

Summary
Single-cell RNA sequencing (scRNA-seq) is an evolving technology used to elucidate the cellular architecture of adult organs. Previous scRNA-seq on breast tissue utilized reduction mammoplasty samples, which are often histologically abnormal. We report a rapid tissue collection/processing protocol to perform scRNA-seq of breast biopsies of healthy women and identify 23 breast epithelial cell clusters. Putative cell-of-origin signatures derived from these clusters are applied to analyze transcriptomes of ~3,000 breast cancers. Gene signatures derived from mature luminal cell clusters are enriched in ~68% of breast cancers, whereas a signature from a luminal progenitor cluster is enriched in ~20% of breast cancers. Overexpression of luminal progenitor cluster-derived signatures in HER2+, but not in other subtypes, is associated with unfavorable outcome. We identify TBX3 and PDK4 as genes co-expressed with estrogen receptor (ER) in the normal breasts, and their expression analyses in >550 breast cancers enable prognostically relevant subclassification of ER+ breast cancers.
Keywords: normal breasts, single-cell analyses, epithelial cell clusters, cell of origin, breast cancer
Graphical abstract
Highlights
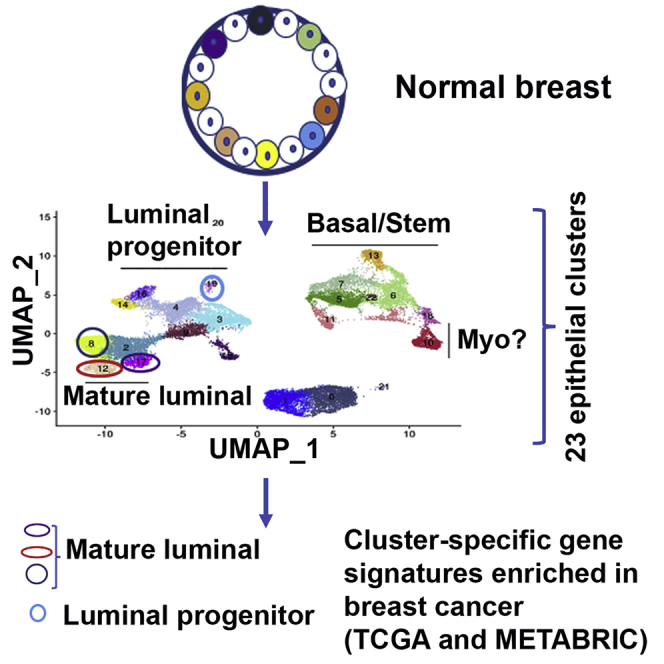
Healthy breast contains 23 subclusters of epithelial cells
Breast cancers may originate from 3 luminal mature and 1 progenitor subclusters
TBX3 and PDK4, co-expressed with estrogen receptor (ER), subclassify ER+ breast cancers
Bhat-Nakshatri et al. describe the single-cell atlas and document 23 epithelial cell subclusters in the healthy breast. Although experimentally validating cell of origin of a tumor is technically challenging, overlap analysis of subcluster-enriched gene signatures with breast tumor transcriptomes revealed dominant representation of differentiated luminal subclusters-derived signatures in breast cancers.
Introduction
Breast cancers are subclassified into multiple subtypes based on gene expression analyses and genomic aberrations.1,2 Among these classifications, intrinsic subtype classification based on gene expression, which classifies breast cancer into luminal-A, luminal-B, HER2+, basal, normal-like, and claudin-low, is suggested to reflect cell of origin of breast cancer.3 Flow-cytometry-based marker profiling and gene expression portraits have identified three major epithelial cell types in the breast, including basal/stem (CD49f+/EpCAM−), luminal progenitors (CD49f+/EpCAM+), and mature luminal (CD49f−/EpCAM+) cells.4,5 Cell-type enriched transcription factor networks, such as TP63/NFIB, ELF5/EHF, and FOXA1/ESR1, control gene expression patterns in basal/stem, luminal progenitor, and mature luminal cells, respectively.6 It is suggested that, although the claudin-low subtype of breast cancers originates from basal/stem cells, luminal progenitors are the source of basal-like breast cancers.5,7,8 Although HER2+ breast cancers may originate from luminal progenitors and mature luminal cells, luminal-A/B breast cancers likely originate from mature luminal cells.3 However, it is acknowledged that heterogeneity exists within basal/stem, luminal progenitors, and mature luminal cells as defined by CD49f/EpCAM cell surface marker profiling.9,10
A recent integrative analysis of 10,000 tumors from 33 types of cancer emphasized the dominant role of cell-of-origin patterns in cancers.11 Because normal tissue itself is composed of multiple cell types, fine mapping of these cell types and identifying potential cancer-vulnerable cell populations in normal tissues would aid in characterization of organ-specific cell of origin of cancers. However, experimentally validating the cell of origin is technically challenging.12 Recent advances in single-cell techniques, including single-cell RNA sequencing (scRNA-seq), scEpigenetics, scDNA-seq, and scProteomics-atlas, are enabling further refinement of cell types within normal and diseased tissues.13 For example, using reduction mammoplasty samples and cells flow sorted based on CD49f/EpCAM, Nguyen et al.14 identified three epithelial cell types in the normal breasts. Using the same technique and mouse mammary tissues at different developmental stages, Pal et al.15 described seven epithelial cell types in the mouse mammary gland. However, Bach et al.16 observed 15 epithelial cell types in the mouse mammary gland. A concern has been raised about the reproducibility of data, which is likely influenced by the types of tissues used, duration between tissue collection and sequencing, and dissociation protocols.13 Although these issues can be standardized for studies involving mouse tissues, standardizing is difficult for studies that utilize human tissues collected after a surgical procedure. In this regard, Lim et al.13 recently proposed the need to establish a rapid tissue dissociation program to advance single cell technology for clinical applications.
A decade ago, our institution established a normal breast tissue bank where healthy women donate breast biopsies for research purposes. This resource has enabled others and us to demonstrate clear differences between our “normal” and both reduction mammoplasty and tumor-adjacent normal tissues, which have been the most common sources of “normal controls” for breast cancer studies in the literature, including the single-cell transcriptome studies. We and others demonstrated clear histologic and molecular abnormalities in these surrogate sources of normal breast tissue.17, 18, 19 For example, only 12% of reduction mammoplasty samples were histologically normal compared to 65% of breast tissues in our tissue collection.19
In this study, we first performed scRNA-seq of five freshly collected samples that included 18,704 cells and 20,647 genes. Results were analyzed at both single sample levels as well as in an integrative manner. To confirm the results of first sequencing, we repeated integrated single-cell analyses of five new cryopreserved samples covering 7,582 cells and 25,842 genes. Using the expression patterns of CD49f and EpCAM as well as basal/stem, luminal progenitor, and mature luminal cell transcription factor networks, we performed refined analyses of epithelial cells. Epithelial-cluster-specific gene signatures were then applied on The Cancer Genome Atlas (TCGA) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) datasets to determine the impact of putative “cell of origin” on breast cancer outcomes.2,20 Because there is limited subclassification of estrogen receptor positive (ER+) breast cancers and it is difficult to characterize ER+ breast epithelial cells from the normal breasts to identify genes co-expressed with ER in normal and tumor cells,21 we performed additional studies on TBX3 and PDK4, two genes that are co-expressed at different levels in ER+ clusters of the normal breasts.
Results
Establishment of rapid tissue procurement and single-cell analyses protocol
Although the primary intention of establishing the Susan G. Komen Tissue Bank (KTB) at IU Simon Cancer Center was to provide a source of healthy breast tissue to be used as normal controls for research, we took advantage of the easily accessible tissue collection procedure in clinic rather than collecting tissue in the operating room, so as to limit time between tissue collection and utilization of tissues for research that is typically associated with collection during surgical procedures. Because these “collection events” have 1:2 donor:volunteer ratio, we were abundantly staffed and able to reduce time from tissue collection to placement in media or cold ischemia time (for cryopreservation) to ~6 min. All tissues used have undergone histologic characterization and are free of abnormalities. Because specimen cellularity varied between individuals, 50% of fresh or cryopreserved tissues provided high-quality data with respect to number of viable cells and minimal ambient RNA contamination of single-cell data. Table S1 provides information about donors. Nine samples were from white women, one was from an Asian woman, and one was from an African American woman. Genetic ancestry mapping has been performed using 41-SNP genetic ancestry informative markers. Two out of 11 women were nulliparous, and two out of 11 were post-menopausal. Five donors had a family history of breast cancer. Based on Tyrer-Cuzick risk scores,22 only three donors had increased lifetime risk of developing breast cancer (>20%; Table S1).
