

Abstract
Summary: A case of cerebral amyloid angiopathy is presented with MR imaging findings of high intense signal on T2-weighted sequences at the level of the white and gray matter of both hemispheres in the absence of neuroradiologic signs of cerebral hemorrhage. The biopsy specimen revealed deposition of amyloid in the walls of the intracranial arterial branches and focal ischemic changes and gliosis in the gray and white matter. We consider this presentation to be very unusual in patients affected by cerebral amyloid angiopathy.
Amyloid is an eosinophilic, insoluble extracellular protein that was first discovered in the brain in 1909 (1). Intracranially, the most common sites of amyloid deposition are blood vessel walls. Deposits of amyloid have been discovered also in senile plaques, in the spongiform encephalitides of kuru, in association with Jakob-Creutzfeldt disease, and in association with other pathologic conditions affecting the white matter (2–4).
Recent literature has reported primary solitary amyloidosis (amyloidoma) as a rare subset of amyloidosis in which the amyloid deposit is isolated and not related to a systemic process or plasma cell dyscrasia (5–7). Cerebral amyloid angiopathy (CAA) is characterized by deposition of homogeneous eosinophilic material in the media and adventitia of arterioles and small arteries of the cortex and leptomeninges (8). CAA is mostly found at autopsy in elderly or demented patients or in cases of massive isolated or multi-compartmental intracranial hemorrhage. CAA is also found in association with several different pathologic conditions, such as arteriovenous malformation (9), radiation necrosis (10), demyelinating disease (11), adult mongolism, dementia pugilistica, chronic vasculitis, and familial cerebral hemorrhage (12). The most common presentation of CAA is intracranial hemorrhage (Table).
MR findings in cerebral amyloid angiopathy

Gray et al (13), in 1985, reported the coexistence of subacute or chronic edematous lesions of the white matter, in patients with hemorrhagic CAA, in regions corresponding to hypodensity on premortem CT scans. They postulated the possibility of concomitant leukoencephalopathy caused by a mechanism of hypoperfusion of the distal white matter. Loes et al (14), in 1990, reported four cases of biopsy-proved CAA with diffuse white matter hyperintensity on T2-weighted MR imaging sequences, and they attributed the signal abnormality to a hypoperfusion of distal white matter resulting from vascular disease. Of the four described cases, only one presented without intracerebral hemorrhage. We present the case of a patient affected by CAA with MR imaging findings of diffuse signal intensity abnormality at the level of gray and white matter of both hemispheres and in the absence of hemorrhage.
Case Report
A 41-year-old patient presented with a history of seizure disorder in June 1999. He stated that he was in good health until 1 week before admission, when he experienced a generalized tonicoclonic seizure. Before that, he had experienced on-and-off headaches for 5 months with no nausea or vomiting and no focal symptoms. The patient's medical history was remarkable for insulin-dependent diabetes mellitus and recent left toe amputation.
At the time of admission, cardiac and abdominal statuses were unremarkable. A neurologic examination revealed an alert and oriented patient. He had normal strength in all limbs and normal sensation and reflexes (+1 in all limbs). Cranial nerves were normal bilaterally.
The patient underwent MR imaging three times during a period of 1 year. The first MR imaging examination, performed in May 1999, showed diffuse signal intensity abnormality in the right temporo-occipital region, with mild mass effect, effacement of the sulci, and moderate compression of the right temporal ventricle. Smaller lesions were also noted in the left occipital lobe, in the anterior aspect of the right insula, and in the right frontoparietal region (Fig 1). At the time, the MR findings were judged to be consistent with multifocal glioma. On June 29, 1999, the patient underwent a right temporal craniotomy for biopsy of the lesion.
fig 1.

Images obtained at the time of the first hospital admission. Axial view turbo spin-echo T2-weighted sequence.
A, and B, Diffuse high intensity signal involving the cortex and the white matter of the right temporo-parieto-occipital region. Note also the involvement of the right frontal and left parieto-occipital regions.
C, High intense signal can be seen in the occipital lobes bilaterally, involving both the gray and white matter.
A histopathologic examination of the specimen revealed numerous abnormal vessels with focal ischemic changes and gliosis. Some vessels were completely occluded with an eosinophilic material deposited in the vessel wall. Congo red stain produced, in many of the vessels, the typical apple green birefringence, and immunohistochemistry for beta amyloid was strongly positive. Immunohistochemistry for glial fibrillary acidic protein showed areas of focal gliosis surrounding the occluded or partially occluded blood vessels, in keeping with the likely ischemic damage to the cortex (Fig 2). The pathology report excluded the presence of tumor.
fig 2.

