

Abstract
HIV infection and antiretroviral therapy have been linked to mitochondrial dysfunction. The role of platelet mitochondrial dysfunction in thrombosis, immunoregulation and age-related diseases is increasingly appreciated. Here, we studied platelet mitochondrial DNA content (mtDNApl) and mitochondrial function in people living with HIV (PLHIV) and related this to platelet function. In a cohort of 208 treated PLHIV and 56 uninfected controls, mtDNApl was quantified, as well as platelet activation, platelet agonist-induced reactivity and inflammation by circulating factors and flow cytometry. In a subgroup of participants, the metabolic activity of platelets was further studied by mitochondrial function tests and the Seahorse Flux Analyzer. PLHIV had significantly lower mtDNApl compared to controls (8.5 copies/platelet (IQR: 7.0–10.7) vs. 12.2 copies/platelet (IQR: 9.5–16.6); p < 0.001), also after correction for age, sex and BMI. Prior zidovudine-use (n = 46) was associated with a trend for lower mtDNApl. PLHIV also had reduced ex vivo platelet reactivity and mean platelet volume compared to controls. MtDNApl correlated positively with both platelet parameters and correlated negatively with inflammatory marker sCD163. Mitochondrial function tests in a subgroup of participants confirmed the presence of platelet mitochondrial respiration defects. Platelet mitochondrial function is disturbed in PLHIV, which may contribute to platelet dysfunction and subsequent complications. Interventions targeting the preservation of normal platelet mitochondrial function may ultimately prove beneficial for PLHIV.
Subject terms: Medical research, HIV infections, Cardiovascular diseases
Introduction
Mitochondrial dysfunction is a well-known phenomenon in people living with HIV (PLHIV), which has been linked with the use of nucleoside reverse transcriptase inhibitors (NRTIs)1–5. The main mechanism underlying NRTI toxicity is inhibition of mitochondrial DNA polymerase γ and increased oxidative stress, resulting in mitochondrial DNA (mtDNA) depletion6–8, and mitochondrial dysfunction at the tissue level7,9. These adverse effects were greatly reduced when the older NRTIs stavudine, zidovudine (AZT) and didanosine were replaced by the newer NRTIs tenofovir (TDF) and abacavir (ABC). However, these newer NRTIs may still impair mitochondrial function, albeit to a lesser degree10–13. More recently, mitochondrial dysfunction has also been reported in people living with HIV (PLHIV) naive for combined antiretroviral therapy (cART)3,14–16, including in elite-controllers 17. The factors responsible for cART-independent mtDNA depletion are less well defined and may involve persistent immune activation18,19. Mitochondrial dysfunction has been suggested to contribute to non-AIDS related co-morbidities such as cardiovascular diseases, diabetes, cancer and dementia in PLHIV20,21.
Platelets are the second most numerous blood cells that are, unlike red blood cells, equipped with mitochondria with mtDNA22,23. Human platelets lack a nucleus and mitochondria are essential in maintaining platelet health and lifespan, as recently reviewed23. MtDNA copy number is considered to reflect mitochondrial function24. Healthy platelets contain between 5 and 8 mitochondria, which serve important processes such as platelet activation, ATP production and platelet viability25–29. Platelets are traditionally known for their role in hemostasis, but an increasing body of evidence supports their role in key processes beyond hemostasis, including inflammation and immunoregulation23. With the importance of mitochondria in platelet metabolism, it is no surprise that the health consequences of abnormalities in platelet mitochondrial DNA and function has received increased attention, and platelet mtDNA has been proposed to serve as biomarker for different diseases22,30–32. Data on platelet function in PLHIV is contradictory, with some studies reporting increased 33–37, but other reduced agonist-induced platelet reactivity28,38,39.
We hypothesized that mitochondrial dysfunction in PLHIV is associated with reduced platelet mtDNA copies and platelet dysfunction. Here, we show that platelet mtDNA copies are lower in PLHIV on long-term cART and this was validated in a subgroup of PLHIV using mitochondrial functional assays.
Results
Cohort characteristics
Between December 2016 and February 2017, a total of 208 virally suppressed PLHIV on long-term cART and 56 healthy controls (sampled twice) were concurrently enrolled. Baseline characteristics are shown in Table 1. PLHIV were older compared to controls (52 years (IQR: 45.8–59.0) vs 30 years (IQR: 25.8–53), respectively, p < 0.001). Median duration of cART use was 6.6 years (IQR: 4.2–11.9).
Table 1.
Baseline characteristics.
| PLHIV (n = 208) | Healthy controls (n = 56) | |
|---|---|---|
| Sex (% Female) | 17 (8.2) | 22 (39.2)* |
| Age (years, median [IQR]) | 52.0 [45.8, 59.0] | 30.0 [25.8–53.0]* |
| BMI (median [IQR]) | 24.1 [22.0, 26.0] | 23.8 [21.5–25.6] |
| HIV infection duration (years, median [IQR]) | 8.5 [5.0, 14.2] | |
| Way of transmission (%) | ||
| Heterosexual | 8 (3.8) | |
| IDU | 3 (1.4) | |
| MSM | 158 (76.0) | |
| Other/unknown | 39 (18.8) | |
| CD4 nadir (median [IQR]) | 250.0 [135.0, 362.5] | |
| CD4 count (median [IQR]) | 660.0 [480.0, 812.5] | |
| Undetectable HIV load, n (%) | 208 (100) | |
| CD4/CD8 ratio (median [IQR]) | 0.8 [0.6, 1.1] | |
| cART duration (years; median [IQR]) | 6.6 [4.1, 11.8] | |
| cART regimen | ||
| NRTI-use (%) | 200 (96.2) | |
| NtRTI-use (%) | 97 (46.6) | |
| NNRTI-use (%) | 61 (29.3) | |
| PI-use (%) | 32 (15.4) | |
| Maraviroc-use (%) | 3 (1.4) | |
| INSTI-use (%) | 140 (67.3) | |
| ABC (%) | 93 (44.7) | |
| DTG (%) | 86 (41.3) | |
| EVG (%) | 15 (7.2) | |
| RAL (%) | 38 (18.3) | |
| Smoking (%) | 59 (28.4) | |
| Pack years (median [IQR]) | 13.8 [0.0, 28.0] | |
| Hypercholesterolemia (%) | 56 (26.9) | |
| Hypertension (%) | 40 (19.2) | |
| Diabetes Mellitus (%) | 9 (4.3) | |
| No cardiovascular risk factors (%) | 50 (24.0) | |
| Statins (%) | 56 (26.9) | |
| Aspirin (%) | 18 (8.7) | |
| Metformin (%) | 9 (4.3) | |
BMI body mass index, cART combination antiretroviral therapy, NRTI nucleoside reverse transcriptase inhibitor, NtRTI nucleotide reverse transcriptase inhibitor, NNRTI non-nucleoside reverse transcriptase inhibitor, PI protease inhibitor, INSTI integrase inhibitor, ABC abacavir, DTG dolutegravir, EVG elvitegravir, RAL raltegravir.
*Significantly different between cohorts.
