

Abstract
Triple-negative breast cancers (TNBCs) are aggressive cancers, which currently do not have effective treatment options. Migration and establishment of metastatic colonies require dynamic cytoskeletal modifications characterized by polymerization and depolymerization of actin. Studies have demonstrated a direct molecular link between the integrin-focal adhesion kinase (FAK) pathway and cytoskeletal modifications. Nimbolide, a major bioactive compound present in neem leaves, shows promising anti-cancer effect on various cancers. In this study, we have demonstrated the growth and metastasis inhibitory potential of nimbolide on TNBC cells. Nimbolide inhibited cell proliferation, migratory, and invasive abilities of TNBC cells and also changed the shape of MDA-MB-231 cells, which is correlated with cytoskeletal changes including actin depolymerization. Furthermore, analysis revealed that integrins αV and β3, ILK, FAK, and PAK levels were downregulated by nimbolide. Even in cells where Rac1/Cdc42 was constitutively activated, nimbolide inhibited the formation of filopodial structures. Immunofluorescence analysis of phosphorylated p21 activated kinase (pPAK) showed reduced expression in nimbolide-treated cells. Nimbolide significantly reduced the metastatic colony formation in lung, liver, and brain of athymic nude mice. In conclusion, our data demonstrate that nimbolide inhibits TNBC by altering the integrin and FAK signaling pathway.
Keywords: nimbolide, triple-negative breast cancer, metastasis, integrin-FAK, Rac1/Cdc42, actin depolymerization, actin cytoskeleton
Graphical Abstract
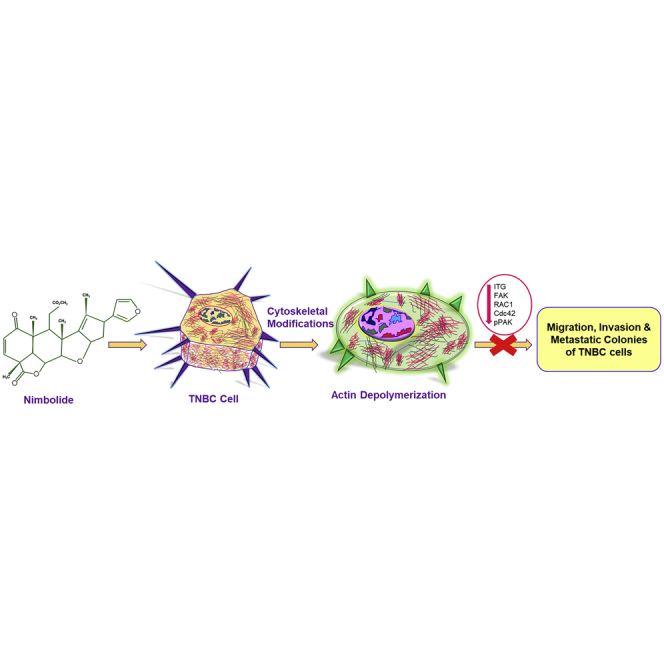
Nimbolide effectively inhibits the growth and metastasis of triple-negative breast cancer by altering the actin cytoskeleton through the integrin and FAK signaling pathway. These findings demonstrate a novel mechanism that could be targeted to inhibit triple-negative breast cancer.
Introduction
Triple-negative breast cancers (TNBCs) are highly aggressive cancers that frequently metastasize,1,2 which is the major cause of mortality. Despite their initial response to chemotherapeutic drugs, a major fraction of TNBCs develop chemoresistance.1,3 Hence, it is essential to identify new therapeutic targets/agents to reduce metastasis and TNBC-associated mortality.
Metastatic progression requires migration, invasion, and colony establishment.2,4 Migration and establishment of metastatic colonies requires dynamic cytoskeletal modifications.5,6 Actin cytoskeletal modifications are initiated and regulated mainly by extracellular matrix (ECM) factors through integrin-focal adhesion kinase (ITG-FAK) signaling.7, 8, 9, 10, 11 In response to binding with ECM, ITG-FAK forms complexes with ILK/parvin-B/PINCH to activate, Rac1/Cdc42 signaling.8, 9, 10, 11 This phosphorylates PAK1, which leads to inactivation of cofilin, which results in actin depolymerization.12 The ITG-FAK-mediated cofilin inactivation is critical for migration and metastatic colonization.
Nimbolide is a terpenoid lactone molecule, which is rich in leaves and flowers of Azadirachta indica, widely known as neem. Several lines of studies have established the anticancer effects of nimbolide in various cancers.13, 14, 15, 16, 17, 18, 19, 20 Anticancer properties of nimbolide are attributed to the induction of reactive oxygen species stimulation of apoptosis and inhibition of proliferation. Nimbolide treatment inhibits cancer cell invasion and migration through various mechanisms, including inhibition of PI3K/AKT signaling, ERK1/2 inhibition, suppression of MMPs, and induction of RECK in various cancers.13, 14, 15, 16, 17, 18, 19, 20 In the present study, we have evaluated the inhibitory potential of nimbolide on TNBC cell growth and metastasis. Many cancer cells overexpress FAK, and it plays a vital role in cancer progression and metastasis. Further, a variety of cellular functions that are essential for cancer progression and metastasis are impacted by integrins and Rho GTPases and Cdc42. These findings indicate the significant role played by this signaling axis in cancer. Though nimbolide has been studied as an anticancer agent, its influence on the ITG/FAK/Rac1/Cdc42 axis has not been investigated. Our study indicates that nimbolide inhibited an array of molecular pathways, in particular FAK-integrin-Rac1/Cdc42 signaling, to suppress tumor growth and metastasis.
