

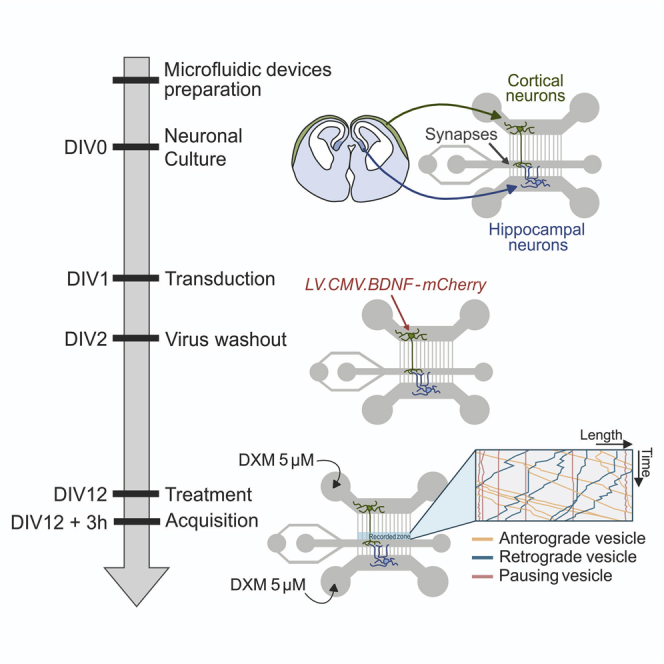
Summary
BDNF levels are reduced in the chronically stressed brain, in the area of hippocampus. Part of the hippocampal BDNF is provided by neuronal projection of the entorhinal cortex. Studying the cortico-hippocampal transport of BDNF in vivo is technically difficult. Here, we describe a protocol that reproduces mouse cortico-hippocampal circuit in vitro by plating neurons on the microfluidic devices and infecting the neurons with virus-encoding BDNF-mCherry, which allows investigation of the effects of elevated corticosterone levels on BDNF axonal transport.
For complete details on the use and execution of this protocol, please refer to Agasse et al. (2020).
Subject areas: Cell biology, Cell culture, Cell isolation, Cell-based assays, Neuroscience
Graphical abstract
Highlights
-
•
A detailed protocol to produce microfluidic devices modeling neuronal circuit
-
•
Preparation and plating of mouse neurons into microfluidic devices
-
•
Dexamethasone application to mimic chronic stress in neuronal cultures
-
•
Guidelines to record and analyze the dynamics of BDNF vesicles in axons
BDNF levels are reduced in the chronically stressed brain, in the area of hippocampus. Part of the hippocampal BDNF is provided by neuronal projection of the entorhinal cortex. Studying the cortico-hippocampal transport of BDNF in vivo is technically difficult. Here, we describe a protocol that reproduces mouse cortico-hippocampal circuit in vitro by plating neurons on the microfluidic devices and infecting the neurons with virus-encoding BDNF-mCherry, which allows investigation of the effects of elevated corticosterone levels on BDNF axonal transport.
Before you begin
Timing: 0.5–4 h
The original protocol to recreate mature neuronal circuits was published in (Virlogeux et al., 2018). The device has been modified from previous microfluidic chambers to allow independent access to and manipulation of synapses and their presynaptic and postsynaptic compartments (Taylor et al., 2010).
Chronic stress in humans and animals causes elevated levels of cortisol or corticosterone (CORT) respectively. CORT normally acts on two receptors, the mineralocorticoid receptor and the glucocorticoid receptor, but under conditions of stress (high CORT levels), CORT acts predominantly on the glucocorticoid receptor. Therefore, we used dexamethasone, a glucocorticoid receptor agonist, to capture this specificity in primary neuronal cultures.
-
1.
Gather the epoxy replicas made from master molds as described in (Virlogeux et al., 2018). The molds are available upon request (Lead contact). The polydimethylsiloxane (PDMS) microfluidic devices used in this protocol were described in (Virlogeux et al., 2018). The microfluidic device consists of two neuronal chambers, each containing either cortical or hippocampal neurons, connected via an intermediate synaptic chamber through microchannels of different lengths (500 μm for cortical neurons and 75 μm for hippocampal neurons) (Figure 1). Axons extend through the microchannels and form synaptic contacts. PDMS microfluidics can be replicated from these epoxy resins.
-
2.
Make sure to obtain a pregnant C57BL/6 mouse at stage E15.5 the day of the neuronal culture (day 1). One pregnant C57BL/6 mouse generally carries 6 to 9 embryos. One embryo yields 4 to 5 million of cortical neurons and 250 000 to 500 000 of hippocampal neurons. Each compartment of the microfluidic device requires 100 000 cells.
Note: We use the C57BL/6 mouse in this protocol because most of our knock-in mouse models were generated in this background. However, it is possible to use other mouse strains, such as CD1, as described in (Virlogeux et al., 2018). Another advantage of the CD1 mouse is the large number of embryos generated per pregnancy (12 to 14 embryos).
