

Abstract
BACKGROUND AND PURPOSE: Diffusion-weighted MR imaging has emerged as a noninvasive tool for the detection of regional neuronal damage. We hypothesize that changes in diffusion-weighted images will correlate with pathophysiologic alterations caused by pilocarpine-induced status epilepticus.
METHODS: MR images of brain tissues were examined in vivo by use of T2- and diffusion-weighted imaging at 3, 6, 12, and 24 hours after pilocarpine-induced seizures. Histologic verification of neuronal damage was also performed after imaging to assess the extent and the time course of neuronal cell death.
RESULTS: The piriform cortex, amygdala, and retrosplenial (and somatosensory) cortex displayed significant apparent diffusion coefficient (ADC) decreases 12 hours after seizure initiation. In contrast, an ADC rise of 19% was observed in the hippocampus 24 hours after seizure induction. Histologic data from the piriform cortex and amygdala confirmed severe neuronal loss, whereas hippocampal damage was much less pronounced at 12 hours. Interestingly, very little histologic damage was seen in the retrosplenial cortex.
CONCLUSION: This study capitalized on diffusion-weighted imaging as a sensitive technique for the early identification of seizure-induced neuronal damage and differentiation of regional severity of these alterations. Hippocampal neuropathology is slower and longer in duration (∼7 days), while the piriform cortex and amygdala exhibit very rapid neurodegenerative alterations (∼24 hours) after pilocarpine-induced status epilepticus. These histologic changes are reflected in opposing ADC values within these regions.
Human temporal lobe epilepsy (TLE) involves the limbic structures of the brain, including the hippocampal formation (Ammon's horn, dentate gyrus, subiculcum), entorhinal cortex, and piriform cortex (1). Mesial temporal sclerosis, including hippocampal sclerosis, is the most common pathologic abnormality found in TLE, and is characterized by the loss of specific neurons in the hippocampus, parahippocampal gyrus, and amygdala, with subsequent glial proliferation (2, 3).
MR imaging can detect seizure-induced brain lesions that are visible on T1- and T2-weighted images (4–6). Diffusion-weighted imaging, available on most clinical scanners, sensitizes the MR signal to the Brownian motion of water molecules. The high sensitivity of diffusion-weighted imaging to neuronal damage has resulted in its use for detecting seizure-related brain alterations (7, 8). Diffusion-weighted imaging has been used in several studies with the kainic acid (an excitatory amino acid) model of TLE. As early as 1 hour, at 5 hours (8), and 12 hours (7) after kainic acid injection, the apparent diffusion coefficient (ADC) decreased in the piriform cortex and amygdala. The lowest ADC values were observed at 24 hours, after which they returned to control levels by 7 days (7). In the hippocampus, ADC values decreased transiently at 24 hours.
Obenaus et al (9), using the phenotypically similar pilocarpine model of epilepsy, observed significant ADC decreases in the amygdala and piriform cortex at 24 hours post pilocarpine injection, similar to those previously reported (7). However, in contrast to the kainic acid studies, an increased ADC was observed at 24 hours in the hippocampus (7, 9).
Lacking in published diffusion-weighted imaging studies of epileptic damage is the systematic examination of the diffusional changes during the acute phase of seizure-induced damage (7–11). Furthermore, a comprehensive comparison of the pathophysiological changes associated with the acute diffusional changes seen on diffusion-weighted images is needed. Although there are ample data describing histologic alterations caused by pilocarpine-induced seizures (12–16), no correlative studies comparing MR with histologic analysis have been reported. To our knowledge, this is the first study that describes MR and histologic changes that occur during the acute phase of status epilepticus (SE). These findings can provide valuable information to clinicians regarding the neuropathologic characteristics associated with seizures, as viewed with MR imaging.
This study used diffusion-weighted imaging to visualize the neuroanatomic changes occurring from 3 to 24 hours after the induction of SE by pilocarpine administration. We hypothesize that changes visualized using diffusion imaging will correlate with the acute progression of seizure-induced neuronal damage in the hippocampus and piriform cortex seen in histologic sections.
Methods
Animals and Seizure Assessment
Sustained seizures were induced in young adult male Sprague-Dawley rats (200–250 g; Harlan) by the administration of freshly dissolved pilocarpine hydrochloride (380 mg/kg i.p.; Sigma, St Louis, MO), a cholinergic agonist. The injection protocols were similar to those previously described (12, 16, 17). Peripheral cholinergic effects were minimized by injection of scopolamine methyl nitrate (1 mg/kg i.p.; Sigma, St Louis, MO) 30 minutes prior to pilocarpine administration (18, 19). After injection of pilocarpine hydrochloride, animals were placed in an observation box, and their behavior was monitored. Only rats that demonstrated robust behavioral seizures were included in the present study. These animals were then studied with MR imaging and neuroanatomic methods and at 3, 6, 9, 12, and 24 hours after pilocarpine-induced seizures.
MR Imaging
Rats were anesthetized with ketamine hydrochloride (100 mg/kg s.c.; Ayerst, Guelph, Ontario, Canada) and Xylazine (0.01 cc/rat s.c.; Bayer, Etobicoke, Ontario, Canada). MR control scans were performed on each rat prior to the injection of pilocarpine. Additional saline control scans were performed at 6 and 12 hours post injection to confirm that no ADC changes had occurred because of animal handling and injections. Imaging was performed on a 1.5-T SP Magnetom unit (Siemens, Erlangen, Germany) with a 150-mm-diameter, small field-of-view surface coil. Scout images were obtained in the coronal, axial, and transverse planes to position the slices accurately. Seven coronal slices, each with 2-mm thickness and 2-mm separation (center to center) were positioned on the transverse scout image at the level of the hippocampal formation and piriform cortex. After slice positioning, three interleaved spin-echo sequences were implemented: 1) unweighted diffusion, 2200/111 (TR/TE), b = 0, 2) diffusion-weighted, 2200/111, b = 1228 s/mm2; and 3) multi-echo T2-weighted, 2000/20–245, 16 echoes. The constant “b” is a composite derived from the magnitude of the diffusion gradients. Data were collected in a 128 × 128 matrix, and the images were reconstructed after applying a Fermi filter to enhance the signal-to-noise ratio. The diffusion gradient was applied in the z direction, normal to the coronal plane of the stereotaxically positioned rat, thus controlling anisotropic variance. The unweighted sequence refers to an image collected where the diffusion-encoding gradients have zero amplitude. These images contain T2 contrast imposed by the echo time (TE = 111 ms). In weighted images, the diffusion-encoding gradient amplitude is set to achieve the desired diffusion contrast. The gradient amplitude and duration are used to compute the weighting factor, “b.” In these experiments, weighted images contain both diffusion and T2 contrast. T2- and diffusion-weighted maps were then calculated from the images.