Results
Nimbolide inhibits TNBC cell proliferation
Treatment with different concentrations of nimbolide significantly reduced the growth of the TNBC cells (Figure 1A). The IC50 concentration of nimbolide was ∼7.5 μM and ∼8.5 μM for MDA-MB-231 and MDA-MB-468, respectively, and we used this concentration for further experiments (Figure 1A). Nimbolide caused significant reduction in the S phase of the cell cycle (Figures 1B and 1C). Analysis of cell cycle regulatory proteins revealed that nimbolide increased the accumulation of cyclin E and decreased CDK2 phosphorylation (Figures S1A and S1B). Further, nimbolide treatment increased γH2AX and 14-3-3σ levels, indicating the occurrence of DNA damage and cell cycle arrest21 (Figures S1A and S1B). Nimbolide inhibited the activation of AKT and mTOR and its downstream molecular targets p70S6K and 4EBP1 as early as 3 h after treatment (Figure 1D). Further, nimbolide inhibited the activation of ERK1/2 and increased the phosphorylation of JNK as early as 30 min until 6 h (Figure 1E). These data demonstrate that nimbolide inhibits TNBC cell proliferation by inhibiting cell cycle progression and also by inhibiting AKT/mTOR signaling. Further, elevated levels of 14-3-3σ and inhibition of pERK1/2 could be another reason for inhibition of cell proliferation by nimbolide.
Figure 1.

Nimbolide inhibits the growth of TNBC cells
(A) Measurement of cell viability. MDA-MB-231, MDA-MB-468, and MCF10A cells were treated with indicated concentrations of nimbolide for 24 h and the cell viability was measured using MTS assay. Data are the mean ± SD of three independent experiments (with 5 replicates) (n = 3). ∗p < 0.05 versus UT. (B and C) Cell cycle analysis. MDA-MB-231 cells were treated with nimbolide (7.5 μM) for 12 h and 24 h and stained with propidium iodide, and we analyzed the DNA content in the flow cytometer. Data are the mean ± SD of three independent experiments. ∗p < 0.05 versus control (n = 3). (D and E) Immunoblotting was performed to analyze the mTOR, p70S6K, 4EBP1, pAKT, and MAPK signaling in MDA-MB-231 cells treated with nimbolide for the indicated time points (n = 3).
Nimbolide induced apoptosis and inhibited invasion and migration
Our immunofluorescence studies revealed that nimbolide treatment drastically increased the expression of active caspase 3 (Figure 2A). Flow cytometric analysis of MDA-MB-231 cells treated with nimbolide showed 15.6% increase in apoptotic cell death (Figure S1C). Further, molecular markers of apoptotic cell death like cleaved PARP and cleaved caspase 3 levels were elevated in the nimbolide-treated cells at 6 h and kept rising until 24 h, whereas the pro-caspase 3 levels started to decline as early as 30 min (Figure 2B). Pro-apoptotic proteins Bax, Bak, and Bim levels were increased in response to nimbolide treatment, and anti-apoptotic proteins Bcl2 and Bcl-xl levels were reduced (Figure 2C). Collectively, these data indicate that nimbolide induces apoptosis in TNBC cells through both extrinsic13,14 and intrinsic pathways.
Figure 2.

Nimbolide inhibits the migration and invasion of MDA-MB-231 cells
(A) MDA-MB-231 cells were treated with nimbolide for 6 h and analyzed for cleaved caspase 3 expression by immunofluorescence analysis using confocal microscopy. Cl-Casp3, cleaved caspase 3. (B) MDA-MB-231 cells were treated with nimbolide for indicated time points, and the lysates were analyzed for the expression of cleaved PARP, pro, and cleaved caspase 3. (C) Cell lysates from MDA-MB-231 cells treated with nimbolide for indicated times were analyzed by immunoblotting using indicated pro and anti-apoptotic protein-specific antibodies. (D and E) Nimbolide inhibits invasion and migration of MDA-MB-231 cells. Cells were treated with nimbolide for 24 h, and cell invasion and migration were evaluated by using Matrigel invasion assay and scratch assay, respectively. (F) Nimbolide altered the expression of EMT markers in MDA-MB-231 cells. Immunoblotting of cells treated with nimbolide was performed to analyze the expressions of E-cadherin, Slug, Zeb1, and β-catenin (n = 3).
Our results indicated that there was a 10-fold reduction in both invasion and migration of nimbolide-treated cells, clearly indicating the inhibitory effect of nimbolide (Figures 2D and 2E). Further, investigation of epithelial-to-mesenchymal (EMT) markers indicated that nimbolide drastically suppressed the EMT-associated transcription factors slug and zeb1 along with β-catenin (Figure 2F). In contrast, expression of epithelial marker E-cadherin level was increased by nimbolide, indicating suppression of EMT (Figure 2F).
Nimbolide modifies cellular cytoskeleton by inducing actin depolymerization
We noticed a drastic change in the cell morphology in response to nimbolide treatment. The elongated MDA-MB-231 cells underwent morphological changes and became round in shape rapidly, in 30 min after nimbolide treatment (Figure 3A). Almost 90% of the cells acquired round morphology by 3 h of nimbolide treatment (Figure 3A). Cellular shape is primarily regulated by cytoskeletal proteins, and the morphological changes observed due to nimbolide treatment prompted us to investigate changes in the cytoskeletal proteins. The immunofluorescent imaging of actin indicated that the wild-type MDA-MB-231 cells have a long, elongated shape with a well-defined network of actin stress fibers. Nimbolide treatment disrupted the actin network in a dose-dependent manner (Figure S2A). We also observed the initiation of apoptosis at early time points due to nimbolide treatment (Figure S2B). The live observation of actin filaments (F-actin) continuously for 100 min after nimbolide treatment indicated that nimbolide-induced actin cytoskeletal modification started as early as 10 min and affected almost all the cells by 90 min (Figure 3B; Video S1). Further, we noticed that the cells that are long and elongated in structure are impacted first and the most (Video S1). Live cell imaging analysis also indicated that actin filaments underwent rapid depolymerization post nimbolide treatment. Our results further indicated that there was a drastic increase in actin depolymerization after the addition of nimbolide. The depolymerization caused by nimbolide increase with time (30 min [180%], 1 h [196%], and 3 h [278%]) compared to controls (Figures 3C and 3D). Furthermore, F-actin levels declined in nimbolide-treated cells at different time points (30 min [70%], 1 h [56%], and 3 h [36%]) compared to controls (Figure 3E). Our confocal microscopy data indicated that in the control cells the expression and co-localization of α-actinin was primarily found inside the cells and mainly at the base of lamellipodia structures (Figure 3F; Video S2). Predominantly, in the nimbolide-treated cells, the localization of α-actinin was at the surface, and most of the α-actinin was co-localized with actin, indicating the strong stress fiber formation at the cell boundary (Figure 3F; Video S3). Collectively, the data demonstrated that nimbolide altered the cellular cytoskeleton by inducing actin depolymerization and disrupted the actin cytoskeletal network of the cells leading to change in cellular morphology.
