

Abstract
Certain mutations in the ALDH7A1 gene cause pyridoxine-dependent epilepsy (PDE), an autosomal recessive metabolic disease characterized by seizures, and in some cases, intellectual disability. The mutational spectrum of PDE is vast and includes over 70 missense mutations. This review summarizes the current state of biochemical and biophysical research on the impact of PDE missense mutations on the structure and catalytic activity of ALDH7A1. Paradoxically, some mutations that target active site residues have a relatively modest impact on structure and function, while those remote from the active site can have profound effects. For example, missense mutations targeting remote residues in oligomer interfaces tend to strongly impact catalytic function by inhibiting formation of the active tetramer. These results shows that it remains very difficult to predict the impact of missense mutations, even when the structure of the wild-type enzyme is known. Additional biophysical analyses of many more disease-causing mutations are needed to develop the rules for predicting the impact of genetic mutations on enzyme structure and catalytic function.
Keywords: aldehyde dehydrogenase, ALDH7A1, pyridoxine-dependent epilepsy, missense mutations, protein oligomerization, inherited metabolic disease
Graphical Abstract
1. Introduction
The enzyme aldehyde dehydrogenase 7A1 (ALDH7A1)1 catalyzes the last step of lysine catabolism – the NAD+-dependent oxidation of α-aminoadipate semialdehyde (AASAL) to α-aminoadipate (AA) (Fig. 1A). Also known as AASAL dehydrogenase and antiquitin, ALDH7A1 belongs to the ALDH superfamily, a large group of enzymes that catalyze the NAD(P)+-dependent oxidation of aldehydes to carboxylic acids [1–3]. The human ALDH superfamily consists of 19 enzymes, which oxidize numerous substrates, including small aldehydes such as acetaldehyde, amino acid derivatives, and lipids. ALDHs play important roles in the detoxification of reactive aldehydes, amino acid metabolism, embryogenesis and development, neurotransmission, oxidative stress, and cancer [3]. Some ALDHs are biomarkers of cancer stem cells and can mediate resistance to chemotherapeutic agents [4–7], which has motivated efforts to develop inhibitors for cancer therapy [8]. Mutations in ALDH genes are associated with inherited metabolic diseases, [3] including the subject of this review, pyridoxine-dependent epilepsy (PDE).
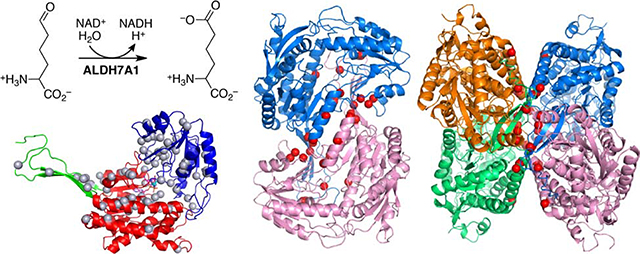
Fig. 1.

Reactions and protein fold of ALDH7A1. (A) The reaction catalyzed by ALDH7A1 (top) and the reaction of P6C with PLP (bottom). (B) The fold of ALDH7A1, with the domains colored red (NAD+-binding), blue (catalytic), and green (oligomerization). NAD+ and AA are shown in magenta and cyan sticks, respectively. The gray spheres mark the residues affected by pathogenic missense mutations (Table 1). (C) Missense mutations affecting active site residues mapped onto the protein structure (gold spheres).
Certain mutations in the ALDH7A1 gene cause PDE, an autosomal recessive metabolic disease characterized by seizures, and in some cases, intellectual disability [9, 10]. The mutational spectrum of PDE spans over 100 different mutations, including over 70 missense mutations [9, 11–13] (Fig. 1B). Missense mutations have been identified in all 18 exons of the ALDH71A gene, except exons 3, 8, and 11 [12]. Exons 5, 6, 10, and 15 harbor over 60% of the known pathogenic missense mutations. The estimated carrier frequency of ALDH7A1 mutations is 1:127, while the incidence of ALDH7A1 deficiency is estimated to be 1:64,352 births [12].