Platelet mtDNA copies are reduced in PLHIV
Platelet mtDNA copies (mtDNApl) were significantly lower in PLHIV compared to healthy controls (Fig. 1A; median 8.5 copies/platelet (plt) (IQR 7.0–10.7) vs 12.2 copies/plt (IQR: 9.5–16.6), p < 0.001). mtDNApl copies correlated inversely with age (Fig. 1B; Pearson’s R = − 0.24, p < 0.001). MtDNApl remained significantly associated with HIV status after correction for age, sex and body-mass index (BMI) in a linear regression model (B = 0.77 SE: 0.14, P < 0.0001). When analysis was restricted to individuals of 40 years and above (Supplemental Table S1; PLHIV: n = 173 vs HC: n = 22), mtDNApl remained significantly lower in PLHIV compared to controls (Supplemental Fig. S1; PLHIV: 8.4 copies/plt (IQR: 6.6–10.2) vs HC: 10.5 copies/plt (IQR: 8.9–14.7), p = 0.001). Similarly, the differences in mtDNApl between PLHIV and controls remained when analysis was restricted to males only (Supplemental Table S2; PLHIV: 8.5 copies/plt (IQR: 6.6–10.7) vs HC: 11.6 copies/plt (IQR: 9.3–14.7), p < 0.001). MtDNApl levels were neither associated with duration of cART nor with current CD4 count, CD4 nadir or CD4/CD8 ratio in univariate analysis (Supplemental Table S3). Furthermore, no differences in mtDNApl were found between PI, NNRTI and INSTI-based regimens (all p > 0.1; Supplemental Fig. S2).
Figure 1.

Platelet mtDNA content. Mitochondrial DNA content in platelets (mtDNApl) was determined using qPCR after normalization for platelet count in isolated platelets. (A) Dotplot showing mtDNA copies per platelet in people living with HIV (PLHIV) and controls (HC); the line indicates median and error bars the interquartile range (IQR). Student’s t-test was used after inverse rank-based normalization. (B) Normalized (inverse rank based) mtDNApl versus age. (C) Normalized mtDNApl versus soluble CD163 (a macrophage scavenger receptor). Correlations were analyzed using Pearson’s correlation coefficients.
Although abacavir (ABC)-use has been shown to affect platelet function40, current ABC use (p = 0.89) or cumulative ABC exposure (in days) were not associated with mtDNApl copies (Supplemental Fig. S2). Conversely, PLHIV with prior zidovudine-use (prior AZT-use, n = 46/184; 25%) showed a trend towards lower copies of mtDNApl (AZT-use: 7.8 copies/plt [IQR: 6.0, 9.4] vs never-AZT: 8.6 copies/plt [IQR: 6.9, 10.8], p = 0.055). After correcting for age in a linear regression model, a non-significant negative correlation remained (B: − 0.07 (− 0.15 to 0.01, p = 0.069). Neither cumulative days on AZT (median 2069 days; IQR 792–3180 days; R = − 0.2, p = 0.12), nor total NRTI exposure (median 4520 days; IQR: 2734–7955; R = 0.05) correlated with mtDNA-use. Use of metformin (p = 0.079) or antihypertensive drugs (p = 0.061) showed a tendency for a lower mtDNApl copies, whereas use of statins (p = 0.74) or acetylsalicylic acid (p = 0.21) did not (Supplemental Fig. S3).
Mitochondrial dysfunction is associated with inflammatory diseases such as atherosclerosis and sepsis in HIV-negative individuals22. Hence, we explored the association of known markers of persisting immune activation, a known driver of non-AIDS related comorbidities20,21, with mtDNApl copy number. Overall, we observed higher plasma concentrations of immune activation markers soluble CD14 (sCD14), soluble CD163 (sCD163) and high-sensitive CRP (hsCRP) in PLHIV than in controls (Table 2). Among participants older than 40 years, only sCD163 remained significantly different between groups (Supplemental Table S1; HIV 741 ng/mL (IQR: 547–906) vs controls 517 ng/mL (IQR: 443–684), p = 0.01). sCD163 correlated negatively with mtDNApl copies (R = − 0.23, p < 0.001, Fig. 1C), whereas sCD14 (R = − 0.073, p = 0.27) and hsCRP (R: − 0.087, p = 0.2) did not (Supplemental Fig. S4).
Table 2.
MPV: mean platelet volume.
| Platelet indices | PLHIV (n = 208) | Healthy controls (n = 56) | p-value |
|---|---|---|---|
| Platelet count (109/L) | 260 [200, 310] | 270 [210, 320] | 0.126 |
| MPV (fL) | 10.1 [9.7, 10.7] | 10.8 [10.3, 11.3] | < 0.001 |
| IPF (%) | 3.3 [2.5, 4.6] | 3.7 [2.8, 5.9] | 0.018 |
| Unstimulated fibrinogen binding (MFI) | 2.0 [1.6, 2.2] | 2.1 [1.6, 2.4] | 0.072 |
| Unstimulated P-selectin expression (MFI) | 2.7 [2.1, 3.2] | 2.6 [2.2, 3.0] | 0.158 |
| Plasma CCL5 (ng/ml) | 2.66 [1.64, 4.29] | 2.61 [1.69, 4.18] | 0.841 |
| Plasma CXCL4 (ng/mL) | 570.0 [290.0, 723.3] | 492.9 [306.1, 839.5] | 0.201 |
| Plasma CXCL7 (ng/mL) | 289.4 [180.3, 465.1] | 306.6 [194.2, 609.8] | 0.192 |
| sCD14 (ng/mL) | 2139.6 [1778.2, 2661.5] | 1789.0 [1502.7, 2071.6] | < 0.001 |
| sCD163 (ng/mL) | 716.5 [528.7, 899.4] | 517.3 [410.7, 578.1] | < 0.01 |
| hsCRP (ng/mL) | 1423.3 [608.8, 2726.3] | 651.2 [205.9, 1179.2] | < 0.001 |
Unstimulated platelet aggregation measured as Fibrinogen binding by flowcytometry in median fluorescence intensity (MFI). Unstimulated platelet degranulation measured as P-selectin expression by flow cytometry by MFI.
Data were analyzed using Mann–Whitney U test.
IPF immature platelet fraction as a percentage of platelet count (Sysmex, Kobe, Japan); sCD14 serum levels of CD14, a marker of monocyte activation; sCD163 serum levels of CD163, a monocyte- and macrophage-specific scavenger receptor; hsCRP high sensitive C-reactive protein.