Figure 3.

Nimbolide induced cellular reorganization through actin depolymerization
(A) Phase contrast microscopic image of MDA-MB-231 cells treated with nimbolide for 30 min, 1 h, and 3 h showing change in cellular morphology. (B) Live cell fluorescence microscopic images of MDA-MB-231 cells treated with nimbolide showing reduction in actin cytoskeletal network with increase in time. MDA-MB-231 cells were incubated with SiR-actin (fluorescent, cell membrane permeable molecule binds specifically to F-actin) for 1 h and treated with nimbolide for further 90 min. The cellular events were captured every 10 s using Leica microscope (with 63× objective) (F-actin, red). (C) Actin depolymerization assay was performed using MDA-MB-231 cell lysates treated with nimbolide for 30 min, 1 h, and 3 h. Cell lysates were added to the pyrene-labeled actin filaments and change in fluorescence intensity was recorded using BMG microplate reader. (D) Actin depolymerization was analyzed quantitatively by analyzing the reduction in pyrene fluorescence using ClARIOstar software version V5.61. Data represent mean ± SD of three independent experiments. ∗p < 0.05 versus control. (E) Nimbolide treatment reduced the expression of F-actin. F-actin was isolated from MDA-MB-231 cells treated with nimbolide at indicated time points, run on SDS-PAGE, and probed with actin antibody. The expression of F-actin was analyzed using densitometry; the data represent mean ± SD of three independent experiments. ∗p < 0.05 versus control. (F) Immunofluorescent analysis of α-actinin and actin in MDA-MB-231 cells after treatment with nimbolide (n = 3).
Nimbolide treatment induced actin cytoskeletal modification started as early as 10 minutes and affected almost all the cells by 90 minutes.
Confocal microscopy data indicated that in the control cells the expression and co-localization of α-actinin was primarily found inside the cells and mainly at the base of lamellipodia structures.
Nimbolide treatment resulted in localization of α-actinin at the cell surface and most of the α-actinin was co-localized with actin indicating the strong stress fiber formation at the cell boundary.
Nimbolide inhibits FAK signaling
FAK signaling is a key signaling pathway that influences invasion and migration and also controls actin dynamics.22 Our FAK-focused PCR array data indicated that ITG-FAK signaling pathway-associated components were downregulated in the nimbolide treatment, including integrin αV (−6.22), β3 (−7.43), ILK (−3.95), FAK (−6.53), and PAK (−4.72) (Figure 4A). Our immunoblot analysis also revealed that integrins αV, β3, and β4 levels were reduced 3 h after nimbolide treatment, and the reduction was prominent after 1 h (Figure 4B). Further, diminished levels of ILK and pFAK were also observed at early time points (Figure 4B). The major downstream effect of ITG-FAK signaling is Rac1/Cdc42 activation, and our results showed that active Cdc42 levels were drastically decreased in the nimbolide-treated cells as early as 15 min (Figure 4B). Phosphorylation of PAK and cofilin, molecular targets of active Rac1/Cdc42, was declined drastically at 15 min after nimbolide treatment (Figure 4B). Additionally, nimbolide treatment strikingly downregulated the expression of pFAK, and the actin networks were not complete (Figure S3; Videos S4 and S5). Collectively, these data suggest that nimbolide inhibited the activation of FAK, Rac1/Cdc42, PAK, and cofilin. We next investigated the co-localization status of FAK and paxillin in the cells in response to nimbolide treatment. In the control cells, expression of FAK and paxillin was high and also co-localized completely, whereas in the nimbolide-treated cells the expression of FAK was low and the co-localization of FAK and paxillin was also markedly reduced (Figure 4C). Vasodilator stimulated phosphoproteins (VASPs) are known to inhibit Arp2/3-induced actin branching and thereby inhibit lamellipodia formation. Immunoblot data showed that nimbolide treatment increased the levels of phosphorylated VASP (pVASP) (S157), pVASP (S239), and Diaphanous1 (Diaph1) (Figure 4D). Our confocal microscopy results showed that in the control cells the expression of pVASP was very low and confined to the middle of the cells (Figure 5A). In the nimbolide-treated cells, the expression of pVASP increased and was distributed all over the cell (Figure 5A). Immunofluorescence data show that diaph1 levels were moderate in the control cells but were spread throughout the cell, indicating that diaph1 could also be a reason for active actin network formation (Figure 5B). At 30 min after nimbolide treatment, we observed that diaph1 was mainly localized to the cell membrane, and this could be the cause of increased stress fiber formation at 3 h of nimbolide treatment (Figure 5B).
Figure 4.

Nimbolide inhibits the expression of integrin and associated FAK signaling
(A) Focal adhesion kinase pathway-focused PCR array of cells treated with nimbolide (6 h) was performed. The differential expression of genes over 2-fold compared to control were represented in the bar diagram. The data represent mean ± SD of three replicates. (B) Immunoblotting of MDA-MB-231 cells treated with nimbolide for indicated times reveals the inhibition of integrin-FAK signaling axis. (C) Co-localization of actin and paxillin (focal adhesion point protein) was evaluated by immunofluorescence analysis (FAK, focal adhesion kinase; PAX, paxillin). (D) Immunoblot analysis of MDA-MB-231 cells treated with nimbolide at different time points shows that phosphorylated vasodilator stimulated phosphoprotein (pVASP) (both S157 and S239) and Diaphanous 1 (Diaph1) were increased (n = 3).