The biochemical basis of PDE is a decrease of the ubiquitous enzyme cofactor pyridoxal 5´-phosphate (PLP). PDE-causing mutations that cause a loss of function of ALDH7A1 lead to a buildup of the substrate AASAL. The susceptibility of aldehydes, such as AASAL, to nucleophilic attack makes them highly reactive in the context of cells. One fate of AASAL is the intramolecular nucleophilic attack by the amino group, resulting in cyclization to Δ1-piperideine-6-carboxylic acid (P6C) with loss of water (Fig. 1A). The cellular pool of PLP is depleted via the irreversible reaction of P6C with PLP [14] (Fig. 1A). Treatment with pyridoxine (vitamin B6) addresses the PLP deficiency and typically provides adequate seizure control, yet 75% of individuals with PDE have intellectual disability and developmental delay [9, 11].
The effects of mutations on the structure and catalytic activity of ALDH7A1 is an important aspect of the molecular basis of PDE. This biochemical/biophysical information allows for the prediction of symptom-severity and aids the development of patient-specific medical treatments. Herein, we summarize the current state of biochemical and biophysical research on the impact of PDE missense mutations on the structure and catalytic activity ALDH7A1.
2. A note about residue numbering
Two conventions for numbering the amino acid residues of ALDH7A1 are found in the literature. One refers to isoform 1 (UniProt P49419–1), which includes a mitochondrial transit peptide and contains 539 amino acid residues. This convention is recommended by the Human Genome Variation Society [15] and typically is used in articles on the genetics of PDE and patient case studies. The other convention refers to the isoform 2 (UniProt P49419–2), which is generated by alternative splicing and lacks residues 1 – 28 of isoform 1. This convention is used in some structural studies, and the crystal structures of ALDH7A1 downloaded from the Protein Data Bank [16] will typically have this numbering scheme. The essential point to remember is to add 28 to the crystal structure residue numbers to obtain the HGVS convention. We use the HGVS numbering here.
3. Catalytic mechanism of ALDH7A1
ALDH7A1 follows a covalent catalytic mechanism, which is conserved by ALDH superfamily enzymes [17, 18]. The mechanism begins with the binding of the aldehyde substrate to the enzyme-NAD+ complex. Nucleophilic attack by the catalytic cysteine (Cys330 in ALDH7A1) on the aldehyde produces a hemithioacetal intermediate. The negatively-charged O atom of the intermediate is stabilized by hydrogen bonds with a conserved asparagine residue (Asn195 In ALDH7A1) and the backbone N-H group of the catalytic cysteine (“oxyanion hole”). Hydride transfer from the hemithioacetal intermediate to NAD+ generates NADH and the acyl-enzyme intermediate. Next, hydrolysis of the acyl-enzyme intermediate yields the carboxylic acid product. The hydrolysis step involves a conserved glutamate residue (Glu296 in ALDH7A1), which helps activate the hydrolytic water molecule. Several PDE missense mutations target residues in the binding sites for AASAL and NAD+ (Fig. 1C)
4. 3D structure of ALDH7A1
ALDH7A1 exhibits the fold and oligomeric structure characteristic of the ALDH superfamily [19]. The fold consists of three domains: NAD+-binding, catalytic, and oligomerization (Fig. 1B). The NAD+-binding domain adopts the Rossmann fold. The catalytic domain has an α/β structure and contains the catalytic Cys330 residue. The oligomerization domain is a β-substructure that protrudes from the NAD+-binding domain. The active site is located in the crevice between the NAD+-binding and catalytic domains.
