

Abstract
Esophageal carcinoma (EC) is recognized as one of the most frequently occurring malignancies worldwide, and its high morbidity rate motivates efforts to identify potential therapeutic targets. Notably, forkhead box (FOX) family genes are highlighted as possible biomarkers for diagnostics, prognostics, and therapeutics of various malignancies, including EC. Our present study was performed to investigate the underlying mechanism of FOXO6 on the development of EC. We observed a significant upregulation of FOXO6 in EC tissues, contributing to the migration and proliferation in EC cells through gain- and loss-of-function assays. FOXO6 directly interacted with the ubiquitin-specific processing protease 7 (USP7) gene promoter and enhanced its transcriptional activity, which resulted in suppressed cancer cell apoptosis as revealed by chromatin immunoprecipitation (ChIP)-qPCR. USP7 enhanced the ubiquitination of Jumonji domain-containing protein D3 (JMJD3), elevated JMJD3-promoted growth of EC cells, and transcriptionally activated clusterin (CLU) expression at the promoter region via histone H3 lysine 27 tri-methyl (H3K27me3) demethylation, according to immunoprecipitation and ubiquitination assays. Finally, we verified that FOXO6 mediated effects on the USP7/JMJD3/CLU axis to exert an oncogenic role in vivo, which was blocked by USP7 and JMJD3 inhibitor. Our findings demonstrate an important role of the FOXO6/USP7/JMJD3/CLU pathway in EC progression and thus provide attractive potential therapeutic targets for EC patients.
Keywords: esophageal carcinoma, FOXO6, USP7, JMJD3, CLU, H3K27me3, ubiquitination
Graphical Abstract
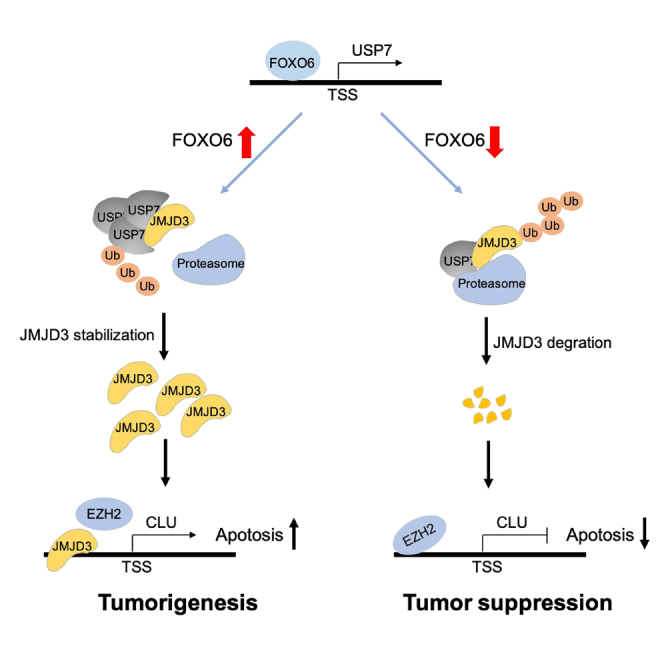
This study aimed at researching the function of FOXO6 on EC. FOXO6 overexpression or JMJD3 overexpression was observed to promote the development of EC. FOXO6 promoted USP7 expression, which enhanced JMJD3 ubiquitination and promoted CLU expression. This study highlights that the FOXO6/USP7/JMJD3/CLU axis may be a novel therapeutic target for EC.
Introduction
Esophageal carcinoma (EC) is the eighth most common cancer worldwide and ranks as the the sixth leading cause of cancer death globally, with a poor 5-year overall survival rate worldwide ranging from 15% to 25%.1,2 EC is the most aggressive disease of all gastrointestinal malignancies, characterized by a high degree of locoregional and distant recurrence after primary surgical resection.3 EC includes two dominant histologic types of esophageal adenocarcinoma and esophageal squamous cell carcinoma (ESCC),1,4 where ESCC accounts for about 90% of EC cases. Concurrent chemoradiotherapy (CCRT) has been recommended as the first-line treatment of advanced ESCC, whether or not with subsequent esophagectomy.3 However, variable treatment responses to CCRT result in uncertain patient outcome, which can include the development of drug resistance and disease progression.5 Therefore, increasing research has focused on possible therapeutic targets and their underlying mechanisms. The alterations of transcription factor expression are associated with the stepwise malignant transition of ESCC,6 including multidrug resistance,7 proliferation, invasion,8 and metastasis.9 In this study, we attempt to explore a novel transcription signaling pathway in the regulation of EC progression.
Forkhead box O6 (FOXO6) is a member of the FOXO proteins group, which is a sub-family of FOX-containing transcription factors.10,11 Previous work suggests that the abnormal expression of FOXO6 may be involved in cancer progression. For example, increased expression of FOXO6 may provoke the development of gastric cancer, and it predicts poor prognosis of afflicted patients.12 Interestingly, FOXO6 inhibits tumor cell proliferation via upregulation of ubiquitin-specific processing protease 7 (USP7) in lung cancer.13 Moreover, USP7 is also overexpressed in ESCC tissues,14 which was reported to exert an inhibitory effect on cell proliferation in ESCC.15
In principle, USP7 promotes the stabilization of Jumonji domain-containing protein D3 (JMJD3) to regulate and control gene expression.16 JMJD3 serves an oncogenic role in gastric cancer,17 and JMJD3 facilitates invasion and migration of non-small cell lung cancer cells.18 The Cancer Genome Atlas (TCGA) database reported that JMJD3 is overexpressed in EC, and it might thus promote cell proliferation in ESCC.19 JMJD3 has been identified as an H3K27 demethylase, which has a context-specific role in cancer development.17 Clusterin (CLU) is generally overexpressed in many human cancers, and its expression correlates closely with chemoradiotherapy resistance and poor prognosis. Overexpressed CLU in castration-resistant prostate cancer was able to promote prostate cancer growth.20 Decreased expression of CLU enhances gemcitabine sensitivity and suppresses tumor growth in pancreatic ductal adenocarcinoma.21 Importantly, CLU epithelial expression is correlated with ESCC resistance to chemoradiotherapy.22 However, the mechanism underlying the role of FOXO6 in regulation of JMJD3 and CLU in EC is so far unclear.
Based on these reports, we speculated that the USP7-mediated JMJD3/CLU axis may be involved in the regulation of FOXO6 in EC. To confirm our hypothesis, in this study, we undertook a series of in vivo and in vitro experiments to explore the interactions among transcription factors FOXO6, USP7, JMJD3, and CLU, as well as to investigate their effects on proliferation, migration, and invasion of EC cells, thus aiming to provide functional evidence of the tumor-promoting role of FOXO6 in EC.
Results
FOXO6 expression was elevated in EC
Recent studies suggest that FOXO6 is overexpressed in multiple carcinomas,23, 24, 25 yet the function of FOXO6 in EC remains unknown. To determine the expression pattern of FOXO6 in EC, we first interrogated the FOXO6 expression in the published Gene Expression Omnibus (GEO: GSE26886) and TCGA EC datasets. We found that FOXO6 was upregulated in EC compared with normal esophageal tissue (Figures 1A and 1B). To validate this finding, we then analyzed FOXO6 expression among 38 EC specimens and paired non-cancerous specimens by immunohistochemistry (IHC), quantitative reverse transcription polymerase chain reaction (qRT-PCR), and western blot analysis. The results showed that the expression of FOXO6 was higher in EC tissues than in non-cancerous tissues (Figures 1C–1F). Furthermore, we compared the expression of FOXO6 between human EC cell lines (Kyse30, Kyse140, Kyse450, Kyse510, Kyse520) and esophageal epithelial cell lines (HEEpiC, HESMC) with western blot analysis. As expected, FOXO6 exhibited higher expression in human EC cell lines compared to that in esophageal epithelial cell lines (Figure 1G). These data indicated that FOXO6 may be involved in EC progression.
