

Abstract
Summary: We present the MR imaging findings in four patients (two pairs of siblings from two unrelated families) with adult Krabbe disease. In the first family, clinical presentation mimicked familial spastic paraplegia. Their MR images showed selective, increased signal intensity on T2-weighted sequences along the corticospinal tracts, most prominently in the proband and barely detectable in her brother. Proton MR spectroscopy showed increased choline and myo-inositol in the affected white matter. In the second family, the clinical presentation differed in that the signs of pyramidal tract involvement were asymmetrical, with concomitant asymmetry on MR images in one. In adults, Krabbe disease may present on MR imaging with selective pyramidal fiber involvement.
Krabbe disease, or globoid cell leukodystrophy, is a demyelinating disorder caused by a genetic deficiency of lysosomal enzyme galactocerebrosidase (GALC), a key component in metabolic pathways of myelin turnover and breakdown. The GALC gene maps to chromosome 14q24.3 to 14q32.1, and a variety of Krabbe disease–causing mutations have been identified (1). GALC deficiency results in the accumulation of galactosylsphingosine, which is considered to be neurotoxic to both the central and peripheral nervous system. The most frequent form of Krabbe disease has an infantile onset, whereas the late-onset form is rare. Here we describe two pairs of siblings with adult Krabbe disease studied at different institutions, one from Italy (cases 1 and 2), the other from India (cases 3 and 4). The siblings of the first family (cases 1 and 2) had spastic paraparesis, increased signal intensity along the corticospinal tracts on MR images, and slightly increased levels of choline and myo-inositol in the affected white matter at proton MR spectroscopy. The two siblings of the second family (cases 3 and 4) had similar but asymmetrical clinical and MR imaging findings. The MR imaging pattern is characteristic and, in the appropriate clinical setting, may suggest the diagnosis.
Case Reports
Case 1
A 33-year-old woman with talipes cavus since childhood and slowly progressive paraparesis was found to have hereditary spastic paraplegia because her brother had a similar, although milder, disorder. On neurologic examination she had talipes cavus with hammer toes, spastic paraparesis, increased deep tendon reflexes in all limbs, Babinski sign, mild wasting, and decreased vibration sense distally in the lower limbs. Nerve conduction studies showed a mild demyelinating sensory neuropathy.
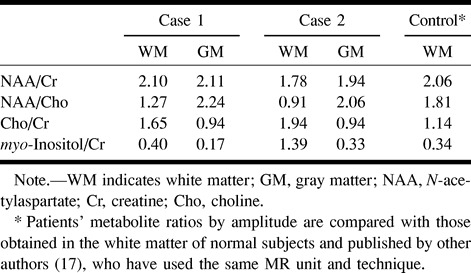
Brain MR imaging revealed symmetrical hyperintensity on proton density–and T2-weighted images along the corticospinal tracts, extending from the subcortical white matter of the upper rolandic area down to the cerebral peduncles. The abnormal signal intensity extended across the corpus callosum at the isthmus, which showed focal thinning (Fig 1). The abnormal signal intensity was well seen on proton density–weighted images, T2-weighted conventional spin-echo and turbo spin-echo images, and fluid-attenuated inversion recovery (FLAIR) images; it was also detectable as hypointensity on T1-weighted images. The precentral gyri were atrophic and were as small as the postcentral gyri (Fig 1). Additional signs included a slight hyperintensity of the posterior periventricular white matter on proton density–and T2-weighted images, cerebral atrophy, and mild asymmetry of the lateral ventricles. Single-section proton MR spectroscopy was performed at the level of the centrum semiovale with a point-resolved spectroscopy (PRESS) technique (TR/TE = 1500/136), 2D phase-encoding (field of view = 160 × 160; matrix = 16 × 16), and a 20-mm section thickness. Time of acquisition was 8 minutes. The peak amplitude was measured for each metabolite and the ratios calculated. Proton MR spectra showed mild bilateral elevation of choline in the anterior and middle third of the centrum semiovale and in the right precentral gyrus. A very mild elevation of an abnormal peak at 3.6 ppm (tentatively attributed to myo-inositol or glycine) was present in a few spectra from the centrum semiovale. In this patient, N-acetylaspartate (NAA) and creatine peaks were normal both in gray and white matter. Metabolite ratios by amplitude, measured from the voxels in the middle third of the centrum semiovale, are reported in the Table.
fig 1.
