

Abstract
BACKGROUND AND PURPOSE: Subacute necrotizing encephalomyelopathy, or Leigh syndrome (LS), is a progressive neurodegenerative disorder characterized by symmetrical spongiform lesions in the brain with onset usually in infancy or early childhood. Little is known of the developing process of the brain lesions in LS that are particularly relevant to the occurrence of fatal respiratory failure. Our purpose was to determine whether fatal respiratory failure can be predicted before death on the basis of clinical characteristics or findings on longitudinal MR images of the brain.
METHODS: Clinical records and serial MR studies of eight patients with LS aged 3 months to 12 years who met the diagnostic criteria for LS were reviewed retrospectively, with special reference to a correlation between loss of respiratory control and MR abnormalities. Both T1- and T2-weighted images were obtained at the onset of disease or when clinical symptoms worsened.
RESULTS: Serial MR images were divided into three groups on the basis of the following findings: 1) symmetrical basal ganglia lesions before brain stem involvement (n = 4); 2) initial involvement of the brain stem (n = 2); and 3) cerebral white matter lesions followed by brain stem lesions (n = 2). Lesions of the lower brain stem were always present when patients had near fatal respiratory failure. However, upper brain stem lesions were transient and were found in parallel to reversible respiratory disorder. Fatal respiratory failure was unpredictable from clinical or neuroradiologic findings.
CONCLUSION: Brain stem lesions are associated with the loss of respiratory control in patients with LS, but the time at which fatal respiratory failure will occur is unpredictable.
Subacute necrotizing encephalomyelopathy, or Leigh syndrome (LS), is a progressive neurodegenerative disorder characterized by psychomotor retardation, feeding difficulties, intermittent abnormalities of the respiratory rhythm, cranial palsies, and ataxia, with onset usually in infancy or early childhood (1). Symmetrical spongiform lesions, a characteristic neuropathologic finding first described by Leigh in 1951 (1), are the result of a vacuolation of the neuropil and a relative preservation of the neurons with capillary proliferation. These lesions occur predominantly in the brain stem and the basal ganglia (1–4).
LS is the result of heterogeneous biochemical or molecular abnormalities in the mitochondria, although 40% to 65% of patients have no definite metabolic derangement (5, 6). Modern imaging techniques are able to provide strong diagnostic support for the diagnosis of LS owing to particular properties of the lesions in the brain and their distribution (7–11). However, little is known of the development of these brain lesions.
In analyzing MR images that corresponded to neuropathologic results, we postulated that clinical deterioration advances in a stepwise manner and that total tissue damage in the brain is cumulative. Thus, most neuropathologic findings in LS probably represent the relatively common findings seen at the end stage of the disease, resulting in the emphasis on fatal brain stem lesions.
In this study, we reviewed the process of developing brain lesions on serial MR images and their contribution to respiratory failure in patients with LS. In addition, we examined whether the fatal respiratory failure is predictable from either characteristic clinical features or MR findings.
Methods
Eight patients with LS, aged 7 months to 12 years, were included in the study. In all, 40 MR images were obtained on a superconducting 0.5-T system. Coronal and transaxial cranial imaging was performed using T2-weighted spin-echo sequences (2000/80/2 [TR/TE/excitations]) and conventional inversion recovery sequences (2000/700/2). MR studies were performed at the onset of disease or when clinical symptoms worsened. The interval between neuroimaging examinations ranged from 1 month to 3 years. All the clinical data and MR images were reviewed retrospectively, with special reference to a correlation between loss of respiratory control and MR abnormalities.
The patients fulfilled the following criteria (5): 1) progressive neurologic disease with motor and intellectual developmental delay; 2) signs and symptoms of brain stem and/or basal ganglia dysfunction; 3) elevated lactate levels in the blood and/or CSF; and 4) one or more of the following: a) characteristic features of LS on neuroimaging (ie, symmetrical hyperintense lesions in the basal ganglia and/or brain stem on T2-weighted MR images); b) typical neuropathologic changes at postmortem examination; or c) typical neuropathologic findings in a similarly affected sibling. Informed consent was obtained from the parents before the MR studies.