Figure 1.

FOXO6 expression was elevated in EC
(A) The expression of FOXO6 in normal esophageal tissues (green) and EC tissues (red) was analyzed in the GEO: GSE26886 dataset. (B) The expression of FOXO6 in normal esophageal tissues (gray) and EC tissues (red) was analyzed in TCGA-ESCA from GEPIA. (C) FOXO6 expression between 38 EC specimens and paired non-cancerous specimens by IHC. (D) FOXO6 expression between 38 EC specimens and paired non-cancerous specimens by qRT-PCR. (E) FOXO6 expression between 38 EC specimens and paired non-cancerous specimens by western blot analysis. (F) FOXO6 protein expression was quantified using ImageJ software normalized to β-actin. (G) FOXO6 protein expression normalized to β-actin in EC cell lines (Kyse30, Kyse140, Kyse450, Kyse510, and Kyse520) and esophageal epithelial cell lines (HEEpiC and HESMC) with western blot analysis. A, adjacent non-cancerous tissues; T, tumor tissues. The levels being reported were relative to the mean expression of three non-cancerous samples (three adjacent non-tumorous esophageal tissues or normal esophageal cells) for each experiment. The data are presented as mean ± standard deviation. The data for tumor and adjacent tissues were analyzed by a Student’s t test; comparison of data of multiple groups was performed by one-way ANOVA. ∗p < 0.05 versus group A or HEEpiC cells.
FOXO6 enhanced EC cell proliferation and migration
Given the abnormal expression of FOXO6 in EC, we then assessed whether FOXO6 expression affects EC progression. To this end, we used Kyse30 to construct a FOXO6 overexpression (oe-FOXO6) stable cell line. As was shown in Figures 2A and 2B, western blot analysis and qRT-PCR results revealed upregulated FOXO6 expression in oe-FOXO6 cells compared with negative control cells (oe-NC). Furthermore, Cell Counting Kit-8 (CCK-8) and bromodeoxyuridine (BrdU) incorporation assay results indicated that the accumulated FOXO6 promoted cell proliferation (Figures 2C and 2D). We also performed transwell and wound healing assays to assess whether FOXO6 plays a role in EC metastasis. As shown in Figures 2E and 2F, overexpression of FOXO6 markedly increased cell migration in transwell assays and improved the capacity of wound healing (Figure 2F). Taken together, FOXO6 promotes the proliferation and migration of EC cells. To better understand the role of FOXO6 in EC proliferation and migration, we next knocked down FOXO6 expression by using two independent small interfering RNAs (siRNAs). Western blot analysis and qRT-PCR revealed that the expression of FOXO6 was efficiently silenced in si-FOXO6-treated cells compared with scrambled si-NC cells (Figures 2G and 2H) while FOXO6 expression maintained an extremely low level in 10 days. As shown in Figures 2I and 2J, FOXO6 deficiency strongly decreased cell growth and reduced cell migration of EC (Figures 2K and 2L). Taken together, these data confirmed that FOXO6 played a significant role in EC cell proliferation and migration.
Figure 2.

FOXO6 enhanced cell proliferation and migration.
(A) FOXO6 protein expression normalized to β-actin detected by western blot analysis. oe-FOXO6 indicates FOXO6 overexpression; oe-NC indicates negative control. (B) FOXO6 mRNA expression detect by qRT-PCR. (C) CCK-8 assays detected the effect of overexpression FOXO6 on the growth of EC cells. (D) BrdU assays detected the effect of overexpression FOXO6 on the proliferation of EC cells. (E) Transwell assays detected the effect of overexpressed FOXO6 on the migration of EC cells. (F) Wound healing assays detected the effect of overexpressed FOXO6 on the migration of EC cells. (G) FOXO6 protein expression normalized to β-actin was examined by western blot analysis. (H) FOXO6 mRNA expression was detected by qRT-PCR. (I) CCK-8 assays detected the effect of FOXO6 knockdown on the growth of EC cells. (J) BrdU assays detected the effect of FOXO6 knockdown on the proliferation of EC cells. (K) Transwell assays detected the effect of FOXO6 knockdown on the migration of EC cells. (L) Wound healing assays detected the effect of FOXO6 knockdown on the migration of EC cells. Measurement data were expressed as mean ± standard deviation and compared by an independent sample t test between two groups and by a two-way ANOVA at different time points. The experiment was repeated three times independently. ∗p < 0.05 versus the oe-NC group or the si-NC group.
Silencing of FOXO6 physically reduced USP7, resulting in promoted EC apoptosis
A previous study13 showed that USP7 transcriptional activation by FOXO6 suppresses lung cancer progression and that USP7 inhibited cell proliferation in EC.14 We hypothesized that FOXO6 initiates the activation of USP7 in EC. According to interrogation of TCGA-Esophageal Carcinoma (ESCA) database with GEPIA software, we found that the expression level of USP7 was positively correlated with FOXO6 expression (Figure 3A). Based on human tissue microarrays of EC, USP7 expression was found to be higher in tumor tissues (Figure 3B). Furthermore, silencing of FOXO6 expression attenuated the mRNA and protein levels of USP7, but it enhanced apoptotic protein cleaved-caspase3 (c-CASP3) expression (Figures 3C and 3D). Flow cytometry analysis indicated that FOXO6 or USP7 inhibition increased the number of apoptotic cells compared with control treatment, whereas simultaneous knockdown of FOXO6 and USP7 expressions dramatically increased the number of apoptotic cells compared with that in the group with the knockdown of a single molecular target (Figure 3E). We then performed chromatin immunoprecipitation (ChIP) assays to examine whether FOXO6 might bind to the USP7 promoter to control its expression. Indeed, FOXO6 complexes were successfully captured at the USP7 promoter, and FOXO6 inhibition significantly attenuated its enrichment to the promoter of USP7 gene compared with control treatment (Figure 3F). Collectively, these data indicated that deficiency of FOXO6 transcriptionally deregulated expression of USP7 and induced USP7-mediated apoptosis.
Figure 3.

Silencing of FOXO6 physically reduced USP7, resulting in promoted apoptosis
(A) The correlation between FOXO6 and USP7 expression was analyzed in TCGA-ESCE database. (B) IHC was used to detect the expression level of USP7 EC tissues and paired non-cancer tissues. (C) The expression level of USP7 was detected by qRT-PCR in FOXO6 knockdown EC cells. (D) Western blot analysis of protein levels of USP7 and c-CASP3 (a splint of CASP3) normalized to β-actin in FOXO6-knockdown EC cells. (E) Flow cytometry analysis was performed to detect the effects of FOXO6 knockdown, USP7 knockdown, or knockdown of both on apoptosis of EC cells. (F) The enrichment of FOXO6 in the USP7 promoter region was detected by ChIP-qPCR. Measurement data are expressed as mean ± standard deviation and compared by a one-way ANOVA among multiple groups. The experiment was repeated three times independently. ∗p < 0.05 versus the si-NC group.