Figure 5.

Nimbolide altered the expression of proteins associated with actin dynamics
(A–C) Immunofluorescence images of MDA-MB-231 cells treated with nimbolide shows the localization of (A) F-actin and pVASP, (B) F-actin and Diaph1, and (C) F-actin and phosphorylated p21 activated kinase (pPAK). (D) Nimbolide-induced actin depolymerization is overcome by Rac1/Cdc42 activation. MDA-MB-231 cells were treated with 1 μg/mL of Rac1/Cdc42 activator (bacterial cytotoxic necrotizing factor [CNF] toxin-based G-switch reagent) for 1 h and followed by nimbolide for indicated times. The cells were analyzed for F-actin and pPAK expression using immunofluorescence analysis (n = 3).
Immunofluorescence showing elongated focal adhesion points and prominent interaction with actin cytoskeleton in control cells.
Nimbolide treatment strikingly down regulates the expression of pFAK and the actin networks are not complete.
Rac1/Cdc42 activation overrides the nimbolide induced actin depolymerization
FAK-induced activation of Rac1/Cdc42 is known to regulate the stability of polymerized actin. Active Cdc42 stabilizes F-actin mainly by phosphorylating the actin-severing protein cofilin, which results in inactivation of cofilin.23,24 To identify the impact of nimbolide on cofilin, we evaluated PAK1, the immediate upstream regulator of cofilin. Phospho PAK1 expression was diminished in the cells treated with nimbolide (Figure 5C). In our next experiment, we constitutively activated Rac1/Cdc42 by using a bacterial cytotoxic necrotizing toxin-based inhibitor and analyzed the expression of active PAK1, a downstream molecule of Rac1/Cdc42 signaling, which directly phosphorylates cofilin and inactivates it. The activation of Rac1/Cdc42 increased the lamellipodia and filopodia formation in the control cells, while nimbolide treatment inhibited the filopodial structures remarkably (Figure 5D). The phosphorylation of PAK1 was not affected by nimbolide treatment in the Rac1/Cdc42-activated cells, and we also observed prominent, well-structured actin filament networks (Figure 5D), indicating that nimbolide inhibits upstream of PAK1 signaling. Further, we performed Rac/Cdc42 rescue experiments to investigate the effect of nimbolide on proliferation, migration, and invasion. Activation of Rac/Cdc42 diminished the anti-proliferative, anti-migratory, and anti-invasive effect of nimbolide, indicating that nimbolide could be exerting its anticancer effects through Rac/Cdc42 (Figures S4A–S4C).
Nimbolide inhibits in vivo tumor growth and metastatic colony formation
Nimbolide treatment (20 mg/kg body weight [BW]) significantly suppressed the growth of MDA-MB-231 xenograft tumors (Figures 6A and 6B). Xenograft tumors receiving nimbolide showed strong reduction in pAKT, pmTOR, p44/42, and Bcl2 levels, suggesting that cellular proliferation and survival mechanisms are considerably reduced (Figure 6C). On the other hand, apoptotic markers such as cleaved caspase 3 and Bax were increased, demonstrating the induction of apoptosis in these xenograft tumors (Figure 6C). We also analyzed the components of ITG-FAK signaling, which revealed that nimbolide treatment suppressed the expression of integrins β3, β4, pFAK, and ILK (Figure 6C). Further, active Rac1+Cdc42, phosphorylated p21 activated kinase (pPAK1), and pCofilin quantities were also reduced in the nimbolide-treated xenograft tumors, suggesting the inhibition of Rac1/Cdc42 signaling (Figure 6C). We observed a strong spread and establishment of metastatic colonies in the control animals, predominantly in the lungs and a few colonies in the brain and liver (Figures 6D and 6E). However, the metastatic spread of MDA-MB-231 cells was reduced drastically in nimbolide-treated animals (Figures 6D and 6E). Altogether, these results suggest that nimbolide treatment significantly suppresses the aggressiveness of TNBC cells.
Figure 6.

Nimbolide (20 mg/kg BW) inhibits xenograft tumor growth and metastatic colony formation
(A) Tumor volume measurements of control and nimbolide-treated mice showing significant reduction in tumor growth of mice treated with nimbolide. (B) Representative images of tumor-bearing mice treated with vehicle and nimbolide. (C) Xenograft tumors from control and nimbolide-treated mice were subjected to immunoblotting of proteins involved in proliferation, apoptosis, and integrin-FAK signaling. (D) In vivo imaging of mice injected with MDA-MB-231 cells expressing GFP shows the metastatic colonies in lung, liver, and brain of mice from both (control and nimbolide) groups. (E) Representative images and the corresponding number of metastatic colonies of lung, liver, and brain from mice treated with vehicle and nimbolide. Each bar represents the mean ± SD of IHC slides from 5 animals, ∗p < 0.05 (n = 3).
Discussion
In this study, our primary objective was to evaluate the anti-proliferative and anti-metastatic potential of nimbolide against TNBC. Our results demonstrate that nimbolide is cytotoxic to TNBC cells by inhibiting the AKT/mTOR axis, and it also blocks metastasis through inhibition of the ITG-FAK pathway. Studies have shown that the AKT/mTOR pathway is the major signaling pathway that is upregulated in cancer cells.25,26 However, targeting the AKT/mTOR pathway alone is not enough to control cancer progression, because of the activation of alternative proliferative mechanisms, including MAPK signaling. We and others have demonstrated that nimbolide blocks several significant cell survival and proliferative pathways to inhibit the growth of cancers.17, 18, 19 Nimbolide is also shown to activate apoptotic signaling through various mechanisms, including reactive oxygen species (ROS), TRAIL, death receptor, and nuclear factor κB (NF-κB)-mediated pathways.13,14 The ITG/FAK/Rac1/Cdc42 axis plays a major role in cancer progression and metastasis. We attempted to investigate how nimbolide influenced this pathway to inhibit TNBC. The risk of cancer increases with age, and mutations drive the evolution of cancers. Plant products like nimbolide are cytotoxic by causing DNA damage and could influence the mutations that drive cancers. Though they can influence these processes in cancer cells, they might also do the same in some normal cells, which could be the reason for undesirable side effects. Based on our in vivo data, nimbolide is well tolerated, without major side effects. Considering the metastatic progression of cancer cells, FAK signaling is a major player, which arguably enhances the extravasation step by increasing cell migration and invasion. Studies evaluating the effect of nimbolide on these lines are limited. In our studies, we observed prominent alteration in 14-3-3σ and DNA damage marker γH2AX and speculate that this increase in 14-3-3σ could have played a role in induction of cell cycle arrest and subsequent DNA damage.