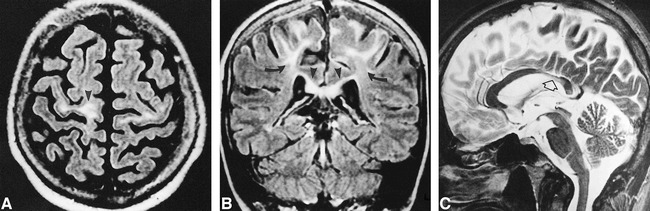
Case 1.
A, Axial FLAIR image (9264/150/2; TI = 2200) shows increased signal intensity of the white matter of the precentral gyri, which are atrophic (arrowhead).
B, Coronal FLAIR image (9264/150/2; TI = 2200) shows the hyperintensity limited to the upper part of the motor cortex (arrows) and extending across the corpus callosum (arrowheads).
C, Midline sagittal turbo spin-echo T2-weighted image (5422/110/2) shows thin, slightly hyperintense isthmus of the corpus callosum (arrow).
Metabolites ratios from proton MR spectroscopy
On biochemical investigation, GALC activity was absent in leukocytes. Using the polymerase chain reaction assay described by Luzi et al (2) the deletion of exons 11 through 17 was found in one of the two GALC alleles. In the other allele, a C to T point mutation was found, affecting nucleotide 1850 and causing a methionine for threonine substitution at codon 617.
Case 2
The 35-year-old brother of patient 1 also had talipes cavus since childhood and abnormal gait that had become evident during the last few years; his spastic paraparesis was milder. He also had a mild demyelinating sensory neuropathy.
He presented with very mild MR imaging abnormalities. A slight hyperintensity on T2-weighted images along the corticospinal tracts was recognizable from the internal capsules to the cerebral peduncles. The corpus callosum showed focal thinning and minimally abnormal signal intensity at the isthmus. The precentral gyri were slightly atrophic. Proton MR spectroscopy in the centrum semiovale showed a moderate bilateral elevation of choline, that was more elevated in the middle third. The abnormal elevation of choline was more prominent than in the other sibling. A mild elevation of an abnormal peak at 3.6 ppm was also present in a few spectra. NAA was mildly reduced in the white matter and in the parasagittal gray matter (Fig 2). Metabolite ratios by amplitude are reported in the Table.
fig 2.
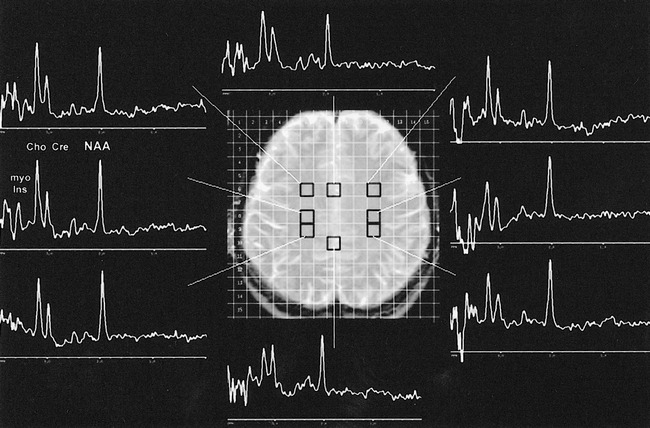
Case 2. Selected proton MR spectra (PRESS, 1500/136/1; nominal voxel size, 2 cm3) from the voxels indicated on the scout spin-echo T2-weighted MR image (2000/90/1; thickness, 10 mm) show moderate choline elevation (3.2 ppm), mild NAA loss (2.02 ppm), normal creatine peak (3.0 ppm), and an abnormally elevated peak at 3.6 ppm (myo-inositol) in the centrum semiovale. Spectra from the parasagittal cortical gray matter show only mild NAA loss. Similar proton MR spectroscopic findings in the centrum semiovale were found in case 1
GALC activity in leukocytes was very low (0.07 nmol/mg per hour; control values, 3.7 ± 1.0). Molecular analysis revealed the same GALC mutation seen in his sister.
Case 3
A 29-year-old woman had insidious onset of disease at age 23, presenting as weakness of the left foot. Gradually, progressive weakness and spasticity developed, with signs of pyramidal tract involvement, primarily on the left side, also extending to the upper extremities. Motor-evoked response was abnormal on the right side.