USP7 sustained JMJD3 stability in EC
Previous research showed that USP7 stabilized JMJD3 in T cells.16 In the present study, we silenced USP7 in EC cells using siRNAs and examined the JMJD3 levels, which revealed that the reduction of JMJD3 mRNA was less pronounced in si-USP7-treated cells (Figure 4A). Since USP7 acts as a deubiquitinating enzyme, we hypothesized that USP7 regulated JMJD3 stability on the protein level. Indeed, the JMJD3 protein level was reduced in si-USP7-treated cells (Figure 4B). Co-immunoprecipitation (coIP) against USP7 showed that numerous JMJD3 complexes were effectively captured in the USP7 IP group compared to the control immunoglobulin G (IgG) group, suggesting that USP7 directly interacted with JMJD3 (Figure 4C). To confirm the role of USP7 in JMJD3 stabilization, we transfected HEK293T cells with expression plasmids FLAG-FOXO6 and hemagglutinin (HA)-ubiquitin with or without Myc-USP7. Ubiquitinated JMJD3 was immunoprecipitated with an anti-FLAG antibody and detected by western blot analysis with anti-HA antibody. As expected, co-expression of USP7 increased the JMJD3 levels but decreased ubiquitinated JMJD3 levels (Figure 4D). In addition, when treated with cycloheximide (CHX), a protein synthesis inhibitor, upregulation of USP7 decreased the degradation of JMJD3 (Figure 4E). Meanwhile, when treated USP7 knockdown cells with MG132, a proteasome inhibitor, the stability of JMJD3 was restored (Figure 4F). These data supported our proposal that USP7 targeted JMJD3 for deubiquitination to improve JMJD3 protein stability.
Figure 4.

USP7 sustained JMJD3 stability in EC
(A) The expression level of JMJD3 was detected by qRT-PCR in USP7 knockdown EC cells. (B) The expression level of JMJD3 normalized to β-actin was detected by western blot analysis in USP7 knockdown EC cells. (C) The internal interaction between USP7 and JMJD3 was detected by coIP assays. (D) Myc-USP7, FLAG-JMJD3, and HA-Ub plasmids were transfected into HEK293T cells, respectively, and then cells were harvested after a 12-h treatment with 10 μM MG132 for the ChIP assay. (E) Empty vector and Myc-USP7 plasmid were transferred into HEK293T cells, respectively. After transfection for 48 h, cells were treated with CHX (100 μg/mL). Western blot analysis was used to detect the expression of corresponding proteins normalized to β-actin. (F) The USP7 knockdown cells were treated with MG132 (10 μM) for 12 h, and the corresponding protein expression was detected by western blot analysis. (G) Expression level of JMJD3 normalized to β-actin in EC and adjacent tissues in TCGA-ESCA database by using the GEPIA platform. (H) Expression levels of JMJD3 in EC (T) and paired non-tumor (A) tissues. (I) Transwell assay was used to detect the effect of overexpression of JMJD3 on the migration of EC cells. Measurement data are expressed as mean ± standard deviation and compared by an independent sample t test between two groups, by a one-way ANOVA among multiple groups, and by a two-way ANOVA at different time points. The experiment was repeated three times independently. ∗p < 0.05 versus the si-NC group, no-load (Vec) group, or si-NC + DMSO group; #p < 0.05 versus the si-NC+ MG132 group.
To dissect the function role of JMJD3 in EC, we analyzed the data from the TCGA-ESCA database, which revealed higher JMJD3 levels in tumor tissues than in normal tissues. This finding was further validated in our clinical specimens (Figures 4G and 4H). We also noticed a marked promotion of EC growth upon JMJD3 compared to the control in a colony formation assay (Figure 4I). Collectively, these data strongly suggested that JMJD3 acted as an oncogene in EC.
CLU transcriptional activation by JMJD3 correlates with methylation
To further investigate whether FOXO6 mediated EC progression by the JMJD3/CLU axis, we silenced FOXO6 in Kyse30 cells. As expected, knockdown of FOXO6 decreased JMJD3 protein levels, yet JMJD3 mRNA expression did not differ significantly (Figure 5A), indicating that the JMJD3 changes were dependent on FOXO6. Previous research showed that CLU expression was positively related with JMJD3,26 so we assumed that CLU, as a downstream target gene of FOXO6, mediated the USP7/JMJD3 pathway in the context of EC progression. We examined the CLU level in si-FOXO6-treated cells and found that FOXO6 inhibition indeed attenuated CLU mRNA expression (Figure 5B). It is well known that JMJD3 acts as demethylase regulating the methylation levels of histone H3 lysine 27 tri-methyl (H3K27me3), which has an established function in transcriptional inhibition. Thus, to explore the mechanism of JMJD3-mediated regulation of CLU expression, we performed ChIP-qPCR, which showed that the CLU promoter was directly immunoprecipitated with anti-H3K27me3 antibody and that depletion of FOXO6 increased the enrichment of H3K27me3 at the CLU promoter compared with vector cells (Figure 5C). In addition, JMJD3 accumulated at the CLU promoter as marked by anti-JMJD3 antibody, but depletion of FOXO6 decreased JMJD3 promotor binding compared with vector cells (Figure 5D). Moreover, overexpression of JMJD3 resulted in downregulation of H3K27me3 and upregulation of JMJD3 at the CLU promoter (Figures 5E and 5F). Enhancer of zeste homolog 2 (EZH2) is known as a methylase of H3K27me3, leading us to hypothesize that EZH2 might competitively bind to JMJD3 at the CLU promoter region. As predicted, ChIP-qPCR results suggested that JMJD3 overexpression markedly decreased the EZH2 enrichment at the CLU promoter (Figure 5G). Taken together, these results demonstrated that FOXO6 regulated the stability of JMJD3 by mediating USP7, and that JMJD3 could specifically bind to EZH2 at the promoter region of CLU, which together contributed to the occurrence and development of EC.
Figure 5.

CLU transcriptional activation by JMJD3 correlates with methylation
(A) Western blot analysis and qRT-PCR of the effect of FOXO6 knockdown on JMJD3 protein (normalized to β-actin) and mRNA levels, respectively, in EC cells. (B) qRT-PCR detection of the effect of FOXO6 knockdown on CLU mRNA level in EC cells. (C) ChIP-qPCR detection of H3K27me3 enrichment in CLU promoter region after FOXO6 knockdown in Kyse30 cells. (D) ChIP-qPCR detection of JMJD3 enrichment in CLU promoter region after FOXO6 knockdown in Kyse30 cells. (E) The enrichment of JMJD3 at the promoter region of CLU was detected by ChIP-qPCR in response to overexpression of JMJD3 in Kyse30 cells. (F) The enrichment of H3K27me3 at the promoter region of CLU was detected by ChIP-qPCR in response to overexpression of JMJD3 in Kyse30 cells. (G) The enrichment of JMJD3 EZH2 at the promoter region of CLU was detected by ChIP-qPCR in response to overexpression of JMJD3 in Kyse30 cells. Measurement data were expressed as mean ± standard deviation and compared by independent sample t test between two groups and by one-way ANOVA among multiple groups. The experiment was repeated three times independently. ∗p < 0.05 versus the si-NC group or oe-NC group.
CLU regulated by JMJD3 correlates with demethylation of H3K27me3
To verify whether JMJD3 regulates CLU by regulating the level of H3K27me3, we knocked down JMJD3 by using siRNAs, which resulted in a significant decrease in the levels of JMJD3 and CLU (Figures 6A and 6B). Furthermore, after knocking down JMJD3 by using clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 in EC cells, the JMJD3 protein level was significantly reduced and the level of H3K27me3 was significantly increased, while the level of CLU was decreased. Of note, transfecting JMJD3-knockdown cells with full-length JMJD3 (JMJD3-fl) plasmids but not mutant JMJD3 (JMJD3-mut) plasmids (H1390/E1392)27 restored CLU expression and reduced H3K27me3 level, and this finding was further validated by ChIP-qPCR (Figures 6C–6E). These data indicated that JMJD3 affected CLU expression by regulating the level of H3K27me3 at the CLU promoter depending on the activity of JMJD3.
Figure 6.