Epithelial cell clusters of the normal breasts
Unlike the previous studies, which purified breast epithelial cells using CD49f/EpCAM markers prior to single-cell analyses,14 we subjected single cells after dissociation directly to RNA sequencing and then used CD49f/EpCAM as well as transcriptional regulators known to specify basal/stem, luminal progenitors, and mature luminal cells to subcluster epithelial cells.6 Uniform manifold approximation and projection (UMAP) plot of combined samples is shown in Figure 1A. As expected, the normal breasts contained a variety of cell types in addition to epithelial cells, including monocytes, T cells, NK cells, endothelial cells, and fibroblast-like cells (Figure 1A). Fibroblast-like and endothelial cells displayed three closely related clusters, suggesting heterogeneity within these cells. Heterogeneity in endothelial cells, driven largely by metabolic plasticity, has been previously described in other organs and disease conditions.23 Similarly, functionally heterogeneous fibroblasts in the normal breast have been described.24
Figure 1.

The normal breast contains 13 epithelial clusters
(A) Integrated analysis of single cells of the normal breast biopsies of five healthy donors. Epithelial cells dominate among cell types.
(B) Subclustering of epithelial cell types using CD49f/EpCAM as well as NFIB, TP63, EHF, ELF5, ESR1, and FOXA1 expression patterns.
(C) Representation of various cell types in each sample. Subclusters in individual sample are shown in Figure S1A.
(D) Hierarchical clustering of top cluster-enriched genes.
Epithelial cell types were dominant. Using CD49f/EpCAM expression pattern as well as TP63/NFIB, ELF5/EHF, and FOXA1/ESR1 as functional markers of basal/stem, luminal progenitors, and mature luminal cells, we performed subcluster analyses of epithelial cells, which revealed 13 different epithelial cells (Figures 1B and 1C). Number of cells in each cluster and average expression value of genes that differentiated these clusters are shown in Table S2. A heatmap of average expression levels of top marker genes of these clusters is shown in Figure 1D. CD49f+/EpCAM− basal/stem cells contained three closely related subclusters (clusters 5, 7, and 9). Each of these clusters within basal/stem cells can be distinguished through expression of specific genes. For example, cluster 5 expressed higher levels of NDUFA4L2, a mitochondrial NADPH dehydrogenase (Figure 2A). This cluster also expressed higher levels of CD36, a lipid transporter associated with breast cancer metastasis, as well as Vimentin, a marker of basal cells.15,25 Cluster 7 expressed ACKR1, a decoy receptor for CCL2 and interleukin-8 (IL-8).26 Cluster 9 is enriched for MECOM (EVI1), a stem-cell-associated transcription factor.27
Figure 2.

Expression patterns of representative cluster-enriched genes
(A) Genes enriched in basal/stem cell clusters.
(B) Genes enriched in various clusters within luminal progenitor cells.
CD49f+/EpCAM+ cells contained two clusters that appeared as a continuum of cells (clusters 0 and 2) and five other well-separated clusters (clusters 6, 8, and 10–12). Although cluster 0 and 2 appeared as a continuum of cells, significant differences in gene expression are evident (Figures 1D and 2B). For example, although all luminal progenitor cells expressed secreted fizzled related protein 1 (SFRP1), a modulator of Wnt signaling,28 genes such as SLP1, ANXA1, RARRES1, KLK5, and KRT15 were enriched in cluster 2. KRT14 and KRT17 expression was enriched in cluster 2, but not in cluster 0. Cluster 6 was enriched for CXCL14 and ACTA2. Cluster 8 was enriched for SCGB2A1 and CALML5. Cluster 10 was KRT14 positive and enriched for the expression of GLYATL2. Cluster 11 was enriched for the expression of multiple genes, including TOP2A, NUSAP1, UBE2C, TPX2, SPC25, MKI67, CDK1, CENPF, and CCNA2 (Figures 1D and 2B). In fact, this cluster displayed a higher number of genes that are differentially expressed than other clusters and constituted a major signaling network associated with regulation of cell cycle, chromosome segregation, and spindle checkpoint, to name a few (Table S2). Cluster 12 was characterized by elevated expression of MEG3, IGF1, and PTGDS.
CD49f−/EpCAM+ mature luminal cells were composed of three clusters, which appeared as a continuum of cells (clusters 1, 3, and 4), although there were distinct differences in gene expression. All three of these clusters expressed ESR1 and pioneering factors FOXA1 and GATA3.29 TBX3 and PDK4 are two other genes that showed variable expression in these clusters. XBP1 and STC2, ESR1 target genes,30 were uniformly expressed at higher levels in all three of these clusters (Figure 3A). In fact, while the expression level of SFRP1 was able to distinguish luminal progenitors from mature luminal cells, expression levels of XBP1 and STC2 were able to distinguish mature luminal cells from luminal progenitors. Cluster 1 showed enrichment of RUNX1 and BATF. Cluster 3 was enriched for ANKRD30A, whereas cluster 4 was enriched for PIP, MUCL1, TAT, and TSPAN8.
Figure 3.

Mature luminal cells are enriched for ESR1 and XBP1, whereas SFRP1 is enriched in luminal progenitor cells
(A) Genes enriched in mature luminal cells. Note that cluster C4 within mature luminal cells is distinctly enriched for MUCL1 and PIP.
(B) Identification of ESR1-expressing subclusters and genes co-expressed with ESR1 in the normal breasts.
(C) Various cell types in the normal breast of a donor.
ER signaling plays a significant role in the development of the breast as well as breast cancer.31 We first did hierarchical clustering and pathway analysis of cluster-enriched genes and found that clusters 1, 3, and 4 were enriched for genes in ER signaling. Hierarchical clustering data from a representative sample that displayed ESR1 transcripts are shown in Figure 3B, and various cell types present in the breast of this donor are shown in Figure 3C. Clusters 1, 3, and 4 expressed ESR1 transcripts. In the integrated analysis of all samples, these three clusters expressed the highest levels of known ER regulators FOXA1, GATA3, and TBX3 as well as lesser known PDK4 (Table S2). Co-expression of ESR1, GATA3, TBX3, and PDK4 is evident in this sample (Figure 3B). Thus, there are three closely related clusters of estradiol-responsive breast epithelial cells.
The distribution pattern of epithelial clusters in the individual samples is shown in Figure S1A, and number of cells per cluster is indicated in Table S2. Almost every sample contained similar levels of most of the clusters; two minor clusters, clusters 11 and 12, showed inter-sample variability.
Reproducibility of cluster analyses
To determine whether epithelial clusters identified in the above analyses can be reproduced using cryopreserved tissues from healthy donors, we isolated cells from five cryopreserved tissues, pooled cells, and analyzed them all together. Because five samples were combined, there were enough cells to divide samples into two and perform cDNA synthesis and library preparation by two independent labs. In addition, we used the latest version of the library preparation from 10X Genomics with improved chemistry, paired-end sequencing, and better efficiency. Because pooled samples contained more lymphocytes, lymphocyte-related cells were removed from the analyses. Without lymphocyte removal, there were 28 clusters (Table S2). Side-by-side comparisons of the second set of pooled samples and re-analyses of first five samples are shown in Figure 4A. Re-analyses of individual samples are shown in Figure S1B, and number of cells in each cluster is shown in Table S2. With increased number of cells and more genes sequenced, further subclassification of epithelial cells became possible. Because the number of clusters identified in this new clustering are different (23) from the first analysis (13), new clusters are named with prefix N (N0–N22). The cluster defining gene lists showing fold differences (value in cluster/value in all other clusters combined) and p values are provided in Table S3.
Figure 4.

Recharacterization of epithelial cells of the normal breasts with additional samples
(A) Combined integrated analyses that included samples in Figure 1, a new sample from an Asian (Chinese), and pooled five new samples. There were 23 clusters of cells, which can be subdivided into three major groups of basal/stem, luminal progenitor, and mature luminal cells. Potential myoepithelial cells (myo) distinct from basal/stem cells are also indicated. The bottom panel shows distribution patterns of cell clusters in five samples of the first set and the five pooled samples of the second set. Clusters in individual samples are shown in Figure S1B. Expression patterns of various markers that are used to subclassify clusters are shown Figure S2.
(B) CD49f, EpCAM, ALDH1A3, and KRT14 expression in various clusters.
