

Summary
Cardiotoxicity, defined as toxicity that affects the heart, is one of the most common adverse drug effects. Numerous drugs have been shown to have the potential to induce lethal arrhythmias by affecting cardiac electrophysiology, which is the focus of current preclinical testing. However, a substantial number of drugs can also affect cardiac function beyond electrophysiology. Within this broader sense of cardiotoxicity, this review discusses the key drug-protein interactions known to be involved in cardiotoxic drug response. We cover adverse effects of anticancer, central nervous system, genitourinary system, gastrointestinal, antihistaminic, anti-inflammatory, and anti-infective agents, illustrating that many share mechanisms of cardiotoxicity, including contractility, mitochondrial function, and cellular signaling.
Keywords: cardiotoxicity, side effects, adverse reactions, cell signaling, mechanisms of toxicity
Graphical abstract
Adverse cardiac effects remain a top reason for drug withdrawal from the market. Mamoshina et al. provide a comprehensive review of drug-protein interactions involved in cardiotoxic drug response, covering anticancer, central nervous system, genitourinary system, gastrointestinal, antihistaminic, anti-inflammatory, and anti-infective agents and identifying avenues into drug discovery, safety pharmacology, and pharmacovigilance studies.
Introduction
Concerns regarding cardiac safety are among the top reasons for drug withdrawal from clinical trials and the market. Strict examination of cardiac safety liabilities has resulted in a sustained increase in attrition rates at all phases of drug development over the last decades.1 This increased attrition rate is illustrated in Figure 1, summarizing by therapeutic area all drugs withdrawn from the market due to cardiac safety concerns, as well as their commercialization lifespans. The latter have been greatly reduced over the last decades. Although this process of continuous pharmacovigilance greatly reassures the safety of new commercialized compounds, it also highlights that many constituent molecular processes underlying cardiotoxicity have yet to be comprehensively understood. This is substantially challenging for several scientific reasons. First, the spectrum of cardiotoxicity is broad, spanning from arrhythmia, to myocardial dysfunction, to terminal heart failure. In addition, the extent of the effects depends on exposure and varies among patients. Finally, adverse drug effects have been shown to depend on the gender, age, and genetic background of the individual.2, 3, 4
Figure 1.

Drugs withdrawn from market due to cardiotoxicity up to May 2020
(A) Number of drugs withdrawn according to therapeutic area.
(B) Lifespan of withdrawn agents.
CNS, central nervous system.
Drug-induced cardiotoxicity thus imposes substantial limits on drug development, as well as in the clinical management of existing drugs.1 Particularly critical across safety liabilities is the risk to induce potentially lethal arrhythmias via direct interactions with cardiac electrophysiology. However, recent studies have shown that a considerable number of drugs in use can also disrupt cardiac function by impairing myocardial metabolism and cardiac structure. These include anticancer therapies associated with myocardial apoptosis,5 neurodegenerative disease agents with severe risk of fibrotic valvular heart disease,6 or antibacterial7 and antiviral8 treatments leading to mitochondrial damage.
Multiple drug classes have the potential to induce cardiovascular adverse effects in patients. In this review, we summarize the drug-protein mechanisms by which drug-induced cardiotoxicity may develop. We provide a detailed review of 78 drugs with known cardiac adverse effects, 27 of which have been withdrawn from the market due to cardiotoxicity (Figure 1; Table 1). We cover adverse effects and cardiotoxic mechanisms of anticancer, central nervous system (CNS), genitourinary system, gastrointestinal, antihistaminic, anti-inflammatory, and anti-infective agents. We illustrate that they have common modes of action on cardiac function and on adverse cardiac events. Even though many of these points could be discussed in detail, we expect these findings will provide reliable guidance in identifying new critical pharmacophores, as well as key assays for the evaluation of drug-induced cardiotoxicity.
Table 1.
List of compounds with their targets and cardiac side effects
| Drug | Introduced–withdrawn | Mechanism of action with targets | Side effects on cardiac function | Mechanism of cardiac toxicity | References |
|---|---|---|---|---|---|
| Antineoplastic agents | |||||
| 5-fluorouracil | 2000–NA | DNA cross-linking | arrhythmias, myocardial ischemia, heart failure | TXA2 activation leading to cardiac remodeling, electrolyte imbalances | 9,10 |
| Arsenic trioxide | 2000–NA | inhibition of TXNRD1; activation of IKBKB, JUN, MAPK3, MAPK1 | QT prolongation, tachycardia | hERG trafficking inhibition | 11,12 |
| Bevacizumab | 2016–NA | blocker of VEGFA | heart failure | disruption of cardiomyocyte survival via VEGF signaling inhibition | 13 |
| Bortezomib | 2003–NA | inhibition of PSMB5, PSMB1 | heart failure, arrhythmia | hERG trafficking inhibition | 14,15 |
| Cisplatin | 1978–NA | DNA cross-linking | arrhythmias, myocardial ischemia, heart failure | TXA2 activation leading to cardiac remodeling, electrolyte imbalance | 9,16 |
| Cytarabine | 1966–NA | DNA intercalation, inhibition of DNA polymerase β | bradycardia, heart failure, ischemia | unknown | 17,18 |
| Daunorubicin | 1980–NA | inhibition of TOP2B | arrhythmias, heart failure | mitochondrial toxicity, oxidative stress leading to apoptosis | 19,20 |
| Dasatinib | 2016–NA | Bcr-Abl kinase, EPHA2, LCK, YES1 | heart failure | disruption of cardiomyocyte survival via VEGF signaling inhibition | 21 |
| Docetaxel | 1991–NA | inhibition of β subunit of tubulin, Bcl-2 | bradycardia, myocardial ischemia, heart failure | apoptosis of cardiac endothelial cells | 22,23 |
| Doxorubicin | 1978–NA | DNA intercalation, inhibition of TOP2B | arrhythmias, heart failure | mitochondrial toxicity, oxidative stress leading to apoptosis | 19,24 |
| Idarubicin | 1989–NA | inhibition of TOB2B | arrhythmias, heart failure | mitochondrial toxicity, oxidative stress leading to apoptosis | 19 |
| Imatinib | 2016–NA | Bcr-Abl kinase, KIT, RET | systolic heart failure, heart failure, left ventricular dysfunction | release of Bcl-2 proteins leading to mitochondrial toxicity, oxidative stress leading to apoptosis, disruption of cardiomyocyte survival via VEGF signaling inhibition | 21 |
| Ipilimumab | 2011–NA | inhibition of CTLA-4 | lethal myocarditis | unknown | 25,26 |
| Lapatinib | 2004–NA | blocking of EGFR, HER2 | left ventricular ejection fraction, congestive heart failure | disruption of cardiomyocyte survival via EGF signaling inhibition | 27 |
| Nilotinib | 2005–NA | inhibition of ABL1, blocking of KIT | myocardial ischemia | disruption of cardiomyocyte survival via VEGF signaling inhibition | 21 |
| Nivolumab | 2017–NA | inhibition of PD-1 | lethal myocarditis | unknown | 25,26 |
| Paclitaxel | 1995–NA | inhibition of β subunit of tubulin, Bcl-2 | bradycardia, myocardial ischemia, heart failure | apoptosis of cardiac endothelial cells | 28,29 |
| Romidepsin | 2009–NA | inhibition of HDAC1, HDAC2, HDAC4, HDAC6, ABCC1 | QT prolongation, myocardial infarction | hERG trafficking inhibition | 30 |
| Sorafenib | 2006–NA | inhibition of BRAF kinase, FLT1, FLT3, FGFR1, KIT, PDGFRB, RAF1, RET, VEGFR2, VEGFR3 | heart failure, myocardial ischemia, QT prolongation | disruption of cardiomyocyte survival via VEGF signaling inhibition | 31 |
| Sunitinib | 2006–NA | inhibition of PDGFRB, FLT1, FLT3, FLT4, KDR, KIT, CSF1R, PDGFRA | long QT, left ejection fraction, myocardial infarction | disruption of cardiomyocyte survival via VEGF signaling inhibition, disruption of stress response via PDGF signaling inhibition, disruption of energy homeostasis and mitochondrial fusion-fission system via inhibition of AMPK signaling | 32 |
| Trastuzumab | 2016–NA | blocker of HER2 | heart failure, tachycardia | disruption of cardiomyocyte survival via EGF signaling inhibition | 33,34 |
| Vandetanib | 2011–NA | inhibition of VEGFA, EGFR, PTK6, TEK | long QT | disruption of cardiomyocyte survival via VEGF and EGF signaling inhibition | 27,35 |
| Vinblastine | 1960s–NA | inhibition of α, β, and δ subunits of tubulin | myocardial ischemia, heart failure | apoptosis of cardiac endothelial cells | 36 |
| Anti-inflammatory agents | |||||
| Diclofenac | 1986–NA | inhibition of COX-1, COX-2, SCN4A, ASIC1; potentiation of ALOX5 | myocardial infarction | blocking prostacyclin synthase | 37,38 |
| Etoricoxib | 2002–2007 | inhibition of COX-2 | thrombotic events | blocking prostacyclin synthase | 37,38 |
| Ibuprofen | 1978–NA | inhibition of COX-1, COX-2 | myocardial infarction, hypertension | blocking prostacyclin synthase | 37,38 |
| Indomethacin | 1966–NA | inhibition of COX-1, COX-2, PLA2G2A, GLO1; activation of PPARG | myocardial infarction | blocking prostacyclin synthase | 37,38 |
| Naproxen | 1978–NA | inhibition of COX-1, COX-2 | myocardial infarction | blocking prostacyclin synthase | 37 |
| Rofecoxib | 1999–2004 | inhibition of COX-2 | myocardial infarction | blocking prostacyclin synthase | 39 |
| Central nervous system agents | |||||
| Benfluorex | 1972–2009 | blocking of 5-HT2B | valvular heart disease | HTR2B-induced activation of TGF-β signaling | 40 |
| Bupivacaine | 1965–NA | inhibition of SCN10A | ventricular arrhythmias, myocardial depression | inhibition of voltage-gated sodium channel, mitochondrial toxicity | 41 |
| Chlorphentermine | 1966–1969 | blocking of 5-HTs | pulmonary heart disease | HTR2B-induced activation of TGF-β signaling | 42 |
| Clozapine | 1991–NA | blocking of DRD2, HTR2A, DRD1, DRD3, DRD4, HTR1A, HTR1B, HTR1D, HTR1E, HTR2C, HTR3A, HTR6, HTR7, HRH1, HRH4, ADRA1A, ADRA1B, ADRA2A, ADRA2B, ADRA2C, CHRM1, CHRM2, CHRM3, CHRM4, CHRM5 | myocarditis, cardiomyopathy | unknown | 43,44 |
| Cocaine | 1884–NA | inhibition of SLC6A3, SLC6A2, SLC6A4, SCN5A | left ventricular hypertrophy, arrhythmias | inhibition of voltage-gated sodium channel, mitochondrial toxicity | 45 |
| Dexfenfluramine | 1996–1997 | inhibition of SLC6A4 | valvular heart disease | HTR2B-induced activation of TGF-β signaling | 46,47 |
| Ergotamine | 1925–NA | activation of ADRA1A, DRD2, HTR1B, HTR1D, HTR2A | valvular heart disease | induction of fibrosis via HTR2B-induced activation of TGF-β signaling | 48 |
| Fenfluramine | 1973–1997 | inhibition of SLC6A4, blocking of HTR2B | valvular heart disease | HTR2B-induced activation of TGF-β signaling | 47,49 |
| Fluoxetine | 1990–NA | inhibition of SLC6A4 | bradycardia | inhibition of ICaL, IKr; hERG trafficking inhibition | 50 |
| Haloperidol | 1967–NA | blocking of DRD1, DRD2, GRIN2B; inverse activation of DRD3 | QT prolongation, TdP, sudden cardiac death | inhibition of IKr, INa, ICaL | 51 |
| Levomethadyl acetate | 1991–2003 | activation of OPRM1 | QT prolongation, TdP | inhibition of IKr | 52 |
| Lidocaine | 1944–NA | inhibition of SCN10A, SCN9A, SCN5A; blocking of EGFR | bradycardia, cardiac arrest | inhibition of voltage-gated sodium channel, mitochondrial toxicity | 53,54 |
| Methysergide | 1965–NA | blocking of HTR2A, HTR2B, HTR2C, HTR7; activation of HTR1A; binding HTR1B, HTR1E, HTR1F | valvular heart disease | induction of fibrosis via HTR2B-induced activation of TGF-β signaling | 6 |
| Pergolide | 1987–NA | blocking of ADRA1B, ADRA2A, ADRA2B, ADRA2C, DRD1, DRD2, DRD3, DRD4, DRD5, HTR1A, HTR1D, HTR2B, HTR2C | valvular heart disease | induction of fibrosis via HTR2B-induced activation of TGF-β signaling | 6,55 |
| Phentermine | 1959–1997 | inhibition of SLC6A2, SLC6A3, SLC6A4; blocking of MAOA, MAOB | valvular heart disease | HTR2B-induced activation of TGF-β signaling | 47 |
| Propoxyphene | 1957–2010 | activation of OP1, OP2, OP3 | QT prolongation, TdP | inhibition of IKr | 56 |
| Sertindole | 1998a–2014 | blocking of DRD2, HTR2A, HTR2C, HTR6 | QT prolongation, TdP, sudden cardiac death | inhibition of IKr | 57 |
| Sibutramine | 2001–2002 | inhibition of SLC6A4, SLC6A2, SLC6A3 | myocardial infarction | inhibition of IKr | 58,59,60 |
| Thioridazine | 1978–NA | blocking of ADRA1A, ADRA1B, DRD1, DRD2, HTR2A; inhibition of KCNH2 | QT prolongation, TdP, sudden cardiac death | inhibition of INaL, IKr | 51 |
| Venlafaxine | 1986–NA | inhibition of SLC6A4, SLC6A2 | QT prolongation, arrhythmias | inhibition of INa | 61,62 |
| Ziprasidone | 2001–NA | inhibition of ADRA1A, ADRA1B, ADRA2A, ADRA2B, ADRA2C, CHRM1, CHRM2, CHRM3, CHRM4, CHRM5, DRD1, DRD2, DRD3, DRD4, DRD5, HTR2A, HTR1B, HTR1D, HTR1E, HTR2C, HTR3A, HTR6, HTR7, HRH1; activation of HTR1A | QT prolongation, TdP, sudden cardiac death | inhibition of IKr | 51 |
| Gastrointestinal agents | |||||
| Cisapride | 1980–2000 | blocking of HTR2A, HTR3A, HTR4; inhibition of KCNH2 | ventricular arrhythmia, QT prolongation, TdP, cardiac arrest | inhibition of IKr | 63 |