Histopathologic examinations of the lesions.
A, Hematoxylin and eosin stain. Vessel walls are thickened by an amorphous eosinophilic substance, and the lumen of the vessel is partially occluded.
B, Immunohistochemistry for glial fibrillary acidic protein shows perivascular gliosis.
C, Immunohistochemistry for β-amyloid shows the presence of amyloid deposits in the vessel walls.
A second MR imaging examination was performed in July 1999. Except for the evidence of surgical changes, the findings were unchanged compared with the previous findings.
The last MR imaging examination was performed in May 2000, and we observed some changes in the distribution of the lesions. We appreciated a progression of the disease, with more severe involvement of the left parietal and temporal lobes but, at the same time, a moderate decrease of the hyperintense areas in the right parieto-occipital region (Fig 3).
fig 3.
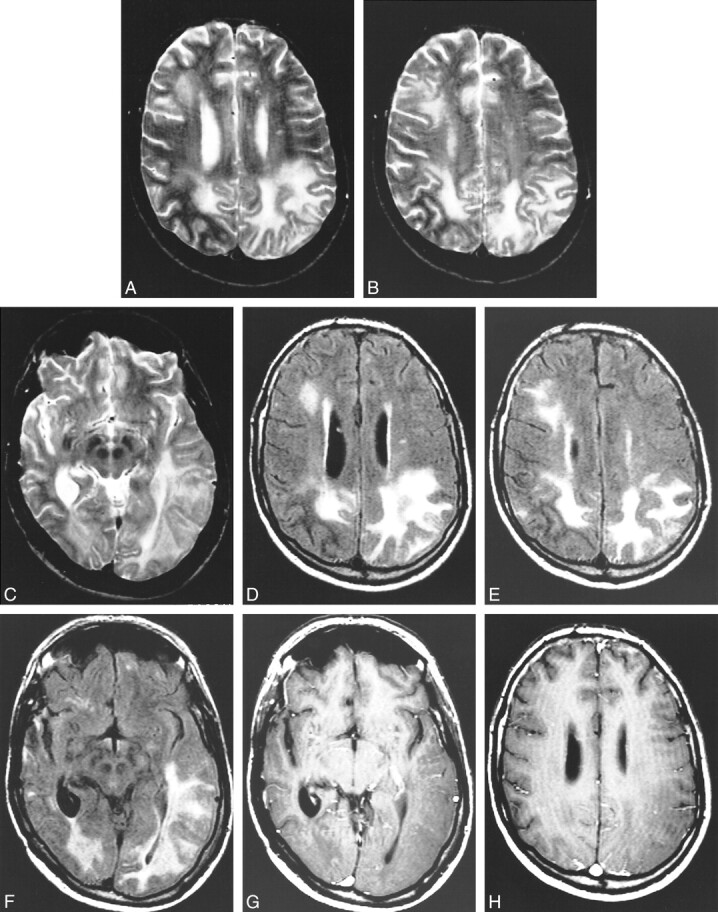
Images obtained at the 1-year follow-up examination.
A, and B, Axial view spin-echo T2-weighted sequence. High intense signal is present in the gray and white matter of the right frontal lobe and of both parietal lobes. Note that compared with figure 1, there is progression of the disease in the left parietal lobe. The right parietal lobe, on the contrary, looks less involved.
C, Axial view spin-echo T2-weighted sequence. The lesion on the left is involving also the temporal lobe.
D, Axial view fluid-attenuated inversion recovery sequence obtained at the same level as that shown in A. The parietal lobes are diffusely hyperintense. High intensity signal is also present in the right frontal lobe. The right parietal lobe appears less involved than on the previous MR image.
E, Axial view fluid-attenuated inversion recovery sequence obtained at the same level as that shown in B. The parietal lobes are diffusely hyperintense. High intensity signal is also present in the right frontal lobe. The right parietal lobe appears less involved than on the previous MR image.
F, Axial view fluid-attenuated inversion recovery sequence obtained at the same level as that shown in C. The parietal lobes are diffusely hyperintense. High intensity signal is also present in the right frontal lobe. The right parietal lobe appears less involved than on the previous MR image.
G, Infused axial view spin-echo T1-weighted sequence. There is no evidence of abnormal contrast enhancement in the diseased regions.
H, Infused axial view spin-echo T1-weighted sequences. There is no evidence of abnormal contrast enhancement in the diseased regions.
