

Abstract
Antibodies with heavy chains that derive from the VH1-2 gene constitute some of the most potent severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-neutralizing antibodies yet identified. To provide insight into whether these genetic similarities inform common modes of recognition, we determine the structures of the SARS-CoV-2 spike in complex with three VH1-2-derived antibodies: 2-15, 2-43, and H4. All three use VH1-2-encoded motifs to recognize the receptor-binding domain (RBD), with heavy-chain N53I-enhancing binding and light-chain tyrosines recognizing F486RBD. Despite these similarities, class members bind both RBD-up and -down conformations of the spike, with a subset of antibodies using elongated CDRH3s to recognize glycan N343 on a neighboring RBD—a quaternary interaction accommodated by an increase in RBD separation of up to 12 Å. The VH1-2 antibody class, thus, uses modular recognition encoded by modular genetic elements to effect potent neutralization, with the VH-gene component specifying recognition of RBD and the CDRH3 component specifying quaternary interactions.
Keywords: COVID-19, multi-donor antibody class, neutralizing antibody, quaternary recognition, RBD, SARS-CoV-2, spike
Graphical abstract
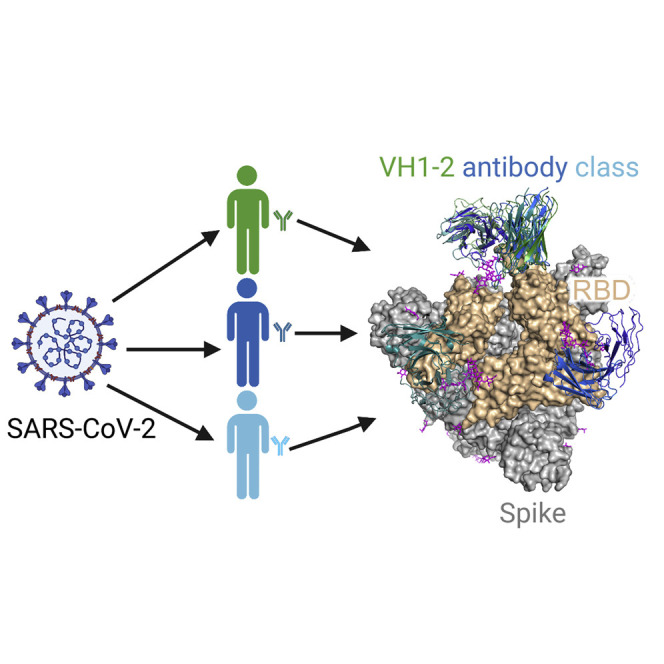
Rapp et al. determine structures of three human VH1-2-derived SARS-CoV-2-neutralizing antibodies and define a prevalent multi-donor antibody class that recognizes the receptor-binding domain (RBD) with convergent heavy- and light-chain modules. They further show the structural basis for RBD-up and RBD-down recognition and quaternary recognition-mediated spike-conformation change.
Introduction
Studies on human antibody responses to viral pathogens, including HIV-1, influenza, Ebola, and malaria, have revealed prominent classes of similar neutralizing antibodies (nAbs), which arise commonly in numerous individuals in response to infection or vaccination (Ehrhardt et al., 2019; Imkeller et al., 2018; Joyce et al., 2016; Kallewaard et al., 2016; Pappas et al., 2014; Zhou et al., 2013, 2015). Such multi-donor antibody classes are defined based on similar V(D)J gene recombination and similar modes of structural recognition—with this combination indicative of a common evolutionary process in antibody development (Kwong and Mascola, 2012). Multi-donor antibody classes are thought to arise based on effective function, combined with genetic accessibility because of class requirements for V(D)J recombination and somatic hypermutations of sufficient frequency to be present in the antibody human repertoire. Some multi-donor antibody classes are potent, broadly neutralizing, and frequent in human antibody repertoire (Imkeller et al., 2018; Joyce et al., 2016; Zhou et al., 2013). A prominent mode of rational vaccine design, “lineage-based vaccine design,” endeavors to elicit such antibody classes by vaccination (Haynes et al., 2012; Jardine et al., 2013; Kwong and Mascola, 2018), and this approach to human vaccination has recently entered clinical assessment (Diemert and McElrath, 2018).
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the causative agent of the ongoing coronavirus disease 2019 (COVID-19) pandemic, has infected more than 80 million people and has claimed over 1 million deaths since the outbreak began in late 2019 (Dong et al., 2020; Zhou et al., 2020; Zhu et al., 2020). Therapies and vaccines are urgently needed to end the pandemic. Many nAbs have now been isolated from COVID-19 convalescent donors, with the most potent nAbs showing promise as prophylactic or therapeutic agents (Brouwer et al., 2020; Chen et al., 2020; Kreer et al., 2020; Liu et al., 2020a; Robbiani et al., 2020; Rogers et al., 2020; Tortorici et al., 2020; Zost et al., 2020). This growing set of nAbs provides an opportunity to identify effective human antibody responses to SARS-CoV-2 common in the population, which will inform therapeutic strategies and help to interpret vaccine readouts.
SARS-CoV-2 nAbs predominantly target the viral spike glycoprotein, which interacts with angiotensin-converting enzyme 2 (ACE2) receptors on the host-cell surface to mediate virus entry (Hoffmann et al., 2020; Wang et al., 2020). The ectodomain of the prefusion spike comprises three copies of both S1 and S2 subunits (Wrapp et al., 2020). The S1 subunit is responsible for ACE2 binding, and the S2 subunit mediates fusion with host-cell membrane. Each S1 subunit comprises an N-terminal domain (NTD) and a receptor-binding domain (RBD). The RBDs are very flexible, adopting either an “up” conformation (open state) or a “down” conformation (closed state), with only the up RBDs capable of interacting with ACE2 (Wang et al., 2020). Currently, many nAbs have been characterized that bind to epitopes on either the up and/or down RBDs (Barnes et al., 2020a; Liu et al., 2020a; Tortorici et al., 2020). These RBD-targeting nAbs neutralize SARS-CoV-2 through mechanisms that include competition with ACE2 for RBD binding and locking the RBDs in the down conformation.
Human SARS-CoV-2 nAbs develop with few somatic hypermutations and strong avidity effects (Barnes et al., 2020b; Kreer et al., 2020; Liu et al., 2020a; Robbiani et al., 2020). Characterization of genetic features of SARS-CoV-2 nAbs identified to date show enrichment of antibody-variable genes, including VH3-53, VH1-2, VH1-69, VH3-66, VH1-58, and VH3-30 (Liu et al., 2020a; Robbiani et al., 2020; Yuan et al., 2020). So far, structural characterization of multiple nAbs have revealed two separate RBD-targeted classes derived from the similar VH3-53 and VH3-66 genes. One of these is characterized by heavy-chain complementarity-determining region 3 (CDRH3) of 15 amino acids or shorter—and recognizes the RBD ridge in the up position on SARS-CoV-2 spike (type I) (Yuan et al., 2020). The second VH3-53/-66 class—with longer CDRH3s—recognizes a similar region of RBD but adopts an approach angle with the heavy- and light-chain orientation rotated 180° (type II), suggesting CDRH3 to be critical for the classification of VH3-53/-66 antibodies (Barnes et al., 2020a; Wu et al., 2020a). Different VH3-30 originated antibodies can recognize at least three different regions of RBDs (Barnes et al., 2020a; Hansen et al., 2020), demonstrating that they do not form a single gene-restricted antibody class. Nevertheless, whether nAbs derived from VH1-2 and other germline genes form gene-restricted classes that represent shared effective antibody responses remains unaddressed. Currently, three VH1-2, potent nAbs (2-4, S2M11, and C121) show similar modes of RBD recognition but differences in quaternary epitope recognition (Barnes et al., 2020a; Liu et al., 2020a; Tortorici et al., 2020). It is, thus, still unclear whether RBD-targeting VH1-2 antibodies form a gene-restricted class. If so, what are the key genetic and structural signatures and determinants of neutralization potency?
Here, we present the structures of three VH1-2-derived nAbs (2-15, 2-43, and H4), revealing that they recognize a SARS-CoV-2 RBD epitope with similar modes of RBD recognition and similar angles of approach. Overall, recognition is modular with RBD predominantly recognized by a VH1-2-gene-encoded module. The second recognition module is represented by the diverse CDRH3s of the VH1-2 antibodies, which mediate quaternary recognition of N343 glycan from an adjacent RBD for a subset of class members. Thus, we define a multi-donor VH1-2 antibody class; members of which can achieve very high neutralization potency, which is prevalent in human responses to SARS-CoV-2. The shared genetic and structural signatures inform strategies to improve members of the VH1-2 antibody class.
Results
VH1-2 antibodies are prevalent in human response to SARS-CoV-2 infection
To identify common features of the human antibody response to SARS-CoV-2, 158 spike-specific antibodies with characterized neutralization potencies were collected from 10 studies (Table S1) (Brouwer et al., 2020; Hansen et al., 2020; Ju et al., 2020; Kreer et al., 2020; Liu et al., 2020a; Robbiani et al., 2020; Rogers et al., 2020; Tortorici et al., 2020; Wu et al., 2020b; Zost et al., 2020). The V(D)J gene usage analysis showed that VH1-2 was the second most frequently used germline gene (Figure 1 A; 25 in total). Comparison of neutralization potencies revealed that 11 of 25 (44%) of the VH1-2 antibodies are potent (half maximal inhibitory concentration [IC50] < 0.1 μg/mL; Figure 1B). Within four studies (Hansen et al., 2020; Liu et al., 2020a; Tortorici et al., 2020; Zost et al., 2020), VH1-2 antibodies ranked the most potent of gene-delimited sets of antibodies (IC50 ranges from 0.015 to 0.0007 μg/mL). All 25 of the VH1-2-derived antibodies have been reported to bind to the SARS-CoV-2 RBD (Brouwer et al., 2020; Hansen et al., 2020; Ju et al., 2020; Kreer et al., 2020; Liu et al., 2020a; Robbiani et al., 2020; Rogers et al., 2020; Tortorici et al., 2020; Wu et al., 2020b; Zost et al., 2020). Sequence alignment of the VH1-2 nAbs showed the heavy chains to carry few somatic hypermutations, with each having a unique CDRH3 with length varying from 11 to 21 amino acids (Figures 1C and S1A; Kabat definition), and using different D genes (Figure S1B). The VH1-2 antibodies used both kappa and lambda genes with enrichment of the IGLV2-14 gene (Figure 1C).
Figure 1.

SARS-CoV-2 infection in humans induces potently neutralizing VH1-2 antibodies
(A) Gene usage of SARS-CoV-2-neutralizing antibodies. SARS-CoV-2 spike-specific VH1-2 antibodies are frequently induced in infected humans. VH1-2 antibodies are more-significantly enriched in the antigen-specific antibody repertoire than they are in healthy donors. Antibody repertoires from 17 healthy donors were used for the analysis.
(B) Many SARS-CoV-2-neutralizing VH1-2 antibodies (red) isolated from human donors achieve high potency, comparable with the most frequent IGHV3-53/-66 antibodies (blue). The half-maximal inhibitory concentration (IC50) of neutralization is shown, except IC100 is shown for antibodies from Kreer et al., (2020). Live virus neutralization potency is shown for antibodies from nine studies, except the Hansen et al. (2020) study. IC50 values greater than 10 μg/mL are set to 10 μg/mL. Neutralizing antibodies targeting both RBD and non-RBD epitopes are included.