MR imaging showed marked hyperintensity on proton density–and T2-weighted images along the corticospinal tract from the precentral gyrus down to the medulla oblongata on the right, and from the internal capsule to the pons on the left. A band of abnormal signal crossing the corpus callosum at the isthmus was also present. Involvement of the pyramidal tract on the right side was not limited to the upper part of the motor strip, but extended laterally, only sparing the opercular area. The right precentral gyrus was atrophic. The ventricles were enlarged, particularly in their posterior aspect. There was no progression of signal changes from 1993 to 1999.
GALC activity was absent. The common 30 kb deletion of exons 11 through 17 was present on one allele, as found in the first two cases. The other allele had an A to G transition at nucleotide (counting from the A of the initiation codon of the complementary DNA) 908, changing tyrosine 303 to cysteine.
Case 4
The 27-year-old brother of patient 3 first noted weakness of the right leg at age 23, with insidious progression of spasticity, more pronounced on the right side than the left. Motor-evoked response was abnormal on the left side.
MR imaging (1996 and 1999) showed signal abnormalities along both corticospinal tracts from the cortex to the pons, slightly prevalent on the left side, and thinning of the isthmus of the corpus callosum, which also appeared hyperintense on T2-weighted images (Fig 3). Both precentral gyri were atrophic. The ventricles were markedly enlarged, particularly in their posterior aspect, with a thin band of hyperintensity on proton density–and T2-weighted images along the temporal and occipital horns. The cerebral sulci were enlarged in the posterior regions.
fig 3.
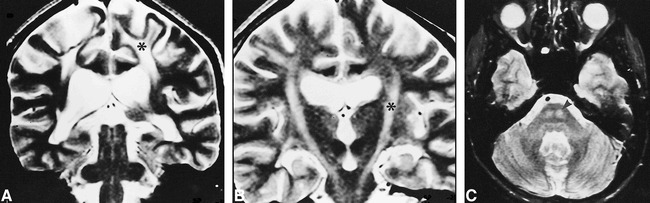
Case 4.
A and B, Coronal T2-weighted images (3648/99/2) of adjacent sections, A posterior to B, show nearly symmetrical involvement of the corticospinal tract from the cortex (A) to the brain stem (B) (asterisks).
C, Axial T2-weighted image (2100/90/2) at pontine level shows symmetrical pyramidal involvement (arrowhead).
GALC activity in leukocytes was severely reduced (0.06 nmol/mg per hour), and molecular analysis revealed the same GALC mutation as in case 3.
Discussion
The most frequent and common form of Krabbe disease is the infantile form, which begins in the first 6 months of life and progresses rapidly, leading to death before age 2. These children with Krabbe disease have rapid psychomotor regression, generalized rigidity, peripheral neuropathy, and increased spinal fluid proteins; they subsequently develop optic atrophy, deafness, and cachexia. MR imaging shows progressive atrophy and diffuse abnormalities of the white matter, which may be difficult to recognize at a very early age, when the white matter is still normally hyperintense on T2-weighted images (3). CT studies may be more helpful, because they may show hyperdense areas in the thalami or in the posterior periventricular regions that likely correspond to the clusters of globoid cells in which the galactosylceramide accumulates; calcium deposits may contribute to the hyperdensity (4). A recent review of MR imaging studies in 22 patients with Krabbe disease showed that in early-onset cases there is a frequent involvement of deep gray matter and cerebellar white matter, and nearly constant involvement of the pyramidal tract (5).
The late-onset forms of Krabbe disease, which also include some cases of adult-onset disease, are clinically more heterogeneous and progress more slowly; various clinical signs have been described, including hemiparesis, spastic paraparesis, intellectual impairment, cerebellar ataxia, visual failure, peripheral polyneuropathy, and talipes cavus. On MR images, these patients have constant corticospinal tract abnormalities (5). They also have hyperintense signal on proton density–and T2-weighted images in the white matter, mostly in the posterior regions, with frequent involvement of the splenium of the corpus callosum (3, 5–8). This pattern is similar to that observed in adrenoleukodystrophy (ALD). Loes et al (5) concluded that pyramidal tract involvement is the most characteristic finding in both early- and late-onset Krabbe disease.
To our knowledge, only three reports have described selective involvement of corticospinal tracts associated with focal atrophy of the corpus callosum in adult patients with Krabbe disease, presenting as progressive spastic paraparesis (9–11). The consistency of the clinical and radiologic picture, confirmed by the findings in our patients, implies that the measurement of GALC activity in leukocytes is diagnostically indicated under these circumstances. In adult Krabbe disease, the type of molecular defect has been considered for relevance to the clinical and radiologic presentation (11). However, in cases 1 and 2, mutations were in close vicinity to those reported by Satoh et al (11), whereas in cases 3 and 4 they were located in a different part of the GALC gene.