Discussion
The usual neurologic presentation of CAA is spontaneous cerebral hemorrhage. The incidence of CAA in patients with cerebral hemorrhage varies from 4% to 10% (15), whereas cerebral hemorrhage has been reported in 40% of autopsy-proved cases of CAA (4). Microscopic changes within blood vessels are characteristically reported. Amyloid deposition in the walls of large leptomeningeal arteries and superficial cortex arteries shows uniform and diffuse infiltration of the entire wall. In the same affected vessels, a superimposed fibrinoid degeneration is documented, which usually causes microaneurysmal dilation of the wall, sometimes concomitant with luminal stenosis (4). The presence of microaneurysms could be assumed to be responsible for massive intraparenchymal hemorrhages in patients with CAA, and the luminal stenosis could be responsible for the hypoxic-ischemic changes. Even though the most common presentation of CAA is spontaneous cerebral hemorrhage, a few reports have been published of cases of non-hemorrhagic masses (16) and of multifocal non-hemorrhagic lesions (14).
Okazaki et al (4) were the first to describe the presence of ischemic cerebral lesions varying in number and size, in their report of 23 autopsied cases of CAA, mostly in the cerebral cortex and immediate subcortical areas. Gray et al (13) examined 12 brains with diffuse hemorrhagic CAA, and in eight brains, they found severe white matter lesions consisting of both diffuse and focal areas of demyelination in the hemispheric white matter, with sparing of U fibers and corpus callosum. For these eight patients, microscopy revealed spongiosis, swollen oligodendroglia, widening of the perivascular spaces with edema, hyalinization of blood vessels walls, myelin loss with partial neuronal loss, and gliosis. Congophilic vessels were not found in the white matter of these patients, and the authors postulated that amyloid deposits in the meningocortical arteries can cause hypoperfusion and ischemic changes at the level of the white matter. Two of the four cases of CAA reported by Loes et al (14) showed white matter signal abnormalities on MR images in the absence of hemorrhage. One of the patients experienced hemorrhage 1 year after undergoing MR imaging. Even in the absence of pathologic correlation, the authors suspected that the white matter lesions observed in patients with and without hemorrhage were related to CAA, so they concluded that the white matter lesions associated with CAA are not specific to this disorder but rather reflect hypoperfusion of distal white matter resulting from vascular disease.
In our case, we observed severe changes in the signal of white and gray matter in agreement with previous literature data (14, 16); moreover, with 1 year of MR imaging follow-up, we appreciated an evolution of the MR pattern that until now has never been reported or postulated. We observed a progression of the disease but, at the same time, a regression of the abnormality in other regions of the brain. This allows us to postulate the possibility of the coexistence of two different pathologic changes in the white and gray matter responsible for the high signal intensity on T2-weighted sequences. We attribute the stability of the lesion over time to myelin loss and astrocytic gliosis (13), whereas the observed partial regression of the disease at the level of the right parieto-occipital region could be due to variation in the degree of vasogenic edema. This double condition supports the hypoxic theory, justifying the signal intensity abnormality's lack of correlation to hemorrhage in cases of CAA. The clinical history of the patient included complicated insulin-dependent diabetes. Diabetes is known to be a risk factor for cerebrovascular disease, causing arteriosclerotic changes in the walls of the vessels. The lesions that can cause cerebrovascular disease in diabetes are various: atherosclerosis of the aorta and large extracranial arteries, atherosclerosis of large intracranial arteries, intracranial atheromatous branch disease, degenerative changes such as lipohyalinosis, and fibrinoid changes within penetrating arteries branches (responsible for subcortical brain infarcts) (17). For our patient, digital subtraction angiography of the carotid arteries and intracranial major arteries showed no evidence of atherosclerotic changes; the biopsy specimen revealed, in the walls of the intracranial arterial branches, deposition of acellular eosinophilic material consistent with amyloid and the absence of lesions fitting with any definite diagnostic category related to the diabetes mellitus. These findings make the changes in the white and gray matter less likely related to the diabetes.
After the administration of contrast material, we did not observe contrast enhancement, which meant that the blood-brain barrier had remained intact (Fig 3G and H). This finding was in agreement with the absence, in the pathology, of inflammatory process, infarction, and neovascular proliferation related to tumoral growing in the regions of the biopsy. Gradient-echo sequences were not performed because at the time of the examination, we did not suspect the possibility of CAA. The presence of micro-hemorrhage may have been overlooked.
Amyloid depositions have been reported also in association with several pathologic conditions involving the brain, such as solitary amyloidoma, spongiform encephalitis, Alzheimer's disease, and other neurodegenerative disorders. Differential diagnosis was obtained in our case by the clinical presentation's exclusion of a neurodegenerative disease and was confirmed by the histologic findings; amyloid deposition was discovered only in the vessel walls causing thrombosis and not in the brain parenchyma. This condition is unique of CAA.