(C) SARS-CoV-2-neutralizing VH1-2 antibodies use diverse light-chain genes.
(D) Sequence alignment of the heavy chain of six VH1-2 antibodies. Antibodies with structures reported in this study are highlighted in red. Residues identical to germline gene are shown as dots.
As described below, we determined structures of three nAbs: 2-15, 2-43, and H4, with the highly potent antibodies 2-15 and 2-43 isolated from donor 2 of our previous study (Liu et al., 2020a), whereas H4 was from a different donor (Wu et al., 2020b). The 2-15, 2-43, and H4 nAbs neutralize authentic SARS-CoV-2 “live” virus with an IC50 of 0.0007, 0.003, and 0.896 μg/mL, respectively. These three antibodies derived from different heavy- and light-chain gene recombinations and, hence, three different B cell lineages (Figure S1B). Both 2-43 and 2-15 use the DH2-15∗01 gene and have a long CDRH3 of 20 amino acids (Figure S1C), but they have different HJ gene origins (JH6∗03 and JH3∗02, respectively). The light chains of 2-43 and 2-15 were derived from recombination of a novel allele of the VL2-14 gene with JL3∗02 and JL1∗01, respectively (Figure S1D). H4 used DH2-2∗01 and JH2∗01 genes and had a CDRH3 of 17 amino acids (Wu et al., 2020b). The H4 light chain was derived from VK2-40∗01 and JK4∗01.
Structures of antibodies 2-15, 2-43, and H4 in complex with SARS-CoV-2 spike or RBD
To understand SARS-CoV-2 spike recognition by the VH1-2-derived antibodies, we used single-particle cryo-electron microscopy (cryo-EM) to produce high-resolution 3D-reconstructions of antigen-binding fragments (Fabs) from 2-15, 2-43, and H4 in complex with the SARS-CoV-2 spike (Table S2). The reconstruction of the 2-43 complex with spike, refined to a global resolution of 3.60Å (Figures S2A–S2E), is significantly improved than in our previous study (5.8Å resolution) (Liu et al., 2020a). The reconstruction revealed a predominant class with three Fab molecules bound per spike trimer (Figure 2 A). Each 2-43 Fab used the heavy-chain-variable domain to bind one primary RBD, with an orientation similar to the previously published antibody 2-4 (Liu et al., 2020a), with all RBDs in the down conformation (Figure 2A). The 2-43 heavy and light chains also recognize a quaternary epitope from the adjacent RBD. The three-dimensional (3D) classification revealed less-populated states with 1 and 2 Fabs bound, but in every case, the Fab was bound to a down RBD. This dependence upon the down conformation is likely due to extensive interactions between the Fab CDRH3 loop and the N-linked glycosylation at residue N343 on the adjacent RBD (Figure S2E). This suggests a neutralization mechanism by which the Fab simultaneously occludes ACE2 binding and locks the RBDs in the down conformation.
Figure 2.

Structures of three SARS-CoV-2-neutralizing VH1-2 antibodies reveal both “RBD-down” and “RBD-up” modes of spike recognition
(A) Side and top views of three 2-43 antibody Fabs bound to the prefusion SARS-COV-2 spike in the closed state. Color schemes are 2-43, green; RBD, salmon; NTD, yellow; N-linked glycans, magenta; other spike regions, gray.
(B) Side and top views of one 2-15 antibody Fab in complex with the prefusion SARS-COV-2 spike in the closed state. 2-15 is colored blue.
(C) Overview of the crystal structure of 2-15 in complex with SARS-CoV-2 RBD. Residue segments missing in the structure are shown as dashed lines.
(D) Side and top views of one H4 Fab recognizing an up RBD on the prefusion SARS-COV-2 spike. H4 is colored cyan.
For the complex of 2-15 with spike (Figure 2B), approximately 56% of the particles were bound to an RBD in the up conformation, and 44% bound to RBD in the down conformation (Figures S2F–S2I), differing from antibody 2-43, which bound only to down RBDs. Because of increased conformational heterogeneity of the RBD-up class, the RBD-down class was the focus of our structural analysis (Figure 2B). In our initial attempt, a 9-fold molar excess of Fab was incubated with the S trimer. However, this resulted in spike disassembly (Figure S2J). To overcome this issue for structural determination, we found that a 1:1 molar ratio left spike-complexes intact, although some spike disassembly was still observed (Figure S2K). Although we were unable to resolve the 2-15-to-spike interaction at an atomic resolution, the cryo-EM analysis did reveal the overall orientation of the Fab, along with the position of the peptide backbone for several of the CDR loops.
To understand the binding mode of the 2-15 at an atomic resolution, we determined the crystal structure of the 2-15 in complex with isolated RBD. The structure was determined by molecular replacement at 3.18-Å resolution to a final crystallographic Rwork/Rfree of 18.6%/23.8% with good overall geometry (Table S3). The electron density for RBD (corresponding to S1-subunit residues 333–527) was well defined for residues 344–518, with missing internal stretches of four (362–366) and seven (383–390) residues, with the C-terminal 18 residues (519–537) disordered (Figure 2C). For Fab 2-15, all residues were visualized in density, with the exception of heavy-chain residues 142–145 in the Fc region. We then docked the crystal structure to the 2-15-spike complex cryo-EM density map by superposing RBD from the crystal structure on RBD from the cryo-EM map, with subsequent rigid body fitting to density.
Overall, the conformation of the 2-15 in the crystal structure agrees with the density observed in the cryo-EM map of the complex with the spike—particularly in the V-gene-encoded CDR regions. However, the conformation of the long CDRH3 loop differs and is far more elongated in the crystal structure (Figure S2L). In the cryo-EM map, it appears that the CDRH3 loop is pushed away from the N343 glycan and the helix encompassing residues 364–371 from the adjacent down RBD. The two light chains exhibit more-significant conformational differences. Save for CDRL3, which bends slightly away from RBD in the crystal structure, the conformations of the loops are quite similar, but the chain is shifted down closer to the RBD.
Finally, a reconstruction of Fab H4 revealed spike complexes with a single Fab bound to RBD in the up conformation (Figures 2D and S2M–S2P). Interestingly, no Fab was seen bound to a down RBD. The conformational flexibility of the RBD in the up conformation made a high-resolution cryo-EM reconstruction unattainable, and the map was refined to an overall resolution of 5.08 Å. Similar to Fab 2-15, the peptide backbone for many CDR loops was observed, and a homology model of the Fab variable domain was docked into the density. The superimposition of modeled H4 to the 2-43/spike complex showed that H4 adopts an RBD approaching angle similar to that of 2-43 (Figure S2Q). However, the light chain of H4 rotates toward the interface between RBDs such that the long CDRL1 of H4 clashes with N343 glycan from an adjacent down RBD (Figure S2R), which suggests a possible explanation for the apparent lack of H4 binding to down RBDs.
Conserved mode of RBD recognition
To understand the similarity in RBD recognition, we characterized the epitope and paratope interactions of 2-43 and 2-15 and compared them with the three published VH1-2 antibodies: 2-4, S2M11, and C121 (Barnes et al., 2020a; Liu et al., 2020a; Tortorici et al., 2020). Overall, 2-43 interacts predominantly with the receptor-binding motif (RBM; residues 438–508) on one down RBD protomer and simultaneously binds the N343 glycan from an adjacent down RBD protomer (Figure 3 A). We describe the N343 glycan interaction below in the last section focused on quaternary interactions. The interaction between 2-43 and the “primary” RBD bound through its RBM buries a total of 756-Å2 paratope body surface area (BSA); 83% of which is contributed by heavy chain (Figures 3A and S3A). Heavy-chain framework 1 (FWH1), CDRH1, CDRH2, and CDRL3 of 2-43 form hydrogen-bond networks with three RBD regions. In the first region, residues in FWH1 and the DE-loop in FWH3 form a groove to hold Y449RBD. Hydrogen bonds are observed between FWH1 residues and Y449RBD and Q498RBD (Figure 3A, second panel). In the second region, involving the “flat” region of RBM, T30HC forms a hydrogen bond with S494RBD. In the third region or the RBD ridge region, Y33HC, N52HC, and S54HC form a hydrogen-bond network with E484RBD. Light-chain residues Y91LC and S95LC further the hydrogen bond with G485RBD and F486RBD. In addition, Y30LC, Y32LC, Y91LC, and Y100jHC also form a hydrophobic pocket to hold F486RBD (Figure S3B), with π-π stacking observed between Y30LC and F486RBD (Figure 3A, right panel).
Figure 3.

Common epitope-paratope interactions define a VH1-2 antibody class
(A) Overview of the 2-43 epitope (left panel) and close-up view of the hydrogen bond networks between 2-43 and the SARS-CoV-2 RBD (right three panels). The RBD epitope recognized by 2-43 heavy (forest) and light (lime green) chains are colored wheat and orange, respectively (RBDA, light gray). Epitope residues interacting with both heavy and light chains are colored lemon. 2-43 also binds to the N343 glycan (magenta) from a neighboring RBD (RBDB, dark gray). Hydrogen bonds and π-π stacking are shown as black dashed lines.
(B) Overview of the epitope of 2-15 (left panel) and close-up view of the hydrogen bond networks and π-π stacking between 2-15 and the SARS-CoV-2 RBD (right three panels). 2-15 heavy and light chains are colored marine and blue, respectively. Epitope residues interacting with both heavy and light chains are colored lemon.
(C) Overview of the epitope of 2-4 (left panel) and close-up view of the hydrogen-bond networks between 2-4 and the SARS-CoV-2 RBD (right three panels). The heavy and light chains of 2-4 are colored chocolate and salmon, respectively. Epitope residues interacting with both heavy and light chains are colored lemon.
See also Figure S3.
The crystal structure of 2-15 complexed with SARS-CoV-2 RBD revealed 2-15 to use similar heavy- and light-chain regions to recognize the RBM (Figure 3B, left panel). Similar to 2-43, 2-15 forms convergent hydrogen bonds to Y449RBD, S494RBD, E484RBD, and G485RBD as well as π-π stacking with F486RBD (Figure 3B, right three panels). Nonetheless, 2-15 interactions that are different from 2-43 are also observed at the RBM flat and ridge regions (Figure 3B, right two panels).
Compared with 2-43 and 2-15, the published VH1-2 antibodies, 2-4, S2M11, and C121, showed a similar RBM approach angle and binding mode (Figures 3C, 3D, S3C, and S3D). Typically, interactions between the VH1-2 antibodies and the RBD are mediated predominantly by the heavy chain (RBDA column in Figure S3C). For all antibodies, the VH1-2-gene-encoded residues form a module for RBM recognition. Despite these antibodies having different light-chain gene origins, convergent tyrosine residues in CDRL1 and CDRL3 pack against F486RBD (Figure S3B), which constitutes another module for RBD recognition. Because each antibody has a unique CDRH3, no conserved polar or hydrophobic interaction is observed (Figures 3 and S3B–S3D), albeit CDRH3 contributes the most BSA among the CDRs, except in 2-15 (Figure S3A). Altogether, the germline gene residues from the VH1-2 gene as well as light-chain V genes anchor the antibodies to the RBM in a similar mode with the heavy-chain V region having a dominant role in determining the mode of recognition.