ALDH7A1 has a notably flexible C-terminus, which is part of the oligomerization domain. The C-terminus exhibits open and closed conformations [19]. The closed conformation is associated with the binding of the aldehyde substrate AASAL. Upon substrate binding, the C-terminus moves into the active site to complete the AASAL site, creating a protected environment for catalysis to occur. The C-terminus then opens to allow release of the product AA. The involvement of the C-terminus in catalysis is notable because the genetic deletion c.1512delG found in some PDE patients affects the C-terminus. This deletion likely abolishes catalytic activity, based on the observation that ALDH7A1 variants with mutations in the C-terminus exhibit profound kinetic defects and are deficient in formation of the active tetrameric form of the enzyme [20].
ALDH7A1 exists in solution as an equilibrium between dimeric and tetrameric forms [21]. The dimer is an interlinked assembly in which the oligomerization domain of one protomer packs intimately against the catalytic domain of the other protomer, forming an inter-molecular β-sheet (Fig. 2B). Two of these dimers assemble into the tetramer such that their oligomerization domains come into contact (Fig. 2D).
Fig. 2.

Pathogenic missense mutations affecting residues in the oligomeric interfaces of ALDH7A1. (A) The fold of ALDH7A1, with the domains colored red (NAD+-binding), blue (catalytic), and green (oligomerization). NAD+ and AA are shown in magenta and cyan sticks, respectively. The pink spheres indicate residues in the dimer interface targeted by pathogenic missense mutations. (B) Two views of the ALDH7A1 dimer. The red spheres indicate dimer interface residues that are targeted by pathogenic missense mutations. (C) The purple spheres indicate residues in the tetramer interface that are targeted by pathogenic missense mutations. (D) Tetramer of ALDH7A1. The red spheres indicate tetramer interface residues that are targeted by pathogenic missense mutations.
The position of the dimer-tetramer equilibrium is sensitive to both enzyme concentration [21] and the presence of NAD+ [22]. Increasing either the concentration of total enzyme or that of NAD+ favors the tetramer. This is significant, because the tetramer is the active form of ALDH7A1 [22]. Thus, mutations that affect NAD+ binding are expected to be especially deleterious for function because they not only reduce affinity for the essential cofactor, but also disfavor formation of the active oligomer.
5. Impact of missense mutations in the ALDH7A1 gene on enzyme structure and catalytic function
Over 70 missense mutations have been identified as being related to PDE [12] (Table 1). Mapping the mutations onto the 3D structure of the enzyme can provide insight into the possible impact of the mutations on enzyme structure and function. The affected residues belong to all three structural domains of ALDH7A1 (Fig. 1B).
Table 1.
Structural context and biochemical phenotypes of pathogenic missense variants of ALDH7A1a
| Variant | Domainb | AASAL sitec | NAD+ sitec | Dimer interfacec | Tetramer interfacec | Internald | Biochemical phenotype |
|---|---|---|---|---|---|---|---|
| p.A40G | N | ||||||
| p.V80G | N | X | |||||
| p.P106L | N | X | Insoluble [21] | ||||
| p.G111E | N | X | Insoluble [21] | ||||
| p.R122W | N | X | Mild [12] | ||||
| p.G143R | N | X | |||||
| p.E149K/D | N | X | X | ||||
| p.C154R | N | X | |||||
| p.Y156C | N | X | |||||
| p.A157P | N | X | Severe [21] | ||||
| p.S161P | N | X | |||||
| p.G165V | O | X | Severe [21] | ||||
| p.G165E | O | X | |||||
| p.G166V | O | X | Severe [21] | ||||
| p.A177E | O | X | Severe [21] | ||||
| p.A177P | O | X | |||||
| p.G189E | N | X | |||||