Diffusion and T2 Map Generation
ADC was determined by the equation:
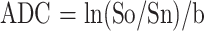 |
where Sn is the mean intensity for a diffusion-weighted image, and So is the mean intensity for the corresponding unweighted diffusion image (20). ADCs were calculated for each pixel in the map. High ADC values were represented as bright on diffusion-weighted maps. ADCs for regions of interest (ROIs) were calculated as the mean of the ADC for all pixels in the specified area.
T2 maps were generated from 16 echo T2 sequences. T2 relaxation constants were calculated for each pixel by nonlinear least squares curve fit to the data by using the equation:
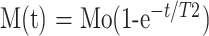 |
where Mo is the initial magnetization value before decay, t is the echo time (ms) and T2 is the spin-spin relaxation time.
Image Analysis
Image analysis was performed for each rat on a single slice immediately anterior to the slice where the hippocampus can be seen curling inferiorly. This position corresponded approximately to bregma −3.60 mm and maximized the cross-sectional area of each ROI (Fig 1) (21). Cheshire image processing software (Hayden Image Processing Group, Waltham, MA) was used to outline and analyze the ROIs that were confirmed by a second researcher. The bilateral ROIs included the amygdala (and associated nuclei), piriform cortex (including part of the entorhinal and perirhinal cortices), hippocampus, retrosplenial cortex (including motor and somatosensory cortices), and thalamus (Fig 1). The thalamic ROI was used as an intra-rat control, because it showed little signal change on diffusion-weighted images after seizure induction. A 2-pixel width separated the hippocampi and retrosplenial ROIs. A line drawn across the bottom of both hippocampi that extended across the cortex demarcated the inferior border of the retrosplenial ROI. The piriform and amygdala ROIs abutted each other and extended the same distance superiorly and inferiorly. Medially, 2 to 4 pixels separated the thalamus from the amygdaloid ROI to minimize signal contribution from the lateral ventricle (Fig 1B). A 5 × 5 pixel square was centered within the thalamus.
fig 1.
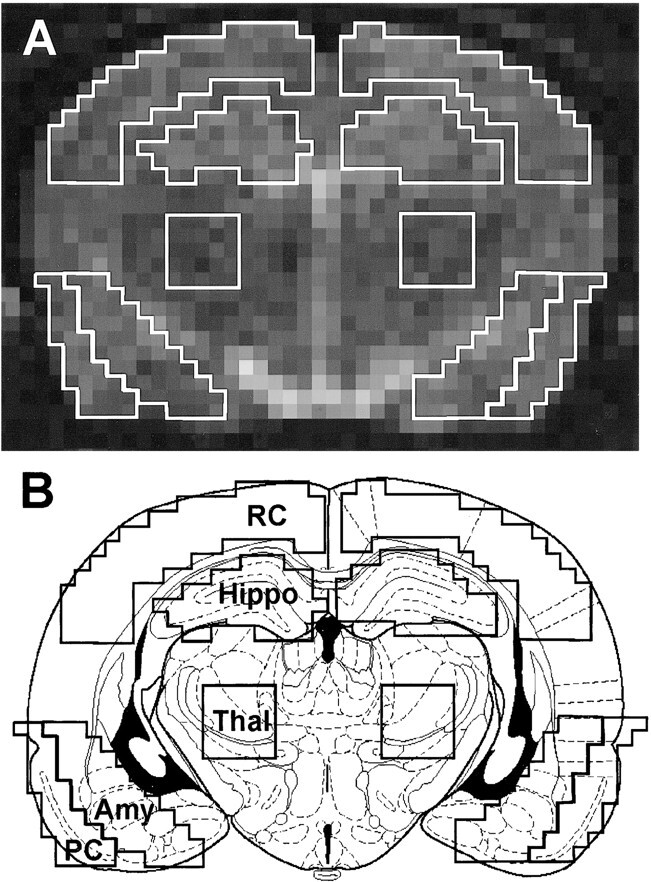
ROIs used for quantitative analysis. A, Representative control image on which ROIs were outlined using Chesire image processing software. B, Schematic drawing of a rat brain at similar level with the identical ROIs superimposed, illustrating the accuracy of our ROIs. ROIs were defined as: retrosplenial cortex (RC), hippocampus (Hippo), thalamus (Thal), amygdala (Amy), piriform cortex (PC)
Statistical Analysis of the MR Data
Left and right comparison of the bilateral ROIs was performed for each animal by use of a one-way analysis of variance (ANOVA) (P < .05). No significant differences were found, so comparison of all rats in each treatment group was performed using a one-way ANOVA (P < .05 for ADCs, P < .01 for T2 values). No significant differences were found between treatment groups and data were averaged for all rats in their respective experimental and timed groups. A two-tailed Student's t test (significant at P < .05, highly significant at P < .01) was then performed to compare the control values with the experimental values at each time point post pilocarpine injection. Saline injected controls, performed at 6 and 12 hours, were compared with untreated controls by use of a one-way ANOVA (P < .05).
Tissue Preparation and Histologic Methods
Immediately after completion of the imaging sequence, the animals were perfused intracardially with a fixative solution of 4% paraformeldehyde in 0.12 M Millonig's phosphate buffer (pH 7.3). Rats received 1 mL of fixative per 1 g of body weight. After perfusion, the brains were processed, as described previously (16).
Every 10th section was stained with cresyl violet to determine the general histologic characteristics of the tissue and the location of the sections within the rostrocaudal extent of the hippocampal formation. Sections at similar levels were then processed for neuronal degeneration by using silver impregnation stain. Silver impregnation methods were a modification of Gallyas (22), and have been described in detail (16, 23).
Cresyl violet and silver stained sections were qualitatively examined to determine general neuronal loss in the ROIs and for the density of argoyphilic neurons that are in the process of degenerating, indicating neuronal cell death.
Results
Animals
Injection of pilocarpine in 65 rats resulted in 20 animals (31%) having robust behavioral seizures for inclusion in the present study (Table 1). Twenty-four control scans obtained prior to injection were used for the T2 maps (one experimental rat lacked a control scan). Twenty-three control scans were gathered for the ADC maps (20 prescans from pilocarpine-injected rats and three scans from rats used for control histology). Saline controls were performed on six rats, and no behavioral abnormalities were observed after injection. One experimental rat demonstrated severe ventricular hypertrophy, and was removed from the study. Behavioral seizures often began 20 to 40 minutes after pilocarpine injection, and were similar to previous reports (13, 19).
