

Abstract
BACKGROUND AND PURPOSE: Sphenoid dysplasia associated with neurofibromatosis type 1 is classically thought to be primarily related to abnormal development of the sphenoid bone. We investigated the possibility that these changes may be progressive.
METHODS: We conducted a retrospective review of sphenoid bone changes in all patients with craniofacial neurofibromatosis type 1 who had undergone CT (31 patients) and MR imaging (seven patients) at our facility. A review of repeat images of 20 patients permitted analysis of progressive sphenoid bone changes.
RESULTS: Eighteen patients had abnormalities of the sphenoid wings, 13 of whom also had enlargement of the middle cranial fossa compatible with descriptions of classic sphenoid dysplasia. All the patients with sphenoid dysplasia had neurofibromas in the ipsilateral superficial temporal fossa that were often contiguous with a radiologically abnormal temporo-squamosal suture. All except one had tumor infiltration in the deep orbit, contiguous with the sphenoid wings. Four patients had radiologic evidence of progressive sphenoid bone changes over time.
CONCLUSION: The origin of sphenoid bone changes may be multifactorial. A modified concept of sphenoid dysplasia is proposed that emphasizes interaction between neurofibromas and sphenoid bone during skull development.
Neurofibromatosis type 1 (NF1) is an autosomal dominant disorder caused by mutations of the neurofibromin gene located on the long arm of chromosome 17. Neurofibromin is a tumor suppressor gene, the malfunction of which results in part in benign and occasionally malignant sensory nerve tumors, often in utero or early in life (1). Sphenoid dysplasia is a prominent but not entirely pathognomonic facial feature of NF1 (1), with radiologic characteristics described broadly before the era of CT to include defects in the greater sphenoid wing and enlargement of the middle cranial fossa (2).
Some early surgical explorations of patients with NF1 with cranial bone defects failed to reveal tumor, and a congenital neuroectodermal and mesodermal maldevelopment hypothesis was proposed to explain these profound bone changes (3). Current neuroimaging has dramatically improved the ability to resolve subtle soft-tissue and bone changes due to neurofibroma infiltration of orbital nerves, sclera, choroid, extraocular muscles, and even the optic nerve sheath (4). Radiologic and clinical reports (5) have now shown that neurofibromas are frequently present near NF1 facial bone changes (6), implying that some bone changes might be caused by contiguous tumor. The report by Macfarlane et al (7) of a young patient with progressive sphenoid bone changes shown on CT studies performed 6 years apart has raised the question of whether the abnormalities of sphenoid dysplasia associated with NF1 occur primary to abnormal bone formation or secondary to the presence of adjacent tumor. We have reviewed the neuroimaging experience at this institution in the hope of resolving some of the issues regarding the cause and progression of sphenoid dysplasia.
Methods
Group 1 consisted of all 31 patients with the diagnosis of NF1 who had CT scans on record in the Diagnostic Imaging Department. These patients had been diagnosed as having NF1 based on National Institutes of Health clinical criteria (all patients) and biopsy of plexiform neurofibromas (20 patients). Seven patients had MR images on record as well. After institutional review board approval, an attempt was made to recall each group 1 patient with a scan and/or image obtained more than 2 years previously for clinical examination and repeat CT. Fourteen patients were evaluated in this fashion and were included in group 1a, which consisted of 20 patients from group 1 who had undergone more than one neuroimaging study during a period of 2 or more years, the results of which could be analyzed for possible progressive sphenoid bone changes. Demographics of group 1 and group 1a were compared by using the Student’s t test.
Most CT studies (initial and repeat) were performed using the same scanning protocol, including 5-mm-thick sections obtained at the level of the posterior fossa structures and upper cranium and 1.5-mm contiguous sections for the orbits and optic pathways with neuro-ocular incidence. Several patients had additional coronal views. Bone and soft-tissue algorithm reconstructions were available for review. MR imaging studies were performed with 1.5-T units in the Riyadh community, by using a variety of imaging protocols.
The radiographic criteria for sphenoid dysplasia consisted of remodeling or decalcification of a sufficient segment of the sphenoid wings so that an anatomic variant could be ruled out and anteroposterior enlargement of the middle cranial fossa. These assessments were made by using the contralateral anatomic structures for comparison. When the disorder was bilateral, overall deformity of the skull was also taken into account.