| p.T192M | N | X | |||||
| p.N195S | N | X | X | Moderate [23] | |||
| p.F196I | N | X | X | ||||
| p.P197S | N | X | Moderate [23] | ||||
| p.A199V | N | X | Severe [23, 29] | ||||
| p.G202V | N | X | Insoluble [23] | ||||
| p.G202D | N | X | |||||
| p.W203G | N | X | X | Mild [23] | |||
| p.T222A | N | ||||||
| p.G274E | N | X | |||||
| p.T276P | N | X | |||||
| p.G283D | N | X | Severe [21] | ||||
| p.F290S | N | X | |||||
| p.G291E | N | Severe [21] | |||||
| p.G299R | C | X | X | ||||
| p.N301I | C | X | Severe [29] | ||||
| p.D308G | C | ||||||
| p.S317L | C | X | Severe [12] | ||||
| p.A318P | C | ||||||
| p.A322V | C | ||||||
| p.T325K/R | C | Severe [29] | |||||
| p.Q328R | C | X | |||||
| p.R335Q | C | X | Severe [29] | ||||
| p.Y354C | C | X | |||||
| p.P371L | C | X | |||||
| p.V395G | C | Moderate [30] | |||||
| p.V395M | C | ||||||
| p.G398R | C | ||||||
| p.G406R | C | ||||||
| p.P411L | C | ||||||
| p.A421V | C | ||||||
| p.T426P | C | ||||||
| p.E427Q/G/De | C | X | Severe [24] | ||||
| p.P431L | C | ||||||
| p.F438L | C | X | Moderate [30] | ||||
| p.N448K | C | X | |||||
| p.N449K | C | ||||||
| p.Q453R | C | Severe [30] | |||||
| p.L455P | C | X | X | ||||
| p.S457I | C | X | |||||
| p.S458N | C | X | Severe [29] | ||||
| p.I459F | C | X | Severe [29] | ||||
| p.R469H | C | ||||||
| p.S476L | C | X | |||||
| p.D477N | C | X | |||||
| p.C478S | C | X | Moderate [30] | ||||
| p.G489E | C | X | X | ||||
| p.G494R | N | X | |||||
| p.A495T | N | X | |||||
| p.G505R | N | Severe [29] | |||||
| p.D511N | N | X | |||||
| p.Y516C | N | X | Moderate [12] | ||||
| p.R518G | O | X | |||||
| p.R519K | O | X | |||||
| p.S520P | O | X | |||||
| p.D529N | O | X |
List of pathogenic variants was obtained from Coughlin et al. [12].
Domain of ALDH7A1: C, catalytic; N, NAD+-binding, O, oligomerization.
Structural context based on 4.5 Å distance cutoff.
Defined as residues that are > 97% buried in the context of a monomer.
Missense mutations of Glu427 account for ~30% of the mutated alleles in PDE patients [12].
Only a small subset of the known missense mutations affect residues that contact either the aldehyde substrate or NAD+ cofactor (Fig. 1C). These mutations might be expected to have a large impact on function; however, detailed structural and biochemical studies of the purified enzyme variants shows this is not necessarily true. For example, two mutations that affect residues of the AASAL binding site were found to have mild (p.W203G) or moderate (p.N195S) effects on catalytic activity, and neither mutation significantly disrupted the enzyme structure [23]. The moderate biochemical phenotype N195S is especially interesting, considering that Asn195 is conserved in the ALDH superfamily and plays a role in catalysis by stabilizing the negative charge of the hemithioacetal intermediate. A priori, one might have predicted that this mutation would have a devastating effect on activity. The moderate biochemical phenotype of N195S underscores the necessity of performing biochemical and structural analysis of disease variants.
Some mutations to active site residues profoundly affect catalytic function. For example, mutation of Glu427 abolishes catalytic activity and severely disrupts the structure of the NAD+ binding site [24]. This is significant, because missense mutations of Glu427, especially p.Glu427Gln, account for ~30% of the mutated alleles in PDE patients [12]. The severe biochemical phenotype reflects the crucial role Glu427 plays in positioning the NAD+ nicotinamide for hydride transfer and the fact that this residue is invariant in the ALDH superfamily. This phenotype also reflects the shift in the self-association equilibrium away from formation of the active tetramer.
