

Abstract
BACKGROUND AND PURPOSE: Despite the benign histology of optic pathway glioma (OPG) (low-grade astrocytoma), its biological behavior is unpredictable, and it is unclear whether specific morphologic or anatomic patterns may be predictive of prognosis. It is also unclear whether OPG associated with neurofibromatosis (NF) is a distinct entity from non–NF-OPG. Our purpose was to describe the MR imaging features of OPG, compare the findings between patients with and those without NF, and identify prognostic imaging signs.
METHODS: MR examinations of 91 patients with OPG (47 with NF and 44 without) were reviewed at presentation and during follow-up. The images were evaluated for size and extension of tumor, and imaging parameters. Statistical bivariate analysis was used to compare the patients with and those without NF, and Pearson correlation was used to evaluate the correlation between the different imaging parameters and prognosis. Kappa values were calculated to determine intraobserver and interobserver variability.
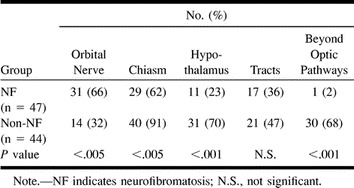
RESULTS: The most common site of involvement in the NF group was the orbital nerve (66%), followed by the chiasm (62%). In the non-NF group, the chiasm was the most common site of involvement (91%); the orbital nerves were involved in only 32%. Extension beyond the optic pathway at diagnosis was uncommon in the NF group (2%) but frequent in the non-NF group (68%). In the NF group, the tumor was smaller and the original shape of the optic pathways was preserved (91% vs. 27% in the non-NF group). The presence of cystic components was significantly more common in the non-NF patients (66% vs. 9% in the NF group). During follow-up, half the NF patients remained stable, in contrast to 5% of the non-NF group. No statistical correlation was found between imaging features and biological behavior of the tumor.
CONCLUSION: NF-OPG is a separate entity from non–NF-OPG, with different imaging features and prognosis, thereby warranting a specific diagnostic, clinical, and therapeutic approach.
Optic pathway gliomas (OPGs) account for approximately 5% of all brain tumors (1) and 10% to 15% of supratentorial tumors (2) in children. The tumor can arise anywhere along the optic pathway, from just behind the globe to the occipital cortex. The diagnosis of OPG also covers cases of hypothalamic chiasmatic glioma, in which it is difficult to distinguish the site of origin (3). In children, OPGs are almost always low-grade astrocytomas (1, 2). Ten percent to 70% of children with OPG have neurofibromatosis (NF) (3).
OPGs are associated with a high rate of morbidity and mortality. About 20% to 30% of patients experience visual impairment, neurologic deficits, and even death (3). Although there are no specific clinical, histologic, or neuroimaging features to differentiate aggressive from indolent OPGs (1), the prognosis is reportedly much better in children with NF than in those without (1). Treatment consists of surgery, chemotherapy, and irradiation. There are no strict criteria for starting treatment.
Most of the large neuroimaging studies of OPG were conducted at least a decade ago and were based on CT findings (4). Since the advent of MR imaging, most of the studies have focused on OPG in NF. The purpose of the present study was to describe the features of OPG on MR images, to compare the findings between patients with and without NF, and to identify prognostic imaging signs that could help in the decision for treatment.
Methods
The computerized radiology, oncology, and hospital records of the Hospital for Sick Children in Canada and the Schneider Children's Medical Center of Israel were searched for all cases of OPG between 1985 and 1996. Only patients for whom neuroimaging was available at presentation, before initiation of any kind of treatment or surgical intervention, were included in the study.
Ninety-one eligible patients were identified, 71 in the first institution and 20 in the second. These included 47 patients with NF and 44 without. In the patients without NF, the diagnosis of OPG was based on biopsy findings in 41 and on the characteristic involvement of the optic pathways in three. In the patients with NF, the diagnosis of OPG was based solely on the imaging findings.