The VH1-2 antibody class and similarity to other SARS-CoV-2 RBD targeting antibodies
To gain an overall understanding of the similarity in the binding orientations of the VH1-2 antibodies, we superposed RBD and antibody complexes and calculated pairwise root-mean-square deviation (RMSD) to compare relative binding orientations of the six VH1-2 antibodies (2-43, 2-15, H4, 2-4, C121, and S2M11) as well as the 31 additional RBD-targeting antibodies originated from other VH genes. Overall, the VH1-2 antibodies have similar binding orientations (Figures 4 A, 4B, and S4A). Their epitopes overlap with the ACE2 binding site (Figures S4B and S4C). The similarity in their genetic origin, details of their interactions with RBD (unavailable for H4), and their angles of approach, suggests that these six antibodies form a VH1-2 antibody class.
Figure 4.

The VH1-2 antibody class
(A) Overview of modes of RBD recognition by the VH1-2 class antibodies: 2-43, 2-15, 2-4, H4, S2M11, and C121. The RBD and antibody complexes are superimposed on RBDs.
(B) Approach angles of SARS-CoV-2 RBD-targeting antibodies originated from different VH genes. Members of the VH1-2 antibody class have similar approach angles. The VH3-53 class antibody C144 has an angle of approach similar to that of the VH1-2 antibody class.
(C) Overview of VH1-2 signature motifs (green) and type II VH3-53 class (orange) on the RBD surface. The RBD is colored with the conservation score calculated from the circulating SARS-CoV-2 strains. The 2-43 structure is used to generate the conservation plot. Residues in gray have no mutation, whereas residues colored red have relatively high mutation frequency.
(D) Signature motifs and compatible germline V genes of the VH1-2 antibody class.
See also Figures S1 and S4.
The structural, recombination, and somatic hypermutation analyses (described below in the next section) presented here revealed critical residues that determine the specificity and binding affinity of the VH1-2 class antibodies. We define residues forming the conserved side chain polar and/or hydrophobic interactions as genetic signatures. The heavy-chain signatures include a T30-x-x-Y33 motif in CDRH1 and an [NS]52-x-[NIV]-S54 motif (x represents any amino acid) in CDRH2 (Figure 4C), with no conserved motif observed in CDRH3 (Figure S1A). Searches of germline gene databases using this signature showed only alleles of the VH1-2 gene to match both motifs (Figure 4E), suggesting that the antibody class is likely to be restricted to VH1-2.
For light chain, residues Y32LC and Y91LC are signature residues that interact with both the primary RBD (Figures 3 and S3B) and the N343 glycan from adjacent RBDs (described below in the quaternary recognition section). However, because the light-chain interaction signatures can be found in many germline genes (Figures 4D and S4D), the VH1-2 antibody class may not be restricted with respect to light-chain origin gene.
To understand the conservation of the VH1-2 antibody epitope, we calculated the conservation score for each RBD residue and observed that the flat region is highly conserved in the natural SARS-CoV-2 reservoir. The four RBD residues critical for VH1-2 antibody recognition—Y449RBD, E484RBD, F486RBD, and Q493RBD—showed low mutation frequencies (approximately 0, 3, 7, and 7 per 10,000 sequences, respectively). Nonetheless, SARS-CoV-2 strains with mutations at the four positions are resistant to C121 and S2M11 of the VH1-2 antibody class (Barnes et al., 2020a; Tortorici et al., 2020).
In addition, the VH1-2 antibodies show high similarity in approach angle and epitope to antibody C144 (Figures 4C and S4A), a type-II VH3-53/-66 antibody. However, C144 uses different CDRH1 and CDRH2 motifs for interacting with the conserved residues in the flat region and ridge of RBD (e.g., the N32-Y33 and [T/S]54-G-G-[T/S]57 motifs in the VH3-53/-66 antibodies) (Figure 4C) (Barnes et al., 2020a; Wu et al., 2020a). The CDRH2 of C144 shifts toward the RBD ridge, perhaps because the CDRH2s of the VH3-53/-66 genes are one residue shorter than the VH1-2 gene. Nonetheless, both antibody classes use similar light-chain genes, including VL2-14 and VL2-23, which provide key Tyr residues that interact with F486RBD.
Somatic hypermutations and avidity improve antibody potency
To understand the effects of somatic hypermutation (SHM) on binding affinity and neutralization potency, we reverted SHMs in the paratope regions of 2-43, 2-15, and 2-4 to their germline residues individually and in combination. The 2-43 only has one SHM in the paratope region, S76THC (Figure S1A); the reversion of which reduces the immunoglobulin G (IgG)-apparent binding affinity and pseudovirus neutralization potency by about 6-fold and 5-fold, respectively (Figures 5 A, 5B, and S4E). For 2-15, three paratope SHMs were observed: N53IHC, G55DHC, and Y32FLC. The reversion of N53IHC and Y32FLC individually reduced binding affinity by 56-fold and 12-fold, respectively (Figure 5A), as well as neutralization potency by 217-fold and 42-fold, respectively (Figure 5B). The results suggested that the N53IHC and F32YHC mutations are critical for the affinity maturation of 2-15. Structural analysis showed that the N53IHC mutation allows the Ile side chain to fit a hydrophobic pocket on RBD (Figures 3B and S5A). A convergent mutation is also observed in S2M11 (N53IHC) and C121 (N53VHC). We then introduced the N53IHC mutation to 2-43 and 2-4 and observed significant improvements of both binding affinity and neutralization potency (both IC50 and maximum potency) (Figure 5). These results suggested that mutation to a hydrophobic residue at 53HC can be used to improve members of the VH1-2 antibody class. The crystal structure further showed that 2-15 32LC does not interact with RBD (Figure S3B); the Y32FLC SHM may alter the interaction with RBD indirectly. For 2-4, the SHM A60THC generates an N-glycosylation site at position N58HC. The N58HC glycan interacts with the RBD ridge (Figure 3C). The reversion of this SHM reduces binding affinity and neutralization by about 6-fold and 9-fold, respectively (Figure 5). In summary, somatic hypermutation analysis revealed that the precursors of these antibodies could bind antigens with nanomolar-apparent binding affinity, suggesting that the precursors of these antibodies can be activated efficiently by the SARS-CoV-2 spike protein. Nonetheless, SHMs significantly improve both the binding affinity and the neutralization potency of these antibodies. Because the observed SHMs are frequently generated by the somatic hypermutation machinery (Figure S1A), we anticipate that requirements for somatic hypermutations are unlikely to present a significant barrier for affinity maturation of this antibody class.
Figure 5.

N53I mutation improves many VH1-2 class members
(A) Apparent binding affinities of 2-43, 2-15, and 2-4 IgGs and revertants. Somatic hypermutations improve the binding affinities of the three antibodies. N53IHC was introduced to 2-43 and 2-4 and showed a significantly increased binding affinity. The numbers in parentheses represent standard errors in the last integer.
(B) Pseudovirus neutralization profiles of 2-43, 2-15, 2-4, and their somatic hypermutation revertants. Data represent means ± SEM of technical triplicates.
See also Figures S1, S3, and S5.
In addition, previous studies showed that bivalent binding (avidity) is critical for certain RBD-directed antibodies to achieve high potency (Barnes et al., 2020b; Liu et al., 2020b). We, therefore, examined whether avidity contributes to the observed high potency of the VH1-2 antibody class. Comparison of neutralization potency between IgGs and Fabs revealed that the Fabs of 2-43, 2-15, and 2-4 have potencies of about 140-, 95-, and 14-fold less than their IgG forms, respectively (Figure S5C), suggesting that avidity effects are critical for achieving high-neutralization potency by the VH1-2 class antibodies.
Recognition of quaternary epitopes modulates spike conformation
Our previous study with a low-resolution cryo-EM structure showed that 2-43 recognizes a quaternary epitope (Liu et al., 2020a). However, details on the quaternary interactions and their functional relevance have not yet been characterized. Here, high-resolution cryo-EM reconstructions revealed atomic-level interactions between 2-43 and the quaternary epitope. Overall, 2-43 interacts comprehensively with N343 glycan as well as helix 364–371 from an adjacent down-RBD protomer (RBDB) (Figures 6 A and S6A), burying a total of 999-Å BSA (Figure S3A). The quaternary interaction is predominantly mediated by the long CDRH3, which is held by the two branches of the N343 glycan; the structure of which is highly flexible and has not been observed in previous studies. The tip of one N343 glycan branch is further stabilized by hydrogen bonding with the 2-43 CDRL1 and the ridge of RBDA (Figure S6A). Altogether, 2-43 forms strong quaternary interactions with the adjacent down RBD, which is critical for locking RBDs in the all-down conformation. In contrast to 2-43, the cryo-EM density maps of the 2-15 and H4 spike complexes revealed no quaternary contacts (Figures 2D, S2L, and 6B).
Figure 6.

Subset of the VH1-2 antibody class uses an elongated CDRH3 to recognize the glycan N343 from a neighboring RBD, a quaternary interaction that expands the SARS-CoV-2 spike
(A) Close-up view of the quaternary epitope of 2-43. The 2-43 CDRH3 forms two hydrogen-bond networks (black dashed lines) with an adjacent RBD (RBDB) N343 glycan and also interacts with helix 364–371 of RBDB.
(B) Close-up view of potential quaternary interactions between 2-15 and the neighboring RBD (RBDB). 2-15 reconstructed from cryo-EM data shows a minor quaternary interaction.
(C) Close-up view of the interaction between 2-4 and the N343 glycan of the neighboring RBD.
(D) Distance between antibody-free and antibody-bound trimeric RBDs. Antibody binding induces significantly larger expansion of the trimeric RBDs. The three RBDs are gray. Residues at the RBD interfaces are cyan, light blue, and blue for protomers RBDA, RBDB, and RBDC. respectively. The distances between RBD protomers was measured using Cα of position 503. Note: because of low resolution, the interface residues between RBDs were not shown for the 2-15 bound spike. 2-15 (dashed circle) binds to protomer RBDC.
(E) Distance of Cα of position 503 between the trimeric down RBDs. Antibody-free spikes have a shorter distance than the antibody-bound spikes. For the boxplots, the middle lines are medians. The lower and upper hinges correspond to the first and third quartiles, respectively. The Mann-Whitney U test was used. Outliers are shown as gray triangles.
See also Figures S2, S3, and S6.