Many of the pathogenic missense mutations target residues in the oligomer interfaces of ALDH7A1. These residues can be classified as either in the dimer interface (Figs. 2A and 2B) or in the tetramer interface (Figs. 2C and 2D). These residues are outside of the active site (Figs. 2A and 2C), so their impact cannot be appreciated unless they are viewed within the context of the active tetramer. Specialized biophysical techniques are required to determine the oligomeric state of an enzyme in solution, such as small-angle X-ray scattering [25] and analytical ultracentrifugation [26]. In addition, we have found that negative stain electron microscopy is ideally suited for identifying the oligomeric structure of the active enzyme, because it can be performed at the low enzyme concentrations typically used in activity assays, whereas other biophysical techniques tend to require higher enzyme concentrations [22, 27, 28].
Most oligomer interface missense variants studied to date exhibit severe biochemical phenotypes [21]. In most cases, the mutated enzymes are defective in tetramer formation and exhibit no detectable catalytic activity. In one case, p.P106L, the enzyme expressed in Escherichia coli was insoluble and could not be analyzed.
Some of the residues affected by missense mutations are not easily classified as being in the active site or oligomer interfaces. To some extent, this reflects the fact that these “geographical” definitions are based on distance cutoffs, and a residue can fall just outside of the cutoff. A few examples include Gly111 and Ala157, which are close to the tetramer interface, and Pro197, Ala199, and Gly202, which are near the AASAL binding site. Interestingly, these five residues are examples of “internal” residues, defined as those buried in the context of the monomer. This makes sense, because residues that are clearly in the substrate binding sites or oligomer interfaces are by definition solvent exposed when viewed in the context of the monomer.
Missense mutations targeting buried residues can be highly deleterious. For example, G111E and G202V are insoluble when expressed in E. coli, suggesting a folding defect. Others such as A157P and A199V display no catalytic activity. The catalytic defect of A157P results from an inability to form the active tetramer, while that of A199V is due to the bulky valine side chain preventing closure of the active site. Interestingly, in the four cases studied where the mutation of an internal residue impaired either folding or catalytic activity, the mutation increased the size of the side chain (G111E, G202V, A157P, A199V). That such mutations are disruptive reflects the fact that the interior of proteins is very tightly packed.
5. Conclusions
The promise of personalized medicine is to tailor medical treatment based on the specific characteristics of the individual. This is especially suited to the treatment of inherited metabolic diseases, where the development of genotype-phenotype relationships may be tractable, as opposed to polygenetic diseases where this may be more difficult. Structural biology and biophysics can play an important role in this effort by determining the impact of genetic variations on the molecular properties of the protein, such as catalytic function and 3D structure. Biochemical experiments on purified enzyme variants provides quantitative information on catalytic activity, which may allow for prediction of disease severity. This may be especially useful for understanding the effects of novel (private) missense mutations in both symptomatic and asymptomatic patients.
Biophysical studies of PDE variants reinforce the idea that it is very difficult to predict the impact of a missense mutation, even when the structure of the wild-type enzyme is known. Some mutations that target active site residues can have a relatively mild impact on structure and function, while those remote from the active site can have profound effects. Additional biophysical analyses of many more disease-causing mutations are needed to develop computational methods for predicting the impact of genetic mutations on enzyme structure and catalytic function.
Highlights.
Certain mutations in the ALDH7A1 gene cause pyridoxine-dependent epilepsy
The impact of missense mutations on enzyme function is very challenging to predict
Biophysical analysis of enzyme variants yields deep insight into disease pathology
3D protein structure analysis is an enabling technology of personalized medicine
Acknowledgments
Funding
This work was supported by the NIGMS of the National Institutes of Health under award number R01GM093123.