CLU regulated by JMJD3 correlates with demethylation of H3K27me3
(A) The effect of JMJD3 knockdown on CLU protein levels normalized to β-actin in EC cells was detected by western blot analysis. (B) The effect of JMJD3 knockdown on CLU mRNA expression in EC cells was detected by qRT-PCR. (C) Western blot analysis of the effect of JMJD3 knockdown on JMJD3 protein levels normalized to β-actin in EC cells. (D) In WT and JMJD3 knockout (KO) EC cells, Vec, full-length JMJD3 (JMJD3-fl), and JMJD3-mut plasmids were transferred, respectively, and the expression of proteins was detected by western blot analysis. (E) The enrichment of H3K27me3 in the CLU promoter region was detected by ChIP-qPCR. Measurement data are expressed as mean ± standard deviation and compared by one-way ANOVA among multiple groups. The experiment was repeated three times independently. ∗p < 0.05 versus the si-NC group or WT + Vec group; #p < 0.05 versus the WT + JMJD3-mut group or KO + Vec group.
FOXO6 exerted pro-tumor function by the USP7/JMJD3/CLU pathway
To verify whether FOXO6 promoted the progression of EC through JMJD3/CLU by mediating USP7, we first constructed FOXO6 overexpression cell lines combined with or without si-USP7 plasmid. As shown in Figure 7A, overexpression of FOXO6 enhanced USP7, JMJD3, and CLU levels, but it decreased the level of H3K27me3, and these effects of FOXO6 could be offset by silencing USP7. In addition, downregulation of JMJD3 by siRNA treatment in oe-FOXO6-treated cells reduced CLU expression but had no corresponding effect on USP7 expression, as for USP7 regulated by FOXO6 (Figure 7B). A flow cytometry assay revealed that overexpression of FOXO6 decreased the number of apoptotic cells, but it increased apoptotic cells when the overexpression was accompanied with silencing of USP7 or JMJD3 (Figure 7C). Transwell and wound healing assays revealed that overexpression FOXO6 in conjunction with silencing USP7 or JMJD3 reduced cell migration capacity (Figures 7D and 7E).
Figure 7.

FOXO6 exerted pro-tumor function by the USP7/JMJD3/CLU pathway
(A) Western blot analysis of FOXO6, USP7, JMJD3, H3K27me3, and CLU normalized to β-actin in si-USP7- or si-NC-treated EC cells. (B) Western blot detection of the corresponding protein expression normalized to β-actin in FOXO6 overexpressed EC cells after knocking down USP7 or JMJD3, respectively. (C) A flow cytometry assay was used to detect the apoptosis of EC cells. (D) Transwell assays detected the migration of EC cells. (E) Wound healing assays detected the migration of EC cells. (F) Tumor volume growth curves were presented. (G) Tumor weights were examined. (H) Western blot analysis validation results were obtained from the knocked down effect of FOXO6 normalized to β-actin in the transplanted tumor. (I) Growth of the transplanted tumor. (J) Weight of the transplanted tumor. Measurement data are expressed as mean ± standard deviation and compared by an independent sample t test between two groups, by a one-way ANOVA among multiple groups, and by a two-way ANOVA at different time points. The experiment was repeated three times independently. ∗p < 0.05 versus the si-NC + Vec group, sh-NC group, or oe-NC + DMSO group; #p < 0.05 versus the si-usp7.1 + Vec group, si-NC + FOXO6 group, si-usp7.1 + Vec group, or oe-FOXO6 + DMSO group; &p < 0.05 versus the si-USP7.2 + Vec group or si-JMJD3.1 + Vec group. n = 8.
Finally, to validate the above results in vivo, we injected shNC or stably expressing FOXO6 cells into the subcutaneous tissues of nude mice. The results showed that sh-FOXO6 significantly inhibited the xenograft tumor growth (Figures 7F and 7G), whereas western blot analysis results validated that sh-FOXO6 effectively knocked down FOXO6 in transplanted tumor (Figure 7H). However, when oe-FOXO6 xenograft mice were treated with USP7 or JMJD3 inhibitors, the growth of tumors was significantly inhibited, suggesting that USP7 and JMJD3 were involved in EC progression (Figures 7I and 7J). Collectively, these data indicated that FOXO6 promoted the proliferation and angiogenesis of EC by mediating USP7 expression via the JMJD3/CLU pathway.
Discussion
Recent research has given compelling evidence that the concerted expression of FOXO6, USP7, JMJD3, and CLU is involved in cancer progression.12,14,16,22 In the current study, we made efforts to explore the functional interaction between FOXO6 and USP7/JMJD3 in the development of EC, thus clarifying the role of FOXO6 in EC. Collectively, the experimental data demonstrated that the transcription factor FOXO6 promotes the tumorigenesis of EC by mediating effects on the deubiquitinase USP7-regulated JMJD3/CLU axis. Understanding how this feedback loop modulates tumor cell growth and how the cellular level of FOXO proteins can regulate the cancer development may thus reveal important new therapeutic opportunities.
FOXO6 was found to be highly expressed in EC according to the GEO: GSE26886 dataset, which showed similar results as in TCGA-ESCA database. A significant correlation between FOXO6 expression and tumor growth was reported, which revealed that FOXO6 expression promotes tumor cell proliferation and migration through suppression of epithelial-mesenchymal transition (EMT) by transcriptionally regulating expression of Sirt6 in breast cancer.28 Alternatively, the activation of FOXO6 in lung cancer delayed the apoptotic response induced by counteracting the pro-apoptotic effects of epidermal growth factor receptor inhibition.29 Consistent with this picture, our current experimental results showed that the expression level of transcription factor FOXO6 was significantly higher in patient EC tissues than that of adjacent tissues. Additionally, expression of FOXO6, detected by western blot analysis, in EC cells (Kyse30, Kyse140, Kyse450, Kyse510, and Kyse520) was significantly higher than that of normal esophageal cells (HEEpiC and HESMC). Furthermore, we found that FOXO6 is highly expressed in EC and is positively correlated with the degree of cancerization and malignant transition of EC tissues.
For exploration purpose, we established Kyse30 EC cells with oe-FOXO6 treatment. Experimental results revealed an increased proliferation ability of the Kyse30 cells with oe-FOXO6, with distinctly enhanced invasion and superior wound healing. Conversely, FOXO6 knockdown was also found to inhibit the migration of EC cells in our experiment, indicating that FOXO6 may promote the proliferation and migration of EC cells, while playing a similar role in gastric cancer.30 Moreover, the FOXO6/USP7 molecular network has been demonstrated previously to play an important role in the regulation of lung cancer development,13 while USP7 can promote breast carcinogenesis.31 Consistent with this scenario, we found in our study that the expression of USP7 in EC was significantly increased to a degree positively correlating with the expression of FOXO6. In addition, FOXO6 knockdown suppressed the expression of USP7 and that of apoptosis-related protein c-CASP3. The above evidence revealed that FOXO6 upregulated the expression of USP7 in EC to inhibit the apoptosis of tumor cells and promote their proliferation. Notably, it was reported previously that USP7 controls cancer cell growth in leukemia through stabilizing the expression levels of NOTCH1 and JMJD3 histone demethylase.16 Similarly, our further mechanistic investigations demonstrated that overexpression of USP7 increased the protein level of JMJD3 and its ubiquitin modification level. Impressively, our experimental data elucidated that overexpressed USP7 significantly inhibited the degradation rate of JMJD3.
In the context of several controversies in oncological research, JMJD3 plays contradictory roles in both tumor suppression and tumorigenesis.32 JMJD3 was shown to serve as a potential tumor suppressor in different types of cancers.26 Meanwhile, in other investigations, JMJD3 was suggested to promote invasion and migration of lung cancer cells, which was significantly correlated with shortened patient survival in lung cancer and gastric cancer.18,33 Additionally, it was also suggested that JMJD3 expression could regulate carcinoma cell growth in ESCC.19 In our current investigation, JMJD3 protein was expressed at a high level in EC tissues, while tumor cells possessed enhanced tumorigenic ability when JMJD3 was overexpressed, thus indicating that JMJD3 plays an oncogenic role in EC.