Results
Group 1 consisted of 31 patients (18 male and 13 female) with an average age of 14.0 years (age range, 1–40 years). Demographics of group la were not statistically different.
Group 1 Analysis
Eighteen patients had decalcification or remodeling of the greater or lesser wing of the sphenoid bone. This process included some remodeling of the sphenoid body and expansion of orbital foramina due to neurofibroma infiltration of orbital nerves. All except one of these patients had extra-axial tumor present in the posterior orbit and pterygoid fossa, contiguous with abnormal bone (Fig 1A). This type of radiologic change was also noted in other areas of the skull, including the jaw, frontal and temporal bones (Fig 1B), and even occipital bone (Fig 1C).
Fig 1.

Series of axial CT scans.
A, Infiltration and decalcification of the sphenoid wing underlying neurofibromas.
B, Infiltration and decalcification of the temporal bone underlying neurofibromas.
C, Infiltration and decalcification of the occipital bone underlying neurofibromas.
Thirteen of these 18 patients also had enlargement of the middle cranial fossa in primarily the anteroposterior dimension, with an appearance of the greater sphenoid wing typical of classic sphenoid dysplasia (Fig 2). Enlargement of the middle cranial fossa was always associated with tumor in the ipsilateral superficial temporal fossa, and some of these patients also had radiologically abnormal squamosa and temporal squamosal sutures (Fig 3). One patient had discrete expansion of the middle cranial fossa with no tumor detected near the sphenoid bone (Fig 4), but 12 of the 13 patients had tumor contiguous to altered sphenoid bone. The temporal lobe did not herniate forward behind the distorted greater sphenoid wing in patients with expanded middle cranial fossae because every patient had an arachnoid cyst between the anterior temporal lobe and the anterior wall of the middle cranial fossa. Tumor was present in the pterygoid fossa of nine of the 13 patients with sphenoid dysplasia.
Fig 2.
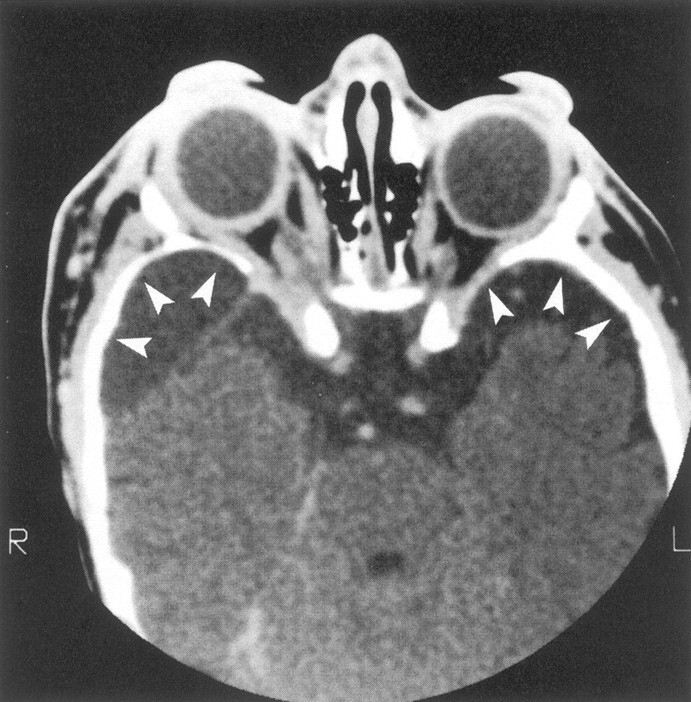
Axial contrast-enhanced CT scan shows bilateral facial soft-tissue tumor infiltration, bilateral enlarged middle cranial fossae (arrowheads), bilateral large globes (buphthalmos), and infiltration of both optic nerve sheaths.
Fig 3.