TABLE 1:
Number of animals used at each of the imaging time points

MR Assessment of Neuronal Alteration
T2 Images and Maps
T2 images, acquired to identify edematous brain regions, were processed to generate T2 maps as a quantitative measure of these regions. Signal changes on the T2-weighted images were observed from 3 to 24 hours after pilocarpine administration and were similar to those for the T2 maps.
Significant increases (P < .05) in T2 values were seen at 3 hours in the piriform cortex, amygdala, and thalamus (Table 2). Also, the piriform cortex and amygdala had highly significant increases at 12 and 24 hours (P <.01), and the hippocampus demonstrated highly significant increases at 24 hours. The mean T2 value for the retrosplenial cortex was significantly elevated at 12 hours (103.6 ± 4.1 ms), with a return to control level at 24 hours (92.7 ± 4.3 ms). Elevated T2 relaxation constants were seen in most brain regions at all experimental time points (Table 2).
TABLE 2:
Mean T2 relaxation constants (SEM; ms) from the regions of interest after pilocarpine injection

Diffusion-weighted Imaging
ADC values were translated into gray-scale values on ADC maps for spatial representation (Fig 2). No significant changes were observed in control animals 6 or 12 hours after saline injection. Three to 9 hours after pilocarpine-induced seizures, there was a trend toward decreasing ADC values in the amygdala and piriform and retrosplenial cortices. No overt ADC changes were observed in the hippocampus at these time points. Twelve hours after seizure initiation, significant decreases in ADC values were detected in the piriform cortex (48% of control ADC), amygdala (33%), and retrosplenial cortex (37%) (Figs 2 and 3). At 24 hours, the mean ADC from the piriform cortex and amygdala remained well below control values, whereas the mean ADC calculated from the retrosplenial cortex returned to control levels (Table 3). Only at 24 hours was there a significant increase in the mean hippocampal ADC (19%) (Figs 2 and 3).
fig 2.
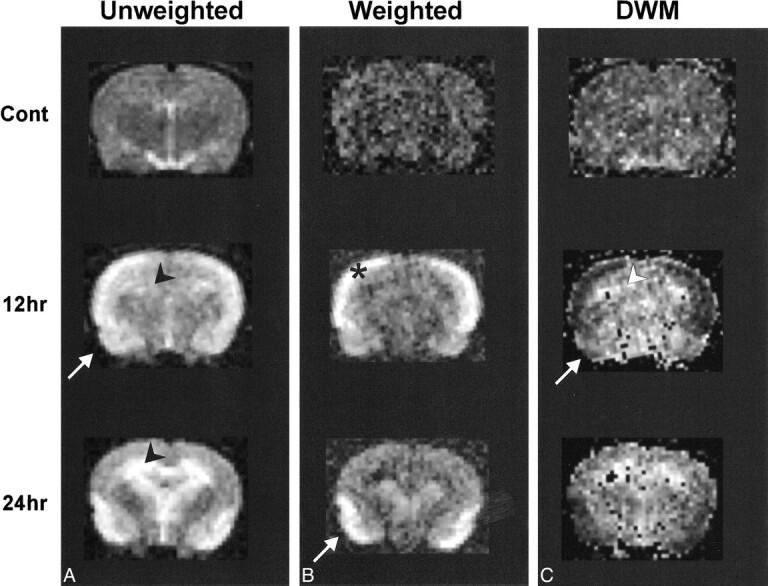
Diffusion maps.
Diffusion maps were generated from unweighted (b = 0) and weighted (b = 1228 s/cm2) images. The b factor refers to the effective diffusion weighting; it is a function of the amplitude of the gradients and their duration (see Methods for details). It is readily apparent that at 12 and 24 hours there are widespread changes in signal intensity in the hippocampus and piriform cortex. These alterations are quantified by an increase in ADC in the hippocampus (arrowheads), whereas there is a decrease in the piriform cortex (arrows) and retrosplenial cortex (*). Note that the retrosplenial cortex returns to control ADC levels by 24 hours.
TABLE 3:
Mean ADC values (SEM; × 10−7 cm2/s) in the regions investigated after status epilepticus
Histologic Analysis
Cresyl Violet Staining
Cresyl violet staining of brain tissue from controls revealed normal neuronal morphology in all of the brain regions examined. However, different rates of neuronal degeneration were observed between the hippocampus and piriform cortex. A previous study described the loss of hippocampal neurons primarily within the hilar region of the dentate gyrus, whereas CA1–3 was much less involved (16). The same study showed that most of the neuronal degeneration was complete by 7 days after pilocarpine-induced seizures.
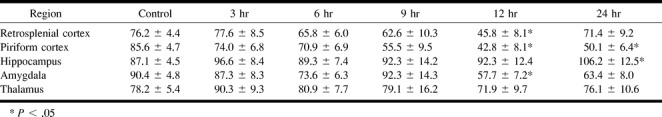
In the present study, histologic assessment using cresyl violet staining to determine neuronal loss revealed no cell loss at 3 hours in any subregion of the hippocampus. At 6 hours, cell density appeared to decrease within the hilar region of the dentate gyrus and CA3 regions with no change apparent in the CA1 region. Marked cell loss in the hilar region, with some cellular thinning in the CA1 region and medial and lateral regions of CA3, were observed at 12 hours after seizure initiation. Cell density continued to decrease in the hilar region and CA3 at 24 hours (Fig 4).
fig 4.
Cresyl violet and silver impregnation stain of the hilar region of the dentate gyrus.
A, Cresyl violet–stained sections in a control rat after imaging showing no loss of neurons. (M-molecular region, G-granule cell layer, H-hilus, CA3-pyramidal cells of the hippocampal CA3 layer).
B, Twenty-four hours after the induction of seizures, there is a pronounced loss of neurons in the hilus seen in cresyl violet–stained sections.
C, The silver degeneration stain in an adjacent section, from the same control animal in A, confirms the lack of neuronal loss within the hilus of the dentate gyrus. Neuronal degeneration can be visualized as darkly stained neurons; this control section lacks any darkly stained neurons.
D, The widespread loss of hilar neurons is confirmed after staining with silver degeneration methods. Arrowheads denote several neurons that are clearly in the process of degenerating. Note that the other regions, including the CA3 region, did not have significant neuronal degeneration. (Scale bar: 100 microns)
At higher resolution, microscopic examination revealed shrunken, condensed nuclei lacking a nucleolus in hilar neurons at all time points except 3 hours. However, on cresyl violet–stained sections, pyknotic nuclei were difficult to identify in CA1 and CA3 regions (Fig 4).