Satoh et al (11) proposed that the selective vulnerability of the corticospinal tracts in patients with late-onset Krabbe disease may depend on their myelin metabolism. These authors suggested that these tracts may be particularly active as compared with the white matter of other parts of the brain, where residual GALC activity may be sufficient to maintain a normal or nearly normal status of the myelin.
A finding observed both in our cases and in those reported previously (9–11) is that the site of the lesions is in the upper part of the motor strip, which explains the involvement of the lower extremities. The lower part of the motor strip, corresponding to the areas of representation of the hand and face, was affected to a lesser extent (cases 3 and 4) or not at all (cases 1 and 2). A peculiar MR finding in case 3 was the asymmetry of the corticospinal tract involvement, which correlated with the asymmetry of the neurologic signs. The absence of progression of MR signal changes over a 6-year interval observed in one of our cases may be explained by the extremely slow progression of the disease.
From the clinical and neuroradiologic points of view, a few other aspects are worth mentioning. In patients with familial spastic paraplegia, the clinical diagnosis of Strümpell-Lorrain disease should be considered. Pathologic changes in the corticospinal tracts below the pons or the medulla oblongata are found in this disease, and a review of 16 cases studied with MR imaging revealed no signal abnormalities in the brain (12).
Another disease that can present with spastic paraparesis is adrenomyeloneuropathy (AMN), which can be diagnosed by levels of very long chain fatty acids in plasma and skin fibroblasts. In AMN, MR images of the spinal cord show atrophy. MR images of the brain may be entirely normal or show mild cerebellar atrophy or very limited signal changes, similar to those of ALD, but confined to focal areas, such as the splenium of the corpus callosum (3). Among the leukodystrophies, ALD typically involves the corticospinal tracts, usually below the internal capsule, but often only in the brain stem, from the cerebral peduncles to the pyramids (13). This segmental involvement of corticospinal tracts may be the first MR imaging manifestation of CNS abnormality in asymptomatic patients with ALD.
The involvement of the corticospinal tracts in an adult patient may suggest the possibility of amyotrophic lateral sclerosis (ALS). However, aside from the different clinical presentation, the MR signal changes in patients with ALS are very faint (14), similar to those observed in our case 2, and do not usually reach the intensity seen in the other patients we observed or in those reported previously (9–11).
To our knowledge, proton MR spectroscopic studies of adult patients with Krabbe disease have not been reported in the literature. In our patients, proton MR spectroscopy showed abnormal choline elevation in the centrum semiovale beyond the areas with abnormal signal on T2-weighted images. Frahm and Hanefeld (15) studied two cases of the infantile form of Krabbe disease and found elevated choline, creatine, and myo-inositol associated with moderate NAA reduction and mild lactate accumulation. Although elevation of choline-containing compounds and myo-inositol may reflect accumulation of myelin breakdown products, they are more likely accounted for by changes in glial membrane composition and microglia activation for a synthesis of phospholipid membranes (15). Postmortem findings in patients with Krabbe disease have shown a remarkable increase of activated microglial cells with large macrophages, containing myelin breakdown products. These MR spectroscopic findings are not specific for Krabbe disease. Abnormal elevation of choline and myo-inositol has also been found in active demyelinating lesions in ALD, metachromatic leukodystrophy, Alexander disease (15), and multiple sclerosis (16). Elevated choline has been shown to return to normal within a couple of months in a few multiple sclerosis lesions (13). Myo-inositol found early in acute multiple sclerosis lesions is probably permanent and no return to normal was documented during a follow-up of 9 months (16). Elevation of myo-inositol is best detected at short TEs (<50 ms), thus the elevated myo-inositol in these two cases may have been underestimated by the intermediate TE (136 ms) used in our studies. NAA, a molecule found only in neurons and their axons, was mildly decreased.
Conclusion
In adult patients with slowly progressive spastic paraparesis, the differential diagnosis should include late-onset Krabbe disease, especially when brain MR images reveal distinctly increased signal intensity along the corticospinal tracts.
Acknowledgments
We thank Professor Giancarlo Dal Pozzo, Florence, for referring patient 1.
Footnotes
Supported in part by NIH grant DK 38795 (D.A.W.).