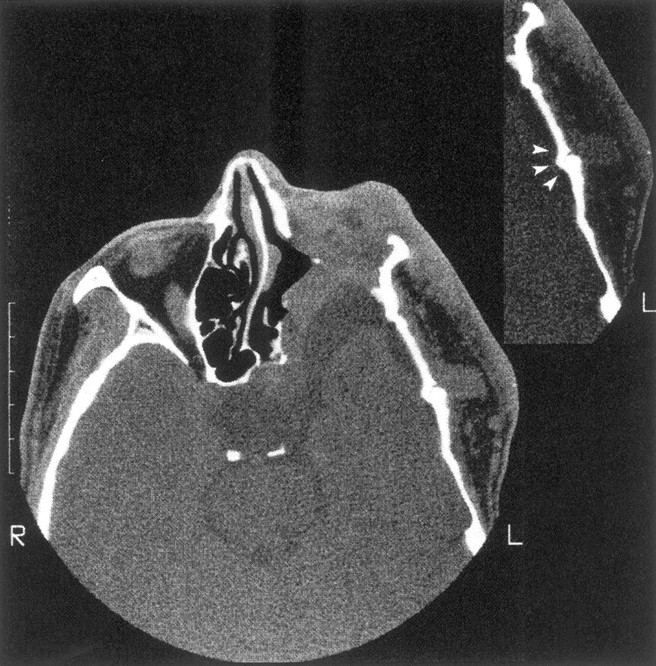
Enlargement of the left middle cranial fossa with temporal arachnoid cyst, absence of the left sphenoid wing, and flattening of the temporal bone. There is tumor invasion of the orbit with reduced orbital volume, and the left eye was enucleated. The insert shows an abnormal temporal squamosa suture (arrowheads) underlying tumor in the left superficial temporal fossa.
Fig 4.
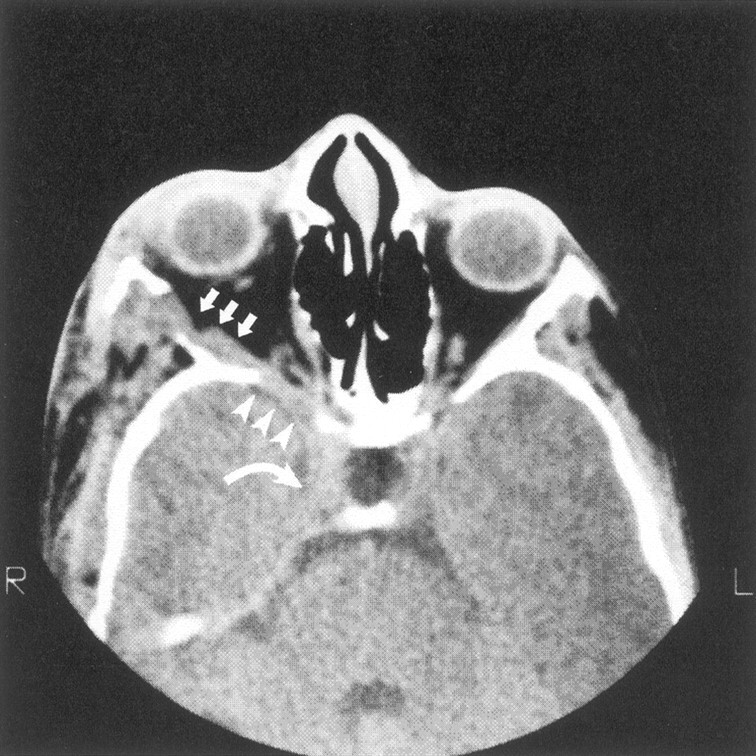
Axial CT scan shows decalcification of sphenoid bone (arrowheads) adjacent to neurofibroma infiltration of the lateral rectus muscle (arrows) in the absence of middle cranial fossa enlargement. Tumor also enlarges the superior orbital fissure and extends into the cavernous sinus (curved arrow).
Five of the 18 patients had decalcification of various portions of the sphenoid bone with contiguous extra-axial tumor but without expansion of the middle cranial fossa (Fig 5). One patient had a smaller (rather than larger) middle cranial fossa on the affected side, with flattened sphenoid and temporal bones (Fig 6).
Fig 5.