In our experiments, we noticed a drastic change in cell shape and structure in response to nimbolide, which prompted us to investigate its influence on cytoskeletal modifications and eventually on metastatic progression. Nimbolide induced actin depolymerization by inhibiting ITG-FAK and Rac1/Cdc42 pathways. These are key pathways that are involved in the metastatic colonization immediately after extravasation.27,28 ITG-FAK signaling primarily regulates actin dynamics through focal adhesion points, where FAK binds to and regulates actomyosin stress fibers.29,30 Disruption of ITG-FAK signaling and subsequent induction of actin depolymerization is a promising approach to reduce metastasis. Phosphorylation of FAK was blocked by nimbolide inside the cell. Though there was stress fiber formation in the edges, it did not form a complete actin filament network like control cells (Videos S4 and S5). Nimbolide, by disrupting FAK activation, dismantles the actin cytoskeletal network, which in turn inhibits migration, invasion, and ensuing metastasis.
Activation of PAK by Rac1/Cdc42 induces formation of lamellipodia, filopodia, membrane ruffles, stress fibers, and focal adhesion points.31,32 Nimbolide treatment drastically reduced the levels of active Rac1/Cdc42. Enhanced levels of active (unphosphorylated) cofilin due to decreased pPAK indicates that nimbolide induces actin depolymerization through inhibition of Rac1/Cdc42/PAK signaling. In our experiments, constitutive activation of Rac1/Cdc42 increased the number of filopodia-like structures, while the nimbolide treatment drastically reduced these structures. On the other hand, nimbolide treatment did not promote actin depolymerization in Rac1/Cdc42-overexpressing cells. This suggests that nimbolide inhibits the upstream regulators of Rac1/Cdc42 signaling. Further, our data also suggested that activation of Rac1/Cdc42 was effective in reducing the anticancer effect of nimbolide, indicating that nimbolide could be exerting its action through the Rac1/Cdc42-signaling axis.
VASPs are critical regulators of actin dynamics, specifically at the membrane protrusions. Studies indicate that VASPs can inhibit the actin cytoskeleton by binding to actin polymerizing enzymes such as formins, profilins, and Arp2/3.33,34 Nimbolide increased the phosphorylation of VASP at S157 and S239. VASP mainly binds to the actin monomer and blocks the binding of actin polymerizing enzymes at the lamellipodial structures and inhibits actin polymerization.35 We speculate that nimbolide, by increasing pVASP, blocked actin polymerization, and this could be the reason for inhibition of Rac1/Cdc42-induced filopodial structures. Molecular analysis of tumor tissues displayed that active forms of major signaling molecules of the integrin-FAK pathway were downregulated. Though this is not considered as direct evidence, the downregulation of FAK signaling is also a factor in nimbolide-induced suppression of TNBC metastasis. We and others have also shown that nimbolide can influence its anticancer properties through ROS signaling.19,36,37 In addition, it has also been demonstrated that nimbolide impacts various signaling pathways, including the NF-κB pathway,14,38 the Wnt/β-catenin pathway,14 and the STAT-3-signaling pathway.36
To summarize, our findings demonstrate that nimbolide inhibits TNBC growth by inhibiting AKT/mTOR and MAPK signaling and induces apoptosis. Nimbolide also exerts its anti-metastatic effects through inhibition of ITG-FAK and Rac1/Cdc42-signaling pathways to stimulate actin depolymerization. Thus, our results provide evidence for the anti-cancer and anti-metastatic effects of nimbolide against TNBCs.
Materials and methods
Cell lines and cell culture
TNBC cell lines MDA-MB-231, MDA-MB-468, and non-malignant breast epithelial cell line MCF10A were obtained from the American Type Culture Collection. MDA-MB-231 and MDA-MB-468 cell lines were maintained in RPMI 1640 medium supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin antibiotic mixture. MCF10A cells were cultured in mammary epithelial basal media (MEBM) supplemented with SingleQuots. Cells were cultured at 37°C, in an atmosphere of 95% air and 5% CO2.
Cell viability assay
Cell viability was evaluated using the CellTiter 96 AQueous One Solution Cell Proliferation Assay kit as per the manufacturer’s protocol. Briefly, cells were seeded in a 96-well plate at a density of 3 × 103 cells/well and incubated at 37°C overnight. Then the cells were treated with various concentrations of nimbolide (2.5–10 μM) for 24 h. Rac1/Cdc42 rescue experiments were performed by using Rac/Cdc42 activator I. Control cells were treated with equal amount of DMSO, and the amount of DMSO added was less than 0.05% of total volume. At the end of the experimental period, AQueous One reagent was added to each well and incubated for 1 h, and the optical density (OD) was measured at 490 nm using a microplate reader.
Cell death assay
Cell death analysis was performed using annexin V-fluorescein isothiocyanate (FITC) apoptosis detection kit. Briefly, cells (2 × 105) were plated in 6-well plates and incubated with IC50 concentration of nimbolide for 24 h and 48 h. At the end of the treatment period, cells were collected and stained with annexin V-FITC and propidium iodide. Stained cells were analyzed in a C6 Accuri tabletop flow cytometer.