Note: E14 to E15.5 cortices and hippocampi consist of pure neuronal population. Later embryonic stages are inappropriate, as astrocytes will contaminate the cultures.
-
3.
Produce BDNF-mCherry (LV.CMV.BDNF-mCh) lentivirus as in (Salmon and Trono, 2007). The lentiviral plasmid is available upon request (Lead contact). An equivalent of 1 × 107 infectious units (IU/mL) are used to transduce the cortical neurons in the presynaptic chamber.
-
4.
Prepare stock solutions that can be stored at 4°C and/or aliquoted at −20°C, namely: the dissociation medium (DM), 10× kynurenic acid/magnesium (Ky/Mg), 10 mM dexamethasone (DXM), poly-D-lysine (0.1 mg/mL), poly-D-lysine (0.1 mg/mL) - laminin (10 μg/mL), and OptiMEM glucose. See Materials and equipment for detailed recipes.
Figure 1.

Schematic representation of the microfluidic device allowing fluidic isolation of cortical and hippocampal neuronal cultures
Chambers are connected via microchannels and an intermediate synaptic chamber. Neurons are seeded in rounded chambers. The addition (input) and withdrawal (output) of liquids (culture medium, coating, virus, and drugs) in the chambers are represented by arrows. The microchannels differ in length to allow axons from cortical neurons and dendrites from hippocampal neurons to reach the synaptic chamber. A coating gradient is used to keep hippocampal axons out of the synaptic chamber. The trafficking of BDNF-mCherry containing vesicles is recorded in the distal part of axons of cortical neurons.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Lentivirus: LV.CMV.BDNF-mCh | Lead contact | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Dow Corning Sylgard 184 Silicone Elastomer (contains silicon elastomer and curing agent) | Ellsworth | 0002-05-000007 |
| Neurobasal medium | Thermo Fisher Scientific | 21103049 |
| B27 | Thermo Fisher Scientific | 17504044 |
| Glutamax | Thermo Fisher Scientific | 35050038 |
| Penicillin/streptomycin | Thermo Fisher Scientific | 15140122 |
| Poly-D-lysine | Merck | P6407-5MG |
| Laminin | Merck | L2020 |
| 10× PBS | Thermo Fisher Scientific | Cat#14040-091 |
| 96% Ethanol | VWR | 20824.366 |
| Dexamethasone | Tocris Bioscience | 1126 |
| OptiMEM | Thermo Fisher Scientific | 31985047 |
| Glucose | Merck | G8769 |
| Na2SO4 | Merck | S6547 |
| K2SO4 | Merck | P9458 |
| MgCl2 | Merck | M8266 |
| CaCl2 | Merck | C1016 |
| HEPES | Merck | 83264 |
| Phenol red | Merck | 1072410005 |
| NaOH | Merck | S5881-M |
| Kynurenic acid | Merck | K3375 |
| Trypan blue solution | Merck | T8154 |
| Experimental models: organisms/strains | ||
| E15.5 pregnant C57/BL6J mouse | Charles River | N/A |
| Software and algorithms | ||
| ImageJ/Fiji | NIH | https://imagej.nih.gov/ij/download.html |
| Plugin Kymo Toolbox | https://github.com/fabricecordelieres/IJ-Plugin_KymoToolBox | N/A |
| CorelDRAW | https://www.coreldraw.com/ | N/A |
| GraphPad Prism 7.0 | https://www.graphpad.com/ | N/A |
| MetaMorph software | Molecular devices | https://www.moleculardevices.com/ |
| Other | ||
| FluoroDish sterile culture dish 35 mm | WPI | FD35100 |
| Epoxy replicas | Lead contact | N/A |
| 145 × 20 mm Petri dish | Greiner | 639160 |
| Plasma cleaner | Diener Electronic | Femto |
| Ultrasound cleaning bath | VWR | USC300TH |
| Centrifuge | N/A | N/A |
| Oven | Memmert | N/A |
| Whatman paper | Dutscher | 1607465 |
| Compressed air blow gun | Prevost | 27202-MTL |
| 0.22 μm syringe filter unit | Dutscher | 146611 |
| Stericup | Merk | S2GPU02RE |
| Inverted microscope | Zeiss | Axio Observer |
| Spinning disk confocal system | Yokogawa | CSU-W1-T3 |
| Wide field electron-multiplying CCD camera | Princeton Instrument | ProEM+1024 |
| Dumont #5 forceps, 11 cm, 0.05 × 0.02 mm straight tips | Fine science tools | 11295-10 |
| Surgical scissors, serrated, 40 cm | Fine science tools | 14007-14 |
| Surgical scissors straight tips, 11.5 cm | Fine science tools | 14060-11 |
| 50 mL conical tube | Dutscher | 352070 |
| 15 mL conical tube | Dutscher | 352096 |
| 1.5 mL Eppendorf tubes | Dutscher | 33290 |
| 2.5 mm Biopsy punch | Dutscher | 30733 |
| 3 mm Biopsy punch | Dutscher | 30735 |
| Scalpel blade | Fischer | 11728363 |
| Malassez cell | Dutscher | 140501 |
Materials and equipment
Dissociation medium (DM)
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Na2SO4 | 82 mM | 20.5 mL |
| 0.5 M K2SO4 | 30 mM | 15 mL |
| 1 M MgCl2 | 5.8 mM | 1.45 mL |
| 1 M CaCl2 | 252 μM | 63 μL |
| 1 M HEPES (pH 7.3–7.4) | 1 mM | 0.25 mL |
| 2.5 M Glucose | 20 mM | 2 mL |
| 0.5% Phenol red | 0.001% | 0.5 mL |
| 1 N NaOH pH 7.4 | 200–240 μN | 50–60 μL |
| dH2O | n/a | Up to 210 mL |
| Total | n/a | 250 mL |
We filter sterilize the DM under the cell culture hood. Store at 4°C. The DM can be used up to 1 year if properly stored.