In tissue from control or saline-treated animals, cresyl violet staining revealed a dense normal band of pyramidal cells in layer II of the piriform cortex (Fig 5A). Layer III also contained numerous cresyl violet–stained neurons, although the staining pattern was more diffuse. Clusters of darkly stained neurons could be also seen in the amygdala, which corresponded with tissue nuclei of this region.
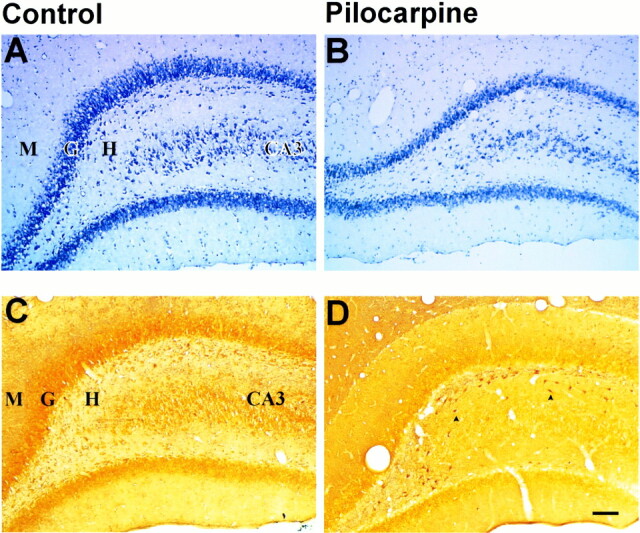
fig 5.
Histologic assessment of neuronal damage in the piriform cortex and amygdala 12 hours after pilocarpine-induced seizures.
A, Cresyl violet–stained section from a control animal. Note dark neuronal staining in layers II-III and in the nuclei of the amygdala.
B, There is marked neuronal loss and vacuolization of layer II-III in the piriform cortex (arrows).
C, Silver degeneration staining confirms the lack of neuronal loss in this section from a control animal.
D, Twelve hours after the induction of SE, there are numerous darkly stained neurons in layer II-III of the piriform cortex (arrowheads) and some nuclei of the amygdala. Arrows indicate widespread loss of pyramidal cells in the piriform cortex. (Scale bar: 100 microns)
As early as 3 hours after SE, tissue changes were evident within the piriform cortex. Many pyknotic nuclei were evident along the entire length of cortical layers II and III at 3 to 24 hours after seizures (Fig 5B). Decreasing neuronal density was observed in layers II and III at 6 and 12 hours, and by 24 hours few cells remained within the piriform cortex (Fig 5). This decreasing neuronal density corresponded with an increasing concentration of neuronal debris (Fig 5). The amygdaloid nuclei followed a similar temporal progression of damage, demonstrating severe neuronal loss by 24 hours.
Examination of control cortical tissue stained with cresyl violet revealed a reduced band of darkly stained neurons in layer II of retrosplenial (and somatosensory) cortex. More diffuse staining of pyramidal cells was observed in layers III and VI. No pyknotic or shrunken neurons were observed in control tissue sections (Fig 6B).
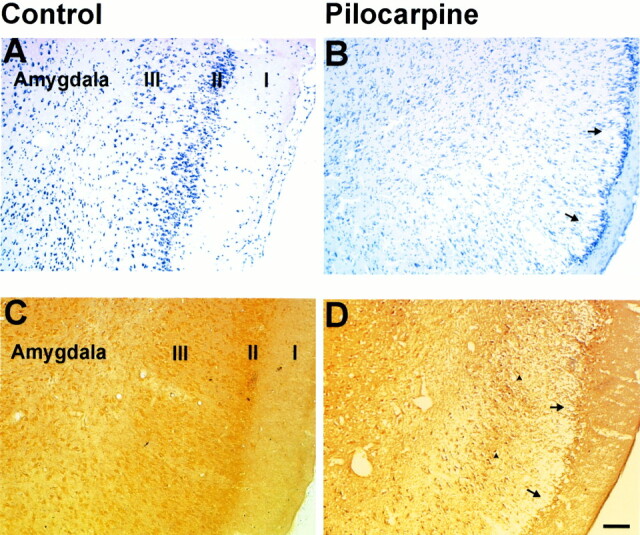
fig 6.
Silver and cresyl violet staining of retrosplenial cortex.
Histologic assessment of the retrosplenial cortex following pilocarpine-induced seizures. A, Control (A), 12-hour (B), and 24-hour (C) cresyl violet–stained sections after pilocarpine-induced seizures. Note the apparent swelling in cortical layers III-VI (arrow) at 12 hours, which appears to remit to control levels by 24 hours.
Control sections (D) stained for silver degeneration show no neuronal degeneration, whereas 12-hour tissue sections (E) depict moderate neuronal death (arrowheads). By 24 hours (F), there is no overt neuronal degeneration. There are occasional neurons that appear to be undergoing neuronal death (arrowheads) at this time point. (Scale bar: 100 microns)
Histologic changes in the retrosplenial cortex were observed exclusively at 12 hours. A band of decreased staining intensity and scattered pyknotic cellular nuclei incorporating cortical layer IV–VI stretched along the entire retrosplenial ROI. Surprisingly, 12 hours later, the neurons in the band exhibited normal staining characteristics and cellular morphology, although occasional pyknotic neuronal nuclei could be seen (Fig 6). This is in concordance with the MR findings where the mean ADC for the retrosplenial cortex also returns to normal at 24 hours after a significant decrease at 12 hours (Fig 3).
fig 3.

Normalized ADC values after pilocarpine-induced seizures.
Contrasting ADC (with standard error of the mean) changes were found between different limbic regions. The piriform cortex (and amygdala) show significant ADC decreases as early as 12 hours that are still present at 24 hours. Conversely, the hippocampus displays a slow increase in ADC that is significantly elevated at 24 hours. The retrosplenial cortex shows a decrease in ADC that peaks at 12 hours but returns to control levels by 24 hours.
No observable histologic changes in the thalamus were seen at any of the experimental time points.
Silver Impregnation Methods
A modified Gallyas (22) silver degeneration technique was used to impregnate neurons in the process of cellular degeneration. “Development” of the tissue reveals dying neurons stained darkly as black- or dark brown–filled cells (Fig 4) (22).