Results
Patients
A summary of the clinical findings in each patient with LS is presented in Table 1. Patients 2 and 3 were male siblings, possessing complete defects in cytochrome c oxidase (COX) activity in the skeletal muscles. Patients 5 and 8 had a partial enzyme defect of COX, histochemically. Patient 8 carried a mitochondrial DNA point mutation, nucleotide 3243A→G. The remaining four patients had neither known functional mutations at nt8993, 8344, 3243, or 3271 in mitochondrial DNA nor mitochondrial enzyme defects in the skeletal muscles or peripheral lymphocytes. Patients 6 and 8 died of sudden respiratory failure at 16 months and 12 years of age, respectively; both had postmortem examinations.
TABLE 1:
Clinical features and results of investigations in Leigh syndrome

An initial diagnosis of LS was made in patients 1, 2, 3, and 6 on the basis of typical clinical features or biochemical defects. In the other four patients, MR findings of multiple infarctions, progressive diffuse atrophy, and white matter involvement without putamenal lesions suggested initial diagnoses of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke (MELAS); Alpers disease; chronic progressive external ophthalmoplegia (CPEO); and myoclonus epilepsy associated with ragged-red fibers (MERRF). In these patients, sudden respiratory dysrhythmia, brain stem lesions on MR images, or autopsy results were key factors leading to a retrospective diagnosis of LS.
Distribution of Brain Lesions on MR Images
The most common characteristic finding on T2-weighted MR images was a bilateral, symmetrical, focal hyperintensity in the brain. The distribution of lesions on the final MR imaging study before death was as follows: basal ganglia (n = 8), upper brain stem (n = 8), lower brain stem (n = 7), cerebellar white matter (n = 5), cerebellar cortex (n = 2), cerebral white matter (n = 4), and spinal cord gray matter (n = 2). Patient 1 had a right parietal infarction (Fig 1) and patient 4 had multifocal infarctions in the subcortical white matter with marked progressive diffuse cerebral atrophy.
fig 1.

Patient 1: group BG lesion pattern. Axial T2-weighted image (2000/80/2) at the age of 7 months during an acute strokelike episode shows a hyperintense lesion in the right parietotemporal region (arrowheads)
Serial MR Imaging Findings
Abnormal MR findings before brain stem involvement were divided into the following three categories.
Group BG (Fig 2): The basal ganglia was involved before the appearance of brain stem lesions in patients 1, 2, 3, and 4. Initially, T1 and T2 prolongation appeared symmetrically in the putamina, globi pallidus, caudate nuclei, and thalami. After 4 to 78 months, the substantia nigra, periaqueductal gray matter, inferior colliculus, inferior olivary nuclei, and medullary reticular formation were also involved, as in group BS, described below. Although some abnormalities on MR images and the patients' respiratory failure were transient, irreversible lesions in the lower brain stem appeared later, accompanied by fatal respiratory failure. Patient 4 had progressive multiple infarctions in the subcortical white matter before the appearance of the basal ganglia lesion. Two patients had definite central white matter involvement at the advanced stage of the disease.
fig 2.
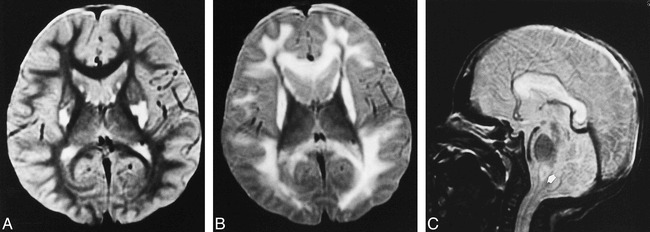
Patient 2: group BG lesion pattern.
A, Axial T2-weighted image (2000/80/2) at the age of 3 years shows increased signal intensity in the bilateral putamina and caudate heads, but no detectable brain stem lesion. Clinical findings included mental retardation, muscle weakness, and failure to thrive.