Eighty-seven patients underwent MR imaging as the initial radiologic examination and four underwent CT; 34 patients had both. Follow-up examinations were almost exclusively performed with MR imaging. Owing to the retrospective nature of the study, the MR examinations were performed with different systems (GE Sigma 1.5 T, Siemens 1.5 T, or Elscint 0.5 and 2.0 T) and different protocols. All patients underwent at least one T1-weighted and one T2-weighted MR sequence. A detailed study of the optic pathways, either axial or coronal, was always included. Contrast agent (gadopentetate dimeglumine 0.1 to 0.2 mmol/kg) was used in 73 cases (80%).
For the purposes of the study, the neuroimaging scans were evaluated twice. The initial assessment was performed in Canada by a pediatric radiologist in consultation with a CAQ-certified pediatric neuroradiologist, and in Israel by three pediatric radiologists. The images were evaluated for multiple parameters: extension of the OPG, relation of the tumor to the blood vessels, calcifications, presence of cysts, peritumoral edema, and enhancement after contrast material injection. To evaluate the extent of the tumor, we defined seven locations: intraorbital optic nerves, retroorbital prechiasmatic optic nerves, chiasm, hypothalamus, retrochiasmatic proximal optic tracts, distal optic tracts, and extension outside the optic tracts (eg, into the frontal or temporal regions or the prepontine cistern). The optic nerves were categorized as enlarged or normal. The chiasm was categorized as thickened if it was enlarged but had preserved its original shape. Pronounced enlargement of the chiasm (greater than 1 cm) was measured in three planes, and the values multiplied to estimate volume. The hypothalamus was considered involved when it was expanded or included in the mass, or if it exhibited signal intensity changes; we did not attempt to determine the origin of the tumor other than to note whether the hypothalamus, chiasm, or both were involved. The posterior optic tracts were considered involved if they were hyperintense on T2-weighted images or enhanced after contrast injection, or if they contained a cystic lesion. Optic radiations were considered involved only in cases with direct extension of the mass in that region. The presence of hydrocephalus at diagnosis was noted.
The images obtained before initiation of any kind of treatment were evaluated for interval changes in the size and extension of the tumor. Four states were defined: 1) stable, no change in size or extent of involvement of the visual pathways; 2) minimally enlarged, unequivocal enlargement in one plane only or an increase of less than 25% in two planes; 3) enlarged, an increase in size of over 25% in two planes or further extension; and 4) reduced, shrinkage of more than 25% in two planes.
The charts were also reviewed for symptoms at diagnosis, interval between symptoms and diagnosis, length of follow-up, and method of treatment. The serial imaging studies were reviewed to determine the outcome of treatment (medical or surgical), and the imaging findings at presentation were correlated with the long-term findings. The histologic reports were also reviewed.
The second evaluation was performed 2 years after the first by the same radiologists and by a fellow in pediatric radiology in Canada, and by two of the radiologists and a fellow in pediatric radiology in Israel. Data for 82 patients were available for reevaluation. For 30 patients, the films from the diagnosis were not found, so they were included only in the analysis of interobserver variability. The review was performed independently and reviewers were blinded to the results of the initial assessment. A second evaluation was done of the imaging studies obtained at presentation or at the earliest available examination and of the last follow-up scans obtained in the study period. Tumor extension along the optic pathways, bilaterality, shape, and size were defined according to the same criteria as in the initial evaluation. The size and extension of the tumor were compared between the first and the last examinations. Kappa values were calculated for every parameter to determine intraobserver and interobserver variability.
The data were analyzed with BMPD software (5). Differences between groups for the continuous variables were determined with analysis of variance, and the association between discrete variables was calculated using Pearson's χ2-test and Fisher's exact test. All significance tests were two-sided; P values less than .05 were considered statistically significant.