In control tissue from all brain regions examined, it was rare to see any darkly stained neurons after silver impregnation. However, in the hippocampus after pilocarpine-induced seizures, a moderate number of hilar neurons impregnated with silver were observed from 3 to 24 hours (Fig 4C and D). Similar profiles of degenerating neurons, starting at 6 hours and persisting until 24 hours after SE induction, were also present in the CA3 region. In the CA1 region, degenerating neurons were only observed in significant quantity at 12 hours.
In the piriform cortex, few degenerating neurons were observed up to 6 hours, but by 12 hours, the majority of neurons began to degenerate in layer II. Layer III of the piriform cortex and all amygdaloid nuclei demonstrated moderate numbers of dying neurons at 6 to 12 hours (Fig 5C and D). Twenty-four hours after seizure onset, only a moderate number of dying neurons were observed in the piriform cortex and amygdala, as this region now appeared to be largely devoid of neurons. Decreased staining intensity in the inner cortical layers of the piriform cortex was also seen at 12 hours, indicating possible tissue edema with concomitant neuronal loss (Fig 5).
The retrosplenial cortex followed a unique progression of cellular death. Occasional scattered dying neurons were evident at 6 hours in layers II and III. However, at 12 hours after seizure induction, a band of edematous tissue, indicated by decreased background staining and increased distance between neurons, incorporating layer IV–VI, was observed in the silver-stained sections, similar to those evident on the cresyl violet–stained sections (Fig 6B and E). Few degenerating neurons inhabited this band, but a moderate proportion of the neurons in layer III were argyophilic (Fig 6E). At 24 hours, the edema was resolved, and few dying neurons were seen in this region (Fig 6C and F). Thus, by 24 hours, this tissue appeared similar to control levels from imaging and histologic perspectives.
At all experimental time points, occasional scattered thalamic neurons were observed to be in the process of degeneration (data not shown).
Discussion
The novel findings of the present report are: 1) a decrease in mean ADC in the amygdala/piriform cortex region temporally associated with neuronal death, 2) an increase in ADC within the hippocampus by 24 hours, and 3) a large decrease in ADC in the retrosplenial cortex at 12 hours that recovered by 24 hours without a corresponding decrease in neuronal density. Diffusion-weighted imaging proved to be a sensitive MR technique for the early identification of regional seizure-induced neuronal damage.
Pilocarpine Model of SE
The pilocarpine model of epilepsy, similar to human TLE, is associated with neuronal loss in the hippocampus, specifically the CA3, CA1, and hilar region, with subsequent glial proliferation (24); for review, (16, 25). Furthermore, axonal remodeling in the supragranular region of the dentate gyrus, in conjunction with the neuronal loss, is seen in both the pilocarpine model (16, 26) and human TLE (27). Neuronal damage in human TLE and in the pilocarpine model is not restricted to the hippocampus, but often extends to extrahippocampal limbic structures as well.
The pilocarpine model was used in the present study because, 1) the chemoconvulsant (pilocarpine) is easy to administer, 2) when animals have prolonged seizures, consistent neuronal damage is observed, 3) neuronal damage within the hippocampus is not extensive, but tends to be more selective for dentate hilar neurons, 4) previous work has provided a suitable basis for undertaking the current study, and 5) a large percentage of animals exhibit recurrent seizures many months after the original SE event.
Piriform Cortex/Amygdala
We saw a significant ADC drop in the amygdala and piriform cortex at 12 hours that coincided with an increase in the number of degenerating neurons and a moderate decrease in neuronal density. Tissue edema, as indicated by increasing mean T2 relaxation constants, accompanied the ADC reduction (Table 2). At 24 hours, ADCs remained low in the amygdala/piriform cortex and were similar to values cited in previous reports (Figs 2 and 3) (8, 9).
To our knowledge, this is the first MR study to evaluate neuronal damage at acute time points of less than 24 hours after pilocarpine-induced SE. Two previous studies examined the acute phase of seizure-induced damage after kainic acid injection (7, 8). The similarity of damage induced by both kainic acid and pilocarpine in this region allows some comparison of the MR findings. Our findings of early ADC decreases and severe neuronal damage in the amygdala/piriform cortex are similar to kainic acid–induced seizures (Figs 3 and 5) (7, 8, 28).
As previously reported, we found that the limbic structures were highly sensitive to pilocarpine-induced SE (12, 15, 19). Of particular sensitivity are the piriform cortex and amygdala. Several characteristics of these structures account for their high sensitivity. First, the neurons of piriform cortical layer have electrophysiological properties that allow for rapid depolarization and cell firing. Secondly, kindling studies have shown a rapid progression to seizures in the amygdala, and in vitro electrophysiological studies of the piriform cortex have demonstrated neuronal foci with a high propensity for epileptiform discharges (29–31). Finally, cholinergic fibers from the olfactory bulbs terminate in the amygdala and piriform cortex. Pilocarpine administration after removal of the olfactory bulbs prevents all neuropathologic sequalae normally associated with pilocarpine-induced seizures (32).
Hippocampal Formation
The diffusional changes seen in the hippocampal formation (CA1, CA3, hilus, and dentate gryus) are a novel finding of this study. The rise in ADC within the hippocampal formation at 24 hours correlated with moderate numbers of degenerating neurons in the hilar and CA3 regions and resulted in a minimal overall decrease in neuronal numbers (Figs 3 and 4). Wang et al (7) reported a significant decrease in hippocampal ADC at 24 hours in the kainic acid model used to induce SE. While at 3 and 6 hours, occasional degenerating neurons were observed, general neuronal density in the hippocampus was unchanged. This translated into no significant change in ADC values, but a significant rise in the mean T2 relaxation constants (Tables 2 and 3). Thus, the increased T2 relaxation constants do not necessarily reflect neuronal damage. For example, in the thalamus, T2 relaxation constant increases were not associated with any significant tissue damage.
However, the increased ADC in the hippocampus at 24 hours found in the present study is at odds with the kainic acid studies in which a decreased ADC was observed in the same region. Several differences between these models could account for these opposing results. First, this discrepancy could be due to the different mechanisms employed in the generation of SE. Kainic acid, an excitatory amino acid similar to glutamate, selectively effects neurons expressing the kainic acid receptor. Concomitant activation of N-methyl-d-aspartate (NMDA) receptors by glutamate results in excitotoxic neuronal cell death (33–36) in hippocampal CA1 and CA3 pyramidal cells, which are particularly sensitive to kainic acid due to a high density of kainic acid and NMDA receptors (37). This increased receptor density results in significantly more pyramidal cell loss (CA3, CA1) than that seen in the pilocarpine model (15). Cell loss in the kainic acid model is so dramatic that the CA3 region is often totally ablated.