Consistent with our earlier report and a recent report on organoid-derived single-cell data, there were inter-individual differences in the proportion of cells in each cluster.17,21 The UMAP cell embeddings and cell cluster information generated from Seurat analysis were imported into 10X Genomics Loupe Browser. By checking the expression of various genes with the Loupe Browser, we first assigned the subdivided clusters into basal/stem, luminal progenitor, and luminal mature cells. Based on CD49f and EpCAM expression patterns (Figure 4B), clusters N5–7, N11, N13, N18, and N22 were basal; N3, N4, N9, N14, N16, N19, and N20 were luminal progenitors; and N2, N8, N12, and N17 were mature luminal cells. ALDH1A3 expression, which has been suggested to identify breast cancer stem cells,32 was expressed mainly in N4, N14, and N16 clusters of luminal progenitor cells (Figure 4B). Among genes that define basal/stem, luminal progenitor, and luminal mature cells, as expected, CD117 (KIT) expression was restricted to luminal progenitor cells,5 whereas ESR1 and FOXA1 expression was restricted to luminal mature cell clusters (Figure S2). Although ESR1 expression was widespread across mature luminal subclusters, with cluster 12 displaying stronger signals, the expression of its target gene PGR was much more restricted within mature luminal cells, suggesting the natural existence of ER+/PR+ and ER+/PR− cells, similar to the features of luminal A and luminal B breast cancers.33 Expression of the best-studied ER target gene GREB1 overlapped with PGR expression, suggesting ER has cell-type-specific targets within the normal breast. Consistent with an earlier report,34 RANK (TNFRSF11A) expression was restricted to a few luminal progenitor cells, whereas its ligand RANKL (TNFSF11) expression was observed in progesterone-receptor-positive mature luminal cells (Figure S2). EHF and ELF5 expression showed strong signals in subclusters N14 and N16, which could be alveolar progenitor cells, as these two transcription factors play a major role in alveolar differentiation during pregnancy.35 However, expression of NFIB and TP63 did not correlate with prior assessment,6 as NFIB expression was not restricted to basal/stem cells, whereas TP63 expression was observed only in cluster 15. Cluster 15 is likely composed of myoepithelial cells, as this cluster expressed higher levels of ACTA2 and KRT17, previously described markers of myoepithelial cells (Figure S2).14 Although basal cells are expected to express KRT14, we found its expression predominantly in a subpopulation of luminal progenitor cells and in N15 with myoepithelial characteristics (Figure 4B). KRT18 and KRT19 expression was found equally in luminal progenitor and mature luminal subclusters (Figure S2).
To further document reproducibility, we analyzed a surgical sample from a 33-year-old Hispanic BRCA1 mutation carrier and a core biopsy of a healthy Asian (Chinese) woman. The BRCA1 sample was analyzed from cryopreserved tissue, and we included duplicate samples because of availability of large starting material. One sample was prepared as above, involving both enzymatic and mechanical disruption, whereas another sample utilized only digestion with a gentle hyaluronidase/collagenase cocktail from STEMCELL Technologies. Sample preparation using gentle hyaluronidase/collagenase yielded lower numbers of basal cells compared to the method that involved both enzymatic and mechanical disruption. Nonetheless, we did not observe any clusters unique to the BRCA1-mutated sample (Figure S1B). The breast tissue from the Asian/Chinese donor showed a disproportionately higher number of cells with basal/stem characteristics compared to other samples.
Gene expression overlap in clusters of two sets of analysis
We next determined similarities in the clusters of two sets of analysis by overlapping gene expression between clusters of the two sets and evaluating their statistical significance. Table S4 provides a summary of this analysis. With the exception of N9, N15, and N16, all the other clusters in the new analysis classified similarly as mature luminal, luminal progenitor, or basal-cell-type clusters. For example, gene expression in N2, N8, N12, and N17 overlapped with gene expression in C1, C3, and C4, which are all mature luminal clusters in both analyses. N0, N1, N3, N4, N14, and N19 were similar to C0, C2, C6, C11, and C12, which are all luminal progenitor clusters in both analyses. N5, N6, N7, N10, N11, N13, N18, N20, N21, and N22 overlapped with C5, C7, and C9, which are all basal cell clusters in both analyses. These results indicate reproducibility of single-cell sequencing and data analyses.
Cluster 12 in the first analysis was relatively minor, but its counterpart in the second N0/N1 was relatively large and represented 15% of cells (Figure 4; Table S2). In addition, cluster 11 of the first analysis, despite being minor, expressed several cell-cycle-related genes, such as MKI67 and CDK1. Figure 5 shows the expression patterns of C11- and C12-enriched genes in the second analysis. Similar to C11, N19 expressed MKI67, BIRC5, and PCLAF. Similar to C12, N0 and N1 expressed PTGDS and IGF1. N0 and N2 are likely enriched for stemness-associated genes as well, as these cells expressed higher levels of EGFR and CD44 and low levels of various keratins,36 and likely different from the rest of the luminal progenitor cells, as they expressed very low levels of CD49f and EpCAM. A fraction of these cells, as well as the cluster N6 cells among the basal/stem cell group, were PROCR+, which is another mammary stem cell marker.37 Note that none of the 23 clusters expressed mesenchymal stem cell markers, such as CD90, CD73, and CD105.38
Figure 5.

Gene expression in clusters N19 and N0–N1 of Figure 4 overlap with unique genes in C11 and C12, respectively
(A) MKI67, BIRC5, and PCLAF, which are all overexpressed in cluster C11 (Figure 1D), are enriched in N19.
(B) PTGDS and IGF1, which are overexpressed in cluster C12 (Figure 1D), are enriched in N0–N1 clusters. This cluster also expresses ZEB1 and EGFR.
The majority of breast cancers are enriched for the expression of genes in the mature luminal cell clusters
Cell of origin, adaptive cell signaling, and mutational landscape determine the transcriptome of tumors. Although it is not possible to definitively link cancer to its cell of origin,12 a recent pan-cancer analysis revealed cell-of-origin gene signatures are dominant in tumors.11 To determine whether such a relationship exists between signatures of normal breast epithelial cell clusters and breast tumors, we compared gene scores of each epithelial cluster with METABRIC and TCGA breast cancer gene expression datasets.2,20 In the TCGA dataset, 795 tumors were ER+ and 237 were ER−; 160 tumors were HER2+ and 558 were HER2−. In addition, in this dataset, 151 and 932 samples came from deceased and living donors, respectively. In the METABRIC dataset, 1,221 samples were ER+ and 683 were ER−; 188 samples were HER2+ and 1,716 samples were HER2−. In this dataset, 1,102 and 801 samples came from deceased and live donors, respectively. We used cluster classification of the second analysis, because this cluster classification was more robust with higher number of cells per cluster compared to the first analysis.
In both TCGA and METABRIC datasets, gene expression in the majority of breast cancers overlapped with gene expression in mature luminal clusters (Table S4; Figure 6A). For example, gene expression in the highest number of breast tumors (~68%) overlapped with gene expression in clusters N8 and N12. N8 and N12 are closely related clusters, both expressing ESR1 (Figure S2). The next highest overlap between breast tumors and epithelial cell clusters was with the N19 luminal progenitor cluster, particularly in the METABRIC dataset (Figure 6B).
Figure 6.

Breast-cancer-subtype-specific expression of cluster-signature genes
(A) Breast cancer gene expression data in TCGA (left) and METABRIC datasets were analyzed for enrichment of cluster-specific genes described in Table S3.
(B) PAM50 intrinsic subtype classifiers were used to subdivide breast cancers into luminal A, luminal B, HER2, basal, and claudin-low subtypes. Enrichment of cluster-specific genes in these subtypes of breast cancer were further analyzed.
(C) Kaplan-Meier curves show overall survival based on overlap in gene expression between specific clusters and specific subtypes of breast cancer. Additional data can be found in Figure S3.
To gain insight into the putative cell of origin of ER− breast cancers, we overlapped our cluster signatures with gene expression in only ER− breast cancers. The highest number of ER− breast cancers showed gene expression overlap with mature luminal N17 and luminal progenitor N19 clusters in the TCGA dataset (Figure 6B). In the METABRIC dataset, ER− breast cancers were represented in N8/N12 mature luminal clusters and luminal progenitor N19. Similar to ER− breast cancers, gene expression in HER2+ breast cancers of the METABRIC dataset overlapped with gene expression in the N8/N12 and N19 clusters (Figure 6B).
We used the PAM50 classifier to subclassify breast cancers into intrinsic subtypes luminal A, luminal B, basal, and claudin-low and then did gene expression overlap analysis. Cluster N8 still appears to be dominantly represented in all tumor subtypes, followed by N19 and N12 (Figure 6B).
We generated Kaplan-Meier curves of tumors with gene expression overlapping with specific clusters either globally or in a subtype-specific manner. In the global analysis, tumors with gene expression patterns overlapping N19 clusters displayed better outcome compared to tumors with gene expression patterns overlapping either N8 or N12 (Figure S3). In subtypewise comparisons, tumors with gene expression overlapping N19 displayed better outcome compared to N8 or N12, with the exception of the HER2+ subtype (Figures 6C and S3). In HER2+ cases, tumors with gene expression overlapping N8 displayed better outcome than those with N19 cluster gene expression overlap (Figure S3). Thus, gene expression signatures derived from normal breast epithelial cell clusters can be developed into prognostic signatures using well-annotated datasets.