Platelet mitochondrial dysfunction in PLHIV
Energy demand for platelet ATP production and other metabolic processes that are essential for platelet activation is met by the combined actions of glycolysis and mitochondrial OXPHOS41. To validate our findings that the lower platelet mtDNA content is associated with platelet mitochondrial dysfunction, and to investigate platelet glycolysis activity, we assessed the metabolic activity of washed platelets of five PLHIV and five age-sex matched controls to assess membrane potential (Δψm), mitochondrial superoxide production (ROSm) and real-time glycolysis and mitochondrial respiration using the Seahorse Extracellular Flux Analyzer41. Platelet Δψm, as assessed by Tetramethylrhodamine ethyl ester (TMRE) fluorescence, was lower in PLHIV compared to controls (1669 ΔMFI (IQR: 911–2233) vs. 4545 ΔMFI (IQR: 2249–5168) respectively, p = 0.02; Fig. 2E, t-test). In line with reduced Δψm, there was a small increase in ROSm in PLHIV at basal conditions (Fig. 2F; 7654 MFI (IQR: 6777–7798) vs 4956 MFI (IQR: 3840–6284), p = 0.007, t-test). Next, using the Seahorse Extracellular Flux Analyzer, mitochondrial respiration and glycolytic capacity were investigated by assessing the oxygen consumption rate (OCR; measure for mitochondrial respiration/OXPHOS) and the acidification rate (ECAR; measure for glycolysis). There was a trend towards lower baseline platelet mitochondrial respiration (OCR in pmol/min) in PLHIV compared to controls (Fig. 2D; 47.4 pmol/min (SD: 11.3) vs 55.1 pmol/min (SD: 12.2), p = 0.084). Additionally, PLHIV had a smaller increase in OCR after ex vivo platelet stimulation with both CRP-XL (108% (SD: 16) vs 119% (SD: 16), p = 0.056, Fig. 2B and Supplemental Fig. S5) and Thrombin receptor activating peptide (TRAP; 139% (SD: 14) vs 167% (SD: 13), p = 0.037; Fig. 2A–C) compared to matched controls.
Figure 2.

Platelet energy phenotype determined by the Seahorse extracellular flux analyzer and mitochondrial membrane potential and ROS production. Samples were measured with five replicates, 1 × 107 washed platelets per well. Five age-and sex-matched donors per group. (A) Real-time mitochondrial respiration depicted as oxygen consumption rate (OCR) at 18 timepoints. Four agonists or inhibitors were added in the following order: (1) Thrombin Receptor Activator Peptide-6 (TRAP-6; 50 µM) or medium causing platelet activation; (2) Oligomycin which inhibits cellular ATP production; (3) Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP), an uncoupling agent causing maximum oxygen consumption through complex IV, and (4) Antimycin A which inhibits all mitochondrial respiration (CRP-XL can be found in Supplemental Fig. 9). (B) Energymap showing the change in mitochondrial respiration (as change in OCR) and glycolysis (extracellular acidification rate (ECAR) from basal conditions after ex vivo stimulation with platelet agonists. ((A)) Depicts the real-time ECAR after (1) medium or TRAP-6 (2) oligomycine, (3) FCCP and (4) antimycin A. Samples were measured with five replicates, 1 × 107 washed platelets per well. Five age-and sex-matched donors per group. (D) Basal OCR as mean of first three measurements. (E) Membrane potential was determined using Tetramethylrhodamine ethyl ester (TMRE) staining with the uncoupler FCCP disrupting mitochondrial membrane potential as a negative control for every sample. Data depicted as delta geometric mean fluorescence intensity (MFI). (F) Mitochondrial ROS production with MitoSox staining in MFI. Data are depicted as mean ± standard error of the mean (SEM) and analyzed using the unpaired student’s T-test.
The fraction of the maximal mitochondrial capacity (maximal OCR after stimulation with the uncoupling agent FCCP) used after ex vivo platelet stimulation with TRAP was high in both groups (Supplemental Fig. S6d; p > 0.1). This suggests that maximal mitochondrial capacity is a limiting factor in platelet activation. Conversely, there was no difference between groups in ECAR (glycolysis) at baseline (Fig. 2B; HIV: 16.0 mpH/min (SD: 3.4) vs HC: 14.7 mpH/min (SD: 2.4), p > 0.1) or after ex vivo stimulation. The increase in mitochondrial respiration (OCR) after ex vivo stimulation significantly correlated with mtDNApl (R = 0.77, p = 0.045, n = 7, Fig. 3A) and a similar trend was found for basal mitochondrial respiration of platelets (R = 0.61, p = 0.14, n = 7, Fig. 3B). Taken together, our data suggest that the lower platelet mtDNA in PLHIV in associated with a concurrent reduction in mitochondrial respiration capacity (OXPHOS) without a compensatory increase in glycolysis.
Figure 3.

Platelet mitochondrial DNA vs mitochondrial respiration measured by Oxygen consumption rate (OCR in pmol/min). (A) Correlation plot showing platelet mitochondrial DNA (mtDNA) with change in oxygen consumption rate (mitochondrial respiration) measured by Seahorse extracellular flux analyzer after ex vivo stimulation with thrombin (TRAP 50 μM). mtDNA was normalized using inverse rank-based transformation and Pearson’s correlation coefficient is shown. (B) mtDNA in platelets vs basal OCR in pmol/min (an average of first three measurements). OCR was measured in triplo and for 5 PLHIV and 2 controls both mtDNA in platelets and real-time measurements of mitochondrial respiration were available and included in the analysis.
HIV infection is associated with platelet dysfunction
Next, we explored the possible consequences of lower mtDNApl and mitochondrial dysfunction for platelet parameters and function. Platelet counts were similar between PLHIV and healthy controls (Table 2), but PLHIV had smaller platelets (mean platelet volume: PLHIV:10.1 fL (IQR: 9.7:10.7) vs 10.8 fL (IQR: 10.3–11.3), p < 0.0001), as well as a lower immature platelet fraction (IPF; a marker for freshly released platelets from the bone marrow) compared to controls (Table 2; 3.3% (IQR: 2.5–4.6) vs 3.7% (IQR: 2.8–5.9) respectively, p = 0.018). Platelet size (mean platelet volume) correlated positively with mtDNApl (Fig. 5C).
Figure 5.

Association of platelet mitochondrial DNA with platelet reactivity and mean platelet volume (MPV). (A, B) correlation of normalized mitochondrial DNA content in platelets (mtDNApl) and the binding of fibrinogen to the activated integrin αIIbβ3 after ex vivo platelet stimulation with ADP (125uM) in PLHIV (A) and healthy controls (B). (C, D) correlation of normalized mitochondrial DNA content in platelets (mtDNApl) and MPV in PLHIV (C) and healthy controls (D). All correlations were analyzed using Pearson’s correlation coefficients.
Next, platelet activation and function and function were determined using multiple methods. First, plasma markers of in vivo platelet activation (chemokines released from alpha-granules; CCL5, CXCL4, CXCL7) were comparable between PLHIV and controls (Table 2). Second, using flow cytometry, the activation status of circulating platelets, as well as their reactivity to ex vivo stimulation by platelet agonists was assessed. In unstimulated platelets, the expression of the alpha-granule marker P-selectin (measure of platelet degranulation) and the binding of fibrinogen to the activated integrin αIIbβ3 (measure of aggregation; Table 2) were also similar across groups. When analysis was restricted to individuals above 40 years of age or male only, unstimulated platelet activation was lower in PLHIV compared to controls (Supplemental Tables S1, S2). In line with this observation, fibrinogen binding to αIIbβ3 in response to stimulation by adenosine diphosphate (ADP)- and collagen related peptide (CRP-XL) was reduced in PLHIV-induced) (Fig. 4A). Differences in P-selectin reactivity across the groups were smaller with only a significant difference with a high dose (125 µM) of ADP stimulation showed a significant difference between PLHIV and controls (Fig. 4B). We observed no correlations with cART regimens containing either NNRTI, PI or INSTI-use as well as ABC and platelet reactivity indices (all P > 0.15). In addition, persistent immune activation did not correlate with platelet indices in this cohort (Supplemental Table S4). In summary, these data show that platelet reactivity, and especially αIIbβ3 activation is reduced in PLHIV.