Cell cycle analysis
Cells were treated with IC50 concentration of nimbolide for 12 h and 24 h. After the treatment, cells were harvested in cold PBS, fixed in 70% ethanol, and stained with propidium iodide for 30 min. The cell cycle was assessed using LSR II FACScan analyzer. The data were analyzed, and the distribution of cells in different phases of the cell cycle were calculated using FlowJo software.
Immunoblot analysis
MDA-MB-231 cells were incubated with IC50 concentration of nimbolide at different time points (0–6 h). After each time point, cells were lysed using mammalian protein extract reagent. Total proteins were resolved by gel electrophoresis and transferred to PVDF membranes. Blots were probed with the respective primary antibodies (Table S1). The blots were re-probed with β-actin antibody (Sigma-Aldrich) as a loading control. Blots were exposed with enhanced chemiluminescence (ECL) reagent, and the signals were captured using Fujifilm LAS-4000 luminescent image analyzer.
Migration assay
MDA-231 cells (3 × 105 cells/well) were cultured in a monolayer in each well of a 6-well plate. Scratches were made using a 200 μL pipette tip, then washed with PBS to remove dead cells and also the cells in the scratch area. Cells were treated with 5 μM concentration of nimbolide or with DMSO as a control. The 6-well plate was placed in a BioStation CT that was programed to capture images every 2 h. Cell migration was analyzed by calculating the distance migrated after 48 h.
Matrigel invasion assay
The invasiveness was measured using the Matrigel-coated Transwell chambers as described previously.39 The effect of 5 μM nimbolide on percentage of invaded cells was calculated by analyzing the images using NIS-Elements AR software.
FAK pathway-focused PCR array and real-time RT-PCR
Total RNA was extracted from MDA-MB-231 cells using Trizol. The concentration of RNA was determined using NanoDrop 2000, and 1 μg of total RNA was converted to cDNA using the single-strand synthesis kit and used for the PCR array. Real-time PCR was performed, and the fold change in gene expression was calculated using the SABiosciences web-based PCR array analysis software.
Live cell imaging
Live cell imaging of F-actin was performed using the SiR-actin Kit. MDA-MB-231 cells (1 × 104 cells/chamber) were plated in a Nunc Lab-Tek four-chamber slide. 1 μM SiR-actin in RPMI 1640 medium containing 10% FBS was added to the cells and incubated at 37°C for 1 h. Cells were then treated with nimbolide and live cell images were recorded at every 10 s using DMI 6000 B confocal microscope. LAS X software was used to analyze the images and make movies.
Immunofluorescence
MDA-MB-231 cell (1 × 104 cells/chamber) were seeded on a coverslip and treated with IC50 concentration of nimbolide for 30 min, 1 h, and 2 h. After treatment, the cells were fixed with 4% paraformaldehyde in PBS for 15 min. Cells were permeabilized with 0.1% Triton X-100 and blocked with 3% BSA for 30 min. Indicated primary antibodies were added and incubated for 1 h, washed, and probed with appropriate secondary antibody. The coverslips were mounted using DAPI-containing mounting medium. For staining F-actin, acti-stain-488 or acti-stain-555 was used. Cells were observed using a confocal microscope.
Actin depolymerization assay
The effect of nimbolide on actin polymerization was evaluated using the Actin Polymerization Biochem Kit. MDA-MB-231 cells were treated with IC50 concentration of nimbolide for 30 min, 1 h, and 3 h, and the cells were extracted with 20 mM HEPES and 20 mM NaCl containing protease inhibitors. Pyrene F-actin was prepared using G-actin stock. Buffer and F-actin were added to a 96-well plate and change in fluorescence was monitored every 60 s using ClARIOstar fluorimeter until the signal was stable. Protein extract from control and nimbolide-treated samples was added to the plate and change in fluorescence was monitored for 40 min. The fluorescence intensity was analyzed using MARS data analysis software.
Cellular F-actin assay
Cellular F-actin levels were analyzed using G-Actin/F-Actin In Vivo Assay Biochem Kit. Cells were collected and lysed in the F-actin stabilization buffer and centrifuged at 100,000 × g for 1 h at 37°C to collect the F-actin pellet. Then F-actin was depolymerized with F-actin depolymerization solution, run on SDS-PAGE, and transferred to polyvinylidene fluoride (PVDF) membrane. The blot was then probed with anti-actin rabbit primary antibody followed by anti-rabbit-horseradish peroxidase (HRP) secondary antibody. Blot was exposed with ECL reagent, and the chemiluminescent signals were captured using Fujifilm LAS-4000 luminescent image analyzer.
Xenograft tumor growth assay and metastatic colony formation assay
All animal experiments were performed in accordance with guidelines and regulations approved by Texas Tech University Health Sciences Center Animal Care and Use Committee. MDA-MB-231 cells (2 × 106) were mixed with 50 μL of Matrigel (Corning) and injected into the flanks of 7-week-old athymic nude mice purchased from Harlan Laboratories. The tumors were allowed to grow up to ∼50 mm3 and the animals were divided into 2 groups as control and nimbolide treated (n = 6). Control animals received DMSO, and the experimental group of mice were intraperitoneally administered with 20 mg/kg BW nimbolide, twice/week. The growth of the tumors was monitored for 5 weeks, and the tumor volume was calculated using the formula 4/3π × r12 × r2. At the end of the experiment, mice were euthanized, and tumors were excised and used for molecular analysis.
For metastatic colony formation assay, MDA-MB-231 cells expressing GFP plasmid (Cell Biolabs) were used. Briefly, 1 × 106 cells were injected into the mice through tail vein. The mice were divided into 2 groups; the control mice received DMSO and the experimental mice received 20 mg/ kg BW of nimbolide intraperitoneally, twice/week for 3 weeks. The formation of metastatic colonies was monitored using IVIS imaging system. After 3 weeks of treatment, the mice were euthanized; liver, lung, and brain were excised and fixed in 0.1% paraformaldehyde for 10 min and perfused in OCT medium for immunofluorescence analysis. Images were taken using DMI 6000 B confocal microscope and were processed using LAS X software.