TBX3 and PDK4 expression patterns determine subtypes of ER+ breast cancers
We observed expression of ESR1 in three mature luminal clusters (C1, C3, and C4) of the normal breasts, which are characterized by expression of TBX3, PDK4, and GATA3 in the first analysis (Figure 3C). In the second analysis, mature luminal clusters N2, N8, N12, and N17 expressed variable levels of ESR1 (Figure S2). Among these four clusters, N2 and N8, but not N12 and N17, expressed FOXA1, GATA3, and TBX3 (Table S3). Because FOXA1 and GATA3 serve as pioneer factors and regulate ER activity through chromatin accessibility,29 ER activity is likely regulated differently in N12 and N17 clusters compared to N2 and N8. In this study, we focused our attention on PDK4 and TBX3, given that their expression is linked to anti-estrogen response.39,40 To determine whether there is a relationship between clinical progression and ER/TBX3/PDK4 status, we immunostained a 586 breast tumor containing tissue microarray (TMA) with 15 years of follow up for TBX3 and PDK4. As expected, although TBX3 staining was predominantly nuclear, PDK4 expression was cytoplasmic (Figure 7A).
Figure 7.

PDK4 and TBX3 enable further classification of ER+ breast cancers
(A) Immunohistochemistry of breast TMA for PDK4 and TBX3.
(B) ER+ breast cancers expressing lower levels of PDK4 compared to tumors with higher PDK4 and not received endocrine therapy were associated with poor disease-free survival (DFS). Similarly, ER+ tumors expressing lower levels of both TBX3 and PDK4 compared to tumors expressing higher levels of PDK4 and TBX3 were associated with poor DFS.
PDK4 expression levels correlated with ER+/PR+/HER2− status (p = 0.0113). However, higher PDK4 H-score correlated with lower overall survival (hazard ratio 1.382; p = 0.0431). In multivariable models treating the PDK4 H-score as dichotomous, H-score category was significant for tumors that are ER+ and for ER+ tumors where the patient was on endocrine therapy (Table S5). In multivariable models treating the H-score as a continuous variable, the H-score was significant in the subset of patients who were ER+ on endocrine therapy and for patients who were ER+/PR+/HER2−. Kaplan-Meier curves of disease-free survival analyses are shown in Figure 7B.
In the case of TBX3, expression levels correlated with tumor grades (p < 0.0001) and stage (p = 0.0063). Higher grade/stage tumors had higher TBX3 expression compared to lower grade/stage tumors. However, higher TBX3 H-score correlated with better overall survival (hazard ratio 0.721; p = 0.033). In multivariable models treating the H-score as dichotomous, H-score category was significant for patients with tumors that were ER+ and not on endocrine therapy and patients whose tumors were not ER+/PR+/HER2− (Table S5). In multivariable models treating the H-score as continuous, the H-score was significant in patients whose cancers were not ER+/PR+/HER2−.
Because our TMA had >300 ER+ cases, we were able to perform subgroup analyses that included all three markers: ER; TBX3; and PDK4. The analyses included ER+/TBX3+/PDK4+, ER+/TBX3+/PDK4low, ER+/TBX3low/PDK4high, and ER+/TBX3low/PDK4low. Although we did not observe any difference in overall survival between these groups, disease-free survival was shorter for patients with the tumors displaying ER+/TBX3low/PDK4low expression patterns compared to ER+/TBX3+/PDK4+ expression patterns (Figure 7B). Among 399 ER+ tumors, 138 displayed ER+/TBX3+/PDK4+ characteristics and 57 showed ER+/TBX3low/PDK4low characteristics. These results indicate that ER+ breast cancers can be subclassified into at least four distinct subtypes based on TBX3 and PDK4 expression patterns, potentially representing four different cells of origin of ER+ breast cancers.
Discussion
In this study, we present evidence for the presence of 23 different clusters of epithelial cells in the normal breast. We also show that the gene expression patterns of a majority of breast cancers overlap with gene expression signatures from four clusters; three of them are mature luminal and one is a luminal progenitor cluster. It is possible that cells in these four clusters are the cancer-prone population of normal breast epithelial cells. We acknowledge that the number of samples that gave quality single-cell data is relatively small, which is mainly due to the use of breast core biopsies with different cellularity instead of surgical specimen for single-cell analysis. Nonetheless, findings from this study may permit breast cancer classification based on cell of origin of tumors. Although each intrinsic subtype of breast cancer ostensibly has a distinct cell of origin in the breast stem-progenitor-mature cell hierarchy,3 we observed a cluster-enriched signature being represented in more than one intrinsic subtype and an intrinsic subtype being represented in more than one cluster of epithelial cells. Existing technologies do not permit experimental validation of cell of origin of tumors, but the use of techniques such as scRNA-seq may allow further refinement of cancer classification based on presumptive cell of origin.
Complexities in breast epithelial cell types: past and the present
Because scRNA-seq technology is still an evolving field requiring constant improvement, starting from source of tissues to dissociation protocols, sequencing techniques, and bioinformatics tools,13 it is likely that clusters that we identified here will undergo further refinement in the future. Thus, it is appropriate to compare what has been done in the past to the current data. There has been a limited number of studies that utilized scRNA-seq technology to subclassify breast epithelial cells. Two publications, to our knowledge, utilized tissues from reduction mammoplasty samples, and cells were either purified by flow cytometry or grown under organoid cultures prior to single-cell sequencing.14,21 Although our source of tissue and methodology differed significantly from these studies, as we used breast tissues from healthy women and were able to prepare single-cell cDNA within 2 h of tissue collection, there were several overlapping observations. For example, the L2 luminal differentiated cell cluster described by Nguyen et al.,14 luminal differentiated cluster C3 in our first analysis, and cluster N8 of our second analysis are enriched for ANKRD30A (p = 1.67E−31; Table S2). Similarly, luminal progenitor cluster L1 in that study and our luminal progenitor subcluster C10 are enriched for the expression of SLPI and ANXA1. Similar to that study, our luminal mature cell subcluster 4 was enriched for PIP. A basal subcluster identified by Nguyen et al.14 and our cluster 5 (N5 and N22 of the second analysis; Table S3), which is basal, both expressed TCF4. There were a few differences. Nguyen et al.14 suggested that ACTA2, which codes for α-SMA, distinguishes basal/myoepithelial cell from other cell types. Although we observed expected enrichment of ACTA2 in basal subclusters, it is also expressed in a distinct subcluster of luminal progenitor cells (cluster C6 of the first analysis and N15 of the second analysis). Both cluster C6 and the corresponding N15 are enriched for MYLK, an actin binding protein, regulated by ZEB1/miR-200 feedback loop associated with epithelial-to-mesenchymal transition.41 It is possible that cluster C6 (N15) cells correspond to naturally occurring luminal/basal hybrid cells that can trans-differentiate based on environmental cues or to mixed luminal/basal lineage cells described in the mouse mammary gland.15
Three studies have described distinct cell types in the mouse mammary gland during different stages of development and lactation.15,16,42 Similar to differences in the number of epithelial subgroups identified in two human studies (three by Nguyen et al.14 and 23 by us), Bach et al.16 identified 15 clusters of mouse mammary epithelial cells through single-cell sequencing of sorted EpCAM+ cells. Pal et al.15 identified seven clusters of epithelial cells. There is some overlap in genes expressed in specific clusters of the mouse mammary gland and human tissue identified in our study. Similar to our results, Pal et al.15 showed ACTA2 expression in both basal cells and two small subclusters of luminal cells. CXCL14 expression was found in a subset of luminal progenitor cells and basal cells (N0, N15, and N22). Gene expression in hormone-sensing cells that included ESR1 and FOXA1 showed similarity in expression between Bach et al.16 and our studies. Pal et al.15 identified SFRP1 as a marker of pre-pubertal mammary epithelial cells, which decreased after puberty. In our analysis, SFRP1-expressing cells were luminal progenitor cells.
Basal, luminal progenitor, and mature luminal cells are defined using cell surface markers CD49f and EpCAM.4 However, these markers are not ideal for in situ estimation of the three cell types. A closer look at genes enriched in each cluster and their signature genes revealed that XBP1 is expressed predominantly in luminal mature cells, whereas SFRP1 is expressed predominantly in luminal progenitor cells. XBP1 expression pattern is interesting, as its expression is linked to estrogen independence and anti-estrogen resistance in breast cancer,43,44 and differences in its basal expression levels between luminal mature cells may determine hormone dependency of cells. Multiple genes are enriched in basal cell clusters, including CLDN5 (N5 and N7), CD36 (N7), and CD93 (N5, N13, and N22). These genes can be used in the future for in situ estimation of composition of the breast.
Gene signatures of epithelial clusters and their relevance to breast cancer
Although prior reports suggested that the majority of breast cancers originate from luminal progenitors, gene expression signatures of only the N19 luminal progenitor cluster showed overlap with gene expression in ~20% of breast cancer. This cluster as well as its counterpart in the first analysis (C9), although representing <2% of epithelial cells, is characterized by expression of cell cycle markers MKI67 and TK1 (p = 3.83E−13) and mitotic spindle checkpoint protein ZWINT (p = 4.62E−17). Distinct cell cycle regulatory pathways may predispose cells of this cluster for aberrant chromosome segregation and mutations.