Results
The study group included 45 boys and 46 girls aged 0.5 to 14.5 years. The NF subgroup included 22 boys and 25 girls aged 0.5 to 13.0 years (mean age, 5.4 years), and the non-NF group had 24 boys and 20 girls aged 0.5 to 14.5 years (mean age, 4.6 years). There was no significant difference between the patients with and without NF in regard to age or sex.
Symptoms were present at diagnosis in all the children without NF (100%) but in only 32% of those with NF (P < .0001). The specific symptoms are listed in Table 1. Within the NF group, the age of the symptomatic children was significantly younger (4.3 ± 2.1 years) than that of the asymptomatic children (6.0 ± 3.3 years) (P < .05). The mean interval between onset of symptoms and diagnosis in the non-NF group was 7 ± 11 months (range, 0 to 48 months), whereas in the NF group, it was almost zero (0.5 ± 0.09) (range, 0 to 1.5 months) (P < .005).
TABLE 1:
Symptoms at presentation of children with OPG

Involvement of the optic pathways by site at diagnosis is summarized in Table 2. The most common site of involvement in the NF group was the orbital nerve, noted in 66% of the patients, as compared with only 32% in the non-NF group (P < .005). Among the non-NF children, the chiasm was involved in 91%, as compared with 62% in the NF group (P < .005). Extension beyond the optic tract at diagnosis was uncommon in the NF group (1 patient, 2%) but frequent in the non-NF group (30 patients, 68%). Involvement of a single site was rare: involvement of only the orbital nerve was noted in three patients (6%) in the NF group, and involvement of only the chiasm was observed in three patients (7%) in the non-NF group. Bilateral optic nerve involvement was present in 11 patients (24%) with NF and in three patients (7%) without NF. The average number of sites involved was 3.0 ± 1.4 in the NF group and 3.7 ± 1.6 in the non-NF group; this difference was of borderline significance (P = .054). In the non-NF group, there was a significant correlation between the presence of a chiasmatic-hypothalamic mass and the occurrence of nonvisual symptoms (P < .0016). No correlation was found between the presence of orbital involvement and visual symptoms. In the NF group, no correlation was found between the location of the tumor and the type of symptoms.
TABLE 2:
Involvement of optic tract by site
The presence of cystic components in the tumor was significantly more common in the non-NF patients (66% vs. 9% in the NF group, P < .0001); in nine patients (20%), they accounted for more than 50% of the tumoral mass.
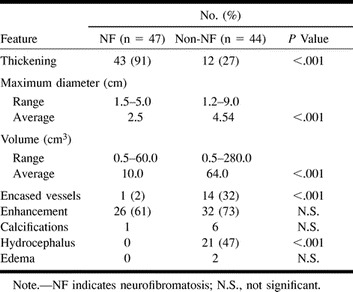
In most of the NF patients (43/47, 91%) the tumor did not affect the original shape of the optic pathway and appeared on images as a thickening of the optic nerve and/or chiasm (Fig 1). In the non-NF group, this configuration was present in only 12 patients (27%) (P < .0001). Both the maximal diameter and the volume of the tumor were significantly greater in the non-NF group. Hydrocephalus was found in none of the children with NF but in 47% of those without NF. There was no correlation between the size of the mass and the presence of hydrocephalus. Note that although the blood vessels may have been encased by tumor, the anterior or middle cerebral artery frequently caused only an indentation in the mass, without being completely entrapped (13% in the NF patients, 34% in the non-NF patients) (Fig 2). Additional imaging findings are summarized in Table 3.
fig 1.

14-year-old boy with NF. A and B, Sagittal unenhanced (A) and coronal contrast-enhanced (B) T1-weighted MR images show typical findings of OPG in patients with NF: thickened retroorbital optic nerves and chiasm, with preservation of original contour. The tumor is small and homogeneous, without any cystic components
fig 2.