B and C, Axial (B) and sagittal (C) T2-weighted images (2000/80/2) at the age of 9 years show static and atrophic lesions in the bilateral putamina and progressive lesions in the cerebral white matter, corpus callosum, and medial medulla oblongata (arrow, C). Clinical findings included irregular breathing, lethargy, and inability to feed.
Group BS (Fig 3): Brain stem lesions appeared with or without basal ganglia and white matter lesions in patients 5 and 6. Patient 6 had initial lesions of the substantia nigra, periaqueductal gray matter within the midbrain, and thalami, which expanded to the periaqueductal gray matter and reticular formation in the medulla and cervical gray matter 4 months later. Patient 5 had progressive ventriculomegaly during the 42 months of his life, followed by a sudden appearance of symmetrical periaqueductal gray matter lesions throughout the brain stem.
fig 3.
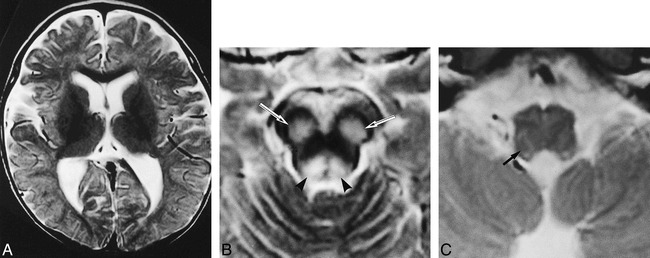
Patient 6: group BS lesion pattern.
A–C, Axial T2-weighted images (2000/80/2) at the age of 11 months show lesions in the substantia nigra (arrows, B) and periaqueductal region (arrowheads, B), and subtle increased signal intensity within the medullary reticular formation (arrow, C) but not in the basal ganglia. Clinically, the patient had episodic unexplained tachypnea.
Group WM (Fig 4): Patients 7 and 8 had initial lesions in the cerebral white matter that were followed by brain stem lesions. Both patients showed T2 prolongation in the retrotrigonal white matter, extending to the peripheral white matter and leading to a diffuse demyelination without basal ganglia lesions. In addition, both patients had periaqueductal gray matter involvement after 29 and 90 months, respectively.
fig 4.
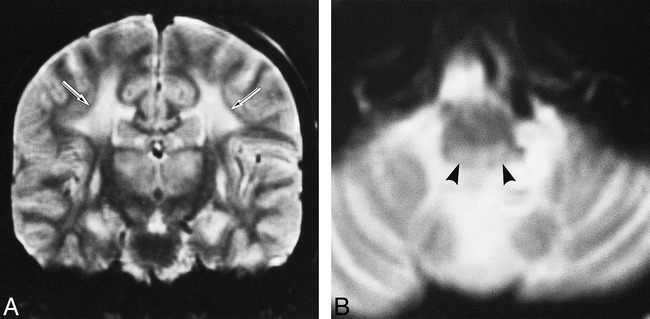
Patient 8: group WM lesion pattern.
A and B, Coronal (A) and axial (B) T2-weighted images (2000/80/2) at the age of 12 years show symmetrical hyperintensity in the cerebral white matter (arrows, A), thalamus, basal ganglia, and medullary tegmentum (arrowheads, B). Clinically, the patient had acute respiratory failure.
Brain Stem Lesions and Respiratory Failure
A chart showing the correlation between the appearance of brain stem lesions on MR images and abnormal respiration is presented in Figure 5. Seven patients who went on to have abnormal respiratory patterns and a deterioration in consciousness had lesions of the lower brain stem on MR images, especially in the periaqueductal gray matter and the reticular formation of the medulla oblongata. Patients 6 and 8 died suddenly; patients 4 and 5 were placed on permanent mechanical ventilation; and patients 1, 2, and 7 died within 1 year after acute or subacute neurologic deterioration. All the patients had upper brain stem lesions limited to the pons and midbrain. Isolated upper brain stem lesions, which were never directly correlated to fatal respiratory failure, progressed to the lower brain stem after 5 to 65 months, with an average of 27 months in patients 2, 4, and 6.
fig 5.