| Loperamide | 1976–NA | blocking of OPRM1, OPROD1, OPRK; inhibition of POMC; modulation of CALM1 | cardiac arrest, QT prolongation, ventricular tachycardia, TdP | inhibition of voltage-gated calcium channels | 64,65 |
| Omeprazole | 1987–NA | inhibition of ATP4A | acute myocardial infarction, heart failure | disruption of NO synthesis via ADMA production | 66 |
| Tegaserod | 1997–2007b | blocking of HTR2A, HTR2B, HTR2C, HTR4 | ischemia | inhibition of IKr | 67 |
| Genitourinary system agent | |||||
| Terodiline | 1975–1991 | blocking of muscarinic acetylcholine receptors | ventricular tachycardia, cardiac death | inhibition of IKr, blocking of calcium cycling | 68,69 |
| Antiallergic agents | |||||
| Astemizole | 1992–1999 | blocking of HRH1, inhibition of KCNH2 | long QT syndrome, TdP | inhibition of IKr | 70,71 |
| Diphenhydramine | 1946–NA | blocking of HRH1, CHRM2 | QT prolongation | inhibition of IKr | 72,73 |
| Terfenadine | 1985–1997 | blocking of HRH1 | QT prolongation, TdP | inhibition of IKr | 74 |
| Anti-infective agents | |||||
| Azidothymidine | 1989–NA | inhibition of Pol, TERT | dilated cardiomyopathy | mitochondrial toxicity | 75 |
| Azithromycin | 1988–NA | inhibition of 23S rRNA, rpID, rpIV, PADI4 | QT prolongation, TdP, cardiac death | mitochondrial toxicity | 76 |
| Clarithromycin | 1993–NA | inhibition of rpIJ, SLCO1B1, SLCO1B3 | QT prolongation, myocardial infarction, arrhythmias, cardiac death | mitochondrial toxicity | 77,78 |
| Erythromycin | 1955–NA | inhibition of 23S rRNA, MLNR, KCNH2, ALB | QT prolongation, ventricular tachycardia, TdP, ventricular fibrillation | mitochondrial toxicity | 7 |
| Grepafloxacin | 1998–1999 | inhibition of gyrA, parC | QT prolongation | inhibition of IKr | 79 |
| Sofosbuvir | 2013–NA | inhibition of NS5b | bradycardia | unknown | 80 |
| Sparfloxacin | 1997–2001 | inhibition of parC, gyrA, TOP2A | QT prolongation | inhibition of IKr | 79 |
| Pentamidine | 1975–NA | inhibition of RNA transfer | QT prolongation, arrhythmias | hERG trafficking inhibition | 81,82, 83, 84 |
| Cardiovascular agents | |||||
| Buflomedil | 1970sc–2011 | blocking of ADRA1A, ADRA2A | QT prolongation, cardiac arrest | unknown | 85,86 |
| Dofetilide | 2000–2004 | inhibition of KCNH2, KCNK2, KCNJ12 | QT prolongation, TdP | inhibition of IKr | 87 |
| Encainide | 1987–1991 | inhibition of SCN5A | QT prolongation, TdP | inhibition of IKr | 88 |
| Lidoflazine | 1973–1989 | blocking of calcium channels | QT prolongation | inhibition of IKr | 89 |
| Mibefradil | 1997–1998 | inhibition of CACNA1G, CACNA1H, CACNA1C, CACNA1D, CACNA1F, CACNA1I, CACNA1S, CACNB1, CACNB2, CACNB3, CACNB4 | QT prolongation | inhibition of Kir | 90,91 |
| Orciprenaline | 1965–2011 | activation of ADRB2 | tachycardia, palpitations | activation of ADRB2 | 92 |
| Prenylamine | 1960s–1988 | blocking of MYLK2, CALM | QT prolongation, sudden cardiac death, ventricular tachycardia, TdP | inhibition of IKr | 85 |
| Probucol | 1995–2009d | inductor of LDL catabolism | QT prolongation, arrhythmias | hERG trafficking inhibition | 82 |
| Other agents | |||||
| Alogliptin | 2013–NA | inhibition of DPP4 | heart failure | unknown | 93 |
| Clobutinol | 1963–2007 | inhibition of GABA receptors | QT prolongation | inhibition of IKr | 94 |
| Rosiglitazone | 1994–NA | activation of PPARG | heart failuree | unknown | 95 |
| Saxagliptin | 2009–NA | inhibition of DPP4 | heart failure | unknown | 96 |
TdP, torsade de pointes.
Australia approved.
Available only under a restricted distribution program.
EMA approved only.
Removed from the US market.
FDA lifted restrictions for rosiglitazone and confirmed its safety.
Main molecular mechanisms of drug cardiotoxicity
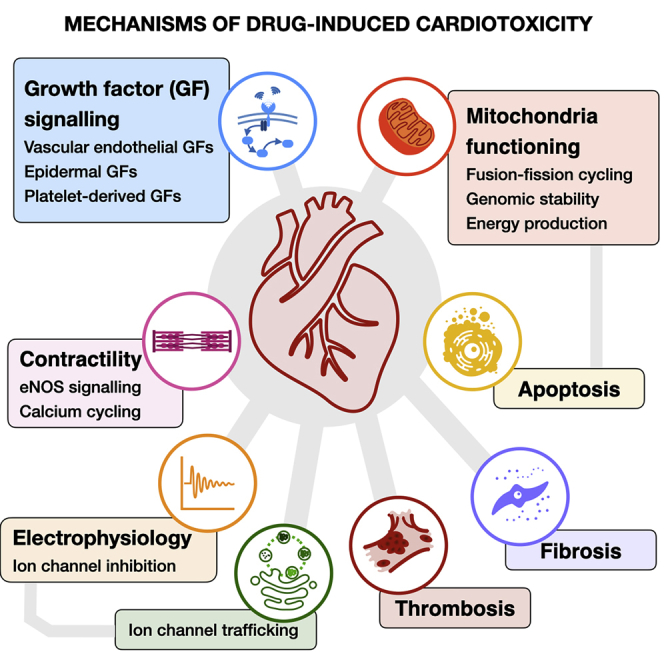
Cardiotoxicity generally results from the simultaneous interruption of key myocardial functions and viabilities. In this section, we focus on adverse drug effects via the disruption of electrophysiology, contractility, mitochondrial toxicity, growth factor, and cytokine regulation. These mechanisms of drug cardiotoxicity are summarized in Figure 2, together with the main types of therapeutic agents manifesting such risks. Importantly, the mechanisms underlying adverse cardiac events are often multifactorial, exerting their action through complex cell signaling pathways. The main signaling pathways underlying drug cardiotoxicity, further developed in the subsequent sections of this review paper, are illustrated in Figure 3.
Figure 2.

Mechanisms of drug-induced cardiotoxicity
Antibiotics and antiviral therapies can induce mitochondrial dysfunction, leading to an impaired fusion-fission cycle. Local anesthetics increase mitochondrial permeability, affecting function. Cardiotoxicity of tyrosine kinase inhibitors and monoclonal antibodies is primarily linked to inhibition of major signaling pathways for cardiomyocyte survival and maintenance. Different CNS and antidiabetic agents have been associated with the emergence of cardiac fibrosis. Proton pump inhibitors can affect cardiac contractility via inhibition of NO synthesis. Similarly, tyrosine kinases inhibitors have been linked to calcium cycling dysregulation. Multiple neural, cardiovascular, and anti-infective agents also have been shown to interact with cardiac electrophysiology. Equivalently, cardiac adverse effects of fluoxetine, HDAC inhibitors, and bortezomib are primarily linked to affecting ion channel trafficking rather than acute channel block. CV, cardiovascular; NSAIDs, nonsteroidal anti-inflammatory drugs; HDAC, histone deacetylase; eNOS, endothelial nitric oxide synthase.
Figure 3.

Overview of mechanisms of cardiotoxicity
Abnormalities in action potential duration or conduction velocity are associated with a direct block of ionic currents or inhibition of their trafficking from nucleus to cell membrane. Multiple pathways can trigger apoptosis, including VEGFR or EGFR inhibition (cardiomyocyte survival), PDGF inhibition (compensatory stress response), DR-induced TNF signaling activation, mitochondrial damage, or elevated ROS levels. α-Adr, VEGFR signaling, or eNOS inhibition affects calcium cycling. AMPK signaling inhibition affects both the mitochondrial fusion-fission cycle and the production of ATP. Serotonin-induced activation of the TGF-β pathway is primarily linked to cardiac fibrosis induction. IKr, rapid delayed rectifier current; INa, inward sodium current; INaL, late inward sodium current; ICaL, L-type calcium current; α-Adr, alpha-adrenergic receptor; VEGFR, vascular endothelial growth factor receptor; EGFR, epidermal growth factor receptor; DRs, death receptors; PDGF, platelet-derived growth factor; 5-HT2β, 5-hydroxytryptamine receptor (serotonin receptor 2B); TNF, tumor necrosis factor; TGF-β, transforming growth factor beta; ROS, reactive oxygen species; CaMKII, calcium/calmodulin-dependent protein kinase II; AMPK, AMP-activated protein kinase; ATP, adenosine triphosphate.
Electrophysiology
Direct block
hERG-encoded channels, carrying the rapid delayed rectifier current (IKr), are main determinants of cardiac repolarization and proarrhythmic events (Figure 3, top left). Therefore, in the last decades, preclinical testing for cardiac adverse events has primarily focused on screening assays of hERG channel inhibition.97 However, the biological role of hERG channels is not limited to their electrophysiological function. hERG-encoded channels are also involved in cell proliferation and malignant cell apoptosis,98 and reducing hERG expression in gliomas has been proposed as a target for antineoplastic therapy.99 Beyond IKr block, drugs often interact with multiple channels. Recent work on multichannel action and its connection to adverse effects has resulted in significantly improved toxicity prediction.100,101 Evidence also suggests that the electrophysiological implications of drug-induced block of hERG channels could be alleviated by interactions with other channels.102 Adverse electrophysiological effects can be accentuated by several factors, especially heart disease. For example, diabetes has been shown to abate the amplitude of multiple ionic currents,103 enhancing susceptibility to adverse effects.
Trafficking inhibition
In addition to direct hERG channel block, multiple pharmacological agents can cause hERG deficiency (with hERG channel block or independently) by the inhibition of its biogenesis and trafficking. A detailed schematic of the subcellular processes involved in hERG biogenesis, trafficking, and degradation, together with established pathways of drug-induced IKr deficiency, is presented in Figure 4. Drugs with reported effects on hERG trafficking impairment are listed in Table 1. Compared with the fast action of acute inhibition, hERG trafficking impairment manifests with timescales of hours to days.
Figure 4.

Schematic of hERG biogenesis, trafficking, and degradation and pathways of drug-induced IKr deficiency
Subcellular processes modulating hERG expression on the cellular membrane include (1) mRNA synthesis, (2) synthesis of polypeptide chain, (3) transport of polypeptide from endoplasmic reticulum to Golgi apparatus with Hsp70 and Hsp90 chaperones attached, (4) transport of the polypeptide in the COPII-mediated vesicle, (5) glycosylation, (6) exocytosis, (7) endocytosis, (8) ubiquitination, and (9) degradation of hERG.
For example, arsenic trioxide (Figure 4), extensively used in cancer treatment, interacts with chaperones Hsp70 and Hsp90, altering the folding process of hERG. As a result, it has been shown to affect hERG maturation in HEK293 cell lines.104 Folding inhibition also has been proposed as a mechanism of the dual tumor suppression and carcinogenicity of arsenic trioxide.105 Similarly, the antidiarrheal berberine and its derivative, dihydroberberine (Figure 4), have been shown to impair hERG folding in both HEK293 cell lines and guinea pig ventricular cardiomyocytes.106,107 The inhibition of folding by those drugs results in the reduction of the mature glycosylated form of 155 kDa hERG, which can be easily measured along with immature hERG (135 kDa). Accumulation of unfolded proteins activates the unfolded protein response pathway, as shown in HEK293 cells incubated with berberine107 and in mouse vascular endothelium cell lines incubated with arsenic trioxide.108 For the anti-infective drug pentamidine (Figure 4), mature hERG deficiency was identified as the result of reduced immature transport in HEK293 cell lines and neonatal rat cardiomyocytes.81
Cardiac glycosides (Figure 4), specifically Na+/K+-ATPase inhibitors with known QT prolongation risk, reduce hERG trafficking in HEK293 lines and in guinea pig ventricular cardiomyocytes.109 Incubation with digitoxin, digoxin, and ouabain resulted in a significant reduction of mature hERG. Later evidence showed that digoxin-induced hERG trafficking inhibition is a result of the low intracellular potassium concentration caused by Na+/K+-ATPase inhibition.110 Moreover, digoxin has been reported to generally enhance protein degradation through a lysosomal pathway.111
The lipid-lowering compound probucol also causes QT prolongation without a direct block of hERG channels (Figure 4). This is believed to result from trafficking disruption, as shown in neonatal rat ventricular cardiomyocytes112 and later in HEK293 cells, in which enhanced degradation of mature hERG via neddylation was indicated as a mechanism.113 Meanwhile, arsenic trioxide showed no effect on neddylation of hERG.113 Gene expression inhibition and alteration of the acetylation of proteins involved in ion channel trafficking and degradation also have been proposed as possible cardiotoxic mechanisms of histone deacetylase (HDAC) inhibitors.114
In addition, a substantial number of drugs are known to bind to the hERG channel, simultaneously inhibiting it and disrupting trafficking of the mature channel. One example is ketoconazole (Figure 4), an anti-infective agent that causes clinical QT prolongation and has been shown to co-inhibit IKr and hERG trafficking in HEK293 cells.115 Interestingly, reduced mature hERG was observed in both wild-type and nonbinding mutant channels, suggesting trafficking impairment as main mechanism. However, this was not studied in detail, and biological assays showed inconsistent results with inhibition of the Hsp90 chaperone in yeast strains, but not in cancer or rabbit cell lysate Hsp90. Similar effects in HEK293 cells were shown by atazanavir, another anti-infective agent with known arrhythmic risk,116 and by the antidepressants fluoxetine and norfluoxetine.117 In contrast, the inhibition and trafficking mechanisms of the antidepressant desipramine (Figure 4) have been well characterized.118 Like probucol, desipramine accelerates degradation of mature hERG in HEK293 cells without interfering with Hsp90 and Hsp70 chaperones. A study of celastrol in HEK293 cells expressing hERG, KCNJ2, and KCNB1 genes showed it inhibits both the function and the trafficking of Kir, encoded by KCNJ2, and hERG channels, but not Kv2.1, encoded by KCNB1.119 hERG trafficking effects were 5- to 10-fold more potent than the acute block of the channel.