Further mechanistic investigations showed that the mRNA level of JMJD3 remained steady after knockdown of USP7, but that the protein level of JMJD3 decreased significantly. This phenomenon is plausible because USP7 is a deubiquitinating enzyme and thus a regulator at the protein level rather than the mRNA level. The subsequent experiments showed that FOXO6 knockdown significantly decreased the abundance of JMJD3 as well as that of CLU. JMJD3 is a family member of the H3K27me3-specific demethylases,26 and it plays carcinostatic or carcinogenic roles in many types of cancer through activating key target genes by demethylation.34 Additionally, JMJD3 is widely known as a transcription inhibitor.26 Therefore, we speculated that JMJD3 may regulate the expression of CLU by regulating the level of H3K27me3. The evidence that the accumulation of H3K27me3 in the CLU promoter region, after FOXO6 knockdown, was increased contrary to that of JMJD3 verified our speculation.
The molecular mechanism investigations showed that JMJD3 knockdown by siRNA decreased transcription and expression of CLU. Moreover, these experimental data demonstrated that JMJD3 controlled CLU expression in EC through regulating the accumulation of H3K27me3 in the CLU promoter region, and that this regulation strictly depended on the demethylase activity of JMJD3. Meanwhile, CLU was also directly repressed by EZH2,35 which is another member of the H3K27me3-specific demethylase family.36 Downregulation of USP7 promotes the stabilization of EZH2 protein.36 Current experimental data showed that JMJD3 overexpression reduced the accumulation of EZH2 in the CLU promoter region, indicating that FOXO6 was able to mediate USP7 to regulate the stability of JMJD3, which is a competitor for EZH2 in binding with the promoter region of CLU, the downstream target gene, and then participate in regulating the tumorigenesis and development of EC.
In breast cancer, FOXO6 suppressed cell migration, invasion, and proliferation of cancer cells by suppressing EMT through regulation of Sirt6.27 In colorectal cancer, FOXO6 knockdown inhibited cell proliferation, migration, invasion, and glycolysis by regulation of phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway.24 In gastric cancer, FOXO6 promoted tumorigenicity of cancer cells through upregulation of C-myc.23 In the present study, we reported that in FOXO6 overexpressed EC cells, the protein expression levels of USP7, JMJD3, and CLU all increased significantly. Additionally, JMJD3 and CLU expression was not increased by knockdown of USP7 in the FOXO6 overexpressed cells, whereas CLU expression was decreased by knockdown of JMJD3, and USP7 expression could still be regulated by FOXO6 simultaneously. This proved that FOXO6 upregulates the expression of JMJD3 and CLU by mediating USP7, which was confirmed by observations of neoplasia growth in vivo.
To conclude, our findings elaborated that FOXO6 serves as a promotor of tumorigenesis and development of EC. FOXO6 increases the expression of deubiquitinating enzyme USP7 by binding to its promoter region. USP7 then stabilizes the expression of JMJD3 and promotes the expression of CLU by binding to the CLU promoter region, inducing tumorigenesis and progression of EC. The mechanistic model of the tumorigenesis-promotor function of FOXO6 is depicted in Figure 8. Present findings provide new insights into the role of FOXO6 in EC, suggesting that FOXO6 might be potential target as a biomarker for diagnosis or treatment of EC. However, it remains to be elucidated whether the promotor role of FOXO6 acts via regulation of the JMJD3/CLU axis in vivo. Further investigations are required to better establish the impact of FOXO6 overexpression in tumor cellular metabolism.
Figure 8.

Mechanistic illustration of tumorigenesis promotor function of FOXO6 in EC
Materials and methods
Bioinformatics analysis
The FOXO6 expression profile of EC dataset GEO: GSE26886 was downloaded from the GEO database (https://www.ncbi.nlm.nih.gov/geo/). The RNA sequencing (RNA-seq) raw data of FOXO6 and JMJD3 were downloaded from TCGA analyzed by GEPIA software (http://gepia.cancer-pku.cn) as described previously.37
Patient samples
Thirty-eight EC tissues and adjacent non-tumorous esophageal tissues (5 cm away from tumor tissues serving as normal controls) were collected from The Fourth Hospital of China Medical University between July 2018 and September 2019.14 All patients were diagnosed in accordance with the American Joint Committee on Cancer manual criteria and signed consent approving the use of their tissues. The study protocol for the tissues was approved by the Ethics Committee of The Fourth Hospital of China Medical University. The tissue samples were snap-frozen in liquid nitrogen after surgery and stored at −80°C until further use. All experiments were performed complied with the Code of Ethics of the World Medical Association (Declaration of Helsinki).
Cell culture and reagent
The human normal esophageal cells (HEEpiC and HESMC), EC cells (Kyse30, Kyse150, Kyse450, Kyse510, and Kyse520), and human embryonic kidney cells (HEK293T) were cultured in Roswell Park Memorial Institute medium 1640 or Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (Sigma-Aldrich, St Louis, MO, USA), penicillin (100 U/mL), and 1% streptomycin (100 mg/mL) at 37°C in a 5% CO2 humidified incubator. HEEpiC, HESMC, Kyse30, Kyse150, Kyse450, Kyse510, Kyse520, and HEK293T cells were purchased from The Cell Bank of Type Culture Collection of Chinese Academy of Sciences. In addition to USP7 inhibitor (P22077, ab273383, Abcam), JMJD3 inhibitor (S7281) and MG132 (S2619) were purchased from Selleck Chemicals (Houston, TX, USA). CHX was purchased as a 300 mM sterile-filtered solution (100 mg/1.185 mL) from Sigma-Aldrich (#5087390001).
Plasmids
The plasmids pCMV-HA-U, pHAGE-puro-HA, pHAGE-puro-HA-FOXO6, pcDNA5-FLAG-JMJD3, pCMV-Myc-USP7, pLKO.1-Puro, pLKO.1-Puro-shNC, pLKO.1-Puro-shFOXO6.1, and pLKO.1-Puro-FOXO6.2 were constructed by GenePharma (Shanghai, P.R. China).
Lentivirus packaging and transfection
Lentiviral particles contain target-specific constructs, and lentiviral packaging plasmids (pMD2G, psPAX2) were transfected to HEK293T cells by Lipofectamine 2000 (11668-019, 1.5 mL, Invitrogen, Carlsbad, CA, USA). After transfection for 48 h, supernatants containing lentivirus particles were collected for further use. Stable cell lines were established by selections with puromycin (50 μg/μL, Selleck Chemicals, S7417) for 48 h, and the target gene expression levels were monitored using western blot analysis and qRT-PCR. On the 3rd, 7th, and 10th days after si-FOXO6 transfection, FOXO6 expression was measured to determine whether Kyse30 cells could stably express si-NC or si-FOXO6.
siRNA transfections
The siRNAs targeting human FOXO6 mRNA, USP7 mRNA, JMJD3 mRNA, and NC were synthesized by GenePharma.14 The 5 × 105 Kyse30 cells were seeded in 60-mm plates transfected using Lipofectamine 2000 (CAT). The siRNA sequences were as follows: si-NC, 5′-UUCUCCGAACGUGUCACGU-3′; siFOXO6.1, 5′-CAGTGACGAAATGGACTTCAA-3′; siFOXO6.2, 5′-CCTCGCCACTCATGTACCCAAC-3′; siUSP7.1, 5′-ACCCUUGGACAAUAUUCCU-3′; siUSP7.2, 5′-AGUCGUUCAGUCGUCGUAU-3′; siJMJD3.1, 5′-AGATTCTTTCTATGGGCTTTA-3′; and siJMJD3.2, 5′-GATCTCTATGCATCCAATATT-3′.