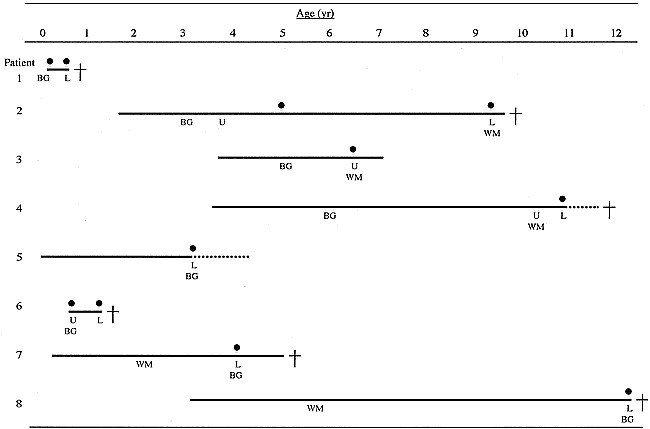
Brain stem lesions on MR images and abnormal respiration. Patient numbers correspond to those in Table 1. L indicates lower brain stem lesion; U, upper brain stem lesion; BG, basal ganglia lesion; WM, cerebral white matter lesion; solid line, duration of the disease; solid circle, acute respiratory failure; dotted line, permanent mechanical ventilation; cross, death
Fatal respiratory failure had no fixed prodromal symptoms. Some patients had irregular breathing, deep sighing respiration, unexplained hyperventilation, or hiccups with lethargy and serious feeding problems lasting for a few days to several weeks. Other patients ran a rapidly fatal course from irregular respiration to coma, convulsions, and paralysis of cranial nerves.
Neuropathologic Findings in Patients 6 and 8
In patient 6, axial sections of the brain revealed symmetrical necrosis in the thalamus, substantia nigra, brain stem tegmentum, and spinal cord gray matter. The necrosis was accompanied by mild edema and lateral ventricular dilatation. Microscopic examination showed spongy changes with hypervascularity and a relative preservation of the neurons in these lesions, compatible with the neuropathologic features of LS.
The brain of patient 8 showed a similar spongy change in the cerebral cortex, putamen, globus pallidus, thalamus, tegmentum of brain stem, and the substantia nigra. The cerebral white matter, which was the initial site of predominant lesions on MR images, showed spongy changes and mild demyelination, as did the other areas of involvement. Hyperintense foci on T2-weighted MR images were compatible with the topography of the spongy changes in the brain.
Discussion
Acute respiratory failure is a frequent symptom of LS, found in 64% to 72% of patients, and is a common cause of death (4, 5, 8, 12, 13). In this study, lesions in the lower brain stem were common by the end stages of LS, but their occurrence was unpredictable from longitudinal MR imaging studies. Furthermore, our results suggest an association between lower brain stem lesions and near fatal respiratory failure, whereas patients with reversible, limited, upper brain stem lesions did not necessarily suffer any respiratory disorder.
Previously, a large autopsy study revealed the involvement of the brain stem tegmentum in 98% of patients with LS (2). However, 15 of 84 patients with respiratory failure had no abnormal findings in the medulla oblongata (4). Rather than signifying a poor clinicopathologic correlation, three possibilities should be considered. First, several extramedullary areas that are functionally important in the formation of respiratory rhythm are frequently involved in LS, such as the pontomedullary reticular formation, the inferior olivary nuclei, and the pons tegmentum (2–4, 14). Second, the characteristic responses of the brain tissue may not be detectable by MR imaging until after a longer period of time, although functional deficits are apparent. The youngest reported patient with LS, who died at the age of 4 days, lacked significant changes in vascular proliferation and glial responses in the brain tissue; however, vascular congestion and sponginess of the neuropil were observed (15). And third, limited MR sensitivity cannot be excluded as the reason for a lack of abnormal findings. MR imaging may fail to detect early pathologic changes in the brain tissue, such as a necrotic neuropil, a small amount of cellular proliferation, and edema (7, 15). Considering these difficulties, the detection of lower brain stem lesions at the onset of respiratory abnormalities might be specific for LS.