Contractility
Drugs have also been observed to affect contractility, the most fundamental heart function. Cardiomyocytes connect electromechanically with one another and the extracellular matrix via gap junctions, key molecular regulators for intercellular communication and coordinated contraction. In addition, calcium cycling, a complex process involving multiple regulatory molecules, is essential for contraction.
Specifically, the calcium/calmodulin-dependent protein kinase II (CaMKII) regulates calcium cycling via phosphorylation of its targets on both the cellular membrane and the sarcoplasmic reticulum (Figure 3, center). Accordingly, CaMKII inhibition has been shown to have potential efficacy for antiarrhythmic therapy.120 Nitric oxide (NO) further regulates intracellular calcium, promotes vascular relaxation, and inhibits platelet aggregation. In cardiomyocytes, NO is primarily synthesized by endothelial nitric oxide synthase (eNOS), in turn regulated by CaMKII. Protein phosphatases, which act in opposition to CaMKII, are believed to regulate cardiac gap junction communication.121
Depressing either contractility or heart rate is a mechanism of action of many cardiac drugs, such as calcium-channel blockers that bind to L-type calcium channels and beta blockers that block beta adrenoceptors. Cardiac glycosides, which inhibit the Na+/K+ pump, can also depress heart rate, despite their well-known positive inotropic effects. The safe administration of cardiac glycosides is regarded as a difficult task because of their narrow safety margins,122 and the most potent inotropic agents generally have the lowest toxic-to-therapeutic ratios.123
However, some medications have been reported to disrupt contractility on other levels. The destabilization of the homeostatic calcium system has been proposed as the cardiotoxic mechanism of proton pump inhibitors. These have been suggested to inhibit dimethylarginine dimethylaminohydrolase (DDAH), an enzyme responsible for eliminating asymmetric dimethylarginine (ADMA), resulting in excess of ADMA impeding NO synthesis.124 Similarly, antineoplastic agents such as tyrosine kinases have been linked to upregulated CaMKII expression and activity.125
Mitochondrial toxicity
Mitochondria play an important role in cardiac function, mainly by satisfying the immense energy requirements of contraction. As such, failure to replace malfunctioning mitochondria is highly injurious. Altered cardiac metabolism has been linked to cardiac disease development, such as ischemia,126 and conditions that increase the risk thereof, such as diabetes.127 Further demonstrating their association, contractility and the mitochondrial fusion-fission system were recently shown to be closely coupled (Figure 3): the fusion-fission cycle depends on calcium homeostasis, and fusion-fission abnormalities can lead to aberrant contraction.128
As a by-product of their metabolic function, mitochondria produce reactive oxygen species (ROS) that, as shown in Figure 3, play an important role in proapoptotic signaling.129 ROS also engage dynamically in the mitochondrial fusion-fission cycle to respond to environmental stressors. Although mitochondrial fusion and fission help to reduce cellular stress under mild environmental stressors, these processes can also result in apoptosis and tissue necrosis in response to extreme stressors.130
Several pharmaceutical agents have been found to produce or facilitate mitochondrial toxicity, including in the heart.131 These inimical drugs span multiple classes, including anthracyclines, antivirals, antidepressants, and local anesthetics. The mitochondrial toxicity of anthracyclines is primarily linked to the inhibition of their direct target, topoisomerase (DNA) II beta (TOP2B), required for mitochondrial DNA replication.132 Antiretroviral drugs, often used to treat HIV, directly target and inhibit reverse transcriptase. However, antiretrovirals have been found to also inhibit mitochondrial DNA polymerase gamma (DNA Pol-γ), depleting mitochondrial DNA and disrupting mitochondrial function.133 Moreover, anti-HIV therapies have also been shown to inhibit the mitochondrial fusion-fission cycle.8 Local anesthetics have been suggested to interact with phospholipids on the mitochondrial membrane, which often result in increased membrane permeability, electron transport chain disruption, and calcium accumulation.134
Growth factors and cytokine signaling
Growth factors and cytokines are biologically active molecules that act directly on many cellular functions, such as adhesion, proliferation, and migration. Consequently, growth factor and cytokine signaling broadly affect tissue and organ function (Figure 3, top right).
Vascular endothelial growth factor (VEGF) is a signal protein responsible for the regulation of vascular formation, angiogenesis, cardiomyocyte development and proliferation, and myocardial regeneration. VEGF is also a major antiapoptotic factor that promotes cardiomyocyte survival in response to environmental stress or disease.135,136 Because angiogenesis is closely related to neoplastic metastasis and malignancy, controlling angiogenesis via inhibition of VEGF signaling is an attractive target.137 However, VEGF-inhibitory therapies are known to produce many adverse side effects, including cardiotoxicity.138 Closely related to VEGF, the platelet-derived growth factor (PDGF) is also key in myocardial development,139 in the angiogenic cardiovascular compensatory stress response,140 and in cardiac fibrosis.141 Inhibition of PDGF by sunitinib has been linked to its cardiotoxicity.138
The epidermal growth factor receptor (EGFR) belongs to the epidermal growth factor (EGF) family of protein kinases, whose members include HER2, HER3, and HER4, and is essential to almost every known cellular process. HER2 is particularly crucial for cardiomyocyte differentiation and embryonic cardiac development.142 EGFR signaling is antiapoptotic, indicating it could lead to uncontrolled growth or intensified oncogenesis. Sufficient evidence suggests that EGF-activated VEGF signaling may enable the regulation of both angiogenesis and eNOS signaling.143 Taking these factors together, targeting EGFR to inhibit neoplastic growth has become an attractive avenue for potential antineoplastic therapies. Unfortunately, EGF inhibitors, such as lapatinib or trastuzumab, have recently been linked to adverse cardiac effects.33 Activation of AMP-activated protein kinase (AMPK), which controls cellular energy and survival homeostasis and is required for mitochondrial fission in response to stress,144 could underlie the relatively low toxicity rate of lapatinib33 compared with the myocardial metabolism disruption associated to AMPK attenuation by other tyrosine kinase inhibitors.138
Main classes of drugs causing clinical cardiotoxicity
Antineoplastic agents
Although significant improvements have arisen in recent years in chemotherapy safety, cardiovascular damage remains one of the most common adverse side effects of antineoplastic agents, often resulting in diminished life expectancy for cancer patients. Two types of chemotherapy-induced cardiotoxicity have been established: irreversible (type I) and reversible (type II), respectively characterized by cellular damage and dysfunction.
Antimitotics
Anthracyclines
Anthracyclines have been shown to be effective for the widest range of antineoplastic applications. They have four mechanisms of action by which they target actively proliferating neoplastic cells: DNA-RNA intercalation, TOP2B inhibition, iron-mediated generation of free radicals, and induction of the eviction of histone from chromatin.24
The cumulative and dose-related cardiotoxicities of anthracyclines are primarily linked to ROS formation, mitochondrial dysfunction, and cardiomyocyte apoptosis145 (Figure 3). Doxorubicin, a member of this class of agents, also has been shown to upregulate the expression of tumor necrosis factor (TNF)-mediated death receptors, involved in apoptosis in cardiomyocytes derived from induced pluripotent stem cells.5 Although cumulative cardiotoxicity is frequent among anthracyclines, the manufacturers of several claim no evidence of induced cardiotoxicity. For example, this is the case with amrubicin, approved by the Food and Drug Administration (FDA) of the United States of America, and marketed as Calsed in 2011 for lung cancer treatment.
Antimetabolites
Antimetabolites suppress the division of potentially cancerous, quickly dividing cells via interference with DNA replication. Because of their mechanism of action, their most frequent side effects are observed in highly proliferative tissues, such as the gastrointestinal tract, skin, or hair. Although less common, if cardiotoxicity arises, it generally does so within a week, with severity ranging from symptomatic arrhythmias to sudden cardiac death.9 The proposed mechanisms include multifactorial effects on the cardiovascular system: for example, 5-fluorouracil and cisplatin activate thromboxane A2 (TXA2) formation and platelet aggregation, potentially leading to cardiac remodeling and ischemia.146,147 Both drugs are also known to cause electrolyte imbalances, which play an important role in gastrointestinal toxicity,148 and are believed to be involved in their cardiotoxicity.149 Cytarabine is another antimetabolite with known cardiotoxicity;17,18 however, an understanding of its cardiotoxic mechanisms is still lacking.
Antitubulins
Vinca alkaloids and taxanes eliminate tumors by binding to microtubules. Like anthracyclines and antimetabolites, they target cell division and are primarily linked to gastrointestinal toxicity, although several antitubulins have been connected to specific adverse cardiac events. For example, this is the case with vinblastine, additionally associated with myocardial ischemia and infarction.36 Similarly, paclitaxel and docetaxel have been linked with bradycardia, ischemia, and heart failure.22 One of their proposed cardiotoxic mechanisms involves the inhibition of actively proliferating cardiac endothelial cells.150 However, given the sequential or combinatorial nature of cancer treatment, tubulin inhibitors are frequently administered after anthracyclines, which are often involved in cardiovascular adverse events. Therefore, the precise cardiotoxic role of tubulin inhibitors remains open for debate.
Tyrosine kinase inhibitors
Rather than targeting proliferating tissues, tyrosine kinase inhibitors exert their action by inhibiting tyrosine kinases as main enzymes responsible for the activation of signaling cascades in the synthesis of proteins. Despite reduced rates of side toxicity, some tyrosine kinase inhibitors have been associated with cardiovascular system damage. Cardiotoxicity is primarily linked to inhibition of major signaling pathways responsible for cardiomyocyte survival and maintenance, such as in the case of sorafenib or vandetanib, which are VEGF signaling inhibitors.138 However, lapatinib, an EGFR signaling inhibitor, presents low cardiotoxicity rates. As previously mentioned, this presumably results from its activation of the AMPK signaling pathway (Figure 3), which mobilizes cardiomyocytes and increases ATP synthesis and storage.33 In contrast, the tyrosine kinase inhibitor sunitinib, whose targets include vascular endothelial growth factor receptor (VEGFR), also inhibits the AMPK signaling pathway and potentially inhibits energy metabolism and PDGF signaling (Figure 3), all involved in the cardiomyocyte mechanical stress response.138,32 Different cardiotoxic mechanisms have been proposed for other tyrosine kinase inhibitors. Imatinib inhibits the chimeric oncogene bcr-abl, the protein constructed by the fusion of a breakpoint cluster region (bcr) with an Abelson tyrosine kinase (abl). Its cardiotoxicity is primarily linked to the release of B cell lymphoma 2 (Bcl-2) proteins, which cause mitochondrial damage.138 Interestingly, imatinib can inhibit the proliferation and apoptosis of neoplastic cells via hERG inhibition. Sunitinib and imatinib have demonstrated the ability to activate CaMKII expression and activity in vitro but without affecting myocardial contractility.125 In addition, sunitinib and imatinib induce high ROS levels (see also Figure 3), leading to reduced cell viability.151 Other bcr-abl inhibitors, such as dasatinib and nilotinib, are also known to induce cardiac adverse events,21 presumably linked to VEGF signaling inhibition.152
Monoclonal antibodies
Monoclonal antibodies, identical to antibodies produced by the immune system, specifically bind to extracellular and cell surface proteins, activating cellular apoptosis and blocking tumor proliferation. The adverse effects of this class of agents are primarily linked to their targets. HER2 inhibition by monoclonal antibodies is principally associated with cardiac dysfunction.33 Trastuzumab, a vascular endothelial growth factor A (VEGFA) inhibitor, is also known to downregulate Neuregulin-1, a signaling molecule in cardiac homeostasis and development.34 Its most common side effect is hypertension, but myocardial infarction may also occur.13 Bevacizumab, another monoclonal antibody, also negatively affects the coagulation system, probably because of its VEGFR inhibition effects.13
HDAC inhibitors
HDACs are a class of enzymes that remove acetyl groups from an amino acid on a histone. HDAC inhibitors, used for neurological disease, have recently been proposed as a powerful new class of antineoplastic agents. However, growing concern regarding their cardiac safety has slowed their progress into clinical trials. For example, romidepsin, approved for clinical use in 2009, has been suggested to produce diverse cardiac adverse effects, including QT prolongation, torsade de pointes arrhythmias, and sudden cardiac death.153 Its cardiac adverse events are primarily linked to hERG trafficking inhibition rather than direct channel block.114 To date, 17 HDAC inhibitors exist, classically divided into 4 classes. However, cardiotoxicity is not class specific. HDAC inhibitors of classes I, II, and IV are generally used as antineoplastics, whereas class III (known as sirtuins) could potentially be used as a cardioprotective agent by mainly reducing the risk of thrombosis, atherosclerosis, and endothelial dysfunction.154
Other antineoplastic agents
Bortezomib is a proteasome inhibitor approved for myeloma treatment that inhibits the ability of malignant cells to escape apoptosis. Potential side effects include neutropenia, thrombocytopenia, and cardiotoxicity. Cardiac adverse events are considered reversible; however, bortezomib has been reported to cause arrhythmias and to even lead to heart failure.14,15 Cardiotoxicity is linked to its primary target, the ubiquitin-proteasome system, which is essential for cardiomyocyte and mitochondria function and controls hERG trafficking.155
Recent reports have also raised concerns over the cardiac safety of immune checkpoint inhibitors, newer and highly effective anticancer therapies.25,26 Given their target, checkpoint inhibitors have been mainly associated with immune-related complications, such in the case of vitiligo.156 Lethal myocarditis has also been reported, although the underlying cardiotoxic mechanism remains unknown.
Used in leukemia, arsenic trioxide is another example of an anticancer drug with undesired cardiac complications, such as ventricular and supraventricular tachycardias.11,12 Its hERG trafficking inhibition (Figure 4) results in IKr deficiency and action potential prolongation in both animal and human ventricular cells.157
CNS agents
The electrophysiological function of both myocardial and nervous tissues requires the propagation of action potentials, a process triggered by external and intracellular mechanisms and involving the depolarization and repolarization of cellular membranes. Given the similarities in electrophysiology, CNS agents that target neuronal electrophysiology can also affect cardiac tissue. Consequently, drugs that cause neurotoxicity may also result in cardiotoxicity, although neurotoxicity is typically exhibited at lower doses.158 Other CNS agents have alternative mechanisms of cardiotoxicity that do not involve alterations of the electrophysiology.