Abbreviations
- AA
α-aminoadipate
- AASAL
α-aminoadipate semialdehyde
- ALDH
aldehyde dehydrogenase
- ALDH7A1
aldehyde dehydrogenase 7A1
- HGVS
Human Genome Variation Society
- P6C
Δ1-piperideine-6-carboxylic acid
- PDE
pyridoxine-dependent epilepsy
- PLP
pyridoxal 5′-phosphate
Footnotes
Conflict of Interest
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Sophos NA, Vasiliou V, Aldehyde dehydrogenase gene superfamily: the 2002 update, Chem. Biol. Interact, 143–144 (2003) 5–22. [DOI] [PubMed] [Google Scholar]
- [2].Vasiliou V, Nebert DW, Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family, Hum. Genomics, 2 (2005) 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vasiliou V, Thompson DC, Smith C, Fujita M, Chen Y, Aldehyde dehydrogenases: from eye crystallins to metabolic disease and cancer stem cells, Chem. Biol. Interact, 202 (2013) 2–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ma I, Allan AL, The role of human aldehyde dehydrogenase in normal and cancer stem cells, Stem. Cell. Rev, 7 (2011) 292–306. [DOI] [PubMed] [Google Scholar]
- [5].Muzio G, Maggiora M, Paiuzzi E, Oraldi M, Canuto RA, Aldehyde dehydrogenases and cell proliferation, Free Radic. Biol. Med, 52 (2012) 735–746. [DOI] [PubMed] [Google Scholar]
- [6].Abdullah LN, Chow EK, Mechanisms of chemoresistance in cancer stem cells, Clin. Transl. Med, 2 (2013) 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Januchowski R, Wojtowicz K, Zabel M, The role of aldehyde dehydrogenase (ALDH) in cancer drug resistance, Biomed. Pharmacother, 67 (2013) 669–680. [DOI] [PubMed] [Google Scholar]
- [8].Muralikrishnan V, Hurley TD, Nephew KP, Targeting Aldehyde Dehydrogenases to Eliminate Cancer Stem Cells in Gynecologic Malignancies, Cancers (Basel), 12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].van Karnebeek CD, Tiebout SA, Niermeijer J, Poll-The BT, Ghani A, Coughlin CR 2nd, Van Hove JL, Richter JW, Christen HJ, Gallagher R, Hartmann H, Stockler-Ipsiroglu S, Pyridoxine-dependent epilepsy: An expanding clinical spectrum, Pediatr Neurol, 59 (2016) 6–12. [DOI] [PubMed] [Google Scholar]
- [10].Stockler S, Plecko B, Gospe SM Jr., Coulter-Mackie M, Connolly M, van Karnebeek C, Mercimek-Mahmutoglu S, Hartmann H, Scharer G, Struijs E, Tein I, Jakobs C, Clayton P, Van Hove JL, Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up, Molecular genetics and metabolism, 104 (2011) 48–60. [DOI] [PubMed] [Google Scholar]
- [11].Stockler S, Plecko B, Gospe SM Jr., Coulter-Mackie M, Connolly M, van Karnebeek C, Mercimek-Mahmutoglu S, Hartmann H, Scharer G, Struijs E, Tein I, Jakobs C, Clayton P, Van Hove JL, Pyridoxine dependent epilepsy and antiquitin deficiency: clinical and molecular characteristics and recommendations for diagnosis, treatment and follow-up, Mol. Genet. Metab, 104 (2011) 48–60. [DOI] [PubMed] [Google Scholar]
- [12].Coughlin CR 2nd, Swanson MA, Spector E, Meeks NJL, Kronquist KE, Aslamy M, Wempe MF, van Karnebeek CDM, Gospe SM Jr., Aziz VG, Tsai BP, Gao H, Nagy PL, Hyland K, van Dooren SJM, Salomons GS, Van Hove JLK, The genotypic spectrum of ALDH7A1 mutations resulting in pyridoxine dependent epilepsy: A common epileptic encephalopathy, Journal of inherited metabolic disease, 42 (2019) 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Scharer G, Brocker C, Vasiliou V, Creadon-Swindell G, Gallagher RC, Spector E, Van Hove JL, The genotypic and phenotypic spectrum of pyridoxine-dependent epilepsy due to mutations in ALDH7A1, J. Inherit. Metab. Dis, 33 (2010) 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Mills PB, Struys E, Jakobs C, Plecko B, Baxter P, Baumgartner M, Willemsen MA, Omran H, Tacke U, Uhlenberg B, Weschke B, Clayton PT, Mutations in antiquitin in individuals with pyridoxine-dependent seizures, Nat. Med, 12 (2006) 307–309. [DOI] [PubMed] [Google Scholar]