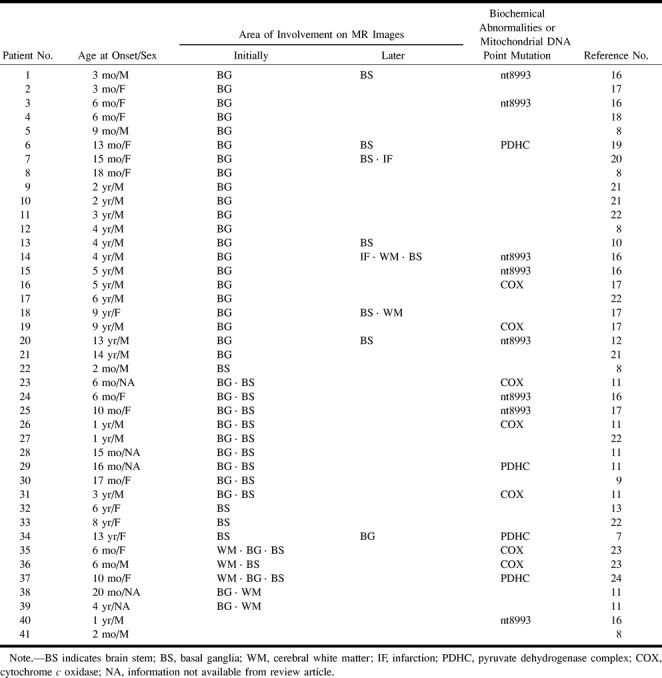
The organization of brain lesions as detected by MR imaging in patients with LS, both in the literature (7–13, 16–24) and in this study, can be divided into three patterns: groups BG, BS, and WM (Table 2). Forty-one patients, aged 2 months to 14 years, have been found to possess either genetic or biochemical derangements, including eight with mitochondrial gene abnormalities, four with a pyruvate dehydrogenase deficiency, seven with a COX deficiency, and 22 of unknown origin. Of these 41 patients, 21 (51%) had lesions with a group BG pattern, 16 (39%) with a group BS pattern, and two (5%) with a group WM pattern. Clinical course, age at onset, and basic metabolic defects were not correlated with any group. Two patients had normal MR imaging findings. One died at the age of 2 months and was found to have a lesion in the basal ganglia at autopsy. The other patient had a sibling with LS, but possessed only a mild mental delay. Two patients had infarction in either the parietal or parietooccipital lobe (16, 20), and one patient showed progressive ventriculomegaly (18). The proportion of patients with group BS lesions exceeded that found in this study by a substantial margin, because information regarding early brain lesions of the patients reported in the literature was very limited.
TABLE 2:
MR imaging findings in 41 patients with Leigh syndrome reported in the literature
Patients with group BS lesions have the most typical longitudinal MR findings of LS. Small focal brain stem lesions have been reported to occur in MELAS also, and are probably found more often in MERRF (15). CPEO and Kearns-Sayre-Shy syndrome, often associated with extensive lesions in both cerebral gray and white matter (25, 26), are accompanied by rare, symmetrical brain stem signal abnormalities caused by spongy myelinopathy in the midbrain and dorsal pons. Putamenal involvement, seen in patients with group BG lesions, has been documented as highly characteristic of LS neuroradiologically (10, 11), but it is relatively common in other mitochondrial disorders as well, such as MELAS or related conditions (25, 26). A large autopsy study revealed basal ganglia involvement in 55% to 67% of LS patients, but only in 20% of the patients in the first year of life (2, 15). Therefore, infantile-onset LS, as observed in two patients in this study, may differ from the characteristics of group BG.