Local anesthetics
Both heart and nervous tissues depend on sodium channels for action potential triggering. Local anesthetics frequently block sodium channels (Figure 3, top left) and therefore directly affecting both systems. The most cardiotoxic and neurotoxic local anesthetic is cocaine, first used in 1884.45 The next generation of local anesthetics, such a bupivacaine and lidocaine, was intentionally developed to overcome this toxicity. However, clinical evaluations demonstrated that bupivacaine (markedly more cardiotoxic than other local anesthetics) causes hemodynamic and electrophysiological disturbance, which may result in hypoxia, arrhythmia, and even cardiac arrest. Lidocaine and ropivacaine have also been shown to cause dose-dependent adverse cardiac reactions.41,53 A hypothesized mechanism of cardiotoxicity involves interactions between local anesthetics and mitochondrial membranes (Figure 3) by increasing the permeability of cardiolipin, thus interrupting function and potentially inducing apoptosis.134
Antidepressants
Available evidence emphasizes antidepressants as a CNS class with significant cardiovascular side effects. Indeed, several antidepressants were withdrawn from the market or restricted for use because of cardiac adverse reactions. Tricyclic antidepressants and neuroleptics directly interact with sodium, calcium, and potassium channels (Figure 3, top left) and can cause fatal arrhythmias and hypotension in overdose.159 The next generation of antidepressants is considered of lower risk, with fewer cardiac adverse effects reported, but they too inhibit cardiac sodium, calcium, and potassium channels.160 As an example, the selective serotonin inhibitor fluoxetine inhibits the L-type calcium current (ICaL) and IKr in addition to its primary targets. It has also been shown to disrupt hERG trafficking,117 and long-term use may result in mild bradycardia. The potentially more arrhythmic venlafaxine, another selective serotonin inhibitor, has been shown to block the inward sodium current (INa) in both humans and animals.72,161
Antipsychotics
Psychiatric disorders are known to be accompanied by an increased risk of cardiovascular disease.162 In addition, many antipsychotics have been found to cause QT prolongation and cardiotoxicity.57 Clozapine, a highly effective treatment for schizophrenia, has been linked to serious complications such as myocarditis, an inflammatory cardiac muscle disease.43,44 Therefore, it is generally considered the last resort if other antipsychotics are ineffective. However, the mechanism underlying clozapine-induced myocarditis is unknown. The antipsychotic sertindole was removed from market in 1998 due to QT prolongation and sudden cardiac death, primarily because of its high-affinity IKr block.163 However, it was relaunched on the European market in 2005 with required cardiac monitoring. Other antipsychotics for schizophrenia and bipolar disorder, such as thioridazine, ziprasidone, and haloperidol, can lead to QT prolongation, torsade de pointes arrhythmias, or even sudden cardiac death.51 Ziprasidone has been shown to block IKr, thioridazine blocks both late inward sodium current (INaL) and IKr, and haloperidol blocks IKr, INa, and ICaL without affecting INaL.160,164 Ziprasidone and clozapine also have been found to block alpha-adrenergic receptors,165 involved in calcium cycling regulation in cardiomyocytes.
Neurodegenerative disease agents
At present, neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease are incurable, attracting immense interest in the development of prospective therapies. Among them is pergolide, a dopamine receptor antagonist with potential application in Parkinson’s disease. However, pergolide has been associated with the emergence of fibrosis in multiple tissues, including the heart.6 This is believed to stem from pergolide activating serotonin HTR2B receptors (Figure 3, right), inducing the transforming growth factor beta (TGF-β) pathway.55,166 Pergolide use was consequently restricted because of increased risk of fibrotic valvular heart disease.
Other CNS agents
Further applications of CNS agents include their use in pain or appetite control. Methysergide and ergotamine, serotonin 5-hydroxytryptamine (5-HT) receptor inhibitors (Figure 3, right side) used for migraines, are known to share adverse cardiac effects with pergolide and to cause cardiac fibrosis.48 In addition, several appetite suppressants, including dexfenfluramine, fenfluramine and phentermine, chlorphentermine, and benfluorex, were lifted from the market due to cardiac fibrosis. The primary mechanism of fibrosis induction is believed to involve the activation of the TGF-β pathway through 5-HT2B receptors. Another discontinued appetite suppressant drug, sibutramine, was linked to myocardial infarction. Its underlying mechanism remains undescribed, but in vitro experiments showed that sibutramine blocks the IKr channel.58
Opioid analgesics also have been shown to affect cardiac electrophysiology.56 This led to the withdrawal of levomethadyl acetate in 2001 from the European market and propoxyphene in 2010. Although the FDA reported that levomethadyl acetate had no safety concerns, it was also lifted from the US market in 2003.
Genitourinary system agents
Muscarinic receptor antagonists are a class of drugs approved for treatment of incontinence. Although generally considered safe, they have a well-known potential to affect cardiac function and may increase heart rate, prolong QT, and increase cardiovascular risk when used concomitantly with other medications.167 Especially relevant within this class, terodiline, a muscarinic receptor antagonist, was withdrawn from the market due to serious cardiac side effects, such as torsade de pointes and even cardiac arrest among older people.68 The toxicity of terodiline is linked to the inhibition of the hERG-encoded channel and the disruption of calcium cycling, with its cardiotoxic effects depending on concentration and genetic background.2 The withdrawal of terodiline has since raised concerns about potential cardiac adverse reactions by the whole group of muscarinic receptor antagonists.
Gastrointestinal agents
As in the heart and nervous tissues, the function of the gastrointestinal system is heavily controlled by cellular electrophysiology. 5-HT4 receptors, located in the alimentary tract, heart, or CNS, can be pharmacologically modulated to control the release of neurotransmitters. Cisapride, a drug that increases motility in the upper gastrointestinal tract, was designed to block serotonin 5-HT4, but it also inhibits hERG, in addition to 5-HT2A and 5-HT3A serotonin receptors.168 Its marked hERG inhibition produced cardiac arrhythmias and torsade de pointes, leading to its withdrawal from the market in most countries. Tegaserod, a nonselective 5-HT4 inhibitor, was also withdrawn by the FDA in 2007 despite absence of reports showing hERG channel inhibition.67 New versions of selective 5-HT4 blockers, such as clebopride and mosapride, are considered cardiac safe.169
Other types of receptors are also present in the gastrointestinal system. For example, loperamide, an opioid antagonist approved for treating diarrhea symptoms, blocks the mu-opioid receptor. Loperamide has also been shown to inhibit voltage-gated calcium channels and may produce cardiotoxic effects in humans.64,68 In 2016, the FDA issued an announcement indicating that high doses of loperamide, or its misuse, could cause serious cardiac adverse events, including lethal heart attacks.
Proton pump inhibitors, such as omeprazole, are another class of gastrointestinal agents labeled as cardiotoxic. Although a direct connection between omeprazole and cardiotoxicity has never been fully established and the mechanism of action remains unknown,66 it may arise because of ADMA production, which interferes with NO synthesis (Figure 3) and accelerates endothelial cellular senescence.124,170
Antihistamines
H1 antihistamines reduce allergic responses by blocking histamine H1 receptors on the cell surface. The biological role of histamines includes participation in the immune response to pathogens and allergens by increasing capillary permeability. Within the cardiovascular system, the stimulation of H1 receptors results in the constriction of coronary blood vessels, inducing a positive chronotropic effect. However, two second-generation antihistamines, terfenadine and astemizole, were withdrawn from the market due to QT prolongation and torsade de pointes linked to their direct block of IKr.70 Diphenhydramine, the first antihistamine introduced to the market, also demonstrated a QT prolongation signature upon overdose.73
Anti-inflammatory agents
Nonsteroidal anti-inflammatory drugs (NSAIDs) are a broad class of agents that target one or both prostaglandin synthases (COX-1 and COX-2) and hence display anti-inflammatory properties. NSAID side toxicities, such as gastrointestinal toxicity, primarily result from COX-1 inhibition, and new generations of NSAIDs selectively target COX-2. However, both selective COX-2 inhibitors and high doses of nonselective COX inhibitors can display cardiotoxic side effects. In 2004, the FDA withdrew the COX-2 inhibitor rofecoxib, one of the most widely used drugs ever withdrawn from the market. Although its significant cardiotoxicity seems to be linked to its unique metabolism,171 in 2007, the FDA issued a nonapproval letter for the selective COX-2 inhibitor etoricoxib due to cardiotoxic concerns. In response to these concerns, the FDA strengthened the safety warning on NSAID labels in 2015. Long-term administration of both selective COX-2 inhibitors, such as diclofenac, and nonselective COX-1/COX-2 inhibitors, such as ibuprofen, naproxen, and indomethacin, has been shown to increase the risk of cardiac arrest.37 The cardiotoxic mechanism of selective COX-2 inhibitors is primarily linked to blocking prostacyclin synthase without affecting TXA2 synthesis, leading to an increased risk of thrombosis.172 In contrast to antimetabolites that induce TXA2 formation, increasing the risk of ischemia, evidence suggests that COX-2 inhibitors decrease the risk of ischemia because of their ability to reduce inflammation.173
Anti-infective agents
Anti-infective agents are compounds with selective toxicity against pathogens, such as bacteria, viruses, or other microorganisms. Anti-infective agents vary by the mechanism of action and side effects.
Antibiotics
Antibiotics, among the most prescribed drugs worldwide, primarily disrupt the bioactive processes of pathogens, such as cell wall construction, protein and nucleic acid metabolism, and repair. Because human cells share some functions with prokaryotic pathogens, some antibiotics (such as anthracyclines) are also used as antineoplastic agents. The cardiotoxicity of cytostatic antibiotic agents has been discussed. Macrolides, which target bacterial protein synthesis via inhibition of prokaryotic ribosomal subunits and are widely used for respiratory infections, have also been shown to block hERG.174 Macrolides such as erythromycin, azithromycin, and clarithromycin are considered arrhythmogenic.7 It has also been shown that macrolides cause mitochondrial toxicity (Figure 3) by inhibiting protein synthesis in mitochondria.175 Fluoroquinolones are another group of antibiotics that can affect hERG and cause torsade de pointes.79 Because of this effect, use of fluoroquinolones such as grepafloxacin and sparfloxacin was discontinued.
Antivirals
The design of safe antivirals is complicated, because viruses use host cell structures to replicate. A major concern of antiviral therapy is mitochondrial toxicity in liver, skeletal muscle, and heart tissues.133 Azidothymidine, an antiretroviral HIV treatment, has been shown to induce mitochondrial dysfunction, leading to mitochondrial fragmentation and an impaired fusion-fission cycle8 (Figure 3). Azidothymidine inhibits mitochondrial DNA polymerase alongside its main target, reverse transcriptase.133 Sofosbuvir, recently reported as cardiotoxic, is an inhibitor of RNA polymerase nonstructural protein 5B used to treat hepatitis C. Although the drug exhibited a cardiac-safe profile during clinical trials, several cases of severe bradycardia were reported post-marketing.80 The mechanism by which sofosbuvir induces cardiac adverse effects is still unclear.
Other anti-infective agents
Pentamidine, used to treat leishmaniasis, trypanosomiasis, and pneumonia, has been associated with QT prolongation and ventricular arrhythmias in intravenous treatment.82 In vitro experiments showed that despite being a poor direct blocker of IKr at therapeutic concentrations,83 pentamidine inhibits hERG trafficking (Figure 4), causing action potential prolongation in both animal and human cells.81,84
Cardiovascular agents
Paradoxically, the worsening of arrhythmic risk is a serious side effects of some antiarrhythmic agents. Several highly potent drugs were lifted from the market or restricted in use because of proarrhythmic effects, including dofetilide and encainide. Some nonselective calcium blockers, such as lidoflazine and prenylamine, were also suspended because of their life-threatening QT prolongation. This side effect is primarily linked to their high-affinity block of hERG channels.176 Buflomedil, an alpha-adrenoceptor antagonist, was withdrawn from the European market due to its unfavorable cardiac safety profile. The selective beta-adrenoceptor blocker orciprenaline was removed from the market due to life-threatening cardiac side effects.92
The efficacy and safety of cardiovascular agents have been investigated by landmark clinical trials, such as the Cardiac Arrhythmia Suppression Trial (CAST) focused on the class I antiarrhythmic agents encainide and flecainide.88 This study reported increased mortality rates caused by arrhythmia and myocardial infarction shock in patients treated with flecainide and encainide compared with placebo groups. Follow-up studies suggested existing structural heart disease, with ischemic and electrical instability present, to be the major risk factor for developing adverse reactions to flecainide.177 Later, the Survival with Oral d-Sotalol (SWORD) study raised concerns regarding the use of class III antiarrhythmic agents and demonstrated post-myocardial infarction patients at a higher risk of developing drug-induced arrhythmias.178 The same was shown for dronedarone, another class III antiarrhythmic, in the Antiarrhythmic Trial with Dronedarone in Moderate to Severe CHF Evaluating Morbidity Decrease (ANDROMEDA)179 and in the Permanent Atrial Fibrillation Outcome Study Using Dronedarone on Top of Standard Therapy (PALLAS).180
The cholesterol-lowering agent probucol, despite association with QT prolongation and arrhythmias,82 was discontinued from the US market due to lack of efficacy against coronary artery disease. Probucol affects hERG trafficking (Figure 4), and it was shown to cause electrophysiological abnormalities in neonatal rat ventricular cardiomyocytes.112
The antihypertension drug mibefradil is another example of a cardiovascular drug removed from the market, in 1998, because of safety concerns.90 Mibefradil blocks calcium channels and was shown to cause abnormal QT prolongation.91
Other agents
A negative effect of antidiabetic drugs is their increased risk of heart failure, which has been confirmed in different studies.181 Considering such a risk, the European Medicines Agency (EMA) and FDA issued restrictions in 2010 for different drugs containing rosiglitazone, a thiazolidinedione-class antidiabetic drug. After a thorough examination of large clinical trials, the FDA removed those restrictions and eliminated the risk evaluation and mitigation strategy for rosiglitazone. Concerns regarding the safety of saxagliptin and alogliptin were also raised.
In 2007, the cough suppressant clobutinol was lifted from the market by its manufacturer due to risk of QT prolongation. Animal experiments suggested that clobutinol inhibits hERG channels and can induce torsade de pointes.94
Conclusions
In this review, we have presented a comprehensive discussion of drug-protein mechanisms underlying clinical drug-induced cardiotoxicity. This significantly broadens other studies by covering adverse effects and cardiotoxic mechanisms of cardiovascular, anticancer, CNS, genitourinary system, gastrointestinal, antihistaminic, anti-inflammatory, and anti-infective agents beyond their direct interactions with cardiac electrophysiology. As a result, we illustrate that many of these drug classes share modes of action on cardiac function and on adverse cardiac events, with cardiotoxicity frequently resulting from the simultaneous interruption of key myocardial functions and viabilities (mitochondrial dysfunction, inhibition of major signaling pathways responsible for cardiomyocyte survival and maintenance, fibrosis, NO synthesis, calcium cycling, and cellular trafficking).