With respect to ER+ breast cancers, these cancers can originate from both luminal mature and luminal progenitor cells, as gene expression in the majority of luminal A and luminal B breast cancers overlapped with gene expression in the N8 and N12 mature luminal clusters and the N19 luminal progenitor cluster. Although ESR1 expression in luminal progenitor cells is undetectable compared to that in mature luminal cells (Figure S2), it is possible that luminal A and luminal B breast cancers with luminal progenitor cell origin may acquire ESR1 expression during transformation. The N8 cluster expresses the highest level of ESR1 as well as its pioneer factors FOXA1, GATA3, and also TBX3. Using an independent TMA, we were able to further subclassify mature luminal cell-derived ER+ tumors based on TBX3 and PDK4 expression. Although the role of TBX3 in ER activity and its mutations in ER+ lobular carcinomas have been described in the literature,39,45 there are no studies that functionally linked PDK4 to ER. PDK4 is a cytoplasmic kinase involved in the citric acid cycle and induces metabolic changes in transformed cells.46 Whether it also modulates transcription by targeting transcription machinery needs further investigation.
scRNA-seq as well as single-cell protein sequencing have been used to subclassify breast cancers and to identify treatment-resistant populations. For example, Karaayvaz et al.47 described an aggressive disease-associated gene signature related to glycosphingolipid metabolism by scRNA-seq study of six triple-negative breast cancers (TNBCs). However, pathway analyses of our cluster-enriched genes did not identify a normal breast epithelial cluster enriched for this pathway. Similarly, signatures derived through scRNA-seq of a chemoresistant subpopulation of TNBCs did not show overlap with any of our normal cell clusters.48 Genes in the breast-cancer-specific RNA signature that detect circulating tumor cells did not show overlap with any specific cluster, but genes like CXCL14 (N0, N15, and N22) and SFRP2 (N0, N1, N21, and N22), which are markers of circulating tumor cells, are enriched in distinct clusters.49 Thus, genomic aberrations and transcription programming rather than cell of origin may have given rise to drug-resistant and metastatic subpopulations of tumor cells.
From a basic research point of view, results presented here provide an opportunity to determine whether a gene is truly differentially expressed in tumors compared to normal, as the expression pattern of a specific gene in the tumor could be a reflection of its cell of origin. Using single-cell RT-PCR of tumor-adjacent normal and tumor cells from the same individual, we had previously demonstrated that elevated expression of few genes in tumor can be attributed to cell of origin of tumor instead of a tumor-specific genomic aberration.10 Resources created here, which will be made available to researchers, can be mined for expression patterns of specific genes in various epithelial clusters of the normal breasts using a tool such as Loupe Browser of 10X Genomics. This approach would also allow streamlining of drug discovery efforts by focusing on targets that are truly differentially expressed in tumors due to genomic aberrations.
Limitations of study
scRNA-seq studies, including one reported here, have technical limitations, as the current scRNA-seq methods accurately sample only 10%–40% of all transcripts of a cell and that several of the transcriptional events identified by RNA-seq are not replicated in the proteome. Thus, interpreting these results in the context of biological functions needs some caution. Because our study involved breast biopsies of healthy women, tissue is very limited in quantity, and breast-region-specific differences in the cell types could not be ascertained. Recent studies have identified different biology and marker profiles in fibroblasts of the ductal and lobular regions of the breast,24 and it will not be a surprise if transcriptome of epithelial cells in the ductal and lobular regions are different, which the current approaches cannot address. Lastly, it is difficult to account for inter-individual differences in transcriptome due to genetic diversity in the human population.50 An extensive study that includes tissues from multiple regions of the breast from several donors of different age groups and genetic ancestry is required to generate a comprehensive single-cell map of the normal breasts. Nonetheless, we establish that it is feasible to map the breast of healthy women at single-cell level through a standardized and rapid tissue collection and processing procedure.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PDK4 | Abcam | Cat # Ab71240; RRID: AB_1269709 |
| TBX3 | Abcam | Cat# Ab99302; RRID: AB_10861059 |
| Biological samples | ||
| Human Breast tissues | Komen tissue bank and IUSCCC tissue bank | N/A |
| Human Breast Cancer Tissue Microarray | IUSCCC tissue bank | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Cryopreservation media | LONZA | 12-132A |
| ROCK Inhibitor Y-27632 | TOCRIS | 1254 |
| Gentle Collagenase/Hyaluronidase | StemCell technologies | 07919 |
| Tumor dissociation kit (human) | Miltenyi Biotech | 130-095-929 |
| Red cell lysis buffer | Miltenyi Biotech | 130-094-183 |
| Debris removal kit | Miltenyi Biotech | 130-109-398 |
| Critical commercial assays | ||
| Chromium Single cell 3′reagents | 10X Genomics | CG00052 Rev B or CG000183 Rev C |
| Bioanalyzer HSDNA CHIP | G2943CA | Agilent |
| Deposited data | ||
| Single cell RNA-seq | GEO | GSE164898 |
| Experimental models: organisms/strains | ||
| Breast tissues from healthy women | Komen Tissue Bank and IU Simon Cancer Center Tissue Bank with the approval from the Institutional Review Board. | Table S1 for details |
| Software and algorithms | ||
| CellRanger 2.1.0 or 3.0.2 | 10X Genomics | https://support.10xgenomics.com |
| Loupe Browser | 10X Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/installation |
| SAS Version 9.4 | SAS Analytical Software | https://www.sas.com |
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Harikrishna Nakshatri (hnakshat@iupui.edu)
Materials availability
This study did not generate unique reagents.
Data and code availability
The accession number for the single cell sequence data reported in this paper is GEO GSE164898.
Experimental model and subject details
Normal breast tissues
All breast tissues from healthy women were collected by the Komen Normal Tissue Bank with informed consent and with the approval from the institutional review board. International Ethical Guidelines for Biomedical Research Involving Human subjects were followed. Standard operating procedure for tissue collection is described on the Komen Tissue Bank website. Per standard operating procedures, the normal breast biopsies were always collected from the upper outer quadrant of the breasts. Within an average of six minutes from the time of biopsy, tissues were either placed in a growth media and transported to the lab for immediate single cell sequencing or cryopreserved for single cell sequencing at a later date. Our cryopreservation protocol has been described previously.17 Briefly, tissue was minced and placed in one ml of 50% growth media and 50% Lonzo freezing media with 2 μM ROCK inhibitor. Vials with tissues were placed in CoolCell Containers (Nalgene) and placed in a −80°C freezer overnight and then in liquid nitrogen. Tissue specimens were thawed rapidly at 37°C and then washed extensively in growth media prior to dissociation. Tissue specimens represented women of different race, age, parity, menstrual phase, Tyrer-Cuzick score, and BMI (Table S1).
Method details
Tissue dissociation procedure, cDNA library preparation and sequencing
We used the human tumor dissociation kit from Miltenyi Biotech to generate single cells from tissue specimens. Red blood lysis buffer and debris removal solution were used as needed to improve purity of single cells. Viability and single cell status were determined via trypan blue staining and phase contrast microscopy. Samples with 80% or more viability were utilized for the subsequent steps. Cells were suspended at ~100-800 cells/μl depending on sample and subjected to cDNA library generation using 10 X Genomics V2 (initial study) or V3 (second set of samples) Chromium Single Cell 3′ Reagents (CG00052 Rev B and CG000183 Rev C, respectively). We used HSDNA Chips on the Bioanalyzer from Agilent technologies (G2943CA) to quantify cDNA. cDNA was amplified using Chromium TM Single cell Library kit v2 or v3. The resulting libraries were sequenced on Illumina NovaSeq 6000 to a read depth of ~50,000 reads per cell. 26 bp of cell barcode and UMI sequences, and 91 bp RNA reads were generated for the libraries made with the V2 kit; and 28 bp plus 91 bp paired-end for the libraries with the V3 kit.
Breast cancer TMA and immunostaining for TBX3 and PDK4
The breast cancer TMA with ~15-years of follow up has been described previously.51 All tissue samples were collected following a detailed IRB approved protocol, informed patient consent, and HIPAA compliance protocol. Tissues were fixed overnight at room temperature in 10% NBF. A pathologist (GES) utilized light microscopy (Leica) to evaluate the staining in each tissue core (range from 0 to +3) to make sure there was no over staining and/or extensive background staining. The slides were imaged using the Aperio Scanscope CS. Computer-assisted morphometric analysis of digital images was performed using the Aperio Image Analysis software that came with the Aperio Whole Slide Digital Imaging System. The Positive Pixel Count algorithm was used to quantify the amount of a specific stain present in a scanned slide image. A range of color (range of hues and saturation) and three intensity ranges (weak, positive, and strong) were masked and evaluated. The algorithm counted the number and intensity-sum in each intensity range, along with three additional quantities: average intensity, ratio of strong/total number, and average intensity of weak positive pixels.
