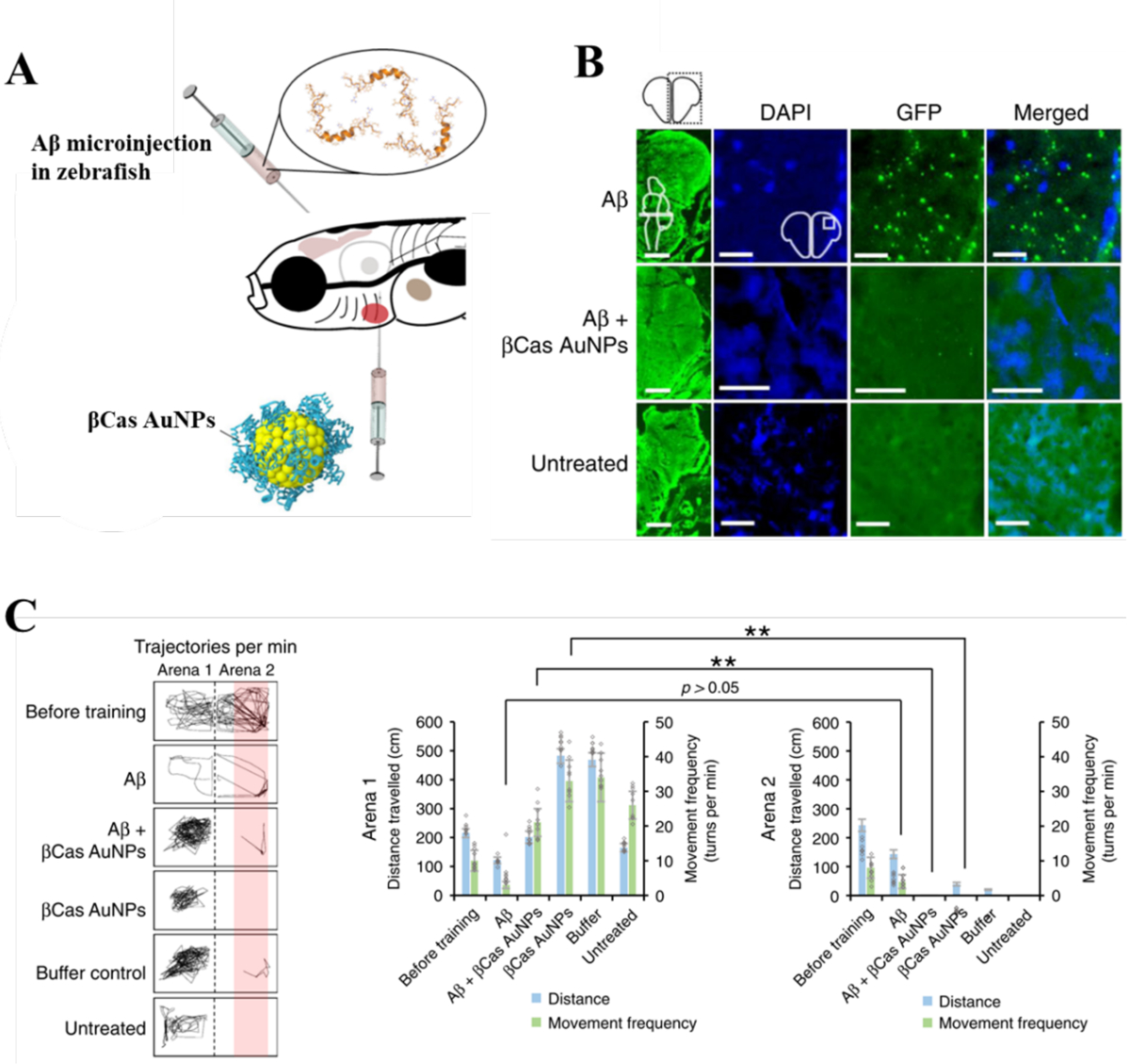
(A) Adult zebrafish (10 months old) were microinjected (cerebroventricular) with Aβ (1 μL, 50 μM). βCas AuNPs were microinjected (retro-orbital, 1 μL, 0.5 mM) 2 h prior to Aβ treatment. (B) Immunohistochemistry was performed on adult zebrafish brain sections to image the Aβ deposition. The first column represents the right cerebral brain of adult zebrafish in the GFP channel (Scale bars: 200 μM). DAPI, GFP and merged images at higher magnifications revealed Aβ plaque deposition in Aβ treated but not in Aβ + βCas AuNPs, or untreated control (Scale bars: 20 μM). (C) Cognitive behavior of adult zebrafish was analyzed. The movement trajectories of the fish in arena 1 vs. arena 2 are presented in the left panel. Comparative analysis of distance traveled and movement frequency of the fish in arena 1 vs. arena 2 revealed cognitive dysfunction of the Aβ-treated fish that were unable to avoid arena 2. Reproduced with permission from Ref. 24. Copyrights 2015 The Royal Society of Chemistry.
Polymeric nanocomposites possess versatile physicochemical properties, which can be utilized to render nanoparticle-biomolecular composites for biomedical applications.[158] By altering their lipid concentration and composition, polymethacrylate-copolymer (PMA) encased lipidnanodiscs (~10 nm) effectively regulated Aβ fibrillization to exhibit less neurotoxicity.[159] Wan et al. developed high generations of phenyl-modified carboxyl-terminated polyamidoamine (PAMAM) dendrimers, with the capability of inhibiting Aβ42 aggregation and altering the ultrastructure of Aβ42 aggregates via the hydrophobic binding-electrostatic repulsion (HyBER) theory.[160] Aβ-targeting ligand conjugation on nanoparticles is an efficient approach for disrupting the amyloidogenic process. Aβ-binding peptide KLVFF integrated with polymeric nanocomposites (NC-KLVFF) remarkably eliminated toxic Aβ aggregates, regained endocranial microglia’s capability to phagocytose the peptide, and protected hippocampal neurons against apoptosis in AD mice (Fig. 8).[22] In addition, the inhibitory potency of biomimetic nanomaterials against amyloidosis has been recently demonstrated, further displaying satisfactory biocompatibility and biodegradability (Table 3).[161–162] A nanostructure αNAP-GM1-rHDL, comprised of GM1-modified reconstituted high-density lipoprotein (GM1-rHDL) and a neuroprotective peptide NAPVSIPQ (NAP), was found effective in protecting neurons from Aβ42 oligomers/glutamic acid-induced cytotoxicity in vitro.[163] The biomimetic nanocomposite also showed the ablity to reduce Aβ deposition, ameliorate neurologic changes, and rescue memory loss efficiently in AD mice. Exploiting the membrane affinity of Aβ, GM1-rHDL displayed an antibody-like strong binding affinity for Aβ, thereby facilitating Aβ degradation and Aβ efflux across the BBB. The efficient loading of NAP further enabled the nanoparticulate drug delivery system for achieving AD therapy.[163] Smart nanoparticles, namely dcHGT NPs, were designed to not only inhibit and reduce Aβ aggregation but also simultaneously regulate acetylcholine imbalance.[164] Comprised of clioquinol (metal-ion chelating agent) and donepezil (acetylcholinesterase inhibitor), transcriptional activator protein, GM1 and human serum albumin (HSA), dcHGT NPs significantly inhibited Aβ aggregation while relieving acetylcholine-related inflammation in microglial cells. Furthermore, dcHGT NPs reduced Aβ deposition, ameliorated morphological neuron changes, rescued memory deficits, and significantly improved acetylcholine regulation in vivo.
Figure 8. Nanocomposite-mediated neurotoxicity mitigation and Aβ removal in an AD mouse model.