Stock solution of kynurenic acid/magnesium (Ky/Mg) 10×
| Reagent | Final concentration | Amount |
|---|---|---|
| Kynurenic acid | 10 mM | 378 mg |
| 0.5% Phenol red | 0.0025% | 1 mL |
| 1 N NaOH | 9 mM | 1.8 mL |
| 1 M HEPES | 5 mM | 1 mL |
| 1 M MgCl2 | 100 mM | 20 mL |
| dH2O | n/a | 170 mL |
| Total | n/a | 200 mL |
Adjust the pH to 7.4 and filter sterilize the 10× Ky/Mg under the culture hood. Aliquots of 15 mL can be stored at −20°C for up to 2 years.
Growth medium
| Reagent | Final concentration | Amount |
|---|---|---|
| Neurobasal medium | n/a | 48 mL |
| Glutamax | 1% (v/v) | 0.5 mL |
| B27 | 2% (v/v) | 1 mL |
| Penicillin/streptomycin | 1% (v/v) | 0.5 mL |
| Total | n/a | 50 mL |
Keep at 4°C for up to 1 week.
Other solutions
| Name | Reagents |
|---|---|
| Dexamethasone (10 mM) | Dissolved in DMSO, aliquoted (5 μL) and stored at −20°C for up to 1 month |
| OptiMEM glucose | Mix 496 mL of OptiMEM with 4 mL of 2.5 M glucose. Filter sterilized under the cell culture hood. Store at 4°C for up to 6 months |
| DM with Ky/Mg (DM Ky/Mg) | Mix 27 mL DM with 3 mL of 10× Ky/Mg stock solution. Filter sterilize under the cell culture hood. It cannot be stored, prepare fresh solution for each culture |
| Poly-D-lysine (0.1 mg/mL) | Mix 5 mg of poly-D-lysine in 50 mL autoclaved distilled water. Aliquot 20 mL by 500 μL and store at −20°C for up to 1 year |
| Poly-D-lysine (0.1 mg/mL) - laminin (10 μg/mL) | Mix 30 mL of poly-D-lysine (0.1 mg/mL) with 300 μL of laminin at 1 mg/mL. Aliquot by 500 μL and store at −20°C for up to 1 year |
Step-by-step method details
Microfluidic devices (day 0)
Note: the duration of the preparation depends on the number of microfluidic devices to be prepared. Three microchambers are prepared per experimental conditions. A full day is required to prepare 24 microchambers. We usually perform the preparation in two half days.
-
1.Preparation of the PDMS:
-
a.Weigh 50 g of silicon elastomer (PDMS) and 5 g of curing agent (ratio 10:1) and add both to a disposable plastic cup.
-
b.Stir vigorously for 2–3 min and transfer the mixture into two 50 mL conical tubes.
-
c.Centrifuge for 2 min at 210 × g (15°C–25°C).
-
d.Fill the epoxy resins with the PDMS mixture.
-
e.Remove air bubbles by incubating the filled resins under vacuum for 1 h (15°C–25°C).
-
f.Allow the PDMS to polymerize for 3 h at 60°C in the oven.
-
a.
CRITICAL: It is important to remove any bubbles from the PDMS before polymerization in the oven.
Note: Epoxy resins containing polymerized PDMS can be stored for up to 1 year in a cupboard to protect them from dust (15°C–25°C).
-
2.Unmold and punch the microfluidic chambers.
-
a.Recover the PDMS pattern from the epoxy resin using a micropipette tip (Figure 2A).
-
b.Fill the epoxy resin as in the step 1.Note: Epoxy resins must always be filled with PDMS. After removing the pattern, refill the epoxy resins. These filled resins can be stored indefinitely until the next preparation is needed.