The algorithm was applied to an image by using the TMA Lab algorithm. This program allowed us to select each core, specify the input parameters, run the algorithm, and view/save the algorithm results. When using the Image Scope program, a pseudo-color markup image is also shown as an algorithm result. The H score was calculated using the Aperio TMA software algorithm. Formula is:
Approximately 80 to 90 breast biopsies in each of the 14 breast TMA immunostain were evaluated with TBX3 and PDK4 antibodies. Anti-PDK4 (ab71240) and anti-TBX3 (ab99302) antibodies were obtained from Abcam. The normal tissue controls (TMA orientation cores) were normal liver, cecum, kidney, spleen, tonsil, and heart.
With TBX3, immunostaining was seen in both the cytoplasm and nucleus in most tumor cells, and within few stromal cells in a few cases. In cases with inflammation, several of the lymphocytes (subset) were strongly stained. Staining patterns in tumor cells ranged from weak to moderate to strong. The two cores from the same patient in the arrays were often similar depending on the amount of fat and /or stroma in the core. Little to no background staining was seen in the other tissues in the core (vascular endothelial cells, smooth muscle cells, adipocytes, and fibroblasts).
With PDK4, immunostaining was seen in the cytoplasm of most tumor cells and in some cases, that of a few stroma cells. Lymphocyte staining was seen only in cases with inflammation. The tumor cells were weak to moderate to strong in staining with two cores from the same patient. This was consistent in all arrays with minimal background staining in the other tissues in the core (vascular endothelial cells, smooth muscle cells, adipocytes, and fibroblasts).
Quantification and statistical analysis
Analysis of scRNA-seq sequence data
CellRanger 2.1.0 or 3.0.2 (https://support.10xgenomics.com/) was utilized to process the raw sequence data generated. Briefly, CellRanger used bcl2fastq (https://support.illumina.com/) to demultiplex raw base sequence calls generated from the sequencer into sample-specific FASTQ files. The FASTQ files were then aligned to the human reference genome GRCh38 with RNA-seq aligner STAR. The aligned reads were traced back to individual cells and the gene expression level of individual genes were quantified based on the number of UMIs (unique molecular indices) detected in each cell.
The filtered gene-cell barcode matrices generated with CellRanger were used for further analysis with the R package Seurat version 2.3.1 and development version 3.0.0.9000 with R studio version 1.1.453 and R version 3.5.1.52,53 Quality control (QC) of the data was implemented as the first step in our analysis. We first filtered out genes that were detected in less than five cells and cells with less than 200 genes. To further exclude low-quality cells in downstream analysis we used the function isOutlier from R package scater together with visual inspection of the distributions of number of genes, UMIs, and mitochondrial gene content.54 Cells with extremely high or low number of detected genes/UMIs were excluded. In addition, cells with high percentage of mitochondrial reads were also filtered out. After removing likely doublets/multiplets and low-quality cells, the gene expression levels for each cell were normalized with the NormalizeData function in Seurat. To reduce variations sourced from different number of UMIs and mitochondrial gene expression, we used the ScaleData function to linearly regress out these variations. Highly variable genes were subsequently identified.
To integrate the single cell data from individual donor samples, functions FindIntegrationAnchors and IntegrateData from Seurat v3 were implemented. The integrated data was then scaled and PCA was performed. Clusters were identified with the Seurat functions FindNeighbors and FindClusters. The FindConservedMarkers function was subsequently used to identify canonical cell type marker genes. Cell cluster identities were manually defined with the cluster-specific marker genes or known marker genes. The cell clusters were visualized using the UMAP plots. To help interactively explore various gene expression pattern across cell clusters, the UMAP cell embeddings and cell cluster information generated from Seurat analysis were imported into 10X genomics Loupe Browser (https://support.10xgenomics.com/single-cell-gene-expression/software/visualization/latest/installation). R packages ggplot2 and Seurat FeaturePlot were used to generate feature plots to visualize specific gene expression across clusters. R package ComplexHeatmap was used to generate the heatmaps.55,56
TCGA and METABRIC dataset analyses
3186 genes in all 23 clusters of the second analyses are considered as signatures (Table S3). Centroids of clusters were generated for Prediction Analysis of Microarray (PAM) algorithm using the 3186 genes.57,58 Expression, clinical, and mutation data of METABRIC and TCGA BRCA were retrieved from cBioportal.11,59, 60, 61, 62, 63, 64, 65, 66, 67 Expression data are median centered and applied to the PAM classifier based on the 3186 genes using Spearman’s rank correlation as distance. Each sample in METABRIC and TCGA BRCA data was assigned to one of the 23 clusters. Relationship of each sample’s cluster membership with intrinsic subtypes was analyzed. Survival analysis of METABRIC samples between clusters were analyzed using R package survminer v0.4.6 in R.55,68,69
Statistical analyses of TMA data
For subjects with multiple tumor samples available, we included only the sample with the highest PDK4 or TBX3 H-score. Wilcoxon Rank Sum and Kruskal-Wallis tests were used to determine if PDK4 or TBX3 H-scores correlated with other tumor markers. Cox proportional hazards regression models were used to determine whether H-scores and other variables were related to overall and disease-free survival either univariately or in multivariable models. In these analyses, TBX3 H-scores were divided into low and high categories at the score of 27.91721 for overall survival (time from surgery to death or censoring) and disease-free survival (time from surgery to first recurrence or censoring, excluding patients with M1 stage at surgery). PDK4 H-scores were divided into low and high categories at the score of 19.41508 for overall survival (time from surgery to death or censoring) and 34.05692 for disease-free survival (time from surgery to first recurrence or censoring, excluding patients with distant metastatic diseases at surgery). These cutoff values were determined by using the maximum chi-square value for all score values between the 25th and 75th percentile as described previously.51 PDK4 high/low and TBX3 high/low was included in all multivariable models. As a double check on the direction of the hazard ratio and as a more powerful test if the H-score effect was truly linear, we also fit multivariable models with the H-score as continuous.
We conducted subgroup analyses on overall survival using the ER-positive subgroup, endocrine therapy group, ER-positive on endocrine therapy, ER-negative, and ER+/PR+/HER2-. First, log-rank tests were done with the dichotomous H-score variable. Second, multivariable models with the H-score as dichotomous and then continuous were fit similar as was done in the main analyses. Analyses were conducted using SAS Version 9.4. An α level of 5% was used to determine statistical significance.
Acknowledgments
We thank the countless number of women who donated normal and malignant breast tissues for research. We also thank the volunteers who facilitated this tissue collection. Special thanks to members of the Komen Tissue Bank, including Ms. Jill Henry, Alison Hughes, Pam Rockey, Julia Rose von Arx, Rana German, and Dr. Natascia Marino, as well as the IU Simon Cancer Center tissue procurement facility for providing tissues and related data. The Catherine Peachy Fund of the Heroes Foundation family (H.N.), Breast Cancer Research Foundation (H.N.), Chan-Zuckerberg Initiative Human Atlas Project (H.N., A.M.S., and Y.L.), Susan G. Komen for the Cure OGKTB1301 (A.M.S.), and Vera Bradley Foundation for Breast Cancer Research (IUSM). Walther Cancer Institute provided support to Cancer Bioinformatics Core. We worked to ensure gender balance in the recruitment of human subjects. We worked to ensure ethnic or other types of diversity in the recruitment of human subjects. We worked to ensure that the study questionnaires were prepared in an inclusive way. The author list of this paper includes contributions from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
Author contributions
Conceptualization, H.N.; methodology, P.B.-N., P.C.M., X.X., L.S., J.W., G.S., H.G., Y.L., and H.N.; validation, P.B.-N., H.G., Y.L., and H.N.; formal analysis, P.B.-N., P.C.M., A.C., G.S., and H.N.; investigation, P.B.-N., A.M.S., H.G., Y.L., L.S., J.W., and H.N.; resources, H.N. and A.M.S.; data curation, H.G., G.S., S.K.A., and A.C.; writing – original draft, H.N.; writing – review and editing, P.B.-N., H.G., L.S., P.C.M., X.X., J.W., Y.L., S.K.A., A.C., G.S., A.M.S., and H.N.; visualization, H.G., L.S., S.K.A., and H.N.; supervision, H.N.; project administration, H.N.; funding acquisition, Y.L., A.M.S., and H.N.; reviewed and approved final version of manuscript, all authors.
Declaration of interests
The authors declare no competing interests.
Published: March 16, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xcrm.2021.100219.
Supplemental information
: Number of cells per cluster of the first five samples. Sheet 2: Number of cells per cluster in the second analysis; Sheet 3: Average expression levels of various genes in clusters of the first analysis. Sheet 4: Signaling network generated using cluster 11 enriched genes.