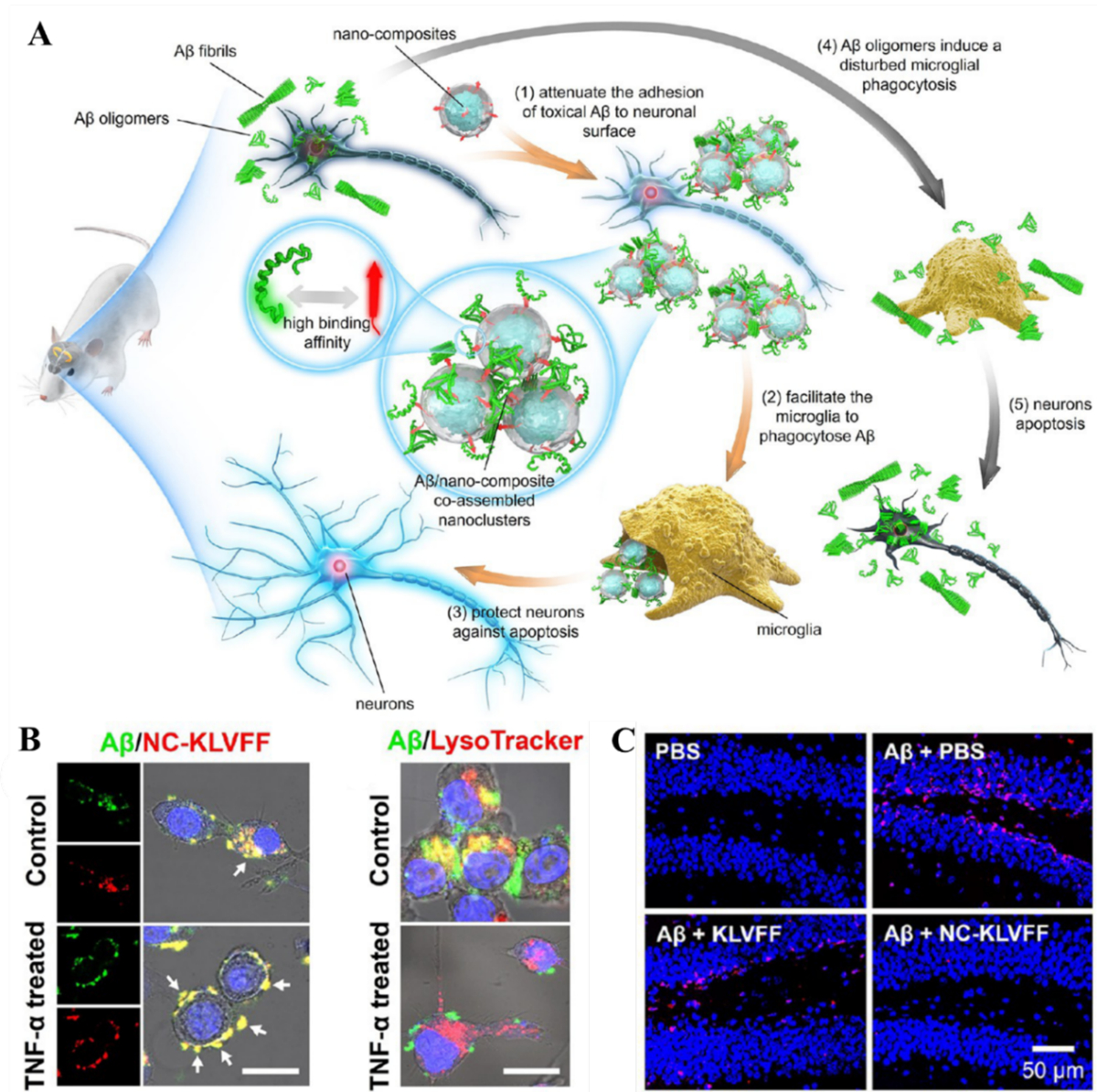
(A) The Aβ-nanocomposite interaction entailed mitigation of Aβ aggregation and associated neurotoxicity. (B) Fluorescence images showing the Aβ/NC-KLVFF clusters elimination by BV-2 cells pretreated with or without TNF-α. Aβ was labeled with FITC (green), while NC-KLVFF was labeled with rhodamine B (red). Scale bar: 25 μm. (C) The nanocomposite mitigated the neurotoxicity elicited by Aβ aggregation in an AD mouse model. In situ neuronal apoptosis in hippocampal dentate gyrus subregion was labeled red. Reproduced with permission from Ref. 22. Copyrights 2019 American Chemical Society.
5. AD nanomedicines and cell membranes
This section describes different routes for the transport of anti-AD nanomedicines across the BBB and how Aβ-membrane affinity may be exploited in the context of nanomedicine design. Nanomedicines that mimic the physicochemical properties of cell membranes may be crafted by either surface modification with lipid moieties or by cloaking them with cellular membranes. Such membrane-philic nanomedicines may hold the key for the effective BBB delivery of anti-AD therapies.
5.1. Nanoparticle-BBB interaction
The BBB is a highly selective semipermeable endothelial layer reinforced by tight junctions. The BBB separates the brain parenchyma from the lumens of blood vessels and protects the CNS from the entrance of xenobiotics from the periphery. The mechanisms a nanoparticle may employ to cross the BBB include passive diffusion of the nanoparticle through cellular membranes, hitchhiking the luminal surface transporters of the BBB for essential solutes, or via the endocytotic pathways.[169] In addition to the BBB being a major hurdle to AD nanomedicines, hematic proteins and immunity cells in the blood may further sequester nanomedicines from circulation. Among the myriad types of nanomaterials which have been tested over the years, inorganic nanoparticles, dendrimers, liposomes, polymeric nanoparticles and nanogels have been the most employed for the delivery of therapeutic agents and probes to the brain.[170–171] Recently, cell-mediated transport has been demonstrated as a promising strategy, where biomolecular-nanoparticle complexes were entrapped inside immune cells to reach the diseased tissues by chemotaxis.[169] Chemotactic drug delivery further enabled the formulation of a self-propelling system to shuttle across tissues driven by a glucose concentration gradient.[172] Alternatively, nanoparticles may be modified to mimic activated leukocytes, or functionalized with polymers or cellular membrane derivatives. These strategies disguise nanoparticles to freely circulate through the blood and effectively translocate across the BBB. Furthermore, the gaps between tight junctions may be transiently widened by ultrasound, or by downregulating the expression of occludin and claudin-5 that are components of the tight junctions,[173–174] thereby allowing the paracellular passage of nanoparticles.
5.2. AD nanomedicines exploiting cell membranes and the BBB
Although the pathophysiological mechanisms and pharmacological targets of AD remain under debate, there is a consensus that the primary cause of neuronal damage is induced by the membrane-association of small Aβ oligomers (n<10) that have a β-sheet rich structure with a tertiary hairpin.[175–177] The membrane affinity of the Aβ monomers is far less than the oligomers or a mixture of the monomers and oligomers.[178] The neuronal membranes also contribute to the structural transition of the Aβ monomers to oligomers. Indeed, the oligomers formed in buffer conditions and in the presence of cell membranes show different secondary structures. Kakio et al. used amide I and II band deconvolution of FTIR and demonstrated that the Aβ oligomers formed in buffer possessed in-register parallel β-sheets with strong interchain interactions. However, the Aβ oligomers formed in the presence of raft-like membranes presented anti-parallel β-sheets with weak interchain interactions.[179] One explanation for this difference is based on the fact that, in buffer conditions, the hydrophobic amyloidogenic region of Aβ is consumed in self-assembly, while in close proximity to cell membranes the peptide hydrophobic region is engaged in interactions with lipid rafts or gangliosides.[180] Exploiting this affinity-based interactions, AD nanomedicines have been devised by ascribing them with membrane-like surfaces, which can sequester the Aβ oligomers and prevent their interactions with cell membranes. In this context, coating of lipid molecules on the surface of metal nanoparticles provided a promising strategy to shut-down Aβ fibrillization. Suga et al. synthesized AuNPs of 100 nm in diameter that were coated with 1,2-dimylistoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dimyristoyl-sn-glycero-3-phospho-L-serine (DMPS).[181] The AuNPs were kept in their monodisperse form by the addition of NaCl at physiological concentrations. Exposure of the AuNPs to Aβ resulted in the displacement of NaCl and deposition of an Aβ corona on the nanoparticle surface. The Aβ species further triggered agglomeration of the AuNPs via their concurrent interactions with multiple AuNPs. Similarly, Bhowmik et al. coated silver nanoparticles (AgNPs) of 60 nm in size with a lipid bilayer of POPC, POPG (1-palmitoyl-2-oleoyl-sn-glycero-3 phosphoglycerol) and cholesterol.[182] These lipid-coated AgNPs sequestered Aβ oligomers onto their surfaces to reveal conformational changes of the membrane-bound Aβ oligomers. Raman spectroscopy indicated that the β-turns in the membrane-bound Aβ oligomers were flanked by anti-parallel β-sheets. Exploiting the membrane affinity of Aβ, Song et al. designed apolipoprotein E (ApoE3) containing high-density lipoproteins (HDL) nanodiscs that captured both the Aβ40 and Aβ42 monomers as well as oligomers.[162] Based on TEM, the Aβ40 oligomers were found to bind with the lateral side of the nanodiscs via ApoE3. The Aβ42 oligomers, on the other hand, presented diffuse binding with nanodiscs to render larger nanostructures. These ApoE3-HDL nanodiscs crossed the BBB at 0.4% ID/g (injected dose per gram of tissue), suppressed the microgliosis and supported the immunological clearance of the Aβ monomers and oligomers from the brain of an AD mouse model. This ApoE3-HDL nanosystem was further utilized as a drug carrier against Aβ fibrillization. α-Mangostin, a polyphenolic compound, was loaded inside the ApoE3-HDL nanocarriers to translocate across the BBB (Fig. 9).[161] These nanocarriers, once loaded with α-Mangostin (ANC-α-M), targeted Aβ with an even greater affinity and accumulated specifically around the Aβ plaques in the AD mouse model. In addition to the membrane-mimicking nanoparticles, another delivery strategy which can benefit from the Aβ-membrane affinity is cell membrane-based delivery systems,[183] which use the whole cell or shredded cell exosomes. Another alternative is to coat polylactic-glycolic acid (PLGA) nanoparticles and AuNPs with cell membranes.[184–185] Specifically, purified red blood cells (RBCs) were extruded together with the nanoparticles multiple times through a polycarbonate membrane, rendering a 7–8 nm coating layer of RBC membranes onto the NPs. The RBC membranes were in the right-side-out orientation that provided a negative zeta potential (−15 mV) and immune compatibility to the nanoparticles.
Figure 9. ApoE3-HDL nanomedicine for the mitigation of Aβ pathology.