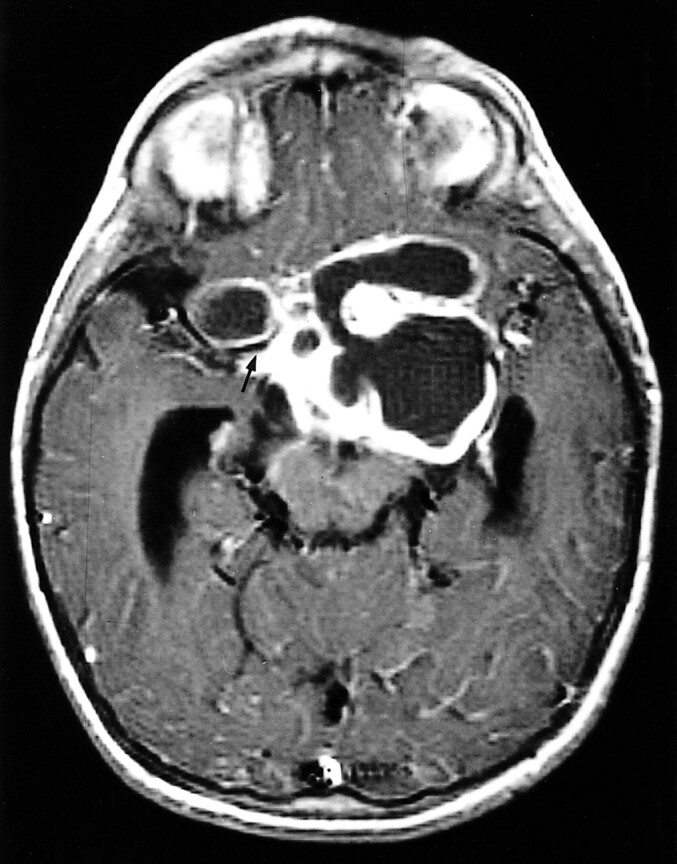
4-year-old girl without NF. Axial contrast-enhanced T1-weighted image shows typical findings of optic glioma in patients with NF: a large mass, with cystic components, that does not respect the boundaries of the optic pathways. The temporal horns are enlarged, and the right middle cerebral artery courses in a fissure of the mass (arrow)
TABLE 3:
Imaging features of OPG
Follow-up data were available for 81 patients, 90% in the NF group (maximum follow-up period, 10 years; average follow-up period, 4 years 3 months) and 90% of the non-NF group (maximum follow-up period, 11 years; average follow-up period, 4 years 2 months). Findings obtained during the follow-up period are shown in Table 4. About half (21/41) the NF patients remained stable, six (15%) had tumor enlargement, six had minimal enlargement, and three (7%) had a spontaneous reduction in tumoral mass. In the non-NF group, only two (5%) of 40 patients remained stable; 15 had tumor enlargement (P < .001 compared with the NF group) and none had a reduction in tumor size.
TABLE 4:
Size and extension of optic pathway glioma at follow-up
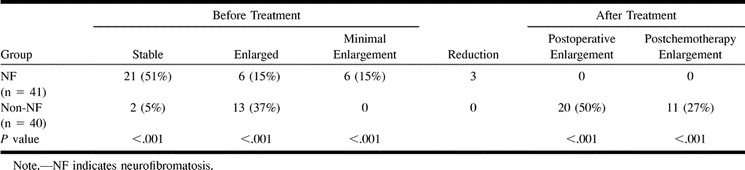
In the NF group, eight patients were treated, six because of visual deterioration and two because of failure to thrive. All had chemotherapy (one with additional radiation); two also underwent biopsy and two also had surgery. Chemotherapy included vincristine with actinomycin D, which was later changed to vincristine and VP-16 and thereafter to vincristine and carboplatin. After treatment, none of the patients had tumor enlargement. Since the presence of a lesion is an indication for treatment in non–NF-OPG, in this group, all 40 patients (with follow-up data) were treated: 32 (80%) with surgery, 25 (62%) with chemotherapy, 11 (27%) with radiation; most (55%) received multiple forms of treatment, and eight (20%) underwent multiple operations. The chemotherapy regimen was the same in the NF group, although a few unresponsive patients received other combinations. The details of the treatment regimen are beyond the scope of this study. Twenty patients (50%) had tumor enlargement after surgery and 11 (27%) after chemotherapy (Table 4). Leptomeningeal spread was noted in three.