Fold change in expression and p values in a given cluster compared to average expression in rest of the clusters combined is given.
References
- 1.Sotiriou C., Neo S.Y., McShane L.M., Korn E.L., Long P.M., Jazaeri A., Martiat P., Fox S.B., Harris A.L., Liu E.T. Breast cancer classification and prognosis based on gene expression profiles from a population-based study. Proc. Natl. Acad. Sci. USA. 2003;100:10393–10398. doi: 10.1073/pnas.1732912100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Curtis C., Shah S.P., Chin S.F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y., METABRIC Group The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prat A., Perou C.M. Mammary development meets cancer genomics. Nat. Med. 2009;15:842–844. doi: 10.1038/nm0809-842. [DOI] [PubMed] [Google Scholar]
- 4.Visvader J.E., Stingl J. Mammary stem cells and the differentiation hierarchy: current status and perspectives. Genes Dev. 2014;28:1143–1158. doi: 10.1101/gad.242511.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim E., Vaillant F., Wu D., Forrest N.C., Pal B., Hart A.H., Asselin-Labat M.L., Gyorki D.E., Ward T., Partanen A., kConFab Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- 6.Pellacani D., Bilenky M., Kannan N., Heravi-Moussavi A., Knapp D.J.H.F., Gakkhar S., Moksa M., Carles A., Moore R., Mungall A.J. Analysis of normal human mammary epigenomes reveals cell-specific active enhancer states and associated transcription factor networks. Cell Rep. 2016;17:2060–2074. doi: 10.1016/j.celrep.2016.10.058. [DOI] [PubMed] [Google Scholar]
- 7.Prat A., Parker J.S., Karginova O., Fan C., Livasy C., Herschkowitz J.I., He X., Perou C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Proia T.A., Keller P.J., Gupta P.B., Klebba I., Jones A.D., Sedic M., Gilmore H., Tung N., Naber S.P., Schnitt S. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. 2011;8:149–163. doi: 10.1016/j.stem.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Colacino J.A., Azizi E., Brooks M.D., Harouaka R., Fouladdel S., McDermott S.P., Lee M., Hill D., Madden J., Boerner J. Heterogeneity of human breast stem and progenitor cells as revealed by transcriptional profiling. Stem Cell Reports. 2018;10:1596–1609. doi: 10.1016/j.stemcr.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anjanappa M., Cardoso A., Cheng L., Mohamad S., Gunawan A., Rice S., Dong Y., Li L., Sandusky G.E., Srour E.F., Nakshatri H. Individualized breast cancer characterization through single-cell analysis of tumor and adjacent normal cells. Cancer Res. 2017;77:2759–2769. doi: 10.1158/0008-5472.CAN-16-3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V., Cancer Genome Atlas Network Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304.e6. doi: 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta P.B., Pastushenko I., Skibinski A., Blanpain C., Kuperwasser C. Phenotypic plasticity: driver of cancer initiation, progression, and therapy resistance. Cell Stem Cell. 2019;24:65–78. doi: 10.1016/j.stem.2018.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim B., Lin Y., Navin N. Advancing cancer research and medicine with single-cell genomics. Cancer Cell. 2020;37:456–470. doi: 10.1016/j.ccell.2020.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen Q.H., Pervolarakis N., Blake K., Ma D., Davis R.T., James N., Phung A.T., Willey E., Kumar R., Jabart E. Profiling human breast epithelial cells using single cell RNA sequencing identifies cell diversity. Nat. Commun. 2018;9:2028. doi: 10.1038/s41467-018-04334-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pal B., Chen Y., Vaillant F., Jamieson P., Gordon L., Rios A.C., Wilcox S., Fu N., Liu K.H., Jackling F.C. Construction of developmental lineage relationships in the mouse mammary gland by single-cell RNA profiling. Nat. Commun. 2017;8:1627. doi: 10.1038/s41467-017-01560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bach K., Pensa S., Grzelak M., Hadfield J., Adams D.J., Marioni J.C., Khaled W.T. Differentiation dynamics of mammary epithelial cells revealed by single-cell RNA sequencing. Nat. Commun. 2017;8:2128. doi: 10.1038/s41467-017-02001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakshatri H., Anjanappa M., Bhat-Nakshatri P. Ethnicity-dependent and -independent heterogeneity in healthy normal breast hierarchy impacts tumor characterization. Sci. Rep. 2015;5:13526. doi: 10.1038/srep13526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakshatri H., Kumar B., Burney H.N., Cox M.L., Jacobsen M., Sandusky G.E., D’Souza-Schorey C., Storniolo A.M.V. Genetic ancestry-dependent differences in breast cancer-induced field defects in the tumor-adjacent normal breast. Clin. Cancer Res. 2019;25:2848–2859. doi: 10.1158/1078-0432.CCR-18-3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degnim A.C., Visscher D.W., Hoskin T.L., Frost M.H., Vierkant R.A., Vachon C.M., Shane Pankratz V., Radisky D.C., Hartmann L.C. Histologic findings in normal breast tissues: comparison to reduction mammaplasty and benign breast disease tissues. Breast Cancer Res. Treat. 2012;133:169–177. doi: 10.1007/s10549-011-1746-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas N., Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rosenbluth J.M., Schackmann R.C.J., Gray G.K., Selfors L.M., Li C.M., Boedicker M., Kuiken H.J., Richardson A., Brock J., Garber J. Organoid cultures from normal and cancer-prone human breast tissues preserve complex epithelial lineages. Nat. Commun. 2020;11:1711. doi: 10.1038/s41467-020-15548-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tyrer J., Duffy S.W., Cuzick J. A breast cancer prediction model incorporating familial and personal risk factors. Stat. Med. 2004;23:1111–1130. doi: 10.1002/sim.1668. [DOI] [PubMed] [Google Scholar]
- 23.Rohlenova K., Goveia J., García-Caballero M., Subramanian A., Kalucka J., Treps L., Falkenberg K.D., de Rooij L.P.M.H., Zheng Y., Lin L. Single-cell RNA sequencing maps endothelial metabolic plasticity in pathological angiogenesis. Cell Metab. 2020;31:862–877.e14. doi: 10.1016/j.cmet.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 24.Morsing M., Kim J., Villadsen R., Goldhammer N., Jafari A., Kassem M., Petersen O.W., Rønnov-Jessen L. Fibroblasts direct differentiation of human breast epithelial progenitors. Breast Cancer Res. 2020;22:102. doi: 10.1186/s13058-020-01344-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pascual G., Avgustinova A., Mejetta S., Martín M., Castellanos A., Attolini C.S., Berenguer A., Prats N., Toll A., Hueto J.A. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature. 2017;541:41–45. doi: 10.1038/nature20791. [DOI] [PubMed] [Google Scholar]
- 26.Davis M.B., Walens A., Hire R., Mumin K., Brown A.M., Ford D., Howerth E.W., Monteil M. Distinct transcript isoforms of the atypical chemokine receptor 1 (ACKR1)/duffy antigen receptor for chemokines (DARC) gene are expressed in lymphoblasts and altered isoform levels are associated with genetic ancestry and the duffy-null allele. PLoS ONE. 2015;10:e0140098. doi: 10.1371/journal.pone.0140098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sato T., Goyama S., Kataoka K., Nasu R., Tsuruta-Kishino T., Kagoya Y., Nukina A., Kumagai K., Kubota N., Nakagawa M. Evi1 defines leukemia-initiating capacity and tyrosine kinase inhibitor resistance in chronic myeloid leukemia. Oncogene. 2014;33:5028–5038. doi: 10.1038/onc.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baharudin R., Tieng F.Y.F., Lee L.H., Ab Mutalib N.S. Epigenetics of SFRP1: the dual roles in human cancers. Cancers (Basel) 2020;12:445. doi: 10.3390/cancers12020445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zaret K.S., Carroll J.S. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McBryan J., Howlin J., Kenny P.A., Shioda T., Martin F. ERalpha-CITED1 co-regulated genes expressed during pubertal mammary gland development: implications for breast cancer prognosis. Oncogene. 2007;26:6406–6419. doi: 10.1038/sj.onc.1210468. [DOI] [PubMed] [Google Scholar]
- 31.Ali S., Coombes R.C. Estrogen receptor alpha in human breast cancer: occurrence and significance. J. Mammary Gland Biol. Neoplasia. 2000;5:271–281. doi: 10.1023/a:1009594727358. [DOI] [PubMed] [Google Scholar]
- 32.Marcato P., Dean C.A., Pan D., Araslanova R., Gillis M., Joshi M., Helyer L., Pan L., Leidal A., Gujar S. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 2011;29:32–45. doi: 10.1002/stem.563. [DOI] [PubMed] [Google Scholar]
- 33.Sørlie T., Perou C.M., Tibshirani R., Aas T., Geisler S., Johnsen H., Hastie T., Eisen M.B., van de Rijn M., Jeffrey S.S. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nolan E., Lindeman G.J., Visvader J.E. Out-RANKing BRCA1 in mutation carriers. Cancer Res. 2017;77:595–600. doi: 10.1158/0008-5472.CAN-16-2025. [DOI] [PubMed] [Google Scholar]