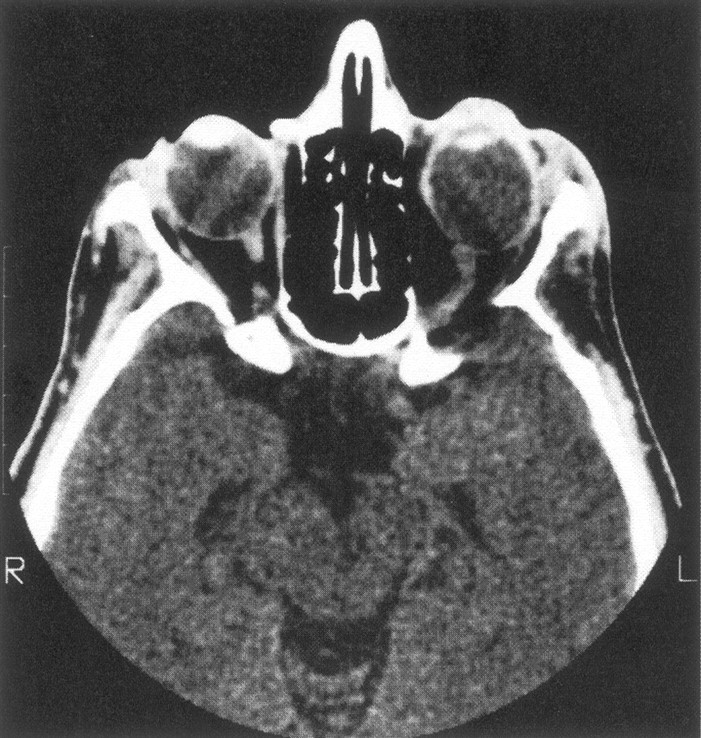
CT scan of our one patient with minimal expansion of the middle cranial fossa and abnormally large decalcification of the ipsilateral sphenoid wing, with no detectable adjacent tumor. The left globe is also enlarged.
Fig 6.
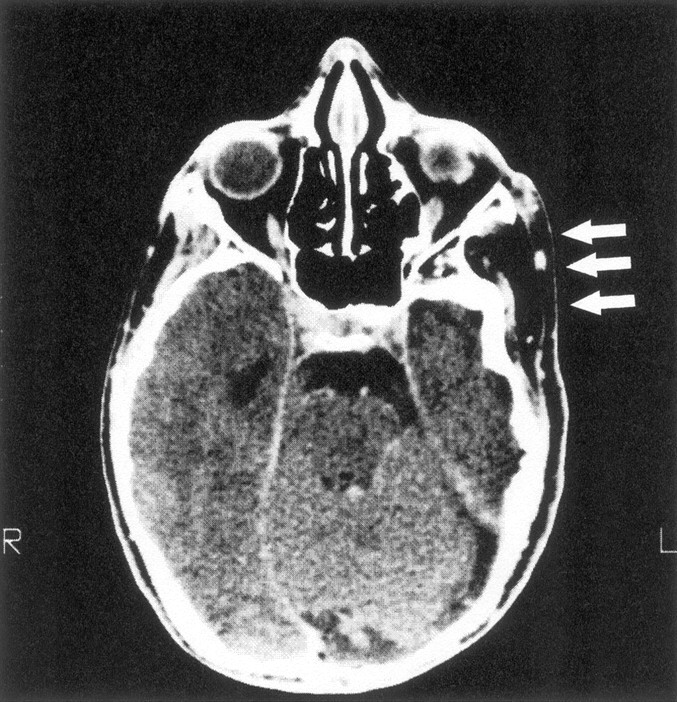
Axial CT scan of a patient with smaller middle cranial fossa and flattened temporal bone ipsilateral to extracranial tumor (arrows).
Group 1a Analysis
Twenty patients with repeat CT scans were analyzed in group 1a. Four patients who had expansion of the middle cranial fossa with plexiform neurofibromas (PNFs) contiguous to the remodeled sphenoid bone showed progression over time. Progressive sphenoid bone changes in these patients might be attributable either to abnormal sphenoid development or to tumor-associated decalcification or both (Fig 7). Imaging changes were apparent on CT scans obtained between 2 and 12 years apart. The short interval in some instances implied that bone alteration might be rapid on occasion.
Fig 7.
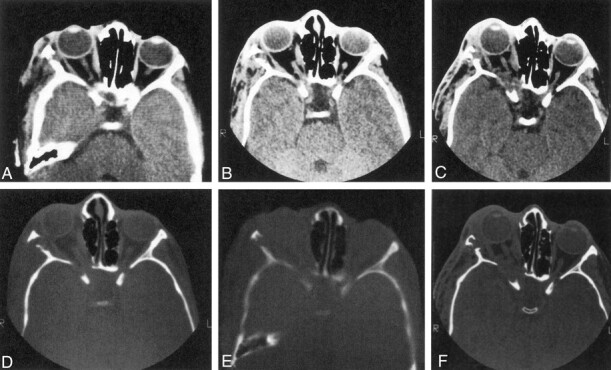
Series of axial CT scans (soft-tissue and bone windows) obtained during three scanning sessions over several years show progression of sphenoid bone abnormalities over time. Note that the soft-tissue mass increases over time (original, A and D; 5 years later, B and E; and 10 years later, C and F). and the ipsilateral middle cranial fossa becomes progressively more enlarged.