Kappa values for the revised parameters at presentation were 0.84 to 1.0, except for involvement of the distal optic tracts, which yielded values of 0.61 to 0.63 for both intraobserver and interobserver variability. However, the P value remained significant, indicating the validity of the observation.
Repeated measurements of the maximal tumor diameter and calculated volume were not statistically significantly different from the initial evaluation (P < .05). Kappa values for the latest examination were 0.75 to 1.0.
In the NF group, we tried to define prognostic neuroimaging factors by comparing the multiple clinical and imaging features with the biological behavior of the mass on follow-up studies. No statistically significant correlations were found. In the non-NF group, the small number of patients with stable tumors precluded a statistical analysis.
Histologic examination was available in 41 non-NF patients. One tumor was an anaplastic astrocytoma, the other 40 were low-grade astrocytomas, 21 of pilocytic type.
Discussion
Most studies to date have failed to distinguish between OPG in patients with and those without NF (2, 4, 6–8), and the few studies that have methodically compared these two groups relied mostly on CT findings (9–11). Therefore, it remains unclear whether there are specific morphologic or anatomic patterns that can help predict the prognosis of OPG, and whether NF-OPG is a distinct entity from non–NF-OPG.
The present study yielded several morphologic features that distinguish NF-OPG from non–NF-OPG. In the patients with NF, the most common site of involvement was the orbital nerve. The tumor was smaller than in the non-NF patients, the original shape of the optic pathway was preserved, and cystic components were uncommon. In the non-NF group, the chiasm and hypothalamus were the most common sites of involvement, the tumor was masslike, and cystic components were frequently seen, as was extension beyond the optic pathways. The prognosis was also significantly different: half the NF patients remained stable compared with only 5% of the non-NF patients. However, no correlation was found between imaging features and prognosis.
Studies in the literature seem to indicate that OPG patients with and those without NF differ significantly by tumor location (9–11), with involvement of the optic nerve being much more common in the NF group (1, 10, 12). Hoffman et al (13) found that virtually all their patients with orbital tumors had NF, and Listernick et al (10) reported that isolated involvement of the optic nerve occurred only in the patients with NF. However, the optic nerves may be involved also in non-NF patients (11, 14), and not all patients with NF have thickened optic nerves (9). In our sample, the optic nerve was involved in only 66% of the patients with NF. Fewer data are available for non-NF patients. In some studies, extension into the optic nerves was observed in 3%, 6%, and 53% of patients without NF (9, 11, and 14, respectively); and in our study, an extension was seen in 32% of patients without NF (P < .005 relative to those with NF). The chiasm is involved more often in non–NF-OPG. Deliganis et al (11) reported a 75% rate (vs. 60% in the NF group; N.S.), and we found a 91% rate in the present study (vs. 62% in the NF group; P < .005). Involvement of the chiasm alone is also more likely in the absence of NF (9–11). Indeed, in the various series of OPG with NF, involvement of the chiasm alone was rare (9–12, 15, 16). Extension into the hypothalamus is reported to be more common in non–NF-OPG (9, 11), and this was also true in our sample (70% versus 23%; P < .0001). We found that patients with NF apparently have less diffuse involvement of the optic pathways (ie, fewer sites along the optic apparatus), although the difference from the non-NF group was not significant. However, isolated optic nerve or chiasmatic involvement was rare in both the NF (n = 3) and non-NF (n = 1) groups. Therefore, on the basis of MR findings, we suggest that localized gliomas may be less common than previously thought. Several studies refer only to involvement of the optic tracts, with some reporting a higher prevalence in patients with NF (9, 10, 17, 18), and others describing a higher occurrence in those without NF (11). We did not find any significant difference between the NF and the non-NF groups. Assessment of involvement of the optic tracts by tumor is difficult, because the increased signal intensity on T2-weighted MR images can be caused by edema as well as by direct tumor extension (2). Indeed, evaluation of this segment of the optic pathway in our study yielded the worst kappa values (0.61 to 0.63; N.S.).