-
c.Punch and cut the PDMS microchambers according to the pattern (Figure 2B).
-
i.Cut around the pattern with a scalpel blade.
-
ii.Punch the three synaptic wells with the 2.5 mm punch.
-
iii.Punch the other wells with the 3 mm punch.
-
i.
-
a.
Pause point: After this step, the microchambers can be stored at room temperature for several weeks (15°C–25°C).
-
3.Preparation of microfluidics devices:
-
a.Wash the PDMS microchambers with 96% ethanol.
-
b.Place the microchambers in a 50 mL conical tube (up to 6 microchambers per tube) filled with 25 mL of 96% ethanol.
-
c.Sonicate the microchambers contained within the conical tube (3 × 15 s) using an ultrasound cleaning bath to remove grease and dust.Note: The microchambers can stick to each other. Gently shake the conical tube between the three rounds of sonication.
-
d.Wash three times with autoclaved Milli-Q water.
-
e.Dry the microchambers with a compressed air blow gun and incubate at 60°C for 30 min in the oven (Figure 3A).
-
f.Place the PDMS microchambers (8–10), pattern side up, and glass-bottom Petri dishes (one per microchamber), without the lid, into a plasma cleaner under vacuum for 30 s (Figures 3B and 3C).
-
g.Stick the PDMS microchambers, with the pattern side facing down, to the glass at the bottom of the Petri dish (one chamber per Petri dish) to form an irreversible tight seal (Figure 3D) (Troubleshooting 1). Close the Petri dishes with their lids.
-
h.Incubate in the oven at 60°C for 15 min.
-
i.Place the lids of the Petri dishes face down and the micro chambers stuck to the Petri dishes in the plasma cleaner for 10 s under vacuum (Figure 3E).
-
j.Then, quickly close the Petri dishes with their lids.
-
k.Sterilize the microchambers contained in the Petri dishes for 20 min under the UV lamp of a culture hood (Figure 3F). Maintain the Petri dish lids closed during this step.
-
l.At the same time, sterilized a 12 cm Petri dish containing a piece of Whatman filter paper under the hood. This 12 cm Petri dish serves as a humidified chamber for up to eight microchamber-containing small Petri dishes.
-
m.Under a culture hood, humidify the Whatman filter paper with sterile water and place the small Petri dishes in the 12 cm Petri dish (Figure 3G).
-
a.
Note: it is critical to keep the Whatman filter paper humid to avoid the evaporation of the medium in the microchambers.
Note: Carefully perform all the steps of sterilization (Troubleshooting 2).
-
4.Coating of the microfluidic devices:
-
a.Under a culture hood, coat the upper and synaptic chambers with 20 μL of poly-D-lysine (0.1 mg/mL).
-
b.Fill the postsynaptic chamber with 20 μL of a mix of poly-D-lysine (0.1 mg/mL) and laminin (10 μg/mL). This asymmetric coating step generates a gradient of laminin that limits the number of hippocampal axons reaching the synaptic chamber (Troubleshooting 3).
-
c.Close the Petri dish lids and incubate the microchambers 12 h at 4°C for coating.
-
a.
Figure 2.

From the epoxy master mold to the microfluidic device
Four molds are filled with PDMS. After polymerization in the oven, the solid PDMS patterns are unmolded with a pipette tip (A) and punched to generate the wells (B), as indicated in Figure 1.
Figure 3.

Cleaning and sterilization of the microfluidic devices
The various steps necessary for the sterilization of the microfluidic devices include: drying the microchambers with a compressed air blow gun (A), passing the PDMS microchambers and small Petri dishes in a plasma cleaner to remove impurities (B and C), sticking the PDMS microchambers to the bottom of the small glass Petri dishes, with the motif side facing down (one chamber per Petri dish) to form an irreversible tight seal (D), repeating a cycle of plasma cleaning with the Petri dishes containing the microchambers and their lids (E). The microchambers contained in the small Petri dishes are sterilized under a UV lamp (F) and placed in a 12-cm Petri dish containing a piece of humidified Whatman filter paper, serving as a humidified chamber (G).
Primary culture of neurons (day 1)
Neurons are isolated from cortices and hippocampi of mouse embryos and plated into the microfluidic devices.
-
5.Rinse the coating of the microfluidic devices:
-
a.Under a culture hood, wash the microchambers three times with filtered Neurobasal medium.
-
b.Fill the microchambers with growth medium and place them at 37°C in a tissue-culture incubator.
-
a.
-
6.Prepare the following solutions under the cell culture hood:
-
a.50 mL of growth medium (Troubleshooting 2) and 30 mL of DM Ky/Mg and OptiMEM glucose. Filter sterilize the solutions.
-
b.Fill a 35 mm Petri dish with DM Ky/Mg. Close the lid and place the Petri dish on ice. This Petri dish serves for the dissected cortices and hippocampi.