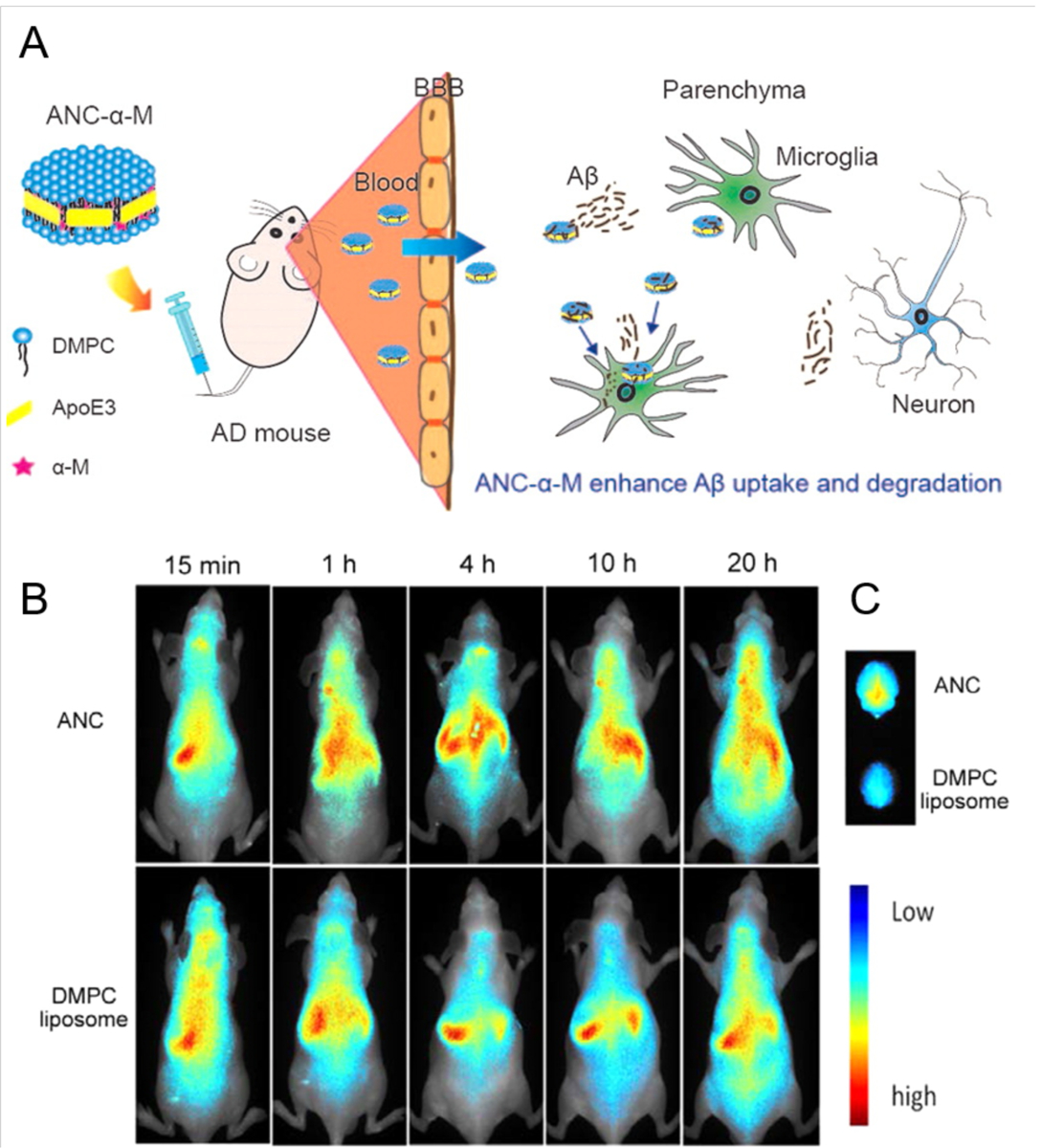
(A) α-Mangostin loaded ApoE3-HDL nanocarriers (ANC-α-M) were able to translocate across the BBB of SAMP8 mice to promote Aβ clearance and rescue memory loss. (B) In vivo distribution in nude mice was studied after conjugating the ANC and DMPC liposomes, as control, with DIR fluorescence dye and administering to the mice via intravenous tail injection. (C) The ANCs were distributed to the brain with higher affinity, due to the presence of ApoE3. The figure is reproduced with permission from Ref. 161. Copyrights 2016 American Chemical Society.
A general criterion for nanoparticles to permeate across the BBB is that the nanoparticles should be in the size range of 5–200 nm and possess neutral to negative charge.[186] In the context of AD nanomedicine, three types of organic nanoparticles, i.e., dendrimers, polymeric nanoparticles and liposomes, have demonstrated their BBB translocation capacity. Lightweight, hyperbranched dendrimers displayed a native inhibitory activity against Aβ fibrillization.[187] They also exhibited a neuronal anti-inflammatory potential that can be utilized to develop a suitable drug delivery system against AD.[188] Moscariello et al. used PAMAM dendrimer conjugated with streptavidin to study their mode of BBB translocation in a porcine-based in vitro model.[189] The dendrimers navigated from the luminal to abluminal side of blood vessels via endocytosis (Fig. 10). The intercellular tight junctions remained intact indicating no paracellular transport.
Figure 10. Permeation of dendritic polymers across the BBB.
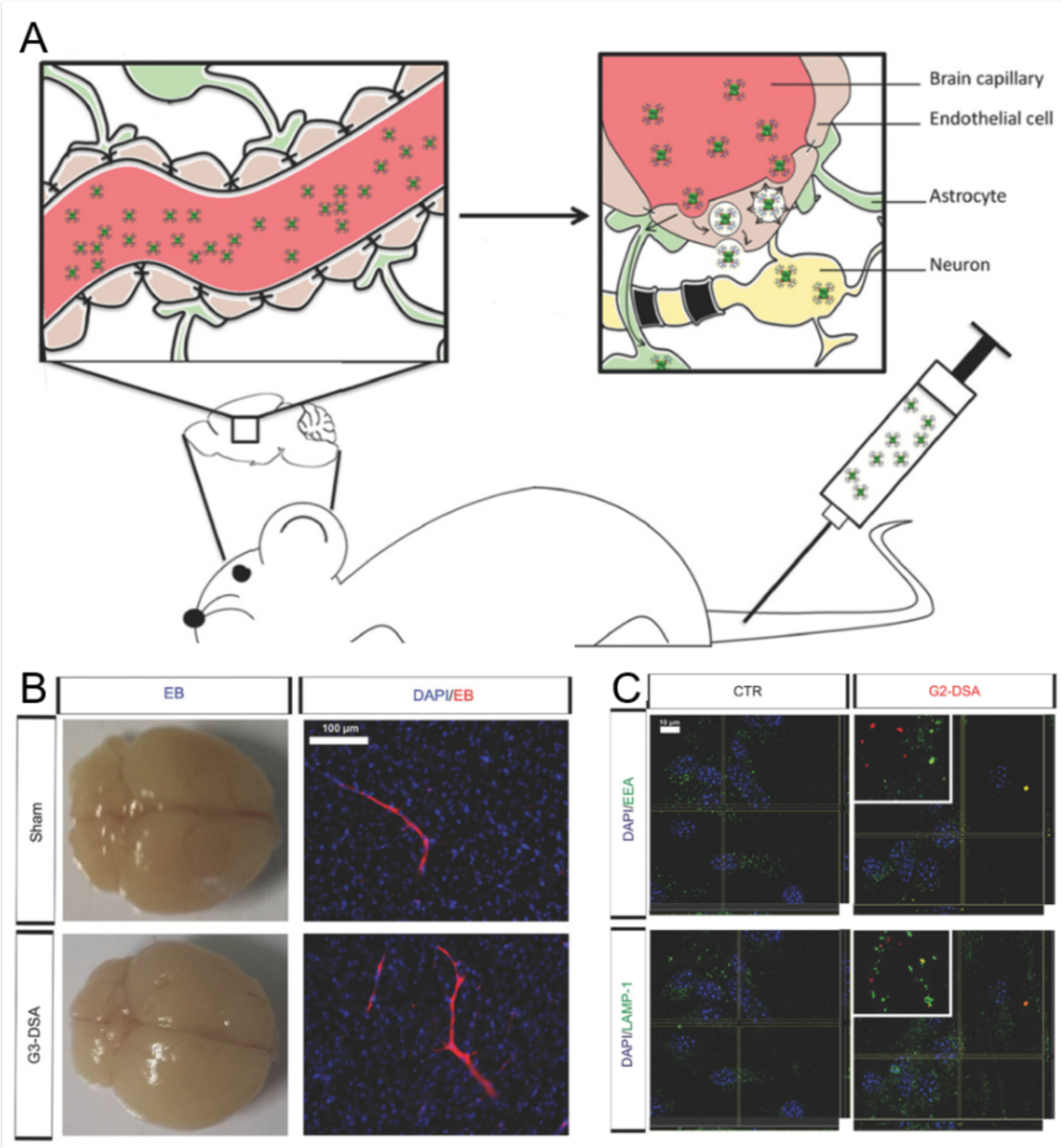
(A) Streptavidin-conjugated PAMAM dendrimers were able to permeate through the BBB via transcytosis in P21 mice. (B) Generation-3 DSA (succinamic acid dendrimer with curcumin) did not disrupt the BBB integrity. No extravasation of Evans blue (EB) dye was observed in whole brain epifluorescence, after administration of G3-DSA and Sham (positive control). (C) Endosomal trafficking of G2-DSA (generation-2 dendrimers with different length and charge of the dendron molecule) through bEnd.3 mouse neuronal cells. Colocalization of DSA positive vesicles (red) and early endosome antigen (EEA; green) for early endosomes and lysosomal-associated membrane protein (LAMP1; green) for late endosomes was observed. Figure is reproduced from Ref. 189, an open access publication from Advanced Science. Copyrights 2018 John Wiley & Sons.
In addition to dendritic nanoparticles, tween 80 coated poly(n-butylcyanoacrylate) nanoparticles were able to deliver an anti-AD drug.[186, 190] Specifically, the coating of tween 80 facilitated anchorage of plasma ApoE3 on the surface of the nanoparticles to enable their transcytosis across the BBB. Liposomes, after conjugation with cell-penetrating peptides or ApoE3, exhibited significant BBB permeation largely based on transcytosis.[191–192] In the case of inorganic nanoparticles, small (5–20 nm) sized AuNPs capped with casein, HDL or transferrin antibodies have demonstrated BBB translocation in vitro and in vivo.[24, 193–194] Overall, the use of cellular membrane mimicking or specific ligands can aid in the BBB permeation of anti-AD nanomedicines and their subsequent affinity-based interactions with toxic Aβ species.