Comparison of quaternary interactions among the five VH1-2-derived antibodies revealed two structural groups. Group 1 includes 2-43, 2-4, and S2M11, which bind predominantly down RBDs. Group 2 includes 2-15, H4, and C121, which bind both up and down or only up RBDs. For group 1, the quaternary recognition is mediated mostly by CDRH3, followed by CDRL1 and CDRL2 (Figures 6C and S3A). Differences between each antibody-specific CDRH3 determine that each antibody has a unique quaternary epitope (Figures 6A–C, S3B, and S6B). Different from other class members, which bind both the spike and isolated RBDs (Liu et al., 2020a; Robbiani et al., 2020; Tortorici et al., 2020; Wu et al., 2020b), the quaternary interaction is indispensable for 2-43 (Liu et al., 2020a). The quaternary epitope of 2-4, only part of which appears to be visible in the cryo-EM map (Liu et al., 2020a), comprises only the N343 glycan from the adjacent down-RBD protomer (Figure 6C), likely because 2-4 has a short CDRH3. The quaternary epitope of S2M11 includes the N343 glycan and helices 339–343 and 364–371 of the adjacent RBD (Figure S6B). Altogether, accommodation of the N343 glycan from the adjacent down RBD is a common feature of quaternary recognition by group-1 antibodies. The diverse CDRH3s of the group-1 antibodies form a module for quaternary recognition. In contrast, group-2 antibodies 2-15 and C121 show minor interactions with neighboring down RBDs (Figures 2C, 6B, S2R, and S3A), which may explain why they do not lock the trimeric RBDs in an all-down conformation.
To understand whether antibody binding induces conformation change in the SARS-CoV-2 spike protein, we compared the antibody-bound and ligand-free spike structures in the closed (all down) prefusion conformation. These comparisons showed that the binding of the VH1-2 antibodies (except S2M11), as well as other RBD-directed antibodies leads to significantly larger distances and less contacts between the trimeric down RBDs (Figures 6D and 6E). Each of these antibodies has a role in bridging the interactions between RBDs as well as in reorienting the conformation of the N343 glycan. However, antibodies like 2-15, which does not form strong interactions with the adjacent RBD, may disassemble the spike when three Fabs bind to the RBDs (Figures S2J and S2K). In addition, superposition of 2-15 on the down RBD in the ligand-free spike revealed the light chain to significantly clash with an adjacent up RBD (Figure S6D), suggesting members of the VH1-2 antibody class to either bind only down RBDs when a neighboring down RBD is available or push the neighboring up RBD away to bind down RBDs. Consistent with the first mechanism, we did not observe a neighboring up RBD adjacent to 2-15, and 2-4 bound down RBDs in the cryo-EM data. In contrast, C121 adopts the second mechanism through light-chain interaction with the adjacent up RBD (Figure S6D) (Barnes et al., 2020a). In summary, the quaternary interaction and angle of approach determine that the VH1-2 antibody class modulates the SARS-CoV-2 spike conformation when binding to a down RBD.
Discussion
In this study, we determined structures of three nAbs: 2-43, 2-15, and H4, which revealed a VH1-2 antibody class with a common RBD-binding mode and similar angles of approach. The VH1-2 antibody class uses two modules for spike recognition with the VH1-2-gene-encoded module for recognition of RBD and CDRH3 module for quaternary recognition. The VH1-2 antibody class has little or no restraint on CDRH3 length. The prevalence of the VH1-2 antibody class in numerous donors (Brouwer et al., 2020; Hansen et al., 2020; Ju et al., 2020; Kreer et al., 2020; Liu et al., 2020a; Robbiani et al., 2020; Rogers et al., 2020; Tortorici et al., 2020; Wu et al., 2020b; Zost et al., 2020) suggests it to be a common component of the effective antibody response to SARS-CoV-2, which can include highly potent neutralizing antibodies.
For some members of the VH1-2 class, recognition of a quaternary epitope can lock RBDs in the down conformation, providing an additional mechanism by which to achieve high neutralization potency. The quaternary epitopes of the VH1-2 class include the N343 glycan and helix 364–371 of an adjacent RBD. A similar quaternary epitope is recognized by the type-II VH3-53/-66 class antibody C144 (Barnes et al., 2020a). Thus, this quaternary epitope appears to represent a supersite at which antibodies lock the RBDs in the all-down conformation. We also observe that the quaternary interaction can induce distinct conformational changes of the trimeric down RBDs. The quaternary interaction observed in S2M11 does not significantly alter the distance between spike protomers (Figure 4E) (Tortorici et al., 2020). In contrast, the quaternary interactions of 2-43 and 2-4, mediated predominantly by CDRH3, increase the distance of the trimeric RBDs (Figure 4). For 2-15, the cryo-EM structure of one Fab-bound spike revealed a CDRH3 conformation moving away from the adjacent down RBD to avoid a steric clash. Despite that, 2-15 also increases the distance among RBDs, which may be the result of mobile quaternary contacts that cannot be observed in the low-resolution cryo-EM reconstruction. We anticipate that the interactions among RBDs could be further weakened when all three protomers are bound by 2-15, which may be the cause of the observed spike disassembly by 2-15 (Figures S2J and S2K). The disassembly of the spike may be another mechanism for 2-15 to achieve ultrapotent neutralization. In addition, the crystal structure of 2-15 in complex with isolated RBDs showed 2-15 to have another stable binding mode in the absence of a quaternary contact. We hypothesize that this binding mode may be observed when 2-15 binds to up RBDs and cannot form quaternary interactions.
The structural information we present also provides clues for further optimization of the VH1-2 antibody class. Despite members of the class achieving high potency with germline gene-mediated interactions, we observe that somatic hypermutations can further improve this class significantly. The conserved mode of RBD recognition implies that members of the antibody class will be sensitive to similar viral escape mutations. In particular, K417NRBD, L452RRBD, E484KRBD, and N501YRBD mutations are observed in emerging SARS-CoV-2 variants (e.g., B.1.351, B.1.1.7, and B.1.429) with transmission rates dramatically higher than that of preexisting strains (Davies et al., 2021; Tegally et al., 2020). We found that positions 452 and 484 are within the VH1-2 antibody epitopes (Figure S3E). Mutation E484KRBD, but not K417NRBD and N501YRBD, impairs both binding affinity and neutralization potency of the VH1-2 antibody class (Barnes et al., 2020a; Tortorici et al., 2020; Wang et al., 2021). We anticipate that L452RRBD will also impair the recognition by the VH1-2 antibody class, but further investigation is required to examine the hypothesis.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 2-43 | Liu et al., 2020b | N/A |
| 2-15 | Liu et al., 2020b | N/A |
| 2-4 | Liu et al., 2020b | N/A |
| H4 | Wu et al., 2020b | N/A |
| anti-VSV-G antibody | ATCC | Cat#CRL-2700; RRID:CVCL_G654 |
| Bacteria and virus strains | ||
| SARS-CoV-2 pseudovirus | Liu et al., 2020b | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| polyethylenimine | Polysciences, Inc. | Cat# 23966 |
| Turbo293 | Speed BioSystems | Cat#PXX1002 |
| HRV-3C protease | MilliporeSigma | Cat# SAE0045 |
| SARS-CoV-2 spike S2P | Wrapp et al., 2020 | N/A |
| SARS-CoV-2 spike | provided by Dr. Peihui Wang, China | N/A |
| SARS-CoV-2 RBD | This study | N/A |
| Critical commercial assays | ||
| Strep-Tactin XT Superflow 50% | Zymo research | Cat#P2004-1-5 |
| Sensor Chip CM5 | Cytiva | Cat#BR100030 |
| His Capture Kit | Cytiva | Cat#28995056 |
| Glycine 1.5 | Cytiva | Cat# BR100354 |
| HBS-EP+ Buffer | Cytiva | Cat# BR100826 |
| QuikChange II site directed mutagenesis kit | Agilent | Cat# 200524 |
| rProtein A Sepharose | GE | Cat#17-1279-01 |
| NI-NTA | GE Health care | Cat# 17-0921-09 |
| Superdex 200 Increase 10/300 GL | Cytiva | Cat# 28990945 |
| Pierce Fab Preparation Kit | Thermo fisher | Cat# 44985 |
| Deposited data | ||
| New IGVL2-14 gene allele (IGVL2-14∗0X) | This study | ENA: PRJEB31020 |
| Cryo-EM structures: SARS-CoV-2 spike | Walls et al., 2020 | PDB ID: 6VXX |
| Cryo-EM structures: SARS-CoV-2 spike | Henderson et al., 2020 | PDB ID: 6X2C |
| Cryo-EM structures: SARS-CoV-2 spike | Herrera et al., 2020 | PDB ID: 6X6P |
| Cryo-EM structures: SARS-CoV-2 spike | Henderson et al., 2020 | PDB ID: 6X29 |
| Cryo-EM structures: SARS-CoV-2 spike | McCallum et al., 2020 | PDB ID: 6X79 |
| Cryo-EM structures: SARS-CoV-2 spike | Zhou et al., 2020 | PDB ID: 6XLU |
| Cryo-EM structures: SARS-CoV-2 spike | Zhou et al., 2020 | PDB ID: 6XM5 |
| Cryo-EM structures: SARS-CoV-2 spike | Cai et al., 2020 | PDB ID: 6XR8 |
| Cryo-EM structures: SARS-CoV-2 spike | Toelzer et al., 2020 | PDB ID: 6ZB4 |
| Cryo-EM structures: SARS-CoV-2 spike | Wrobel et al., 2020 | PDB ID: 6ZGE |
| Cryo-EM structures: SARS-CoV-2 spike | Wrobel et al., 2020 | PDB ID: 6ZGI |
| Cryo-EM structures: SARS-CoV-2 spike | Xiong et al., 2020 | PDB ID: 6ZOX |
| Cryo-EM structures: SARS-CoV-2 spike | Xiong et al., 2020 | PDB ID: 6ZOY |
| Cryo-EM structures: SARS-CoV-2 spike | Xiong et al., 2020 | PDB ID: 6ZOZ |
| Cryo-EM structures: SARS-CoV-2 spike | Xiong et al., 2020 | PDB ID: 6ZP0 |
| Cryo-EM structures: SARS-CoV-2 spike | Xiong et al., 2020 | PDB ID: 6ZP1 |
| Cryo-EM structures: SARS-CoV-2 spike | Xiong et al., 2020 | PDB ID: 6ZP2 |
| Cryo-EM structures: SARS-CoV-2 spike | Bangaru et al., 2020 | PDB ID: 7JJI |
| Cryo-EM structures: SARS-CoV-2 spike | Wrobel et al., 2020 | PDB ID: 6ZGG |
| Crystal structure: SARS-CoV-2 RBD | Wu et al., 2020b | PDB ID: 7BZ5 |
| Crystal structure: CH65 UCA | Schmidt et al., 2013 | PDB ID: 4HK0 |
| Cryo-EM structure: 2-4 | Liu et al., 2020b | PDB ID: 6XEY |