-
c.Fill two 1.5 mL Eppendorf tubes with 1 mL of DM Ky/Mg, label them “cortex” and “hippocampus,” and place them on ice.
-
d.100 mL of 1× PBS from 10× PBS and fill a 12 cm Petri dish. Close the lid and place the Petri dish on ice. This Petri dish serves for washing of the uterine horns of the mouse.
-
a.
-
7.
Prepare the dissection bench, wipe the bench clean with 70% ethanol, immerse cleaned forceps and surgical scissors into 70% ethanol, and wipe clean the stereological microscope platform with 70% ethanol.
-
8.
Anesthetize the mouse by intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine. Check the absence of reflexes by pitching the tip of the tail.
-
9.
Place the animal in dorsal decubitus position and disinfect the lower abdomen part of the mouse with 70% ethanol.
-
10.Harvest the uterine horns:
-
a.Holding the skin with tweezers, make a 1–2 cm mid ventral incision in the skin with surgical scissors, the round tip facing the muscular plane.
-
b.Then, similarly incise the abdominal muscle fibers, the round tip facing the abdominal cavity.
-
c.Extract the uterine horns by cutting and cut the extremities.
-
d.Rinse the uterine horns in 1× PBS contained in a 12 cm Petri dish and incise them with fine straight-tip scissors to clear the embryos from their amniotic sac and the placenta.
-
a.
Note: The entire dissection procedure is performed at 15°C–25°C. After uterine horn removal, the female mouse is sacrificed by exsanguination.
-
11.Dissect the brain tissue.
-
a.Transfer the rinsed embryos into the Petri dish containing the DM Ky/Mg previously chilled at 4°C.
-
b.Under a stereological microscope, decapitate the fetuses and remove the skin and skull with a tweezers.
-
c.Remove the meninges and dissect out the cortex and the hippocampus using fine straight-tip forceps as described in (Seibenhener and Wooten, 2012)
-
d.Transfer the cortices and hippocampi into their respective Eppendorf tubes maintained on ice. The next steps are performed under the tissue-culture hood.
-
a.
Note: Dissecting the cortices and hippocampi from E15.5 embryos takes up to 1 h if all the offspring are dissected. One pregnant C57BL/6 mouse generally carries 6 to 9 embryos. Both cortical and hippocampal tissues are dissected from the same brain and one embryo yields 4 to 5 million of cortical neurons and 250 000 to 500 000 of hippocampal neurons.
-
12.Prepare the cortical and hippocampal cells suspension:
-
a.Transfer the brain structures into a 15 mL conical tube.
-
b.Wait for the structures to sediment, discard the supernatant, and add 2 mL optiMEM glucose.
-
c.Repeat the previous step two times.
-
d.Mechanically dissociate the structures in optiMEM glucose by repeated pipetting with a Pasteur pipette (Troubleshooting 4).
-
e.Count the cells using a hemocytometer.Note: We use a Malassez hemocytometer. Trypan blue (50% v/v) can be used to discriminate dead from living cells.
-
f.Pipette the volume corresponding to 5 × 106 neurons.
-
g.Centrifuge the cells at 60 × g for 5 min and remove the supernatant.
-
h.Resuspend the neurons in 100 μL growth medium.
-
i.Plate the neurons on the microfluidics device (Figure 4).
-
i.Remove the growth medium contained in the microchambers.
-
ii.Add growth medium to the synaptic chamber (6 μL in each well).
-
iii.Add 2 μL of the cortical neuron suspension to the presynaptic compartment.
-
iv.Add 2 μL of the hippocampal cell suspension to the postsynaptic compartment.Note: As shown in Figure 1, the medium is added to the left wells of the microchambers, whereas it is removed from the right wells of the microchambers.CRITICAL: Always fill the synaptic chamber first. The generated flux impedes the subsequently plated neurons from entering the microchannels.
-
v.Incubate the devices for 1 h in the incubator to let the cells sediment.
-
i.
-
j.After 1 h, fill all compartments with up to 20 μL of growth medium.
-
a.
Figure 4.

Freshly plated cortical and hippocampal neurons in a microfluidic device
Transmission light microscopy image of cortical neurons (left) and hippocampal neurons (right) freshly plated in a microchamber. The two cultures are separated by the microgrooves and the synaptic chamber. Scale bar, 100 μm.
Virus infection (day 1 and day 2)
Cells are transduced with the virus 24 h after plating. The virus is removed after a further 24 h.
Note: Warm up growth medium to 37°C before addition to the cells.
-
13.The day after the plating (DIV1) infect the neurons with lentivirus (LV).
-
a.Dilute 0.2 μL virus in 15 μL growth medium per microchamber.
-
b.Remove the medium of the presynaptic compartment of the microchambers.
-
c.Add 15 μL of LV.CMV.BDNF-mCherry to the presynaptic compartment and incubate for 24 h.
-
a.