6. Conclusion and future prospect
AD is a primary cause of death for the global ageing population. While the pathobiological origin of the disease remains debatable and AD treatment remains a formidable challenge in modern medicine, it has been generally accepted that neurodegeneration in AD involves the central process of Aβ amyloidosis, exacerbated by its downstream events in tau accumulation and neuroinflammation (e.g., dysregulation of immune activation and ApoE expression in the brain, etc.).[195] As the scope of AD physiopathology is vast, while the field of AD nanomedicine is nascent, in this review we have focused our attention on the structural origin and conformational susceptibility of the amyloidogenic peptide Aβ, and summarized the interplay between the peptide, its membrane environments (i.e., membrane mimetics, cell membranes, the BBB, and more), as well as its nanoparticulate inhibitors in silico, in vitro and in vivo. We wish to demonstrate through this review that our knowledge accumulated over the past two decades concerning Aβ-membrane, nanoparticle-membrane, and Aβ-nanoparticle interactions is highly relevant and beneficial to our understanding and manipulation of the membrane axis of Aβ amyloidogenesis. Briefly, the association of Aβ with cell membranes is understood to trigger conformational changes of the peptide to give rise to the toxic oligomers, which may subsequently induce membrane poration, lipid extraction, ROS production and cell death. In comparison, the adsorption of nanoparticles onto cell membranes may enable the translocation or permeation of the nanoparticles via endocytosis or NanoEL, which may elicit toxicity in the host depending on the size, shape, surface charge and functionalization of the nanoparticles as well as their protein corona. The interaction between Aβ and nanoparticles, on the other hand, may inhibit or accelerate the amyloid aggregation of the peptide and ascribe an amyloid protein corona on the nanoparticle surface.[147] In vivo, the peptide aggregates themselves may also acquire a dynamic protein corona depending on their changing hydrophobicity, mediated by their nonspecific interactions with the myriad biomolecular species in the host media.[196–198] While much remains to be understood concerning the interplay between cell membrane, Aβ, and nanoparticle inhibitors, an effective strategy is to elevate Aβ-nanoparticle interaction over Aβ/nanoparticle-membrane interaction by tailoring the nanoparticle surface chemistry to trap the peptide in its monomeric state, or sequester the toxic peptide aggregates via the assembly of functional-pathogenic protein coronae.[199] It is our hope that advanced nano-biomedical research may address many unanswered questions in this interdisciplinary area to facilitate the development of future AD nanomedicines.
Acknowledgements
This work was supported by the Australian Research Council Project No. CE140100036 (Davis), NSF CAREER CBET-1553945 (Ding) and NIH MIRA R35GM119691 (Ding).
List of Abbreviations
- AD
Alzheimer’s disease
- AC
Alternating current
- AFM
Atomic force microscopy
- AgNPs
Silver nanoparticles
- AgTNPs
Silver triangular nanoplates
- ANC
ApoE-reconstituted high-density lipoprotein nanocarrier
- ANC-α-M
α-M loaded ANC
- Apo-A1
Apolipoprotein A1
- ApoE
Apolipoprotein E
- ApoE3-rHDL
Apolipoprotein E3-reconstituted high-density lipoprotein
- APP
Amyloid precursor protein
- AuNCs
Gold nanocubes
- AuNPs
Gold nanoparticles
- AuNRs
Gold nanorods
- Aβ
Amyloid beta
- Aβm
Monomeric Aβ
- Aβo
Aβ oligomers
- BBB
Blood-brain barrier
- BP
Black phosphorus
- BP@BTA
4-(6-methyl-1,3-benzothiazol-2-yl) cphenylamine-modified black phosphorus
- BSA
Bovine serum albumin
- BTA
4-(6-methyl-1,3-benzothiazol-2-yl) phenylamine
- BTLE
Brain total lipid extract
- CeONP
Cerium oxide nanoparticles
- CeONP@POMs
POMs coated CeONP
- CG
Coarse-grained
- CGA
Chlorogenic acid
- CLIC
Clathrin-independent carriers
- CNpNC
N-terminal β-strand faced the water-filled pore and C-terminal β-strand faces the bilayer
- CNS
Central nervous syste
- CNTs
Carbon nanotubes
- CPNPs
Fluorescent conjugated polymer nanoparticles
- CQDs
Carbon quantum dots
- CTAB
Cetyltrimethylammonium bromide
- DC
Direct current
- dcHGT NPs
Donepezil and clioquinol co-encapsulated human serum albumin nanoparticles
- DFT
Density functional theory
- DHAEE
Docosahexaenoic acid etyl-ester
- DHLA
Dihydrolipoic acid
- DHPC
1,2-dihexanoyl-snglycero-3-phosphocholine
- DLPC
Dilauroyl phosphatidylcholin
- DM
Dodecylmaltoside, decylmaltoside
- DMPC
1,2-dimylistoyl-sn-glycero-3-phosphocholine
- DMPG
1,2-Dimyristoyl-sn-glycero-3-phosphoglycerol
- DMPS
1,2-dimyristoyl-sn-glycero-3-phospho-L-serine
- DOEPC
1,2-Dioleoyl-sn-glycero-3-ethylphosphocholine
- DOPC
1, 2-dioleoylsn-glycero-3-phosphocholine
- DOPG
1,2-dioleoyl-snglycero-3-phospho-(10-rac-glycerol)
- DOPS
Dioleylphosphatidylserine
- DPC
Dodecylphosphocholine
- DPD
Dissipative particle dynamics
- PDMP
D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol
- DPPC
Dipalmitoylphosphatidylcholine
- DSA
Succinamic acid dendrimer
- EB
Evans blue
- E. coli
Escherichia coli
- EL
Endothelial leakiness
- FA
Folic acid
- fBLM
Floating bilayer lipid membrane
- FO
Fibrillar oligomers
- FPS-ZM1
4-Chloro-N-cyclohexyl-N-(phenylmethyl)benzamide
- FTIR
Fourier-transform infrared spectroscopy
- GEEC
Glycosylphosphotidylinositol- anchored protein enriched compartment
- GM1
Monosialotetrahexosylganglioside
- GO
Graphene oxide
- GQDs
Graphene quantum dots
- HDL
High-density lipoproteins
- HSA
Human serum albumin
- HyBER
Hydrophobic binding-electrostatic repulsion
- IAPP
human islet amyloid polypeptide
- IL-β
Interleukin-1β
- KLVFF
Aβ binding peptides KLVFF (Aβ16–20, Lys-Leu-Val-Phe-Phe)
- LDAO
Lauryldimethylamine-N-oxide
- L-GO
Long-graphene oxide
- L-MWNTs
Long-multi walled nano tubes
- L-SWNTs
Long-single walled nano tubes
- LTP
Long term potentiation
- LUVs
Large unilamellar vesicles
- MCM-41
Mesoporous silica material name
- MD
Molecular dynamics
- MDCK
Madin-Darby Canine Kidney
- MoS2
Molybdenum disulfide
- MSNs
Mesoporous silica nanoparticles
- MW
Molecular weight
- nAChRs
Nicotinic acetylcholine receptors
- NanoEL
Nanoparticles caused endothelial cells leakiness
- NAP
NAPVSIP, a femtomolar-acting neuroprotective peptide
- NC-KLVFF
Nanocomposite with KLVFF
- NCpCN
C-terminal β-strand faced the solvated pore
- NET
Neutrophil extracellular traps
- NFO
Globular nonfibrillar oligomers
- NF-κB
Nuclear factor kappa B
- NIR
Near-infrared
- NMDAR
N-methyl-D-aspartate receptor
- NOM
Natural organic matter
- NPPs
Nanoporous polymer particles
- NPs
Nanoparticles
- NR2B
N-methyl D-aspartate receptor subtype 2B
- NRs
Nanorods
- OAEE
Oleic acid ethyl-ester
- OG
Octyl glucoside
- PAMAM
Carboxyl-terminated polyamidoamine dendrimer
- PC
Phosphatidylcholine
- PD
Parkinson’s disease
- PDC
Diallyldimethylammonium chloride
- PDMP
D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol
- PE
Phosphatidylethanolamine
- PEG
Poly(ethylene oxide)
- PLGA
Polylactic-glycolic acid
- PMA
Polymethacrylate-copolymer
- PMASH
poly-(methacrylic acid)
- PMF
Potential of mean force
- POEPC
1-palmitoyl-2-oleoyl-3-ethylphosphocholine
- POMs
Metalsubstituted polyoxometalates
- POPC
1- palmitoyl-2-oleoyl phosphatidylcholine
- POPE
Palmitoyloleoylphosphatidylethanolamine
- POPG
Palmitoyloleoylphosphatidylglycerol
- POPS
1-palmitoyl-2-oleoyl-3-phosphoserine
- PrPC
Cellular Prion Protein
- PS
Phosphatidylserine
- PS
Poly-styrene
- PVA
Poly(vinyl alcohol-co-N-vinylamine)
- PVP
Polyvinylpyrrolidone
- QCM-D
Quartz crystal microbalance-dissipation monitorin
- QDs
Quantum dots
- RAGE
Receptor for advanced glycation end products
- RBC
Red blood cell membrane
- rHDL
ApoE3-reconstituted high-density lipoprotein
- ROS
Reactive oxygen species
- SBA-15
Santa Barbara Amorphous-15 (MSNs)
- SCA-7
Stably transfected CV-1 cells expressing PrPC
- SeNPs
Selenium nanoparticles
- S-GO
Short-graphene oxide
- SM
Sphingomyelin
- S-MWNTs
Short-multi walled nano tubes
- SOPC
1-stearoyl,2-oleoylphosphatidylcholine
- SPIONs
Superparamagnetic iron oxide nanoparticles
- SPR
Surface plasmon resonance
- S-SWNTs
Short-single walled nano tubes
- SV2
Synaptic vesicle protein 2
- SWNT-OH
Hydroxylated single-walled carbon nanotubes
- T2D
Type 2 diabetes
- TEM
Transmission electron microscopy
- TLR4
Toll-like receptor 4
- TNF-α
Tumor necrosis factor α
- VE-cadherin
Vascular endothelial cadherin
- α7nAChR
Neuronal α7 nicotinic acetylcholine receptor
- α-M
α-Mangostin
- αNAP-GM1-rHDL
NAP-loaded GM1-rHDL
- αS
Alpha synuclein
- βCas AuNPs
β-casein coated-gold nanoparticles
- p-τ
Phosphorylated τ-protein
Footnotes
Declaration of Competing Interest
There are no conflicts to declare.