CRISPR knockout cells
A Cas9-2A-PURO plasmid was purchased from Addgene (plasmid 48139). Two guide RNAs (gRNAs) targeting exon 17 of mouse JMJD3 were designed by means of an online software (https://zlab.bio/guide-design-resources) and then cloned into Cas9-2A-PURO plasmid using the BbsI restriction enzyme sites. Kyse30 cells were transfected using Lipofectamine 2000 (CAT). After 24 h, cells were cultured in fresh medium containing 1 μg/mL puromycin for 48 h. Seven days later, colonies were selected and JMJD3 knockout efficiency was detected by western blot analysis.38 The sequences were as follows: JMJD3-gRNA1.1, 5′-CACCTGTGGATGTTACCCGCATGA-3′; JMJD3-gRNA1.2, 5′-AAACTCATGCGGGTAACATCCACA-3′; JMJD3-gRNA2.1, 5′-CACCGTCCCTGGCAGCCGAACGCC-3′; and JMJD3-gRNA2.2, 5′-AAACGGCGTTCGGCTGCCAGGGAC-3′.
IHC
A human EC tissue array (OD-CT-DgEso01) was obtained from Shanghai Outdo Biotech (Shanghai, P.R. China). Specific antibody (1:50 dilution) of FOXO6, USP7, and JMJD3 was used for IHC staining following procedures as previously described.14
RNA extraction and qRT-PCR
Total cellular RNA was extracted using a TRIzol reagent kit (Invitrogen) according to the manufacturer’s protocol and then quantified by NanoDrop 1000 (ND-1000, NanoDrop, Wilmington, DE, USA).14,24 cDNA was synthesized from 400 ng of RNA by a PrimeScript RT reagent kit (Takara, Dalian, Liaoning, P.R. China), and real-time PCR was performed on thermal cycler dice real-time system (TP800, Takara) with SYBR Premix Ex Taq formed on Tli RNase H Plus (Takara) according to the manufacturer’s instructions. The relative expression of mRNA was calculated by the 2−ΔΔCT with glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control. The primers were purchased from Ribobio (Guangzhou, P.R. China). The sequences of the primers used are listed in Table1.
Table 1.
qRT-PCR primer sequences
| Gene | Forward (5′→3′) | Reverse (5′→3′) |
|---|---|---|
| GAPDH | ACTCCACTCACGGCAAATTC | TCTCCATGGTGGTGAAGACA |
| FOXO6 | GGCCGCGCTCGTGTACC | TACACGAGCGCGGCCG |
| JMJD3 | GGTCTGTTGTACCCCACTGC | GTCTCCGCCTCAGTAACAGC |
| CLU | GACTCTGCTGCTGTTTGTGG | CATTGTCTGAGACCGTCTGG |
| USP7 | TGGAAGCGGGAGATACAGAT | CGGTGTTGTGTCCATCACTC |
| USP7-pro | CAGCTTCGACGTCAC | GACTGCAACATTGCACT |
| CLU-pro | GTGCAGAGAAGGGGTCTCAG | GTGCCAGGCCTTTATATCCA |
qRT-PCR, quantitative reverse transcriptase polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; FOXO6, forkhead box O6; JMJD3, Jumonji domain-containing protein D3; CLU, clusterin; USP, ubiquitin-specific processing protease 7.
Western blot analysis
Radio immunoprecipitation assay lysis buffer (C0481, Sigma-Aldrich) was used for total protein extraction.23 The protein sample was quantified using a bicinchoninic acid kit (Beyotime, Shanghai, P.R. China), separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, and electrotransferred onto polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA). Immunoblots were incubated with primary antibodies overnight at 4°C and then incubated with horseradish peroxidase-conjugated secondary antibodies (ab99702, Abcam, Cambridge, UK). Immunoreactive bands were visualized by enhanced chemiluminescence (Baomanbio, Shanghai, P.R. China), and protein expression was normalized against β-actin. Antibodies against the following proteins were used (all from Abcam): β-actin (ab115777), FOXO6 (ab48730), c-CASP3 (ab13847), USP (ab4080), JMJD3 (ab38113), HA (ab9110), FLAG (ab1162), and Myc (ab32072). The levels being reported were relative to the mean expression of three non-cancerous samples (three adjacent non-tumorous esophageal tissues or normal esophageal cells) for each experiment.
CCK-8 and colony formation assays
A CCK-8 assay was used to evaluate cell proliferation with a CCK-8 kit (Beyotime) according to the manufacturer’s instructions. In brief, Kyse30 cells were seeded in 96-well culture plates (3 × 103 cells/well) and then incubated with CCK-8 reagent. The optical density value in the medium was measured at 24, 48, and 72 h by determining the absorbance at 450 nm. For colony formation assays, 500 cells were plated in six-well plates and cultured at 37°C. After 12 days, cells were fixed with 4% paraformaldehyde and stained with gentian violet (C0121, Beyotime). Each colony was counted with Adobe Photoshop software.
BrdU assay
A BrdU assay was performed as described previously.39,40 In brief, cells were cultured in a 24-well plate and incubated with rat primary antibody against BrdU (1:300, ab6326, Abcam) for 1 h, followed by incubation with goat anti-rat IgG secondary antibody (ab150116, Abcam) and nuclear staining with 4′,6-diamidino-2-phenylindole (300 nM). At least eight microscopic fields were recorded for quantitation of positive cells (Nikon 80i, Tokyo, Japan) for BrdU-positive rate calculation.
Transwell and wound healing assays
For transwell migration assays, 1 × 104 cells suspended in serum-free medium were seeded in the upper chamber membranes of 8-μm wells (Millipore).27 Then, the medium with serum was added to the lower chamber. After 24 h, cells in the bottom of the upper chamber were fixed with methanol and stained with 0.1% crystal violet. Cells in the inner chamber were scraped away using a cotton swab. Then, stained cells were counted under a microscope in five random fields for each well. For the wound healing assay, cells were seeded in 12-well plates. When 100% confluence was reached, a 200-μm pipette tip was used to scratch the cell monolayer. Then, the healing wound was imaged at 0 and 24 h, respectively. The scratch rate (%) was calculated as scratch distance at 24 h/scratch distance at 0 h × 100% to evaluate cell migration ability. Moreover, 5 μM mitomycin C was added into the medium to inhibit the effect of cell proliferation on migration. ImageJ software was introduced for statistical analysis.
Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide staining and flow cytometry analyses
The cell apoptosis assay was performed according to protocol described previously41 In brief, 1 × 106 cells were washed with cold phosphate-buffered saline and stained using an annexin V-FITC apoptosis detection kit (C1062M, Beyotime) in the dark for 15 min. A flow cytometer (FACSVerse/FACSCalibur/FACSAria II SORP, BD Biosciences, USA) was to detect the ratio of apoptotic cells.
ChIP assays
A commercial kit (Millipore) was used for ChIP assays. A total of 1 × 107 cells were fixed by using 1% paraformaldehyde and then quenched with glycine (2.5 mM).42, 43 Protein-bound chromatin was fragmented by sonication for the ChIP assay of JMJD3 and EZH2, whereas MNase (USB) was used for the ChIP assay of H3K27me3. Chromatin was immunoprecipitated at 4°C overnight with anti-JMJD3, anti-EZH2, anti-H3K27me3, or normal IgG as a NC. Following extensive washing the immunoprecipitated DNA was treated with protein agarose/Sepharose, and the crosslinking was relieved by incubation at 65°C. The DNA fragments were purified and confirmed by qRT-PCR.