Figure 4.
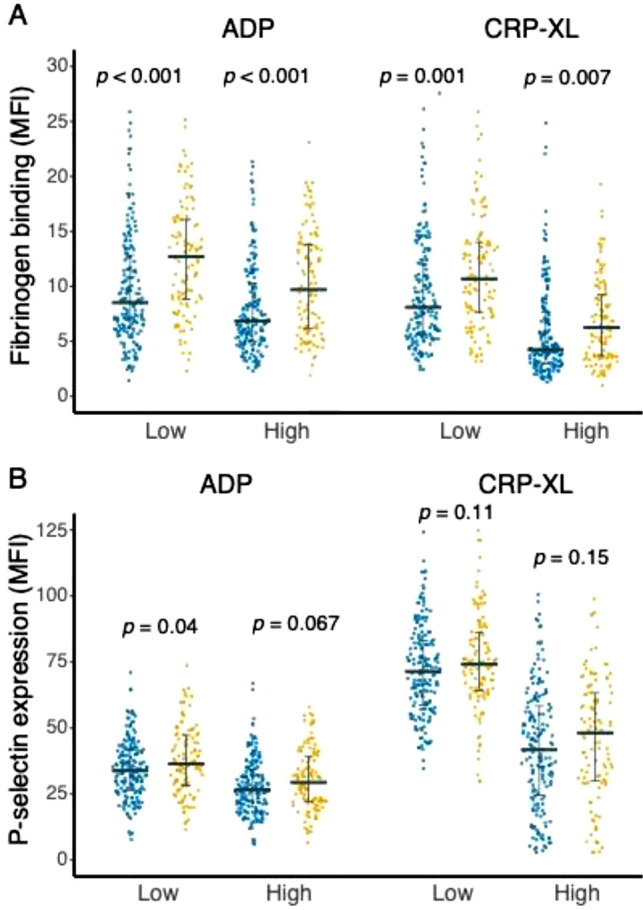
Platelet reactivity after collagen (CRP-XL) and ADP stimulation. (A) Platelet fibrinogen binding is depicted as mean fluorescence intensity (MFI) after ex vivo stimulation with platelet agonist collagen-related peptide (CRP-XL; 33 ng/mL; 625 ng/mL) and adenosine diphosphate (ADP; 1.2 μM, 125 μM). (B) Platelet degranulation measured by P-selectin expression is depicted as mean fluorescence intensity (MFI) after ex vivo stimulation with CRP-XL (33 ng/mL; 625 ng/mL) and ADP (1.2 μM, 125 μM). Data are shown as dotplot with error bars median and interquartile range (IQR). Data were analyzed using unpaired Student’s T-test. People living with HIV (PLHIV); healthy controls (HC).
Association of mtDNApl with platelet function
Next, given the role of platelet mitochondria in platelet function, we assessed associations of mtDNApl with platelet reactivity in PLHIV and controls. MtDNApl copies did neither correlate with ADP-induced P-selectin expression, nor with binding of fibrinogen to platelets or mean platelet volume (MPV) in PLHIV (Fig. 5A,B). In controls, MtDNApl copies were significantly correlated with MPV (Fig. 5C) and a positive trend was observed with fibrinogen binding (Fig. 5D).
To further explore the link between platelet reactivity and platelet activation, we performed a principal component analysis (PCA) to summarize both platelet activation (plasma markers of platelet activation and unstimulated P-selectin expression and fibrinogen binding) and platelet reactivity (P-selectin expression and fibrinogen binding after ex vivo ADP and CRP-XL stimulation). This PCA showed that Principal component (PC) 1 mainly represented platelet reactivity (P-selectin expression and fibrinogen binding after stimulation) whereas PC2 mainly represented in vivo platelet activation (plasma markers and unstimulated P-selectin expression and fibrinogen binding; Supplemental Fig. S7a,b). We used these derivatives to correlate mtDNApl with platelet reactivity (coordinates on PC1) and platelet activation (coordinates on PC2). mtDNApl correlated with platelet reactivity (Supplemental Fig. S7c; PC1 of platelet parameters vs mtDNApl, R = 0.14, p = P = 0.024), but not with in vivo platelet activation (Supplemental Fig. S7d; PC2 of platelet parameters vs mtDNApl, R = 0.05, P = 0.41). These data suggest that mtDNA depletion is associated with platelet dysfunction with a reduced platelet reactivity capacity, but not with increased platelet activation status.
Whereas mitochondrial dysfunction can result in platelet dysfunction, platelet activation itself may also contribute to loss of mitochondria from platelets through the formation of platelet microparticles (PMP) formation42. We therefore studied PMP in a subgroup of 20 PLHIV and age-sex matched controls. No differences in total PMP number (Supplemental Fig. S8a), nor PMPs containing mitochondria (Supplemental Fig. S8b) were observed across both groups.
Discussion
The present data show that PLHIV on long-term cART have reduced platelet mitochondrial content (mtDNApl) which was associated with platelet mitochondrial dysfunction and reduced energy supply. Platelet mitochondria play a key role in platelet metabolism, ATP production and platelet activation and lifespan, and we propose that the observed abnormalities in mtDNApl and platelet mitochondrial function contribute to platelet dysfunction in PLHIV.
The literature on platelet function in cART treated individuals is contradictory, with some studies reporting increased platelet reactivity33–37, while others reporting reduced reactivity28,38,39. This heterogeneity in study results may be partly explained by differences in the characteristics of study participants, including the enrolment of PLHIV with detectable plasma HIV-RNA, the degree of persistent immune activation, cART regimens and timing of treatment initiation, factors that all have changed considerably over the years. In accordance with our present findings, Mesquita et al.28 recently reported decreased platelet reactivity and platelet mitochondrial dysfunction in 36 PLHIV on stable cART. In contrast to our findings, platelets in the PLHIV in their cohort exhibited increased P-selectin expression. In the present study, not only platelet P-selectin expression, but also soluble markers of platelet activation were similar in PLHIV and controls. In addition, PLHIV exhibited a lower immature platelet fraction and a similar number of mitochondria-containing platelet microparticles. Together, these findings argue against excessive platelet activation being primarily responsible for reduced platelet reactivity in PLHIV. Whether mitochondrial depletion contributes to the observed platelet dysfunction in PLHIV remains uncertain. A recent study reported that chemotherapy-associated platelet hyporeactivity was caused by mitochondrial dysfunction and subsequent reduction in mitochondrial respiration27. Consistent with these observations, the lower platelet mtDNA content in PLHIV was associated with a reduction in mitochondrial respiration capacity, which may negatively impact platelet reactivity. In our study, however, mtDNApl levels in PLHIV did not correlate with ex vivo platelet reactivity, whereas a positive trend was observed in HIV uninfected controls. The fact that all PLHIV exhibited reduced mtDNApl with little variation in the absolute values may explain the absence of a correlation with platelet reactivity measures. Future studies focusing on platelet dysfunction should incorporate mitochondrial dysfunction to corroborate our findings.