| Cryo-EM structure: S2M11 | Tortorici et al., 2020 | PDB ID:7K43 |
| Cryo-EM structure: C121 | Barnes et al., 2020a | PDB ID: 7K8X |
| Cryo-EM structure: C002 | Barnes et al., 2020a | PDB ID: 7K8T |
| Cryo-EM structure: C144 | Barnes et al., 2020a | PDB ID: 7K90 |
| Cryo-EM structure: BD23 | Cao et al., 2020 | PDB ID: 7BYR |
| Cryo-EM structure: B38 | Wu et al., 2020b | PDB ID: 7BZ5 |
| Cryo-EM structure: S2H13 | Tortorici et al., 2020 | PDB ID: 7JV2 |
| Cryo-EM structure: S309 | Pinto et al., 2020a | PDB ID: 6WPS |
| Cryo-EM structure: C119 | Barnes et al., 2020a | PDB ID: 7K8W |
| Cryo-EM structure: C102 | Barnes et al., 2020a | PDB ID: 7K8M |
| Cryo-EM structure: C104 | Barnes et al., 2020a | PDB ID: 7K8U |
| Cryo-EM structure: C110 | Barnes et al., 2020a | PDB ID: 7K8V |
| Cryo-EM structure: S2E12 | Tortorici et al., 2020 | PDB ID:7K45 |
| Cryo-EM structure: C135 | Barnes et al., 2020a | PDB ID: 7K8Z |
| Cryo-EM structure: COVA1-16 | Liu et al., 2020a | PDB ID: 7JMW |
| Cryo-EM structure: P17 | Yao et al., 2021 | PDB ID: 7CWO |
| Crystal structure: BD-236 | Du et al., 2020 | PDB ID: 7CHB |
| Crystal structure: BD-368-2 | Du et al., 2020 | PDB ID: 7CHE |
| Crystal structure: BD-629 | Du et al., 2020 | PDB ID: 7CH5 |
| Crystal structure: P2C-1F11 | Wang et al., 2020 | PDB ID: 7CDI |
| Crystal structure: P2D-1A3 | Wang et al., 2020 | PDB ID: 7CDJ |
| Crystal structure: EY6A | Zhou et al., 2020 | PDB ID: 6ZER |
| Crystal structure: CV07-250 | Kreye et al., 2020 | PDB ID: 6XKQ |
| Crystal structure: CV07-270 | Kreye et al., 2020 | PDB ID: 6XKP |
| Cryo-EM structure: S309 | Pinto et al., 2020b | PDB ID: 6WPT |
| Crystal structure: P2B-2F6 | Ju et al., 2020 | PDB ID: 7BWJ |
| Crystal structure: CB6 | Shi et al., 2020 | PDB ID: 7C01 |
| Cryo-EM structure: BD-23 | Cao et al., 2020 | PDB ID: 7BYR |
| Crystal structure: B38 | Wu et al., 2020b | PDB ID: 7BZ5 |
| Crystal structure: COVA2-39 | Wu et al., 2020a | PDB ID: 7JMP |
| Crystal structure: COVA2-04 | Wu et al., 2020a | PDB ID: 7JMO |
| Crystal structure: H014 | Lv et al., 2020 | PDB ID: 7CAH |
| Crystal structure: CR3022 | Huo et al., 2020 | PDB ID: 6YM0 |
| Crystal structure: CV30 | Hurlburt et al., 2020 | PDB ID: 6XE1 |
| Cryo-EM structure: REGN10933 | Hansen et al., 2020 | PDB ID: 6XDG |
| Cryo-EM structure: REGN10987 | Hansen et al., 2020 | PDB ID: 6XDG |
| Cryo-EM structure: C105 | Barnes et al., 2020b | PDB ID: 6XCN |
| Cryo-EM structure: CC12.1 | Yuan et al., 2020 | PDB ID: 6XC2 |
| Cryo-EM structure: CC12.3 | Yuan et al., 2020 | PDB ID: 6XC4 |
| Crystal structure: 2-15 | This study | PDB ID: 7L5B |
| Cryo-EM structure: 2-43 | This study | PDB ID: 7L56; EMDB-23165 |
| Cryo-EM structure: 2-15 | This study | PDB ID: 7L57; EMDB-23166 |
| Cryo-EM structure: H4 | This study | PDB ID: 7L58; EMDB-23167 |
| Experimental models: Cell lines | ||
| Expi293 | ThermoFisher | Cat#A14527; RRID:CVCL_D615 |
| HEK293T/17 | ATCC | Cat# CRL-11268; RRID:CVCL_1926 |
| Vero E6 | ATCC | Cat# CRL-1586; RRID:CVCL_0574 |
| Expi293F GnTI- Cells | Thermo Fisher | Cat# A39240 |
| Recombinant DNA | ||
| pVRC8400 vector | https://www.addgene.org | Cat#63160; RRID:Addgene_63164 |
| gWiz | Aldeveron | Cat# 5008 |
| pcDNA3.4 | ThermoFisher | Cat# A14697 |
| Software and algorithms | ||
| GraphPad Prism 7.01 Software | GraphPad Prism Software, Inc. | N/A |
| The PyMol Molecular Graphics System, v1.8.6 | Schrödinger, LLC | https://pymol.org/2/ |
| IMGT/V-Quest | IMGT | http://www.imgt.org/IMGT_vquest/vquest |
| SONAR | Schramm et al., 2016 | https://github.com/scharch/SONAR |
| Muscle | Edgar, 2004 | https://drive5.com/usearch/ |
| CLUSTALO | Sievers and Higgins, 2018 | http://clustal.org/omega/ |
| Python v3.8.3 | https://www.python.org/downloads/ | N/A |
| cryoSPARC, v2.15.0 | Punjani et al., 2017 | https://cryosparc.com/ |
| UCSF Chimera, v1.14 | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| UCSF ChimeraX, v1.1 | Goddard et al., 2018 | https://www.cgl.ucsf.edu/chimerax/ |
| Schrödinger Release 2020-2:BioLuminate | Zhu et al., 2014 | https://www.schrodinger.com/products/bioluminate |
| ISOLDE, v.1.0.1 | Croll, 2018 | https://isolde.cimr.cam.ac.uk/ |
| COOT, 0.9 | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix, v1.18 | Adams et al., 2010 | https://www.phenix-online.org/; |
| Leginon, v.3.5beta | Suloway et al., 2005 | N/A |
| XDS | Kabsch, 2010 | https://xds.mpimf-heidelberg.mpg.de/ |
| CCP4i (Aimless) | Winn et al., 2011 | https://www.ccp4.ac.uk |
| PDB-redo | Joosten et al., 2014 | https://pdb-redo.eu/ |
| MolProbity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| Other | ||
| Free style Media | Invitrogen | Cat # 12338026 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zizhang Sheng (zs2248@cumc.columbia.edu).
Materials availability
Expression plasmids generated in this study for expressing SARS-CoV-2 proteins and antibody mutants will be shared upon request.
Data and code availability
Cryo-EM maps and fitted coordinates of 2-43, H4, and 2-15 were deposited to the EMDB (2-43: EMDB-23165; H4: EMDB-23167; 2-15: EMDB-23166) and RCSB (2-43: 7L56; H4: 7L58; 2-15: 7L57 and 7L5B) respectively. The new IGVL2-14 gene allele (IGVL2-14∗0X) was deposited to European Nucleotide Archive (ENA): PRJEB31020. All deposited data will be available upon publication of the study.
Experimental model and subject details
Expi293 cells and Expi293F GnTI- Cells were from ThermoFisher Scientific Inc (ThermoFisher, cat#A14527 and cat# A39240 respectively). HEK293T/17(cat# CRL-11268) and Vero E6 cells (cat# CRL-1586) were from ATCC.
Method details
Expression and purification of SARS-CoV-2 spike and RBD proteins
The mammalian expression vector that encodes the ectodomain of the SARS-CoV-2 S trimer was kindly provided by Jason McLellan (Wrapp et al., 2020). SARS-CoV-2 S trimer expression vector was transiently transfected into Expi293TM cells using 1 mg/mL of polyethylenimine (Polysciences). Five days post transfection, the S trimer was purified using Strep-Tactin XT Resin (Zymo Research).
The SARS-CoV-2 RBD (residues 331-528), was cloned into the pVRC-8400 mammalian expression plasmid, with a C-terminal 6X-His-tag and an intervening HRV-3C protease cleavage site. Expression vector was transiently transfected into Human Embryonic Kidney (HEK) 293 GnTI- Freestyle cells suspension culture in serum-free media (Invitrogen) using polyethyleneimine (Polysciences). Media was harvested 4 days after transfection and the secreted protein purified using Ni-NTA IMAC Sepharose 6 Fast Flow resin (GE Healthcare) followed by size exclusion chromatography (SEC) on Superdex 200 (GE Healthcare) in 10 mM, Tris pH 8.0, 150 mM NaCl. Peak fractions containing RBD were pooled and HRV-3C protease (Thermo fisher, cat# SAE0045) was added in a mass ratio 1:100 relative to RBD, followed by incubation for 24 h at 20°C, to remove the C-terminal histidine-tag. The protein solution was again passed through the NI-NTA column to remove the His-tag and any traces of uncleaved protein. Protein purity was analyzed by SDS-PAGE and buffer exchanged to SEC buffer and concentrated to ∼5 mg/mL and used for crystallization experiments.
Antibody production and mutagenesis
For each antibody, the variable genes were optimized for human cell expression and synthesized by GenScript. VH and VL were inserted separately into plasmids (gWiz or pcDNA3.4) that encoding the constant region for heavy chain and light chain respectively. Monoclonal antibodies were expressed in Expi293 (ThermoFisher, A14527) by co-transfecting heavy chain and light chain expressing plasmids using polyethylenimine (PEI, Linear, MV∼25,000, Polysciences, Inc. Cat. No. 23966) and culture in 37.0C degree shaker at 125RPM and 8% CO2. Supernatants were collected on day 5, antibodies were purified by rProtein A Sepharose (GE, 17-1279-01) affinity chromatography.
For H4, the antibody expression constructs were synthesized (Gene Universal Inc, Newark DE) and subcloned into corresponding pVRC8400 vectors. To express the antibodies, equal among of heavy and light chain plasmid DNA were transfected into Expi293F cells (Life Technology) by using Turbo293 transfection reagent (Speed BioSystems). The transfected cells were cultured in shaker incubator at 120 rpm, 37°C, 9% CO2 for 5 days. Culture supernatants were harvested and purified over Protein A (GE Health Science) resin in columns. Each antibody was eluted with IgG elution buffer (Pierce), immediately neutralized with one tenth volume of 1M Tris-HCL pH 8.0. The antibodies were then buffer exchanged in PBS by dialysis.
Antibody gene mutations were introduced by QuikChange II site directed mutagenesis kit (Agilent, cat#200524)
Production of Fab from IgG
Fab fragments were produced from purified IgGs of monoclonal antibodies 2-15, 2-43 and H4 by digestion with Papain in the presence of the reducing agent 30 mM cysteine and were purified by affinity chromatography on protein A following the manufacturer’s protocols (Thermo fisher). The purity of the resultant Fabs was analyzed by SDS-PAGE, and buffer exchanged into SEC buffer (10 mM Tris pH 7.4 and 150 mM NaCl) for crystallization experiments.
IgG Surface Plasmon Resonance binding experiments
SPR binding assays for IgGs binding to spike, were performed using a Biacore T200 biosensor, equipped with a Series S CM5 chip (cat# BR100030, Cytiva, MA), in a running buffer of 10mM HEPES pH 7.4, 150mM NaCl, 3 mM EDTA, 0.05% P-20 (HBS-EP+ buffer, Cytiva, MA) at 25°C. SARS-CoV-2 spike protein was captured through its C-terminal his-tag over an anti-his antibody surface. These surfaces were generated using the His-capture kit (Cat#: 28995056, Cytiva, MA) according to the instructions of the manufacturer, resulting in approximately 10,000 RU of anti-his antibody over each surface. Spike protein was captured over a single flow cell at a capture level of 125-200 RU. An anti-his antibody surface was used as a reference flow cell to remove bulk shift changes from the binding signal.