Understanding of the mechanisms of drug-induced toxicity and differences in cardiac safety profiles of therapies is important for the development of new compounds and of safety assays for preclinical testing. In the clinical scenario, the drug-induced mechanisms leading to adverse cardiac events are generally considered multifactorial in origin, and they often remain poorly understood. Therefore, a refined understanding of the multifaceted components of cardiotoxicity can lead to the development of new cardioprotective agents, with the potential of reducing the manifestation and damage of otherwise-effective but risky drugs. As an exemplar, statins have been proposed to lower anthracycline-induced harm via a reduction of ROS signaling and regulation of TOP2B.182 Similarly, the activation of the AMPK pathway by lapatinib may underlie its lower cardiotoxicity compared with other tyrosine kinase inhibitors. Other AMPK activators, such as metformin, have been shown to be protective for several heart conditions.183 Nevertheless, it is important to remember that cardiotoxicity highly depends on exposure levels (especially overdose) and the duration of treatment; thus, only some of the discussed adverse effects may be involved in clinical practice. For clinical applications such as oncology treatments, the risk-benefit ratios often overpower the consequences of potential cardiotoxic events.
This review focuses on mechanisms of drug-induced cardiac adverse effects, centering its scope on drug-protein interactions. Although certain mechanisms of action, such as tyrosine kinase inhibition or mitochondrial damage, could affect other cell types in the organism, in this review, we have centered our efforts on summarizing their known clinical manifestations on cardiac side effects. Drug-drug and drug-food interactions are also known to promote adverse cardiovascular effects, usually linked to competition among enzymes participating in drug metabolism or alterations in drug metabolism.184,185 In addition, recent advances in the annotation of RNA-RNA interactions suggest they might be of even higher importance in cardiac disease, such as heart failure and hypertrophy.186,187
All of these mechanisms of drug-induced cardiotoxicity constitute important and promising prospects for future research into drug discovery, safety pharmacology, and pharmacovigilance studies. Therefore, their integration as a priority into multi- and interdisciplinary approaches across academia, industry, and healthcare settings, for the complex characterization of human heart physiology,188,189 is expected to yield major advances for the analysis and prediction of adverse drug reactions and cardiotoxicity in humans, both in health and in disease.
Acknowledgments
This work was funded by a British Heart Foundation (BHF) Intermediate Basic Science Fellowship (FS/17/22/32644 to A.B.-O.), a Wellcome Trust Fellowship in Basic Biomedical Sciences (214290/Z/18/Z to B.R.), an Infrastructure for Impact Award from the National Centre for Replacement Refinement and Reduction of Animals in Research (NC/P001076/1), and the Oxford BHF Centre of Research Excellence (RE/13/21/30181 and RE/13/2/30182).
Author contributions
P.M., B.R., and A.B.-O. conceived the study approach and planning. P.M. performed the literature review and wrote the manuscript. B.R. and A.B.-O. provided critical discussion. All authors revised and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
References
- 1.Ferri N., Siegl P., Corsini A., Herrmann J., Lerman A., Benghozi R. Drug attrition during pre-clinical and clinical development: understanding and managing drug-induced cardiotoxicity. Pharmacol. Ther. 2013;138:470–484. doi: 10.1016/j.pharmthera.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Ford G.A., Wood S.M., Daly A.K. CYP2D6 and CYP2C19 genotypes of patients with terodiline cardiotoxicity identified through the yellow card system. Br. J. Clin. Pharmacol. 2000;50:77–80. doi: 10.1046/j.1365-2125.2000.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Pavia P., Kim Y., Restrepo-Cordoba M.A., Lunde I.G., Wakimoto H., Smith A.M., Toepfer C.N., Getz K., Gorham J., Patel P. Genetic Variants Associated With Cancer Therapy-Induced Cardiomyopathy. Circulation. 2019;140:31–41. doi: 10.1161/CIRCULATIONAHA.118.037934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sloan J.A., Goldberg R.M., Sargent D.J., Vargas-Chanes D., Nair S., Cha S.S., Novotny P.J., Poon M.A., O’Connell M.J., Loprinzi C.L. Women experience greater toxicity with fluorouracil-based chemotherapy for colorectal cancer. J. Clin. Oncol. 2002;20:1491–1498. doi: 10.1200/JCO.2002.20.6.1491. [DOI] [PubMed] [Google Scholar]
- 5.Zhao L., Zhang B. Doxorubicin induces cardiotoxicity through upregulation of death receptors mediated apoptosis in cardiomyocytes. Sci. Rep. 2017;7:44735. doi: 10.1038/srep44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Waller E.A., Kaplan J. Pergolide-associated valvular heart disease. Compr. Ther. 2006;32:94–101. doi: 10.1385/comp:32:2:94. [DOI] [PubMed] [Google Scholar]
- 7.Albert R.K., Schuller J.L., COPD Clinical Research Network Macrolide antibiotics and the risk of cardiac arrhythmias. Am. J. Respir. Crit. Care Med. 2014;189:1173–1180. doi: 10.1164/rccm.201402-0385CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nomura R., Sato T., Sato Y., Medin J.A., Kushimoto S., Yanagisawa T. Azidothymidine-triphosphate impairs mitochondrial dynamics by disrupting the quality control system. Redox Biol. 2017;13:407–417. doi: 10.1016/j.redox.2017.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Polk A., Vaage-Nilsen M., Vistisen K., Nielsen D.L. Cardiotoxicity in cancer patients treated with 5-fluorouracil or capecitabine: a systematic review of incidence, manifestations and predisposing factors. Cancer Treat. Rev. 2013;39:974–984. doi: 10.1016/j.ctrv.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 10.Rateesh S., Luis S.A., Luis C.R., Hughes B., Nicolae M. Myocardial infarction secondary to 5-fluorouracil: not an absolute contraindication to rechallenge? Int. J. Cardiol. 2014;172:e331–e333. doi: 10.1016/j.ijcard.2013.12.274. [DOI] [PubMed] [Google Scholar]
- 11.Ducas R.A., Seftel M.D., Ducas J., Seifer C. Monomorphic ventricular tachycardia caused by arsenic trioxide therapy for acute promyelocytic leukaemia. J. R. Coll. Physicians Edinb. 2011;41:117–118. doi: 10.4997/JRCPE.2011.204. [DOI] [PubMed] [Google Scholar]
- 12.Ohnishi K., Yoshida H., Shigeno K., Nakamura S., Fujisawa S., Naito K., Shinjo K., Fujita Y., Matsui H., Takeshita A. Prolongation of the QT interval and ventricular tachycardia in patients treated with arsenic trioxide for acute promyelocytic leukemia. Ann. Intern. Med. 2000;133:881–885. doi: 10.7326/0003-4819-133-11-200012050-00012. [DOI] [PubMed] [Google Scholar]
- 13.Economopoulou P., Kotsakis A., Kapiris I., Kentepozidis N. Cancer therapy and cardiovascular risk: focus on bevacizumab. Cancer Manag. Res. 2015;7:133–143. doi: 10.2147/CMAR.S77400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meseeha M.G., Kolade V.O., Attia M.N. Partially reversible bortezomib-induced cardiotoxicity: an unusual cause of acute cardiomyopathy. J. Community Hosp. Intern. Med. Perspect. 2015;5:28982. doi: 10.3402/jchimp.v5.28982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao Y., Yin J., Wei J., Shang Z. Incidence and risk of cardiotoxicity associated with bortezomib in the treatment of cancer: a systematic review and meta-analysis. PLoS ONE. 2014;9:e87671. doi: 10.1371/journal.pone.0087671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fukunaga T., Soejima H., Sugamura K., Kojima S., Sugiyama S., Sakamoto T., Yoshimura M., Tanoue T., Kageshita T., Ono T., Ogawa H. Acute myocardial infarction induced by cisplatin-based combination chemotherapy for malignant melanoma: a case report. J. Cardiol. 2006;47:191–195. [PubMed] [Google Scholar]
- 17.Conrad M.E. Cytarabine and cardiac failure. Am. J. Hematol. 1992;41:143–144. doi: 10.1002/ajh.2830410219. [DOI] [PubMed] [Google Scholar]
- 18.Wayangankar S.A., Patel B.C., Parekh H.D., Holter J.L., Lazzara R. High-dose cytosine arabinoside-induced symptomatic bradycardia. J. Cardiovasc. Med. (Hagerstown) 2015;16(Suppl 1):S38–S41. doi: 10.2459/JCM.0b013e328341d0e5. [DOI] [PubMed] [Google Scholar]
- 19.Bloom M.W., Hamo C.E., Cardinale D., Ky B., Nohria A., Baer L., Skopicki H., Lenihan D.J., Gheorghiade M., Lyon A.R., Butler J. Cancer Therapy-Related Cardiac Dysfunction and Heart Failure: Part 1: Definitions, Pathophysiology, Risk Factors, and Imaging. Circ. Heart Fail. 2016;9:e002661. doi: 10.1161/CIRCHEARTFAILURE.115.002661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Druhan L., Fasan O., Copelan O.R. Acute Heart Failure in a Patient with Acute Myeloid Leukemia following Daunorubicin Treatment: a Case Report. J. Leuk. 2015;3:2. [Google Scholar]
- 21.Xu Z., Cang S., Yang T., Liu D. Cardiotoxicity of tyrosine kinase inhibitors in chronic myelogenous leukemia therapy. Hematol. Rev. 2009;1:e4. [Google Scholar]
- 22.Suter T.M., Ewer M.S. Cancer drugs and the heart: importance and management. Eur. Heart J. 2013;34:1102–1111. doi: 10.1093/eurheartj/ehs181. [DOI] [PubMed] [Google Scholar]
- 23.Shimoyama M., Murata Y., Sumi K.I., Hamazoe R., Komuro I. Docetaxel induced cardiotoxicity. Heart. 2001;86:219. doi: 10.1136/heart.86.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitry M.A., Edwards J.G. Doxorubicin induced heart failure: Phenotype and molecular mechanisms. Int. J. Cardiol. Heart Vasc. 2016;10:17–24. doi: 10.1016/j.ijcha.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johnson D.B., Balko J.M., Compton M.L., Chalkias S., Gorham J., Xu Y., Hicks M., Puzanov I., Alexander M.R., Bloomer T.L. Fulminant myocarditis with combination immune checkpoint blockade. N. Engl. J. Med. 2016;375:1749–1755. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Varricchi G., Marone G., Mercurio V., Galdiero M.R., Bonaduce D., Tocchetti C.G. Immune Checkpoint Inhibitors and Cardiac Toxicity: An Emerging Issue. Curr. Med. Chem. 2018;25:1327–1339. doi: 10.2174/0929867324666170407125017. [DOI] [PubMed] [Google Scholar]
- 27.Moy B., Goss P.E. Lapatinib-associated toxicity and practical management recommendations. Oncologist. 2007;12:756–765. doi: 10.1634/theoncologist.12-7-756. [DOI] [PubMed] [Google Scholar]
- 28.Dermitzakis E.V., Kimiskidis V.K., Lazaridis G., Alexopoulou Z., Timotheadou E., Papanikolaou A., Romanidou O., Georgiadis G., Kalogeras K.T., Tsiptsios I. The impact of paclitaxel and carboplatin chemotherapy on the autonomous nervous system of patients with ovarian cancer. BMC Neurol. 2016;16:190. doi: 10.1186/s12883-016-0710-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Esber C., Breathett K., Sachak T., Moore S., Lilly S.M. Acute Myocardial Infarction in Patient With Triple Negative Breast Cancer After Paclitaxel Infusion: A Case Report. Cardiol. Res. 2014;5:108–111. doi: 10.14740/cr325w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivers Z.T., Oostra D.R., Westholder J.S., Vercellotti G.M. Romidepsin-associated cardiac toxicity and ECG changes: A case report and review of the literature. J. Oncol. Pharm. Pract. 2018;24:56–62. doi: 10.1177/1078155216673229. [DOI] [PubMed] [Google Scholar]
- 31.Brose M.S., Frenette C.T., Keefe S.M., Stein S.M. Management of sorafenib-related adverse events: a clinician’s perspective. Semin. Oncol. 2014;41(Suppl 2):S1–S16. doi: 10.1053/j.seminoncol.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 32.Force T., Kolaja K.L. Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 2011;10:111–126. doi: 10.1038/nrd3252. [DOI] [PubMed] [Google Scholar]
- 33.Pondé N.F., Lambertini M., de Azambuja E. Twenty years of anti-HER2 therapy-associated cardiotoxicity. ESMO Open. 2016;1:e000073. doi: 10.1136/esmoopen-2016-000073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zeglinski M., Ludke A., Jassal D.S., Singal P.K. Trastuzumab-induced cardiac dysfunction: A ‘dual-hit’. Exp. Clin. Cardiol. 2011;16:70–74. [PMC free article] [PubMed] [Google Scholar]
- 35.Grande E., Kreissl M.C., Filetti S., Newbold K., Reinisch W., Robert C., Schlumberger M., Tolstrup L.K., Zamorano J.L., Capdevila J. Vandetanib in advanced medullary thyroid cancer: review of adverse event management strategies. Adv. Ther. 2013;30:945–966. doi: 10.1007/s12325-013-0069-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bergeron A., Raffy O., Vannetzel J.M. Myocardial ischemia and infarction associated with vinorelbine. J. Clin. Oncol. 1995;13:531–532. doi: 10.1200/JCO.1995.13.2.531. [DOI] [PubMed] [Google Scholar]
- 37.Sondergaard K.B., Weeke P., Wissenberg M., Schjerning Olsen A.-M., Fosbol E.L., Lippert F.K., Torp-Pedersen C., Gislason G.H., Folke F. Non-steroidal anti-inflammatory drug use is associated with increased risk of out-of-hospital cardiac arrest: a nationwide case-time-control study. Eur. Heart J. Cardiovasc. Pharmacother. 2017;3:100–107. doi: 10.1093/ehjcvp/pvw041. [DOI] [PubMed] [Google Scholar]