A noteworthy finding in the present study was the different shape and size of the tumor in the two groups. In the patients with NF, the involved chiasm was thickened although its contour was preserved (91%), whereas in the non-NF group, the tumor was masslike (73%) (P < .0001) and larger (P < .0001), with a higher rate of cystic components (P < .0001) and extension beyond the optic pathways (P < .001). As bulky tumors are referred for surgery, these data are important to the neurosurgeon (13, 17). Most of the earlier studies did not correlate the size or shape of the tumor to the presence of NF. Sutton (17) mentioned that globular chiasmatic-hypothalamic gliomas are less likely to appear in patients with NF, and Wisoff (8) noted that diffuse enlargement of the chiasm with variable extension into the optic nerves and chiasm is more likely in patients with NF.
The symptomatology of the two groups also differed significantly. All the non-NF patients were referred for examination because of symptoms, whereas only 32% of the NF patients were symptomatic at presentation (P < .0001). The smaller dimensions and the relative sparing of the hypothalamus in the NF patients may account for this finding. The lag between the onset of symptoms and the imaging study was much longer in the non-NF group (up to 4 years), whereas NF patients were examined immediately (P < .0005). The lower rate of occurrence of symptoms combined with the shorter interval between symptom onset and diagnosis in the NF group may reflect a bias in referral patterns, with a lower threshold of suspicion in NF patients.
In the NF group, 20% presented with visual symptoms and 15% with nonvisual symptoms. No significant correlation between site of tumor and type of symptoms was found. In the non-NF group, 84% had visual symptoms and 61% had other symptoms (Table 1). A significant correlation was found between hypothalamic involvement and the occurrence of endocrinologic symptoms (such as failure to thrive and precocious puberty) and signs of increased intracranial pressure. Interestingly, orbital involvement did not correspond to visual symptoms, possibly because infants and even young children tend not to complain of visual disturbances.
Hydrocephalus as a presenting symptom was found exclusively in the non-NF group (P < .0001), as reported also by Listernick et al (10). In the latter study, precocious puberty was associated only with NF, but in our study and in others (16–18), it appeared as the presenting sign in both groups.
One of the most striking features differentiating NF-OPG from non–NF-OPG is the long-term behavior of the tumor. In our study, 15% of the NF group showed minimal tumor enlargement, 15% showed enlargement, and 51% remained stable; in the non-NF group, there was a clear propensity for growth in 95% of the patients (P < .001). Although earlier studies (4, 6, and Ellsworth cited in [9], as well as the 1994 review by Sutton [17]) reported that the presence of NF has no influence on prognosis of OPG, nowadays it is accepted that NF acts as a protective factor (1, 10, 11, 13, 19–21). OPGs have been found to enlarge in 9%, 12%, 13%, and 22% of patients with NF (10, 16, 19, and 20, respectively), and in 54% if isolated intraorbital lesions are excluded (9), as compared with 63%, 68%, and 100% of patients without NF (9, 10, and 20, respectively). Others have observed that the presence of NF also improves the mean time to tumor progression (11, 21). It is of interest that spontaneous regression of the tumor was observed in three of our patients with NF. This has been described previously in NF patients and also in non-NF patients (22, 23). At the same time, despite the low-grade histologic findings in three of our patients, there was leptomeningeal spread (1).