References
- [1].Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, Patton RL, Luehrs DC, Daugs ID, Kuo Y-M, Emmerling MR, Soares H, Quinn JF, Kaye J, Connor DJ, Silverberg NB, Adler CH, Seward JD, Beach TG, Sabbagh MN, Alzheimer’s & Dementia 2009, 5, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hardy J, Selkoe DJ, Science 2002, 297, 353. [DOI] [PubMed] [Google Scholar]
- [3].Österlund N, Luo J, Wärmländer SKTS, Gräslund A, Biochim. Biophys. Acta Proteins Proteom 2019, 1867, 492. [DOI] [PubMed] [Google Scholar]
- [4].Fabiani C, Antollini SS, Front. Cell. Neurosci 2019, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Laurén J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM, Nature 2009, 457, 1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Arispe N, Rojas E, Pollard HB, Proc. Natl. Acad. Sci. U. S. A 1993, 90, 567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serra-Batiste M, Ninot-Pedrosa M, Bayoumi M, Gairí M, Maglia G, Carulla N, Proc. Natl. Acad. Sci. U. S. A 2016, 113, 10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cabaleiro-Lago C, Quinlan-Pluck F, Lynch I, Lindman S, Minogue AM, Thulin E, Walsh DM, Dawson KA, Linse S, J. Am. Chem. Soc 2008, 130, 15437. [DOI] [PubMed] [Google Scholar]
- [9].Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, Wanker EE, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bieschke J, Herbst M, Wiglenda T, Friedrich RP, Boeddrich A, Schiele F, Kleckers D, Lopez del Amo JM, Grüning BA, Wang Q, Schmidt MR, Lurz R, Anwyl R, Schnoegl S, Fändrich M, Frank RF, Reif B, Günther S, Walsh DM, Wanker EE, Nat. Chem. Biol 2012, 8, 93. [DOI] [PubMed] [Google Scholar]
- [11].Mahmoudi M, Akhavan O, Ghavami M, Rezaee F, Ghiasi SM, Nanoscale 2012, 4, 7322. [DOI] [PubMed] [Google Scholar]
- [12].Palhano FL, Lee J, Grimster NP, Kelly JW, J. Am. Chem. Soc 2013, 135, 7503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gao N, Sun H, Dong K, Ren J, Duan T, Xu C, Qu X, Nat. Commun 2014, 5, 3422. [DOI] [PubMed] [Google Scholar]
- [14].Li M, Zhao A, Dong K, Li W, Ren J, Qu X, Nano Res. 2015, 8, 3216. [Google Scholar]
- [15].Gurzov EN, Wang B, Pilkington EH, Chen P, Kakinen A, Stanley WJ, Litwak SA, Hanssen EG, Davis TP, Ding F, Ke PC, Small 2016, 12, 1615. [DOI] [PubMed] [Google Scholar]
- [16].Chen Q, Du Y, Zhang K, Liang Z, Li J, Yu H, Ren R, Feng J, Jin Z, Li F, Sun J, Zhou M, He Q, Sun X, Zhang H, Tian M, Ling D, ACS Nano 2018, 12, 1321. [DOI] [PubMed] [Google Scholar]
- [17].Kim D, Yoo JM, Hwang H, Lee J, Lee SH, Yun SP, Park MJ, Lee M, Choi S, Kwon SH, Lee S, Kwon SH, Kim S, Park YJ, Kinoshita M, Lee YH, Shin S, Paik SR, Lee SJ, Lee S, Hong BH, Ko HS, Nat. Nanotechnol 2018, 13, 812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Luo Q, Lin YX, Yang PP, Wang Y, Qi GB, Qiao ZY, Li BN, Zhang K, Zhang JP, Wang L, Wang H, Nat. Commun 2018, 9, 1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wang J, Liu L, Ge D, Zhang H, Feng Y, Zhang Y, Chen M, Dong M, Chem. Eur. J 2018, 24, 3397. [DOI] [PubMed] [Google Scholar]
- [20].Wang M, Sun Y, Cao X, Peng G, Javed I, Kakinen A, Davis TP, Lin S, Liu J, Ding F, Ke PC, Nanoscale 2018, 10, 19995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sahoo BR, Genjo T, Cox SJ, Stoddard AK, Anantharamaiah GM, Fierke C, Ramamoorthy A, J. Mol. Biol 2018, 430, 4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhao Y, Cai J, Liu Z, Li Y, Zheng C, Zheng Y, Chen Q, Chen H, Ma F, An Y, Xiao L, Jiang C, Shi L, Kang C, Liu Y, Nano Lett. 2019, 19, 674. [DOI] [PubMed] [Google Scholar]
- [23].Kakinen A, Xing Y, Hegoda Arachchi N, Javed I, Feng L, Faridi A, Douek AM, Sun Y, Kaslin J, Davis TP, Higgins MJ, Ding F, Ke PC, Nano Lett. 2019, 19, 6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Javed I, Peng G, Xing Y, Yu T, Zhao M, Kakinen A, Faridi A, Parish CL, Ding F, Davis TP, Ke PC, Lin S, Nat. Commun 2019, 10, 3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Sahoo BR, Genjo T, Nakayama TW, Stoddard AK, Ando T, Yasuhara K, Fierke CA, Ramamoorthy A, Chem. Sci 2019, 10, 3976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ke PC, Pilkington EH, Sun Y, Javed I, Kakinen A, Peng G, Ding F, Davis TP, Adv. Mater 2020, 32, 1901690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lemkul JA, Bevan DR, FEBS J 2009, 276, 3060. [DOI] [PubMed] [Google Scholar]
- [28].Lemkul JA, Bevan DR, Biochemistry 2013, 52, 4971. [DOI] [PubMed] [Google Scholar]
- [29].Tofoleanu F, Brooks BR, Buchete N-V, ACS Chem. Neurosci 2015, 6, 446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Tofoleanu F, Buchete N-V, J. Mol. Biol 2012, 421, 572. [DOI] [PubMed] [Google Scholar]
- [31].Miyashita N, Straub JE, Thirumalai D, J. Am. Chem. Soc 2009, 131, 17843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Poojari C, Kukol A, Strodel B, Biochim. Biophys. Acta. Biomembr 2013, 1828, 327. [DOI] [PubMed] [Google Scholar]
- [33].Best RB, Curr. Opin. Struct. Biol 2017, 42, 147. [DOI] [PubMed] [Google Scholar]
- [34].Hall BA, Chetwynd AP, Sansom MSP, Biophys. J 2011, 100, 1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liu Y, Ren B, Zhang Y, Sun Y, Chang Y, Liang G, Xu L, Zheng J, Biochim. Biophys. Acta. Biomembr 2018, 1860, 1906. [DOI] [PubMed] [Google Scholar]
- [36].Nasica-Labouze J, Nguyen PH, Sterpone F, Berthoumieu O, Buchete N-V, Coté S, De Simone A, Doig AJ, Faller P, Garcia A, Laio A, Li MS, Melchionna S, Mousseau N, Mu Y, Paravastu A, Pasquali S, Rosenman DJ, Strodel B, Tarus B, Viles JH, Zhang T, Wang C, Derreumaux P, Chem. Rev 2015, 115, 3518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tofoleanu F, Buchete N-V, Prion 2012, 6, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Press-Sandler O, Miller Y, Biochim. Biophys. Acta. Biomembr 2018, 1860, 1889. [DOI] [PubMed] [Google Scholar]
- [39].Sciacca MFM, Tempra C, Scollo F, Milardi D, La Rosa C, Biochim. Biophys. Acta. Biomembr 2018, 1860, 1625. [DOI] [PubMed] [Google Scholar]
- [40].Davis CH, Berkowitz ML, Biophys. J 2009, 96, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Meng F, Bellaiche MMJ, Kim J-Y, Zerze GH, Best RB, Chung HS, Biophys. J 2018, 114, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Xu Y, Shen J, Luo X, Zhu W, Chen K, Ma J, Jiang H, Proc. Natl. Acad. Sci. U. S. A 2005, 102, 5403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Davis CH, Berkowitz ML, J. Phys. Chem. B 2009, 113, 14480. [DOI] [PubMed] [Google Scholar]
- [44].Davis CH, Berkowitz ML, Proteins 2010, 78, 2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Brown A, Bevan D, Biophys. J 2016, 111, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Dies H, Toppozini L, Rheinstädter MC, PLOS One 2014, 9, e99124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Owen MC, Kulig W, Poojari C, Rog T, Strodel B, Biochim. Biophys. Acta. Biomembr 2018, 1860, 1709. [DOI] [PubMed] [Google Scholar]
- [48].Yu X, Zheng J, J. Mol. Biol 2012, 421, 561. [DOI] [PubMed] [Google Scholar]