- 38.Arfè A., Scotti L., Varas-Lorenzo C., Nicotra F., Zambon A., Kollhorst B., Schink T., Garbe E., Herings R., Straatman H., Safety of Non-steroidal Anti-inflammatory Drugs (SOS) Project Consortium Non-steroidal anti-inflammatory drugs and risk of heart failure in four European countries: nested case-control study. BMJ. 2016;354:i4857. doi: 10.1136/bmj.i4857. [DOI] [PubMed] [Google Scholar]
- 39.Loewen P.S. Review of the selective COX-2 inhibitors celecoxib and rofecoxib: focus on clinical aspects. CJEM. 2002;4:268–275. doi: 10.1017/s1481803500007508. [DOI] [PubMed] [Google Scholar]
- 40.Weill A., Païta M., Tuppin P., Fagot J.-P., Neumann A., Simon D., Ricordeau P., Montastruc J.-L., Allemand H. Benfluorex and valvular heart disease: a cohort study of a million people with diabetes mellitus. Pharmacoepidemiol. Drug Saf. 2010;19:1256–1262. doi: 10.1002/pds.2044. [DOI] [PubMed] [Google Scholar]
- 41.Graf B.M. The cardiotoxicity of local anesthetics: the place of ropivacaine. Curr. Top. Med. Chem. 2001;1:207–214. doi: 10.2174/1568026013395164. [DOI] [PubMed] [Google Scholar]
- 42.Rothman R.B., Ayestas M.A., Dersch C.M., Baumann M.H. Aminorex, fenfluramine, and chlorphentermine are serotonin transporter substrates. Implications for primary pulmonary hypertension. Circulation. 1999;100:869–875. doi: 10.1161/01.cir.100.8.869. [DOI] [PubMed] [Google Scholar]
- 43.Hatton J.L., Bhat P.K., Gandhi S. Clozapine-induced myocarditis: recognizing a potentially fatal adverse reaction. Tex. Heart Inst. J. 2015;42:155–157. doi: 10.14503/THIJ-13-3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Layland J.J., Liew D., Prior D.L. Clozapine-induced cardiotoxicity: a clinical update. Med. J. Aust. 2009;190:190–192. doi: 10.5694/j.1326-5377.2009.tb02345.x. [DOI] [PubMed] [Google Scholar]
- 45.Schwartz B.G., Rezkalla S., Kloner R.A. Cardiovascular effects of cocaine. Circulation. 2010;122:2558–2569. doi: 10.1161/CIRCULATIONAHA.110.940569. [DOI] [PubMed] [Google Scholar]
- 46.Cannistra L.B., Davis S.M., Bauman A.G. Valvular heart disease associated with dexfenfluramine. N. Engl. J. Med. 1997;337:636. doi: 10.1056/nejm199708283370912. [DOI] [PubMed] [Google Scholar]
- 47.Malak J., Deprez P., Adam J.F. Valve disease associated with chronic intake of fenfluramines. J. Lab. Clin. Med. 1998;131:475. doi: 10.1016/s0022-2143(98)90149-4. [DOI] [PubMed] [Google Scholar]
- 48.Andrejak M., Tribouilloy C. Drug-induced valvular heart disease: an update. Arch. Cardiovasc. Dis. 2013;106:333–339. doi: 10.1016/j.acvd.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 49.Connolly H.M., Crary J.L., McGoon M.D., Hensrud D.D., Edwards B.S., Edwards W.D., Schaff H.V. Valvular heart disease associated with fenfluramine-phentermine. N. Engl. J. Med. 1997;337:581–588. doi: 10.1056/NEJM199708283370901. [DOI] [PubMed] [Google Scholar]
- 50.Kurdyak P.A., Manno M., Gomes T., Mamdani M.M., Juurlink D.N. Antidepressants, metoprolol and the risk of bradycardia. Ther. Adv. Psychopharmacol. 2012;2:43–49. doi: 10.1177/2045125311433580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beach S.R., Celano C.M., Noseworthy P.A., Januzzi J.L., Huffman J.C. QTc prolongation, torsades de pointes, and psychotropic medications. Psychosomatics. 2013;54:1–13. doi: 10.1016/j.psym.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 52.Wieneke H., Conrads H., Wolstein J., Breuckmann F., Gastpar M., Erbel R., Scherbaum N. Levo-alpha-acetylmethadol (LAAM) induced QTc-prolongation—results from a controlled clinical trial. Eur. J. Med. Res. 2009;14:7–12. doi: 10.1186/2047-783X-14-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horáček M., Vymazal T. Lidocaine not so innocent: Cardiotoxicity after topical anaesthesia for bronchoscopy. Indian J. Anaesth. 2012;56:95–96. doi: 10.4103/0019-5049.93362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Elsaie M.L. Cardiovascular collapse developing after topical anesthesia. Dermatology. 2007;214:194. doi: 10.1159/000098877. [DOI] [PubMed] [Google Scholar]
- 55.Antonini A., Poewe W. Fibrotic heart-valve reactions to dopamine-agonist treatment in Parkinson’s disease. Lancet Neurol. 2007;6:826–829. doi: 10.1016/S1474-4422(07)70218-1. [DOI] [PubMed] [Google Scholar]
- 56.Adler A., Viskin S., Bhuiyan Z.A., Eisenberg E., Rosso R. Propoxyphene-induced torsades de pointes. Heart Rhythm. 2011;8:1952–1954. doi: 10.1016/j.hrthm.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 57.Leucht S., Cipriani A., Spineli L., Mavridis D., Orey D., Richter F., Samara M., Barbui C., Engel R.R., Geddes J.R. Comparative efficacy and tolerability of 15 antipsychotic drugs in schizophrenia: a multiple-treatments meta-analysis. Lancet. 2013;382:951–962. doi: 10.1016/S0140-6736(13)60733-3. [DOI] [PubMed] [Google Scholar]
- 58.Yun J., Chung E., Choi K.H., Cho D.H., Song Y.J., Han K.M., Cha H.J., Shin J.S., Seong W.K., Kim Y.H., Kim H.S. Cardiovascular safety pharmacology of sibutramine. Biomol. Ther. (Seoul) 2015;23:386–389. doi: 10.4062/biomolther.2015.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eroglu E., Gemici G., Bayrak F., Kalkan A.K., Degertekin M. Acute myocardial infarction in a 24 year-old man possibly associated with sibutramine use. Int. J. Cardiol. 2009;137:e43–e45. doi: 10.1016/j.ijcard.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 60.James W.P.T., Caterson I.D., Coutinho W., Finer N., Van Gaal L.F., Maggioni A.P., Torp-Pedersen C., Sharma A.M., Shepherd G.M., Rode R.A., Renz C.L., SCOUT Investigators Effect of sibutramine on cardiovascular outcomes in overweight and obese subjects. N. Engl. J. Med. 2010;363:905–917. doi: 10.1056/NEJMoa1003114. [DOI] [PubMed] [Google Scholar]
- 61.Howell C., Wilson A.D., Waring W.S. Cardiovascular toxicity due to venlafaxine poisoning in adults: a review of 235 consecutive cases. Br. J. Clin. Pharmacol. 2007;64:192–197. doi: 10.1111/j.1365-2125.2007.02849.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson E.M., Whyte E., Mulsant B.H., Pollock B.G., Weber E., Begley A.E., Reynolds C.F. Cardiovascular changes associated with venlafaxine in the treatment of late-life depression. Am. J. Geriatr. Psychiatry. 2006;14:796–802. doi: 10.1097/01.JGP.0000204328.50105.b3. [DOI] [PubMed] [Google Scholar]
- 63.Quigley E.M.M. Prokinetics in the Management of Functional Gastrointestinal Disorders. Curr. Gastroenterol. Rep. 2017;19:53. doi: 10.1007/s11894-017-0593-6. [DOI] [PubMed] [Google Scholar]
- 64.Upadhyay A., Bodar V., Malekzadegan M., Singh S., Frumkin W., Mangla A., Doshi K. Loperamide Induced Life Threatening Ventricular Arrhythmia. Case Rep. Cardiol. 2016;2016:5040176. doi: 10.1155/2016/5040176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swank K.A., Wu E., Kortepeter C., McAninch J., Levin R.L. Adverse event detection using the FDA post-marketing drug safety surveillance system: Cardiotoxicity associated with loperamide abuse and misuse. J Am Pharm Assoc (2003) 2017;57(2S):S63–S67. doi: 10.1016/j.japh.2016.11.011. [DOI] [PubMed] [Google Scholar]
- 66.Juurlink D.N., Dormuth C.R., Huang A., Hellings C., Paterson J.M., Raymond C., Kozyrskyj A., Moride Y., Macdonald E.M., Mamdani M.M. Proton pump inhibitors and the risk of adverse cardiac events. PLoS ONE. 2013;8:e84890. doi: 10.1371/journal.pone.0084890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Food and Drug Administration . Basel; 2007. Drug Safety and Availability—Zelnorm (tegaserod maleate)https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/021200s014lbl.pdf [Google Scholar]
- 68.Thomas S.H., Higham P.D., Hartigan-Go K., Kamali F., Wood P., Campbell R.W., Ford G.A. Concentration dependent cardiotoxicity of terodiline in patients treated for urinary incontinence. Br. Heart J. 1995;74:53–56. doi: 10.1136/hrt.74.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stewart D.A., Taylor J., Ghosh S., Macphee G.J., Abdullah I., McLenachan J.M., Stott D.J. Terodiline causes polymorphic ventricular tachycardia due to reduced heart rate and prolongation of QT interval. Eur. J. Clin. Pharmacol. 1992;42:577–580. doi: 10.1007/BF00265918. [DOI] [PubMed] [Google Scholar]
- 70.Jo S.-H., Hong H.-K., Chong S.H., Lee H.S., Choe H. H(1) antihistamine drug promethazine directly blocks hERG K(+) channel. Pharmacol. Res. 2009;60:429–437. doi: 10.1016/j.phrs.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 71.Bishop R.O., Gaudry P.L. Prolonged Q-T interval following astemizole overdose. Arch. Emerg. Med. 1989;6:63–65. doi: 10.1136/emj.6.1.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khalifa M., Daleau P., Turgeon J. Mechanism of sodium channel block by venlafaxine in guinea pig ventricular myocytes. J. Pharmacol. Exp. Ther. 1999;291:280–284. [PubMed] [Google Scholar]
- 73.Shah A., Yousuf T., Ziffra J., Zaidi A., Raghuvir R. Diphenhydramine and QT prolongation—A rare cardiac side effect of a drug used in common practice. J. Cardiol. Cases. 2015;12:126–129. doi: 10.1016/j.jccase.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Monahan B.P., Ferguson C.L., Killeavy E.S., Lloyd B.K., Troy J., Cantilena L.R., Jr. Torsades de pointes occurring in association with terfenadine use. JAMA. 1990;264:2788–2790. [PubMed] [Google Scholar]
- 75.Carr A., Cooper D.A. Adverse effects of antiretroviral therapy. Lancet. 2000;356:1423–1430. doi: 10.1016/S0140-6736(00)02854-3. [DOI] [PubMed] [Google Scholar]
- 76.Ray W.A., Murray K.T., Hall K., Arbogast P.G., Stein C.M. Azithromycin and the risk of cardiovascular death. N. Engl. J. Med. 2012;366:1881–1890. doi: 10.1056/NEJMoa1003833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Berni E., de Voogd H., Halcox J.P., Butler C.C., Bannister C.A., Jenkins-Jones S., Jones B., Ouwens M., Currie C.J. Risk of cardiovascular events, arrhythmia and all-cause mortality associated with clarithromycin versus alternative antibiotics prescribed for respiratory tract infections: a retrospective cohort study. BMJ Open. 2017;7:e013398. doi: 10.1136/bmjopen-2016-013398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wong A.Y.S., Root A., Douglas I.J., Chui C.S.L., Chan E.W., Ghebremichael-Weldeselassie Y., Siu C.W., Smeeth L., Wong I.C.K. Cardiovascular outcomes associated with use of clarithromycin: population based study. BMJ. 2016;352:h6926. doi: 10.1136/bmj.h6926. [DOI] [PubMed] [Google Scholar]
- 79.Anderson M.E., Mazur A., Yang T., Roden D.M. Potassium current antagonist properties and proarrhythmic consequences of quinolone antibiotics. J. Pharmacol. Exp. Ther. 2001;296:806–810. [PubMed] [Google Scholar]
- 80.Durante-Mangoni E., Parrella A., Vitrone M., Rago A., Pafundi P.C., Nigro G., Utili R., Russo V. Electrophysiological Adverse Effects of Direct Acting Antivirals in Patients With Chronic Hepatitis C. J. Clin. Pharmacol. 2017;57:924–930. doi: 10.1002/jcph.872. [DOI] [PubMed] [Google Scholar]
- 81.Dennis A.T., Wang L., Wan H., Nassal D., Deschenes I., Ficker E. Molecular determinants of pentamidine-induced hERG trafficking inhibition. Mol. Pharmacol. 2012;81:198–209. doi: 10.1124/mol.111.075135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haverkamp W., Breithardt G., Camm A.J., Janse M.J., Rosen M.R., Antzelevitch C., Escande D., Franz M., Malik M., Moss A., Shah R. The potential for QT prolongation and pro-arrhythmia by non-anti-arrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Cardiovasc. Res. 2000;47:219–233. doi: 10.1016/s0008-6363(00)00119-x. [DOI] [PubMed] [Google Scholar]
- 83.Katchman A.N., Koerner J., Tosaka T., Woosley R.L., Ebert S.N. Comparative evaluation of HERG currents and QT intervals following challenge with suspected torsadogenic and nontorsadogenic drugs. J. Pharmacol. Exp. Ther. 2006;316:1098–1106. doi: 10.1124/jpet.105.093393. [DOI] [PubMed] [Google Scholar]
- 84.Gibson J.K., Yue Y., Bronson J., Palmer C., Numann R. Human stem cell-derived cardiomyocytes detect drug-mediated changes in action potentials and ion currents. J. Pharmacol. Toxicol. Methods. 2014;70:255–267. doi: 10.1016/j.vascn.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 85.Grenadier E., Keidar S., Alpan G., Marmor A., Palant A. Prenylamine-induced ventricular tachycardia and syncope controlled by ventricular pacing. Br. Heart J. 1980;44:330–334. doi: 10.1136/hrt.44.3.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Legras A., Piquemal R., Furet Y., Dequin P.F., Perrotin D. Buflomedil poisoning: five cases with cardiotoxicity. Intensive Care Med. 1996;22:57–61. doi: 10.1007/BF01728332. [DOI] [PubMed] [Google Scholar]
- 87.Shantsila E., Watson T., Lip G.Y.H. Drug-induced QT-interval prolongation and proarrhythmic risk in the treatment of atrial arrhythmias. Europace. 2007;9(Suppl 4):iv37–iv44. doi: 10.1093/europace/eum169. [DOI] [PubMed] [Google Scholar]
- 88.Echt D.S., Liebson P.R., Mitchell L.B., Peters R.W., Obias-Manno D., Barker A.H., Arensberg D., Baker A., Friedman L., Greene H.L. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N. Engl. J. Med. 1991;324:781–788. doi: 10.1056/NEJM199103213241201. [DOI] [PubMed] [Google Scholar]
- 89.Hanley S.P., Hampton J.R. Ventricular arrhythmias associated with lidoflazine: side-effects observed in a randomized trial. Eur. Heart J. 1983;4:889–893. doi: 10.1093/oxfordjournals.eurheartj.a061417. [DOI] [PubMed] [Google Scholar]