Tumor xenograft model in nude mice
Sixty-four female nude mice (3–5 weeks of age, weighing 12–16 g) were housed under specific pathogen-free conditions with controlled relative humidity (40%–60%) at 24°C–26°C, and provided with sterilized food and water.14 Kyse30 with stably overexpressing FOXO6 cells or control cells was injected subcutaneously into nude mice, which were treated with USP7 inhibitor (10 mg/kg) or JMJD3 inhibitor (50 mg/kg) on a 3 days off schedule (n = 8). Tumor diameter was measured every 5 days as follows: tumor volume (mm3) = (length × width2)2. After 30 days, mice were sacrificed and the tumors were removed, imaged, and weighed. Animal experiments were approved by the Institutional Ethics Committees of The Fourth Hospital of China Medical University, and all procedures were conducted consistent with the guidelines of the Institutional Animal Care and Use Committees.
Statistical analysis
Statistical analysis was performed SPSS 21.0 software (IBM, Armonk, NY, USA). For three independent experiments, all data are presented as the mean ± standard deviation. A Student’s t test and Dunnett’s t test were used to analyze the differences between two groups. For cell activity, data at different time points were compared using two-way analysis of variance (ANOVA), and tumor volume data were compared with a repeated-measures ANOVA. p <0.05 indicates statistical significance.
Acknowledgments
The authors would like to acknowledge the helpful comments on this paper received from the reviewers.
Author contributions
N.L. and Z.Z. conceived and designed research. P.L. and Y.Z. performed experiments and interpreted results of experiments. S.C. and Y.S. analyzed data and prepared figures. B.W. drafted the paper. Z.Z. edited and revised the manuscript. All authors read and approved the final version of the manuscript.
Declarations of interest
The authors declare no competing interests.
References
- 1.Murphy G., McCormack V., Abedi-Ardekani B., Arnold M., Camargo M.C., Dar N.A., Dawsey S.M., Etemadi A., Fitzgerald R.C., Fleischer D.E. International cancer seminars: a focus on esophageal squamous cell carcinoma. Ann. Oncol. 2017;28:2086–2093. doi: 10.1093/annonc/mdx279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Watanabe M., Otake R., Kozuki R., Toihata T., Takahashi K., Okamura A., Imamura Y. Recent progress in multidisciplinary treatment for patients with esophageal cancer. Surg. Today. 2020;50:12–20. doi: 10.1007/s00595-019-01878-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shapiro J., van Lanschot J.J.B., Hulshof M.C.C.M., van Hagen P., van Berge Henegouwen M.I., Wijnhoven B.P.L., van Laarhoven H.W.M., Nieuwenhuijzen G.A.P., Hospers G.A.P., Bonenkamp J.J., CROSS study group Neoadjuvant chemoradiotherapy plus surgery versus surgery alone for oesophageal or junctional cancer (CROSS): long-term results of a randomised controlled trial. Lancet Oncol. 2015;16:1090–1098. doi: 10.1016/S1470-2045(15)00040-6. [DOI] [PubMed] [Google Scholar]
- 4.Abnet C.C., Arnold M., Wei W.Q. Epidemiology of esophageal squamous cell carcinoma. Gastroenterology. 2018;154:360–373. doi: 10.1053/j.gastro.2017.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuo I.Y., Huang Y.L., Lin C.Y., Lin C.H., Chang W.L., Lai W.W., Wang Y.C. SOX17 overexpression sensitizes chemoradiation response in esophageal cancer by transcriptional down-regulation of DNA repair and damage response genes. J. Biomed. Sci. 2019;26:20. doi: 10.1186/s12929-019-0510-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogerson C., Britton E., Withey S., Hanley N., Ang Y.S., Sharrocks A.D. Identification of a primitive intestinal transcription factor network shared between esophageal adenocarcinoma and its precancerous precursor state. Genome Res. 2019;29:723–736. doi: 10.1101/gr.243345.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu H., Chen X., Chen B., Chen B., Fan J., Song W., Xie Z., Jiang D., Li Q., Zhou M. Activating transcription factor 4 mediates a multidrug resistance phenotype of esophageal squamous cell carcinoma cells through transactivation of STAT3 expression. Cancer Lett. 2014;354:142–152. doi: 10.1016/j.canlet.2014.07.044. [DOI] [PubMed] [Google Scholar]
- 8.Li Z., Li X., Li C., Su Y., Fang W., Zhong C., Ji W., Zhang Q., Su C. Transcription factor OCT4 promotes cell cycle progression by regulating CCND1 expression in esophageal carcinoma. Cancer Lett. 2014;354:77–86. doi: 10.1016/j.canlet.2014.07.049. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y., Zhang C., Chen J., Zhang B., Zhang H., Yang X., Liu J., Wu Q. Expression of transcription factor 21 (TCF21) and upregulation its level inhibits invasion and metastasis in esophageal squamous cell carcinoma. Med. Sci. Monit. 2018;24:4128–4136. doi: 10.12659/MSM.909138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deng Y., Wang F., Hughes T., Yu J. FOXOs in cancer immunity: knowns and unknowns. Semin. Cancer Biol. 2018;50:53–64. doi: 10.1016/j.semcancer.2018.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma J., Matkar S., He X., Hua X. FOXO family in regulating cancer and metabolism. Semin. Cancer Biol. 2018;50:32–41. doi: 10.1016/j.semcancer.2018.01.018. [DOI] [PubMed] [Google Scholar]
- 12.Wang J.H., Tang H.S., Li X.S., Zhang X.L., Yang X.Z., Zeng L.S., Ruan Q., Huang Y.H., Liu G.J., Wang J., Cui S.Z. Elevated FOXO6 expression correlates with progression and prognosis in gastric cancer. Oncotarget. 2017;8:31682–31691. doi: 10.18632/oncotarget.15920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu H.J., Zhang L.G., Wang Z.H., Guo X.X. FoxO6 inhibits cell proliferation in lung carcinoma through up-regulation of USP7. Mol. Med. Rep. 2015;12:575–580. doi: 10.3892/mmr.2015.3362. [DOI] [PubMed] [Google Scholar]
- 14.Hu T., Zhang J., Sha B., Li M., Wang L., Zhang Y., Liu X., Dong Z., Liu Z., Li P., Chen P. Targeting the overexpressed USP7 inhibits esophageal squamous cell carcinoma cell growth by inducing NOXA-mediated apoptosis. Mol. Carcinog. 2019;58:42–54. doi: 10.1002/mc.22905. [DOI] [PubMed] [Google Scholar]
- 15.Xu Y., Lu S. Metformin inhibits esophagus cancer proliferation through upregulation of USP7. Cell. Physiol. Biochem. 2013;32:1178–1186. doi: 10.1159/000354517. [DOI] [PubMed] [Google Scholar]