Extensive cerebral white matter involvement, similar to leukodystrophy, has been reported in a few patients with COX deficiency (23), and massive myelin loss in the centrum semiovale has frequently been observed at autopsy in one third of patients with LS (15). Atypical neuroimaging findings at early stages of LS, such as cerebral white matter involvement, progressive cerebral atrophy, or infarction, make it difficult to establish the correct diagnosis.
Conclusion
LS, similar to a number of enzyme defects that might involve mitochondrial function and, specifically, electron transport, represents a spectrum of diseases with highly variable clinical, imaging, and pathologic presentations. It typically presents in the first two years of life, so that four of our patients were somewhat atypical in that clinical presentation occurred at a later age. Our results, as well as those reported by others, suggest that brain stem lesions associated with the loss of respiratory control are the most suggestive finding in LS, but fatal respiratory failure is unpredictable from clinical or neuroradiologic findings. Biochemical or genetic abnormalities have been found in fewer than 35% to 60% of patients with LS. Further identification of the functional relevance of these abnormalities may elucidate a pathogenic mechanism that defines the topography of the pathologic changes in the brain and that may enable an early diagnosis of LS. Close monitoring of patients for respiratory disturbances and the application of a variety of tests for assessing brain stem dysfunction, including auditory brain stem responses, short-latency somatosensory evoked potentials, electrically elicited blink reflexes, and polysomnography, may prevent sudden death among patients in whom LS has been diagnosed at an early stage.
Acknowledgments
We thank S. Takashima, M. Makino, Y. Goto, and I. Nonaka of the National Institute of Neuroscience, National Center of Neurology and Psychiatry, and T. Horie of the Chiba Children's Hospital for their helpful suggestions and technical expertise.
References
- 1.Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry 1951;14:216-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Montpetit VJA, Andermann F, Carpenter S, Fawcett JS, ZborowskaSluis D, Giberson HR. Subacute narcotizing encephalomyelopathy. Brain 1971;94:1-30 [DOI] [PubMed] [Google Scholar]
- 3.Sparaco M, Bonilla E, DiMauro S, Powers J. Neuropathology of mitochondrial encephalomyopathies due to mitochondrial DNA defects. J Neuropathol Exp Neurol 1993;52:1-10 [DOI] [PubMed] [Google Scholar]
- 4.Lyon G, Adams RD, Kolodny EH. Neurology of Hereditary Metabolic Diseases of Children.. New York: McGraw-Hill 1996;94-100
- 5.Rahman S. Blok RB, Dahl HHM, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996;39:343-351 [DOI] [PubMed] [Google Scholar]
- 6.Makino M. Horai S, Goto Y. Nonaka I. Confirmation that a T-to-C mutation at 9176 in mitochondrial DNA is an additional candidate mutation for Leigh syndrome. Neuromuscul Disord 1998;8:149-151 [DOI] [PubMed] [Google Scholar]
- 7.Heckman JM, Eastman R, Handler L, Wright M, Owen P. Leigh disease (subacute necrotizing encephalomyelopathy): MR documentation of the evolution of an acute attack. AJNR Am J Neuroradiol 1992;14:1157-1159 [PMC free article] [PubMed] [Google Scholar]
- 8.Greenberg SB, Faerber EN, Riviello JJ, Leon G, Capitanio MA. Subacute necrotizing encephalomyelopathy (Leigh disease): CT and MRI appearances. Pediatr Radiol 1990;21:5-8 [DOI] [PubMed] [Google Scholar]