Age, history of zidovudine (AZT)-use and innate immune activation were all associated with decreased mtDNApl. NRTI-use is a well-known cause of mitochondrial dysfunction and mtDNA depletion6–8. We found that prior AZT-use was a possible risk factor for reduced mtDNApl, a trend that remained after correcting for age and CD4 nadir, whereas total duration of cART-use or duration of HIV infection were not. As platelets are short-lived, it is conceivable that mitochondrial mass is reduced during thrombopoiesis and that the known bone marrow toxicity of AZT 1 is still present even after switching to newer NRTIs such as TDF or ABC. Even though these NRTIs are known to have lower mitochondrial toxicity1, it is unclear whether long-term treatment does not exert any cumulative reduction in mtDNApl too. While ABC-use has been linked to platelet perturbations in multiple studies43–45, others could not confirm ABC associated platelet dysfunction35,46. In our study, neither mtDNApl content nor platelet function were associated with current or prior ABC-use. Unfortunately, we could not dissect the link between the mtDNApl, overall NRTI exposure and duration of HIV infection itself, as exposure to NRTIs was high in the total study group. Still, as mtDNApl content was lower in PLHIV than in controls, it is plausible that both NRTIs and HIV itself exert long-term changes in mitochondrial function2. This possible long-term NRTI effect on mitochondrial function in platelets supports recent efforts to implement NRTI sparing regimens as viable treatment options for long-term HIV treatment47. It would be interesting to assess mtDNApl content, as well as platelet function, in PLHIV who are switched to a NRTI sparing regimen.
Mitochondrial dysfunction and depletion have been associated with many diseases such as dementia, neuropsychiatric diseases, and cardiovascular diseases22,23,30,48. In PLHIV, these (non-AIDS related) co-morbidities have also been linked to persistent inflammation20,21. In our cohort, we indeed found increased levels of hsCRP, sCD14 and sCD163, but only the latter parameter was associated with mtDNA levels in platelets. Importantly, sCD163 was shown to be independently correlated with overall mortality in PLHIV and the incidence of non-AIDS related comorbidities49,50.
Targeting mitochondrial dysfunction and inflammation may help reduce excess mortality and morbidity that is associated with HIV infection, even when treated successfully with cART22,27. Reducing inflammation, besides reducing NRTI exposure, could indirectly reduce oxidative stress and mitochondrial dysfunction while treatment with ROS scavengers may also have beneficial effects in reducing non-AIDS related co-morbidities.
Multiple methods of mtDNA quantification have been used in whole blood or peripheral blood mononuclear cell (PBMC) fractions in HIV and other diseases14,32. However, different methods of quantification, and heterogeneity of the cell composition of whole blood or PBMCs may hamper its interpretation32,51. It is conceivable that platelet mtDNA content could mirror mitochondrial toxicity in other cell types as well. As a single cell-type source of mtDNA, platelet mtDNA may indeed serve as a possible biomarker for mitochondrial toxicity and non-AIDS related comorbidities associated with mitochondrial dysfunction, such as neurocognitive impairment and cardiovascular disease22.
Our study has limitations associated with its cross-sectional design. Even though sample size was large enough to explore the link between inflammation, cART-use, mtDNApl and platelet function, it lacked power to confirm a possible link between mtDNApl levels and clinical outcomes such as non-AIDS co-morbidities and NRTI-related adverse events. Although we observed reduced oxygen consumption in individuals with reduced mtDNApl, the overall reduction of mtDNApl in PLHIV prevented to investigate functional consequences of mtDNApl depletion in PLHIV. Furthermore, we did not perform immunofluorescence confocal or transmission electron microscopy experiments in the current study. In addition, age and sex differed substantially between cohorts, with an effect of age on mtDNA content in the uninfected cohort. We explored multiple methods to account for these differences using adjusted models and subgroup analyses. The subgroup of above > 40 years revealed a significant difference between PLHIV and controls supporting the independent correlation of HIV infection in the age-sex adjusted model. Finally, our study included mostly Caucasian men limiting generalization of the findings to women and non-Caucasians.
In conclusion, PLHIV under long-term cART have reduced platelet mtDNA content and abnormalities in platelet mitochondrial respiration, which may possibly contribute to platelet dysfunction. Given the key role of platelets and mitochondria in the pathophysiology of long-term complications of HIV, interventions targeting platelet mitochondria, such as introducing NRTI sparing regimens, should be considered.
Methods
Patient selection
This cross-sectional, single center, prospective study was performed at the Radboud university medical center, a tertiary teaching hospital in The Netherlands. This study is part of the Human Functional Genomics Project (HFGP; www.humanfunctionalgenomicsproject.org) and was conducted in accordance with the Declaration of Helsinki after approval of the ethics committee (CMO Arnhem-Nijmegen, The Netherlands; NL42561.091.12, 2012/550). No animal experiments were performed in the current study. Adult HIV-1-infected individuals receiving cART for at least six months were included after providing written informed consent. Other inclusion criteria were a suppressed viral load (< 200 copies/mL). Exclusion criteria included, use of P2Y12 receptor antagonists (platelet inhibitor), an active hepatitis B or C infection and/or signs of other active intercurrent infection other than HIV-1 (e.g. fever in last week or antibiotic-use in last 4 weeks). Healthy controls were concurrently included throughout the duration of inclusion of PLHIV. Exclusion criteria were use of medication (excluding oral contraceptives or paracetamol) and/or signs of an active infection in the last month. Clinical data was collected by extracting data from electronic medical record (Epic Systems, Verona, WI, USA). History of cART use was extracted from the Dutch HIV registry (Stichting HIV-monitoring). In a separate validation experiment for mitochondrial function, five virally suppressed male PLHIV (45–60 years) who were not using statins or acetylsalicylic acid (ASA), were enrolled together with five age and sex matched controls.
Platelet count and function
Platelet count and parameters were determined using an automated hematology analyzer (Sysmex, Kobe, Japan). Platelet reactivity was determined in citrated whole blood (3.2% sodium citrate, Becton Dickinson, Franklin Lakes, NJ, USA) using a flow cytometry based assay as previously described between 1 and 3 h after blood collection52. Platelets were ex vivo stimulated with ADP (1.2 and 125 μM; Sigma-Aldrich, Zwijndrecht, The Netherlands) and CRP-XL (a kind gift from Prof. Farndale, Cambridge, UK) for 20 min at room temperature. Platelets were stained using anti-CD61 (Beckman Coulter, Brea. CA, USA), anti-P-selectin (Biolegend, San Diego, CA, USA) and anti-fibrinogen (DAKO, Santa Clara, CA) antibodies and fixated in 0.2% paraformaldehyde. Platelets were identified based on Size (FSC), granularity (SSC) and their expression of CD61. Degranulation was determined as the membrane expression of α-granule protein P-selectin and platelet aggregation was quantified as the amount of fibrinogen binding to the activated integrin αIIbβ3. Platelet reactivity was measured on a FC500 flow cytometer (Beckman Coulter, Brea, USA). Data were extracted using Kaluza 2.1 (Beckman Coulter), normalized against quality controls to ensure measurement stability and are expressed as median fluorescence intensity (MFI). A gating strategy is provided in Supplemental Fig. S1.