Secondly, pilocarpine, a cholinergic agonist, activates cholinergic inputs to the granule cells of the dentate gyrus. In the hippocampus, the excitatory cholinergic and glutaminergic activation is filtered by the granule cells of the dentate gyrus, which are highly resistant to SE-induced death. However, the secondary excitation of CA3 and CA1 neurons is not sufficient in the pilocarpine model for initiating large-scale neuronal damage often seen in the kainic acid model. The neurons within the hilus are very sensitive to seizure-induced activation, and in virtually all models of epilepsy these cells are found to die (Fig 4) (16, 19, 28, 38).
Finally, large numbers of hippocampal neurons (CA1, CA3, hilus) in the kainic acid model die quickly over a relatively short period. This contrasts considerably with the pilocarpine model of SE, where there is neuronal death to a limited degree in selected regions, and the time course of this neuronal death is much longer, often taking 7 to 14 days to complete (Fig 4) (16). This limited and slower neuronal death would result in a decreased macrophage and astroglial response within the hippocampal formation compared with that in the piriform cortex. This limited gliosis, at least during the first several days, would decrease the tortuosity, allowing increased bulk flow of water, thus leading to increased ADC within the hippocampal regions.
In summary, the increased ADC in the hippocampus seen in our study compared with the decreased ADC seen in the kainic acid reports likely reflects quantitative differences in the degree of neuronal loss within the hippocampus. Kainic acid administration often results in large-scale neuronal degeneration in the CA1 and CA3 regions, whereas in the pilocarpine model, only small numbers of neurons are affected in this region.
Retrosplenial Cortex
Interestingly, in the retrosplenial cortex, a significant decrease in ADC and T2 values contrasted with the normal (control) values seen at all other experimental time points (Fig 3, Table 2 and 3). At 12 hours, the silver degeneration stain revealed a limited number of dying neurons in cortical layers IV through VI. In cresyl violet– and silver-stained sections, there appeared to be large distances between neurons, which was reflected as a decreased staining intensity of the tissue (Fig 6), suggesting that edema in layers IV–VI was present at 12 hours. No edema and little cell degeneration that coincided with the ADC values and T2 relaxation constants returning to control levels were observed at 24 hours. These novel findings illustrate the importance of serial measurements of diffusion-weighted imaging in clinical settings for temporal and regional changes.
In our study, as in previous reports, neocortical damage was significantly less than in limbic structures (14, 15).
Putative Mechanisms for ADC Changes
Currently there is a debate over mechanisms that may contribute to shifts in the ADC after tissue damage resulting from seizures. This is due, in part, to a lack of data comparing histologic changes with the associated ADC fluctuations. Previous studies of diffusion-weighted imaging have focused primarily on MR changes (7, 8). We attempted to do a systematic comparison of diffusion-weighted images and associated histopathologic changes to provide a cohesive view of seizure-induced alterations within the rodent brain. In the amygdala/piriform cortex, the decreased ADC was associated with significant neuronal dropout and ongoing cellular death (Figs 3 and 5). In animal models of focal brain ischemia, ADC decreases within 10 to 30 minutes of the initial infarct and is thought to be due mostly to a rapid shift of water from the extracellular to the intracellular compartment (39). The intracellular compartment is more restrictive to water movement, and thereby decreases ADC (39, 40). Epileptic-induced lesions have different characteristics than those of ischemia: there is no cessation of perfusion, energy depletion, or decrease in tissue temperature that might, in part, cause the ADC decreases seen in ischemia (41). However, in both cases, there is decreased water mobility, but the mechanisms mediating the ADC changes are likely to be different.
Multiple factors can contribute to seizure-induced decreases in ADC seen within the amygdala/piriform cortex. Neuronal cell death has been observed to begin as early as 3 hours after the onset of SE. This neurodegeneration leads to macrophage invasion and swelling of both astrocytes and the remaining neurons, resulting in an increase in total water (edema) seen on the T2 maps. Swelling of the neuronal population is likely caused by cytotoxic edema due to long-term excitation. Taken together, these mechanisms would contribute to reduce the extracellular space (42). Neuronal density was markedly decreased after 12 hours of initiation of SE. Thus, one would postulate that decreased density of neurons would actually lead to an increase in the extracellular space, and therefore increase water mobility; however, this is not the case. In response to neuronal cell death, a large number of macrophages and then astrocytes proliferate and hypertrophy. We propose that these mechanisms contribute to the decreased water mobility. Injury-related changes, including release of amino acids and ions, result in pulsatile or long-term glial and astrocytic swelling. This in turn leads to compensatory decrease in the extracellular volume and increased tortuosity (43). Additionally, tortuosity, which reflects both the hindrances to movement imposed by the cellular structures of the brain and the connectivity of the extracellular space, could play a significant role (42). Thus glial hypertrophy, proliferation, and swelling may increase both extracellular and intracellular tortuosity, resulting in a decreased ADC.
A different mechanism would appear to account for increased hippocampal ADC. The degree and onset of hippocampal neuronal damage is very much less pronounced than in the amygdala/piriform cortex. Neuronal degeneration within the hippocampus occurs more slowly, and continues for 7 to 14 days after seizures (16). Thus, slow but continuous neuronal degeneration within the hilus of the dentate gyrus could reduce the amount of hypertrophy and proliferation of glial cells (Fig 4). Water still accumulates, as indicated by the increased T2 signal, but we speculate that glial and astrocytic membranes are likely not present in large enough numbers to restrict the water movement. This results in a progressive increase in ADC that may return to control levels as gliosis starts to occur in the hippocampus (Fig 3) (9, 16).
The mechanisms underlying the decrease in ADC seen in the retrosplenial cortex are unique. The moderate and transient neuronal activation from seizures that may manifest at 12 hours are a result of transient swelling of the neuropil that increase the tortuosity of the cortex (Fig 3, 6). The tissue edema at 12 hours appears to resolve by 24 hours after SE and is reflected by the ADC returning to control values. This finding, to our knowledge, has not been previously described in the literature.
Clinical Implications
Human TLE involves the limbic structures of the brain, including the hippocampal formation (Ammon's horn, dentate gyrus, subiculcum), entorhinal cortex, and piriform cortex. Forty percent of TLE cases are considered intractable to pharmacologic therapy, and many of these patients must undergo surgical resection of the seizure foci to control their seizures (1). MR imaging is the diagnostic technique of choice for noninvasive detection of these brain abnormalities. Seizure-induced brain lesions are visible on T1- and T2-weighted MR images, whereas gross remodeling is detectable by use of hippocampal volumetry (4–6).