- 90.Billups S.J., Carter B.L. Mibefradil withdrawn from the market. Ann. Pharmacother. 1998;32:841. doi: 10.1345/aph.17356. [DOI] [PubMed] [Google Scholar]
- 91.Meinertz T. Mibefradil—a drug which may enhance the propensity for the development of abnormal QT prolongation. Eur. Heart J. 2001;3(Suppl.):K89–K92. [Google Scholar]
- 92.Medicines and Healthcare products Regulatory Agency . 2009. Orciprenaline sulphate (Alupent): withdrawal due to unfavourable benefit-risk profile.https://www.gov.uk/drug-safety-update/orciprenaline-sulphate-alupent-withdrawal-due-to-unfavourable-benefit-risk-profile [Google Scholar]
- 93.White W.B., Cannon C.P., Heller S.R., Nissen S.E., Bergenstal R.M., Bakris G.L., Perez A.T., Fleck P.R., Mehta C.R., Kupfer S., EXAMINE Investigators Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N. Engl. J. Med. 2013;369:1327–1335. doi: 10.1056/NEJMoa1305889. [DOI] [PubMed] [Google Scholar]
- 94.Takahara A., Sasaki R., Nakamura M., Sendo A., Sakurai Y., Namekata I., Tanaka H. Clobutinol delays ventricular repolarization in the guinea pig heart: comparison with cardiac effects of HERG K+ channel inhibitor E-4031. J. Cardiovasc. Pharmacol. 2009;54:552–559. doi: 10.1097/FJC.0b013e3181bfb17c. [DOI] [PubMed] [Google Scholar]
- 95.Nissen S.E., Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N. Engl. J. Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 96.Scirica B.M., Bhatt D.L., Braunwald E., Steg P.G., Davidson J., Hirshberg B., Ohman P., Frederich R., Wiviott S.D., Hoffman E.B., SAVOR-TIMI 53 Steering Committee and Investigators Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013;369:1317–1326. doi: 10.1056/NEJMoa1307684. [DOI] [PubMed] [Google Scholar]
- 97.Friedrichs G.S., Patmore L., Bass A. Non-clinical evaluation of ventricular repolarization (ICH S7B): results of an interim survey of international pharmaceutical companies. J. Pharmacol. Toxicol. Methods. 2005;52:6–11. doi: 10.1016/j.vascn.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 98.Babcock J.J., Li M. hERG channel function: beyond long QT. Acta Pharmacol. Sin. 2013;34:329–335. doi: 10.1038/aps.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Arcangeli A., Becchetti A. hERG Channels: From Antitargets to Novel Targets for Cancer Therapy. Clin. Cancer Res. 2017;23:3–5. doi: 10.1158/1078-0432.CCR-16-2322. [DOI] [PubMed] [Google Scholar]
- 100.Mirams G.R., Cui Y., Sher A., Fink M., Cooper J., Heath B.M., McMahon N.C., Gavaghan D.J., Noble D. Simulation of multiple ion channel block provides improved early prediction of compounds’ clinical torsadogenic risk. Cardiovasc. Res. 2011;91:53–61. doi: 10.1093/cvr/cvr044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Passini E., Britton O.J., Lu H.R., Rohrbacher J., Hermans A.N., Gallacher D.J., Greig R.J.H., Bueno-Orovio A., Rodriguez B. Human in silico drug trials demonstrate higher accuracy than animal models in predicting clinical pro-arrhythmic cardiotoxicity. Front. Physiol. 2017;8:668. doi: 10.3389/fphys.2017.00668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Britton O.J., Bueno-Orovio A., Virág L., Varró A., Rodriguez B. The electrogenic Na+/K+ pump is a key determinant of repolarization abnormality susceptibility in human ventricular cardiomyocytes: A population-based simulation study. Front. Physiol. 2017;8:278. doi: 10.3389/fphys.2017.00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Turan B., Dhalla S.N. Springer Science & Business Media; 2014. Diabetic Cardiomyopathy. [Google Scholar]
- 104.Ficker E., Kuryshev Y.A., Dennis A.T., Obejero-Paz C., Wang L., Hawryluk P., Wible B.A., Brown A.M. Mechanisms of arsenic-induced prolongation of cardiac repolarization. Mol. Pharmacol. 2004;66:33–44. doi: 10.1124/mol.66.1.33. [DOI] [PubMed] [Google Scholar]
- 105.Zhang X., Yu D., Geng H., Li F., Lv L., Zhao L., Yan C., Li B. Dual effects of arsenic trioxide on tumor cells and the potential underlying mechanisms. Oncol. Lett. 2018;16:3812–3820. doi: 10.3892/ol.2018.9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yu D., Lv L., Fang L., Zhang B., Wang J., Zhan G., Zhao L., Zhao X., Li B. Inhibitory effects and mechanism of dihydroberberine on hERG channels expressed in HEK293 cells. PLoS ONE. 2017;12:e0181823. doi: 10.1371/journal.pone.0181823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang K., Zhi D., Huang T., Gong Y., Yan M., Liu C., Wei T., Dong Z., Li B., Yang B. Berberine induces hERG channel deficiency through trafficking inhibition. Cell. Physiol. Biochem. 2014;34:691–702. doi: 10.1159/000363034. [DOI] [PubMed] [Google Scholar]
- 108.Weng C.-Y., Chiou S.-Y., Wang L., Kou M.-C., Wang Y.-J., Wu M.-J. Arsenic trioxide induces unfolded protein response in vascular endothelial cells. Arch. Toxicol. 2014;88:213–226. doi: 10.1007/s00204-013-1101-x. [DOI] [PubMed] [Google Scholar]
- 109.Wang L., Wible B.A., Wan X., Ficker E. Cardiac glycosides as novel inhibitors of human ether-a-go-go-related gene channel trafficking. J. Pharmacol. Exp. Ther. 2007;320:525–534. doi: 10.1124/jpet.106.113043. [DOI] [PubMed] [Google Scholar]
- 110.Apaja P.M., Foo B., Okiyoneda T., Valinsky W.C., Barriere H., Atanasiu R., Ficker E., Lukacs G.L., Shrier A. Ubiquitination-dependent quality control of hERG K+ channel with acquired and inherited conformational defect at the plasma membrane. Mol. Biol. Cell. 2013;24:3787–3804. doi: 10.1091/mbc.E13-07-0417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hubbi M.E., Hu H., Kshitiz, Ahmed I., Levchenko A., Semenza G.L. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J. Biol. Chem. 2013;288:10703–10714. doi: 10.1074/jbc.M112.414771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Guo J., Massaeli H., Li W., Xu J., Luo T., Shaw J., Kirshenbaum L.A., Zhang S. Identification of IKr and its trafficking disruption induced by probucol in cultured neonatal rat cardiomyocytes. J. Pharmacol. Exp. Ther. 2007;321:911–920. doi: 10.1124/jpet.107.120931. [DOI] [PubMed] [Google Scholar]
- 113.Shi Y.-Q., Yan C.-C., Zhang X., Yan M., Liu L.-R., Geng H.-Z., Lv L., Li B.-X. Mechanisms underlying probucol-induced hERG-channel deficiency. Drug Des. Devel. Ther. 2015;9:3695–3704. doi: 10.2147/DDDT.S86724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spence S., Deurinck M., Ju H., Traebert M., McLean L., Marlowe J., Emotte C., Tritto E., Tseng M., Shultz M., Friedrichs G.S. Histone Deacetylase Inhibitors Prolong Cardiac Repolarization through Transcriptional Mechanisms. Toxicol. Sci. 2016;153:39–54. doi: 10.1093/toxsci/kfw104. [DOI] [PubMed] [Google Scholar]
- 115.Takemasa H., Nagatomo T., Abe H., Kawakami K., Igarashi T., Tsurugi T., Kabashima N., Tamura M., Okazaki M., Delisle B.P. Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome. Br. J. Pharmacol. 2008;153:439–447. doi: 10.1038/sj.bjp.0707537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Han S.N., Sun X.Y., Zhang Z., Zhang L.R. The protease inhibitor atazanavir blocks hERG K(+) channels expressed in HEK293 cells and obstructs hERG protein transport to cell membrane. Acta Pharmacol. Sin. 2015;36:454–462. doi: 10.1038/aps.2014.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rajamani S., Eckhardt L.L., Valdivia C.R., Klemens C.A., Gillman B.M., Anderson C.L., Holzem K.M., Delisle B.P., Anson B.D., Makielski J.C., January C.T. Drug-induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br. J. Pharmacol. 2006;149:481–489. doi: 10.1038/sj.bjp.0706892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Dennis A.T., Nassal D., Deschenes I., Thomas D., Ficker E. Antidepressant-induced ubiquitination and degradation of the cardiac potassium channel hERG. J. Biol. Chem. 2011;286:34413–34425. doi: 10.1074/jbc.M111.254367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sun H., Liu X., Xiong Q., Shikano S., Li M. Chronic inhibition of cardiac Kir2.1 and HERG potassium channels by celastrol with dual effects on both ion conductivity and protein trafficking. J. Biol. Chem. 2006;281:5877–5884. doi: 10.1074/jbc.M600072200. [DOI] [PubMed] [Google Scholar]
- 120.Mustroph J., Neef S., Maier L.S. CaMKII as a target for arrhythmia suppression. Pharmacol. Ther. 2017;176:22–31. doi: 10.1016/j.pharmthera.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 121.Hood A.R., Ai X., Pogwizd S.M. Regulation of cardiac gap junctions by protein phosphatases. J. Mol. Cell. Cardiol. 2017;107:52–57. doi: 10.1016/j.yjmcc.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wasserstrom J.A., Aistrup G.L. Digitalis: new actions for an old drug. Am. J. Physiol. Heart Circ. Physiol. 2005;289:H1781–H1793. doi: 10.1152/ajpheart.00707.2004. [DOI] [PubMed] [Google Scholar]
- 123.Karagueuzian H.S., Katzung B.G. Relative inotropic and arrhythmogenic effects of five cardiac steroids in ventricular myocardium: oscillatory afterpotentials and the role of endogenous catecholamines. J. Pharmacol. Exp. Ther. 1981;218:348–356. [PubMed] [Google Scholar]
- 124.Yu L.-Y., Sun L.-N., Zhang X.-H., Li Y.-Q., Yu L., Yuan Z.-Q.-Y., Meng L., Zhang H.-W., Wang Y.-Q. A Review of the Novel Application and Potential Adverse Effects of Proton Pump Inhibitors. Adv. Ther. 2017;34:1070–1086. doi: 10.1007/s12325-017-0532-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mooney L., Skinner M., Coker S.J., Currie S. Effects of acute and chronic sunitinib treatment on cardiac function and calcium/calmodulin-dependent protein kinase II. Br. J. Pharmacol. 2015;172:4342–4354. doi: 10.1111/bph.13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rosano G.M.C., Fini M., Caminiti G., Barbaro G. Cardiac metabolism in myocardial ischemia. Curr. Pharm. Des. 2008;14:2551–2562. doi: 10.2174/138161208786071317. [DOI] [PubMed] [Google Scholar]
- 127.Williams L.J., Nye B.G., Wende A.R. Diabetes-Related Cardiac Dysfunction. Endocrinol. Metab. (Seoul) 2017;32:171–179. doi: 10.3803/EnM.2017.32.2.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Eisner V., Cupo R.R., Gao E., Csordás G., Slovinsky W.S., Paillard M., Cheng L., Ibetti J., Chen S.R.W., Chuprun J.K. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc. Natl. Acad. Sci. USA. 2017;114:E859–E868. doi: 10.1073/pnas.1617288114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ott M., Gogvadze V., Orrenius S., Zhivotovsky B. Mitochondria, oxidative stress and cell death. Apoptosis. 2007;12:913–922. doi: 10.1007/s10495-007-0756-2. [DOI] [PubMed] [Google Scholar]
- 130.Youle R.J., van der Bliek A.M. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Varga Z.V., Ferdinandy P., Liaudet L., Pacher P. Drug-induced mitochondrial dysfunction and cardiotoxicity. Am. J. Physiol. Heart Circ. Physiol. 2015;309:H1453–H1467. doi: 10.1152/ajpheart.00554.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.McGowan J.V., Chung R., Maulik A., Piotrowska I., Walker J.M., Yellon D.M. Anthracycline Chemotherapy and Cardiotoxicity. Cardiovasc. Drugs Ther. 2017;31:63–75. doi: 10.1007/s10557-016-6711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lewis W., Day B.J., Copeland W.C. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat. Rev. Drug Discov. 2003;2:812–822. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- 134.Tsuchiya H., Mizogami M. Interaction of local anesthetics with biomembranes consisting of phospholipids and cholesterol: mechanistic and clinical implications for anesthetic and cardiotoxic effects. Anesthesiol. Res. Pract. 2013;2013:297141. doi: 10.1155/2013/297141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Taimeh Z., Loughran J., Birks E.J., Bolli R. Vascular endothelial growth factor in heart failure. Nat. Rev. Cardiol. 2013;10:519–530. doi: 10.1038/nrcardio.2013.94. [DOI] [PubMed] [Google Scholar]
- 136.Zhao T., Zhao W., Meng W., Liu C., Chen Y., Gerling I.C., Weber K.T., Bhattacharya S.K., Kumar R., Sun Y. VEGF-C/VEGFR-3 pathway promotes myocyte hypertrophy and survival in the infarcted myocardium. Am. J. Transl. Res. 2015;7:697–709. [PMC free article] [PubMed] [Google Scholar]
- 137.Quesada A.R., Medina M.Á. Angiogenesis in cancer: Therapeutic targets and angiogenesis inhibitors. In: Chatterjee M., editor. Angiogenesis & Therapeutic Targets In Cancer. Bentham Science Publishers; 2010. pp. 45–67. [Google Scholar]
- 138.Chen Z.I., Ai D.I. Cardiotoxicity associated with targeted cancer therapies. Mol. Clin. Oncol. 2016;4:675–681. doi: 10.3892/mco.2016.800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bloomekatz J., Singh R., Prall O.W., Dunn A.C., Vaughan M., Loo C.-S., Harvey R.P., Yelon D. Platelet-derived growth factor (PDGF) signaling directs cardiomyocyte movement toward the midline during heart tube assembly. eLife. 2017;6:e21172. doi: 10.7554/eLife.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chintalgattu V., Ai D., Langley R.R., Zhang J., Bankson J.A., Shih T.L., Reddy A.K., Coombes K.R., Daher I.N., Pati S. Cardiomyocyte PDGFR-beta signaling is an essential component of the mouse cardiac response to load-induced stress. J. Clin. Invest. 2010;120:472–484. doi: 10.1172/JCI39434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gallini R., Lindblom P., Bondjers C., Betsholtz C., Andrae J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 2016;349:282–290. doi: 10.1016/j.yexcr.2016.10.022. [DOI] [PubMed] [Google Scholar]