Statistical analysis
All studies were performed as three independent experiments with 6 experimental replicates and are represented as the mean ± SD. Six animals per group were used to study the effect of nimbolide in vivo. Differences between the control and experimental groups were analyzed by unpaired Student’s t test, using GraphPad Prism, version 7.0 (GraphPad Prism Software, San Diego, CA, USA). p values < 0.05 were considered statistically significant.
Acknowledgments
This work was supported by Texas Tech University Health Sciences Center El Paso funds.
Author contributions
Conception and design: A.A. and R.L.; development of methodology: A.A., R.S., and R.L.; acquisition of data (provided animals, provided facilities, etc.): A.A., R.S., and R.L.; analysis and interpretation of data: A.A., R.L., and R.S.; writing, review, and/or revision of the manuscript: A.A., R.L., and R.S.; administrative, technical, or material support: R.L.; study supervision: R.L.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2021.02.014.
Supplemental information
References
- 1.Ovcaricek T., Frkovic S.G., Matos E., Mozina B., Borstnar S. Triple negative breast cancer - prognostic factors and survival. Radiol. Oncol. 2011;45:46–52. doi: 10.2478/v10019-010-0054-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neophytou C., Boutsikos P., Papageorgis P. Molecular Mechanisms and Emerging Therapeutic Targets of Triple-Negative Breast Cancer Metastasis. Front. Oncol. 2018;8:31. doi: 10.3389/fonc.2018.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 4.Chiang A.C., Massagué J. Molecular basis of metastasis. N. Engl. J. Med. 2008;359:2814–2823. doi: 10.1056/NEJMra0805239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fife C.M., McCarroll J.A., Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br. J. Pharmacol. 2014;171:5507–5523. doi: 10.1111/bph.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langley R.R., Fidler I.J. The seed and soil hypothesis revisited--the role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer. 2011;128:2527–2535. doi: 10.1002/ijc.26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gkretsi V., Stylianopoulos T. Cell Adhesion and Matrix Stiffness: Coordinating Cancer Cell Invasion and Metastasis. Front. Oncol. 2018;8:145. doi: 10.3389/fonc.2018.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang Y., Tu Y., Gkretsi V., Wu C. Migfilin interacts with vasodilator-stimulated phosphoprotein (VASP) and regulates VASP localization to cell-matrix adhesions and migration. J. Biol. Chem. 2006;281:12397–12407. doi: 10.1074/jbc.M512107200. [DOI] [PubMed] [Google Scholar]
- 9.Böttcher R.T., Lange A., Fässler R. How ILK and kindlins cooperate to orchestrate integrin signaling. Curr. Opin. Cell Biol. 2009;21:670–675. doi: 10.1016/j.ceb.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Horton E.R., Astudillo P., Humphries M.J., Humphries J.D. Mechanosensitivity of integrin adhesion complexes: role of the consensus adhesome. Exp. Cell Res. 2016;343:7–13. doi: 10.1016/j.yexcr.2015.10.025. [DOI] [PubMed] [Google Scholar]
- 11.Horton E.R., Byron A., Askari J.A., Ng D.H.J., Millon-Frémillon A., Robertson J., Koper E.J., Paul N.R., Warwood S., Knight D. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat. Cell Biol. 2015;17:1577–1587. doi: 10.1038/ncb3257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Legate K.R., Fässler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J. Cell Sci. 2009;122:187–198. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- 13.Gupta S.C., Prasad S., Reuter S., Kannappan R., Yadav V.R., Ravindran J., Hema P.S., Chaturvedi M.M., Nair M., Aggarwal B.B. Modification of cysteine 179 of IkappaBalpha kinase by nimbolide leads to down-regulation of NF-kappaB-regulated cell survival and proliferative proteins and sensitization of tumor cells to chemotherapeutic agents. J. Biol. Chem. 2010;285:35406–35417. doi: 10.1074/jbc.M110.161984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kavitha K., Vidya Priyadarsini R., Anitha P., Ramalingam K., Sakthivel R., Purushothaman G., Singh A.K., Karunagaran D., Nagini S. Nimbolide, a neem limonoid abrogates canonical NF-κB and Wnt signaling to induce caspase-dependent apoptosis in human hepatocarcinoma (HepG2) cells. Eur. J. Pharmacol. 2012;681:6–14. doi: 10.1016/j.ejphar.2012.01.024. [DOI] [PubMed] [Google Scholar]
- 15.Roy M.K., Kobori M., Takenaka M., Nakahara K., Shinmoto H., Isobe S., Tsushida T. Antiproliferative effect on human cancer cell lines after treatment with nimbolide extracted from an edible part of the neem tree (Azadirachta indica) Phytother. Res. 2007;21:245–250. doi: 10.1002/ptr.2058. [DOI] [PubMed] [Google Scholar]
- 16.Bodduluru L.N., Kasala E.R., Thota N., Barua C.C., Sistla R. Chemopreventive and therapeutic effects of nimbolide in cancer: the underlying mechanisms. Toxicol. In Vitro. 2014;28:1026–1035. doi: 10.1016/j.tiv.2014.04.011. [DOI] [PubMed] [Google Scholar]
- 17.Lin H., Qiu S., Xie L., Liu C., Sun S. Nimbolide suppresses non-small cell lung cancer cell invasion and migration via manipulation of DUSP4 expression and ERK1/2 signaling. Biomed. Pharmacother. 2017;92:340–346. doi: 10.1016/j.biopha.2017.05.072. [DOI] [PubMed] [Google Scholar]