Note: It is also possible to use adeno-associated virus (AAV) or to electroporate the neurons with plasmids before plating. Selective infection of the postsynaptic or presynaptic compartments can also be performed.
-
14.
After 24 h (DIV2), remove the virus and fill the microchambers with growth medium (20 μL).
-
15.
Allow the axons and dendrites to grow and form synapses for 10 days.
-
16.
Regularly check the levels of medium in the microchambers, as they are prone to dessication. Typically, 10 μL fresh medium is added once per week to maintain the 20 μL volume of growth medium.
Note: Axons reach the synaptic chamber between DIV4 and DIV5.
Treatment and acquisition (DIV12)
Cells are treated for 3 h with 5 μM of DXM and imaged using a spinning disk confocal system.
Note: Before performing this step, carefully inspect the microchambers for contamination. Select uncontaminated cultures with healthy neurons.
-
17.
Under a culture hood, prepare DXM at 50 μM by diluting 1 μL of 10 mM DXM in 200 μL growth medium. Warm up to complete dissolution and agitate.
-
18.
Add 2 μL of 50 μM DXM to the left wells feeding the pre- and postsynaptic compartments. Control microchambers consist of cells treated with DMSO prepared similarly.
-
19.
Incubate the treated cells for 3 h in a CO2 incubator.
-
20.
Record live cells using an inverted microscope coupled to a spinning disk confocal system connected to a wide field electron-multiplying CCD camera. Maintain cells at 37°C and 5% CO2. Acquire images every 200 ms for 30 s to study BDNF-mCherry trafficking (63× oil-immersion objective, 1.46 NA). Perform five acquisitions per microchamber and three microchambers per condition. The acquisitions are performed using MetaMorph software (Troubleshooting 5).
Note: See the Figure 1 for the acquisition zone.
Note: After treatment it also possible to fix neurons in 4% paraformaldehyde and perform an immunostaining.
Note: Presynaptic and postsynaptic compartments are treated to mimic systemic stress in all brain structures.
Expected outcomes
Each recording consists of 151 images. Videos are obtained using MetaMorph software and reconstituted by importing the nd files with the MetaMorph Stack builder function in Image J. An example is provided in Methods video S1.
Scale bar, 10 μm.
Kymographs are generated from the videos using the KymoToolBox plugin for ImageJ (Zala et al., 2013) with a length of 100 μm (x axis) and a total time of 30 s (y axis). Several kinetics parameters can be extracted from these kymographs. Here, we provide a range of values obtained under control and DXM conditions (Table 1; Agasse et al., 2020).
Table 1.
Representative values obtained in a standard experiment
| Kymograph quantification | Mean anterograde velocity (μm/s) ± SEM | Mean retrograde velocity (μm/s) ± SEM | Mean number of anterograde vesicles (/100 μm/30 s) ± SEM | Mean number of retrograde vesicles (/100 μm/30 s) ± SEM | Net flux (μm/30 s) ± SEM | Linear flow (μm/30 s) ± SEM | Number of axons analyzed |
|---|---|---|---|---|---|---|---|
| DMSO | 2.32 ± 0.09 | −1.73 ± 0.07 | 5.18 ± 0.36 | −3.56 ± 0.25 | 6.46 ± 1.34 | 21.63 + 1.83 | 76 (4 different cultures) |
| DXM | 1.97 ± 0.07 | −1.72 ± 0.07 | 5.43 ± 0.34 | −4.10 ± 0.26 | 3.30 ± 0.69 | 18.60 + 1.19 | 94 (4 different cultures) |
The current protocol describes the establishment of cortico-hippocampal circuits but various types of neurons can be plated, thus creating a wide range of circuits. We have successfully reconstituted wild-type and HD cortico-striatal circuits (Virlogeux et al., 2018). We also reconstituted a cortico-striatal circuit mimicking Rett syndrome through silencing of the MeCp2 protein (Ehinger et al., 2020) and a cortico-cortical circuit allowing us to investigate the trafficking of amyloid-precursor protein to the synapse (Bruyere et al., 2020).
Quantification and statistical analysis
The parameters obtainable from the kymographs are calculatable as follows:
- Anterograde velocity:
- Vma (μm/s) = Anterograde Distance (μm) / Time (s)
- Retrograde velocity:
- Vmr (μm/s) = Retrograde Distance (μm) / Time (s)
- Global velocity:
- Vg (μm/s) = Anterograde Distance (μm) + | Retrograde Distance (μm) | / Time (s)
- Number of anterograde vesicles per 100 μm:
- Na (anterograde vesicles/100 μm) = na (anterograde vesicles) / Axon length (100 μm)
- Number of retrograde vesicles per 100 μm:
- Nr (retrograde vesicles/100 μm) = nr (retrograde vesicles) / Axon length (100 μm)
- Number of pausing vesicles per 100 μm:
- Np (pausing vesicles/100 μm) = np (pausing vesicles) / Axon length (100 μm)
- Pausing time
- Pt (%) = Np / (Na + Nr + Np) ∗100
- Linear Flow Rate:
- Q (μm/s) = | Vma | ∗ na + | Vmr | ∗ nr
- Net Flux:
- D (μm/s) = | Vma |∗ na − | Vmr ∗ nr
One should consider only fluorescent puncta whose sizes are ranging from 50 nm (synaptic vesicles and precursors of synaptic vesicles) to 150–200 nm (dense core vesicles, DCVs or secretory vesicles). BDNF vesicles are usually around 150 nm (DCVs). These vesicles can be motile or static. Very large puncta above 300 nm -that usually do not move- should not be considered.