- [49].Lemkul JA, Bevan DR, Protein Sci. 2011, 20, 1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ciudad S, Puig E, Botzanowski T, Meigooni M, Arango AS, Do J, Mayzel M, Bayoumi M, Chaignepain S, Maglia G, Cianferani S, Orekhov V, Tajkhorshid E, Bardiaux B, Carulla N, Nat. Commun 2020, 11, 3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Strodel B, Lee JWL, Whittleston CS, Wales DJ, J. Am. Chem. Soc 2010, 132, 13300. [DOI] [PubMed] [Google Scholar]
- [52].Jang H, Teran Arce F, Ramachandran S, Kagan BL, Lal R, Nussinov R, Chem. Soc. Rev 2014, 43, 6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nguyen PH, Campanera JM, Ngo ST, Loquet A, Derreumaux P, J. Phys. Chem. B 2019, 123, 3643. [DOI] [PubMed] [Google Scholar]
- [54].Jang H, Zheng J, Nussinov R, Biophys. J 2007, 93, 1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Jang H, Teran Arce F, Ramachandran S, Capone R, Lal R, Nussinov R, J. Phys. Chem. B 2010, 114, 9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Jang H, Arce FT, Capone R, Ramachandran S, Lal R, Nussinov R, Biophys. J 2009, 97, 3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jang H, Arce FT, Ramachandran S, Capone R, Azimova R, Kagan BL, Nussinov R, Lal R, Proc. Natl. Acad. Sci. U. S. A 2010, 107, 6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R, J. Mol. Biol 2010, 404, 917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Connelly L, Jang H, Teran Arce F, Capone R, Kotler SA, Ramachandran S, Kagan BL, Nussinov R, Lal R, J. Phys. Chem. B 2012, 116, 1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Connelly L, Jang H, Teran Arce F, Ramachandran S, Kagan BL, Nussinov R, Lal R, Biochemistry 2012, 51, 3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cheignon C, Tomas M, Bonnefont-Rousselot D, Faller P, Hureau C, Collin F, Redox Biol. 2018, 14, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ke PC, Zhou R, Serpell LC, Riek R, Knowles TPJ, Lashuel HA, Gazit E, Hamley IW, Davis TP, Fändrich M, Otzen DE, Chapman MR, Dobson CM, Eisenberg DS, Mezzenga R, Chem. Soc. Rev 2020, 49, 5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Vander Zanden CM, Wampler L, Bowers I, Watkins EB, Majewski J, Chi EY, Langmuir 2019, 35, 16024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Hong S, Ostaszewski BL, Yang T, O’Malley TT, Jin M, Yanagisawa K, Li S, Bartels T, Selkoe DJ, Neuron 2014, 82, 308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG, J. Biol. Chem 2004, 279, 46363. [DOI] [PubMed] [Google Scholar]
- [66].Bode DC, Freeley M, Nield J, Palma M, Viles JH, J. Biol. Chem 2019, 294, 7566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Matsuzaki K, Int. J. Alzheimers Dis 2011, 2011, 956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Williams TL, Serpell LC, FEBS J 2011, 278, 3905. [DOI] [PubMed] [Google Scholar]
- [69].Korshavn KJ, Satriano C, Lin Y, Zhang R, Dulchavsky M, Bhunia A, Ivanova MI, Lee YH, La Rosa C, Lim MH, Ramamoorthy A, J. Biol. Chem 2017, 292, 4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Meker S, Chin H, Sut TN, Cho N-J, Langmuir 2018, 34, 9548. [DOI] [PubMed] [Google Scholar]
- [71].Shabestari MH, Meeuwenoord NJ, Filippov DV, Huber M, J. Biol. Phys 2016, 42, 299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Mrdenovic D, Majewska M, Pieta IS, Bernatowicz P, Nowakowski R, Kutner W, Lipkowski J, Pieta P, Langmuir 2019, 35, 11940. [DOI] [PubMed] [Google Scholar]
- [73].Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG, Science 2003, 300, 486. [DOI] [PubMed] [Google Scholar]
- [74].Sepulveda FJ, Parodi J, Peoples RW, Opazo C, Aguayo LG, PLoS One 2010, 5, e11820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Bode DC, Baker MD, Viles JH, J. Biol. Chem 2017, 292, 1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sciacca Michele F. M., Kotler Samuel A., Brender Jeffrey R., Chen J, Lee D.-k., Ramamoorthy A, Biophys. J 2012, 103, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Ko M, Hattori T, Abdullah M, Gong J-S, Yamane T, Michikawa M, Brain Res. 2016, 1642, 376. [DOI] [PubMed] [Google Scholar]
- [78].Habchi J, Chia S, Galvagnion C, Michaels TCT, Bellaiche MMJ, Ruggeri FS, Sanguanini M, Idini I, Kumita JR, Sparr E, Linse S, Dobson CM, Knowles TPJ, Vendruscolo M, Nat. Chem 2018, 10, 673. [DOI] [PubMed] [Google Scholar]
- [79].Yanagisawa K, Odaka A, Suzuki N, Ihara Y, Nat. Med 1995, 1, 1062. [DOI] [PubMed] [Google Scholar]
- [80].Yanagisawa K, Ihara Y, Neurobiol. Aging 1998, 19, S65. [DOI] [PubMed] [Google Scholar]
- [81].Yamamoto N, Matsubara T, Sato T, Yanagisawa K, Biochim. Biophys. Acta. Biomembr 2008, 1778, 2717. [DOI] [PubMed] [Google Scholar]
- [82].Evangelisti E, Cascella R, Becatti M, Marrazza G, Dobson CM, Chiti F, Stefani M, Cecchi C, Sci. Rep 2016, 6, 32721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Fernandez-Perez EJ, Sepulveda FJ, Peoples R, Aguayo LG, Biochim. Biophys. Acta Mol. Basis Dis 2017, 1863, 3105. [DOI] [PubMed] [Google Scholar]
- [84].Re F, Airoldi C, Zona C, Masserini M, Ferla BL, Quattrocchi N, Nicotra F, Curr. Med. Chem 2010, 17, 2990. [DOI] [PubMed] [Google Scholar]
- [85].Di Scala C, Yahi N, Boutemeur S, Flores A, Rodriguez L, Chahinian H, Fantini J, Sci. Rep 2016, 6, 28781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, Pericak-Vance MA, Goldgaber D, Roses AD, Proc. Natl. Acad. Sci. U. S. A 1993, 90, 9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD, Proc. Natl. Acad. Sci. U. S. A 1993, 90, 8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM, Nat. Neurosci 2012, 15, 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kostylev MA, Kaufman AC, Nygaard HB, Patel P, Haas LT, Gunther EC, Vortmeyer A, Strittmatter SM, J. Biol. Chem 2015, 290, 17415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Gomes LA, Hipp SA, Rijal Upadhaya A, Balakrishnan K, Ospitalieri S, Koper MJ, Largo-Barrientos P, Uytterhoeven V, Reichwald J, Rabe S, Vandenberghe R, von Arnim CAF, Tousseyn T, Feederle R, Giudici C, Willem M, Staufenbiel M, Thal DR, Acta Neuropathologica 2019, 138, 913. [DOI] [PubMed] [Google Scholar]
- [91].Hughes C, Choi ML, Yi J-H, Kim S-C, Drews A, George-Hyslop PS, Bryant C, Gandhi S, Cho K, Klenerman D, Commun. Biol 2020, 3, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Wang H, Chen F, Du YF, Long Y, Reed MN, Hu M, Suppiramaniam V, Hong H, Tang SS, Neuropharmacology 2018, 131, 143. [DOI] [PubMed] [Google Scholar]
- [93].Barykin EP, Garifulina AI, Kruykova EV, Spirova EN, Anashkina AA, Adzhubei AA, Shelukhina IV, Kasheverov IE, Mitkevich VA, Kozin SA, Hollmann M, Tsetlin VI, Makarov AA, Cells 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].De S, Wirthensohn DC, Flagmeier P, Hughes C, Aprile FA, Ruggeri FS, Whiten DR, Emin D, Xia Z, Varela JA, Sormanni P, Kundel F, Knowles TPJ, Dobson CM, Bryant C, Vendruscolo M, Klenerman D, Nat. Commun 2019, 10, 1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Mrdenovic D, Su Z, Kutner W, Lipkowski J, Pieta P, Nanoscale Adv. 2020, 2, 3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Henry S, Vignaud H, Bobo C, Decossas M, Lambert O, Harte E, Alves ID, Cullin C, Lecomte S, Biomacromolecules 2015, 16, 944. [DOI] [PubMed] [Google Scholar]
- [97].Qiao R, Roberts AP, Mount AS, Klaine SJ, Ke PC, Nano Lett. 2007, 7, 614. [DOI] [PubMed] [Google Scholar]