Another favorable prognostic factor mentioned in the literature is older age. In our study, 43% of the patients were 5 years old or younger, but we did not find any association with worse prognosis or bulkier masses. Listernick (10) noted that symptomatic OPGs are seen primarily in very young children, regardless of the presence of NF. Others have stated that in younger children the tumors are larger (8), behave more aggressively (1, 8), and have a poorer prognosis (1, 8, 14); however, they disagree as to the specific age, with some specifying under 2 years (8, 14) and others under 5 years (1).
The location of the tumor has also been reported to be related to long-term outcome. In our study, as single-site involvement was rare, no such correlation could be found. In an extensive review (based on CT and pre-CT imaging), Dutton (4) concluded that mortality is significantly lower when there is orbital rather than hypothalamic involvement. Others found better prognosis for orbital involvement than for chiasmatic-hypothalamic involvement (8).
When CT became available 30 years ago, the routine management of NF patients included serial neuroimaging examinations (6, 10). The advent of MR imaging raised questions regarding its presymptomatic use in children with NF. According to the 1997 recommendations of Listernick et al (24), based on controlled longitudinal data, MR imaging is indicated only in children with optic nerve dysfunction at ophthalmologic evaluation. This was supported by the guidelines of the National Neurofibromatosis Foundation (25). Furthermore, in children with known or symptomatic OPG, if the tumor has been stable, the interval between MR studies can be gradually increased (20).
The NF1 gene has been located at chromosome 17, and its protein product, neurofibromin, characterized (25). The association of both benign pilocytic and diffuse astrocytomas with NF points to a role for the NF1 gene in the pathogenesis of gliomas in these patients. Neurofibromin is believed to act as a tumor suppressor. As for sporadic astrocytic brain tumors, recent studies have provided initial evidence that the NF1 gene is not primarily involved in astrocytomas in non-NF patients (26).
Conclusion
We found no correlation between the imaging parameters analyzed and the biological behavior of OPG associated with NF. This observation may indicate that neuroimaging is not contributory to the prediction of outcome in this group of patients. OPG in patients without NF differs significantly from NF-OPG in both imaging features and prognosis. Non–NF-OPG and NF-OPG are apparently distinct entities, each warranting a specific diagnostic, clinical, and therapeutic approach.
Acknowledgments
We thank Gloria Ginzach and Marian Propp for their editorial and secretarial assistance.
Footnotes
Address reprint requests to Liora Kornreich, MD, Imaging Department, Schneider Children's Medical Center of Israel, Petah Tiqva 49202, Israel.
References
- 1.Alshail E, Rutka JE, Becker LE, Hoffman JH. Optic chiasmatic- hypothalamic glioma. Brain Pathol 1997;7:799-806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barkovich JA. Pediatric Neuroimaging. Philadelphia: Lippincott Williams & Wilkins; 1997:507–509
- 3.Luh GH, Bird CR. Imaging of brain tumors in the pediatric population. Neuroimaging Clin 1999;9:691-716 [PubMed] [Google Scholar]
- 4.Dutton JJ. Gliomas of the anterior visual pathway. Surv Ophthalmol 1994;38:427-452 [DOI] [PubMed] [Google Scholar]
- 5.Dixon WJ. BMDP Statistical Software. Berkeley: University of California Press; 1992
- 6.McCullough DC, Epstein F. Optic pathway tumors: a review with proposal for clinical staging. Cancer 1985;56:1789-1791 [DOI] [PubMed] [Google Scholar]