IgGs were tested using a three-fold dilution series of IgGs with concentrations ranging from 33.3 nM to 1.2 nM. The association and dissociation rates were each monitored for 55 s and 300 s respectively, at 50 μL/min. The bound spike/IgG complex was regenerated from the anti-his antibody surface using 10 mM Glycine pH 1.5. Blank buffer cycles were performed by injecting running buffer instead of IgG to remove systematic noise from the binding signal. The resulting data was processed and fit to a 1:1 binding model using Biacore Evaluation Software.
Pseudovirus neutralization assays
Recombinant Indiana VSV (rVSV) expressing SARS-CoV-2 spikes were generated as previously described (Han et al., 2021; Liu et al., 2020b). HEK293T cells were grown to 80% confluency before transfection with pCMV3-SARS-CoV-2-spike (kindly provided by Dr. Peihui Wang, Shandong University, China) using FuGENE 6 (Promega). Cells were cultured overnight at 37°C with 5% CO2. The next day, medium was removed and VSV-G pseudo-typed ΔG-luciferase (G∗ΔG-luciferase, Kerafast) was used to infect the cells in DMEM at an MOI of 3 for 1 hr before washing the cells with 1X DPBS three times. DMEM supplemented with anti-VSV-G antibody (I1, mouse hybridoma supernatant from CRL-2700; ATCC) was added to the infected cells and they were cultured overnight as described above. The next day, the supernatant was harvested and clarified by centrifugation at 300 g for 10 min and aliquots stored at −80°C.
Neutralization assays were performed by incubating pseudoviruses with serial dilutions of antibodies, and scored by the reduction in luciferase gene expression. In brief, Vero E6 cells were seeded in a 96-well plate at a concentration of 2 × 104 cells per well. Pseudoviruses were incubated the next day with serial dilutions of the test samples in triplicate for 30 mins at 37°C. The mixture was added to cultured cells and incubated for an additional 24 hr. The luminescence was measured by Britelite plus Reporter Gene Assay System (PerkinElmer). IC50 was defined as the dilution at which the relative light units were reduced by 50% compared with the virus control wells (virus + cells) after subtraction of the background in the control groups with cells only. The IC50 values were calculated using non-linear regression in GraphPad Prism.
Antibody gene assignments and genetic analyses
The 158 SARS-COV-2 neutralizing antibodies were collected from ten publications. We annotated these antibodies using IgBLAST-1.16.0 with the default parameters (Ye et al., 2013). For antibodies which have cDNA sequences deposited, the V and J genes were assigned using SONAR version 2.0 (https://github.com/scharch/sonar/) with germline gene database from IMGT (Lefranc et al., 2009; Schramm et al., 2016). For each antibody, the N-addition, D gene, and P-addition regions were annotated by IMGT V-QUEST (Brochet et al., 2008). To identify somatic hypermutations, each antibody sequence was aligned to the assigned germline gene using MUSCLE v3.8.31 (Edgar, 2004). Somatic hypermutations were identified from the alignment. In addition, the analysis of single cell antibody repertoire sequencing data of SARS-CoV-2 patient 2 from Liu et al. (2020b), from which 2-15 and 2-43 were isolated, showed that 29 of the 38 unique transcripts assigned to IGLV2-14∗01 share nucleotide mutations G156T and T165G. These mutations lead to amino acid mutations E50D and N53K. Both nucleotide mutations are also observed in 82 of 90 unique IGLV2-14 transcripts from patient 1 of the same study. Because these transcripts having different VJ recombination and paired with different heavy chain genes, the chances that the two convergent mutations are the results of somatic hypermutation are very low. Thus, we suspect that both donors contain a new IGVL2-14 gene allele (IGVL2-14∗0X), which was deposited to European Nucleotide Archive (ENA) with project accession numbers: PRJEB31020. 2-43 and 2-15 were assigned to the IGLV2-14∗0X allele.
Cryo-EM data collection and processing
For mAb 2-43, SARS-CoV-2 spike protein at a concentration of 2 mg/ml was incubated with six-fold molar excess per spike trimer of the antibody Fab fragments for 30 minutes in 10 mM sodium acetate pH 5.5, 150 mM NaCl, and 0.005% n-dodecyl-β-D-maltoside (DDM). 2μL of the sample was incubated on C-flat 1.2/1.3 carbon grids for 30 s and vitrified using a Leica EM GP. Data were collected on a Titan Krios electron microscope operating at 300 kV, equipped with a Gatan K3 direct electron detector and energy filter, using the Leginon software package (Suloway et al., 2005). A total electron fluence of 51.69 e-/Å2 was fractionated over 40 frames, with a total exposure time of 2.0 s. A magnification of 81,000x resulted in a pixel size of 1.058 Å, and a defocus range of −0.4 to −3.5 μm was used.
For mAb 2-15, SARS-CoV-2 spike protein at a concentration of 1 mg/ml was incubated with a molar ratio of 1:1 Fab fragments to spike trimer for 30 minutes in 10 mM sodium acetate pH 5.5, 150 mM NaCl, and 0.005% % DDM. 2μL of the sample was incubated on C-flat 1.2/1.3 carbon grids for 30 s and vitrified using a Leica EM GP. Data were collected on a Titan Krios electron microscope operating at 300 kV, equipped with a Gatan K3 direct electron detector and energy filter, using the Leginon software package (Suloway et al., 2005). A total electron fluence of 52.40 e-/Å2 was fractionated over 60 frames, with a total exposure time of 3.0 s. A magnification of 81,000x resulted in a pixel size of 1.070 Å, and a defocus range of −0.8 to −3.4 μm was used.
For mAb H4, SARS-CoV-2 spike protein at a concentration of 1 mg/ml was incubated with eight-fold molar excess per spike trimer of the antibody Fab fragments for 30 minutes in 10 mM sodium acetate pH 5.5, 150 mM NaCl, and 0.005% % DDM. 2μL of the sample was incubated on C-flat 1.2/1.3 carbon grids for 30 s and vitrified using a Leica EM GP. Data were collected on a Titan Krios electron microscope operating at 300 kV, equipped with a Gatan K3 direct electron detector and energy filter, using the Leginon software package. A total electron fluence of 42.00 e-/Å2 was fractionated over 40 frames, with a total exposure time of 3.0 s. A magnification of 81,000x resulted in a pixel size of 1.070 Å, and a defocus range of −0.5 to −3.5 μm was used.
All processing was done using cryoSPARC v2.15.0 (Punjani et al., 2017). Raw movies were aligned and dose-weighted using patch motion correction, and the CTF was estimated using patch CTF estimation. A small subset of approximately 200 micrographs were picked using blob picker, followed by 2D classification and manual curation of particle picks, and used to train a Topaz neural network. This network was then used to pick particles from the remaining micrographs, which were extracted with a box size of 384 pixels.
For the mAb 2-43 dataset, 2D classification followed by ab initio modeling and 3D heterogeneous refinement revealed 61,434 particles with three Fabs bound, one to each RBD. A reconstruction of these particles with imposed C3 symmetry resulted in a 3.78 Å map, as determined by gold standard Fourier shell correlation (FSC). Symmetry expansion followed by masked local refinement was used to obtain a 3.81 Å map of the Fab and RBD. The remainder of the S trimer was subjected to local refinement to obtain a 3.61 Å map. These two separate local refinements were aligned and combined using the vop maximum function in UCSF Chimera (Pettersen et al., 2004). This was repeated for the half maps, which were used along with the refinement mask from the global non-uniform refinement to calculate the 3D FSC (ref) and obtain an estimated resolution of 3.60 Å. All maps have been submitted to the EMDB with the ID EMDB-23165.
For the mAb 2-15 dataset, 2D classification followed by ab initio modeling and 3D heterogeneous refinement revealed 16,590 particles with one Fab bound to an RBD in the ‘down’ conformation and 21,456 particles with one Fab bound to an RBD in the ‘up’ conformation. The particles with Fab bound to RBD down were refined using Non-uniform refinement and C1 symmetry to a global resolution of 5.73 Å as determined by gold standard FSC. The RBD and Fab were masked and subjected to local refinement to obtain a map at 6.21 Å. The remainder of the trimer was also refined locally to 5.63 Å. A consensus map was obtained as described for mAb 2-43, with a resolution of 5.87 Å. All maps have been submitted to the EMDB with the ID EMDB-23166.
For the mAb H4 dataset, 2D classification followed by ab initio modeling and 3D heterogeneous refinement revealed 102,290 particles with one Fab bound to an RBD in the ‘up’ conformation. No classes with Fab bound to ‘down’ RBD were identified. 3D Variability Analysis was used to visualize the significant conformational heterogeneity of the ‘up’ RBD. Using the first and last frames of the reaction coordinate as reference maps, representing the extremes of the orientations adopted by the RBD, heterogeneous refinement was repeated to separate the Fab-bound spikes into more homogeneous classes. 56,080 particles adopted a more stable conformation and were refined to 4.78 Å using homogeneous refinement and C1 symmetry. Like the previously described datasets, the Fab and RBD were refined locally to 5.03 Å, with the remainder of the S trimer being refined to 4.32 Å. The final consensus map was estimated to have a resolution of 5.07 Å. All maps have been submitted to the EMDB with the ID EMDB-23167.
Cryo-EM model building
An initial homology model of the 2-43 Fab was built using Schrödinger Release 2020-2:BioLuminate (Zhu et al., 2014) and of H4 using ABodyBuilder (Leem et al., 2016). For mAb 2-15, the crystal structure determined here (PDB: 4HK0) was used as a starting model for the Fab variable domain and the associated RBD. For all models, the S trimer was modeled using the coordinates from PDB ID 6XEY. These models were docked into the consensus map using Chimera. The model was then fitted interactively using ISOLDE 1.0.1 (Croll, 2018) and COOT 0.8.9.2 (Emsley and Cowtan, 2004) and using real space refinement in Phenix 1.18 (Adams et al., 2004). For Fab 2-43, in cases were side chains were not visible in the experimental data, they were truncated to alanine except for residues very close to the RBD:Fab interface. Both H4 and 2-15 were built as poly-alanine models due to the low resolution of the experimental data. Validation was performed using Molprobity (Davis et al., 2004) and EMRinger (Barad et al., 2015). Models were submitted to the PDB with the following IDs: mAb 2-43 is 7L56, mAb H4 is 7L58, and 2-15 is 7L57. Figures were prepared using UCSF ChimeraX (Goddard et al., 2018).