Vesicles are considered to be motile if their velocity is above 0.12 μm/s. Each condition is tested using 12 chambers from four independent cultures. In each chamber, five fields containing at least four axons are analyzed to reach a minimum number of 50 axons (n = number of axons).
Easily identifiable and individualized axons (mCherry positive) passing through the synaptic chambers are selected for analysis. It is important to avoid mCherry positive superposed axons that will lead to abnormal values of trafficking (increased number of vesicles/μm).
Limitations
This protocol describes how to reconstruct the cortico-hippocampal neuronal network using microfluidic devices. The entorhinal cortex at embryonic day 15.5 (E15.5) by itself provides an insufficient number of cells for culturing. Thus, we dissected out whole cortices. We have verified that 25% of the cortical neurons are reelin-positive, i.e., cells expressing the chemical type displayed by layer II neurons of the entorhinal cortex that project to the dentate gyrus (Agasse et al., 2020).
Corticosterone, the glucocorticoid secreted upon stress, acts through mineralocorticoid and glucocorticoid receptors. Corticosterone has a higher affinity for the mineralocorticoid receptor and, hence, binds to it preferentially at low concentrations. However, upon stress, corticosterone is chronically secreted at high levels and binds to the glucocorticoid receptor. Stress-related effects of corticosterone are mediated by the glucocorticoid receptor. We therefore used dexamethasone as a specific agonist of the glucocorticoid receptor to specifically address the effects of stress on BDNF transport. If corticosterone is used, it is important to discriminate between the effects induced by stimulation of the mineralocorticoid receptor and those induced by glucocorticoid receptor stimulation. For example, low (100 nM) and high concentrations (100 μM) of corticosterone have been shown to display opposite effects on the proliferation of hippocampal progenitors (Anacker et al., 2013).
Troubleshooting
Problem 1
Axons of neurons grow “below” the microgrooves (step 3g).
Potential solution
This happens when microchambers do not properly adhere to the glass of the Petri dish. In the protocol, the microchambers are sonicated three times in 96% ethanol. This step is indispensable, as it removes grease and impurities that reduce adhesion of the microchamber surface to the glass of the Petri dish. Make sure to perform this step. In addition, when sticking the microchambers to the glass of the Petri dish, it is necessary to gently squeeze the microchamber against the Petri dish surface to make sure that the entire microchamber surface is in contact with the glass. However, be careful to avoid exerting too much pressure. Otherwise, the wells and grooves of the microchambers may become deformed.
Problem 2
Neuronal cultures in the microchambers are contaminated by bacteria and fungi (step 3 Note and step 6a).
Potential solution
Numerous sterilization steps are indicated in the protocol. Plasma cleaner cycles remove organic contaminants from the surface of the microfluidic devices and small Petri dishes. UV sterilization kills microorganisms. Be sure to perform these steps thoroughly. In our experience, contamination often occurs at the step of neuronal culture. We recommend filter sterilizing all media and using autoclaved PBS and autoclaved Milli-Q water. The growth medium contains antibiotics. If necessary, add anti-mycotics, such as amphotericin b (50 μg/mL, Thermo Fisher Scientific, #152900180) or fungin (10 μg/mL, Invivogen, # ant-fn-1), to the growth medium.
Problem 3
Cells are not adherent in the pre and postsynaptic chambers 24 h after plating (step 4b).
Potential solution
Adhesion of cells is usually compromised by an uneven coating. It is critical to remove any bubbles that may form when pipetting the coating medium into the microchambers. Visually inspect the microchambers when the coating medium is applied. Aspirate any bubbles with a vacuum and add again coating medium; alternatively, you can pop any bubbles with a pipette tip.
Problem 4
Massive cell death is observed the day after plating the neurons in the microchambers (step 12d).
Potential solution
Massive cell death is usually due to excessive mechanical dissociation. Mechanical dissociation must be gently performed by repetitive pipetting. If trouble persists, add DNase I (250 U/mL, Sigma D5025) when performing the mechanical dissociation. It will avoid the cells to bind to genomic DNA released by dying cells. The DNA mucus traps cells and excessive pipetting applied to release them, irreversibly damages the cells.
Problem 5
Faint or no fluorescent BDNF-containing vesicles is observed (step 20).