- [15].den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, McGowan-Jordan J, Roux AF, Smith T, Antonarakis SE, Taschner PE, HGVS Recommendations for the Description of Sequence Variants: 2016 Update, Hum Mutat, 37 (2016) 564–569. [DOI] [PubMed] [Google Scholar]
- [16].Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE, The Protein Data Bank, Nucleic Acids Res, 28 (2000) 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Luo M, Gates KS, Henzl MT, Tanner JJ, Diethylaminobenzaldehyde is a covalent, irreversible inactivator of ALDH7A1, ACS Chem. Biol, 10 (2015) 693–697. [DOI] [PubMed] [Google Scholar]
- [18].Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, Vasiliou V, Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application, Pharmacol. Rev, 64 (2012) 520–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Luo M, Tanner JJ, Structural basis of substrate recognition by aldehyde dehydrogenase 7A1, Biochemistry, 54 (2015) 5513–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Korasick DA, Wyatt JW, Luo M, Laciak AR, Ruddraraju K, Gates KS, Henzl MT, Tanner JJ, Importance of the C-Terminus of Aldehyde Dehydrogenase 7A1 for Oligomerization and Catalytic Activity, Biochemistry, 56 (2017) 5910–5919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Korasick DA, Tanner JJ, Henzl MT, Impact of disease-Linked mutations targeting the oligomerization interfaces of aldehyde dehydrogenase 7A1, Chem Biol Interact, 276 (2017) 31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Korasick DA, White TA, Chakravarthy S, Tanner JJ, NAD(+) promotes assembly of the active tetramer of aldehyde dehydrogenase 7A1, FEBS Lett, 592 (2018) 3229–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Laciak AR, Korasick DA, Wyatt JW, Gates KS, Tanner JJ, Structural and biochemical consequences of pyridoxine-dependent epilepsy mutations that target the aldehyde binding site of aldehyde dehydrogenase ALDH7A1, FEBS J, 287 (2020) 173–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Laciak AR, Korasick DA, Gates KS, Tanner JJ, Structural analysis of pathogenic mutations targeting Glu427 of ALDH7A1, the hot spot residue of pyridoxine-dependent epilepsy, Journal of inherited metabolic disease, 43 (2020) 635–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Korasick DA, Tanner JJ, Determination of protein oligomeric structure from small-angle X-ray scattering, Protein Sci, 27 (2018) 814–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhao H, Brautigam CA, Ghirlando R, Schuck P, Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation, Curr Protoc Protein Sci, Chapter 20 (2013) Unit20 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Wyatt JW, Korasick DA, Qureshi IA, Campbell AC, Gates KS, Tanner JJ, Inhibition, crystal structures, and in-solution oligomeric structure of aldehyde dehydrogenase 9A1, Archives of Biochemistry and Biophysics, (2020) 108477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Korasick DA, Campbell AC, Christgen SL, Chakravarthy S, White TA, Becker DF, Tanner JJ, Redox Modulation of Oligomeric State in Proline Utilization A, Biophys J, 114 (2018) 2833–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Coulter-Mackie MB, Li A, Lian Q, Struys E, Stockler S, Waters PJ, Overexpression of human antiquitin in E. coli: enzymatic characterization of twelve ALDH7A1 missense mutations associated with pyridoxine-dependent epilepsy, Molecular genetics and metabolism, 106 (2012) 478–481. [DOI] [PubMed] [Google Scholar]
- [30].Coulter-Mackie MB, Tiebout S, van Karnebeek C, Stockler S, Overexpression of recombinant human antiquitin in E. coli: partial enzyme activity in selected ALDH7A1 missense mutations associated with pyridoxine-dependent epilepsy, Molecular genetics and metabolism, 111 (2014) 462–466. [DOI] [PubMed] [Google Scholar]