X-ray data collection, structure solution, model building and refinement
For determination of the complex of with RBD, the two proteins were mixed at 1:1 molar ratio and incubated at 4.0°C for 60 min. RBD:2-15 complex was then isolated by gel filtration on Superdex-200 (GE Healthcare). Fractions containing complexes were pooled and concentrated to 6.5 mg/ml in SEC buffer. Screening for initial crystallization conditions was carried out in 96-well sitting drop plates using the vapor-diffusion method with a Mosquito crystallization robot (TTP LabTech) using various commercially available crystallization screens. Diffracting crystals were obtained from 0.1 M HEPES pH 7.5 and 70% MPD. Crystals were directly frozen and X-ray diffraction data was collected to 3.18 Å resolution at 100 K from a single flash-cooled crystal on beamline ID-C at the Advance Photon Source (APS) at Argonne National Laboratory. Diffraction data were processed with XDS (Kabsch, 2010) and scaled using AIMLESS (Evans, 2006) from the CCP4 software suite (Collaborative Computational ProjectNumber 4, 1994). Molecular replacement was performed with Phaser (McCoy et al., 2007), with previously reported RBD structure (PDB 7BZ5) and Fab (PDB code, 4HK0) as search models. Manual rebuilding of the structure using COOT (Emsley et al., 2010) was alternated with refinement using Phenix refine (Afonine et al., 2012) and PDB-REDO (Joosten et al., 2014). The Molprobity server was used for structure validation and PyMOL (version 2.1, Schrödinger, LLC) for structure visualization (Chen et al., 2010). A summary of the X-ray data collection and refinement statistics are shown in Table S2. PISA was used to identify paratope-epitope interfaces and to calculate buried surface area (Krissinel and Henrick, 2007). Hydrogen bonds were identified with a cutoff of 3.8Å. 2-15 model was submitted to the PDB with the following IDs: 7L5B.
Calculation of angle of approach
To measure the RBD approaching angle of antibodies, we first identify shared epitope residues among the five members of the VH1-2 antibody class. PyMOL was used to determine the center of mass of the shared epitope residues. We then determined the center of mass for heavy (the center of mass of the conserved Cys at 22 and 92) and light chains (the center of mass of the conserved Cys at 23 and 88). The heavy and light chain approaching angle was determined by linking the chain center of mass to the center of mass of the shared epitope. PyMOL was used to make structure figures. For antibody B38, the epitope center of mass was determined using the epitope residues of the antibody.
Quantification and statistical analysis
The statistical analyses for the pseudovirus neutralization assessments were performed using GraphPad Prism. The SPR data were fitted using Biacore Evaluation Software. Cryo-EM data were processed and analyzed using CryoSparc and Relion. Cryo-EM structural statistics were analyzed with Phenix and Molprobity. Statistical details of experiments are described in Method details or figure legends.
Acknowledgments
Cryo-EM data collection was performed at the National Center for Cryo-EM Access and Training and the Simons Electron Microscopy Center located at the New York Structural Biology Center, supported by the National Institutes of Health (NIH) Common Fund Transformative High Resolution Cryo-Electron Microscopy program (U24 GM129539) and by grants from the Simons Foundation (SF349247) and the NY State Assembly. Some of this work was also performed at the Columbia University Cryo-Electron Microscopy Center. Data analysis was performed at the National Resource for Automated Molecular Microscopy (NRAMM), supported by the NIH National Institute of General Medical Sciences (GM103310). We thank D. Neau, S. Banerjee, and Igor Kourinov for help with synchrotron data collection conducted at the Northeastern Collaborative Access Team 24-ID-C beamline, which is funded by the National Institute of General Medical Sciences from the NIH (P30 GM124165). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract no. DE-AC02-06CH11357. Support for this work was provided by the Intramural Research Program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases (NIAID), NIH. This study was supported by Samuel Yin, Pony Ma, Peggy, Andrew Cherng, Brii Biosciences, Jack Ma Foundation, JBP Foundation, Carol Ludwig, and Roger and David Wu.
Author contributions
M.R. and G.C. determined the cryo-EM structures of 2-43, 2-15, and H4; Y.G. performed bioinformatics analyses; E.R.R. solved the crystal structure of 2-15; L.L. performed surface plasmon resonance (SPR) assay; P.W. performed the neutralization assessment; J.Y. produced antibodies; P.K. estimated binding affinity data from SPR; J.S.B., F.A.B., S.M.M., and B.Z. produced proteins; Y.H. and D.D.H. supervised antibody production, SPR, and neutralization assays; P.D.K. supervised reagents; L.S. supervised cryo-EM and crystal structure studies; Z.S. supervised the informatics studies. L.S. and Z.S. oversaw the project and, with M.R., Y.G., E.R., L.L., P.W., J.Y., and B.Z., wrote the manuscript, with all authors providing revisions and comments.
Declaration of interests
D.D.H., Y.H., J.Y., L.L., and P.W. are inventors of a patent describing antibodies 2-43, 2-4, and 2-15 reported on here.
Published: March 19, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.108950.
Supplemental information
References
- Adams P.D., Gopal K., Grosse-Kunstleve R.W., Hung L.W., Ioerger T.R., McCoy A.J., Moriarty N.W., Pai R.K., Read R.J., Romo T.D., et al. Recent developments in the PHENIX software for automated crystallographic structure determination. J. Synchrotron Radiat. 2004;11:53–55. doi: 10.1107/s0909049503024130. [DOI] [PubMed] [Google Scholar]
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W., et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru S., Ozorowski G., Turner H.L., Antanasijevic A., Huang D., Wang X., Torres J.L., Diedrich J.K., Tian J.H., Portnoff A.D., et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science. 2020;370:1089–1094. doi: 10.1126/science.abe1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad B.A., Echols N., Wang R.Y., Cheng Y., DiMaio F., Adams P.D., Fraser J.S. EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods. 2015;12:943–946. doi: 10.1038/nmeth.3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C.O., Jette C.A., Abernathy M.E., Dam K.A., Esswein S.R., Gristick H.B., Malyutin A.G., Sharaf N.G., Huey-Tubman K.E., Lee Y.E., et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687. doi: 10.1038/s41586-020-2852-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes C.O., West A.P., Jr., Huey-Tubman K.E., Hoffmann M.A.G., Sharaf N.G., Hoffman P.R., Koranda N., Gristick H.B., Gaebler C., Muecksch F., et al. Structures of human antibodies bound to SARS-CoV-2 spike reveal common epitopes and recurrent features of antibodies. Cell. 2020;182:828–842.e16. doi: 10.1016/j.cell.2020.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brochet X., Lefranc M.P., Giudicelli V. IMGT/V-QUEST: the highly customized and integrated system for IG and TR standardized V-J and V-D-J sequence analysis. Nucleic Acids Res. 2008;36 doi: 10.1093/nar/gkn316. W503-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer P.J.M., Caniels T.G., van der Straten K., Snitselaar J.L., Aldon Y., Bangaru S., Torres J.L., Okba N.M.A., Claireaux M., Kerster G., et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369:643–650. doi: 10.1126/science.abc5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y., Zhang J., Xiao T., Peng H., Sterling S.M., Walsh R.M., Rawson S., Rits-Volloch S., Chen B. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020:eabd4251. doi: 10.1126/science.abd4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y., Su B., Guo X., Sun W., Deng Y., Bao L., Zhu Q., Zhang X., Zheng Y., Geng C., et al. Potent Neutralizing Antibodies against SARS-CoV-2 Identified by High-Throughput Single-Cell Sequencing of Convalescent Patients’ B Cells. Cell. 2020;182:73–84.e16. doi: 10.1016/j.cell.2020.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., III, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P., Nirula A., Heller B., Gottlieb R.L., Boscia J., Morris J., Huhn G., Cardona J., Mocherla B., Stosor V., et al. SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N. Engl. J. Med. 2020;384:229–237. doi: 10.1056/NEJMoa2029849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Croll T.I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N., Barnard C.R., Jarvis I.C., Kucharski J.A., Munday D.J., Pearson A.B.C., Russell W.T., Tully D.C., Abbott S., Gimma A., et al. Estimated transmissibility and severity of novel SARS-CoV-2 variant of concern 202012/01 in England. medRxiv. 2021 doi: 10.1101/2021.02.01.21250959. [DOI] [Google Scholar]
- Davis I.W., Murray L.W., Richardson J.S., Richardson D.C. MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32 doi: 10.1093/nar/gkh398. W615-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diemert D., McElrath J. A phase I trial to evaluate the safety and immunogenicity of eOD-GT8 60mer vaccine, adjuvanted. ClinicalTrials.gov. 2018 [Google Scholar]
- Dong E., Du H., Gardner L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020;20:533–534. doi: 10.1016/S1473-3099(20)30120-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du S., Cao Y., Zhu Q., Yu P., Qi F., Wang G., Du X., Bao L., Deng W., Zhu H., et al. Structurally Resolved SARS-CoV-2 Antibody Shows High Efficacy in Severely Infected Hamsters and Provides a Potent Cocktail Pairing Strategy. Cell. 2020;183:1013–1023.e13. doi: 10.1016/j.cell.2020.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R.C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt S.A., Zehner M., Krähling V., Cohen-Dvashi H., Kreer C., Elad N., Gruell H., Ercanoglu M.S., Schommers P., Gieselmann L., et al. Polyclonal and convergent antibody response to Ebola virus vaccine rVSV-ZEBOV. Nat. Med. 2019;25:1589–1600. doi: 10.1038/s41591-019-0602-4. [DOI] [PubMed] [Google Scholar]
- Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 2006;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E. UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci. 2018;27:14–25. doi: 10.1002/pro.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y., Duan X., Yang L., Nilsson-Payant B.E., Wang P., Duan F., Tang X., Yaron T.M., Zhang T., Uhl S., et al. Identification of SARS-CoV-2 inhibitors using lung and colonic organoids. Nature. 2021;589:270–275. doi: 10.1038/s41586-020-2901-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen J., Baum A., Pascal K.E., Russo V., Giordano S., Wloga E., Fulton B.O., Yan Y., Koon K., Patel K., et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science. 2020;369:1010–1014. doi: 10.1126/science.abd0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes B.F., Kelsoe G., Harrison S.C., Kepler T.B. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat. Biotechnol. 2012;30:423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R., Edwards R.J., Mansouri K., Janowska K., Stalls V., Gobeil S.M.C., Kopp M., Li D., Parks R., Hsu A.L., et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020;27:925–933. doi: 10.1038/s41594-020-0479-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera N.G., Morano N.C., Celikgil A., Georgiev G.I., Malonis R.J., Lee J.H., Tong K., Vergnolle O., Massimi A.B., Yen L.Y., et al. Characterization of the SARS-CoV-2 S Protein: Biophysical, Biochemical, Structural, and Antigenic Analysis. ACS Omega. 2020;6:85–102. doi: 10.1021/acsomega.0c03512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo J., Zhao Y., Ren J., Zhou D., Duyvesteyn H.M.E., Ginn H.M., Carrique L., Malinauskas T., Ruza R.R., Shah P.N.M., et al. Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host Microbe. 2020;28:445–454.e6. doi: 10.1016/j.chom.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurlburt N.K., Seydoux E., Wan Y.H., Edara V.V., Stuart A.B., Feng J., Suthar M.S., McGuire A.T., Stamatatos L., Pancera M. Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation. Nat. Commun. 