Platelet isolation
Platelet rich plasma (PRP) was obtained from citrated plasma (Vacutainer, Beckton-Dickinson) after centrifugation 156g for 15 min without brake at room temperature (RT). Samples were processed within 2 h of blood collection. Platelet count in PRP was measured using an automated hematology analyser (Sysmex, Kobe, Japan). Washed platelets were obtained as previously described53. In short, PRP was supplemented with acid citrate dextrose (10%) and prostaglandin I2 and washed twice with Hepes tyrode’s buffer using centrifugation (330g, 20 min).
Platelet microparticles
Peripheral blood was centrifuged at 1000g for 5 min. Plasma was then centrifuged at 1500g for 20 min to obtain platelet poor plasma (PPP). 1 mL of PPP was centrifuged for 30 min at 20,000g to pellet microparticles (MPs). The MP pellet washed once in 500 µL calcium-free HBS complemented with 0.2% bovine serum albumin (pH 7.3) by 45 min centrifugation at 20,000g. MPs were labeled with MitoTracker Deep Red (200 nM; Invitrogen, Breda, The Netherlands) in HBS at room temperature containing calcium and subsequently stained with anti-CD61, anti-CD62p, Annexin-V (all Biolegend), anti-CD45 and anti-CD41 (both Beckman Coulter). MPs were analyzed using a Cytoflex flow cytometer (Beckman Coulter) including the sensitive violet Side Scatter (405 nM; VSSC) and FSC for detection of ultrasmall particles (1 µm)54. Platelet MPs (PMPs) were selected based on aforementioned markers. Counting beads (Sphero Nano fluorescent, Spherotech, Fulda, Germany) of different sizes were included for reference to correct for concentration and size. All solutions for PMP isolation and staining were centrifuged at 20,000g for 20 min to remove (fluorescent) aggregates. A gating strategy is provided in Supplemental Fig. S2.
mtDNA quantification and mitochondrial function
After platelet count measurement of every PRP sample, 500 µL PRP was pelleted by centrifugation (2000g, 10 min at RT without brake) and lysed using Triton-X100 0.1% for mtDNA quantification. MtDNApl was measured by real time Quantitative PCR using Mitotox Quickscan (Primagen, Amsterdam, Netherlands), Advanced SYBR green Super mix (Bio-rad, Hercules, CA, USA) and CFX96 Real time Detection System (Bio-Rad) according to manufacturer’s instructions. The kit includes primers and calibrators for quantification. A calibration curve with known mtDNA concentration was concurrently measured on every plate to ensure stability and quantification. A dilution curve with PRP showed a high correlation coefficient for mtDNA and platelet concentration. mtDNA copies per platelet (mtDNApl) are calculated by dividing mtDNA copy number per well by platelet count in 500 µL of PRP. Contamination of leukocyte and erythrocyte was low < 1:10,000. Samples with low platelet counts in PRP (below 50 × 1012/mL) and erythrocyte/leukocyte contamination were excluded (n = 2). Mitochondrial respiration was quantified using the 96-well format Seahorse extracellular flux analyzer (Agilent, MA, USA). Washed platelets were plated to 60,000 platelets/μL and seeded onto Cell-Tak coated XF96 microplates (Corning, Corning, NY, USA). A mitochondrial stress test was performed as described eslewhere41 using oligomycin (1 μM, ATP synthase inhibitor (complex V), reducing mitochondrial respiration), Carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP; 1 μM, uncoupling agent causing maximum mitochondrial respiration), antimycin A (2.5 μM, Complex III inhibitor inhibiting mitochondrial respiration) and 2-deoxy-d-glucose (2DG; 40 mM, inhibits glycolysis) after ex vivo stimulation (all Sigma). Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were determined before and after injection of platelet agonists. Samples were measured in five replicates with three measurements after every injection. For mitochondrial respiration measurements using Seahorse extracellular flux analyzer, significant outliers were excluded from analysis if measurement was 2× SD below the mean value in every timepoint with a maximum of one of the five replicates.
Mitochondrial membrane potential was determined using Tetramethylrhodamine ethyl ester (TMRE, Sigma; 100 nM at 37 °C for 20 min). A negative control was generated for every sample using FCCP (1 μM at 37 °C for 10 min). Mitochondrial reactive oxygen species (ROSm) were detected using the cationic probe MitoSOX Red (Invitrogen, Carlsbad, CA, USA (2.5 μM at 37 °C for 30 min). Both probes were quantified in CD61+ cells using a Cytoflex flow cytometer (Beckman Coulter).
Plasma markers
Concentrations of three plasma markers of platelet activation: chemokine (C–X–C motif) ligand 4 (CXCL4 also known as platelet factor 4), CXCL7 (also known as beta-thromboglobulin) and chemokine (C–C motif) ligand (CCL5; also known as RANTES) were determined by ELISA (R and D systems, Minneapolis, USA) according to the manufacturer’s instructions in citrated PPP52. In addition, markers of persistent inflammation, high-sensitive C-reactive protein (hsCRP), sCD163 and sCD14, were measured using ELISA (Quantikine, R and D systems) according to the manufacturer’s instructions in EDTA plasma.
Statistical analyses
Data were analyzed by independent T-test or Mann–Whitney U test. Pearson’s correlations coefficient was used for univariate correlation analyses, unless otherwise stated. An inverse rank-based normalization was performed for non-normal data (e.g. mtDNA in platelets). A multivariate linear regression model was used to correct for age, body mass index (BMI) and sex. Several sensitivity analyses using subgroups (males only and above 40 years) were performed to explore for possible confounding. Principal component analysis (PCA) was performed using singular value decomposition to summarize platelet function and correlate with mtDNApl. cART-use was calculated as days on a certain drug, cumulative use of multiple drugs of the same class were combined. R studio (CRAN project) and Graphpad Prism version 5.03 were used for analyses.
Supplementary Information
Acknowledgements
This study was partly supported by an Aidsfonds Netherlands Grant (P-29001). M.G.N. was supported by a Spinoza grant of the Netherlands Organization for Scientific Research.
Author contributions
W.A.H., Q.M. and A.J.V. designed the study. W.A.H., L.W., M.J., R.C. recruited and included the participants. W.A.H., L.W., M.J. and performed the laboratory experiments. L.H., R.R. and R.T.U. provided laboratory support and contributed vital new reagents and analytical tools. W.A.H analyzed the data and interpreted the data together with Q.M., P.G., R.R. and A.J.V. W.A.H. and A.J.V. wrote the manuscript. All authors have read and contributed significantly to the final manuscript.