Address reprint requests to Laura Farina, MD, Department of Neuroradiology, Istituto Nazionale Neurologico “C. Besta,” Via Celoria 11, 20133 Milan, Italy.
References
- 1.Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat 1997;10:268-279 [DOI] [PubMed] [Google Scholar]
- 2.Luzi P, Rafi MA, Wenger DA. Characterization of the large deletion in the GALC gene found in patients with Krabbe disease. Hum Mol Genet 1995;4:2335-2338 [DOI] [PubMed] [Google Scholar]
- 3.Van der Knaap MS, Valk J. Magnetic Resonance of Melin, Myelination, and Myelin Disorders.. 2nd ed. Berlin: Springer 1995:22-30 68–75-129–139 [Google Scholar]
- 4.Percy AK, Odrezin GT, Knowles PD, Rouah E, Armstrong DD. Globoid cell leukodystrophy: comparison of neuropathology with magnetic resonance imaging. Acta Neuropathol 1994;88:26-32 [DOI] [PubMed] [Google Scholar]
- 5.Loes DJ, Peters C, Krivit W. Globoid cell leukodystrophy: distinguishing early-onset from late-onset disease using a brain MR imaging scoring method. AJNR Am J Neuroradiol 1999;20:316-323 [PMC free article] [PubMed] [Google Scholar]
- 6.Demaerel P, Wilms G, Vendru P, Carton H, Baert AL. MR findings in globoid cell leukodystrophy. Neuroradiology 1990;32:520-522 [DOI] [PubMed] [Google Scholar]
- 7.Barone R, Brühl K, Stoeter P, Fiumara A, Pavone L, Beck M. Clinical and neuroradiological findings in classic infantile and late-onset globoid-cell leukodystrophy (Krabbe disease). Am J Med Genet 1996;63:209-217 [DOI] [PubMed] [Google Scholar]
- 8.Bataillard M, Richard P, Rumbach L, Vanier MT, Truttmann M. Paraparésie spastique isolée révélant une maladie de Krabbe à l'âge adulte. Rev Neurol 1997;153:347-350 [PubMed] [Google Scholar]
- 9.Inatomi Y, Tomoda H, Itoh Y, Fujii N, Kobayashi T, Ohnishi A. An adult patient with Krabbe's disease: the first case reported in Japan. Clin Neurol 1993;33:1188-1194 [PubMed] [Google Scholar]
- 10.Turazzini M, Beltramello A, Bassi R, Del Colle R, Silvestri M. Adult onset Krabbe's leukodystrophy: a report of 2 cases. Acta Neurol Scand 1997;96:413-415 [DOI] [PubMed] [Google Scholar]
- 11.Satoh J-I, Tokumoto H, Kurohara K, et al. Adult-onset Krabbe disease with homozygous T1853C mutation in the galactocerebrosidase gene: unusual MRI findings of corticospinal tract demyelination. Neurology 1997;49:1392-1399 [DOI] [PubMed] [Google Scholar]
- 12.Krabbe K, Nielsen JE, Fallentin E, Fenger K, Herning M. MRI of autosomal dominant pure spastic paraplegia. Neuroradiology 1997;39:724-727 [DOI] [PubMed] [Google Scholar]
- 13.Barkovich AJ, Ferriero DM, Bass N, Boyer R. Involvement of the pontomedullary corticospinal tracts: a useful finding in the diagnosis of x-linked adrenoleukodystrophy. AJNR Am J Neuroradiol 1997;18:95-100 [PMC free article] [PubMed] [Google Scholar]
- 14.Waragai M. MRI and clinical features in amyotrophic lateral sclerosis. Neuroradiology 1997;39:847-851 [DOI] [PubMed] [Google Scholar]
- 15.Frahm J, Hanefeld F. Localized proton magnetic resonance spectroscopy of brain disorders in childhood. In: Bachelard XY, ed. Magnetic Resonance Spectroscopy and Imaging in Neurochemistry: Advances in Neurochemistry. New York: Plenum 1997;8:329-402 [Google Scholar]
- 16.Davie CA, Hawkins CP, Barker GJ, et al. Serial proton MR spectroscopy in acute multiple sclerosis lesions. Brain 1994;117:49-58 [DOI] [PubMed] [Google Scholar]
- 17.Tourbah A, Stievenart JL, Iba-Zizen MT, et al. Localized proton magnetic resonance spectroscopy in patients with adult adrenoleukodystrophy. Arch Neurol 1997;54:586-592 [DOI] [PubMed] [Google Scholar]