- [98].Sayes CM, Fortner JD, Guo W, Lyon D, Boyd AM, Ausman KD, Tao YJ, Sitharaman B, Wilson LJ, Hughes JB, West JL, Colvin VL, Nano Lett. 2004, 4, 1881. [Google Scholar]
- [99].Ke PC, Lamm MH, Phys. Chem. Chem. Phys 2011, 13, 7273. [DOI] [PubMed] [Google Scholar]
- [100].Tu Y, Lv M, Xiu P, Huynh T, Zhang M, Castelli M, Liu Z, Huang Q, Fan C, Fang H, Zhou R, Nat. Nanotechnol 2013, 8, 594. [DOI] [PubMed] [Google Scholar]
- [101].Kraszewski S, Tarek M, Treptow W, Ramseyer C, ACS Nano 2010, 4, 4158. [DOI] [PubMed] [Google Scholar]
- [102].Wong-Ekkabut J, Baoukina S, Triampo W, Tang IM, Tieleman DP, Monticelli L, Nat. Nanotechnol 2008, 3, 363. [DOI] [PubMed] [Google Scholar]
- [103].Yang K, Ma YQ, Nat. Nanotechnol 2010, 5, 579. [DOI] [PubMed] [Google Scholar]
- [104].Shi X, von dem Bussche A, Hurt RH, Kane AB, Gao H, Nat. Nanotechnol 2011, 6, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Chen R, Ratnikova TA, Stone MB, Lin S, Lard M, Huang G, Hudson JS, Ke PC, Small 2010, 6, 612. [DOI] [PubMed] [Google Scholar]
- [106].Kang B, Chang S, Dai Y, Yu D, Chen D, Small 2010, 6, 2362. [DOI] [PubMed] [Google Scholar]
- [107].Wang XY, Lei R, Huang HD, Wang N, Yuan L, Xiao RY, Bai LD, Li X, Li LM, Yang XD, Nanoscale 2015, 7, 2034. [DOI] [PubMed] [Google Scholar]
- [108].Frost R, Jonsson GE, Chakarov D, Svedhem S, Kasemo B, Nano Lett. 2012, 12, 3356. [DOI] [PubMed] [Google Scholar]
- [109].Duan G, Zhang Y, Luan B, Weber JK, Zhou RW, Yang Z, Zhao L, Xu J, Luo J, Zhou R, Sci. Rep 2017, 7, 42767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Chen P, Yue H, Zhai X, Huang Z, Ma GH, Wei W, Yan LT, Sci. Adv 2019, 5, eaaw3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Mukherjee SP, Lazzaretto B, Hultenby K, Newman L, Rodrigues AF, Lozano N, Kostarelos K, Malmberg P, Fadeel B, Chem 2018, 4, 334. [Google Scholar]
- [112].Setyawati MI, Tay CY, Chia SL, Goh SL, Fang W, Neo MJ, Chong HC, Tan SM, Loo SC, Ng KW, Xie JP, Ong CN, Tan NS, Leong DT, Nat. Commun 2013, 4, 1673. [DOI] [PubMed] [Google Scholar]
- [113].Peng F, Setyawati MI, Tee JK, Ding X, Wang J, Nga ME, Ho HK, Leong DT, Nat. Nanotechnol 2019, 14, 279. [DOI] [PubMed] [Google Scholar]
- [114].Setyawati MI, Tay CY, Bay BH, Leong DT, ACS Nano 2017, 11, 5020. [DOI] [PubMed] [Google Scholar]
- [115].Zhao Y, Sun X, Zhang G, Trewyn BG, Slowing II, Lin VS, ACS Nano 2011, 5, 1366. [DOI] [PubMed] [Google Scholar]
- [116].Arvizo RR, Miranda OR, Thompson MA, Pabelick CM, Bhattacharya R, Robertson JD, Rotello VM, Prakash YS, Mukherjee P, Nano Lett. 2010, 10, 2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Burnand D, Milosevic A, Balog S, Spuch-Calvar M, Rothen-Rutishauser B, Dengjel J, Kinnear C, Moore TL, Petri-Fink A, Small 2018, 14, e1802088. [DOI] [PubMed] [Google Scholar]
- [118].Wang X, Wang X, Bai X, Yan L, Liu T, Wang M, Song Y, Hu G, Gu Z, Miao Q, Chen C, Nano Lett. 2019, 19, 8. [DOI] [PubMed] [Google Scholar]
- [119].Wang L, Quan P, Chen SH, Bu W, Li YF, Wu X, Wu J, Zhang L, Zhao Y, Jiang X, Lin B, Zhou R, Chen C, ACS Nano 2019, 13, 8680. [DOI] [PubMed] [Google Scholar]
- [120].Ke PC, Lin S, Parak WJ, Davis TP, Caruso F, ACS Nano 2017, 11, 11773. [DOI] [PubMed] [Google Scholar]
- [121].Yan Y, Gause KT, Kamphuis MM, Ang CS, O’Brien-Simpson NM, Lenzo JC, Reynolds EC, Nice EC, Caruso F, ACS Nano 2013, 7, 10960. [DOI] [PubMed] [Google Scholar]
- [122].Shannahan JH, Podila R, Aldossari AA, Emerson H, Powell BA, Ke PC, Rao AM, Brown JM, Toxicol. Sci 2015, 143, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Treuel L, Brandholt S, Maffre P, Wiegele S, Shang L, Nienhaus GU, ACS Nano 2014, 8, 503. [DOI] [PubMed] [Google Scholar]
- [124].Wan S, Kelly PM, Mahon E, Stockmann H, Rudd PM, Caruso F, Dawson KA, Yan Y, Monopoli MP, ACS Nano 2015, 9, 2157. [DOI] [PubMed] [Google Scholar]
- [125].Kokkinopoulou M, Simon J, Landfester K, Mailander V, Lieberwirth I, Nanoscale 2017, 9, 8858. [DOI] [PubMed] [Google Scholar]
- [126].Linse S, Cabaleiro-Lago C, Xue W-F, Lynch I, Lindman S, Thulin E, Radford SE, Dawson KA, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Cedervall T, Lynch I, Lindman S, Berggård T, Thulin E, Nilsson H, Dawson KA, Linse S, Proc. Natl. Acad. Sci. U. S. A 2007, 104, 2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Mahmoudi M, Kalhor HR, Laurent S, Lynch I, Nanoscale 2013, 5, 2570. [DOI] [PubMed] [Google Scholar]
- [129].Wang B, Pilkington EH, Sun Y, Davis TP, Ke PC, Ding F, Environ. Sci. Nano 2017, 4, 1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Brancolini G, Tozzini V, Curr. Opin. Colloid Interface Sci 2019, 41, 66. [Google Scholar]
- [131].Brambilla D, Verpillot R, Le Droumaguet B, Nicolas J, Taverna M, Kóňa J, Lettiero B, Hashemi SH, De Kimpe L, Canovi M, Gobbi M, Nicolas V, Scheper W, Moghimi SM, Tvaroška I, Couvreur P, Andrieux K, ACS Nano 2012, 6, 5897. [DOI] [PubMed] [Google Scholar]
- [132].Mohajeri M, Behnam B, Barreto GE, Sahebkar A, Pharmacol. Res 2019, 143, 186. [DOI] [PubMed] [Google Scholar]
- [133].Yu X, Wang Q, Lin Y, Zhao J, Zhao C, Zheng J, Langmuir 2012, 28, 6595. [DOI] [PubMed] [Google Scholar]
- [134].Andujar SA, Lugli F, Höfinger S, Enriz RD, Zerbetto F, Phys. Chem. Chem. Phys 2012, 14, 8599. [DOI] [PubMed] [Google Scholar]
- [135].Sun Y, Qian Z, Wei G, Phys. Chem. Chem. Phys 2016, 18, 12582. [DOI] [PubMed] [Google Scholar]
- [136].Jana AK, Sengupta N, Biophys. J 2012, 102, 1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Fu Z, Luo Y, Derreumaux P, Wei G, Biophys. J 2009, 97, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Koppel K, Tang H, Javed I, Parsa M, Mortimer M, Davis TP, Lin S, Chaffee AL, Ding F, Ke PC, Nanoscale 2020, 12, 12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Yang Z, Ge C, Liu J, Chong Y, Gu Z, Jimenez-Cruz CA, Chai Z, Zhou R, Nanoscale 2015, 7, 18725. [DOI] [PubMed] [Google Scholar]
- [140].John T, Gladytz A, Kubeil C, Martin LL, Risselada HJ, Abel B, Nanoscale 2018, 10, 20894. [DOI] [PubMed] [Google Scholar]
- [141].Iori F, Di Felice R, Molinari E, Corni S, J. Comput. Chem 2009, 30, 1465. [DOI] [PubMed] [Google Scholar]
- [142].Bellucci L, Bussi G, Di Felice R, Corni S, Nanoscale 2017, 9, 2279. [DOI] [PubMed] [Google Scholar]
- [143].Brancolini G, Bellucci L, Maschio MC, Di Felice R, Corni S, Curr. Opin. Colloid Interface Sci 2019, 41, 86. [Google Scholar]
- [144].Sudhakar S, Kalipillai P, Balaji P, Mani E, J. Phys. Chem C. 2017, 121. [Google Scholar]
- [145].Vácha R, Linse S, Lund M, J. Am. Chem. Soc 2014, 136, 11776. [DOI] [PubMed] [Google Scholar]
- [146].Radic S, Davis TP, Ke PC, Ding F, RSC Adv. 2015, 5, 105489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Chen P, Ding F, Cai R, Javed I, Yang W, Zhang Z, Li Y, Davis TP, Ke PC, Chen C, Nano Today 2020, 35, 100937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Butterfield SM, Lashuel HA, Angew. Chem. Int. Ed. Engl 2010, 49, 5628. [DOI] [PubMed] [Google Scholar]
- [149].Kim Y, Park JH, Lee H, Nam JM, Sci. Rep 2016, 6, 19548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Han Q, Cai S, Yang L, Wang X, Qi C, Yang R, Wang C, ACS Appl. Mater. Interfaces 2017, 9, 21116. [DOI] [PubMed] [Google Scholar]
- [151].Guan Y, Li M, Dong K, Gao N, Ren J, Zheng Y, Qu X, Biomaterials 2016, 98, 92. [DOI] [PubMed] [Google Scholar]