Discussion
This is the largest recently reported radiologic series of patients with craniofacial anomalies due to NF1. Many of these patients underwent surgery of soft-tissue tumors and/or the globe; therefore, we cannot comment on the natural history of radiologic soft-tissue changes or the potential effect on bone of tumor that was resected. No patient received radiation or underwent surgery involving bone reconstruction or removal that was recorded in the charts or was apparent on the radiologic studies.
The most striking skull change was anteroposterior enlargement of the middle cranial fossa with sphenoid bone changes typical of classic sphenoid dysplasia, always with an anterior temporal arachnoid cyst and often with shortening of the orbit. All these patients had historical, clinical, or radiologic evidence of craniofacial PNF present in the skin and subcutaneous tissues of the face and orbit near the time of birth, with tumor apparent clinically and radiologically at the time of the initial examination at our hospital. The patient presented by Macfarlane et al (7) and all 13 of our patients with pathologic middle cranial fossa expansion had PNF in the superficial temporal fossa, often with radiologically abnormal temporal squamosa sutures underlying the tumor. Tumor present in utero or shortly after birth in the superficial temporal fossa might have caused premature closure of nearby sutures that in turn precipitated an abnormal development of the sphenoid bone as the skull expanded during childhood. Tumor in the pterygoid fossa might have contributed to this distorted development by elevating the greater sphenoid wing and the middle cranial fossa.
Anteroposterior middle cranial fossa enlargement and anterior displacement of the greater sphenoid wing are central to the concept of sphenoid dysplasia. However, craniofacial deformities in cases of NF1 are known to be similar to deformities that occur in other circumstances, such as plagiocephaly, Apert syndrome (9), and mucopolysaccharidoses, with which early cranial suture closure occurs and the middle cranial fossa expands. Each of these entities may include anterior displacement of the middle cranial fossa during skull development, causing orbital contents to shift forward and downward (7). These changes illustrate the Virchow law, which states that growing forces may be redistributed parallel to a closed suture (2). If the sphenoid bone is unable to grow adequately along its lateral margin in cases of NF1 or other circumstances, this will lead to secondary distortion of development along the lesser wing and the greater wing anterior to the temporal lobe.
Abnormal development of the skull in association with NF1 may lead to an irregularity of CSF flow that in turn plays a role in the growth pattern of the skull (10). Altered CSF pulsations are a possible causal agent for similar skull changes that occur in association with chronic juvenile subdural hygromas and large temporal arachnoid cysts (11). Many of the processes causing pathologic middle cranial fossa expansion are associated with anterior temporal arachnoid cysts, including the sphenoid bone changes associated with NF1 in our series of patients. This observation raises the possibility that the pathophysiology of this disturbance of skull development may include an abnormality of CSF flow that is facilitated by early suture closure and adjuvant factors such as abnormal meninges or tumor in the pterygoid fossa. Redistribution of growth forces and abnormal CSF pulsations might explain previous reports of skull defects in patients with NF1 without contiguous tumor.
Sphenoid bone changes are unequivocally progressive in some patients with NF1. Miller (12) and Macfarlane et al (7) described individual patients with progressive sphenoid dysplasia and NF1, and the present series included four additional patients with progressive decalcification of the sphenoid bone. Two different processes may result in progressive sphenoid bone changes over time. Anomalies related to early suture closure may progress during infancy and early childhood because of distorted skull growth. This may well be the case in the patient studied by Macfarlane et al (7) near birth and again years later. Progressive bone changes in older persons may be due to the presence of tumor in the orbital apex contiguous to decalcified or remodeled sphenoid bone, as seemed to be the case with the more subtle bone changes documented for the patients reported herein. Orbital and facial neurofibromas occurring in adult life that are not associated with NF1 do not cause sphenoid dysplasia
This review of sphenoid bone and middle cranial fossa abnormalities associated with NF1 does not support the concept of simple dysplasia of the sphenoid bone. Abnormalities of the skull, orbit, and sphenoid bone may be the result of several processes, occurring separately and together, and PNF may play a more important role in these processes than was classically accepted. PNF present early in life in the superficial temporal fossa and deep orbit may well be essential to the development of craniofacial anomalies, including classic sphenoid dysplasia. Tumor in these locations is sometimes difficult to detect clinically or by using traditional radiography, and its importance may have been underestimated in the past.