Potential solution
The preparation of the viral particles is critical. In our hands, successful experiments were performed with 1 × 107 infectious units (IU/mL). Each viral production should be carefully titered and tested on neurons before use.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Sandrine Humbert (sandrine.humbert@univ-grenoble-alpes.fr).
Materials availability
This study did not generate new unique reagents. The BDNF-mCherry lentiviral plasmid is available upon request (lead contact).
Data and code availability
The protocol includes all datasets generated and analyzed during the study.
Acknowledgments
This protocol has been developed by applying the standards of veterinary practice to laboratory mouse surgery. All experimental procedures were performed in the Grenoble Institute of Neurosciences (GIN, INSERM U1216, license #B3851610008) in strict accordance with the local animal welfare committee (Comité Local Grenoble Institute Neurosciences, C2EA-04), EU guidelines (directive 2010/63/EU), and the French National Committee (2010/63) for the care and use of laboratory animals. We thank the staff of the animal facilities (GIN) for technical support. Our current work is funded by grants from the Agence Nationale de la Recherche ANR-18-CE16-0009-01 AXYON (F.S. and S.H.) and ANR-15-IDEX-02 NeuroCoG (F.S. and S.H.) in the framework of the “Investissements d’avenir” program; the Fondation pour la Recherche Médicale (FRM, DEI20151234418, F.S. and équipe labellisée, DEQ20170336752, S.H.); the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (AdG grant agreement no. 834317, F.S.); and the AGEMED program from INSERM (F.S. and S.H.). The Saudou and Humbert laboratories are part of the Grenoble Center of Excellence in Neurodegeneration (GREEN).
Author contributions
This protocol was developed by S.L., A.G., and F.A. The detailed procedure was written by F.A. and S.L., and F.S. and S.H. wrote the manuscript, which was commented on by all authors.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100382.
Contributor Information
Fabienne Agasse, Email: fabienne.agasse@univ-grenoble-alpes.fr.
Frédéric Saudou, Email: frederic.saudou@univ-grenoble-alpes.fr.
Sandrine Humbert, Email: sandrine.humbert@univ-grenoble-alpes.fr.
References
- Agasse F., Mendez-David I., Christaller W., Carpentier R., Braz B.Y., David D.J., Saudou F., Humbert S. Chronic corticosterone elevation suppresses adult hippocampal neurogenesis by hyperphosphorylating huntingtin. Cell Rep. 2020;32:107865. doi: 10.1016/j.celrep.2020.107865. [DOI] [PubMed] [Google Scholar]
- Anacker C., Cattaneo A., Luoni A., Musaelyan K., Zunszain P.A., Milanesi E., Rybka J., Berry A., Cirulli F., Thuret S. Glucocorticoid-related molecular signaling pathways regulating hippocampal neurogenesis. Neuropsychopharmacology. 2013;38:872–883. doi: 10.1038/npp.2012.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruyere J., Abada Y.S., Vitet H., Fontaine G., Deloulme J.C., Ces A., Denarier E., Pernet-Gallay K., Andrieux A., Humbert S. Presynaptic APP levels and synaptic homeostasis are regulated by Akt phosphorylation of huntingtin. eLife. 2020;9:e56371. doi: 10.7554/eLife.56371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehinger Y., Bruyere J., Panayotis N., Abada Y.S., Borloz E., Matagne V., Scaramuzzino C., Vitet H., Delatour B., Saidi L. Huntingtin phosphorylation governs BDNF homeostasis and improves the phenotype of Mecp2 knockout mice. EMBO Mol. Med. 2020;12:e10889. doi: 10.15252/emmm.201910889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmon P., Trono D. Production and titration of lentiviral vectors. Curr. Protoc. Hum. Genet. 2007;Chapter 12 doi: 10.1002/0471142905.hg1210s54. Unit 12 10. [DOI] [PubMed] [Google Scholar]
- Seibenhener M.L., Wooten M.W. Isolation and culture of hippocampal neurons from prenatal mice. J. Vis. Exp. 2012;65:3634. doi: 10.3791/3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.M., Dieterich D.C., Ito H.T., Kim S.A., Schuman E.M. Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron. 2010;66:57–68. doi: 10.1016/j.neuron.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virlogeux A., Moutaux E., Christaller W., Genoux A., Bruyere J., Fino E., Charlot B., Cazorla M., Saudou F. Reconstituting corticostriatal network on-a-chip reveals the contribution of the presynaptic compartment to Huntington's disease. Cell Rep. 2018;22:110–122. doi: 10.1016/j.celrep.2017.12.013. [DOI] [PubMed] [Google Scholar]
- Zala D., Hinckelmann M.V., Yu H., Lyra da Cunha M.M., Liot G., Cordelieres F.P., Marco S., Saudou F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–491. doi: 10.1016/j.cell.2012.12.029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Scale bar, 10 μm.
Data Availability Statement
The protocol includes all datasets generated and analyzed during the study.