Funding
This study was partly supported by an AIDS-fonds Netherlands Grant. M.G.N. was supported by a Spinoza Grant of the Netherlands Organization for Scientific Research.
Competing interests
Q.M., A.J. V., and M.G.N. received research support from ViiV. All mentioned research support was unrelated to the current study.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-85775-5.
References
- 1.Maagaard A, Kvale D. Mitochondrial toxicity in HIV-infected patients both off and on antiretroviral treatment: A continuum or distinct underlying mechanisms? J. Antimicrob. Chemother. 2009;64:901–909. doi: 10.1093/jac/dkp316. [DOI] [PubMed] [Google Scholar]
- 2.Gardner K, Hall PA, Chinnery PF, Payne BA. HIV treatment and associated mitochondrial pathology: Review of 25 years of in vitro, animal, and human studies. Toxicol Pathol. 2014;42:811–822. doi: 10.1177/0192623313503519. [DOI] [PubMed] [Google Scholar]
- 3.Casula M, et al. Infection with HIV-1 induces a decrease in mtDNA. J. Infect. Dis. 2005;191:1468–1471. doi: 10.1086/429412. [DOI] [PubMed] [Google Scholar]
- 4.Brinkman K, Smeitink JA, Romijn JA, Reiss P. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet. 1999;354:1112–1115. doi: 10.1016/S0140-6736(99)06102-4. [DOI] [PubMed] [Google Scholar]
- 5.Brinkman K, ter Hofstede HJ, Burger DM, Smeitink JA, Koopmans PP. Adverse effects of reverse transcriptase inhibitors: Mitochondrial toxicity as common pathway. AIDS. 1998;12:1735–1744. doi: 10.1097/00002030-199814000-00004. [DOI] [PubMed] [Google Scholar]
- 6.Johnson AA, et al. Toxicity of antiviral nucleoside analogs and the human mitochondrial DNA polymerase. J. Biol. Chem. 2001;276:40847–40857. doi: 10.1074/jbc.M106743200. [DOI] [PubMed] [Google Scholar]
- 7.Martin AM, et al. Accumulation of mitochondrial DNA mutations in human immunodeficiency virus-infected patients treated with nucleoside-analogue reverse-transcriptase inhibitors. Am. J. Hum. Genet. 2003;72:549–560. doi: 10.1086/367849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Koczor CA, Lewis W. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert Opin. Drug Metab. Toxicol. 2010;6:1493–1504. doi: 10.1517/17425255.2010.526602. [DOI] [PubMed] [Google Scholar]
- 9.Cote HC, et al. Changes in mitochondrial DNA as a marker of nucleoside toxicity in HIV-infected patients. N. Engl. J. Med. 2002;346:811–820. doi: 10.1056/NEJMoa012035. [DOI] [PubMed] [Google Scholar]
- 10.McComsey GA, et al. Changes in fat mitochondrial DNA and function in subjects randomized to abacavir-lamivudine or tenofovir DF-emtricitabine with atazanavir-ritonavir or efavirenz: AIDS Clinical Trials Group study A5224s, substudy of A5202. J. Infect. Dis. 2013;207:604–611. doi: 10.1093/infdis/jis720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ezinga M, Wetzels JF, Bosch ME, van der Ven AJ, Burger DM. Long-term treatment with tenofovir: Prevalence of kidney tubular dysfunction and its association with tenofovir plasma concentration. Antivir. Ther. 2014;19:765–771. doi: 10.3851/IMP2761. [DOI] [PubMed] [Google Scholar]
- 12.Casado JL, et al. Prevalence and significance of proximal renal tubular abnormalities in HIV-infected patients receiving tenofovir. AIDS. 2016;30:231–239. doi: 10.1097/QAD.0000000000000901. [DOI] [PubMed] [Google Scholar]
- 13.Cez A, et al. Decreased expression of megalin and cubilin and altered mitochondrial activity in tenofovir nephrotoxicity. Hum. Pathol. 2018;73:89–101. doi: 10.1016/j.humpath.2017.12.018. [DOI] [PubMed] [Google Scholar]
- 14.Maagaard A, Holberg-Petersen M, Kvittingen EA, Sandvik L, Bruun JN. Depletion of mitochondrial DNA copies/cell in peripheral blood mononuclear cells in HIV-1-infected treatment-naive patients. HIV Med. 2006;7:53–58. doi: 10.1111/j.1468-1293.2005.00336.x. [DOI] [PubMed] [Google Scholar]
- 15.Miro O, et al. Mitochondrial effects of HIV infection on the peripheral blood mononuclear cells of HIV-infected patients who were never treated with antiretrovirals. Clin. Infect. Diseases Off. Publ. Infect. Diseases Soc. Am. 2004;39:710–716. doi: 10.1086/423176. [DOI] [PubMed] [Google Scholar]
- 16.Miura T, et al. Depletion of mitochondrial DNA in HIV-1-infected patients and its amelioration by antiretroviral therapy. J. Med. Virol. 2003;70:497–505. doi: 10.1002/jmv.10423. [DOI] [PubMed] [Google Scholar]
- 17.Tarancon-Diez L, et al. Immunometabolism is a key factor for the persistent spontaneous elite control of HIV-1 infection. EBioMedicine. 2019 doi: 10.1016/j.ebiom.2019.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casula M, et al. Mitochondrial DNA decline in T cells of HIV-1 seroconverters may be dependent on immune activation. J. Infect. Dis. 2007;196:371–376. doi: 10.1086/519284. [DOI] [PubMed] [Google Scholar]
- 19.Perez-Santiago J, et al. Increased cell-free mitochondrial DNA is a marker of ongoing inflammation and better neurocognitive function in virologically suppressed HIV-infected individuals. J. Neurovirol. 2017;23:283–289. doi: 10.1007/s13365-016-0497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angelidou K, et al. Changes in inflammation but not in T-cell activation precede non-AIDS-defining events in a case-control study of patients on long-term antiretroviral therapy. J. Infect. Dis. 2018;218:239–248. doi: 10.1093/infdis/jix666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nordell AD, et al. Severity of cardiovascular disease outcomes among patients with HIV is related to markers of inflammation and coagulation. J. Am. Heart Assoc. 2014;3:e000844. doi: 10.1161/JAHA.114.000844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang L, et al. Platelet mitochondrial dysfunction and the correlation with human diseases. Biochem. Soc. Trans. 2017;45:1213–1223. doi: 10.1042/BST20170291. [DOI] [PubMed] [Google Scholar]
- 23.Melchinger H, Jain K, Tyagi T, Hwa J. Role of platelet mitochondria: Life in a nucleus-free zone. Front. Cardiovasc. Med. 2019;6:153. doi: 10.3389/fcvm.2019.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Malik AN, Czajka A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion. 2013;13:481–492. doi: 10.1016/j.mito.2012.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Rondina MT, Weyrich AS, Zimmerman GA. Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res. 2013;112:1506–1519. doi: 10.1161/CIRCRESAHA.113.300512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zharikov S, Shiva S. Platelet mitochondrial function: From regulation of thrombosis to biomarker of disease. Biochem. Soc. Trans. 2013;41:118–123. doi: 10.1042/BST20120327. [DOI] [PubMed] [Google Scholar]