- 35.Luk I.Y., Reehorst C.M., Mariadason J.M. ELF3, ELF5, EHF and SPDEF transcription factors in tissue homeostasis and cancer. Molecules. 2018;23:2191. doi: 10.3390/molecules23092191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morel A.P., Ginestier C., Pommier R.M., Cabaud O., Ruiz E., Wicinski J., Devouassoux-Shisheboran M., Combaret V., Finetti P., Chassot C. A stemness-related ZEB1-MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat. Med. 2017;23:568–578. doi: 10.1038/nm.4323. [DOI] [PubMed] [Google Scholar]
- 37.Wang D., Cai C., Dong X., Yu Q.C., Zhang X.O., Yang L., Zeng Y.A. Identification of multipotent mammary stem cells by protein C receptor expression. Nature. 2015;517:81–84. doi: 10.1038/nature13851. [DOI] [PubMed] [Google Scholar]
- 38.Kfoury Y., Scadden D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16:239–253. doi: 10.1016/j.stem.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 39.Razavi P., Chang M.T., Xu G., Bandlamudi C., Ross D.S., Vasan N., Cai Y., Bielski C.M., Donoghue M.T.A., Jonsson P. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34:427–438.e6. doi: 10.1016/j.ccell.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walter W., Thomalla J., Bruhn J., Fagan D.H., Zehowski C., Yee D., Skildum A. Altered regulation of PDK4 expression promotes antiestrogen resistance in human breast cancer cells. Springerplus. 2015;4:689. doi: 10.1186/s40064-015-1444-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sundararajan V., Gengenbacher N., Stemmler M.P., Kleemann J.A., Brabletz T., Brabletz S. The ZEB1/miR-200c feedback loop regulates invasion via actin interacting proteins MYLK and TKS5. Oncotarget. 2015;6:27083–27096. doi: 10.18632/oncotarget.4807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pervolarakis N., Nguyen Q.H., Williams J., Gong Y., Gutierrez G., Sun P., Jhutty D., Zheng G.X.Y., Nemec C.M., Dai X. Integrated single-cell transcriptomics and chromatin accessibility analysis reveals regulators of mammary epithelial cell identity. Cell Rep. 2020;33:108273. doi: 10.1016/j.celrep.2020.108273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gomez B.P., Riggins R.B., Shajahan A.N., Klimach U., Wang A., Crawford A.C., Zhu Y., Zwart A., Wang M., Clarke R. Human X-box binding protein-1 confers both estrogen independence and antiestrogen resistance in breast cancer cell lines. FASEB J. 2007;21:4013–4027. doi: 10.1096/fj.06-7990com. [DOI] [PubMed] [Google Scholar]
- 44.Davies M.P., Barraclough D.L., Stewart C., Joyce K.A., Eccles R.M., Barraclough R., Rudland P.S., Sibson D.R. Expression and splicing of the unfolded protein response gene XBP-1 are significantly associated with clinical outcome of endocrine-treated breast cancer. Int. J. Cancer. 2008;123:85–88. doi: 10.1002/ijc.23479. [DOI] [PubMed] [Google Scholar]
- 45.Ciriello G., Gatza M.L., Beck A.H., Wilkerson M.D., Rhie S.K., Pastore A., Zhang H., McLellan M., Yau C., Kandoth C., TCGA Research Network Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–519. doi: 10.1016/j.cell.2015.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coloff J.L., Brugge J.S. Metabolic changes promote rejection of oncogenic cells. Nat. Cell Biol. 2017;19:414–415. doi: 10.1038/ncb3521. [DOI] [PubMed] [Google Scholar]
- 47.Karaayvaz M., Cristea S., Gillespie S.M., Patel A.P., Mylvaganam R., Luo C.C., Specht M.C., Bernstein B.E., Michor F., Ellisen L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018;9:3588. doi: 10.1038/s41467-018-06052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim C., Gao R., Sei E., Brandt R., Hartman J., Hatschek T., Crosetto N., Foukakis T., Navin N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173:879–893.e13. doi: 10.1016/j.cell.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kwan T.T., Bardia A., Spring L.M., Giobbie-Hurder A., Kalinich M., Dubash T., Sundaresan T., Hong X., LiCausi J.A., Ho U. A digital RNA signature of circulating tumor cells predicting early therapeutic response in localized and metastatic breast cancer. Cancer Discov. 2018;8:1286–1299. doi: 10.1158/2159-8290.CD-18-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lappalainen T., Sammeth M., Friedländer M.R., ’t Hoen P.A.C., Monlong J., Rivas M.A., Gonzàlez-Porta M., Kurbatova N., Griebel T., Ferreira P.G., Geuvadis Consortium Transcriptome and genome sequencing uncovers functional variation in humans. Nature. 2013;501:506–511. doi: 10.1038/nature12531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perkins S.M., Bales C., Vladislav T., Althouse S., Miller K.D., Sandusky G., Badve S., Nakshatri H. TFAP2C expression in breast cancer: correlation with overall survival beyond 10 years of initial diagnosis. Breast Cancer Res. Treat. 2015;152:519–531. doi: 10.1007/s10549-015-3492-2. [DOI] [PubMed] [Google Scholar]
- 52.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018;36:411–420. doi: 10.1038/nbt.4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stuart, T., Butler, A., Hoffman, P., Hafemeister, C., Papalexi, E., Mauck, W.M., 3rd, Hao, Y., Stoeckius, M., Smibert, P., and Satija, R. (2019). Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed]
- 54.McCarthy D.J., Campbell K.R., Lun A.T., Wills Q.F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017;33:1179–1186. doi: 10.1093/bioinformatics/btw777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wickham H. Springer-Verlag; 2016. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- 56.Gu Z., Eils R., Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016;32:2847–2849. doi: 10.1093/bioinformatics/btw313. [DOI] [PubMed] [Google Scholar]
- 57.Tibshirani R., Hastie T., Narasimhan B., Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. USA. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parker J.S., Mullins M., Cheang M.C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009;27:1160–1167. doi: 10.1200/JCO.2008.18.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pereira B., Chin S.F., Rueda O.M., Vollan H.K., Provenzano E., Bardwell H.A., Pugh M., Jones L., Russell R., Sammut S.J. The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat. Commun. 2016;7:11479. doi: 10.1038/ncomms11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ellrott K., Bailey M.H., Saksena G., Covington K.R., Kandoth C., Stewart C., Hess J., Ma S., Chiotti K.E., McLellan M., MC3 Working Group. Cancer Genome Atlas Research Network Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 2018;6:271–281.e7. doi: 10.1016/j.cels.2018.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Taylor A.M., Shih J., Ha G., Gao G.F., Zhang X., Berger A.C., Schumacher S.E., Wang C., Hu H., Liu J., Cancer Genome Atlas Research Network Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33:676–689.e3. doi: 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao Q., Liang W.W., Foltz S.M., Mutharasu G., Jayasinghe R.G., Cao S., Liao W.W., Reynolds S.M., Wyczalkowski M.A., Yao L., Fusion Analysis Working Group. Cancer Genome Atlas Research Network Driver fusions and their implications in the development and treatment of human cancers. Cell Rep. 2018;23:227–238.e3. doi: 10.1016/j.celrep.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V., Cancer Genome Atlas Research Network An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416.e11. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C., Dimitriadoy S., Liu D.L., Kantheti H.S., Saghafinia S., Cancer Genome Atlas Research Network Oncogenic signaling pathways in The Cancer Genome Atlas. Cell. 2018;173:321–337.e10. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bhandari V., Hoey C., Liu L.Y., Lalonde E., Ray J., Livingstone J., Lesurf R., Shiah Y.J., Vujcic T., Huang X. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019;51:308–318. doi: 10.1038/s41588-018-0318-2. [DOI] [PubMed] [Google Scholar]
- 66.Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kassambara A., Kosinski M., Biecek P. 2019. Drawing survival curves using ‘ggplot2’. R package version 0.4.6.https://CRAN.R-project.org/package=survminer [Google Scholar]
- 69.R Development Core Team . R Foundation for Statistical Computing; 2020. R: A language and environment for statistical computing.https://www.R-project.org/ [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
: Number of cells per cluster of the first five samples. Sheet 2: Number of cells per cluster in the second analysis; Sheet 3: Average expression levels of various genes in clusters of the first analysis. Sheet 4: Signaling network generated using cluster 11 enriched genes.
Fold change in expression and p values in a given cluster compared to average expression in rest of the clusters combined is given.
Data Availability Statement
The accession number for the single cell sequence data reported in this paper is GEO GSE164898.