- 142.Negro A., Brar B.K., Lee K.-F. Essential roles of Her2/erbB2 in cardiac development and function. Recent Prog. Horm. Res. 2004;59:1–12. doi: 10.1210/rp.59.1.1. [DOI] [PubMed] [Google Scholar]
- 143.Makki N., Thiel K.W., Miller F.J., Jr. The epidermal growth factor receptor and its ligands in cardiovascular disease. Int. J. Mol. Sci. 2013;14:20597–20613. doi: 10.3390/ijms141020597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Toyama E.Q., Herzig S., Courchet J., Lewis T.L., Jr., Losón O.C., Hellberg K., Young N.P., Chen H., Polleux F., Chan D.C. Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science. 2016;351:275–281. doi: 10.1126/science.aab4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Geisberg C.A., Sawyer D.B. Mechanisms of anthracycline cardiotoxicity and strategies to decrease cardiac damage. Curr. Hypertens. Rep. 2010;12:404–410. doi: 10.1007/s11906-010-0146-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Iorio-Morin C., Germain P., Roy S., Génier S., Labrecque P., Parent J.-L. Thromboxane A2 modulates cisplatin-induced apoptosis through a Siva1-dependent mechanism. Cell Death Differ. 2012;19:1347–1357. doi: 10.1038/cdd.2012.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Togna G.I., Togna A.R., Franconi M., Caprino L. Cisplatin triggers platelet activation. Thromb. Res. 2000;99:503–509. doi: 10.1016/s0049-3848(00)00294-2. [DOI] [PubMed] [Google Scholar]
- 148.McQuade R.M., Stojanovska V., Abalo R., Bornstein J.C., Nurgali K. Chemotherapy-Induced Constipation and Diarrhea: Pathophysiology, Current and Emerging Treatments. Front. Pharmacol. 2016;7:414. doi: 10.3389/fphar.2016.00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kucharz J., Michalowska-Kaczmarczyk A., Zygulska A.L., Wojtak J., Pawlik W., Herman R.M., Krzemieniecki K. Bradycardia as a rare symptom of cisplatin cardiotoxicity: A case report. Oncol. Lett. 2016;11:2297–2299. doi: 10.3892/ol.2016.4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mikaelian I., Buness A., de Vera-Mudry M.-C., Kanwal C., Coluccio D., Rasmussen E., Char H.W., Carvajal V., Hilton H., Funk J. Primary endothelial damage is the mechanism of cardiotoxicity of tubulin-binding drugs. Toxicol. Sci. 2010;117:144–151. doi: 10.1093/toxsci/kfq189. [DOI] [PubMed] [Google Scholar]
- 151.Doherty K.R., Talbert D.R., Trusk P.B., Moran D.M., Shell S.A., Bacus S. Structural and functional screening in human induced-pluripotent stem cell-derived cardiomyocytes accurately identifies cardiotoxicity of multiple drug types. Toxicol. Appl. Pharmacol. 2015;285:51–60. doi: 10.1016/j.taap.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 152.Moslehi J.J., Deininger M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J. Clin. Oncol. 2015;33:4210–4218. doi: 10.1200/JCO.2015.62.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lancellotti P., Zamorano J.L., Galderisi M. Academic Press, Elsevier; 2016. Anticancer treatments and cardiotoxicity. Mechanisms, diagnostic and therapeutic interventions. [Google Scholar]
- 154.Winnik S., Auwerx J., Sinclair D.A., Matter C.M. Protective effects of sirtuins in cardiovascular diseases: from bench to bedside. Eur. Heart J. 2015;36:3404–3412. doi: 10.1093/eurheartj/ehv290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Nowis D., Ma̧czewski M., Mackiewicz U., Kujawa M., Ratajska A., Wieckowski M.R., Wilczyński G.M., Malinowska M., Bil J., Salwa P. Cardiotoxicity of the anticancer therapeutic agent bortezomib. Am. J. Pathol. 2010;176:2658–2668. doi: 10.2353/ajpath.2010.090690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kourie H.R., Klastersky J. Immune checkpoint inhibitors side effects and management. Immunotherapy. 2016;8:799–807. doi: 10.2217/imt-2016-0029. [DOI] [PubMed] [Google Scholar]
- 157.Yan M., Feng L., Shi Y., Wang J., Liu Y., Li F., Li B. Mechanism of As2O3-Induced Action Potential Prolongation and Using hiPS-CMs to Evaluate the Rescue Efficacy of Drugs With Different Rescue Mechanism. Toxicol. Sci. 2017;158:379–390. doi: 10.1093/toxsci/kfx098. [DOI] [PubMed] [Google Scholar]
- 158.Christie L.E., Picard J., Weinberg G.L. Local anaesthetic systemic toxicity. BJA Educ. 2015;15:136–142. [Google Scholar]
- 159.Thanacoody H.K.R., Thomas S.H.L. Tricyclic antidepressant poisoning: cardiovascular toxicity. Toxicol. Rev. 2005;24:205–214. doi: 10.2165/00139709-200524030-00013. [DOI] [PubMed] [Google Scholar]
- 160.Pacher P., Kecskemeti V. Cardiovascular side effects of new antidepressants and antipsychotics: new drugs, old concerns? Curr. Pharm. Des. 2004;10:2463–2475. doi: 10.2174/1381612043383872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Lenkey N., Karoly R., Lukacs P., Vizi E.S., Sunesen M., Fodor L., Mike A. Classification of drugs based on properties of sodium channel inhibition: a comparative automated patch-clamp study. PLoS ONE. 2010;5:e15568. doi: 10.1371/journal.pone.0015568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Foguet-Boreu Q., Fernandez San Martin M.I., Flores Mateo G., Zabaleta Del Olmo E., Ayerbe García-Morzon L., Perez-Piñar López M., Martin-López L.M., Montes Hidalgo J., Violán C. Cardiovascular risk assessment in patients with a severe mental illness: a systematic review and meta-analysis. BMC Psychiatry. 2016;16:141. doi: 10.1186/s12888-016-0833-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lindström E., Farde L., Eberhard J., Haverkamp W. QTc interval prolongation and antipsychotic drug treatments: focus on sertindole. Int. J. Neuropsychopharmacol. 2005;8:615–629. doi: 10.1017/S1461145705005250. [DOI] [PubMed] [Google Scholar]
- 164.Yang T., Chun Y.W., Stroud D.M., Mosley J.D., Knollmann B.C., Hong C., Roden D.M. Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation. 2014;130:224–234. doi: 10.1161/CIRCULATIONAHA.113.007765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Nasrallah H.A. Atypical antipsychotic-induced metabolic side effects: insights from receptor-binding profiles. Mol. Psychiatry. 2008;13:27–35. doi: 10.1038/sj.mp.4002066. [DOI] [PubMed] [Google Scholar]
- 166.Janssen W., Schymura Y., Novoyatleva T., Kojonazarov B., Boehm M., Wietelmann A., Luitel H., Murmann K., Krompiec D.R., Tretyn A. 5-HT2B receptor antagonists inhibit fibrosis and protect from RV heart failure. BioMed Res. Int. 2015;2015:438403. doi: 10.1155/2015/438403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Arana A., Margulis A.V., McQuay L.J., Ziemiecki R., Bartsch J.L., Rothman K.J., Franks B., D’Silva M., Appenteng K., Varas-Lorenzo C., Perez-Gutthann S. Variation in Cardiovascular Risk Related to Individual Antimuscarinic Drugs Used to Treat Overactive Bladder: A UK Cohort Study. Pharmacotherapy. 2018;38:628–637. doi: 10.1002/phar.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lin J., Guo J., Gang H., Wojciechowski P., Wigle J.T., Zhang S. Intracellular K+ is required for the inactivation-induced high-affinity binding of cisapride to HERG channels. Mol. Pharmacol. 2005;68:855–865. doi: 10.1124/mol.105.012278. [DOI] [PubMed] [Google Scholar]
- 169.Tack J., Camilleri M., Chang L., Chey W.D., Galligan J.J., Lacy B.E., Müller-Lissner S., Quigley E.M.M., Schuurkes J., De Maeyer J.H., Stanghellini V. Systematic review: cardiovascular safety profile of 5-HT(4) agonists developed for gastrointestinal disorders. Aliment. Pharmacol. Ther. 2012;35:745–767. doi: 10.1111/j.1365-2036.2012.05011.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Hazarika S., Annex B.H. Can Endothelial Injury Provide the Passage to Explain the Vascular Effects of Proton Pump Inhibitors? Circ. Res. 2016;118:1858–1860. doi: 10.1161/CIRCRESAHA.116.308978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mason R.P., Walter M.F., Day C.A., Jacob R.F. A biological rationale for the cardiotoxic effects of rofecoxib: comparative analysis with other COX-2 selective agents and NSAids. Subcell. Biochem. 2007;42:175–190. [PubMed] [Google Scholar]
- 172.Chandra M., Kent P.J. Cyclo-oxygenase-2 Inhibitors and Peripheral Thrombosis—A Case Report Demonstrating a Possible Adverse Effect. EJVES Extra. 2009;17:14–16. [Google Scholar]
- 173.Carnieto A., Jr., Dourado P.M.M., Luz P.L., Chagas A.C.P. Selective cyclooxygenase-2 inhibition protects against myocardial damage in experimental acute ischemia. Clinics (São Paulo) 2009;64:245–252. doi: 10.1590/S1807-59322009000300016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Keserü G.M. Prediction of hERG potassium channel affinity by traditional and hologram qSAR methods. Bioorg. Med. Chem. Lett. 2003;13:2773–2775. doi: 10.1016/s0960-894x(03)00492-x. [DOI] [PubMed] [Google Scholar]
- 175.Wang X., Ryu D., Houtkooper R.H., Auwerx J. Antibiotic use and abuse: a threat to mitochondria and chloroplasts with impact on research, health, and environment. BioEssays. 2015;37:1045–1053. doi: 10.1002/bies.201500071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Ridley J.M., Dooley P.C., Milnes J.T., Witchel H.J., Hancox J.C. Lidoflazine is a high affinity blocker of the HERG K(+)channel. J. Mol. Cell. Cardiol. 2004;36:701–705. doi: 10.1016/j.yjmcc.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 177.Anderson J.L., Platia E.V., Hallstrom A., Henthorn R.W., Buckingham T.A., Carlson M.D., Carson P.E. Interaction of baseline characteristics with the hazard of encainide, flecainide, and moricizine therapy in patients with myocardial infarction. A possible explanation for increased mortality in the Cardiac Arrhythmia Suppression Trial (CAST) Circulation. 1994;90:2843–2852. doi: 10.1161/01.cir.90.6.2843. [DOI] [PubMed] [Google Scholar]
- 178.Waldo A.L., Camm A.J., deRuyter H., Friedman P.L., MacNeil D.J., Pauls J.F., Pitt B., Pratt C.M., Schwartz P.J., Veltri E.P. Effect of d-sotalol on mortality in patients with left ventricular dysfunction after recent and remote myocardial infarction. The SWORD Investigators. Survival With Oral d-Sotalol. Lancet. 1996;348:7–12. doi: 10.1016/s0140-6736(96)02149-6. [DOI] [PubMed] [Google Scholar]
- 179.Køber L., Torp-Pedersen C., McMurray J.J.V., Gøtzsche O., Lévy S., Crijns H., Amlie J., Carlsen J., Dronedarone Study Group Increased mortality after dronedarone therapy for severe heart failure. N. Engl. J. Med. 2008;358:2678–2687. doi: 10.1056/NEJMoa0800456. [DOI] [PubMed] [Google Scholar]
- 180.Connolly S.J., Camm A.J., Halperin J.L., Joyner C., Alings M., Amerena J., Atar D., Avezum Á., Blomström P., Borggrefe M., PALLAS Investigators Dronedarone in high-risk permanent atrial fibrillation. N. Engl. J. Med. 2011;365:2268–2276. doi: 10.1056/NEJMoa1109867. [DOI] [PubMed] [Google Scholar]
- 181.Maru S., Koch G.G., Stender M., Clark D., Gibowski L., Petri H., White A.D., Simpson R.J., Jr. Antidiabetic drugs and heart failure risk in patients with type 2 diabetes in the U.K. primary care setting. Diabetes Care. 2005;28:20–26. doi: 10.2337/diacare.28.1.20. [DOI] [PubMed] [Google Scholar]
- 182.Henninger C., Fritz G. Statins in anthracycline-induced cardiotoxicity: Rac and Rho, and the heartbreakers. Cell Death Dis. 2017;8:e2564. doi: 10.1038/cddis.2016.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bromage D.I., Yellon D.M. The pleiotropic effects of metformin: time for prospective studies. Cardiovasc. Diabetol. 2015;14:109. doi: 10.1186/s12933-015-0273-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lee J.W., Morris J.K., Wald N.J. Grapefruit Juice and Statins. Am. J. Med. 2016;129:26–29. doi: 10.1016/j.amjmed.2015.07.036. [DOI] [PubMed] [Google Scholar]
- 185.Wang Y.-C., Hsieh T.-C., Chou C.-L., Wu J.-L., Fang T.-C. Risks of Adverse Events Following Coprescription of Statins and Calcium Channel Blockers: A Nationwide Population-Based Study. Medicine (Baltimore) 2016;95:e2487. doi: 10.1097/MD.0000000000002487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Arunachalam G., Upadhyay R., Ding H., Triggle C.R. MicroRNA Signature and Cardiovascular Dysfunction. J. Cardiovasc. Pharmacol. 2015;65:419–429. doi: 10.1097/FJC.0000000000000178. [DOI] [PubMed] [Google Scholar]
- 187.Chistiakov D.A., Orekhov A.N., Bobryshev Y.V. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction) J. Mol. Cell. Cardiol. 2016;94:107–121. doi: 10.1016/j.yjmcc.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 188.Rodriguez B., Carusi A., Abi-Gerges N., Ariga R., Britton O., Bub G., Bueno-Orovio A., Burton R.A.B., Carapella V., Cardone-Noott L. Human-based approaches to pharmacology and cardiology: an interdisciplinary and intersectorial workshop. Europace. 2016;18:1287–1298. doi: 10.1093/europace/euv320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Mamoshina P., Bueno-Orovio A., Rodriguez B. Dual transcriptomic and molecular machine learning predicts all major clinical forms of drug cardiotoxicity. Front. Pharmacol. 2020;11:639. doi: 10.3389/fphar.2020.00639. [DOI] [PMC free article] [PubMed] [Google Scholar]