- 16.Jin Q., Martinez C.A., Arcipowski K.M., Zhu Y., Gutierrez-Diaz B.T., Wang K.K., Johnson M.R., Volk A.G., Wang F., Wu J. USP7 cooperates with NOTCH1 to drive the oncogenic transcriptional program in T-cell leukemia. Clin. Cancer Res. 2019;25:222–239. doi: 10.1158/1078-0432.CCR-18-1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu Z., Xia Y., Xiao Z., Jia Y., Li L., Jin Y., Zhao Q., Wan L., Yi T., Yu Y. Comprehensive profiling of JMJD3 in gastric cancer and its influence on patient survival. Sci. Rep. 2019;9:868. doi: 10.1038/s41598-018-37340-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li S., Jiang L., He Q., Wei W., Wang Y., Zhang X., Liu J., Chen K., Chen J., Xie D. The prognostic significance of JMJD3 in primary sarcomatoid carcinoma of the lung, a rare subtype of lung cancer. OncoTargets Ther. 2019;12:9385–9393. doi: 10.2147/OTT.S225596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li S.H., Lu H.I., Chen Y.H., Lo C.M., Huang W.T., Tien W.Y., Lan Y.C., Tsai H.T., Chen C.H. JMJD3 expression is an independent prognosticator in patients with esophageal squamous cell carcinoma. Surgery. 2019;165:946–952. doi: 10.1016/j.surg.2018.11.015. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi A., Shiota M., Beraldi E., Thaper D., Takahara K., Ibuki N., Pollak M., Cox M.E., Naito S., Gleave M.E., Zoubeidi A. Insulin-like growth factor-I induces CLU expression through Twist1 to promote prostate cancer growth. Mol. Cell. Endocrinol. 2014;384:117–125. doi: 10.1016/j.mce.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 21.Wang X., Xie J., Lu X., Li H., Wen C., Huo Z., Xie J., Shi M., Tang X., Chen H. Melittin inhibits tumor growth and decreases resistance to gemcitabine by downregulating cholesterol pathway gene CLU in pancreatic ductal adenocarcinoma. Cancer Lett. 2017;399:1–9. doi: 10.1016/j.canlet.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 22.He L.R., Liu M.Z., Li B.K., Rao H.L., Liao Y.J., Zhang L.J., Guan X.Y., Zeng Y.X., Xie D. Clusterin as a predictor for chemoradiotherapy sensitivity and patient survival in esophageal squamous cell carcinoma. Cancer Sci. 2009;100:2354–2360. doi: 10.1111/j.1349-7006.2009.01349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Qinyu L., Long C., Zhen-dong D., Min-min S., Wei-ze W., Wei-ping Y., Cheng-hong P. FOXO6 promotes gastric cancer cell tumorigenicity via upregulation of C-myc. FEBS Lett. 2013;587:2105–2111. doi: 10.1016/j.febslet.2013.05.027. [DOI] [PubMed] [Google Scholar]
- 24.Li Q., Tang H., Hu F., Qin C. Silencing of FOXO6 inhibits the proliferation, invasion, and glycolysis in colorectal cancer cells. J. Cell. Biochem. 2019;120:3853–3860. doi: 10.1002/jcb.27667. [DOI] [PubMed] [Google Scholar]
- 25.Chen H.Y., Chen Y.M., Wu J., Yang F.C., Lv Z., Xu X.F., Zheng S.S. Expression of FOXO6 is associated with oxidative stress level and predicts the prognosis in hepatocellular cancer: a comparative study. Medicine (Baltimore) 2016;95:e3708. doi: 10.1097/MD.0000000000003708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu S.H., Zhu K.Y., Chen J., Liu X.Z., Xu P.F., Zhang W., Yan L., Guo H.Z., Zhu J. JMJD3 facilitates C/EBPβ-centered transcriptional program to exert oncorepressor activity in AML. Nat. Commun. 2018;9:3369. doi: 10.1038/s41467-018-05548-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tumber A., Nuzzi A., Hookway E.S., Hatch S.B., Velupillai S., Johansson C., Kawamura A., Savitsky P., Yapp C., Szykowska A. Potent and selective KDM5 inhibitor stops cellular demethylation of H3K4me3 at transcription start sites and proliferation of MM1S myeloma cells. Cell Chem. Biol. 2017;24:371–380. doi: 10.1016/j.chembiol.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ye H., Duan M. Downregulation of FOXO6 in breast cancer promotes epithelial-mesenchymal transition and facilitates migration and proliferation of cancer cells. Cancer Manag. Res. 2018;10:5145–5156. doi: 10.2147/CMAR.S157661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothenberg S.M., Concannon K., Cullen S., Boulay G., Turke A.B., Faber A.C., Lockerman E.L., Rivera M.N., Engelman J.A., Maheswaran S., Haber D.A. Inhibition of mutant EGFR in lung cancer cells triggers SOX2-FOXO6-dependent survival pathways. eLife. 2015;4:e06132. doi: 10.7554/eLife.06132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim C.G., Lee H., Gupta N., Ramachandran S., Kaushik I., Srivastava S., Kim S.H., Srivastava S.K. Role of forkhead box class O proteins in cancer progression and metastasis. Semin. Cancer Biol. 2018;50:142–151. doi: 10.1016/j.semcancer.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Q., Ma S., Song N., Li X., Liu L., Yang S., Ding X., Shan L., Zhou X., Su D. Stabilization of histone demethylase PHF8 by USP7 promotes breast carcinogenesis. J. Clin. Invest. 2016;126:2205–2220. doi: 10.1172/JCI85747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang S., Yim S., Park H. The cancer driver genes IDH1/2, JARID1C/ KDM5C, and UTX/ KDM6A: crosstalk between histone demethylation and hypoxic reprogramming in cancer metabolism. Exp. Mol. Med. 2019;51:1–17. doi: 10.1038/s12276-019-0230-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y., Hua P.Y., Jin C.Y., Li J.D., Zhang G.X., Wang B. JMJD3 enhances invasiveness and migratory capacity of non-small cell lung cancer cell via activating EMT signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019;23:4784–4792. doi: 10.26355/eurrev_201906_18063. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X., Liu L., Yuan X., Wei Y., Wei X. JMJD3 in the regulation of human diseases. Protein Cell. 2019;10:864–882. doi: 10.1007/s13238-019-0653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang C., Liu Z., Woo C.W., Li Z., Wang L., Wei J.S., Marquez V.E., Bates S.E., Jin Q., Khan J. EZH2 mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res. 2012;72:315–324. doi: 10.1158/0008-5472.CAN-11-0961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zheng N., Chu M., Lin M., He Y., Wang Z. USP7 stabilizes EZH2 and enhances cancer malignant progression. Am. J. Cancer Res. 2020;10:299–313. [PMC free article] [PubMed] [Google Scholar]
- 37.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu H., Dong Z., Wang X., Bai L., Lei Q., Yang J., Li L., Li Q., Liu L., Zhang Y. Dehydrocorydaline inhibits cell proliferation, migration and invasion via suppressing MEK1/2-ERK1/2 cascade in melanoma. OncoTargets Ther. 2019;12:5163–5175. doi: 10.2147/OTT.S183558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu H., Dong Z., Tan P., Zhang Y., Liu L., Yang L., Liu Y., Cui H. Antibiotic drug tigecycline inhibits melanoma progression and metastasis in a p21CIP1/Waf1-dependent manner. Oncotarget. 2016;7:3171–3185. doi: 10.18632/oncotarget.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dong X.D., Yu J., Meng F.Q., Feng Y.Y., Ji H.Y., Liu A. Antitumor effects of seleno-short-chain chitosan (SSCC) against human gastric cancer BGC-823 cells. Cytotechnology. 2019;71:1095–1108. doi: 10.1007/s10616-019-00347-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Naruse C., Shibata S., Tamura M., Kawaguchi T., Abe K., Sugihara K., Kato T., Nishiuchi T., Wakana S., Ikawa M., Asano M. New insights into the role of jmjd3 and Utx in axial skeletal formation in mice. FASEB J. 2017;31:2252–2266. doi: 10.1096/fj.201600642R. [DOI] [PubMed] [Google Scholar]
- 42.Yu J., Liu D., Sun X., Yang K., Yao J., Cheng C., Wang C., Zheng J. CDX2 inhibits the proliferation and tumor formation of colon cancer cells by suppressing Wnt/β-catenin signaling via transactivation of GSK-3β and Axin2 expression. Cell Death Dis. 2019;10:26. doi: 10.1038/s41419-018-1263-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Carey B.W., Finley L.W., Cross J.R., Allis C.D., Thompson C.B. Intracellular α-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature. 2015;518:413–416. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]