- 9.Manzi SV, Hager KH, Murtagh ER, Mazalewski JG. MR imaging in a patient with Leigh's disease (subacute necrotizing encephalomyelopathy). Pediatr Radiol 1990;21:62-63 [DOI] [PubMed] [Google Scholar]
- 10.Koch TK, Yee MHC, Hutchinson HT, Berg BO. Magnetic resonance imaging in subacute necrotizing encephalomyelopathy (Leigh's disease). Ann Neurol 1986;19:605-607 [DOI] [PubMed] [Google Scholar]
- 11.Medina L, Chi TL, DeVivo DC, Hilal SK. MR findings in patients with subacute necrotizing encephalomyelopathy (LS). AJNR Am J Neuroradiol 1990;11:379-384 [PMC free article] [PubMed] [Google Scholar]
- 12.Mak SC, Chi CS, Tsai CR. Mitochondrial DNA 8993 T>C mutation presenting as juvenile Leigh syndrome with respiratory failure. Child Neurol 1998;13:349-350 [DOI] [PubMed] [Google Scholar]
- 13.Martin E, Burger R, Wiestler OD, Caduff R, Boltshauser E, Boesch Ch. Brainstem lesion revealed by MRI in a case of Leigh's disease with respiratory failure. Pediatr Radiol 1990;20:349-350 [DOI] [PubMed] [Google Scholar]
- 14.Bogousslavsky J. Respiratory failure and unilateral caudal brainstem infarction. Ann Neurol 1990;28:668-673 [DOI] [PubMed] [Google Scholar]
- 15.Cavanagh JB, Harding BN. Pathogenic factors underlying the lesions in Leigh's disease. Brain 1994;117:1357-1376 [DOI] [PubMed] [Google Scholar]
- 16.Uziel G, Moroni I, Lamantea E, et al. Mitochondrial disease associated with the T8993G mutation of the mitochondrial ATPase 6 gene: a clinical, biochemical, and molecular study in six families. J Neurol Neurosurg Psychiatry 1997;63:16-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ariga H, Takahashi S, Miyabayashi S, et al. Leigh syndrome: serial CT and MR imaging findings [in Japanese]. Nippon Acta Radiol 1996;56:839-845 [PubMed] [Google Scholar]
- 18.Chi CS, Mac SC, Shian WJ. Leigh syndrome with progressive ventriculomegaly. Pediatr Neurol 1994;10:244-246 [DOI] [PubMed] [Google Scholar]
- 19.Araki S, Hayashi M, Yasaka A, Maruki K. Electrophysiological brainstem dysfunction in a child with Leigh disease. Pediatr Neurol 1997;16:329-333 [DOI] [PubMed] [Google Scholar]
- 20.Narita T, Yamamoto T, Ohno M, Takano T, Ito R, Shimada M. Hypertension in Leigh syndrome: a case report. Neuropediatrics 1998;29:265-267 [DOI] [PubMed] [Google Scholar]
- 21.Erven PMM, Renier WO, Gabreels FJM, Thijssen HOM, Ruitenbeek W, Horstink MWIM. Hypokinesia and rigidity as clinical manifestations of mitochondrial encephalomyopathy: report of three cases. Dev Med Child Neurol 1989;31:81-97 [DOI] [PubMed] [Google Scholar]
- 22.Geyer CA, Sartor KJ, Prensky AJ, Abramson CL, Hodges FJ, Gado MH. Leigh disease (subacute necrotizing encephalomyelopathy): CT and MR in five cases. J Comput Assist Tomogr 1998;12:40-44 [DOI] [PubMed] [Google Scholar]
- 23.Zafeiriou DI, Koletzko B, Wolfgang MF, Paetzke I, Kueffer G, Jensen M. Deficiency in complex IV of the respiratory chain, presenting as a leukodystrophy in two siblings with Leigh syndrome. Brain Dev 1995;17:117-121 [DOI] [PubMed] [Google Scholar]
- 24.Bourgeois M. Deficiency in complex II of the respiratory chain, presenting as a leukodystrophy in two sisters with Leigh syndrome. Brain Dev 1992;14:404-408 [DOI] [PubMed] [Google Scholar]
- 25.Valanne L, Ketonen L, Majander A, Suomalainen A, Pihko H. Neuroradiologic findings in children with mitochondrial disorders. AJNR Am J Neuroradiol 1998;19:369-377 [PMC free article] [PubMed] [Google Scholar]
- 26.Barkovich AJ, Good WV, Koch TK, Berg BO. Mitochondrial disorders: analysis of their clinical and imaging characteristics. AJNR Am J Neuroradiol 1993;14:1119-1137 [PMC free article] [PubMed] [Google Scholar]