- 7.Wong JYC, Uhl V, Wara WM, Sheline GE. Optic gliomas: a reanalysis of the University of California, San Francisco experience. Cancer 1987;60:1847-1855 [DOI] [PubMed] [Google Scholar]
- 8.Wisoff JH. Management of optic pathway tumors of childhood. Neurosurg Clin North Am 1992;3:791-802 [PubMed] [Google Scholar]
- 9.Packer RJ, Bilaniuk LT, Cohen BH, et al. Intracranial visual pathway gliomas in children with neurofibromatosis. Neurofibromatosis 1988;1:212-222 [PubMed] [Google Scholar]
- 10.Listernick R, Darling C, Greenwald M, Strauss L, Charrow J. Optic pathway tumors in children: the effect of neurofibromatosis type I on clinical manifestations and natural history. J Pediatr 1995;127:718-722 [DOI] [PubMed] [Google Scholar]
- 11.Deliganis AV, Geyer JR, Berger MS. Prognostic significance of type 1 neurofibromatosis (von Recklinghausen disease) in childhood optic glioma. Neurosurgery 1996;38:1114-1119 [DOI] [PubMed] [Google Scholar]
- 12.Brown EW, Riccardi VM, Mawad M, Handel S, Goldman A, Bryan RN. MR imaging of optic pathways in patients with neurofibromatosis. AJNR Am J Neuroradiol 1987;8:1031-1036 [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman HJ, Humphreys RP, Drake JM, et al. Optic pathway/hypothalamic gliomas: a dilemma in management. Pediatr Neurosurg 1993;19:186-195 [DOI] [PubMed] [Google Scholar]
- 14.Nishio S, Takeshita I, Fuhiwara S, Fukui M. Optic-hypothalamic glioma: an analysis of 16 cases. Child Nerv Syst 1993;9:334-338 [DOI] [PubMed] [Google Scholar]
- 15.Lund AM, Skovby F. Optic gliomas in children with neurofibromatosis type 1. Eur J Pediatr 1991;150:835-838 [DOI] [PubMed] [Google Scholar]
- 16.Kuenzle C, Weissert M Roulet, et al. Follow-up of optic pathway gliomas in children with neurofibromatosis type 1. Neuropediatrics 1994;25:295-300 [DOI] [PubMed] [Google Scholar]
- 17.Sutton LN. Visual pathway gliomas of childhood. Contemp Neurosurg 1994;16:1-6 [Google Scholar]
- 18.Medlock MD, Madsen JR, Barnes PD, et al. Optic chiasm astrocytomas of childhood. Pediatr Neurosurg 1997;27:121-128 [DOI] [PubMed] [Google Scholar]
- 19.Janss AJ, Grundy R, Cnaan A, et al. Optic pathway and hypothalamic/chiasmatic gliomas in children younger than age 5 years with a 6-year follow-up. Cancer 1995;75:1051-1059 [DOI] [PubMed] [Google Scholar]
- 20.Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr 1994;125:63-66 [DOI] [PubMed] [Google Scholar]
- 21.Chan MY, Foong AP, Heisey DM, Harkness W, Hayward R, Michalsky A. Potential prognostic factors of relapse-free survival in childhood optic pathway glioma: a multivariate analysis. Pediatr Neurosurg 1998;29:23-28 [DOI] [PubMed] [Google Scholar]
- 22.Parazzini C, Triulzi F, Bianchini E, et al. Spontaneous involution of optic pathway lesions in neurofibromatosis type 1: serial contrast MR evaluation. AJNR Am J Neuroradiol 1995;16:1711-1718 [PMC free article] [PubMed] [Google Scholar]
- 23.Gallucci M, Catalucci A, Scheithauer BW, Forbes GS. Spontaneous involution of pilocytic astrocytoma in a patient without neurofibromatosis type 1: a case report. Radiology 2000;214:223-226 [DOI] [PubMed] [Google Scholar]
- 24.Listernick R, Louis DN, Packer RJ, Gutman DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol 1997;41:143-149 [DOI] [PubMed] [Google Scholar]
- 25.Gutman DJ, Aylsworth A, Carey JC, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51-57 [PubMed] [Google Scholar]
- 26.Peters N, Waha A, Wellenreuther R, et al. Quantitative analysis of NF1 and OMPG gene transcripts in sporadic gliomas, sporadic meningiomas and neurofibromatosis type 1-associated plexiform neurofibromas. Acta Neuropathol 1999;97:547-551 [DOI] [PubMed] [Google Scholar]