2020;11:5413. doi: 10.1038/s41467-020-19231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imkeller K., Scally S.W., Bosch A., Martí G.P., Costa G., Triller G., Murugan R., Renna V., Jumaa H., Kremsner P.G., et al. Antihomotypic affinity maturation improves human B cell responses against a repetitive epitope. Science. 2018;360:1358–1362. doi: 10.1126/science.aar5304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardine J., Julien J.P., Menis S., Ota T., Kalyuzhniy O., McGuire A., Sok D., Huang P.S., MacPherson S., Jones M., et al. Rational HIV immunogen design to target specific germline B cell receptors. Science. 2013;340:711–716. doi: 10.1126/science.1234150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R.P., Long F., Murshudov G.N., Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014;1:213–220. doi: 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce M.G., Wheatley A.K., Thomas P.V., Chuang G.-Y., Soto C., Bailer R.T., Druz, A, Georgiev, I.S, Gillespie, R.A, Kanekiyo, M, et al. Vaccine-induced antibodies that neutralize group 1 and group 2 influenza A viruses. Cell. 2016;166:609–623. doi: 10.1016/j.cell.2016.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju B., Zhang Q., Ge J., Wang R., Sun J., Ge X., Yu J., Shan S., Zhou B., Song S., et al. Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature. 2020;584:115–119. doi: 10.1038/s41586-020-2380-z. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallewaard N.L., Corti D., Collins P.J., Neu U., McAuliffe J.M., Benjamin E., Wachter-Rosati L., Palmer-Hill F.J., Yuan A.Q., Walker P.A., et al. Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell. 2016;166:596–608. doi: 10.1016/j.cell.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreer C., Zehner M., Weber T., Ercanoglu M.S., Gieselmann L., Rohde C., Halwe S., Korenkov M., Schommers P., Vanshylla K., et al. Longitudinal isolation of potent near-germline SARS-CoV-2-neutralizing antibodies from COVID-19 patients. Cell. 2020;182:843–854.e12. doi: 10.1016/j.cell.2020.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreye J., Reincke S.M., Kornau H.C., Sánchez-Sendin E., Corman V.M., Liu H., Yuan M., Wu N.C., Zhu X., Lee C.D., et al. A Therapeutic Non-self-reactive SARS-CoV-2 Antibody Protects from Lung Pathology in a COVID-19 Hamster Model. Cell. 2020;183:1058–1069.e19. doi: 10.1016/j.cell.2020.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E., Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007;372:774–797. doi: 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Kwong P.D., Mascola J.R. Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity. 2012;37:412–425. doi: 10.1016/j.immuni.2012.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong P.D., Mascola J.R. HIV-1 vaccines based on antibody identification, B cell ontogeny, and epitope structure. Immunity. 2018;48:855–871. doi: 10.1016/j.immuni.2018.04.029. [DOI] [PubMed] [Google Scholar]
- Leem J., Dunbar J., Georges G., Shi J., Deane C.M. ABodyBuilder: automated antibody structure prediction with data-driven accuracy estimation. MAbs. 2016;8:1259–1268. doi: 10.1080/19420862.2016.1205773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefranc M.P., Giudicelli V., Ginestoux C., Jabado-Michaloud J., Folch G., Bellahcene F., Wu Y., Gemrot E., Brochet X., Lane J., et al. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009;37:D1006–D1012. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Wang P., Nair M.S., Yu J., Rapp M., Wang Q., Luo Y., Chan J.F., Sahi V., Figueroa A., et al. Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike. Nature. 2020;584:450–456. doi: 10.1038/s41586-020-2571-7. [DOI] [PubMed] [Google Scholar]
- Liu H., Wu N.C., Yuan M., Bangaru S., Torres J.L., Caniels T.G., van Schooten J., Zhu X., Lee C.-C.D., Brouwer P.J.M., et al. Cross-neutralization of a SARS-CoV-2 antibody to a functionally conserved site is mediated by avidity. Immunity. 2020;53:1272–1280.e5. doi: 10.1016/j.immuni.2020.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv Z., Deng Y.Q., Ye Q., Cao L., Sun C.Y., Fan C., Huang W., Sun S., Sun Y., Zhu L., et al. Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody. Science. 2020;369:1505–1509. doi: 10.1126/science.abc5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCallum M., Walls A.C., Bowen J.E., Corti D., Veesler D. Structure-guided covalent stabilization of coronavirus spike glycoprotein trimers in the closed conformation. Nat. Struct. Mol. Biol. 2020;27:942–949. doi: 10.1038/s41594-020-0483-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappas L., Foglierini M., Piccoli L., Kallewaard N.L., Turrini F., Silacci C., Fernandez-Rodriguez B., Agatic G., Giacchetto-Sasselli I., Pellicciotta G., et al. Rapid development of broadly influenza neutralizing antibodies through redundant mutations. Nature. 2014;516:418–422. doi: 10.1038/nature13764. [DOI] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Pinto D., Park Y.-J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., et al. Structural and functional analysis of a potent sarbecovirus neutralizing antibody. bioRxiv. 2020 2020.2004.2007.023903. [Google Scholar]
- Pinto D., Park Y.J., Beltramello M., Walls A.C., Tortorici M.A., Bianchi S., Jaconi S., Culap K., Zatta F., De Marco A., et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature. 2020;583:290–295. doi: 10.1038/s41586-020-2349-y. [DOI] [PubMed] [Google Scholar]
- Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017;14:290–296. doi: 10.1038/nmeth.4169. [DOI] [PubMed] [Google Scholar]
- Robbiani D.F., Gaebler C., Muecksch F., Lorenzi J.C.C., Wang Z., Cho A., Agudelo M., Barnes C.O., Gazumyan A., Finkin S., et al. Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature. 2020;584:437–442. doi: 10.1038/s41586-020-2456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers T.F., Zhao F., Huang D., Beutler N., Burns A., He W.T., Limbo O., Smith C., Song G., Woehl J., et al. Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science. 2020;369:956–963. doi: 10.1126/science.abc7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A.G., Xu H., Khan A.R., O’Donnell T., Khurana S., King L.R., Manischewitz J., Golding H., Suphaphiphat P., Carfi A., et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc. Natl. Acad. Sci. USA. 2013;110:264–269. doi: 10.1073/pnas.1218256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm C.A., Sheng Z., Zhang Z., Mascola J.R., Kwong P.D., Shapiro L. SONAR: a high-throughput pipeline for inferring antibody ontogenies from longitudinal sequencing of B cell transcripts. Front. Immunol. 2016;7:372. doi: 10.3389/fimmu.2016.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi R., Shan C., Duan X., Chen Z., Liu P., Song J., Song T., Bi X., Han C., Wu L., et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature. 2020;584:120–124. doi: 10.1038/s41586-41020-42381-y. [DOI] [PubMed] [Google Scholar]
- Sievers F., Higgins D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018;27:135–145. doi: 10.1002/pro.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C.S., Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N., et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. 2020 2020.2012.2021.20248640. [Google Scholar]
- Toelzer C., Gupta K., Yadav S.K.N., Borucu U., Davidson A.D., Kavanagh Williamson M., Shoemark D.K., Garzoni F., Staufer O., Milligan R., et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370:725–730. doi: 10.1126/science.abd3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortorici M.A., Beltramello M., Lempp F.A., Pinto D., Dang H.V., Rosen L.E., McCallum M., Bowen J., Minola A., Jaconi S., et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science. 2020;370:950–957. doi: 10.1126/science.abe3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walls A.C., Park Y.J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Zhang Y., Wu L., Niu S., Song C., Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.Y., et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181:894–904.e9. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P., Liu L., Iketani S., Luo Y., Guo Y., Wang M., Yu J., Zhang B., Kwong P.D., Graham B.S., et al. Increased resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7 to antibody neutralization. bioRxiv. 2021 doi: 10.1038/s41586-021-03398-2. 2021.2001.2025.428137. [DOI] [PubMed] [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A., et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrapp D., Wang N., Corbett K.S., Goldsmith J.A., Hsieh C.L., Abiona O., Graham B.S., McLellan J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrobel A.G., Benton D.J., Xu P., Roustan C., Martin S.R., Rosenthal P.B., Skehel J.J., Gamblin S.J. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat. Struct. Mol. Biol. 2020;27:763–767. doi: 10.1038/s41594-020-0468-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N.C., Yuan M., Liu H., Lee C.D., Zhu X., Bangaru S., Torres J.L., Caniels T.G., Brouwer P.J.M., van Gils M.J., et al. An alternative binding mode of IGHV3-53 antibodies to the SARS-CoV-2 receptor binding domain. Cell Rep. 2020;33:108274. doi: 10.1016/j.celrep.2020.108274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Wang F., Shen C., Peng W., Li D., Zhao C., Li Z., Li S., Bi Y., Yang Y., et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science. 2020;368:1274–1278. doi: 10.1126/science.abc2241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong X., Qu K., Ciazynska K.A., Hosmillo M., Carter A.P., Ebrahimi S., Ke Z., Scheres S.H.W., Bergamaschi L., Grice G.L., et al. CITIID-NIHR COVID-19 BioResource Collaboration A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020;27:934–941. doi: 10.1038/s41594-020-0478-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H., Sun Y., Deng Y.-Q., Wang N., Tan Y., Zhang N.-N., Li X.-F., Kong C., Xu Y.-P., Chen Q., et al. Rational development of a human antibody cocktail that deploys multiple functions to confer Pan-SARS-CoVs protection. Cell Res. 2021;31:25–36. doi: 10.1038/s41422-020-00444-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Ma N., Madden T.L., Ostell J.M. IgBLAST: an immunoglobulin variable domain sequence analysis tool. Nucleic Acids Res. 2013;41 doi: 10.1093/nar/gkt382. W34-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M., Liu H., Wu N.C., Lee C.D., Zhu X., Zhao F., Huang D., Yu W., Hua Y., Tien H., et al. Structural basis of a shared antibody response to SARS-CoV-2. Science. 2020;369:1119–1123. doi: 10.1126/science.abd2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Zhu J., Wu X., Moquin S., Zhang B., Acharya P., Georgiev I.S., Altae-Tran H.R., Chuang G.Y., Joyce M.G., et al. NISC Comparative Sequencing Program Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity. 2013;39:245–258. doi: 10.1016/j.immuni.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T., Lynch R.M., Chen L., Acharya P., Wu X., Doria-Rose N.A., Joyce M.G., Lingwood D., Soto C., Bailer R.T., et al. NISC Comparative Sequencing Program Structural repertoire of HIV-1-neutralizing antibodies targeting the CD4 supersite in 14 donors. Cell. 2015;161:1280–1292. doi: 10.1016/j.cell.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., Si H.R., Zhu Y., Li B., Huang C.L., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K., Day T., Warshaviak D., Murrett C., Friesner R., Pearlman D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins. 2014;82:1646–1655. doi: 10.1002/prot.24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. China Novel Coronavirus Investigating and Research Team A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zost S.J., Gilchuk P., Case J.B., Binshtein E., Chen R.E., Nkolola J.P., Schäfer A., Reidy J.X., Trivette A., Nargi R.S., et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584:443–449. doi: 10.1038/s41586-020-2548-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps and fitted coordinates of 2-43, H4, and 2-15 were deposited to the EMDB (2-43: EMDB-23165; H4: EMDB-23167; 2-15: EMDB-23166) and RCSB (2-43: 7L56; H4: 7L58; 2-15: 7L57 and 7L5B) respectively. The new IGVL2-14 gene allele (IGVL2-14∗0X) was deposited to European Nucleotide Archive (ENA): PRJEB31020. All deposited data will be available upon publication of the study.