This is a case of a very unusual manifestation of congophilic amyloid angiopathy in the absence of intracranial hemorrhage. MR imaging findings revealed the presence of a diffuse signal intensity abnormality involving the gray and white matter of both hemispheres. We postulate, in agreement with previously published data, the presence of hypoxic-ischemic changes in the brain due to hypoperfusion caused by amyloid deposits along vessel walls. The patient underwent a 1-year follow-up examination, and the MR imaging findings revealed both an increase and a decrease in the size of the lesion, with altered signal intensity in the different involved regions of the brain. This leads us to consider two different pathologic changes in the white and gray matter. We attribute the stable lesion to myelin loss and astrocytic gliosis (13) and the lesions in partial regression to vasogenic edema.
Footnotes
This work was supported in part by a grant from the Italian Society of Neuroradiology (to M.C.).
Address reprint requests to Denis Melanson, MD, Department of Neuroradiology, Montreal Neurological Hospital and Institute of McGill University, 3801 rue University, Montreal, Quebec, H3A 2B4 Canada.
References
- 1.Oppenheimen G. Uber drusige nekrozen in der grosshirnrinde. Neurol Centralbi 1909;28:410-413 [Google Scholar]
- 2.Bruni J, Bilbao JM, Pritzker KP. Vascular amyloid in the aging CNS: clinicopathological study and literature review. Can J Neurol Sci 1977;4:239-244 [DOI] [PubMed] [Google Scholar]
- 3.Vanley CT, Aguilar MJ, Kleinhenz RJ, Lagios MD. Cerebral amyloid angiopathy. Hum Pathol 1981;12:609-619 [DOI] [PubMed] [Google Scholar]
- 4.Okazaki H, Reagan TJ, Campbell RJ. Clinicopathological studies of primary cerebral amyloid angiopathy. Mayo Clin Proc 1979;54:22-31 [PubMed] [Google Scholar]
- 5.Cohen M, Lanska D. Amyloidoma of the CNS: I. clinical and pathologic study. Neurology 1992;42:2019-2023 [DOI] [PubMed] [Google Scholar]
- 6.Vidal RG, Kiso J. Amyloidoma of the CNS: II. immunohistochemical and biochemical study. Neurology 1992;42:2024-2028 [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Krol G, Rosenblum M. Primary amyloidoma of the brain: CT and MR presentation. AJNR Am J Neuroradiol 1995;16:712-714 [PMC free article] [PubMed] [Google Scholar]
- 8.Vanley CT, Aguilar MJ, Kleinhenz RJ, Lagios MD. Cerebral amyloid angiopathy. Hum Pathol 198;12:609-616 [DOI] [PubMed] [Google Scholar]
- 9.Peterson EW, Schulz DM. Amyloid in vessels of a vascular malformation in brain. Arch Pathol 1961;72:480-483 [PubMed] [Google Scholar]
- 10.Lowemberg-Scharenberg K, Basset RC. Amyloid degeneration of the human brain following x-ray therapy. J Neuropathol Exp Neurol 1950;9:93-102 [DOI] [PubMed] [Google Scholar]
- 11.Heffner RR, Porro RS, Olson ME, Earle KM. A demyelinating disorder associated with cerebrovascular amyloid angiopathy. Arch Neurol 1976;33:501-506 [DOI] [PubMed] [Google Scholar]
- 12.Cosgrove GR, Leblanc R, Villemure KM, Ethier R. Cerebral amyloid angiopathy. Neurology 1985;35:625-631 [DOI] [PubMed] [Google Scholar]
- 13.Gray F, Dubas F, Roullet E, Escourolle R. Leukoencephalopathy in diffuse hemorrhagic cerebral amyloid angiopathy. Ann Neurol 1985;18:54-59 [DOI] [PubMed] [Google Scholar]
- 14.Loes DJ, Biller J, Yuh W, et al. Leukoencephalopathy in cerebral angiopathy: MR imaging in four cases. AJNR Am J Neuroradiol 1990;11:485-488 [PMC free article] [PubMed] [Google Scholar]
- 15.Mandybur TI, Bates SR. Fatal massive intracerebral hemorrhage complicating cerebral amyloid angiopathy. Arch Neurol 1978;35:246-248 [DOI] [PubMed] [Google Scholar]
- 16.Osumi AK, Tien RD, Felsberg GJ, Rosenbloom M. Cerebral amyloid angiopathy presenting as a brain mass. AJNR Am J Neuroradiol 1995;16:911-915 [PMC free article] [PubMed] [Google Scholar]
- 17.Caplan LR. Diabetes and brain ischemia. Diabetes 1996;45:95-97 [DOI] [PubMed] [Google Scholar]