- 18.Kowshik J., Mishra R., Sophia J., Rautray S., Anbarasu K., Reddy G.D., Dixit M., Mahalingam S., Nagini S. Nimbolide upregulates RECK by targeting miR-21 and HIF-1alpha in cell lines and in a hamster oral carcinogenesis model. Sci. Rep. 2017;7:2045. doi: 10.1038/s41598-017-01960-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subramani R., Gonzalez E., Arumugam A., Nandy S., Gonzalez V., Medel J., Camacho F., Ortega A., Bonkoungou S., Narayan M. Nimbolide inhibits pancreatic cancer growth and metastasis through ROS-mediated apoptosis and inhibition of epithelial-to-mesenchymal transition. Sci. Rep. 2016;6:19819. doi: 10.1038/srep19819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang L., Phan D.D., Zhang J., Ong P.S., Thuya W.L., Soo R., Wong A.L., Yong W.P., Lee S.C., Ho P.C. Anticancer properties of nimbolide and pharmacokinetic considerations to accelerate its development. Oncotarget. 2016;7:44790–44802. doi: 10.18632/oncotarget.8316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laronga C., Yang H.Y., Neal C., Lee M.H. Association of the cyclin-dependent kinases and 14-3-3 sigma negatively regulates cell cycle progression. J. Biol. Chem. 2000;275:23106–23112. doi: 10.1074/jbc.M905616199. [DOI] [PubMed] [Google Scholar]
- 22.Burridge K. Focal adhesions: a personal perspective on a half century of progress. FEBS J. 2017;284:3355–3361. doi: 10.1111/febs.14195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horton E.R., Humphries J.D., James J., Jones M.C., Askari J.A., Humphries M.J. The integrin adhesome network at a glance. J. Cell Sci. 2016;129:4159–4163. doi: 10.1242/jcs.192054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giancotti F.G., Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 25.Follo M.Y., Manzoli L., Poli A., McCubrey J.A., Cocco L. PLC and PI3K/Akt/mTOR signalling in disease and cancer. Adv. Biol. Regul. 2015;57:10–16. doi: 10.1016/j.jbior.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 26.Burris H.A., 3rd Overcoming acquired resistance to anticancer therapy: focus on the PI3K/AKT/mTOR pathway. Cancer Chemother. Pharmacol. 2013;71:829–842. doi: 10.1007/s00280-012-2043-3. [DOI] [PubMed] [Google Scholar]
- 27.Obenauf A.C., Massagué J. Surviving at a Distance: Organ-Specific Metastasis. Trends Cancer. 2015;1:76–91. doi: 10.1016/j.trecan.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen D.X., Bos P.D., Massagué J. Metastasis: from dissemination to organ-specific colonization. Nat. Rev. Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 29.Ross T.D., Coon B.G., Yun S., Baeyens N., Tanaka K., Ouyang M., Schwartz M.A. Integrins in mechanotransduction. Curr. Opin. Cell Biol. 2013;25:613–618. doi: 10.1016/j.ceb.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Humphrey J.D., Dufresne E.R., Schwartz M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014;15:802–812. doi: 10.1038/nrm3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bokoch G.M. Biology of the p21-activated kinases. Annu. Rev. Biochem. 2003;72:743–781. doi: 10.1146/annurev.biochem.72.121801.161742. [DOI] [PubMed] [Google Scholar]
- 32.Szczepanowska J. Involvement of Rac/Cdc42/PAK pathway in cytoskeletal rearrangements. Acta Biochim. Pol. 2009;56:225–234. [PubMed] [Google Scholar]
- 33.Krause M., Gautreau A. Steering cell migration: lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014;15:577–590. doi: 10.1038/nrm3861. [DOI] [PubMed] [Google Scholar]
- 34.Chesarone M.A., Goode B.L. Actin nucleation and elongation factors: mechanisms and interplay. Curr. Opin. Cell Biol. 2009;21:28–37. doi: 10.1016/j.ceb.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Loureiro J.J., Rubinson D.A., Bear J.E., Baltus G.A., Kwiatkowski A.V., Gertler F.B. Critical roles of phosphorylation and actin binding motifs, but not the central proline-rich region, for Ena/vasodilator-stimulated phosphoprotein (VASP) function during cell migration. Mol. Biol. Cell. 2002;13:2533–2546. doi: 10.1091/mbc.E01-10-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J., Ahn K.S., Kim C., Shanmugam M.K., Siveen K.S., Arfuso F., Samym R.P., Deivasigamanim A., Lim L.H., Wang L. Nimbolide-Induced Oxidative Stress Abrogates STAT3 Signaling Cascade and Inhibits Tumor Growth in Transgenic Adenocarcinoma of Mouse Prostate Model. Antioxid. Redox Signal. 2016;24:575–589. doi: 10.1089/ars.2015.6418. [DOI] [PubMed] [Google Scholar]
- 37.Liu J.F., Hou C.H., Lin F.L., Tsao Y.T., Hou S.M. Nimbolide Induces ROS-Regulated Apoptosis and Inhibits Cell Migration in Osteosarcoma. Int. J. Mol. Sci. 2015;16:23405–23424. doi: 10.3390/ijms161023405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Babykutty S., Priya P.S., Nandini R.J., Suresh Kumar M.A., Nair M.S., Srinivas P. Nimbolide retards tumor cell migration, invasion, and angiogenesis by downregulating MMP-2/9 expression via inhibiting ERK1/2 and reducing DNA-binding activity of NF-κB in colon cancer cells. Mol. Carcinog. 2012;51:475–490. doi: 10.1002/mc.20812. [DOI] [PubMed] [Google Scholar]
- 39.Rich R.L., Hoth L.R., Geoghegan K.F., Brown T.A., LeMotte P.K., Simons S.P., Hensley P., Myszka D.G. Kinetic analysis of estrogen receptor/ligand interactions. Proc. Natl. Acad. Sci. USA. 2002;99:8562–8567. doi: 10.1073/pnas.142288199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nimbolide treatment induced actin cytoskeletal modification started as early as 10 minutes and affected almost all the cells by 90 minutes.
Confocal microscopy data indicated that in the control cells the expression and co-localization of α-actinin was primarily found inside the cells and mainly at the base of lamellipodia structures.
Nimbolide treatment resulted in localization of α-actinin at the cell surface and most of the α-actinin was co-localized with actin indicating the strong stress fiber formation at the cell boundary.
Immunofluorescence showing elongated focal adhesion points and prominent interaction with actin cytoskeleton in control cells.
Nimbolide treatment strikingly down regulates the expression of pFAK and the actin networks are not complete.