Although T1- and T2-based imaging protocols can be used to provide anatomic information about chronic brain changes, these MR imaging techniques are limited compared with diffusion-weighted imaging. Diffusion-weighted imaging can make available additional information about ongoing neuropathology associated with acute seizures. Diffusion-weighted imaging is used clinically in the diagnosis and evaluation of ischemia and for acute brain disease (ie, stroke) (44, 45). Recently, several clinical studies using diffusion-weighted imaging have suggested that this MR technique can provide important diagnostic information about acute brain changes as a result of seizures (46). Furthermore, lateralization of seizure foci by use of diffusion-weighted imaging has been beneficial (47). Similar to our findings herein, an increased ADC was found within the hippocampus of patients with refractory epilepsy (48). Thus, we believe that diffusion-weighted imaging can provide useful clinical information for the diagnosis and treatment of patients with epilepsy, with particular reference to neuropathology.
Conclusion
The key finding of the present study is that diffusion-weighted imaging is capable of distinguishing between differing levels of SE-induced damage. After SE, decreased ADC would suggest severe, widespread neuronal damage, as is shown with silver degeneration staining. However, data from the hippocampus in this study suggest that increased ADC actually denotes less severe tissue damage. Thus, we have found diffusion-weighted MR imaging to be a sensitive technique for providing acute indications of evolving in vivo brain tissue alterations.
Acknowledgments
The authors wish to acknowledge the assistance of Jennifer Hadley with the imaging experiments and Brenda Bartnik with the histologic aspects of this study. We also thank Karen Tong, M.D. for helpful discussions regarding the clinical significance of the present study. The University of Saskatchewan College of Medicine provided financial support to Christopher Wall and this study was underwritten in part by a grant from the Health Services Utilization and Research Commission (HSURC) to André Obenaus. André Obenaus is a Medical Research Council of Canada Scholar.
References
- 1.Stears JC, Spitz MC. The imaging of epilepsy. Semin Ultrasound CT MRI 1996;17:221-250 [DOI] [PubMed] [Google Scholar]
- 2.Margerison JH, Corsellis JAN. Epilepsy and the temporal lobes. A clinical, electroencephalographic and neuropathological study of the brain in epilepsy, with particular reference to the temporal lobes. Brain 1966;89:499-530 [DOI] [PubMed] [Google Scholar]
- 3.Mathern GW, Babb TL, Mischel PS, et al. Childhood generalized and mesial temporal epilepsies demonstrate different amounts and patterns of hippocampal neuron loss and mossy fibre synaptic reorganization. Brain 1993;119:965-987 [DOI] [PubMed] [Google Scholar]
- 4.Jack CR, Twomey CK, Zinsmeister AR, Sharbrough FW, Petersen RC, Cascino GD. Anterior temproal lobes and hippocampal formations: normative volumetric measurements from MR images in young adults. Radiology 1989;172:549-554 [DOI] [PubMed] [Google Scholar]
- 5.Jack CR, Sharbrough FW, Twomey CK, et al. Temporal lobe seizures: lateralization with MR volume measurements of the hippocampal formation. Radiology 1990;175:423-429 [DOI] [PubMed] [Google Scholar]
- 6.Jackson GD, Berkovic SF, Duncan JS, Connelley A. Optimizing the diagnosis of hippocampal sclerosis using MR imaging. AJNR Am J Neuroradiol 1993;14:753-762 [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Majors A, Najm I, et al. Postictal alteration of sodium content and apparent diffusion coefficient in epileptic rat brain induced by kainic acid. Epilepsia 1996;37:1000-1006 [DOI] [PubMed] [Google Scholar]
- 8.Nakasu Y, Nakasu S, Morikawa S, Uemura S, Inubushi T, Handa J. Diffusion-weighted MR in experimental sustained seizures elicited with kainic acid. AJNR Am J Neuroradiol 1995;16:1185-1192 [PMC free article] [PubMed] [Google Scholar]
- 9.Obenaus A, Kendall E, Sarty G. Apparent diffusion coefficients following pilocarpine-induced seizures: correlation with anatomical changes. Soc Neurosci Abst 1998;24:719 [Google Scholar]
- 10.Zhong J, Petroff OAC, Prichard JW, Gore JC. Changes in water diffusion and relaxation properties of rat cerebrum during status epilepticus. Magn Reson Med 1993;30:241-246 [DOI] [PubMed] [Google Scholar]
- 11.Tokumitsu T, Mancuso A, Weinstein PR, Weiner MW, Naruse S, Maudsley AA. Metabolic and pathological effects of temporal lobe epilepsy in rat brain detected by proton spectroscopy and imaging. Brain Res 1997;744:57-67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turski L, Cavalheiro EA, Turski WA, Meldrum BS. Excitatory neurotransmission within substantia nigra pars reticulata regulates threshold for seizures produced by pilocarpine in rats: effects of intranigral 2-amino-7-phosphonoheptanoate and N-methyl-D-aspartate. Neurosci 1986;18:61-77 [DOI] [PubMed] [Google Scholar]
- 13.Cavalheiro EA, Leite JP, Bortolotto ZA, Turski WA, Ikonomidou C, Turski L. Long-term effects of pilocarpine in rats: structural damage of the brain triggers kindling and spontaneous recurrent seizures. Epilepsia 1991;32:778-782 [DOI] [PubMed] [Google Scholar]
- 14.Liu Z, Nagao T, Desjardins GC, Gloor P, Avoli M. Quantitative evaluation of neuronal loss in the dorsal hippocampus in rats with long-term pilocarpine seizures. Epilepsy Res 1994;17:237-247 [DOI] [PubMed] [Google Scholar]
- 15.Fujikawa DG. The temporal evolution of neuronal damage from pilocarpine-induced status epilepticus. Brain Res 1996;725:11-22 [DOI] [PubMed] [Google Scholar]