- [152].Sudhakar S, Mani E, Langmuir 2019, 35, 6962. [DOI] [PubMed] [Google Scholar]
- [153].Li Y, Du Z, Liu X, Ma M, Yu D, Lu Y, Ren J, Qu X, Small 2019, 15, e1901116. [DOI] [PubMed] [Google Scholar]
- [154].Wang J, Wang K, Zhu Z, He Y, Zhang C, Guo Z, Wang X, RSC Adv. 2019, 9, 14126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Yang L, Wang N, Zheng G, Nanoscale Res. Lett 2018, 13, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Mirsadeghi S, Shanehsazzadeh S, Atyabi F, Dinarvand R, Mater Sci. Eng. C Mater. Biol. Appl 2016, 59, 390. [DOI] [PubMed] [Google Scholar]
- [157].Dowding JM, Song W, Bossy K, Karakoti A, Kumar A, Kim A, Bossy B, Seal S, Ellisman MH, Perkins G, Self WT, Bossy-Wetzel E, Cell Death Differ. 2014, 21, 1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Andrikopoulos N, Li Y, Cecchetto L, Nandakumar A, Da Ros T, Davis TP, Velonia K, Ke PC, Nanoscale 2020, 12, 14422. [DOI] [PubMed] [Google Scholar]
- [159].Sahoo BR, Genjo T, Bekier M, Cox SJ, Stoddard AK, Ivanova M, Yasuhara K, Fierke CA, Wang Y, Ramamoorthy A, Chem. Commun 2018, 54, 12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Wang Z, Dong X, Sun Y, Langmuir 2019, 35, 14681. [DOI] [PubMed] [Google Scholar]
- [161].Song Q, Song H, Xu J, Huang J, Hu M, Gu X, Chen J, Zheng G, Chen H, Gao X, Mol. Pharm 2016, 13, 3976. [DOI] [PubMed] [Google Scholar]
- [162].Song Q, Huang M, Yao L, Wang X, Gu X, Chen J, Chen J, Huang J, Hu Q, Kang T, Rong Z, Qi H, Zheng G, Chen H, Gao X, ACS Nano 2014, 8, 2345. [DOI] [PubMed] [Google Scholar]
- [163].Huang M, Hu M, Song Q, Song H, Huang J, Gu X, Wang X, Chen J, Kang T, Feng X, Jiang D, Zheng G, Chen H, Gao X, ACS Nano 2015, 9, 10801. [DOI] [PubMed] [Google Scholar]
- [164].Yang H, Mu W, Wei D, Zhang Y, Duan Y, Gao J.-x., Gong X.-q., Wang H.-j., Wu X.-l., Tao H, Chang J, Adv. Sci n/a, 1902906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Ye Z, Wei L, Li Y, Xiao L, Anal. Chem 2019, 91, 8582. [DOI] [PubMed] [Google Scholar]
- [166].Liu Y, Xu LP, Dai W, Dong H, Wen Y, Zhang X, Nanoscale 2015, 7, 19060. [DOI] [PubMed] [Google Scholar]
- [167].Bednarikova Z, Huy PDQ, Mocanu M-M, Fedunova D, Li MS, Gazova Z, Phys. Chem. Chem. Phys 2016, 18, 18855. [DOI] [PubMed] [Google Scholar]
- [168].Liu F, Wang W, Sang J, Jia L, Lu F, ACS Chem. Neurosci 2019, 10, 588. [DOI] [PubMed] [Google Scholar]
- [169].Furtado D, Björnmalm M, Ayton S, Bush AI, Kempe K, Caruso F, Adv. Mater 2018, 30, 1801362. [DOI] [PubMed] [Google Scholar]
- [170].Mulvihill JJ, Cunnane EM, Ross AM, Duskey JT, Tosi G, Grabrucker AM, Nanomedicine. 2020, 15, 205. [DOI] [PubMed] [Google Scholar]
- [171].Poovaiah N, Davoudi Z, Peng H, Schlichtmann B, Mallapragada S, Narasimhan B, Wang Q, Nanoscale 2018, 10, 16962. [DOI] [PubMed] [Google Scholar]
- [172].Joseph A, Contini C, Cecchin D, Nyberg S, Ruiz-Perez L, Gaitzsch J, Fullstone G, Tian X, Azizi J, Preston J, Volpe G, Battaglia G, Sci. Adv 2017, 3, e1700362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Keaney J, Walsh DM, O’Malley T, Hudson N, Crosbie DE, Loftus T, Sheehan F, McDaid J, Humphries MM, Callanan JJ, Brett FM, Farrell MA, Humphries P, Campbell M, Sci. Adv 2015, 1, e1500472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Liu Y, Gong Y, Xie W, Huang A, Yuan X, Zhou H, Zhu X, Chen X, Liu J, Liu J, Qin X, Nanoscale 2020, 12, 6498. [DOI] [PubMed] [Google Scholar]
- [175].Ke PC, Sani M-A, Ding F, Kakinen A, Javed I, Separovic F, Davis TP, Mezzenga R, Chem. Soc. Rev 2017, 46, 6492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Aleksis R, Oleskovs F, Jaudzems K, Pahnke J, Biverstål H, Biochimie 2017, 140, 176. [DOI] [PubMed] [Google Scholar]
- [177].Nag S, Sarkar B, Chandrakesan M, Abhyanakar R, Bhowmik D, Kombrabail M, Dandekar S, Lerner E, Haas E, Maiti S, Phys. Chem. Chem. Phys 2013, 15, 19129. [DOI] [PubMed] [Google Scholar]
- [178].Sarkar B, Das AK, Maiti S, Front. Physiol 2013, 4, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Kakio A, Yano Y, Takai D, Kuroda Y, Matsumoto O, Kozutsumi Y, Matsuzaki K, J. Pept. Sci 2004, 10, 612. [DOI] [PubMed] [Google Scholar]
- [180].Niu Z, Zhang Z, Zhao W, Yang J, Biochim. Biophys. Acta. Biomembr 2018, 1860, 1663. [DOI] [PubMed] [Google Scholar]
- [181].Suga K, Lai Y-C, Faried M, Umakoshi H, Int. J. Anal. Chem 2018, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Bhowmik D, Mote KR, MacLaughlin CM, Biswas N, Chandra B, Basu JK, Walker GC, Madhu PK, Maiti S, ACS nano 2015, 9, 9070. [DOI] [PubMed] [Google Scholar]
- [183].Tan S, Wu T, Zhang D, Zhang Z, Theranostics 2015, 5, 863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Gao W, Hu CMJ, Fang RH, Luk BT, Su J, Zhang L, Adv. Mater 2013, 25, 3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Hu C-MJ, Fang RH, Luk BT, Chen KN, Carpenter C, Gao W, Zhang K, Zhang L, Nanoscale 2013, 5, 2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Wechsler ME, Vela Ramirez JE, Peppas NA, Ind. Eng. Chem. Res 2019, 58, 15079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Klementieva O, Benseny-Cases N. r., Gella A, Appelhans D, Voit B, Cladera J, Biomacromolecules 2011, 12, 3903. [DOI] [PubMed] [Google Scholar]
- [188].Kannan S, Dai H, Navath RS, Balakrishnan B, Jyoti A, Janisse J, Romero R, Kannan RM, Sci. Transl. Med 2012, 4, 130ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Moscariello P, Ng DY, Jansen M, Weil T, Luhmann HJ, Hedrich J, Adv. Sci 2018, 5, 1700897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Wilson B, Samanta MK, Santhi K, Kumar KPS, Paramakrishnan N, Suresh B, Brain Res. 2008, 1200, 159. [DOI] [PubMed] [Google Scholar]
- [191].Re F, Cambianica I, Zona C, Sesana S, Gregori M, Rigolio R, La Ferla B, Nicotra F, Forloni G, Cagnotto A, Nanomedicine. 2011, 7, 551. [DOI] [PubMed] [Google Scholar]
- [192].Salvati E, Re F, Sesana S, Cambianica I, Sancini G, Masserini M, Gregori M, Int. J. Nanomedicine 2013, 8, 1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Johnsen KB, Bak M, Melander F, Thomsen MS, Burkhart A, Kempen PJ, Andresen TL, Moos T, Control J. Release 2019, 295, 237. [DOI] [PubMed] [Google Scholar]
- [194].Chuang ST, Cruz S, Stab J, Wagner S, Von Briesen H, Narayanaswami V, Arterioscler. Thromb. Vasc. Biol 2017, 37, A388. [Google Scholar]
- [195].Long JM, Holtzman DM, Cell 2019, 179, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].Mannini B, Mulvihill E, Sgromo C, Cascella R, Khodarahmi R, Ramazzotti M, Dobson CM, Cecchi C, Chiti F, ACS Chem. Biol 2014, 9, 2309. [DOI] [PubMed] [Google Scholar]
- [197].Nandakumar A, Xing Y, Aranha RR, Faridi A, Kakinen A, Javed I, Koppel K, Pilkington EH, Purcell AW, Davis TP, Faridi P, Ding F, Ke PC, Biomacromolecules 2020, 21, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Pilkington EH, Gustafsson OJR, Xing Y, Hernandez-Fernaud J, Zampronio C, Kakinen A, Faridi A, Ding F, Wilson P, Ke PC, Davis TP, ACS Nano 2018, 12, 6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Javed I, Yu T, Peng G, Sánchez-Ferrer A, Faridi A, Kakinen A, Zhao M, Mezzenga R, Davis TP, Lin S, Ke PC, Nano Lett. 2018, 18, 5797. [DOI] [PubMed] [Google Scholar]