- 27.Baaten C, et al. Impaired mitochondrial activity explains platelet dysfunction in thrombocytopenic cancer patients undergoing chemotherapy. Haematologica. 2018;103:1557–1567. doi: 10.3324/haematol.2017.185165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mesquita EC, et al. Persistent platelet activation and apoptosis in virologically suppressed HIV-infected individuals. Sci. Rep. 2018;8:14999. doi: 10.1038/s41598-018-33403-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kholmukhamedov A, Jobe S. Platelet respiration. Blood Adv. 2019;3:599–602. doi: 10.1182/bloodadvances.2018025155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lindqvist D, et al. Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2018;43:1557–1564. doi: 10.1038/s41386-017-0001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ito S, et al. Functional integrity of mitochondrial genomes in human platelets and autopsied brain tissues from elderly patients with Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 1999;96:2099–2103. doi: 10.1073/pnas.96.5.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurtado-Roca Y, et al. Adjusting MtDNA quantification in whole blood for peripheral blood platelet and leukocyte counts. PLoS ONE. 2016;11:e0163770. doi: 10.1371/journal.pone.0163770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayne E, et al. Increased platelet and microparticle activation in HIV infection: Upregulation of P-selectin and tissue factor expression. J. Acquir. Immune Defic. Syndr. 2012;59:340–346. doi: 10.1097/QAI.0b013e3182439355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O'Brien M, et al. Aspirin attenuates platelet activation and immune activation in HIV-1-infected subjects on antiretroviral therapy: A pilot study. J. Acquir. Immune Defic. Syndr. 2013;63:280–288. doi: 10.1097/QAI.0b013e31828a292c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tunjungputri RN, et al. Reduced platelet hyperreactivity and platelet-monocyte aggregation in HIV-infected individuals receiving a raltegravir-based regimen. AIDS. 2014;28:2091–2096. doi: 10.1097/QAD.0000000000000415. [DOI] [PubMed] [Google Scholar]
- 36.Holme PA, et al. Enhanced activation of platelets with abnormal release of RANTES in human immunodeficiency virus type 1 infection. Faseb. J. 1998;12:79–89. doi: 10.1096/fsb2fasebj.12.1.79. [DOI] [PubMed] [Google Scholar]
- 37.von Hentig N, et al. Platelet-leucocyte adhesion markers before and after the initiation of antiretroviral therapy with HIV protease inhibitors. J. Antimicrob. Chemother. 2008;62:1118–1121. doi: 10.1093/jac/dkn333. [DOI] [PubMed] [Google Scholar]
- 38.Haugaard AK, et al. Discrepant coagulation profile in HIV infection: Elevated D-dimer but impaired platelet aggregation and clot initiation. AIDS. 2013;27:2749–2758. doi: 10.1097/01.aids.0000432462.21723.ed. [DOI] [PubMed] [Google Scholar]
- 39.Satchell CS, et al. Platelet function and HIV: A case–control study. AIDS. 2010;24:649–657. doi: 10.1097/QAD.0b013e328336098c. [DOI] [PubMed] [Google Scholar]
- 40.Satchell CS, et al. Increased platelet reactivity in HIV-1-infected patients receiving abacavir-containing antiretroviral therapy. J. Infect. Dis. 2011;204:1202–1210. doi: 10.1093/infdis/jir509. [DOI] [PubMed] [Google Scholar]
- 41.Ravi S, et al. Defining the effects of storage on platelet bioenergetics: The role of increased proton leak. Biochim. Biophys. Acta. 1852;2525–2534:2015. doi: 10.1016/j.bbadis.2015.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boudreau LH, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood. 2014;124:2173–2183. doi: 10.1182/blood-2014-05-573543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baum PD, Sullam PM, Stoddart CA, McCune JM. Abacavir increases platelet reactivity via competitive inhibition of soluble guanylyl cyclase. AIDS. 2011;25:2243–2248. doi: 10.1097/QAD.0b013e32834d3cc3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor KA, et al. Pharmacological impact of antiretroviral therapy on platelet function to investigate human immunodeficiency virus-associated cardiovascular risk. Br. J. Pharmacol. 2019;176:879–889. doi: 10.1111/bph.14589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Falcinelli E, et al. In vivo platelet activation and platelet hyperreactivity in abacavir-treated HIV-infected patients. Thromb. Haemost. 2013;110:349–357. doi: 10.1160/TH12-07-0504. [DOI] [PubMed] [Google Scholar]
- 46.Diallo YL, et al. Abacavir has no prothrombotic effect on platelets in vitro. J. Antimicrob. Chemother. 2016;71:3506–3509. doi: 10.1093/jac/dkw303. [DOI] [PubMed] [Google Scholar]
- 47.Llibre JM, et al. Efficacy, safety, and tolerability of dolutegravir-rilpivirine for the maintenance of virological suppression in adults with HIV-1: phase 3, randomised, non-inferiority SWORD-1 and SWORD-2 studies. Lancet. 2018;391:839–849. doi: 10.1016/S0140-6736(17)33095-7. [DOI] [PubMed] [Google Scholar]
- 48.Madamanchi NR, Runge MS. Mitochondrial dysfunction in atherosclerosis. Circ. Res. 2007;100:460–473. doi: 10.1161/01.RES.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 49.Knudsen TB, et al. Plasma soluble CD163 level independently predicts all-cause mortality in HIV-1-infected individuals. J. Infect. Dis. 2016;214:1198–1204. doi: 10.1093/infdis/jiw263. [DOI] [PubMed] [Google Scholar]
- 50.Burdo TH, et al. Soluble CD163, a novel marker of activated macrophages, is elevated and associated with noncalcified coronary plaque in HIV-infected patients. J. Infect. Dis. 2011;204:1227–1236. doi: 10.1093/infdis/jir520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maagaard A, et al. Mitochondrial (mt)DNA changes in tissue may not be reflected by depletion of mtDNA in peripheral blood mononuclear cells in HIV-infected patients. Antivir. Ther. 2006;11:601–608. [PubMed] [Google Scholar]
- 52.van der Heijden WA, et al. A switch to a raltegravir containing regimen does not lower platelet reactivity in HIV-infected individuals. AIDS. 2018;32:2469–2475. doi: 10.1097/QAD.0000000000001993. [DOI] [PubMed] [Google Scholar]
- 53.Tunjungputri RN, et al. Differential effects of platelets and platelet inhibition by ticagrelor on TLR2- and TLR4-mediated inflammatory responses. Thromb. Haemost. 2015;113:1035–1045. doi: 10.1160/TH14-07-0579. [DOI] [PubMed] [Google Scholar]
- 54.Wisgrill L, et al. Peripheral blood microvesicles secretion is influenced by storage time, temperature, and anticoagulants. Cytometry A. 2016;89:663–672. doi: 10.1002/cyto.a.22892. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.