- 16.Obenaus A, Esclapez M, Houser CR. Loss of glutamate decarboxylase mRNA-containing neurons in the rat of the dentate gyrus following pilocarpine-induced seizures. J Neurosci 1993;13:4470-4485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cavalheiro EA, Silva DF, Turski WA, Calderazzo-Filho LS, Bortolotto ZA, Turski L. The susceptibility of rats to pilocarpine-induced seizures is age-dependent. Dev Brain Res 1987;37:43-58 [DOI] [PubMed] [Google Scholar]
- 18.Baez LA, Eskridge NK, Schein R. Postnatal development of dopaminergic and cholinergic catalepsy in the rat. Eur J Pharmacol 1976;36:155-162 [DOI] [PubMed] [Google Scholar]
- 19.Turski WA, Cavalheiro EA, Schwarz M, Czuczwar SJ, Kleinrok Z, Turski L. Limbic seizures produced by pilocarpine in rats: a behavioural, electroencephalographic and neuropathological study. Behav Brain Res 1983;9:315-336 [DOI] [PubMed] [Google Scholar]
- 20.Le Bihan D, Breton E, Lalleman D, Grenier P, Cabanis E, Laval-Jeantet M. MR imaging of intravoxel incoherent motions: application to diffusion and perfusion in neurologic disorders. Radiology 1986;161:401-407 [DOI] [PubMed] [Google Scholar]
- 21.Paxinos G, Watson C. The Rat Brain in Stereotaxic Cooridinates.. Vol 4. Toronto: Academic Press 1998;
- 22.Gallyas F, Wolff JR, Bottcher H, Zaborszky L. A reliable and sensitive method to localize terminal degeneration and lysosomes in the central nervous system. Stain Technology 1980;55:299-306 [DOI] [PubMed] [Google Scholar]
- 23.Nadler JV. Use of excitatory amino acids to make axon-sparing lesions of hypothalamus. Methods in enzymology. Academic Press, Inc. 1983;393-400 [DOI] [PubMed]
- 24.Swanson TH. The pathophysiology of human mesial temporal lobe epilepsy. J Clin Neurophysiol 1995;12:2-22 [PubMed] [Google Scholar]
- 25.Obenaus A, Houser CR. Neuronal degeneration in the pilocarpine model of chronic seizures: Differential vulnerability and time course. Epilepsia 1992;33:361370800 [Google Scholar]
- 26.Dudek FE, Obenaus A, Schweitzer JS, Wuarin J-P. Functional significance of hippocampal plasticity in epileptic brain: Electrophysiological changes in the dentate granule cells associated with mossy fiber sprouting. Hippocampus 1994;4:259-265 [DOI] [PubMed] [Google Scholar]
- 27.Babb TL, Pretorius JK, Mello LE, Mathern GW, Levesque MF. Synaptic reorganizations in epileptic human and rat kainate hippocampus may contribute to feedback and feedforward excitation. Epilepsy Res Suppl 1992;9:193-203 [PubMed] [Google Scholar]
- 28.Pollard H, Charriaut-Marlangue C, Cantagrel S, et al. Kainate-induced apoptotic cell death in hippocampal neurons. Neuroscience 1994;63:7-18 [DOI] [PubMed] [Google Scholar]
- 29.Racine RJ, Mosher JM, Kairiss EW. The role of the pyriform cortex in the generation of interictal spikes in the kindled preparation. Brain Res 1988;454:251-263 [DOI] [PubMed] [Google Scholar]
- 30.Haberly LB, Bower JM. Olfactory cortex: model circuit for study of associative memory? TINS 1989;12:258-264 [DOI] [PubMed] [Google Scholar]
- 31.Haberly LB, Sutula TP. Neuronal processes that underlie expression of kindled epileptiform events in the piriform cortex in vivo. J Neurosci 1992;12:2211-2224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millan M, Patel S, Meldrum B. Olfactory bulbectomy protects against pilocarpine-induced motor limbic seizures in rats. Brain Res 1986;398:204-206 [DOI] [PubMed] [Google Scholar]
- 33.Coyle JT. Neurotoxic action of kainic acid. J Neurochem 1983;41:1-11 [DOI] [PubMed] [Google Scholar]
- 34.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci 1987;7:369-379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi DW, Koh J-Y, Peters S. Pharmacology of gluatamate neurotoxicity in cortical cell culture: Attenuation by NMDA antagnonists. J Neurosci 1988;8:185-196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia 1993;34:S37-S53 [DOI] [PubMed] [Google Scholar]
- 37.Miller LP, Johnson AE, Gelhard RE, Insel TR. The ontogeny of excitatory amino acid recpetors in the rat forebrain-II. kainic acid receptors. Neurosci 1990;35:45-51 [DOI] [PubMed] [Google Scholar]
- 38.Lassmann H, Petsche U, Kitz K, et al. The role of brain edema in epileptic brain damage induced by systemic kainic acid injection. Neurosci 1984;13:691-704 [DOI] [PubMed] [Google Scholar]
- 39.Pierpaoli C, Righini A, Linfante I, Tao-Cheng JH, Alger JR, Di Chiro G. Histopathologic correlates of abnormal water diffusion in cerebral ischemia: diffusion-weighted MR imaging and light and electron microscopic study. Radiology 1993;189:439-448 [DOI] [PubMed] [Google Scholar]
- 40.van Gelderen P, de Vleeschouwer MH, DesPres D, Pekar J, van Zijl PCM, Moonen CTW. Water diffusion and acute stroke. Magn Reson Med 1994;31:154-163 [DOI] [PubMed] [Google Scholar]
- 41.Auer RN, Siesjo BK. Biological differences between ischemia, hypoglycemia, and epilepsy. Ann Neurol 1988;24:699-707 [DOI] [PubMed] [Google Scholar]
- 42.Nicholson C, Sykova E. Extracellular space structure revealed by diffusion analysis. TINS 1998;21:207-215 [DOI] [PubMed] [Google Scholar]
- 43.Sykova E. The extracellular space in the CNS: Its regulation, volume and geometry in normal and pathological neuronal function. The Neuroscientist 1997;3:28-41 [Google Scholar]
- 44.Ueda T, Yuh WTC, Taoka T. Clinical application of perfusion and diffusion MR imaging in acute ischemic stroke. J Magn Reson Imag 1999;10:305-309 [DOI] [PubMed] [Google Scholar]
- 45.Armitage PA, Bastin ME, Marshall I, Wardlaw JM, Cannon J. Diffusion anisotropy measurements in ischaemic stroke of the human brain. MAGMA 1998;6:28-36 [DOI] [PubMed] [Google Scholar]
- 46.Bradley WG, Shey RB. MR imaging evaluation of seizures. Radiology 2000;214:651-656 [DOI] [PubMed] [Google Scholar]
- 47.Hugg JW, Butterworth EJ, Kuzniecky RI. Diffusion mapping applied to mesial temporal lobe epilepsy. Preliminary observations. Neurology 1999;53:173-176 [DOI] [PubMed] [Google Scholar]
- 48.Wieshmann UC, Clark CA, Symms MR, Barker GJ, Birnie KD, Shorvon SD. Water diffusion in the human hippocampus in epilepsy. Magn.Res.Imaging 1999;17:29-36 [DOI] [PubMed] [Google Scholar]