A dysgenesis hypothesis also fails to completely explain why sphenoid bone alone is so susceptible to distorted development, why the problem is so often unilateral, why it occurs ipsilateral to extracranial tumor, and why it is progressive over time. Reports have been presented of two patients who seem to have had primary dysplasia of the sphenoid bone occur early during gestation (14), and the question arises regarding why sphenoid dysplasia in a case of NF1 does not display the same spectrum of associated midline anomalies as does this other sphenoid dysplasia. Answers to this and other questions probably lie in observations that brain and meninges are abnormal in cases of NF1, that PNF are tumors that frequently interacts with nearby bone, and that PNF appears in utero or during early childhood in some patients and, therefore, are present to interact with bone while the skull is still developing. Bone abnormalities of the skull and orbit in association with NF1 almost certainly involve an interaction between a number of genetic and developmental factors. The term secondary sphenoid dysplasia might be considered for the sum of the sphenoid bone abnormalities in cases of NF1 described in this report and elsewhere, thereby acknowledging a traditional concept while admitting that current information implies a more complicated pathogenesis.
References
- 1.Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 2000;151:33–40 [DOI] [PubMed] [Google Scholar]
- 2.Newton TH, Potts DG. Radiology of the Skull and Brain. St Louis, Mo: C.V. Mosby;1971. :235–608
- 3.Hunt JC, Pugh D. Skeletal lesions in neurofibromatosis. Radiology 1961;76:1–19 [DOI] [PubMed] [Google Scholar]
- 4.Tada M, Sawamura Y, Ishii N, Chin S, Abe H. Massive plexiform neurofibroma in the orbit in a child with von Recklinghausen’s disease. Childs Nerv Syst 1998;14:210–212 [DOI] [PubMed] [Google Scholar]
- 5.Zimmerman RA, Bilaniuk LT, Metzger RA, Grossman RI, Schut L, Bruce DA. Computed tomography of orbitalfacial neurofibromatosis. Radiology 1983;146:113–116 [DOI] [PubMed] [Google Scholar]
- 6.Grenier N, Guibert-Tranier F, Nicolau A, Caille JM. Contribution of computerized tomography to the study of spheno-orbital dysplasia in neurofibromatosis. J Neuroradiol 1984;11:201–211 [PubMed] [Google Scholar]
- 7.Macfarlane R, Levin AV, Weksberg R, Blaser S, Rutka JT. Absence of the greater sphenoid wing in neurofibromatosis type 1: congenital or acquired: case report. Neurosurgery 1995;37:129–133 [DOI] [PubMed] [Google Scholar]
- 8.National Institutes of Health Concensus Development Conference. Neurofibromatosis: conference statement. Arch Neurol 1988;45:575–578 [PubMed] [Google Scholar]
- 9.Faure C, Bonamy P, Rambert-Misset C. Craniostenosis from premature unilateral fusion of the coronal suture. Ann Radiol 1967;10:32–42 [PubMed] [Google Scholar]
- 10.Havlik RJ, Boaz J. Cranio-orbital-temporal neurofibromatosis: are we treating the whole problem? J Craniofac Surg 1998;9:529–535 [DOI] [PubMed] [Google Scholar]
- 11.Mafee MF. Imaging in Ophthalmology, Part 2. Philadelphia, Pa: W. B. Saunders Company;1987. :781–802
- 12.Miller NR. The phacomatoses. In: Miller NR, ed. Clinical Neuro-Ophthalmology. Philadelphia, Pa: W. B. Saunders Company;1999. :1747–1800
- 13.Shields JA, Shields CL, Lieb WE, Eagle RC. Multiple orbital neurofibromas unassociated with von Recklinghausen’s disease. Arch Ophthalmol 1990;108:80–83 [DOI] [PubMed] [Google Scholar]
- 14.Jacquemin C, Mullaney PB, Bosley TM. Abnormal development of the lesser wing of the sphenoid with clinical microphthalmos and microcephaly. Neuroradiology 2001;43:178–182 [DOI] [PubMed] [Google Scholar]